
Abstract
Despite the improvement of therapeutic options, melanoma patients with advanced metastatic disease are still in high need of durable treatments. Analysis of clinical data from patients receiving targeted and/or immunotherapy, along with genetic and functional studies in preclinical melanoma models, demonstrates the key role of the microphthalmia-associated transcription factor (MITF) throughout disease progression, and provides a solid rationale for its therapeutic inhibition. However, direct targeting of MITF or other basic helix-loop-helix leucine zipper (bHLH-LZ) transcription factors is unprecedented. Here, we report on the discovery of ligands for the DNA binding domain of MITF, using fragment-based screening (FBS) by nuclear magnetic resonance (NMR). Initial fragments, binding the kink pocket of MITF very weakly, are optimized to sub-micromolar affinities by structure-based design enabled by X-ray crystallography and biophysics. Furthermore, NMR experiments and molecular dynamics simulations reveal a dynamic conformational exchange between helices in the asymmetric homodimer, a phenomenon that is perturbed by ligand binding. This work advances our knowledge on direct targeting of bHLH-LZ DNA binding domains and sets the basis to further explore pharmacological inhibition of MITF.
Similar content being viewed by others
Introduction
Melanoma is the most aggressive form of skin cancer, dominated by activating mutations in critical nodes of the mitogen-activated protein kinase (MAPK) pathway1. Approximately 50% of primary melanomas harbor a hyperactivated form of the BRAF oncogene, used as a predictive biomarker of response to combinations of BRAF and MEK kinase inhibitors. Cancers bearing activating mutations in NRAS or carrying other genetic abnormalities are commonly treated with immunotherapy, i.e., inhibitors of the programmed death 1 (PD-1) and cytotoxic T cell antigen 4 (CTLA-4) checkpoints, which have become the standard of care (SoC) for patients with wild-type (WT) BRAF melanomas2. However, despite significant progress, the limited durability or lack of response in a subset of patients underscores the need for additional treatments. They would ideally be orthogonal to existing therapies and active across genetic subtypes, in both primary and metastatic melanomas3.
Microphthalmia-associated transcription factor (MITF), master regulator of melanogenesis, was described more than twenty years ago as a lineage-survival oncogene4, with a critical role in melanoma initiation and progression through regulation of proliferation and apoptosis5,6. While somatic mutations of the MITF gene are rare in cancer7, MITF is transcriptionally overexpressed in skin and uveal melanomas and is found genetically amplified in 10 to 20% of patients with advanced disease8.
As a member of the basic helix-loop-helix and leucine zipper (bHLH-LZ) superfamily of transcription factors (TFs)9, MITF has been largely regarded as undruggable. Except for the DNA-binding domain (DBD), MITF harbors intrinsically disordered regions. X-ray structures of the bHLH-LZ domain, crystallized alone or bound to DNA consensus sequences, are reported in the literature9,10,11. Efforts to directly target MITF have been limited, and the reported MITF inhibitors mostly lack structural and/or biophysical evidence of binding. ML329 can induce downregulation of a subset of MITF-dependent target genes and can negatively impact survival of melanoma cell lines, but the mechanism of action remains unknown12,13. Other compounds, TT-01210,14 and 8-Methoxybutin15, show MITF-dependent preclinical activity that was attributed to disruption of dimerization or to binding of intact homodimers, respectively, based on in silico predictions.
MITF contains a three-amino acid insertion, at position E260-Q261-Q262, inducing the formation of a kink in either LZ helix. The kink represents an important structural element that enables homodimerization, while preventing MITF from heterodimerizing with other related bHLH-LZ TFs of the MYC superfamily (such as MYC, MAX or USF). Regardless, MITF can bind the M-box motif as well as the E-box (CACGTG), which is recognized by MYC/MAX with high affinity. Formation of the kink induces the presence of a small pocket having an approximate volume of 300 Å2 9, which will be referred to as the kink pocket.
In the present work, we describe the discovery of low-molecular-weight (LMW) molecules that selectively bind to the MITF kink pocket, using fragment-based screening (FBS) by nuclear magnetic resonance (NMR) spectroscopy. Fragment screening is a well-established approach to identify chemical starting points and novel binding sites16. Due to its high sensitivity for weak interactions, 19F NMR is the method of choice for screening fluorine-containing fragment libraries17,18. This method was employed to probe the bHLH-LZ domain of MITF for ligandability, leading to the discovery of fragments that could be optimized to sub-micromolar binding affinities through similarity search, fragment merging, and structure-based design. Selected ligands are further characterized by orthogonal biophysical methods that, along with extensive use of X-ray crystallography, provide evidence of unprecedented binding of small molecules to MITF. Genetic studies conducted in MITF-dependent melanoma cell lines suggest a functional role for the kink pocket, warranting further investigation. Taken together, this work indicates a path forward towards the development of therapeutic inhibitors of MITF in the clinic.
Results
Malignant melanoma retains MITF expression following treatment with SoC in the clinic
A clear challenge limiting the efficacy of SoC therapies is represented by inter- and intratumor heterogeneity and cancer cell plasticity, with significant impact on durability of responses and acquisition of resistance19,20. In this context, use of preclinical models has led to discordant conclusions about the link between MITF levels and response to BRAF and MEK kinase inhibitors, pointing to MITF expression as either a positive or negative predictor21,22. Robust preclinical modeling of responses to immunotherapy agents is even more challenging. We therefore sought to interrogate gene expression data from patients enrolled in the COMBI-i trial, a randomized phase-III clinical study that evaluated the impact of Spartalizumab® in combination with Tafinlar® and Mekinist® in advanced BRAF(V600E) mutant melanomas23,24.
A retrospective biomarker analysis was conducted to assess the distribution of MITF at baseline, in relation to the patients’ response to targeted therapy, alone (Fig. 1a) or combined with immunotherapy (Fig. 1b). RNA-seq data from nearly 400 tumor samples clearly indicated that MITF expression can span across a very broad range of levels, within each response group. In fact, baseline MITF has no clear predictive value, and the median expression is significantly higher than the normal reference, not only in responders but also in patients that are refractory to either line of treatment. This population, together with patients relapsing on first- or second line of therapy, represents a persistent unmet need in the current landscape of melanoma treatments.

a, b Distribution of MITF expression at baseline in patients of the COMBI-i trial treated with Tafinlar® (anti-BRAF inhibitor Dabrafenib) and Mekinist® (anti-MEK inhibitor Trametinib), used as dual agent (a) or as triple combination with Spartalizumab® (anti-PD-1 inhibitor PDR001)23,24 (b). Patients are grouped based on clinical responses (according to the RECIST criteria). MITF expression is referenced to levels detected in the GTEx cohort of normal skin tissues (dotted line and gray plot). Sample sizes: a, CR = 33, PR = 92, SD = 40, PD = 18; b, CR = 38, PR = 99, SD = 25, PD = 17; GTEx = 661. Abbreviations: CR, complete response (dark blue); PR, partial response (light blue); SD, stable disease (light red); PD, progressive disease (dark red). c Heatmap representation of MITF and AXL transcriptional programs, based on published signatures25, in matched pair samples from 9 patients of the COMBI-i trial, treated with the triple combination of BRAF, MEK, and PD1 inhibitors. d MITF expression distribution at single-cell level in melanoma tumors collected either before (left) or after (right) treatment with an anti-PD1 inhibitor26. ICI, immune-checkpoint inhibitor.
The same clinical study offered the opportunity to interrogate changes of MITF and of the MITF-dependent transcriptional program induced by exposure to the Tafinlar®/Mekinist®/Spartalizumab® triple combination. Using previously published gene expression signatures25 and consistent with the anticipated heterogeneity across patients, we observed that treatment-induced variations can occur in either direction, with 5 of the 9 matched pair samples having increased MITF target genes, following treatment (Fig. 1c). Consistently, the de novo analysis of an independent dataset, i.e. single-cell RNA-sequencing data published by Jerby-Arnon et al.26 confirmed the co-existence of high and low MITF expressing clones, before and after treatment with an anti-PD1 inhibitor (Fig. 1d).
Collectively, the clinical data indicate that at least a subset of cancer cells express MITF and MITF-activation signatures, regardless of treatment and response efficacy. This suggests that cancer cells may retain growth dependence on the MITF transcriptional program, even if the overall MITF levels decrease.
MITF drives cell survival in preclinical models of drug resistance
Consistent with the role of lineage-survival oncogene, genetic depletion of MITF profoundly affects the growth of melanoma cell lines, as shown by large high-throughput screening campaigns27,28. Cancer cell growth dependence on MITF correlates with the oncogene expression and is unrelated to the BRAF and NRAS mutation status (Supplementary Fig. 1a–c). To validate this concept further, a subset of melanoma models was engineered with doxycycline (dox)-inducible shRNA targeting MITF (shMITF) and the phenotype was assessed in cell lines expressing low-to-high MITF gene levels (Supplementary Fig. 1d). Cell autonomous dependence on the TF was confirmed also in a xenograft model, in which loss of MITF induced tumor regression (Supplementary Fig. 1e, f).
To define whether acquired resistance to MAPK inhibitors affects MITF-dependent cell survival, MITF was knocked-down (KD) in three melanoma models, in which resistance to therapeutically relevant doses of Dabrafenib and Trametinib (DT) was induced through prolonged exposure and cell adaptation to the drugs. MELHO cells, naïve (parental) or resistant to the DT combination, were compared (Fig. 2a, top and bottom, respectively). Notably, even if incomplete, MITF KD induced a profound effect on cell survival and on MITF-dependent target genes, in both sensitive and resistant cell lines (Fig. 2b). Similarly, another BRAF mutant cell model (UACC-257) and the NRAS mutant SKMEL-30 cell line retained a strong growth dependence on MITF expression, after acquisition of resistance to DT or to Trametinib (T) alone, respectively (Fig. 2c and Supplementary Fig. 2).

a Impact of MITF KD on cell survival of MELHO cells, sensitive (S) or resistant to DT (R). Cells were seeded at low density and treated with dox for 10-14 days, before fixing and staining by crystal violet, to enable image acquisition. The impact on cell survival was benchmarked with depletion of Polo-Like Kinase (PLK1). One representative experiment (of 3 biological replicates) is illustrated. A different set of models is shown in Supplementary Fig. 2. b Impact of dox-inducible MITF KD on MITF-M (melanocyte specific transcript) and on selected MITF target genes. MELHO cells (S, R) were treated with dox for 3 days, before being collected for qRT-PCR analysis. Data referring to impact of dox-inducible shMITF is reported in green, for comparison with shNT (non-targeting shRNA), used as control, in gray. c Cell growth dependence on MITF in additional melanoma models. MELHO, UACC-257, and SKMEL-30 cells (either parental or resistant to the indicated treatment) were seeded at 1000 cells/well. Dox was added to the medium to induce MITF depletion and viable cells were quantified using the resazurin assay, after 10 days. Data in b and c is reported as mean ± s.d. of n = 3 independent biological replicates. Source data are provided as a Source Data file. d GSEA from RNA-seq transcriptomic profiling of MELHO cells that are either treatment-naive (S) or resistant to DT combination (R). Differential gene expression analyses represented as heatmaps refer to the following comparisons: MELHO R versus S at baseline (right); MELHO R versus S following dox-inducible MITF KD by shRNA (left). Data in (b–d) is derived from at least 3 experimental replicates.
Next, we looked closer at perturbations of the whole transcriptome by performing RNA-seq in MELHO cells, which were either sensitive (S) or resistant to DT (R), before and after induction of MITF KD by shRNA (Fig. 2d). Interestingly, while acquisition of DT-resistance induces an overall decrease of the MITF transcriptional signature and increase of AXL and markers of the epithelial-mesenchymal transition (R v. S comparison), resistant cells retain expression of MITF and MITF target genes, sufficient to drive cell proliferation. Of note, MITF protein levels do not significantly change, possibly as a sign of cell growth adaptation (Supplementary Fig. 2). In fact, both treatment-naïve and DT-resistant MELHO cells respond to MITF KD by modulating the same gene sets, based on the gene set enrichment analysis (GSEA). They reflect inhibition of the KRAS-MAPK pathway, along with suppression of anti-apoptotic and pro-proliferative signatures associated with MYC and MITF. Impact on the MYC program can be either MITF-dependent (as MYC is transcriptionally activated by MITF) or more generally reflective of an adaptive response. These findings indicate that, while the MITF-dependent transcriptional fingerprint may be decreased in absolute terms, cells adapted to MAPK inhibitors remain equally dependent on MITF, and they are still responsive to MITF-targeted modulation.
FBS by NMR leads to identification of MITF ligands
In order to assess the ligandability of the DNA-binding domain (DBD) of MITF, a FBS campaign was carried out, using the Novartis proprietary library LEF4000 (Local Environment of Fluorine)29. The library contained about 4000 fragments in mixtures of 30 to 32 fragments and was used to screen a protein construct corresponding to the MITF bHLH-LZ DBD(180-296) by ligand-based 19F NMR18. The screening was performed in the absence of DNA, to enable also the identification of DNA-competitive fragments. Following hit triaging and confirmation as individual compounds, the screen resulted in eleven primary hits, nine of which could be validated by protein-observed NMR (initially 1D 1H NMR, later 2D 1H,13C HMQC NMR experiments) (Fig. 3). The hit rate of 0.2% is amongst the lowest that we have observed across multiple FBS campaigns, and it likely reflects the low ligandability of the bHLH-LZ of MITF. Notably, although the LEF4000 library included 1600 CF3-containing fragments, all nine validated hits have a CF motif. The CF-containing fragments have a high 19F chemical shift anisotropy and a large chemical shift difference between free and bound states. This leads to large exchange effects that make them particularly sensitive to weak binding events18.

a Apo structure of MITF(217-296) lacking the basic region (PDB ID 4ATH) highlighting the small pocket, referred to as the kink pocket. The “site points”, shown in white, show the more buried part of the pocket. The three-residue insertion is shown in red. The kinked and straight helices are shown in green and cyan, respectively. b 19F NMR spectra from screening MITF(180-296) with the LEF4000 library. Spectra show a fragment mixture in the absence (black) or presence (red) of MITF. Hit 1 (near -123.5 ppm) is marked by the blue arrow. The signal near -112.5 ppm comes from a different fragment hit. c Validation of 1 by 1D protein observed NMR using MITF(180-296). d Validation of 1 by 2D 1H,13C-HMQC using MITF(217-296).
From the nine validated hits, two chemical series stood out as being particularly attractive, and they are exemplified by LEF hits 1 and 2 (Fig. 4). Both fragments have weak binding affinity: using protein-observed NMR titrations (Supplementary Fig. 3), compound 1 showed a measurable KD of 1 mM, while compound 2 showed a KD larger than 1 mM. Close analogs, not necessarily containing fluorine atoms, were then selected from the internal Novartis compound archive to build structure-activity relationships (SAR). Titration by 2D 1H,13C protein-observed NMR experiments led to the identification of compounds 3 and 4 with an improved KD of 327 and 207 µM, respectively. Of note, the phenyl substituent is in the 3-position of the indole 3, while being an N-substituent in the initial fragment hit 1.

Two hits 1 and 2 from fragment-based screening, analogs 3 and 4 from SAR by archive, and reporter ligand 5 from chemical synthesis are shown. Dissociation constants (KD) were determined by protein-observed NMR titration.
Competition experiments showed that fragment binding was unaffected by the presence of DNA. In fact, the same fragments can bind both MITF(180-296) and MITF(180-296):DNA complex, thus suggesting that the compounds do not bind at the DNA:protein interface. We therefore produced a shorter version of MITF, comprising residues 217-296, that includes the LZ dimerization region without the bHLH DNA-binding motif. MITF(217-296) is able to bind the identified fragments like MITF(180-296) (Fig. 3) and was used to derive NMR resonance assignments and to generate multiple X-ray co-crystal structures.
Crystal structures reveal the binding mode of kink binders
We solved the crystal structures of compounds 3 and 4 in complex with the truncated MITF(217-296) protein, at a resolution of 1.09 Å and 1.94 Å, respectively (Supplementary Fig. 4, Supplementary Table 1). Both compounds bind to the kink pocket, thus providing evidence of ligandability for this particular region (Fig. 5). The indole of compound 3 and the phenyl ring of compound 4 sit in the central region of the kink pocket. The phenyl of compound 3 occupies one of the lateral regions at the entrance of the pocket. In addition, the indole NH of compound 3 forms a hydrogen bond with the side chain of E260(A) of the kinked helix A (colored in green in Fig. 5). The carboxylic acid of 3 makes a 3.3 Å polar contact with the side chain of Q261(B), the straight helix B colored in cyan in Fig. 5, and a long-range ionic contact (3.8 Å) with the flexible K265(B) side chain. Conversely, compound 4 is not involved in any polar contact with the protein, but forms van der Waals interactions with the Q261 and A264 amino acids of both helices.

a, b MITF(217-296) in complex with 3 (PDB ID: 9H7Q, ligand in yellow) or 4 (PDB ID: 9H5H, ligand in orange). The kinked helix A is shown in green, the straight helix B in cyan, and the third helix in purple. The pocket is viewed from the same side in both panels. c, d Ligand interactions diagram for MITF(217-296) in complex with fragments 3 and 4. In (a and b), the chain A, B and C labeled in the diagrams are reported in green, cyan and purple. The hydrogen bonds are shown as purple arrows.
Surprisingly, crystal structures obtained with the truncated MITF(217-296) construct showed the presence of an additional shorter helix (colored in purple in Fig. 5). This is a fragment of the MITF protein, comprising residues Q262 to H295, resulting very likely from partial proteolytic degradation under the crystallographic conditions used. In order to rule out an impact of the third helix on formation of the kink pocket in these crystal structures, we tested a synthetic peptide corresponding to the helix, for binding to MITF(217-296) by NMR (Supplementary Fig. 5). Lack of detectable interaction indicated that the presence of the additional peptide facilitates crystallization of MITF(217-296), by contributing to crystal contacts that are not relevant in solution.
Ligand optimization by fragment merging
Overlay of the two crystal structures of MITF(217–296) in complex with fragments 3 and 4 highlighted the intriguing observation that the 6-membered ring of 3 and the phenyl ring of 4 are coplanar and overlapping (Fig. 6). This is an ideal scenario for a fragment merging approach and prompted us to combine the features of the two hits into one single molecule 6.

a Crystal structure of MITF(217-296) with compound 6 (PDB ID: 9H7R, gray), overlaid to 3 (in yellow) and 4 (in orange) from previously solved structures (Fig. 5). Protein sidechains are omitted for clarity and the overlapping central phenyl ring is highlighted by a blue circle. b Chemical structure of compounds 3, 4, and 6 (overlapping phenyl rings are marked in blue).
In the initial phase of the project, compound affinities were determined by titration using protein-observed NMR. However, compound 6 was too potent for this format since its KD was much smaller than the protein concentration needed for the NMR titration. We therefore established a competition-based displacement assay, using the fluorinated analog 5 as reporter molecule. 5 had a KD of 104 µM, based on a protein-observed NMR titration. The established 19F NMR reporter assay17 could reliably measure compound affinities down to 2 µM (Supplementary Fig. 3).
By using the 19F NMR reporter assay, we determined a Ki of 2.6 µM for compound 6, corresponding to >100-fold improved binding affinity compared to compound 3. This indicated the success of the merging approach, with molecules 6 and 3 having similar ligand efficiency (i.e. 0.28 and 0.25, respectively). This was consistent with the crystal structure shown in Fig. 6, that indicates the almost perfect overlap between compound 6 and the parent fragments 3 and 4. Compound 6 engages in hydrophobic contacts with the central part of the kink pocket through the indole ring, while the phenyl and cyclohexyl groups interact with the two lateral regions that define the access to the pocket. The oxygen of the hydroxyl moiety is placed at 3.58 Å from the carboxylic oxygen of E260 of helix B.
Based on these observations, we designed compound 7 with a methyl substituent to better fit the pocket, compound 8 (with an additional chlorine substituent) aiming at extending the van der Waals contacts with the lateral region, and compound 9 including both features (Fig. 7a). Both modifications led to increased binding affinity and reached the limit of the 19F NMR reporter assay. A competitive NMR HSQC assay30 was therefore developed to enable further optimization and assess binding in the low to sub-µM range of affinities. It allowed us to define the rank order of compounds 6, 7 and 8, with 6 being the weakest and 8 the tightest MITF ligand of those three compounds (Supplementary Fig. 3).

a Summary table of affinities for optimized MITF ligands as measured by NMR, SPR and ITC. b-d ITC of MITF(180-296) with compound 8 performed at 25 °C, showing raw heat peaks (b), the corresponding Wiseman plot with the molar ratio of the ligand divided by the concentration of MITF dimer on the x-axis (c), and thermodynamic signature represented as mean of two experiments (n = 2), corresponding standard deviations (SD) and individual values (d). e Thermal stabilization of MITF(180-296) by compound 8 measured by nanoDSF. f, g SPR sensorgrams (f) and binding curve (g) of compound 8 to MITF(180-296). h Van’t Hoff analysis of compounds 7 and 8 binding to MITF(180-296), based on the SPR binding assay performed at different temperatures. i, j Contributions of enthalpy and entropy to the Gibbs free energy of binding (thermodynamic signatures) at 25 °C for compounds 7 (i) and 8 (j). Source data for panels d, i, and j are provided as a Source Data file.
For quantitative measurement of binding affinities, isothermal titration calorimetry (ITC) experiments were performed. For compound 8, we determined a KD of 0.47 ± 0.17 µM (n = 2) and an enthalpically-driven binding event with unfavorable entropic contributions (ΔH = −49.4 ± 6.5 kJ mol-1, -TΔS = 12.4 ± 8.7 kJ mol-1, ΔG = −36.1 ± 0.88 kJ mol-1) (Fig. 7b–d). Compound 9 had a KD of 0.39 ± 0.20 µM (n = 2) and a similar thermodynamic signature (ΔH = −42.1 ± 6.1 kJ mol-1, -TΔS = 7.6 ± 3.9 kJ mol-1, ΔG = −36.8 ± 1.4 kJ mol-1) (Supplementary Fig. 6g-i). Compounds 6 and 7 had KD values in the low-μM range (Supplementary Fig. 6a–c and d–f, respectively). Nano differential scanning fluorimetry (nanoDSF) experiments indicated a thermal stabilization of MITF by 3.4 °C after binding of 8 (Fig. 7e).
We also developed an SPR assay by immobilizing Avi-tagged and biotinylated MITF(180-296) on a streptavidin-coated chip, to calculate association and dissociation rate constants and binding affinities of kink binders (Fig. 7f,g). Based on this method, compounds 6, 7 and 8 have KD,eq values of 8.0 µM, 4.0 µM and 1.9 µM, respectively. These values are in line with the rank order of the competitive NMR assay, and qualitatively agree with the KD values from ITC, which is considered the gold standard method for quantification of protein-ligand interactions (Fig. 7a). The association and dissociation rate constants of compound 8 (i.e. kon = 1.54×105 L mol-1 s-1 and koff = 0.283 s-1 at 25 °C) translate into a kinetic affinity constant (KD,kin) of 1.8 µM, matching the equilibrium affinity constant. Van ‘t Hoff analysis of SPR measurements at different temperatures (Fig. 7h-j) revealed that compound binding is primarily driven by the enthalpic component, in line with ITC data31.
Notably, while compounds 6 and 7 bind to MITF with low-µM affinities, they induced no NMR chemical shift perturbations in the related bHLH-LZ domain of MYC/MAX, when tested at 50 µM (Supplementary Fig. 7). Since such an assay can detect as little as 10–20% complex formation, one can safely conclude that the KD of 6 and 7 binding to MYC/MAX is greater than 200 µM. This indicates selectivity of the MITF ligands of at least 100-fold over binding to the DBD of MYC/MAX.
To further confirm the identity of the most critical residues in the kink pocket that drive specific binding of the MITF ligands, we generated MITF(180–296) proteins carrying single amino acids replacements of Q261, R263 and A264. Recombinant protein variants were probed by ITC for their ability to bind compound 8. In support of the key role of the side chains of these residues, significant disruption of binding interactions was observed (Supplementary Fig. 8).
Symmetry and dynamics of the MITF homodimer in absence and presence of ligands
As described, MITF homodimers are asymmetrical in the crystal structures, because only one monomer is bent forming a kink in the LZ region. In solution, this asymmetry is averaged out, likely by rapid swapping of the kink between the two monomers, on a time scale that is fast compared to the difference in NMR chemical shifts (Fig. 8a). Therefore, only a single set of peaks is observed for apo MITF in NMR HSQC spectra (Fig. 8b). To characterize the dynamic exchange between kinked and straight helix, we performed molecular dynamics (MD) simulations using the MITF homodimer shown in Fig. 8a, either in the absence or presence of DNA. The MD simulations reproduce the fast (nanosecond scale) dynamic exchange of the two helices and are thus in line with the NMR observations in solution measured in the absence of DNA (Fig. 8c, d).

a Model of MITF(206-296) bound to the M-box DNA sequence and scheme indicating the helices’ exchange. One helix is displayed as a dashed red line, and the other as a solid blue line. Both experience the kinked and straight conformation. b The 1H,13C-HMQC NMR spectrum of MITF(217–296) in the absence of ligands and DNA shows a single set of peaks, indicating rapid conformational exchange of straight and bent helices. Selected resonance assignments are indicated. c MD simulations of apo MITF in the presence of M-box DNA, indicating rapid helical exchange. The distance between the Cα of S250 and L282 for each helix (a) is plotted in blue and red for the two helices.
To monitor the movement of the helices during the simulation, we calculated the distance between the Cα of S250 and L282 of each helix over the 1 μs MD trajectory. Because these two residues are stabilized by other interactions in the bHLH and in the LZ region, respectively, they are less prone to twisting and provide relatively stable points of reference against which to compare helix lengths. This distance is shorter for the kinked helix (ca. 46 Å) than for the straight one (ca. 48 Å), as determined empirically from direct observation of the helix dynamics in simulation. From the simulations, we could quantify the number of times the helices swap between the kinked and straight conformations; the number of swaps was found to be similar in the presence or absence of DNA (Supplementary Fig. 9a). Each simulation condition was run with five replicates to allow for robust statistical analysis (Supplementary Fig. 9).
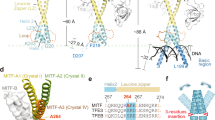
Binding of ligands, such as 7 and 8, leads to line broadening of NMR resonances in the kink region of MITF, indicating conformational exchange on an intermediate, rather than fast, time scale (Fig. 9a). Additional relevant information was derived from X-ray co-crystal structures with compounds 7 and 8. Compound 7 crystallized with the same orientation in the pocket, as compounds 3 and 6, i.e. with the carboxylic acid functionality opposite to the third helix (Supplementary Fig. 10, see also Fig. 5a, Fig. 6a). Interestingly, the crystal structure with compound 8, having only an additional chlorine atom compared to compound 6, shows the ligand flipped 180 degrees in the cavity (Fig. 9b). Structurally, such a flip is equivalent to the swap of one helix from kinked to straight (Fig. 8a). Although we cannot fully exclude an impact from the third helix on this finding, we notice that the protein environment surrounding the chloro-substituted phenyl is either polar or hydrophobic. It is reasonable to believe that the added chlorine of compound 8 prefers the more hydrophobic protein environment, corresponding to the crystallographic pose (in orange in Fig. 9b). In the crystallographic pose a hydrophobic contact (3.7 Å) between the chlorine and the Cδ of L267(B) is observed (Supplementary Fig. 11).

a 1H,13C-HMQC NMR spectra of apo MITF (black) and in the presence of 7 (blue) or 8 (red). Note that for 7 (but not for 8), the signals for L257δ1 and L257δ2 are weaker or absent. b Overlay of crystal structures of MITF in complex with compound 7 (PDB ID: 9H7T, ligand in blue, orientation 2) and compound 8 (PDB ID: 9H7S, ligand in orange, orientation 1). The kinked helix is shown in green and the straight helix in cyan. The additional helix, present in the crystal structures and not used in the MD simulation, is shown in purple. c, d Ligand interactions diagram for MITF(217-296) in complex with 7 and 8, respectively. In panel b the chain A, B and C labeled in the diagrams are reported in green, cyan and purple. The hydrogen bonds are shown as purple arrows. e Number of swaps per trace was calculated as in the Methods, including simulations with and without DNA as the presence or absence of DNA was not found to affect the number of calculated swaps (Supplementary Fig 9a). The “no ligand” condition has n = 5 replicates. Each compound and orientation combination plotted consists of n = 10 replicates, 5 each from simulations with and without DNA, respectively. All box plots shown use the R ggplot geom barplot() defaults, showing the median, two hinges calculated at the 25th and 75th percentiles, and two whiskers that extend from the hinges at most 1.5*(interquartile range of the relevant hinge). All data outside the whiskers are shown as individual dots. The presence of compound 7 significantly decreases the number of observed swaps, whereas presence of 8 only does so in the non-crystallographic pose of orientation 2. Significance was calculated using the two-sided Wilcoxon rank-sum test. Non-significant comparisons are not shown. Multiple comparisons were corrected for using the Benjamini-Hochberg method. ns: p > 0.05; *: p ≤ 0.05; **: p ≤ 0.01; ***: p ≤ 0.001; ****: p ≤ 0.0001. Exact, post-correction p values for comparisons with the “no ligand” condition are provided here: (compound 7, orientation 1): p = 0.003. (compound 7, orientation 2): p = 0.00098. (compound 8, orientation 1): p = 0.971 (compound 8, orientation 2): p = 0.031. Source data for panel e are provided as a Source Data file.
In order to investigate the impact of ligand binding on MITF dynamics, we carried out five MD simulations for both compounds 7 and 8 bound to MITF: DNA, starting from each of the two observed crystallographic orientations (Fig. 9b). The third helix was removed, being a crystallization artifact (as described above). Interestingly, MD shows that the presence of the ligand causes a significant reduction in the number of swap events over the course of a simulation, even accounting for variation between replicates (Fig. 9e). On average, the slowdown is more dramatic for the MITF:DNA:7 simulated complex than for the MITF:DNA:8 complex.
The result of the MD simulations is consistent with the NMR observation that addition of ligands can induce severe NMR line broadening of the L257 side chain methyl resonances, sometimes beyond detection (Fig. 9a). We attribute this line broadening to intermediate conformational exchange of MITF in complex with compound 7 (Fig. 9a). This provides experimental evidence that ligand binding of 7 to the kink pocket slows down the helical exchange, as observed in the MD simulations (Fig. 9c). Intriguingly, compound 8 binding has a much smaller line broadening effect than compound 7 (Fig. 9a), again in line with MD simulations reporting a different swapping behavior between compounds 7 and 8, despite their structural similarity. The nearly identical SPR dissociation rate constants of compounds 7 and 8 (koff = 0.07 s-1 for 7 and 0.03 s-1 for 8, both measured at 8 °C for greater precision) suggests that the NMR line broadening is likely due to the helix exchange in MITF, rather than differences in on-off kinetics of the ligand.
In addition, the MD simulations performed in the presence of the ligands show a larger amplitude of the swaps and a shorter median length of the helices, compared to the MD simulations with the protein alone. This is attributed to a more pronounced bend of the helix to accommodate the ligands.
Targeting the kink pocket may lead to functional MITF inhibition
As previously indicated, the kink pocket is a distinctive structural element that sets MITF apart from other bHLH-LZ family members, which otherwise exhibit high homology within the DBD-LZ domain32. This notion suggests that, besides defining structural constraints that prevent heterodimerization with MYC/MAX9, the kink may have additional functions. We therefore sought to investigate genetically whether residues proximal to the kink pocket have a role in the MITF activity.
Based on the X-ray structures generated in the presence or absence of LMW binders (Fig. 5), we observed that residues Q261, R263 and A264 engage in critical interactions with binders to the kink pocket of MITF. These three amino acid positions were selected to generate a new set of cell lines, derived from UACC-257 and 92.1 models pre-engineered with dox-inducible shMITF (Supplementary Fig. 1d). WT or mutant MITF were reintroduced to replace the endogenous protein and to assess their ability to rescue the MITF-dependent phenotype (Fig. 10). While there is some variability of expression (at least as high as WT MITF), none of the selected MITF mutants (Q261A, R263A and A264C) can fully phenocopy the WT protein. This suggests a functional role of the kink region, which remains to be further explored with more advanced and cellularly active compounds (Fig. 10, Supplementary Fig. 12). DNA binding mutant R217A and dimerization mutant N278D were used as benchmark9. To determine whether introduction of either of the selected kink mutations has a negative impact on protein folding and stability, MITF mutant proteins were generated and characterized by circular dichroism (CD) and nanoDSF. The data confirmed that MITF containing Q261A, R263A or A264C retains helical secondary structures and protein stability against thermal denaturation, to an extent comparable to MITF WT (Supplementary Fig. 13).

a Functional impact of individual MITF mutations in UACC-257 (top) and 92.1 cells (bottom), engineered to express dox-inducible shMITF and constitutive MITF, either WT or mutated in residues N278D, R217A, Q261A, R263A, A264C, respectively. Control cells express only dox-inducible shMITF. Cells were seeded at low density and treated with dox for 14 days (UACC-257) or 7 days (92.1), before fixation, staining by crystal violet and image acquisition. Numbers refer to % of cells detected in the dox-treated samples and quantified using the resazurin assay, compared to the respective control (- dox). The blots displayed are representative of at least 2 independent biological replicates. b Direct binding of (WT or mutated) MITF to an immobilized DNA oligonucleotide containing 6-time repeats of the MITF-specific M-box recognition element. Proteins were expressed using a HeLa-derived in vitro transcription-translation (IVTT) kit and DNA binding was quantified by a customized TransAM assay, adapted to use of an anti-MITF antibody for protein:DNA detection (see Methods). A mutated DNA oligo sequence was used to normalize data based on background signals. The data is reported as mean ± s.d. of n = 3 independent replicates. Mutations in the kink are indicated in blue and validated loss-of-function mutations (DNA binding) are in red. c Direct binding of cellular MITF (WT or A264C) to chromatin around transcription starting sites (TSS) of MITF KD differentially expressed genes (DEG) and MITF signature genes, in engineered UACC-257 cells as in panel (a) (see Methods). d Gene set enrichment analysis of differentially expressed pathways in UACC-257 cells, expressing dox-induced MITF KD (Control) in combination with constitutive MITF (WT or A264C). Each sample is compared to the respective no-DOX UACC-257 cells. e Transcription-factor motif enrichment analyses for MITF peaks within 3Kb of TSS. All peaks were ranked and binned by RNA expression fold change of closest genes in MITF KD. Source data for panels a and b are provided as a Source Data file.
Considering that the kink pocket is distal from the DNA: protein interface (Fig. 3), mutations in this region are unlikely to directly affect DNA binding. This was experimentally confirmed by TransAM to assess MITF binding to synthetic DNA oligonucleotides. All the mutant proteins tested, except the DNA binding mutant R216K, recognize a DNA sequence containing 6 tandem repeats of a native M-box consensus motif, with similar efficiency if compared to WT full-length MITF (Fig. 10b)33.
The MITF(A264C) variant is the one inducing the most profound effect on cancer cell growth, while retaining DNA binding. This observation holds true also for chromatin DNA, as demonstrated by profiling UACC-257 cells by Cut&Tag (Fig. 10c), in which the A264C mutation does not significantly affect MITF ability to interact with native DNA at transcription start sites (TSS). Next, the chromatin binding data were integrated with the gene expression profiling of UACC-257 cells, expressing WT or mutant MITF(A264C) (Fig. 10d).
Interestingly, the RNA-seq experiment showed that, while still able to bind the same promoters, the A264C mutation in the kink fails to fully recapitulate the MITF-dependent transcriptome of UACC-257 cells. In order to define the maximal amplitude of transcriptional changes, gene modulation induced by KD of endogenous MITF (control) and by re-expression of WT MITF were used as reference. At a closer look, DNA binding motifs specific to TFs E2F1 and ETS1 are enriched in gene promoters that are downregulated by MITF KD. The activation state of those genes was not rescued by re-expression of mutant MITF(A264C) (Fig. 10c-e). This may suggest functional cooperation between MITF and E2F1/ETS1, which have been associated with oncogenic functions in various tumors, including melanoma34,35. Although a direct interaction with MITF has not been reported, they can function as cofactors driving at least part of the MITF-dependent transcriptional changes observed in cancer cells.
Discussion
Clinical data is a fundamental source of information that enables retrospective studies on patient responses to treatments and helps define unmet needs. It also gives insights into changes of cancer cells during tumor progression. Transcriptomic analysis of biopsies from melanoma patients enrolled in the phase-III COMBI-i trial23,24 confirmed that the average MITF expression in tumor cells is significantly higher than in normal tissues, consistent with its role as oncogenic driver. This observation is valid irrespective of the clinical response of patients treated with targeted therapy alone or combined with a checkpoint inhibitor. Moreover, although inter- and intra-tumor heterogeneity of MITF distribution is acknowledged, single-cell transcriptomic data, from naïve and treated patients26, indicates the persistent expression of MITF in at least a subpopulation of cancer cells (Fig. 1), providing the rationale for therapeutic MITF inhibition.
Preclinical models with genetic depletion of MITF, before or after acquisition of resistance to Dabrafenib-Trametinib, confirm that melanoma cell growth remains highly dependent on MITF (Fig. 2, Supplementary Fig. 2). While genetic manipulation is a very valuable tool, the future use of advanced and cellularly active MITF inhibitors, and of preclinical models with elements of the tumor microenvironment, will be warranted to address additional critical questions. They include a better understanding of the phenotypes associated with different levels of MITF expression, as well as the clinical implications of cell plasticity and molecular reprogramming36 that can result from direct therapeutic inhibition of MITF.
The use of site-directed mutagenesis and functional experiments was introduced to test new hypotheses on structure-function relationships for the MITF protein in melanoma models. Studying the impact of mutations in the kink pocket suggests a role of the dimerization domain of MITF, beyond its structural properties. The kink has been previously described as a peculiar structural feature, caused by the insertion of three residues (E260-Q261-Q262) in the dimerization domain. It induces a structural asymmetry and formation of a kinked helix, paired with a straight helix9. This phenomenon prevents MITF monomers from heterodimerization with other members of the MYC/MAX superfamily, introducing a critical element of functional selectivity9. Consistently, we demonstrated that binders to the MITF kink pocket do not bind to MYC/MAX, which has a significantly broader tissue expression distribution (Supplementary Fig. 7).
The bHLH-LZ DNA-binding and dimerization domain of this class of TFs has been notoriously difficult to target pharmacologically. Here, we report on the discovery of MITF binders, supported by X-ray co-crystal structures. The binding of LMW molecules was demonstrated by orthogonal biophysical methods and integrated with MD simulation studies in order to further characterize their interaction with the kink pocket in solution.
Fragments were first identified through NMR-based screening of a truncated recombinant MITF protein to assess ligandability. Validated fragments, binding the kink pocket with weak affinities, were progressively improved through iterative cycles of X-ray crystallography, structure-based design, synthetic chemistry, and NMR spectroscopy (Figs. 5, 6). A fragment merging strategy was successfully applied and led to the synthesis of binders with more than 100-fold higher affinity, while retaining ligand efficiency (Fig. 6, Fig. 7a). The most potent molecules described here are compounds 8 and 9, having 0.5 and 0.4 µM affinity to MITF, based on ITC. The thermodynamic parameters recorded for both molecules point to an enthalpy-driven interaction with the kink pocket.
Next, we investigated more closely the impact of compound binding on the structural asymmetry introduced by the kink in MITF homodimers. NMR studies and MD simulations indicate that kinked and straight helices can rapidly interconvert in solution, resulting in averaged asymmetry. Intriguingly, compound binding slows down this rapid dynamic interconversion, as demonstrated by both NMR and MD measurements.
At present, it is unclear what the functional implications of this phenomenon may be. While additional future studies are needed, our data collectively suggest that interfering with the LZ region may impact directly or indirectly MITF functions in cancer cells. The development of more advanced and cellularly active binders will be essential to elucidate the role of the kink pocket and to provide insights into the mechanism of action, required for LMW compounds to inhibit MITF. In fact, their discovery may have several relevant implications, including the use as tools to conduct more extensive exploration of MITF biology, in both in vitro and in vivo preclinical models. However, the molecules described here also represent a blueprint for the development of pharmacological inhibitors of MITF and of other related TFs, sharing structural similarities and similarly considered undruggable, to date. Finally, they can pave the path towards the development of new selective treatments for the most aggressive forms of malignant melanomas by utilizing MITF as a drug target.
Methods
Ethics statement
All animal studies were conducted in accordance with ethics and procedures covered by permit BS-2921 issued by the Kantonales Veterinäramt Basel-Stadt and in strict adherence to guidelines of the Eidgenössisches Tierschutzgesetz and the Eidgenössische Tierschutzverordnung, Switzerland. All animal studies were approved by the internal ethics committee. All animals had access to food and water ad libitum and were identified with transponders. They were housed in a specific-pathogen-free facility in IVC racks under a 12 h–12 h light–dark cycle.
General Information
All reagents and solvents were purchased from commercial suppliers and used without further purification.
Chemistry
The synthesis of compounds 1, 3 and 5 to 9 is described in the Supplementary Information. Compound 2 was purchased from Rieke Metals (AT0278001). Compound 4 was purchased from Asinex (BAS0288013).
Protein expression and purification
MITF-m2 DBD-LZ (180-296) was cloned in pET24 vector with N-terminal His-MBP fusion tag followed by a TEV cleavage site. MITF expression was performed in E. coli BL21(DE3) strain with auto-induction37. Cell pellets were lysed in Tris-HCl buffer pH8.0, 500 mM NaCl and 1 mM TCEP by sonication. Supernatant of cell lysate was loaded on HisTrap Ni column (Cytiva), as a IMAC step to capture His-MBP-tagged MITF. After His-MBP tag cleaved by TEV protease in Ni elution pool, MITF containing sample was diluted to lower salt concentration at 200 mM NaCl and loaded onto SP-HP column (Cytiva). MITF was captured by Heparin column and eluted during the increasing of salt gradient approaching 500 mM. MITF containing solution then was concentrated for size exclusion chromatography (SEC) Surperdex 75 2660 column (Cytiva) equilibrated with 20 mM Tris-HCl pH7.5, 150 mM NaCl and 1 mM TCEP. MITF final sample from SEC was concentrated to the desired concentration for crystallography studies or biophysical NMR and ITC assays accordingly. MITF-m2 (217–296) was expressed and purified using the same method as that of MITF-m2 DBD-LZ (180-296). For NMR experiments, the protein was re-buffered in 25 mM K2HPO4 pH = 6.5, 300 mM NaCl, 0.5 mM TECP. For biotinylated MITF-m2 DBD-LZ (180–296) used in SPR, an avi-tag was engineered at the C-terminus of MITF-m2 DBD-LZ (180–296). The cell lysis method and protein purification process are the same as that of MITF-m2 DBD-LZ (180–296) except adding an in-vitro biotinylation step with BirA38 before the final SEC.
Expression of Isotopically-Labeled Proteins: Medium for 15N-/13C-labeling is 1x M9 salts, 2 mM MgSO4, 1 mL vitamin solution (Sigma R7256), 1x trace elements (made in-house), 1 g 15NH4Cl, 3 g 13C- D-glucose, 250 mg 15N-/13C-Isogrow (Sigma 6068390), antibiotic and sterile water q.s. to 1 L. Medium for 15N-/13C-/2H-4labelling is 1x M9 salts, 2 mM MgSO4, 1 mL vitamin solution, 1x trace elements, 1 g 15NH4Cl, 3 g 13C-/2H- D-glucose, 250 mg 15N-/13C/2H-Isogrow (Sigma 6068297), antibiotic and sterile D2O q.s. to 1 L. In either case, chemically-competent E.coli BL21 (DE3) cells were transformed, plated, and grown overnight. A 4 mL LB + antibiotic seed culture was inoculated with a single colony and incubated at 37 °C for ~6 hours, and for each liter of expression culture, 0.5 mL of the seed culture was used to inoculate a 50 mL starter culture of the respective minimal medium for overnight growth. The following morning, the starter culture was used to inoculate 950 mL of the respective medium, which was incubated at 37 °C until the A600 nm reached ~0.8; at this point protein expression was induced by adding IPTG to 0.25 mM and reducing the temperature to 18 °C. Expression continued overnight ( ~ 18 h), cells were harvested by centrifugation and stored at −80 °C.
NMR spectroscopy
Screening of the LEF4000 fragment library was performed by 19F NMR at 296 K on a 600 MHz Bruker Avance Neo NMR spectrometer equipped with a QCI cryogenic probe. MITF(180-296) was used at a concentration of 10 µM, CF3-containing fragments were screened at a concentration of 20 µM, and CF/CF2-containing fragments were screened at a concentration of 40 µM. The LEF4000 library consisted of 3969 fragments in 131 mixtures (1655 CF3 compounds in 52 mixtures and 2314 CF/CF2 compounds in 79 mixtures) with experimentally confirmed solubility in aqueous buffer solution > 200 µM. Primary hits from the mixtures were re-tested as singles, and those confirmed as singles were validated by 1H,13C-HMQC. 1H,13C-HMQCs of MITF(217-296) were recorded at 296 K on 600 or 800 MHz Bruker Avance III HD NMR spectrometers, using 20-40 µM MITF. The 19F NMR reporter assay was set up using 5 as fluorinated reporter at a concentration of 40 µM, MITF(217-296) at a concentration of 2 µM, and variable concentrations of test compounds.
For backbone and methyl group assignments, samples of (2H,13C,15N) or (13C,15N)-labeled MITF(217-296) were used, respectively, at protein concentrations of 800 µM in 3 mm Shigemi tubes. The buffer was 50 mM KH2PO4, pH 6.8, 250 mM KCl, 0.5 mM d-TCEP, 5% D2O. All spectra were recorded at 296 K on a 800 MHz Bruker Avance III HD NMR spectrometer equipped with four radio-frequency channels and a 5 mm 1H{13C,15N}-triple resonance cryogenic probe. NMR backbone resonance assignments were obtained from 3D 2H-decoupled HNCA (2048(1H) * 128(15N) * 128(13C) complex points / 91.7 * 20.8 * 9.9 ms acquisition time / 8 scans) and 2H-decoupled HNCACB (2048(1H) * 128(15N) * 296(13C) complex points / 91.7 * 20.8 * 9.8 ms acquisition time / 8 scans) triple-resonance experiments39 as well as a 3D 15N-edited NOESY40 (2048(1H) * 88(15N) * 256(1H) complex points / 63.9 * 23.6 * 14.5 ms acquisition time / 8 scans / 120 ms mixing time). Methyl groups were assigned with a 3D HCCH-TOCSY41 (2048(1H) * 128(13C) * 256(13C) complex points / 106.5 * 4.2 * 8.5 ms acquisition time / 8 scans / 22 ms mixing time). The NMR data was processed with the software Topspin 3.6 (Bruker, Switzerland) and analyzed with CCPNMR 2.542 and 3.043. We assigned all expected backbone amide (79/79, 100%) and 13Cα (80/80, 100%) resonances. For 13Cβ, 94% were assigned, except S250, Y253, L274, E275 and N278, due to missing resonances or signal overlap in HNCACB. Additionally, the well-resolved methyl groups in the 2D 1H,13C correlation spectrum were assigned (14/23, 61%). NMR resonance assignments are available at BMRB under accession code 53224. Assigned 1H,15N- and 1H-13C correlation spectra are displayed in Supplementary Figs. 14 and 15. The extent of resonance assignments is displayed in Supplementary Fig. 16.
X-ray structure determination
The MITF(217-296) construct has been crystallized at 12 mg/ml by using the sitting-drop vapor-diffusion method at 20 °C after mixing 400 nL protein complex solution with 200 nL reservoir solution (Mosquito robot dispenser, TTP LabTech). APO crystals have been obtained in 0.1 M MES pH 6.5, 5% PEG400 and 2 M ammonium sulfate conditions. Prior to shock-cooling the crystals for data collectio,n they were soaked in reservoir solution containing 10% glycerol.
To get structures with LMW ligands, the APO crystals have been soaked with 13 mM final concentration of compound in 2.5 M ammonium sulfate, 5% PEG400, 0.1 M MES pH 6.5 and 15% glycerol (8-fold excess ligand with 5% final DMSO) during 20 min to overnight incubation time. These crystals were directly flash-cooled in liquid nitrogen for data collection. X-ray diffraction data were collected at the Swiss Light Source using a PILATUS or EIGER detector by Dectris. Diffraction data were processed with autoPROC (v1.1.7 Global Phasing Ltd). Molecular replacement with Phaser (v2.8.3) yielded a partial structure. Model building was carried out with Coot (v 0.9.8.7). Coordinates of the built structures were refined with Phenix or Buster. Polder maps of the ligands were calculated using the Older module in the Phenix program suite44.
Surface plasmon resonance (SPR)
SPR experiments were performed on a Biacore 8 K instrument (Cytiva, Germany) following an adapted protocol45. The temperature of both the flow cell and the sample compartments was set to 25 °C. A Series S streptavidin functionalized sensor chip was conditioned with 1 M NaCl and 50 mM NaOH using three injections of 60 sec each at 10 µL min-1. Biotinylated MITF(180-296) diluted at 6 µg mL-1 in the immobilization buffer (25 mM Tris-HCl (Invitrogen, USA), 150 mM NaCl (Sigma-Aldrich, USA), 0.05% (v/v) Tween 20 (Sigma-Aldrich, USA), 3 mM EDTA (Sigma-Aldrich, USA), 0.5 mM TCEP (Sigma-Aldrich, USA), pH 7.5) was injected over the sensor surface with a contact time of 140 sec at 10 µL min-1 to obtain the desired capture level (approximately 3000 RU). After capturing, the system was primed with the analysis buffer (25 mM Tris-HCl (Invitrogen, USA), 150 mM NaCl (Sigma-Aldrich, USA), 0.05% (v/v) Tween 20 (Sigma-Aldrich, USA), 3 mM EDTA (Sigma-Aldrich, USA), 0.5 mM TCEP (Sigma-Aldrich, USA), 10 mM L-Arginine Hydrochloride (Merck Millipore, Germany), pH 7.5, supplemented with 3% (v/v) DMSO (Sigma-Aldrich, USA)) and the surface was equilibrated for an hour in this new buffer. For the binding analysis, a 2- or 3- fold concentration series of the ligands was injected over the surface at 60 µL min-1. The association and dissociation were measured for 60 and 150 s, respectively. Data were collected at 10 Hz, referenced against the control surface, and double-referenced using interleaved buffer injections. Sensorgrams were globally fitted to a 1:1 binding model to determine kon, koff and KD using the Biacore Insight Evaluation software (Cytiva, Germany).
Thermodynamic analysis using the van’t Hoff isotherm
Thermodynamic studies were performed at a range of temperatures of 8, 15, 20, 25, 30, 34 and 38 °C. For every temperature, the dissociation constant (KD) was obtained by fitting with a Langmuir binding isotherm (1:1), transformed into an association constant (KA), and plotted in a van’t Hoff plot. The linearity of the points in the complete temperature range indicated that the enthalpy is independent of the temperature and, therefore, the points were fitted to a linear van’t Hoff equation:
The entropy of binding, ΔS calculated from the intercept of the equation, while the enthalpy of binding, ΔH, is calculated from the slope. The bars in the van’t Hoff plot indicate the propagation of the standard error from the KD to the ln KA values plotted in the graph, which was calculated using the following equations:
TΔS and the Gibbs free energy, ΔG, were calculated using the free energy equation:
Nano differential scanning fluorimetry (nanoDSF)
Wild-type MITF(180–296) and the different variants were centrifuged at 16,000 × g and 4 °C for 10 min and the supernatants were collected. The proteins were then diluted to a final concentration of 0.3 mg mL-1 in their respective storage buffers (for the wild-type: 50 mM HEPES (Sigma-Aldrich, USA), 300 mM NaCl (Sigma-Aldrich, USA), 5% (v/v) glycerol (PanReac AppliChem, Germany/Spain), 1 mM TCEP (Sigma-Aldrich, USA), pH 8 / for the variants: 25 mM HEPES (Sigma-Aldrich, USA), 300 mM NaCl (Sigma-Aldrich, USA), 10% (v/v) glycerol (PanReac AppliChem, Germany/Spain), 1 mM TCEP (Sigma-Aldrich, USA), pH 7.5). Compound 8, dissolved and diluted in DMSO (Sigma-Aldrich, USA) to a final concentration of 50 μM, was mixed with the wild-type protein (for the apo conditions, DMSO was used instead of the compound), or with buffer only, using a Mosquito HTS device (TTP LabTech, UK), resulting in a final DMSO concentration of 3% (v/v). The mixtures were incubated under shaking at 450 rpm and RT for 30 min before being centrifuged at 16,000 × g and 4 °C for 10 min. The supernatants were collected and used to load the Prometheus standard capillaries (NanoTemper Technologies GmbH, Germany). Each condition was tested in duplicate. The measurements were performed on a Prometheus Panta instrument (NanoTemper Technologies GmbH, Germany). The samples were heated from 20 °C to 95 °C by 0.5 °C min-1 increments, excited at 280 nm and the intrinsic protein fluorescence at 330 nm and 350 nm was recorded. The data was analyzed using the PR.Panta Analysis (version 1.4.4) software (NanoTemper Technologies GmbH, Germany).
Isothermal titration calorimetry (ITC)
ITC measurements were carried out at 25 °C using a MicroCal PEAQ-ITC instrument (Malvern Panalytical), on a high feedback gain mode. Compounds 6, 7, 8 and 9 were dissolved in DMSO, then diluted into the ITC buffer (25 mM Tris-HCl, 150 mM NaCl, 10 mM L-arginine, 3 mM EDTA and 0.5 mM TCEP, pH 7.5) immediately prior to the experiment and loaded in the injection syringe at concentrations of 220 µM, 200 µM, 200 µM and 182 µM, respectively. Purified, dimeric, wild-type avi-tagged MITF(180-296) was dialyzed twice at 4 °C (for 3 h first and then overnight) against the ITC buffer, before being supplemented with 2% (v/v) DMSO immediately prior to the experiment in order to match the syringe solution to minimize the heats of dilution, and placed in the reaction cell at a concentration of 12 µM. The reference cell was filled with Milli-Q® water. Nineteen successive injections of 2 µL compound solution were then performed at 150-second intervals with a 60 s initial delay and at a stirring speed of 750 rpm. The resulting data was processed with the MicroCal PEAQ-ITC Analysis Software (version 1.41), using the model of one type of binding sites. The automatically adjusted baseline and integration regions were used to minimize the impact of researcher’s arbitrary decision. The experiment was performed in duplicate. The values reported are the average and ± values correspond to standard deviations.
MD simulations
MD simulations were carried out using Amber 202246,47 and PMEMD CUDA48,49,50 using prepared protein structures of human apo MITF, and MITF in complex with compounds 7 and 8. Simulations were performed both with and without the presence of M-box DNA.
Protein Preparation
Because there was no available X-ray structure of human MITF including both the DBD and bHLH domains, a full MITF structure was created by using an available mouse structure of the DBD in complex with DNA (4ATI PDB ID)9 which was then merged with the in-house crystal structure of the human MITF bHLH domain. The mouse MITF DBD is identical to the human MITF DBD. The protein was further prepared using the Structure Preparation workflow in Chemical Computing Group’s Molecular Operating Environment (MOE51, 2024.06 release). Solvent atoms and other co-factors were removed, and the N- and C-termini capped with ACE and NME residues, respectively. Hydrogens were added using the Protonate3D52 application within MOE, with protonation states of all residues set to a pH of 7.4. Resulting rotameric and protonation states were then manually reviewed for consistency of the hydrogen bonding network.
Ligand Preparation
Compounds 7 and 8 were assessed in our MD simulations. Each ligand was parameterized with the general Amber force field (GAFF253 and the OpenEye Quacpac Toolkit54 used to assign partial charges following the AM1-BCC55,56 method.
Full system preparation
Next, the system was assembled for MD simulation. The protein and (where appropriate) ligand and DNA polymers were parameterized using standard Amber forcefields for protein (ff19SB57, DNA (BSC158 and ligands (GAFF249). The DNA polymer was sourced from the same crystal structure of mouse Mitf that was used for the protein structure. The ligands were placed within the kink pocket using alignment to known crystal structures and then adjusting the compound in MOE’s Molecular Builder to produce the correct molecule (e.g. removing a methyl group and adding a chloro group to 7 in order to model 8), to fix the ionization state where necessary, and to rectify any clashes. The system had initial waters placed using 3D-RISM59 and Placevent60. The resulting full system of protein, ligand (where modeled), and DNA (where modeled) was placed in a solvation box of TIP3P61 water molecules with periodic boundary conditions, ensuring a solvent shell of at least 10 Å and that biomolecules did not interact with periodic copies. Finally, Na+ ions were added to neutralize the charge of the system.
Each system was first minimized using constant volume periodic boundaries with subsequent minimizations of 5000 steps of steepest descent followed by 5000 steps of conjugate gradient minimizing, first by restraining the solute using a weak force constant of 0.1 kcal/mol·Å2 and then without restraints. The system was heated to 100 K over 2500 steps (5 ps) using Langevin62 dynamics with constant volume and then warmed from 100 K to 303 K at constant pressure using a time step of 2 fs (total 500 ps). Heating steps were run with fixed solute molecules using a force constant of 5.0 kcal/mol·Å2. Each system was then run at 310 K for 500 ps, in which all Cα atoms, and ligand ions (when present) were restrained using a force constant of 2.0 kcal/mol·Å2. Each experimental condition was run with 5 replicates for a full microsecond of simulation time, using hydrogen mass repartitioning63 to allow for a 4 fs time step, resulting in 10,000 frames per condition (1 nanosecond = 10 frames). Bonds involving hydrogen were constrained using the SHAKE64 algorithm, and pressure maintained at 1 atm using the Monte Carlo (MC) barostat65, with a pressure relaxation time of 5 ps. In all simulations the Particle-Mesh Ewald (PME66,67 method was used to treat long-range electrostatic interactions, with a 9 Å cutoff specified for short-range, nonbonded interactions.
Analysis
MD trajectories were analyzed and visualized using cpptraj68 and R69, with full details and code available at https://doi.org/10.5281/zenodo.17611056. Briefly, cpptraj was used to generate the distance between carbon backbone atoms of Ser 250 and Leu 282. These particular residues were chosen on an empirical basis to reduce the measurement of twisting movements of the helix (as opposed to the flexing/bending movements that were of interest), as it was hypothesized that the leucine zipper in which L282 is placed would have a stabilizing effect. Then, these helix traces were smoothed using a rolling average with a 50 frame window, followed by a calculation of the number of helix crossings (“swaps”) that occurred over the course of each simulation, excluding the first 500 frames (50 nanoseconds of simulation time). The number of swaps was defined as the number of times that the helix traces crossed over each other across the simulation minus some equilibration time. Finally, the significance of the difference in number of swaps between conditions was calculated with the two-sided Wilcoxon rank-sum test70. This test was more appropriate than a t-test because it is non-parametric and suitable for small samples with unknown underlying distributions.
Cell lines and cell tools
Melanoma cancer cell lines were purchased as follows: MELHO from DMSZ, UACC-257 from NCI/DCTD and SKMEL28 and SKMEL30 from ATCC. 92.1 cells were obtained by the Leiden University medical center. All cell lines were maintained in RPMI (Amimed) supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate 10 mM HEPES and 1% Penicillin/Streptomycin.
Resistance to 100 nM Dabrafenib and 3 nM Trametinib was induced in BRAF mutant MELHO and UACC-257 cells, through prolonged exposure to both drugs. Similarly, drug resistance to 3 nM Trametinib in NRAS mutant SKMEL30 cells was obtained following exposure to Trametinib alone, for 4 to 6 weeks’ time of cell adaptation to increasing drug concentrations. Depending on the cell model, a total of 8 to 10 weeks were needed to expand cells that survived the selective pressure, followed by confirmation of drug-resistance by dose-response survival assays.
Cell models selected for conditional knock down (KD) of MITF were generated by transduction with inactivated lentivirus particles bearing pLKO-TET-ON plasmids for the expression of doxycycline (dox)-inducible shRNA targeting MITF (shMITF: 5′- cgggaaacttgattgatcttt-3′. Similarly, cells expressing a non-targeting control (shNT 5′- ccggagaagaagaaatccgtgtgaactcgagttcacacggatttcttcttctttttta-3′) and a pan-lethal control (shPLK1 5′- ccggggtatcagctctgtgataacactcgagtgttatcacagagctgatacctttttaatt-3′) were generated in parallel. Melanoma cancer cell lines pre-engineered with dox-inducible shMITF underwent a second round of virus-mediated transduction to constitutively express wild type (WT) or mutant MITF proteins. Thus, a codon-optimized cDNA sequence encoding for human MITF-M (NP_000239.1) was subcloned into a custom lentiviral vector and expressed under a EF1-alpha promoter.
Cell viability and colony formation assays
Viability assays were performed by seeding cells into 96-well plates in triplicate and read at defined time point following addition of resazurin71. Colony formation assay was performed by seeding cells at low density into 6-well plates and cultivated until colonies appeared. Colonies were fixed by paraformaldehyde and stained with crystal violet as described by Ferretti et al.72
Gene expression and protein analyses
qRT-PCR
RNA isolation was performed using QIAshredder (Qiagen) and RNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s recommendations. Quantitative RT-PCR was performed with QuantStudio 6 Flex (Applied Biosystems) using iTaq Universal Probes One-Step Kit (Bio-Rad) and the following Taqman probes: custom probe to detect endogenous MITF (forward primer 5′- TCTACCGTCTCTCACTGGATT-3′, reverse primer 5’-GGCTTGCTGTATGTGGTACT-3’ and probe /56-FAM/AA TCA CTA T/ZEN/C AGG TGC AGA CCC ACC /3IABkFQ/); BIRC7 (IDT, Hs.PT.58.23314818); IRF4 (IDT, Hs.PT.58.2073651); CDK2 (IDT, Hs.PT.58.302089). Gene expression levels were normalized to Actin or GAPDH (Applied Biosystems).
Western blotting
Cells were harvested and lysed in RIPA buffer supplemented with complete protease inhibitor cocktail (Roche cat. #04906837001) and phosphatase inhibitor cocktail (Roche #04693124001). Following protein quantification by BCA (Thermofisher #23225), protein samples were subjected to SDS-PAGE using 4-15% gels and transferred to PVDF membrane. Protein expression was probed by using the following antibodies: GAPDH (Cell Signaling Technology, CST, cat. #5174), HA (CST cat. #3724, clone C29F4), MITF (Sigma cat. #M6065, clone C5), VINCULIN (Sigma, cat #V9131). HRP-conjugated anti-rabbit or anti-mouse secondary antibodies from CST (cat. #7074 and #7076, respectively) were used for detection.
RNA sequencing
RNA samples were prepared using QIAshredder (Qiagen) and RNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s recommendations. RNA-Seq libraries were prepared from 250 ng of total RNA using the TruSeq Stranded Total RNA Library Prep Gold kit (Illumina, 20020599), according to the manufacturer’s instructions (Illumina TruSeq Stranded mRNA Sample Preparation Guide, 15031047 Rev. E, October 2013). Libraries were sequenced on an Illumina NovaSeq 6000 system.
Cut&Tag
The Cut&Tag (Cleavage Under Targets and Tagmentation) experiments were performed as previously described, using 500,000 cells as input73. The anti-HA (CST cat. #3724, clone C29F4) primary antibody was used at a 1:50 dilution and the secondary guinea-pig anti-rabbit IgG antibody (ABIN101961) was used at 1:100. Libraries were sequenced on the NovaSeq 6000 instrument (Illumina).
Bioinformatic analyses
SALMON aligner (PMID: 28263959) was used to map RNA-seq reads to transcripts in the hg38 reference genome and to quantify read counts for individual transcripts, followed by the estimation of differential expression analysis using edgeR (PMID: 19910308) and gene set enrichment analysis using GSEA (PMID: 16199517). Cut&Tag reads were aligned to human hg38 reference using Bowtie2 software (PMID: 22388286). Alignments were filtered for uniquely mapped reads, and duplicates were removed. Peaks of enrichment were called using MACS2 (PMID: 18798982). Sequence motifs enriched among TSS-proximal MITF peaks were identified using monaLisa (PMID: 35199152). All peaks were ranked and binned by RNA expression fold change of the closest genes in MITF KD.
TransAM assay
cDNA encoding LgBiT-MITF was cloned into pT7 vector by restriction enzyme and individual MITF mutants were generated using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technology). The respective proteins were obtained from these plasmids using 1-Step Human In Vitro Protein Expression Kit (IVT Kit, ThermoFisher) following the manufacturer’s instructions. Protein expression was quantified using Nano-Glo Luciferase Assay System (Promega). Briefly, 0.1 μl of IVT product was mixed with 500 nM HiBiT protein (Genscript) and the resulting luciferase-derived signal was used to quantify and normalize for the expression level of the individual LgBiT-MITF variants. The TransAM assay was performed following the manufacturer’s protocol (TransAM® Flexi NFκB p65, Active Motif), using equivalent amounts of LgBiT-MITF variants (WT and mutant). The sequence of the double-strand oligonucleotides used to coat the experimental plates (i.e. active Biot-6xMbox and negative control 6xMbox-mut) is reported in the Source Data file. An anti-MITF antibody (CST, cat. #97800) was used as the primary antibody for detection.
In vivo experiments
To establish cell line-derived xenograft models, 6 to 7-week-old female athymic nude (Crl:NU(NCr)-Foxn1nu) mice (Charles River Laboratories) were engrafted subcutaneously with 5 million of 92.1 uveal melanoma cells in HBSS (Sigma-Aldrich, H6648) and Matrigel (354234, Corning) ratio 1:1. Dox was dissolved in animal drinking water daily during the treatment. For the efficacy experiment, mice were randomized into groups (n = 6) for a mean tumor size of 250–300 mm3. Dox was dosed orally once daily at 25 mg/kg, and 10 ml/kg for shRNA activation for 7 days. On day 7, satellite animals from each group were sacrificed to analyze MITF KD and PD regulation (n = 3–5). Tumor responses are reported as tumor volumes. To assess PD modulation in 92.1 tumor-bearing mice, dox was administered daily by oral gavage, at 25 mg/kg. Animals were randomized (n = 3–5), and tumor samples were collected 4 h after the last treatment (day 7). Tumors were resected, weighed, frozen in liquid nitrogen and cryogenic dry pulverized with the CryoPrep system (model CP-02, Covaris). Sex of the animals is considered not relevant for the present studies.
Immunoblotting for MITF detection in tumor samples
Cells were lysed in RIPA lysis buffer (Milipore cat. #20-188) supplemented with complete protease inhibitor cocktail (Roche cat. #04906837001) and phosphatase inhibitor cocktail (Roche cat. #04693124001). Total proteins were subjected to SDS-PAGE using 4–15% Criterion TGX gels (BioRad cat. #567-1065) and transferred to nitrocellulose membranes (Amersham cat. #10600002), saturated with Tris Buffered Saline Tween-20 and 5% non-fat milk (BioRad #170-6404), for 1 h. Primary antibodies used for immunoblotting were as follows: GAPDH (Sigma cat. # WH0002597M1, used at 1:5000), MITF (Sigma cat. #M6065, clone C5, used at 1:1000), HA-tag (CST cat. #3724, used at 1:1000). Membranes were incubated overnight with the primary antibodies and 1 h with the HRP-coupled antibodies (rabbit IgG GE Healthcare cat. #NA934V; anti-mouse IgG CST cat. #7076). Following washings with Tris Buffered Saline containing 0.01% Tween-20, protein signal on the membranes was developed using SuperSignal West Femto Maximum Sensitivity Substrate (Pierce cat. #4096) and the fusion FX device (Witec) with the EvolutionCapt software.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper as a Source Data file. Diffraction data and crystallographic coordinates of unliganded MIFT and MITF complexed to compounds 3, 4, 6, 7, and 8 have been deposited at the protein data bank (www.rcsb.org) under accession codes 9H5F (unliganded), 9H7Q (3), 9H5H (4), 9H7R (6), 9H7T (7), and 9H7S (8). NMR resonance assignments have been deposited with the BMRB accession code 53224. All molecular simulations related information, such as initial coordinate and simulation input files, along with a coordinate file of the final output were deposited in a Zenodo repository under the following link: https://doi.org/10.5281/zenodo.17611056. The synthesis of compounds 1, 3 and 5 to 9, including 1H-, 13C- and 19F-NMR analytical data, is described in the Supplementary Information file in the Supplementary Methods. Full scans of all blots and data points of RT-qPCR and cell viability experiments are deposited in the Source Data file. The RNA sequencing and Cut&Tag data has been deposited in GEO under the accession number GSE283857. Source data are provided with this paper.
Code availability
All custom cpptraj and R code used to perform the analyses and generate results in this study is publicly available and has been deposited at the following link: https://doi.org/10.5281/zenodo.17611056, without any restriction for their access and use.
References
Hodis, E. et al. A landscape of driver mutations in melanoma. Cell 150, 251–263 (2012).
Luke, J. J. et al. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 14, 463–482 (2017).
Bai, X. et al. Cell-state dynamics and therapeutic resistance in melanoma from the perspective of MITF and IFNγ pathways. Nat. Rev. Clin. Oncol. 16, 549–562 (2019).
Goding, C. R. et al. MITF–the first 25 years. Genes Dev. 33, 983–1007 (2019).
Garraway, L. A. et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436, 117–122 (2005).
Garraway, L. A. et al. Lineage dependency and lineage-survival oncogenes in human cancer. Nat. Rev. Cancer 6, 593–602 (2006).
Bertolotto, C. et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 480, 94–98 (2011).
Ugurel, S. et al. Microphthalmia-associated transcription factor gene amplification in metastatic melanoma is a prognostic marker for patient survival, but not a predictive marker for chemosensitivity and chemotherapy response. Clin. Cancer Res. 13, 6344–6350 (2007).
Pogenberg, V. et al. Restricted leucine zipper dimerization and specificity of DNA recognition of the melanocyte master regulator MITF. Genes Dev. 26, 2647–2658 (2012).
Liu, Z. et al. A unique hyperdynamic dimer interface permits small molecule perturbation of the melanoma oncoprotein MITF for melanoma therapy. Cell Res 33, 55–70 (2023).
Möller, K. et al. MITF has a central role in regulating starvation-induced autophagy in melanoma. Sci. Rep. 9, 1055 (2019).
Faloon, P. W. et al. A small molecule inhibitor of the MITF molecular pathway. Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD). (Academia, 2010).
Nishikiori, N. et al. Significant and various effects of ML329-induced MITF suppression in the melanoma cell line. Cancers 16, 263–275 (2024).
Silva, C. et al. An exploitable Achilles heel of MITF? Cell Res. 33, 195–196 (2023).
Oh, E. et al. 8-Methoxybutin inhibits α-MSH induced melanogenesis and proliferation of skin melanoma by suppression of the transactivation activity of microphthalmia-associated transcription factor. Biomed. Pharmacother. 152, 113272 (2022).
Erlanson, D. A. et al. Twenty years on: the impact of fragments on drug discovery. Nat. Rev. Drug Discov. 15, 605–619 (2016).
Dalvit, C. et al. Fluorine-NMR experiments for high-throughput screening: theoretical aspects, practical considerations, and range of applicability. J. Am. Chem. Soc. 125, 7696–7703 (2003).
Dalvit, C. et al. Ligand-based fluorine NMR screening: principles and applications in drug discovery projects. J. Med. Chem. 62, 2218–2244 (2019).
Shi, H. et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 4, 80–93 (2014).
Sharma, P. et al. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017).
Boshuizen, J. et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat. Med. 24, 203–212 (2018).
Feng, T. et al. Distinct transcriptional programming drive response to MAPK inhibition in BRAFV600-mutant melanoma patient-derived xenografts. Mol. Cancer Ther. 18, 2421–2432 (2019).
Dummer, R. et al. Combined PD-1, BRAF and MEK inhibition in advanced BRAF-mutant melanoma: safety run-in and biomarker cohorts of COMBI-i. Nat. Med. 26, 1557–1563 (2020).
Tawbi, H. A. et al. Spartalizumab or placebo in combination with dabrafenib and trametinib in patients with BRAF V600-mutant melanoma: exploratory biomarker analyses from a randomized phase 3 trial (COMBI-i). J. Immunother. Cancer 10, e004226 (2022).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Jerby-Arnon, L. et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175, 984–997 (2018).
McDonald, E. R. III et al. Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell 170, 577–592 (2017).
Tsherniak, A. et al. Defining a cancer dependency map. Cell 170, 564–576 (2017).
Vulpetti, A. et al. Design and NMR-based screening of LEF, a library of chemical fragments with different local environment of fluorine. J. Am. Chem. Soc. 131, 12949–12959 (2009).
Zhang, X. et al. Ranking of high-affinity ligands by NMR spectroscopy. Angew. Chem. Int. Ed. 48, 6691–6694 (2009).
Edink, E. et al. The heat is on: thermodynamic analysis in fragment-based drug discovery. Drug Discov. Today Technol. 7, e189–e201 (2010).
Pogenberg, V. et al. Mechanism of conditional partner selectivity in MITF/TFE family transcription factors with a conserved coiled coil stammer motif. Nucleic Acids Res. 48, 934–948 (2020).
Bertolotto, C. et al. Different cis-acting elements are involved in the regulation of TRP1 and TRP2 promoter activities by cyclic AMP: pivotal role of M Boxes (GTCATGTGCT) and of microphthalmia. Mol. Cell. Biol. 18, 694–702 (1998).
Sizemore, G. M. et al. The ETS family of oncogenic transcription factors in solid tumours. Nat. Rev. Cancer 17, 337–351 (2017).
Rouaud, F. et al. E2F1 inhibition mediates cell death of metastatic melanoma. Cell Death Dis. 9, 527 (2018).
Hoek, K. S. & Goding, C. R. Cancer stem cells versus phenotype switching in melanoma. Pigment Cell Melanoma Res. 23, 746–759 (2010).
Studier, F. W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41, 207–234 (2005).
Li, Y. et al. Expression and purification of E. coli BirA biotin ligase for in vitro biotinylation. Protein Expr. Purif. 82, 162–167 (2012).
Yamazaki, T. et al. A suite of triple resonance NMR experiments for the backbone assignment of 15 N, 13C, 2H labeled proteins with high sensitivity. J. Am. Chem. Soc. 116, 11655–11666 (1994).
Fesik, S. W. & Zuiderweg, E. R. P. Heteronuclear three-dimensional NMR spectroscopy. A strategy for the simplification of homonuclear two-dimensional NMR spectra. J. Magn. Reson. 78, 588–593 (1988).
Bax, A. & Clore, G. M. Proton-proton correlation via isotropic mixing of carbon-13 magnetisation, A new three-dimensional approach for assigning proton NMR spectra of carbon-13-enriched proteins. J. Magn. Reson. 88, 425–431 (1990).
Vranken, W. F. et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 (2005).
Skinner, S. P. et al. CcpNmr AnalysisAssign: a flexible platform for integrated NMR analysis. J. Biomol. NMR 66, 111–124 (2016).
Liebschner, D. et al. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Cryst. D. 73, 148–157 (2017).
Malojčić, G. et al. LptE binds to and alters the physical state of LPS to catalyze its assembly at the cell surface. Proc. Natl. Acad. Sci. USA 111, 9467–9472 (2014).
Case, D. A. et al. Amber 2022. http://ambermd.org/doc12/Amber22.pdf (2022).
Case, D. A. et al. AmberTools. J. Chem. Inf. Model. 63, 6183–6191 (2023).
Le Grand, S. et al. SPFP: speed without compromise – A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Comm. 184, 374–380 (2013).
Salomon-Ferrer, R. et al. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 9, 3878–3888 (2013).
Götz, A. W. et al. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 8, 1542–1555 (2012).
Molecular Operating Environment (MOE), 2024.06.01 Chemical Computing Group ULC, 910-1010 Sherbrooke St. W., Montreal, QC H3A 2R7 (MOE, 2025).
Labute, P. Protonate3D: assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins 75, 187–205 (2009).
Wang, J. et al. Development and testing of a general Amber force field. J. Comput. Chem. 25, 1157–1174 (2004).
QUACPAC 2.2.5.1. OpenEye, Cadence Molecular Sciences, Santa Fe, NM, USA. https://www.eyesopen.com.
Jakalian, A. et al. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 21, 132–146 (2000).
Jakalian, A. et al. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 23, 1623–1641 (2002).
Tian, C. et al. ff19SB: amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 16, 528–552 (2020).
Ivani, I. et al. Parmbsc1: a refined force field for DNA simulations. Nat. Methods 13, 55–58 (2016).
Sindhikara, D. J. et al. Analysis of biomolecular solvation sites by 3D-RISM theory. J. Phys. Chem. B 117, 6718–6723 (2013).
Sindhikara, D. J. et al. Placevent: an algorithm for prediction of explicit solvent atom distribution – application to HIV-1 protease and F-ATP synthase. J. Comput. Chem. 33, 1536–1543 (2012).
Jorgensen, W. L. et al. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Pastor, R. W. et al. An analysis of the accuracy of Langevin and molecular dynamics algorithms. Mol. Phys. 65, 1409–1419 (1988).
Hopkins, C. W. et al. Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 11, 1864–1874 (2015).
Ryckaert, J.-P. et al. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
Åqvist, J. et al. Molecular dynamics simulations of water and biomolecules with a Monte Carlo constant pressure algorithm. Chem. Phys. Lett. 384, 288–294 (2004).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Darden, T. et al. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Roe, D. R. et al. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 (2013).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org. R version 4.4.2 (2024-10-31). (2021)
Wilcoxon, F. Individual comparisons by ranking methods. Biometrics Bull. 1, 80–83 (1945).
Chapeau, E. A. et al. Direct and selective pharmacological disruption of the YAP–TEAD interface by IAG933 inhibits Hippo-dependent and RAS–MAPK-altered cancers. Nat. Cancer 5, 1102–1120 (2024).
Ferretti, S. et al. Discovery of WRN inhibitor HRO761 with synthetic lethality in MSI cancers. Nature 629, 443–449 (2024).
Barbosa, I. A. M. et al. Cancer lineage-specific regulation of YAP responsive elements revealed through large-scale functional epigenomic screens. Nat. Commun. 14, 3907 (2023).
Acknowledgements
We thank Eva Altmann, Pascal Rigollier, Krystyna Patora-Komisarska and Andreas Lerchner for medicinal chemistry input and Rita Brauer, Marion Burglin and Fabien Tritsch for compound synthesis; Ayako Honda and Aragen Life Sciences Ltd. and Aurigene Pharmaceutical Services Ltd. for chemical synthesis support; Wassim Abdul Rahman, Patrizia Fontana, Catherine Zimmermann, Marco Meyerhofer and Dirk Erdmann for protein preparation and quality control; Mario Centeleghe for support with running biological experiments; Camilo Velez-Vega and Sepehr Dehghani-Ghahnaviyeh for discussion on MD simulations, and Andreas O. Frank for initial NMR experiments. We thank Sascha Gutmann and Martin Schröder for depositing the crystal structures in the protein data bank and Ulrike Naumann for her support in processing NGS samples. We are grateful to Luca Tordella for critical revision of the manuscript, and to Nicolas Soldermann and Tobias Schmelzle for their scientific guidance.
Author information
Authors and Affiliations
Contributions
K.Y. and J.F. expressed and purified the proteins. C.H., C.D., P.M., C.F., and W.J. designed and performed NMR experiments. E.W. and M.R. solved crystal structures. A.V., R.W., J.H., and N.S. designed compounds. S.P., J.B., S.K., and J.K. synthesized compounds. N.Pi., N.Pa., F.B., and G.M. designed and performed SPR, ITC and nanoDSF experiments. A.V. designed the LEF4000 library. A.V., K.P., and B.R. devised and performed MD simulations. F.J. conducted the statistical analysis of bulk RNA-seq, scRNA-seq and Cut&Tag (from patient samples or preclinical experiments). A.D., F.M.M., M.W., D.M., M.A., F.S., M.K., and A.C.C. generated biology data. D.C., J.H., W.J., M.R., A.V., and G.M. devised the project strategy. D.C., A.V., J.H., G.M., and W.J. wrote the manuscript, with input from other co-authors.
Corresponding authors
Ethics declarations
Competing interests
D.C., A.V., J.H., G.M., W.J., F.J., K.Y., J.F., C.H., C.D., C.F., E.W., N.S., S.P., J.B., S.K., N.Pi., N.Pa., F.B., K.P., A.D., F.M.M., M.W., M.A., F.S., M.K., and A.C.C. are employees and shareholders of Novartis Pharma. P.H.O.M., J.K., R.W., M.R., D.M., and B.R. are former employees of Novartis AG.
Peer review
Peer review information
Nature Communications thanks Pravin Jagtap, Mariusz Hartman and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Castelletti, D., Hinrichs, J., Malojčić, G. et al. Fragment-based discovery enables direct targeting of the melanoma oncogene MITF. Nat Commun 17, 594 (2026). https://doi.org/10.1038/s41467-025-67297-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67297-0
