
Abstract
Social attachments are vital to mammalian health, but the neural microcircuitry underlying their formation remains unknown. Using prairie voles—which form lasting pair bonds—we investigate how the microcircuitry of the nucleus accumbens (NAc) transforms social interaction into attachment. We confirm that accumbal calcium-permeable AMPARs (CP-AMPARs) mediate excitation of fast-spiking interneurons (FSIs) in voles, and find that their blockade prevents pair bond formation. To understand the underlying CP-AMPAR-dependent circuit computations, we combine in vivo calcium imaging with local pharmacology. This reveals that social information is differentially encoded at neuronal and ensemble levels, with bonding leading to an emergence of partner-selective ensembles. CP-AMPAR blockade produces a striking dissociation: it disrupts partner ensemble formation while paradoxically increasing the proportion of partner-selective neurons. Further, blockade selectively impairs ensemble-level decoding, suggesting that temporally structured coactivity is essential for transmitting bonding-related information. Finally, as CP-AMPARs mediate FSI activity, we use in-vivo electrophysiology to show that FSIs dynamically and distributively coordinate medium spiny neuron (MSN) ensemble activity. Our findings delineate an accumbal microcircuit mechanism whereby ensemble formation, gated by CP-AMPARs, transforms social interactions into attachment.
Similar content being viewed by others


Introduction
The transformation of social interactions into enduring attachments is fundamental for many species and profoundly important for physical and mental health. These attachments, whether between mates, social groups, or parents and offspring, shape behavior and well-being throughout life1,2, yet we know little about how the brain accomplishes this transformation. Prairie voles, unlike standard laboratory rodents, form lifelong pair bonds, characterized by a strong and selective preference for their partner over a stranger3,4. Within the nucleus accumbens (NAc), a region of the ventral striatum involved in reward-directed behavior, pair bonds trigger changes in transcription5,6,7,8 and neuromodulatory signaling9,10,11,12,13,14. These molecular and neurochemical biomarkers suggest that the NAc plays a key role in transforming information about a stranger into representations of a bonded partner.
Despite the known importance of the NAc in bonding, the computations that actively encode partner interaction remain poorly understood. Amadei and colleagues15 demonstrated that corticoaccumbal connectivity, measured through local field potentials, modulates affiliative behavior towards partners. Our own prior work16 showed that individual accumbal neurons encode partner interactions and increase activity as a function of bond formation. However, these studies were limited to examining either population-level field potentials or isolated neuronal responses, without resolving the intermediate, microcircuit-level computations.
Calcium-permeable AMPA receptors (CP-AMPARs)17,18,19 are compelling candidates for organizing microcircuit-level computations underlying social attachment. In the NAc, CP-AMPAR upregulation on medium spiny neurons (MSNs) is important for drug-induced synaptic and behavioral plasticity20,21,22,23,24, and CP-AMPARs are also constitutively and preferentially expressed on FSIs25,26,27,28. FSIs provide powerful control over local neuronal activity, and pharmacological blockade of CP-AMPARs in the striatum selectively suppresses FSI but not MSN activity26,28,29,30,31,32,33,34,35,36,37,38. Here, we confirmed this via ex vivo whole cell recordings in the prairie vole NAc. We then found that local bilateral blockade of accumbal CP-AMPARs during initial mate cohabitation impairs partner preference formation, demonstrating that they are required for the formation of social attachment.
To understand the underlying circuit computations, we first characterized how the NAc naturally encodes social interactions throughout attachment formation. Using in vivo calcium imaging combined with local pharmacology, we found that partner interactions are represented at multiple computational levels—from stable individual neurons to emergent and temporally coordinated neuronal ensembles that, unlike the tuning of individual neurons, correlate with bond strength. CP-AMPAR blockade prevented the emergence of partner-encoding ensembles despite increasing the prevalence of individually partner-tuned neurons, suggesting that CP-AMPAR-mediated ensemble coordination is critical for social attachment. This result was replicated by computationally removing ensemble-level information from individual cells in control animals.
Finally, hypothesizing that FSIs gate MSN ensemble formation, we used high-density electrophysiology to record from both FSIs and MSNs in a species with limited genetic tools. We found that FSIs exerted monosynaptic control over MSNs, that FSI bursts were temporally coupled to the activation of multiple MSN ensembles, and that control of individual MSNs was distributed across subsets of FSIs. Together, these findings support a circuit mechanism in which CP-AMPAR-expressing FSIs coordinate ensemble activity, linking receptor function to microcircuit computations that transform social interactions into attachment.
Results
Accumbal CP-AMPARs are present on FSIs and necessary for bond formation
In the absence of genetic tools or enhancer-driven viral constructs that can drive selective expression in specific interneuron populations39,40 in the prairie vole NAc (Fig. S1), we turned to pharmacology to probe fast-spiking interneuron (FSI) function. Single-nucleus RNA sequencing6 from the vole NAc revealed that parvalbumin (PV) interneurons had the lowest GluA2/(GluA1 + A3 + A4) ratio, reflecting relatively low GluA2 expression and a higher potential for calcium-permeable AMPAR composition (Fig. S2), consistent with findings from other species28. We therefore used IEM-1460, a CP-AMPAR antagonist known to robustly and preferentially inhibit FSIs26,36,41,42. To confirm this selectivity in the vole NAc, we performed whole-cell recordings in acute slices. To increase the likelihood of recording from FSIs, we used a viral construct driven by the S5E2 enhancer40 (Fig. S3A), of which ~40% of labeled cells are PV-expressing (Fig. S1). We distinguished FSIs from MSNs on the basis of their intrinsic properties43 (Fig. S3B–D). We then recorded evoked AMPAR-mediated excitatory postsynaptic currents (eEPSCs) before and after bath application of IEM-1460 (50 μM) (Fig. S3E). eEPSCs in FSIs were robustly suppressed by wash-on of IEM-1460 whereas MSNs were unaffected (Fig. S3E, F). Together, these results support preferential CP-AMPAR expression on FSIs in the vole NAc and identify FSIs as a primary functional target of IEM-1460.
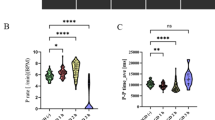
To directly test whether CP-AMPARs are necessary for bond formation, we performed a loss-of-function study, delivering bilateral infusions of IEM-1460 or vehicle into the NAc of prairie voles prior to two cohabitation + mating sessions (Figs. 1A, B and S4A, B). Drug infusion did not affect total locomotion or mating (Figs. 1C and S4C–E), which is facilitatory for bond formation4,9. However, blockade prevented the formation of partner preference, a behavioral proxy for pair bond formation (Fig. 1D, E). While vehicle-treated animals formed partner preferences, IEM-1460-treated animals showed a range of social preferences distributed around no preference, mirroring the spontaneous social preferences of sexually naïve, non-bonded voles44,45 and suggesting that CP-AMPARs are required for social attachment. Total social interaction was unchanged (Fig. S4F), suggesting that CP-AMPAR blockade disrupts the formation of attachment rather than affecting general social interest. Additionally, there was no interaction between sex and the effect on partner preference (Fig. S4H). Because partner preference testing occurred without IEM-1460 present, these results suggest that CP-AMPARs drive a process during cohabitation that is critical for bond formation. The same outcome was observed with infusion of NASPM, another CP-AMPAR antagonist (Fig. S5).

A Timeline for behavioral pharmacology. During cohabitation, two animals interact in a two-chamber apparatus. During the partner preference test (PPT), test animals choose to interact with a partner or stranger animal, tethered on opposite ends of a three-chamber apparatus. B IEM-1460 or vehicle was infused into the NAc. Targeting verified using dye injection. C Effect of infusion on mating bouts, one-sample t(24) = −0.76, p = 0.454. D Vehicle partner vs stranger huddle, paired t(12) = 3.67, p = 0.0032, IEM-1460 paired t(12) = −0.22, p = 0.827. E Partner preference index (partner huddle/total huddle), vehicle vs IEM-1460 t(24) = 3.26, p = 0.003. Data are presented as mean values ± SEM. A, B created partially in BioRender (https://BioRender.com/xiyjkl9).
Organization of neuronal encoding and reversal by CP-AMPAR blockade
To understand how pair bonding transforms accumbal activity and generates partner-specific representations, we developed a novel approach combining cannula-mediated pharmacology with one-photon calcium imaging (Figs. 2A–D and S6A–D). We focally transduced neurons with an adeno-associated virus (AAV) expressing the fluorescent calcium indicator GCaMP6f46 and implanted a gradient refractive index (GRIN) lens above those neurons47, spanning both the medial core and shell (Fig. S6A). During this procedure, we also implanted a chronically-dwelling cannula diagonal to the field of view (FOV) to enable infusions of either vehicle or the CP-AMPAR antagonist IEM-1460 to the imaged neurons. To capture the critical period of bond formation, animals engaged in two 4-h cohabitation sessions on successive days, followed by a partner preference test—mirroring our initial behavioral pharmacology experiment (Figs. 1A and 2A). We collected imaging data during the first cumulative hour of cohabitation (Day 0), the eighth cumulative hour of cohabitation (Day 1), and the first hour of a partner preference test (Day 2). Importantly, infusions occurred prior to each cohabitation session but not before partner preference testing. To examine how CP-AMPARs influence neural encoding without concurrently altering behavior, we performed unilateral infusions. Unlike the bilateral blockade, which abolished partner preference (Fig. 1), unilateral blockade did not affect partner preference or social behavior during the Day 2 partner preference test (Fig. S7A–D), allowing us to examine drug effects on neuronal encoding without altering behavior. Animals in both groups then remained cohoused and unmanipulated until they underwent another partner preference test with no infusions on Day 15.

A Timeline for calcium imaging + pharmacology. B GRIN lens + cannula implant. C Example of implanted hardware. Underside of the skull is shown. D Example histology. E Pseudocolored regions of interest extracted from a representative field of view. Across animals, we extracted 45.1 ± 4.87 neurons per animal. F Example neurons selective for partner and stranger chamber. Gray shading indicates time periods in partner (top) and stranger (bottom) chambers. G Total proportion of neurons (positive and negative) activated by partner interaction during cohabitation on Days 0 and 1. Mixed LM interaction β = –0.163, p = 0.074; post-hoc Day 0, two-sample t(16) = –0.10, p = 0.918; post-hoc Day 1, t(16) = 2.99, p = 0.0086. H Proportions of partner and stranger selective neurons on day 2, mixed LM interaction β = 0.164, p = 0.00002, partner two-sample t(13) = –1.98, p = 0.070, stranger t(13) = 3.97, p = 0.004. I Proportions of partner and stranger selective neurons on day 15, mixed LM interaction β = 0.015, p = 0.798, partner two-sample t(13) = 0.17, p = 0.865, stranger t(13) = 0.45, p = 0.659. J Difference score, calculated as proportion(partner selective) - proportion(stranger selective). Day 2 two-sample t(13) = 5.24, p = 0.0002, day 15 t(13) = –0.24, p = 0.814. Day 15, one-sample t test against 0, vehicle t(7) = –2.03, p = 0.083, expt t(6) = –0.81, p = 0.450. K Representative example of cells registered across days, represented by different shades. L Total cells registered across days. Mixed LM main effect treatment β = −7.795, p = 0.388. M Stability of partner-selective cells across days. Mixed LM treatment x actual-shuffle interaction β = 3.512, p = 0.027. Vehicle: Post-hoc control Day 0-1 two-sample W(8) = 0.0000, p = 0.027, Day 0–2 W(6) = 0.0000, p = 0.043, Day 1-2 W(6) = 6.50, p = 0.787. IEM-1460: Day 0–1 W(7) = 1.000, p = 0.655, day 0–2 W(6) = 1.000, p = 0.655, Day 1–2 W(5) = 0.0000, p = 0.317). ac = anterior commissure. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data are presented as mean values ± SEM. A created in BioRender (https://BioRender.com/l3dztfi). Miniscope image in B reproduced with permission from Daniel Aharoni, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0).
We extracted neuronal Ca2+ activity48,49 (Figs. 2E and S6C) and used area under the receiving operating characteristic curve (auROC)50,51 to identify neurons selective for partner or stranger chamber during partner preference tests (PPTs) (Figs. 2F and S8A, B). Vehicle-treated animals showed a consistent proportion of partner-selective neurons across Days 0–1 (Fig. 2G). During the Day 2 partner preference test, which was not preceded by infusion, we unexpectedly found more stranger-selective than partner-selective neurons (Fig. 2H, J). We also observed a reduction in partner-selective neurons from Days 0–1 to Day 2, likely reflecting that neurons responding broadly to social interaction appeared partner-selective on Days 0–1, but on Day 2—when two social stimuli were present—were no longer categorized as partner-selective.
To examine how single-neuron selectivity depends on CP-AMPARs, we compared vehicle and IEM-1460-treated animals. While partner-selective neuron proportions were initially similar on Day 0, CP-AMPAR blockade reduced partner selectivity on Day 1 (Figs. 2G and S8C, D), suggesting that CP-AMPARs gate an experience-dependent element. Strikingly, during the Day 2 partner preference test, CP-AMPAR blockade reversed normal patterns, increasing partner-selective neurons while decreasing stranger-selective neurons (Figs. 2H, J and S8E). Partner- and stranger-selective neurons were not spatially clustered in either group (Fig. S8G–I). At the Day 15 partner preference test, there was no difference in neuronal selectivity and no predominance of partner or stranger neuronal selectivity in either group (Figs. 2I and S8F), suggesting either diminished NAc involvement post-bonding and/or normalized encoding following drug clearance.
A key question in social attachment is whether individual neurons maintain stable representations of the partner across days. We registered cells across days52 (Fig. 2K) and consistent with recent reports53, found considerable variability with around 45% of neurons co-registered between days in both groups (Fig. 2L). In vehicle-treated animals, partner-selective neurons maintained their functional identity across days above chance levels, but only between Day 0 and later days, and not between Days 1–2 (Fig. 2M1), suggesting that initial partner encounters engage neurons predisposed to stability. In contrast, IEM-1460-treated animals failed to show stable partner selectivity across days (Fig. 2M2). Together, these results indicate that CP-AMPARs regulate the encoding of information into NAc neurons during bond formation, including the establishment of a population of stable neurons that encode the partner across days.
Emergence of partner ensembles is abolished by CP-AMPAR blockade
Beyond single neurons, circuits organize information through neuronal ensembles, or groups of neurons displaying recurrent patterns of coactivity54,55,56. Using PCA-ICA57, we detected ensembles, or groups of neurons that coactivate across time independent of specific behavioral triggers (Fig. 3A). This unsupervised method of ensemble identification captures patterns of coordinated activity rather than experimenter-defined stimulus-locked responses. We found that the majority of neurons (77.7%) participated in ensembles, typically in only one ensemble (Fig. S9A). Blockade of CP-AMPARs did not affect the total number of ensembles extracted (Fig. 3B) or the percentage of neurons participating in ensembles (74.5%) (Fig. S9A). We then used auROC and shuffled controls and found partner-selective ensembles that emerge over the course of bond formation (Fig. 3A, C, D). To determine whether ensemble-level coding reflects attachment and not merely cohabitation or familiarity, we asked whether these physiological measures naturally covary with bond strength. In vehicle-treated animals, the proportion of partner minus stranger ensembles positively correlated with chamber preference (Fig. 3E), while the proportion of partner minus stranger neurons did not (Fig. S9B), suggesting that ensemble-level representations may be a more meaningful neural substrate of attachment.

A Representative identification of ensemble using PCA-ICA, with activity of highly weighted neurons (weights > mean + 2 SD) shown beneath. Ensembles consist of two or more neurons. B Total ensembles across days. Mixed LM treatment β = 0.023, p = 0.159, post-hoc Day 0 two-sample t(16) = –1.10, p = 0.293, Day 1 t(15) = –1.71, p = 0.127, Day 2 t(13) = –0.900, p = 0.388. C Partner-selective ensembles across days. Mixed LM treatment β = –0.022, p = 0.015, post-hoc day 0 two-sample t(16) = 0.491, p = 0.633, Day 1 t(15) = 1.153, p = 0.267, Day 2 t(13) = 2.535, p = 0.0262. D Partner-selective ensembles on Day 2. E Correlation in control animals between % time in partner chamber and partner minus stranger ensembles (divided by total # neurons). Spearman’s r(8) = 0.81, p = 0.0149. F Representative spatial clustering analysis of ensemble neurons. Highly weighted neurons in each ensemble were identified and average pairwise Euclidean distance was computed. G Ensemble clustering across animals. Quantified as a percentile of mean pairwise distances versus null distributions from random cell selections. One-sample t-test versus 50, vehicle t(7) = –2.63, p = 0.034, IEM-1460 t(2) = –11.139, p = 0.00796. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data are presented as mean values ± SEM.
As FSIs receive early afferent input and exert feedforward control over MSNs35,38,58,59, we hypothesized that the formation of ensembles selective for partner chamber depends on CP-AMPAR function. In support of our hypothesis, IEM-1460-treated animals developed fewer partner-selective ensembles during bond formation, as evidenced by the Day 2 partner preference test (Fig. 3C, D). This disruption was specific for partner chamber, as ensemble encoding of stranger chamber was unaffected (Fig. S9C). There were no differences between partner and stranger ensembles in either the control or drug groups by Day 15 (Fig. S9D).
Given MSNs’ capacity to form spatially compact ensembles58,60,61, we assessed the spatial clustering of highly weighted neurons within each partner ensemble (Fig. 3E). In both vehicle and IEM-1460 groups, partner ensemble neurons were more clustered than chance, although highly robust clustering was observed in only a subset of animals (Fig. 3F). Non-partner ensembles showed similar clustering (Fig. S9E). Together, these findings suggest that under native conditions, partner information is encoded by a subset of single neurons as well as the activity of spatially localized ensembles of neurons. CP-AMPAR blockade increases the proportion of individual partner-selective neurons while disrupting partner ensemble formation.
Population variance compresses as a function of cohabitation
Beyond single neurons and ensembles, we next examined how social information is represented at the population level, as cognitively complex information is often encoded in high-dimensional population codes62,63. Throughout pair bond formation, variance in population activity during social bouts was captured by progressively fewer principal components (Fig. 4A), driven by increased variance in the first three PCs (Fig. 4B). This suggests a refinement of neural activity into a lower-dimensional subspace. However, unlike reports of low-dimensional population encoding of social variables in the prefrontal cortex and hypothalamus51,64, partner versus stranger information was not readily distinguishable in PC subspace, with considerable variability across animals (Fig. 4C, D). Despite disrupting single-neuron and ensemble coding, IEM-1460 had no effect on these features (Fig. 4A–D).

A Number of PCs required to explain 95% of variance exhibited during social interaction, normalized by number of neurons. Mixed LM main effect time Day 2 β = –0.269, p = 1.17*10^–8. Main effect treatment β = 0.060, p = 0.149. B Variance during social interaction explained by first 3 PCs. Mixed LM main effect time Day 2 β = 0.125, p = 0.022. Main effect treatment β = 0.009, p = 0.840. Mixed LM, interaction between number of PCs and variance explained by first 3 PCs, β = –0.449, p = 0.0014. C Representative animal, partner and stranger representations in PC space. D Separability of partner and stranger centroids in first 3 PCs, represented by p-values derived from comparing the actual distance to a null distribution. Vehicle one-sample vs 0.5, t(7) = 1.076, p = 0.318, IEM-1460 t(6) = 1.679, p = 0.144. Dashed lines indicate 5th and 95th percentiles. p = 0.0149. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data are presented as mean values ± SEM.
CP-AMPAR blockade dissociates neuronal- and ensemble-level information decodability
To compare how partner and stranger information is represented across organizational levels55, we used linear support vector machines (SVM) to decode partner versus stranger chambers. When using all calcium traces, we could successfully decode the chamber (Fig. 5A, B). Similarly, when using all ensemble activation strengths (as in Fig. 3A), we could successfully decode chamber, suggesting that patterns of temporal coactivity also carry social context information (Fig. 5D).

A Representative animal, SVM prediction. B Performance of Day 2 decoder using calcium traces from all cells. Vehicle paired t(7) = 5.73, p = 0.0007. IEM-1460 paired t(6) = 2.11, p = 0.079. C Similarity of partner-stranger representations. Two-sample t(13) = –2.96, p = 0.011. D Performance of Day 2 decoder using ensemble activations. Vehicle paired t(7) = 5.24, p = 0.0012, IEM-1460 paired t(6) = 0.236, p = 0.821). P = 0.0149. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data are presented as mean values ± SEM. Panel A created partially in BioRender (https://BioRender.com/pqsifny).
IEM-1460 treatment revealed a dissociation: while population-level decoding from calcium traces remained possible in most animals (Fig. 5B), albeit with greater similarity between partner and stranger population vectors (Fig. 5C), decoding from ensemble activations was no longer successful (Fig. 5D). This loss of ensemble-level information was not due to differences between the two groups in the number of total ensembles (Fig. 3B), neurons recorded per animal (Fig. S6C), or highly weighted neurons per ensemble (Fig. S9F). Successful decoding in the IEM-1460-treated animals persisted when using a number of PCs equal to the number of ensembles in each animal (Fig. S9G), indicating that dimensionality reduction didn’t drive the decoding failure. This finding—that following CP-AMPAR blockade, partner information can be read out from the collective activity of all individual neurons but not from ensembles—further suggests that the organization of individual NAc neurons into partner-encoding ensembles may be critical for bond formation.
To explore the behavioral contexts in which ensembles activate, we annotated ensemble activation events with behaviors and found that ensemble activations aligned with a range of behaviors, including social investigation, non-social behavior, and huddling. However, there were no significant differences in the distribution of behaviors associated with ensemble events between control and IEM-1460-treated animals (Fig. S10). These results indicate that, although IEM-1460-treated animals show fewer ensembles tuned to the partner chamber, the behavioral contexts in which ensembles activate remain similar across groups.
Finally, to examine the relationship between individual neurons and ensemble-level activity, we examined how ensemble membership affects single-neuron information. For each neuron belonging to an ensemble, we spliced out epochs of activity during which its ensemble was active (Fig. S11A, B), and quantified partner- versus stranger-selectivity before and after ensemble removal. In vehicle animals, this manipulation revealed enhanced partner selectivity (Fig. S11C), mimicking the effects of CP-AMPAR blockade (Fig. 2H, J). In IEM-1460-treated animals, where ensemble organization was already disrupted, removing ensemble periods produced no systematic change in selectivity (Fig. S11C). This aligns with findings of increased PV interneuron selectivity following CP-AMPAR removal65, suggesting that the broad tuning effect of CP-AMPARs may propagate to local ensembles.
Accumbal fast-spiking interneuron bursts precede and coordinate MSN ensemble activity
We reasoned that if FSIs mediate the effect of CP-AMPAR inhibition on partner ensembles, then they should natively demonstrate a temporal relationship with MSN ensemble activation. Given the absence of enhancer-driven viral targeting specifically of FSIs39,40 (Fig. S1), we directly tested the hypothesis that FSIs coordinate activity in MSN ensembles by using chronically implanted Neuropixels 2.0 probes66 (Fig. 6A, B) to record from both FSIs and MSNs across an identical experimental timeline (Fig. 6C, D). Using data from the Day 2 partner preference test, we isolated putative FSIs based on narrow waveforms (<400 μs) and near-continuous firing (PropISI>2sec < 0.1)32,67 (Figs. 6D–F and S12A).

A Chronic Neuropixels recordings in prairie voles. B Representative probe placement. C Timeline for recording identical as before. D Representative epoch with activity of fast-spiking interneuron (FSI, purple) and medium spiny neuron (green) in close proximity. E FSIs isolated on the basis of waveform durations <= 400 us with inter-spike interval (ISI) ratios below 0.1. Ovals only for visualization. F Waveforms of FSIs, MSNs, and unidentified interneurons (excluded from analysis). G Representative cross-correlogram between FSI and MSN. Red lines indicate 1st and 99th percentiles of surrogate distributions. H Average cross-correlogram across all FSI-MSN pairs demonstrating putative monosynaptic inhibition. Gray shading indicates standard error across pairs. I Average cross-correlogram across all FSI-MSN pairs not demonstrating inhibition. J Average cross-correlogram across all FSI-MSN pairs. K Representative ensemble (top) and highly weighted neurons (bottom). L Decoding partner versus stranger chamber using MSN ensemble activity. Paired t(5) = 7.46, p = 0.00068. M Example of identified epoch of FSI hyperactivity. Top: Z-scored spike rate. Bottom: Raster plot. N Activity of each FSI averaged across all bursts. O Representative MSN ensemble activity time-locked to bursts of individual FSI. Red line over timepoints significantly different from shuffled controls. P MSN ensemble activity across all FSIs. Q Representative epoch of FSI and ensemble activity from a single animal. Top: FSIs. Bottom: MSN ensemble activity. R Lorenz curves for each ensemble based on the prediction of ensemble activity by FSIs. MSN = medium spiny neuron, FSI = fast-spiking interneuron, UI = unidentified interneuron. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. C created partially in BioRender (https://BioRender.com/hthojaq).
Cross-correlograms of nearby (<60 μm) FSI-MSN pairs revealed putative monosynaptic inhibition (Fig. 6G), as well as MSNs demonstrating broadly elevated activity around FSI spikes (Fig. S12B) and more complex dynamics (Fig. S12C), consistent with FSIs exerting complex effects on MSN activity beyond simple inhibition41,68. About 10% of pairs showed inhibition (Fig. 6H–J)—higher than previous reports32,58, likely due to Neuropixels’ dense contact sampling rather than species- or behavior-specific physiology.
Using PCA-ICA, we extracted ensembles of MSNs based on coactivity within 50 ms bins (Fig. 6K) and, as with our calcium imaging data, successfully used ensemble activity to decode partner versus stranger (Fig. 6L). To examine the relationship between FSI and MSN ensemble activity, we identified brief (≥ 50 ms) bursts of individual FSIs (Fig. 6M, N) and time-locked the activity of ensembles of MSNs to these bursts. We found that FSI bursts coincided with activation of MSN ensembles (Fig. 6O, P), with significant activation of ensembles occurring at 0-50, 50–100, and 100–150 ms windows after burst onset, indicating temporally specific coordination. This relationship decreased with lower FSI burst threshold and was present for both partner and non-partner encoding ensembles (Fig. S12D, E). To further test the directionality of FSI-MSN coordination, we applied Granger causality to population firing rates for FSIs and MSNs in each animal with at least 2 FSIs. In all animals, FSI activity significantly predicted MSN activity better than the reverse (Fig. S12F).
Coordination of MSNs is distributed across subsets of FSIs
Given that multiple MSN ensembles, even when spatially distributed, can show activity time-locked to a single FSI (as in Fig. 6O), we suspected that FSIs may exhibit coordinated bursting. Supporting this, we readily observed temporally correlated multi-FSI bursts (Fig. 6Q), although coupling between FSIs appeared idiosyncratic and transient. To quantify the extent to which the effect of FSIs on ensembles is distributed, we measured how removing individual FSIs affected linear models that used FSI activity to predict the activity of a single ensemble of MSNs. Using Gini coefficients to describe the distribution of these contributions, we found that the majority of ensembles showed moderate levels of distribution (Gini coefficients between 0.25–0.75), suggesting that ensembles are typically regulated by subsets of coordinated FSIs rather than individual FSIs or the entire FSI population equally (Fig. 6R). Together, this demonstrates a temporal link between FSI activity and neuronal ensembles, including but not limited to those that encode the partner.
Discussion
In this study, we reveal how accumbal microcircuitry transforms social experience into social attachment. Beyond partner-selectivity in a subset of neurons16, we find that partner information is organized at multiple computational levels: by stable individual neurons that encode the partner across days and by emergent ensembles of coactive neurons. Critically, ensemble-level selectivity correlated with bond strength, whereas single-neuron selectivity did not, pointing to ensembles as a more behaviorally meaningful substrate. These findings demonstrate that ensembles emerge through experience to support naturally occurring attachment behavior55,56. While studies in mice have shown that accumbal neurons activate during general social interaction and can be stably tuned across days53,69, our work demonstrates how accumbal microcircuitry encodes a specific social stimulus in an innate, experience-gated fashion–a phenomenon uniquely accessible in the prairie vole model. Our findings apply specifically to the formation phase of bonding (Days 0–2); once bonds were established (Day 15), we no longer observed clear partner-stranger differences at either single-neuron or ensemble levels.
We find that FSI CP-AMPARs, previously implicated in plasticity in NAc MSNs20,21,22,23,24,70, are necessary for bond formation and orchestrate the transformation of accumbal microcircuitry, providing a novel link between receptor and network function. Following CP-AMPAR blockade, social information was disorganized but still present across individual NAc neurons. However, the pronounced degradation of information at an ensemble level suggests that temporal coactivity of neurons may uniquely encode information necessary for bond formation55. These findings provide one possible framework for understanding ensemble emergence in the NAc and other striatal regions59,61,71, where principal neurons are GABAergic and lack local excitatory architecture.
While we found that CP-AMPAR blockade, as in other studies, is an effective pharmacological tool for silencing FSIs due to their high expression of CP-AMPARs26,36,59, it is not perfectly selective. Low levels of CP-AMPARs can be present in excitatory synapses onto MSNs, particularly for some inputs72,73, and CP-AMPAR blockade can interfere with currents observed at cholinergic interneurons. Thus, we cannot rule out that parts of the observed effects reflect direct or indirect disruption of non-FSI-related physiology. Nevertheless, prior work and our own recordings support FSIs as the dominant CP-AMPAR-expressing population in the NAc, suggesting that the observed effects likely stem primarily from compromised FSI function26,36,38,41,42.
Our electrophysiological data demonstrate that FSIs in the prairie vole NAc can exert monosynaptic control over nearby MSN activity, providing at least a partial exception to the idea that striatal MSNs may require simultaneous input from multiple FSIs to exhibit a noticeable postsynaptic effect32,33,58. Nevertheless, although single FSIs can exert direct monosynaptic control on MSNs, we observed that multi-FSI bursts are dynamically coupled to MSN multi-ensemble activity, indicating that FSIs collectively organize neuronal activity in the NAc.
Inputs to the NAc arrive first at FSIs35,36,59, suggesting that the emergence of partner ensembles—and the effect of CP-AMPAR blockade—may be mediated by FSIs. While the alignment of FSI bursting and activation of MSN ensembles may seem counterintuitive, this is consistent with growing evidence that FSIs can exert bidirectional effects on MSN activity depending on cellular state and circuit context35,36,37,40,58,67. FSIs may support ensemble formation by synchronizing MSN activity, providing the temporal precision needed for coordinated firing in response to their direct afferent drive during social interactions74,75,76,77. In addition, FSIs may inhibit unrelated or weakly driven MSNs and actively suppress competing ensembles6, helping ensure that only the relevant neurons are recruited together. In this way, FSIs may act as local organizers of population activity, shaping when and which neurons fire together. Isolating the role of FSIs in ensemble encoding underlying pair bonding—and differentiating those effects from potential contributions of other CP-AMPAR-expressing neurons or from off-target effects—will require the development of genetic tools allowing for selective targeting of PV interneurons in prairie voles, or for selective editing of AMPAR calcium permeability in FSIs. Similarly, as we do not have access to dopamine D1 receptor-expressing (D1R) and dopamine D2 receptor-expressing (D2R) MSNs in this dataset, such resolution may be critical given evolving models of accumbal function. While early studies suggested that NAc neurons are innately tuned to rewarding and aversive stimuli78, more recent work reveals that D1 MSNs encode valence-free salience, whereas D2 MSNs encode valence-free prediction errors79 and negative outcomes80,81, with activity that dynamically evolves during learning79,82. D1 and D2 MSNs also appear to play reciprocal roles in modulating social interest53. These findings raise the possibility that specific MSN subpopulations—particularly D2 MSNs—may be differentially engaged as a partner becomes increasingly associated with specific cues and outcomes over time, and thus preferentially recruited into ensembles or differentially affected by CP-AMPAR blockade.
CP-AMPAR inhibition produced a paradoxical dissociation: it disrupted partner ensemble formation while increasing the proportion of individual partner-selective neurons. We observed a similar effect in vehicle-treated animals when we subtracted ensemble-associated activity periods from individual neuronal traces. This supports our working model in which disruption of FSI-mediated coordination lowers the threshold for individual MSN activation, permitting isolated partner tuning without structured ensemble membership. One plausible mechanism is interference with spike-timing-dependent plasticity (STDP)31,36,65,74,75,76: FSIs can influence when MSNs spike and when they transition from down to up states, a key determinant of whether afferent inputs produce long-term plasticity83,84. Without this gating, MSN activation may be mistimed relative to inputs83, impairing STDP despite preserved excitatory drive. How striatal plasticity rules are modulated by the inwardly rectifying and anti-Hebbian characteristics of CP-AMPARs25,28 warrants further investigation, especially as CP-AMPAR conductance depends on membrane potential (mV) and may differ for coordinated versus isolated inputs. In sum, we speculate that CP-AMPAR-expressing FSIs gate ensemble recruitment through a self-limiting inhibitory window, filtering weak, asynchronous inputs while allowing strong coincident inputs—as may occur during salient partner encounters—to trigger coordinated MSN activity (Proposed Model in Fig. S13). CP-AMPAR blockade disrupts this gating, increasing individual tuning but impairing ensemble formation.
While one might expect that more neurons tuned to a salient social cue, such as the partner, would improve coding, we find that under control conditions, there are in fact more neurons tuned to the stranger, and that CP-AMPAR blockade, despite disrupting pair bond formation, in fact increases neurons tuned to the partner. These incongruous findings suggest that it is rather the coactivity of neurons at the ensemble level that supports bond formation and that downstream circuits may read out55,56. Consequently, an uncoordinated expansion of partner-selective neurons may be maladaptive. Because the NAc is predominantly GABAergic, ensemble activity likely originates from patterned excitatory afferent inputs15,35,59,71. The spatial clustering of ensemble neurons we observed hints at a topographical organization that may facilitate oscillatory activity involved in local or interregional communication15,58,85. Moreover, this ensemble-level organization may be critical for how downstream structures interpret and act upon accumbal patterns, as it can provide a coherent, temporally-structured code55. Such a code might represent a progressive increase in expected partner value or other partner-specific features of bonding, such as reward associations82 or prediction error79.
Although ensemble activity was often associated with social behaviors, we also observed activity during periods in which no overt social behavior was occurring. This suggests that ensemble activation may not exclusively reflect ongoing social interaction, but could also reflect contextual, anticipatory, or post-experiential signals86. Use of social operant tasks to effectively uncouple partner-directed motivation from consumption of partner access could help clarify which variables are represented at single-neuron versus ensemble levels, and how these codes shape selective social behavior. Whether ensemble coding similarly supports other forms of social bonding, such as same-sex peer bonding6 or other reinforced attachment behaviors such as drug-seeking, remains to be determined. Finally, the role of sex differences in these mechanisms remains to be fully resolved. While electrophysiology experiments were conducted only in females, we included both sexes in our other experiments and did not observe any clear sex differences, although our study was not powered to detect subtle effects.
Altogether, our findings reveal a CP-AMPAR-dependent microcircuit mechanism that orchestrates the formation of neuronal ensembles supporting the transformation of social experience into selective attachment. This mechanism may underlie one of the most profound aspects of human experience—how we form selective, lasting bonds with other individuals. These findings may also provide a broader framework for understanding how experience-gated plasticity enables the brain to form and maintain selective attachments, bridging the gap between receptor signaling and reward-directed behavior.
Methods
Experimental model and subject details
All procedures were conducted in accordance with standard ethical guidelines (National Institutes of Health Guide for the Care and Use of Laboratory Animals) and with approval by the Institutional Animal Care and Use Committee (IACUC) at the University of Colorado Boulder. Prairie voles were bred in-house, initially imported from colonies housed at Cornell University, Emory University, and UC Davis, all of which originated from wild animals captured in Illinois. Animals were maintained at a temperature of 23–26 °C on a 14:10 light:dark cycle. All procedures occurred during the light phase. Animals were given water and rabbit chow ad libitum (5326-3 by PMI Lab Diet). Rabbit chow was supplemented with sunflower seeds and dehydrated fruit bits. Home cages were enriched with cotton nestlets and plastic houses. Cages containing implanted animals were enriched with a wooden block in replacement of a plastic house. At postnatal day 21, animals were weaned and placed into standard static rodent cages (17.5 l. x 9.0 w. x 6.0 h. in.) at a density of 2–4 same sex prairie voles. All animals used for experiments were adults (60–100 days old) at the time of surgery. Following implants, voles were housed with a divider separating them from a single same-sex cage mate. Due to the height of implant fixtures, animals implanted with Neuropixels probes were single-housed in large rat cages (17.5 l. x 10.0 w. x 8.5. h. in.). After being paired with an opposite-sex animal, Neuropixels pairs remained housed in the large cages, while all other pairs were housed in a smaller cage (11.0 l. x 6.5 w. x 5.0 h. in). Both males and females were used for all experiments except for Neuropixels experiments, where only females were used, due to their decreased risk of stranger-directed aggression and subsequent loss of headcap/probe. For all experiments, animals were single-housed three days prior to first introduction to a mate, and remained single-housed between cohabitation sessions and prior to the first partner preference test. To facilitate sexual receptivity, each female, whether a focal animal or paired with a focal male, was primed with estrogen prior to mate introduction. Each female received a subcutaneous injection of estradiol (0.1 mL, 20 µg/mL solution, prepared by diluting a 1 mg/mL ethanol stock in corn oil; Cayman Chemical, MI, USA) once daily for three consecutive days, with the final injection immediately preceding the first cohabitation. Animals used as strangers were never previously pair-bonded and were cohoused with same-sex cage mates.
Surgical procedures
Voles were anesthetized with 1–3% isoflurane at an oxygen flow rate of 1 L/min in a head-fixed stereotactic frame (Kopf Instruments, CA, USA). Body temperature was maintained at 37 °C using a closed-loop heating pad with a rectal thermometer (Kopf Instruments, CA, USA). Eyes were lubricated with ophthalmic ointment (Sterile Lubricant Eye Ointment, Stye, NY, USA). The fur was removed from the incision site using a shaver, and the wound area was disinfected with 70% isopropyl alcohol and betadine. The scalp and any connective tissue close to the planned implant site were removed above the frontal and parietal bones. For cannula and lens implants, one 0.5 mm guide hole was drilled in each of the parietal bones, and anchoring screws were inserted. For Neuropixels implants, an additional 0.9 mm guide hole was drilled in the posterior right parietal bone, followed by insertion of a ground screw soldered to stainless steel wire and a connector socket. The head was leveled in the anterior-posterior plane. For all implant methods described below, Vetbond tissue adhesive (3 M, MN, USA) was used at the end to seal the edges of the skin, and a mixture of triple antibiotic ointment and lidocaine was applied topically around the implant site. Animals recovered in a heated cage. All animals received subcutaneous perioperative injections of extended-release meloxicam (4 mg/kg), enrofloxacin (5 mg/kg) and saline (up to 3 mL), as well as enrofloxacin and saline for three days of postoperative care. All animals with lens or Neuropixels implants also received perioperative dexamethasone (0.2 mg/kg) as well as optional postoperative dexamethasone depending on clinical status and veterinary recommendation. Cannula-only and Neuropixels implants were allowed to recover for at least 10 days prior to initiation of experiments, while lens+cannula implants were allowed to recover for at least 4 weeks to allow for sufficient viral expression.
Cannula implants and drug delivery
0.7 mm guide holes were drilled at + 1.6 mm AP and ±1 mm ML. A 26-gauge bilateral stainless steel guide cannula was inserted to a depth of –1.6 mm DV over 2 min. The cannula and screws were affixed to the skull and one another with an initial layer of Metabond dental cement (Parkell, NY, USA), then with multiple additional layers of Loctite 454 cured with Jet Acrylic Resin Liquid (Lang Dental, IL, USA). Guide cannulas were covered with dummy caps.
Prior to each cohabitation/infusion session, a Hamilton syringe was loaded with either artificial cerebrospinal fluid (ACSF) (Tocris Bioscience, cat. # 3525, Bristol, UK) or IEM-1460 (1 mM in 0.9% saline) (Tocris Bioscience, cat # 1636, Bristol, UK). Polyethylene PE-50 tubing (Fisher Scientific, MA, USA) was attached to the Hamilton syringe and to the injection cannula. The Hamilton syringe was attached to a Kopf Model 5000 Microinjection Unit (Kopf Instruments, CA). Animals were anesthetized as above. Dummy caps were removed, and internal cannulae were inserted through the guide cannulae and seated into place (projection beyond guide of 3.0mm). 500 nL was injected continuously over 2 min, and the cannula was left in place for 5 min, then the procedure was repeated for the other side. At the end of the experiment and prior to infusion, 500 nL of India Ink was infused.
Prior to data analysis, animals were excluded due to preestablished criteria: no mating during cohabitation (n = 2 vehicle, n = 2 IEM-1460), mistargeting (n = 1 vehicle), loss of implant (n = 1 vehicle), and excessive aggression during the partner preference test (n = 1 IEM-1460).
Lens + cannula implants
Procedures for viral injections and lens implants were similar to47. A 1.4 mm hole was drilled at +1.6 mm AP and +1.05 mm ML, then a 0.7 mm hole was drilled at +1.6 mm AP and –2.65 mm ML. The dura was carefully removed from each opening, pressure was applied, and the wound site was irrigated with saline until bleeding stopped. Absorptive spears (Fine Science Tools, CA, USA) were used to remove small amounts of blood as needed. A Nanoject syringe (Drummond Scientific, PA, USA) was lowered, and injection was carried out at a rate of 2 nL s–1. For whole-cell patch-clamp electrophysiology experiments, AAV1/2-S5E2-hM4Di-p2A-dTomato (titer 1.925 * 10^12) (Neurophotonics, custom-made) was injected using 100 nL at each of –4.7, –4.6, –4.5, –4.4, and –4.3 mm DV. For all other experiments, AAV1-hyn-GCaMP6f (titer 7.4 * 10^12) (Addgene, cat # 100837, MA, USA) was injected using one of two methods of viral delivery: 1) 100 nL was injected at each of –4.7, –4.6, –4.5, –4.4, and –4.3 mm DV, and 2) 500 nL was injected at –4.5 mm DV. The syringe was left in place for 8 min following the last infusion. A blunt tip 30-gauge needle was then lowered to –3.6 mm DV and held in place for 8 min. No tissue aspiration was carried out. A GRIN lens (1.0 mm diameter, 9.0 mm length) was then lowered immediately to –3.6 mm, then slowly lowered to a final placement of –4.4 mm DV over 10 min. A layer of Metabond dental cement was applied to affix the lens to the skull, then a unilateral 26-gauge stainless steel guide cannula (P1 Technologies, VA, USA) was inserted to a depth of 2.30 mm DV at an angle of 30 ° through the 0.7 mm hole. The cannula, lens, and screws were affixed to the skull and one another with a layer of dental cement, then with multiple additional layers of Loctite 454 cured with Jet Acrylic Resin Liquid. The lens was covered with a silicone mixture (Body Double, Smooth-On, PA, USA) for protection.
Baseplate attachment and calcium imaging
At least three weeks after completion of surgery, voles were checked for GCaMP expression with a V4 Miniscope (Open Ephys, Lisbon, Portugal). Animals were briefly anesthetized with 1–3% isoflurane with 1 L/min oxygen flow in a head-fixed stereotactic frame. Any remaining silicone was removed, and the lens was cleaned using lens paper soaked in 70% isopropyl alcohol or acetone. A Miniscope V4 baseplate (Open Ephys, Lisbon, Portugal) was attached to a miniscope and lowered over the implanted GRIN lens until the cells and blood vessels appeared maximally sharp. The baseplate was cemented into place using additional Loctite, and the animal was placed into an empty behavioral apparatus to allow for evaluation of fluorescence. Animals with visually sufficient cellular activity and no excessive movement artifact were anesthetized again, and an internal cannula was inserted through the guide cannula, followed by infusion of 500 nL of ACSF as described above. Animals with no pressurized resistance to infusion or clouding of the lens due to suspected bleeding proceeded to experiments.
Prior to start of the experiment, animals were anesthetized and habituated to the scope for 15 min on each of three days, during which the focal plane was selected according to the optimal sharpness as determined visually. Focal plane was maintained and/or adjusted as needed to maintain the same focal plane for all subsequent recording sessions.
For experimental recordings, animals were briefly anesthetized. The internal cannula was inserted through the guide cannula, followed by infusion as described above. Animals were given 10 min to recover prior to the removal of a divider allowing for social access. Open-source UCLA V4 Miniscopes (https://github.com/Aharoni-Lab/Miniscope-v4) and MiniCAMs (Open Ephys, Lisbon, Portugal) were connected to a Miniscope data acquisition board (DAQ) 3.3 via a coaxial cable. The DAQ was then connected to a computer through USB 3.0. Recordings were carried out using Miniscope-DAQ-QT Software v1.11 (https://github.com/Aharoni-Lab/Miniscope-DAQ-QT-Software). Calcium fluorescence videos were recorded at 30 Hz using a gain of 3.5. LED power was selected to allow for sufficient signal-to-noise ratio (SNR) as well as avoidance of pixel saturation and consistency in baseline fluorescence levels across sessions. Cables were looped well above the experimental chamber to allow for minimal experimenter intervention and cable untangling outside of the animals’ FOV.
Prior to data analysis, three animals’ data (n = 2 vehicle, n = 1 IEM-1460) were excluded from day two analyses due to failures of scope PCBs and inability to distinguish signal from artifact, and were removed from the remainder of the experiment. Two animals (n = 2 IEM-1460) were excluded due to the mistargeting of the lens/misalignment of the lens and cannula.
Neuropixels implants and recordings
Procedures for Neuropixels implants were similar to those described in ref. 87. Neuropixels 2.0 four-shank probes were used (Imec, Leuven, Belgium). Prior to implant, the probe was mounted into a custom 3D-printed fixture (Atlas, Leuven, Belgium) and a stainless steel wire was soldered to a socket connector and to the probe ground pad. A single 1.4 mm guide hole was drilled at + 1.72 mm AP and +0.8 mm ML. The dura was carefully removed from the opening, pressure was applied, and the wound site was irrigated with saline until bleeding stopped. Immediately prior to insertion, the probe was dipped in undiluted DiI dye (Thermo Fisher, cat # V22884, MA, USA) three to five times, lowered immediately to 1 mm while ensuring no bending, then lowered at a rate of 1 mm/5 min followed by 1 mm/10 min for the final mm. The tip of the probe was inserted to –5.0 mm DV. While lowering, multiple layers of Metabond dental cement were applied in a circle around the probe. A female pin connected to the skull screw was connected with a male pin on the skull connector to provide an electrical ground. A thin layer of Dowsil 3-4680 (Ellsworth Adhesives, WI, USA) was applied to cover and seal the craniotomy, followed by a layer of KwikCast (World Precision Instruments, FL, USA). Multiple layers of Metabond dental cement and Loctite 454 cured with Jet Acrylic Resin Liquid were applied until the skull and skull connector were thoroughly connected.
One to seven days after surgery, animals were briefly anesthetized, the cable was attached, and the animal was allowed to freely roam a single chamber. Signal quality and neuron yield were assessed using the probe survey feature on SpikeGLX (Janelia Research Campus, VA, USA). All recordings were carried out in a custom-built Faraday cage made of aluminum wire cloth sheets (0.045” opening size) (McMaster-Carr, IL, USA) wrapped around a wooden frame sprayed with wood sealer. Faraday cage, table, camera, and all other equipment were grounded to a single common ground. All recordings were carried out in a custom partner preference chamber, laser-cut and custom-built out of antistatic acrylic sheets (Colorado Plastics, CO, USA) and placed on top of a rubber mat. Recordings were carried out using SpikeGLX, and raw data at 30 kHz were saved to disk.
An Allied Vision Mako G-158C camera (Edmund Optics, cat. # 33-514, NJ, USA) was wired using a breadboard to output a square wave sync pulse with each collected frame (60 Hz). This signal was transmitted to an analog input channel on a National Instruments BNC-2110 (NI, TX, USA) and recorded in SpikeGLX for post-hoc synchronization.
For probe explant, animals were anesthetized and mounted onto a head-fixed stereotactic frame. The main body was unscrewed from the skull connector, then an external retriever was attached to the skull connector and used to lift up the main body. The external retriever was disconnected, and the animal was euthanized. Probes were cleaned in 1% Tergazyme in deionized (DI) water (Alconox, NY, USA) overnight, rinsed in DI water for 5-10 min, dipped 3–5 times in 70% isopropyl alcohol, then reused.
Partner preference tests and cohabitation
For PPTs, partner and stranger animals were tethered to opposite ends of three-chamber acrylic arenas (76.0 cm long, 20.0 cm wide, 30.0 cm tall). Partners and strangers were randomly assigned to arena sides. Tethers consisted of an eye bolt attached to a chain of fishing swivels that slid into the arena wall. Animals were briefly anesthetized with isoflurane and attached to the tether using a zip tie around the neck. Two pellets of rabbit chow were placed in each of the three chambers, and water bottles were secured to each wall, allowing for access while tethered. After tethering partner and stranger animals, experimental animals were placed in the center chamber of the arena. At the start of the test, the opaque dividers between the chambers were removed, allowing the subject to move freely about the arena for 3 h. Overhead cameras (Panasonic WVCP304) were used to video record up to eight tests simultaneously. The movement of all three animals in each test was tracked using TopScan High-Throughput Software v3.0 (Clever Sys Inc, VA, USA). For each apparatus, chambers were manually drawn, and for each frame, contrast-based tracking was used to identify the location of the animal. Partner preference score was calculated as (partner huddle time / [partner huddle time + stranger huddle time]).
To avoid cable interference in PPTs carried out during calcium imaging or Neuropixels, no eye bolts were used, and instead, zip tie tethers were directly inserted through a hole in the corner of the arena. Neural recordings were carried out for an hour, and were terminated early if the non-focal animal exhibited persistent interference and chewing of the cable. Animals were provided water bottles and chow pellets at the conclusion of recording to the continuation of the test, to allow for a 3-h total test time. An overhead MiniCAM was used during miniscope recordings, and a Mako G-158C camera was used during Neuropixels recordings. A single partner preference test was carried out at a time.
For cohabitation, access to one end chamber of a three-chamber PPT apparatus was restricted with a divider, and the two animals were allowed to freely interact. The restricted end chamber was swapped between the two days of cohabitation. During recordings, water bottles and food pellets were removed to prevent cable tangling and chewing artifacts.
During PPTs, social interaction was defined as frames in which the experimental animal was in the partner- or stranger-containing chamber. During cohabitation, social interaction was defined as frames in which the two animals were within 5 cm of one another. These behavioral definitions were used for all neural analyses.
Calcium signal preprocessing
Imaging data were processed using the CaImAn package (v1.9.15) in Python (v3.10.8) (https://github.com/flatironinstitute/CaImAn)49. For each session, one concatenated video was cropped, spatially and temporally downsampled in half (to 15 Hz), then motion-corrected using rigid and, if necessary, piecewise rigid implementations of the NoRMCorre algorithm48. Potential seed pixels were filtered using manually selected correlation and peak-to-noise ratio values, then neuronal activity was extracted using constrained non-negative matrix factorization88. Parameters were kept as constant as possible across sessions for each animal. Putative neurons were manually accepted, rejected, or merged based on manual inspection of calcium traces. Specifically, putative neurons with abnormally shaped cell bodies or with calcium transients visually inseparable from noise were excluded. Traces were converted to ΔF/F by subtracting and dividing by the mean fluorescence of each neuron across the entire session. Prior to downstream analysis, all ΔF/F calcium traces were Z-scored and are presented in units of standard deviation.
For cross-session registration, final cell contours and the maximum intensity projection were converted into Matlab-readable indices and processed through CellReg (https://github.com/zivlab/CellReg)52, an algorithm that aligns fields of view across sessions, then models the similarity distribution of neighboring cell pairs. The outputs were converted back into a Python-readable data structure, and for visualization of cell ROIs, spatial footprints were smoothed using a Gaussian filter (sigma = 2), and thresholded at 50% of the cell’s maximum intensity to generate contours. These contours were pseudocolored for visualization.
In vivo electrophysiological preprocessing and spike sorting
Electrophysiology data were externally referenced (to ground screw) and acquired using SpikeGLX (https://github.com/billkarsh/SpikeGLX). IMec ReadOut tables were created such that on each shank, the 48 electrodes nearest to the tip were recorded. In rare cases in which noise was suspected to be due to ground screw instability, tip referencing was utilized. Preprocessing was carried out using CatGT (https://github.com/billkarsh/CatGT). Artifacts were identified as peak amplitudes (amp) of at least 0.40 mV reaching peak as rapidly as at least 0.1 mV/sample, and were replaced with zeros until the amp decreased back to 0.02 mV. Global common average referencing across all channels was implemented, and action potential data was filtered using a 12th-order Butterworth filter between 300 and 9000 Hz. Individual channels exhibiting abnormal baseline patterns were identified by eye and excluded from common average referencing and downstream spike sorting. Camera sync frames were extracted from the appropriate analog input channel using CatGT’s sync edge extractor to identify any positive deflection rising beyond 0.1 V. These positive deflections were used to synchronize physiology with behavior. The difference between the number of expected frames and the number of detected deflections was ≤3 (50 ms) in all sessions. For one animal, dropped frames were not accurately reflected in the timestamp text file, resulting in unsuccessful synchronization and exclusion from analyses involving behavior.
Preprocessed data was manually inspected for evidence of remaining artifacts. Filtered data was spike-sorted using Kilosort 2.5 using default parameters with the exception of a threshold value of (9,4) (https://github.com/MouseLand/Kilosort). Units were manually and stringently curated in Phy89. Probe trajectories were reconstructed from histology (https://github.com/petersaj/AP_histology) and converted into CCF space. Units from shanks and/or depths not localized to the NAc were removed from analysis.
Spike curation and cell type identification
High-quality single units were defined using the following criteria, and all others were excluded:
-
1.
Units showing <1% of inter-spike intervals below 1 ms.
-
2.
For units with firing rates below 1 Hz, we performed an additional statistical test to ensure the low number of refractory period violations was not due to chance. For each unit, given its firing rate (r) and a refractory period window (t = 1 ms), we calculated the expected probability of a spike occurring within the refractory period of any other spike under a Poisson process. We compared the observed number of refractory period violations to this expected number using a binomial test. Units were included only if their observed violations were significantly lower than the Poisson expectation (p < 0.05).
-
3.
Phy waveform peak-to-trough amp of more than 70 μV.
-
4.
Minimum of 150 spikes.
-
5.
Non-somatic, identified as a significant peak (prominence >1% of maximum waveform amp) preceding the trough with an amp greater than 1.2x the amp of any peak following the trough.
Units passing these criteria were then classified using the following algorithm into MSNs, FSIs, tonically active neurons (TANs), and unidentified interneurons (UINs):
-
1.
Units with firing rates above 30 Hz were automatically classified as FSIs.
-
2.
Units with narrow waveforms (trough-to-peak waveform duration <400 μs) were identified as either FSIs or unidentified interneurons (UINs) based on their propensity for long inter-spike intervals (ISIs).
-
a.
Units with a proportion of ISIs > 2 s exceeding 0.1 were classified as UINs, while those below this threshold were classified as FSIs.
-
a.
-
3.
The remaining units (waveform duration >400 μs) were classified as either MSNs or tonically active neurons (TANs) based on their post-spike suppression duration, measured as the period in the autocorrelation function during which firing rate remained below the baseline (defined as mean rate between 600–900 ms post-spike).
-
a.
Units with post-spike suppression >40 ms were classified as TANs, while those with shorter suppression durations were classified as MSNs.
-
a.
Ex vivo brain slice preparation
Methods for preparing NAc-containing brain slices were adapted from previous protocols90. Female voles (P90-P125) previously injected with AAV1/2-S5E2-hM4Di-p2A-dTomato (viral injection methods described above) were deeply anesthetized using the isoflurane drop method followed by rapid decapitation. Brains were quickly dissected and glued cerebellar side-down on a Leica VT1200 S stage and immersed in an ice-cold, carbogenated (95% O2,–5% CO2) cutting ACSF (in mM: 75 sucrose, 75 NaCl, 2 KCl, 1.25 NaH2PO4, 25 NaHCO3, 15 dextrose, 2.4 Na pyruvate, 1.3 ascorbic acid, 3 MgCl2, 0.5 CaCl2, osmolality: 300–310 mOsm/kg, pH: 7.3–7.4). Serial 300 µm coronal brain slices containing the NAc were prepared and incubated in recording ACSF (in mM: 126 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 dextrose, 1 MgCl2, 2 CaCl2, osmolality: 300–310 mOsm/kg, pH: 7.3–7.4) for 30 min at ~35 °C and at least 30 min at room temperature (RT) (~23 °C) before whole-cell electrophysiology. Slices were used for electrophysiology up to 6 h after sectioning.
Whole-cell electrophysiology
Slices were placed in a submerged slice chamber and perfused with ACSF heated to ~33 °C at a rate of 2 ml/min. Slices were visualized on an Olympus BX51WI microscope equipped with 4x (0.10NA) and 40x (0.80NA) objectives, IR-IC optics, and an Olympus BH2-RFL-T3 mercury burner for fluorescence microscopy. Neurons were visualized using a CoolSNAP EZ Camera (Photometrics) and Micro-Manager 1.4 software (Open Imaging). Whole-cell patch clamp recordings were performed using 1.5 OD borosilicate glass pipettes (3-5MΩ) (Sutter Instrument, BF150-86-10) and filled with a potassium gluconate intracellular solution (in mM: 135 potassium gluconate, 20 KCl, 10 HEPES, 0.1 EGTA, 2 MgATP, 0.3 Na2ATP; osmolality: ∼300 mOsm/kg, pH: 7.3–7.4). Signals were amplified using a MultiClamp 700B amplifier and digitized by an Axon Digidata 1550 system using pCLAMP 10.7 software (Molecular Devices). All signals were sampled at 10 kHz. In current clamp mode, compensation for voltage variations was achieved using a bridge balance circuit. In voltage clamp mode, no series resistance compensation was used, and signals were low-pass filtered at 1 kHz. Access resistance was monitored throughout the experiments using current pulses, and data were discarded with access resistance >30MΩ or where resistance varied by >30% from baseline. Liquid junction potential was calculated using LJPCalc (RRID: SCR_025044).
Intrinsic properties and evoked excitatory postsynaptic currents (eEPSCs)
Active and passive membrane properties were recorded and analyzed, similar to previous studies43. Briefly, after achieving whole-cell configuration, neurons were recorded at rest in current clamp mode (Ihold = 0pA). Following a 3 s baseline period, the holding current was linearly ramped from 0 to 400 pA over 2 s. A total of 5 sweeps of data were collected for each neuron, and the data were used to determine the resting mV and rheobase (pA). The measured resting mV was corrected for the junction potential. Following ramped current injections, neurons were brought to -60mV and voltage responses to a series of square current injections (600 ms square current injections from −200 to +400pA at 25-pA intervals) were recorded. The data collected in these experiments were used to determine if neurons were MSNs or FSIs as previously described43. Briefly, FSIs were identified based on maximum firing rate (Hz) and action potential half-width (ms) following current injection. MSNs were further distinguished from PLTS or cholinergic interneurons based on resting mV, input resistance (MΩ), lack of a prominent voltage sag, and no rebound spikes. PLTS or cholinergic interneurons were excluded.
Following intrinsic property protocols, GABAA receptors and NMDA receptors were blocked via wash-on of bicuculline methiodide (BMI) (20 μM, Sigma Aldrich, Cat#14343) and D-AP5 (50 μM, Cayman Chemical, #14539), respectively, for 5 min. Neurons were held at -70mV throughout the experiment to isolate AMPA currents. AMPA-mediated EPSCs were evoked using a glass stimulating electrode positioned in the striatum dorsally to the recording electrode within 50–200 μm and positioned 50 μm down into the slice from the surface. Input-output (I-O) curves were determined at 0.2 Hz with 1 μA stimuli up to 30 μA. Stimulus intensities that evoked EPSCs (eEPSCs) between 200-400 μA were used for the recording. The tissue was stimulated with 0.2 ms electrical pulses at 0.05 Hz for 10 min to establish a baseline period, followed by the addition of IEM-1460 (50 μM, Cayman Chemical, #15623) for 30 min. Data were binned and averaged across every three sweeps. The final 5 min of baseline were used for normalization.
Single neuron analyses and ROC prediction
For calcium imaging, neurons selective for social interaction were identified using receiver operating characteristics (ROC)50. For each neuron, we systematically varied the threshold from the minimum to the maximum Z-scored ΔF/F value. At each threshold, the calcium signal was binarized and compared to a binary vector representing the occurrence of behavioral events. True positive rate and false positive rate were computed. By computing these values across the range of thresholds, we generated an ROC curve for each neuron. The area under this ROC curve (auROC) served as a measure of how strongly each neuron’s activity was selective for the social condition (e.g., partner or stranger chamber). To determine statistical significance, each neuron’s observed auROC was compared to a null distribution of 1000 auROC values generated by applying the same procedure to randomly and circularly time-shifted versions of the original calcium matrix. A neuron was considered significantly selective if its auROC value exceeded the 95th percentile of the random distribution and significantly inhibited if its auROC value was below the 5th percentile. The same procedure was applied when using ensemble activation strengths.
For electrophysiology, spike times were binned into 50 ms intervals spanning the duration of each session. Firing rates were computed by dividing the spike count in each bin by the bin duration. The resulting rates were smoothed using a Gaussian kernel (sigma = 2 bins) to reduce high-frequency noise while preserving temporal structure.
Ensemble analyses
Ensembles were detected using an unsupervised statistical framework based on principal component analysis (PCA) followed by independent component analysis (ICA). The number of significant cell ensembles was determined based on principal components whose eigenvalues exceeded the threshold as determined by the Marčenko-Pastur distribution. First, we computed the correlation matrix C of the activity matrix Z:
We then performed eigendecomposition of C to obtain its eigenvalues λi and eigenvectors xi. The number of cell assemblies was estimated by comparing the eigenvalues to the upper bound of the Marčenko-Pastur distribution:
where q = Ncolumns / Nrows and σ² = 1 due to Z-score normalization.
The number of eigenvalues exceeding λ max provided an estimate of the number of cell assemblies. To extract ensemble patterns, we projected the calcium matrix onto the subspace spanned by the significant principal components. We then applied the fastICA algorithm to these projections to compute independent components, which represent the ensemble patterns. Ensemble activations were computed by projecting the activity matrix onto these extracted patterns, providing a bin-by-bin activation metric.
For calcium imaging, Z-scored calcium activity (at 15 Hz) was used as the activity matrices. For electrophysiology, Z-scored binned (50 ms, 20 Hz) spike rate matrices extracted only from MSNs were used as activity matrices. For all downstream analyses or visualizations involving ensemble neurons, for each ensemble, neurons were considered to be significant members of the ensemble if their absolute weight (ICA pattern coefficient) exceeded the mean weight plus two standard deviations (of all neurons’ weights).
To evaluate the influence of ensemble activity on decoding in individual neurons, we performed ensemble splicing. For each neuron that participated in one or more ensembles, periods of ensemble activation were identified based on an activation threshold (mean + 2 standard deviations of the ensemble activation trace). For each threshold crossing, a window of ± 35 frames ( ± ~2.33 s) was removed from the neuron’s activity trace. The window size was selected to minimize residual calcium activity. Spliced traces and behaviors were then used for the recalculation of behavior encoding using auROC. This procedure ensured that ensemble-driven activity was excluded while preserving non-ensemble activity for comparison.
To assess whether the number of behaviorally tuned ensembles (or neurons) predicted social preference behavior, we computed the difference in the number of ensembles (or neurons) tuned to the partner and stranger, normalized by the total number of neurons detected in each animal. A Spearman correlation was then computed between each animal’s ensemble tuning metric and its partner chamber preference, defined as the percent of total time each animal spent in the partner’s chamber.
Encoding stability analyses
To analyze stable encoding across days by individual neurons, we used neurons identified by auROC to be partner-selective neurons. Across any two individual sessions, we identified the number of cells encoding the partner in both sessions and divided by the total number of cells encoding the partner in the first of the two sessions. To estimate chance-level overlap, we performed 1000 shuffled iterations, in which cell indices in the second time point were randomly shuffled. This provided a null of the expected overlap percentage occurring by chance. As control and experimental groups could have differing baseline numbers of partner-encoding neurons, control and experimental groups were each compared to their own nulls.
Spatial clustering
To assess the spatial organization of partner or stranger-active neurons, for each cell, we identified its location using the center of mass of its calcium imaging footprint. We then calculated pairwise Euclidean distances between all neurons within each FOV. To determine whether these neurons were spatially clustered or dispersed, we compared the observed mean pairwise distance against a null distribution generated by randomly sampling an equal number of neurons from all available neurons 1000 times. For each animal, we calculated the percentile of the observed mean distance within this null distribution. Animals with fewer than 3 neurons were excluded from this analysis. Group-level statistical significance was assessed using one-sample t-tests comparing these percentiles against a null of 50th percentiles.
To assess the spatial organization of ensemble neurons, we analyzed the spatial organization of highly weighted ensemble neurons by calculating their mean pairwise distances and comparing them to null distributions generated by randomly sampling equivalent numbers of neurons from the field. For each animal, we averaged the percentiles across all ensembles to obtain a single measure of spatial organization for ensemble-participating neurons.
FSI-MSN electrophysiology analyses
To identify putative monosynaptic connectivity between neurons, only FSI-MSN pairs within 60 μm of each other were analyzed30,32,33. Cross-correlograms (CCGs) were computed using ± 50 ms windows with 1 ms bins, centered around FSI spike times. Spikes occurring ± 1 ms of the center were excluded to avoid spike sorting artifacts. Monosynaptic inhibition was established when the counts in the two bins between 1 and 3 ms post-spike were found to be less than 2 standard deviations of the counts across all bins in a ± 20 ms window. CCGs were averaged across pairs classified to exhibit or not exhibit monosynaptic inhibition. For visualization of single-pair error bars, surrogate CCGs were generated by randomly jittering spike times within a ± 50 ms window 1000 times, and the 1st and 99th percentiles of these surrogate distributions were plotted to establish confidence intervals.
For the identification of FSI to MSN ensemble connectivity, FSI bursts were identified using a thresholding procedure. Firing rate was binned into 50 ms intervals as described above, and each FSI’s activity was Z-scored. Bursts were defined as at least 3 consecutive bins in which the FSI’s activity had a Z-score of ≥2.5. To assess the temporal relationship between FSI activity and MSN ensemble activity, ensemble activation patterns were Z-scored, and burst-triggered averages were computed using ± 3 s windows centered around burst onset. To establish statistical significance, surrogate data were generated by circularly shifting each ensemble’s activity pattern by a random amount, preserving temporal structure while breaking the relationship with FSI bursts. This process was repeated 1000 times to generate a null distribution. At each time point, the actual ensemble activity was compared to the 99.9th percentiles of this surrogate distribution to identify periods of significant coordination between FSI bursts and ensemble activity. Consecutive (≥2) significant timepoints are overlaid with a red line. To evaluate directional interactions between FSI and MSN population activity, we performed Granger causality analysis. For each animal with at least 2 FSIs, raw firing rates of FSIs and MSNs were Z-scored, and the mean activity across cells was computed per population. We then applied Granger causality testing (max lag = 10, 500 ms total) in both directions: from FSI to MSN and vice versa. P-values from the F-test of each lag were collected, and the minimum p-value across all lags was used to quantify directional predictiveness. These values were transformed to –log10(p) scores for visualization and comparison. A binomial sign test was performed to determine whether the number of animals showing stronger FSI→MSN predictiveness than the reverse direction exceeded chance levels.
To analyze the contributions of FSIs to ensemble activity, periods of MSN ensemble activation were identified using 50-ms bins of Z-scored ensemble activity patterns. Activation periods were defined as sequences of ≥2 consecutive bins exceeding a Z-score of 2.5. To capture the transition from low to high ensemble activity, activity from the pre-activation and post-activation bins was extracted, and a linear regression model was fitted to predict the ensemble activity using FSI activity. Each FSI’s rate activity was normalized between 0 and 1 to prevent biases arising from differences in firing rates among FSIs. Dropout analyses were then performed, in which each FSI was sequentially excluded from the model, and the percentage change in R2 was calculated. For each ensemble, Lorenz curves were generated to visualize the cumulative percentage contribution of FSIs. Gini coefficients were calculated by comparing the observed Lorenz curve to a perfectly equal distribution.
Decoding and population analyses
Prior to PCA and analyses involving population vectors, calcium traces were temporally smoothed using a sliding mean with a 5-s window to reduce high-frequency noise while preserving the slower timescale of calcium dynamics. To characterize the complexity of neural activity patterns during social interaction, we performed PCA on calcium activity traces during defined behavioral epochs. For each animal, we extracted neural activity during social interaction bouts and computed the average activity pattern for each bout. We then applied PCA to these bout-averaged activity patterns and quantified the dimensionality of the neural representations in two ways: (1) the number of principal components required to explain 95% of the variance, and (2) the cumulative variance explained by the first three principal components. To account for different numbers of neurons across animals, we normalized the number of significant principal components by the total number of recorded neurons. Statistical comparisons were performed using linear mixed-effects models (LMMs) incorporating the relationship between normalized principal components, cumulative variance explained by the first three components, experimental group, and recording day, with individual animals as random effects.
To analyze the separation between neural states during partner versus stranger social interactions in PCA space, for each animal, we projected neural activity during both interaction types into a three-dimensional PCA space and calculated centroids for partner and stranger interaction periods. To assess whether the observed separation between these centroids was significant, we compared the actual Euclidean distance between centroids to a null distribution generated by randomly shuffling the partner/stranger labels 1000 times. This yielded a p-value for each animal based on the rank of the actual distance within the shuffled distribution. Group-level statistical significance was assessed using one-sample t-tests comparing these p-values against 0.5.
For decoding, we used a linear SVM classifier to distinguish neural activity patterns during partner versus stranger social interactions. For neuron-level decoding, population vectors were constructed using Z-scored ΔF/F values for calcium imaging and Z-scored binned spike rate values for electrophysiology, while ensemble-level decoding used ensemble activation strengths and PCA-level decoding used principal component coefficients. For ensemble decoding using electrophysiology, due to the high sampling rate, spike rates were binned into a moving window of 1 s, to better match behavioral timescales and to reduce the influence of zero values on decoding. In all cases, population vectors were constructed by averaging activity across each bout in which the animal was in the partner or stranger chamber. A stratified 10-fold cross-validation approach was used, with the data split into 90% training and 10% test sets. Single-animal example SVM plots were constructed using only the held-out samples that were used for testing. Classification performance was evaluated using F1 score, or the harmonic mean between precision and recall, in part to account for class imbalance observed in partner versus stranger interactions. To establish chance-level performance while preserving the temporal structure of the data, null distributions were generated by randomly circularly shifting the matrix of neural activity (whether neuronal, PCA, or ensemble activity) 1000 times, and repeating the classification process. Significance was assessed by comparing each animal’s decoding performance to its own null performance.
For population similarity, population vectors consisted of one Z-scored fluorescence value per neuron as described above. Cosine similarity was computed between normalized vectors to quantify the similarity of neural representations independent of magnitude and overall activity levels. Similarity was assessed within individual animals and at the group level.
Ensemble-linked behavior analysis
To determine behavioral correlates of ensemble activation, ensemble events were defined as timepoints where a given ensemble’s Z-scored activation exceeded a threshold of 2.0 for at least 0.5 s. For each detected event, we extracted a 1.5-s video segment (0.5 s before to 1 s after the peak frame of activation). These clips were manually annotated using BORIS (Behavioral Observation Research Interactive Software) with an ethogram comprising the following categories: social investigation, huddling, approach, departure, grooming, fighting, and non-social behavior. Social investigation events were further subclassified when clear and possible (e.g., nose-to-nose, nose-to-tail, nose-to-body) using modifier tags; however, due to frequent behavioral blending, only 19% of social investigation events could be reliably subclassified, and subclassifications were grouped for further analyses.
Brain Collection
Upon completion of experimental sessions, voles were transcardially perfused with 4% paraformaldehyde in phosphate-buffered saline (PBS). For calcium imaging, the head was removed and post-fixed for at least 4 days in 4% paraformaldehyde to preserve the lens tract before extracting the brain. For Neuropixels, the brain was immediately extracted. The brain was equilibrated in 30% sucrose, sectioned in 40-50 µM slices using a sliding freezing microtome (Leica, Wetzlar, Germany) and mounted on slides, then coverslipped with Krystalon (Sigma Aldrich, cat. # 64969, MO, USA) or ProLong Gold (Thermo Fisher, cat # P36930, MA, USA). Images were acquired using a fluorescence microscope (Olympus IX83) to confirm the position of lens placement and expression of GCaMP6f, or the position of the probe via the expression of DiI. For lens mapping, implant sites were manually drawn onto corresponding mouse atlas sections.
Immunohistochemistry and cell counting
For immunohistochemistry, brain sections were washed three times in PBS, then incubated in a blocking solution (PBS containing 0.5% Triton X-100 and 10% normal donkey serum) for 90 min at RT. Sections were incubated overnight at 4 °C with primary antibodies diluted in blocking buffer. Antibodies used were rabbit anti-Nkx (1:1000, Abcam cat. # ab76013, Cambridge, UQ) and rabbit anti-PV (1:500, Swant, cat. #PV27, Burgdorf, Switzerland). On the second day, sections were washed three times in PBS, then incubated in secondary antibody for 120 min at RT. Secondary antibody used was donkey anti-rabbit Alexa Fluor 647 (1:500, Invitrogen, cat. # 711-605-152, Waltham, MA). Sections were washed twice in PBS, then mounted on slides and coverslipped with Krystalon Mounting Medium (Sigma Aldrich, cat. # 64969, MO, USA). All ceramic well solutions were carried out at 400 µL per well, and all PBS washes were carried out in plastic wells at 1 mL per well for 10 min.
Using dual-channel images, a standardized grid (70 µm spacing) was applied and ROIs were selected from two slices per animal. Pseudocolors were assigned, and cell counting was performed using the channel capturing the virally-mediated fluorescent reporter. Point selections were then overlaid onto the second channel, and co-localized signals were counted and marked.
Single-nucleus RNA sequencing
Sequencing data was collected as part of a prior experiment6 (GEO # GSE255620). Gria2 ratio was calculated as the number of reads mapped to the Gria2 gene divided by the sum of reads mapped to the Gria1, Gria3, and Gria4 genes for each nucleus. Nuclei with infinite or missing values for the Gria2 ratio were excluded from the analysis. Pairwise comparisons were conducted using one-sided Mann-Whitney U-tests, with the hypothesis that PV interneurons have the lowest Gria2 ratio. P-values were corrected for multiple comparisons using the Bonferroni method.
Statistics
Animals were randomly assigned to vehicle or IEM-1460 groups, and investigators were not blinded to the behavioral condition due to the nature of the experimental design. For pharmacology, data were collected in three replicate groups of animals. For neural data acquisition, given the nature of the experiments, we collected data in groups of 1–3 animals simultaneously. All data were analyzed together upon completion of data acquisition. Target sample sizes were determined by power analysis and prior similar studies. Statistical analyses were performed at the animal level, each serving as a biological replicate (group sizes varied by experiment). All exclusion criteria were preestablished prior to data analysis, and no data were subsequently omitted from analyses.
For analysis requiring significance testing against chance conditions (such as neural encoding, decoding, spatial clustering, PCA centroid separation, burst-triggered ensemble activation), we employed non-parametric shuffling or permutation methods. These methods generate empirical null distributions while preserving temporal autocorrelations and statistical properties. Specific details of each procedure are described in the corresponding Methods sections. For group-level comparisons across conditions and timepoints, statistical significance was primarily assessed using LMMs. Where appropriate, partner/stranger was used as a within-subject factor, vehicle/IEM-1460 as a between-subject factor, and individual animals were included as random effects to account for within-animal variability. Post-hoc comparisons were performed using Welch’s two-sample t-test, one-sample t-tests, or paired t-tests. When normality was not met, Wilcoxon signed-rank tests (for paired data) or Mann-Whitney U tests (for unpaired data) were used. For physiology-behavior correlations, Spearman's rank correlation coefficients were used due to the absence of assumptions about linearity. To assess group differences in the distribution of ensemble-linked behaviors, we used multivariate analysis of variance (MANOVA), which accounts for the fact that behavior proportions within each animal sum to 1. All statistical tests were two-tailed, with a significance threshold of p < 0.05. Significance level is indicated by asterisks: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data are visualized using means ± standard errors of the mean.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data associated with this paper are deposited and publicly available through Dryad, https://doi.org/10.5061/dryad.zgmsbccq2. Source data are provided with this paper.
Code availability
All custom-written code is available at: https://github.com/donaldsonlab/NAcInhib.
References
House, J. S., Landis, K. R. & Umberson, D. Social relationships and health. Science 241, 540–545 (1988).
Snyder-Mackler, N. et al. Social determinants of health and survival in humans and other animals. Science 368, eaax9553 (2020).
Carter, C. S. & Getz, L. L. Monogamy and the prairie vole. Sci. Am. 268, 100–106 (1993).
Williams, J. Development of partner preferences in female prairie voles (Microtus ochrogaster): the role of social and sexual experience. Horm. Behav. 26, 339–349 (1992).
Sadino, J. M. et al. Prolonged partner separation erodes nucleus accumbens transcriptional signatures of pair bonding in male prairie voles. Elife 12, e80517 (2023).
Brusman, L. E. et al. Single nucleus RNA-sequencing reveals transcriptional synchrony across different relationships. Preprint at https://doi.org/10.1101/2024.03.27.587112 (2024).
Duclot, F. Sailer, L. Koutakis, P. Wang, Z. & Kabbaj, M. Transcriptomic Regulations Underlying Pair-bond Formation and Maintenance in the Socially Monogamous Male and Female Prairie Vole. Biol. Psychiatry 91, 141–151 (2022).
Tripp, J. A. et al. Comparative neurotranscriptomics reveal widespread species differences associated with bonding. BMC Genomics 22, 399 (2021).
Williams, J. R., Carter, C. S. & Insel, T. Partner preference development in female prairie voles is facilitated by mating or the central infusion of oxytocin. Ann. N.Y. Acad. Sci. 652, 487–489 (1992).
Keebaugh, A. C., Barrett, C. E., Laprairie, J. L., Jenkins, J. J. & Young, L. J. RNAi knockdown of oxytocin receptor in the nucleus accumbens inhibits social attachment and parental care in monogamous female prairie voles. Soc. Neurosci. 10, 561–570 (2015).
Liu, Y. & Wang, Z. X. Nucleus accumbens oxytocin and dopamine interact to regulate pair bond formation in female prairie voles. Neuroscience 121, 537–544 (2003).
Aragona, B. J. et al. Nucleus accumbens dopamine differentially mediates the formation and maintenance of monogamous pair bonds. Nat. Neurosci. 9, 133–139 (2006).
Borie, A. M. et al. Social experience alters oxytocinergic modulation in the nucleus accumbens of female prairie voles. Curr. Biol. 32, 1026–1037.e4 (2022).
Long, K. L. P. et al. Oxytocin receptor function regulates neural signatures of pair bonding and fidelity in the nucleus accumbens. Preprint at https://doi.org/10.1101/2024.06.23.599940 (2024).
Amadei, E. A. et al. Dynamic corticostriatal activity biases social bonding in monogamous female prairie voles. Nature 546, 297–301 (2017).
Scribner, J. L. et al. A neuronal signature for monogamous reunion. Proc. Natl. Acad. Sci. USA 117, 11076–11084 (2020).
Bernard, V., Somogyi, P. & Bolam, J. P. Cellular, subcellular, and subsynaptic distribution of AMPA-type glutamate receptor subunits in the neostriatum of the rat. J. Neurosci. 17, 819–833 (1997).
Isaac, J. T. R., Ashby, M. C. & McBain, C. J. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54, 859–871 (2007).
Plant, K. et al. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 9, 602–604 (2006).
Conrad, K. L. et al. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454, 118–121 (2008).
Huang, Y. H. et al. In vivo cocaine experience generates silent synapses. Neuron 63, 40–47 (2009).
Koya, E. et al. Silent synapses in selectively activated nucleus accumbens neurons following cocaine sensitization. Nat. Neurosci. 15, 1556–1562 (2012).
Lee, B. R. et al. Maturation of silent synapses in amygdala-accumbens projection contributes to incubation of cocaine craving. Nat. Neurosci. 16, 1644–1651 (2013).
Wolf, M. E. Targeting neuroplasticity in substance use disorders: implications for therapeutics. Annu. Rev. Pharmacol. Toxicol. 65, 259–280 (2025).
Lamsa, K. P., Heeroma, J. H., Somogyi, P., Rusakov, D. A. & Kullmann, D. M. Anti-Hebbian long-term potentiation in the hippocampal feedback inhibitory circuit. Science 315, 1262–1266 (2007).
Gittis, A. H. et al. Selective inhibition of striatal fast-spiking interneurons causes dyskinesias. J. Neurosci. 31, 15727–15731 (2011).
Manz, K. M. et al. Calcium-permeable AMPA receptors promote endocannabinoid signaling at parvalbumin interneuron synapses in the nucleus accumbens core. Cell Rep. 32, 107971 (2020).
Hong, I. et al. Calcium-permeable AMPA receptors govern PV neuron feature selectivity. Nature 635, 398–405 (2024).
Cobb, S. R., Buhl, E. H., Halasy, K., Paulsen, O. & Somogyi, P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature 378, 75–78 (1995).
Koós, T. & Tepper, J. M. Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat. Neurosci. 2, 467–472 (1999).
Wilson, C. J. What Controls the Timing of Striatal Spiny Cell Action Potentials in the Up State? In The Basal Ganglia IX. Advances in Behavioral Biology, Vol. 58 (eds Groenewegen, H., Voorn, P., Berendse, H., Mulder, A. & Cools, A.) 49–61 (Springer, New York, NY., 2009). https://doi.org/10.1007/978-1-4419-0340-2_4.
Gage, G. J., Stoetzner, C. R., Wiltschko, A. B. & Berke, J. D. Selective activation of striatal fast-spiking interneurons during choice execution. Neuron 67, 466–479 (2010).
Berke, J. D. Functional properties of striatal fast-spiking interneurons. Front. Syst. Neurosci. 5, 45 (2011).
Wright, W. J., Schlüter, O. M. & Dong, Y. A feedforward inhibitory circuit mediated by CB1-expressing fast-spiking interneurons in the nucleus accumbens. Neuropsychopharmacology 42, 1146–1156 (2017).
Yu, J. et al. Nucleus accumbens feedforward inhibition circuit promotes cocaine self-administration. Proc. Natl. Acad. Sci. USA 114, E8750–E8759 (2017).
Owen, S. F., Berke, J. D. & Kreitzer, A. C. Fast-spiking interneurons supply feedforward control of bursting, calcium, and plasticity for efficient learning. Cell 172, 683–695.e15 (2018).
Agetsuma, M., Hamm, J. P., Tao, K., Fujisawa, S. & Yuste, R. Parvalbumin-positive interneurons regulate neuronal ensembles in visual cortex. Cereb. Cortex 28, 1831–1845 (2018).
Schall, T. A., Wright, W. J. & Dong, Y. Nucleus accumbens fast-spiking interneurons in motivational and addictive behaviors. Mol. Psychiatry 26, 234–246 (2021).
Dimidschstein, J. et al. A viral strategy for targeting and manipulating interneurons across vertebrate species. Nat. Neurosci. 19, 1743–1749 (2016).
Vormstein-Schneider, D. et al. Viral manipulation of functionally distinct interneurons in mice, non-human primates and humans. Nat. Neurosci. 23, 1629–1636 (2020).
O’Hare, J. K. et al. Striatal fast-spiking interneurons selectively modulate circuit output and are required for habitual behavior. eLife 6, e26231 (2017).
Vollmer, K. M. et al. An opioid-gated thalamoaccumbal circuit for the suppression of reward seeking in mice. Nat. Commun. 13, 6865 (2022).
Gittis, A. H., Nelson, A. B., Thwin, M. T., Palop, J. J. & Kreitzer, A. C. Distinct roles of GABAergic interneurons in the regulation of striatal output pathways. J. Neurosci. 30, 2223–2234 (2010).
Wolff, J., Mech, S., Dunlap, A. & Hodges, K. Multi-male mating by paired and unpaired female prairie voles (Microtus ochrogaster). Behaviour 139, 1147–1160 (2002).
Blocker, T. D. & Ophir, A. G. A preference to bond? Male prairie voles form pair bonds even in the presence of multiple receptive females. Anim. Behav. 122, 89–97 (2016).
Chen, T.-W. et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013).
Resendez, S. L. et al. Visualization of cortical, subcortical and deep brain neural circuit dynamics during naturalistic mammalian behavior with head-mounted microscopes and chronically implanted lenses. Nat. Protoc. 11, 566–597 (2016).
Pnevmatikakis, E. A. & Giovannucci, A. NoRMCorre: an online algorithm for piecewise rigid motion correction of calcium imaging data. J. Neurosci. Methods 291, 83–94 (2017).
Giovannucci, A. et al. CaImAn an open source tool for scalable calcium imaging data analysis. Elife 8, e38173 (2019).
Li, Y. et al. Neuronal representation of social information in the medial amygdala of awake behaving mice. Cell 171, 1176–1190.e17 (2017).
Kingsbury, L. et al. Cortical representations of conspecific sex shape social behavior. Neuron 107, 941–953.e7 (2020).
Sheintuch, L. et al. Tracking the same neurons across multiple days in ca2+ imaging data. Cell Rep. 21, 1102–1115 (2017).
Zhao, P. et al. Accelerated social representational drift in the nucleus accumbens in a model of autism. Preprint at https://doi.org/10.1101/2023.08.05.552133 (2023).
Hebb, D. O. The Organization of Behavior: A Neuropsychological Theory. (Wiley, 1949).
Buzsáki, G. Neural syntax: cell assemblies, synapsembles, and readers. Neuron 68, 362–385 (2010).
Yuste, R., Cossart, R. & Yaksi, E. Neuronal ensembles: building blocks of neural circuits. Neuron 112, 875–892 (2024).
Lopes-dos-Santos, V., Ribeiro, S. & Tort, A. B. L. Detecting cell assemblies in large neuronal populations. J. Neurosci. Methods 220, 149–166 (2013).
Bakhurin, K. I., Mac, V., Golshani, P. & Masmanidis, S. C. Temporal correlations among functionally specialized striatal neural ensembles in reward-conditioned mice. J. Neurophysiol. 115, 1521–1532 (2016).
Trouche, S. et al. A Hippocampus-accumbens tripartite neuronal motif guides appetitive memory in space. Cell 176, 1393–1406.e16 (2019).
Barbera, G. et al. Spatially compact neural clusters in the dorsal striatum encode locomotion-relevant information. Neuron 92, 202–213 (2016).
Klaus, A. et al. The spatiotemporal organization of the striatum encodes action space. Neuron 95, 1171–1180.e7 (2017).
Rigotti, M. et al. The importance of mixed selectivity in complex cognitive tasks. Nature 497, 585–590 (2013).
Tye, K. M. et al. Mixed selectivity: cellular computations for complexity. Neuron 112, 2289–2303 (2024).
Remedios, R. et al. Social behaviour shapes hypothalamic neural ensemble representations of conspecific sex. Nature 550, 388–392 (2017).
Hong, I. et al. Calcium-permeable AMPA receptors govern PV neuron feature selectivity. Nature https://doi.org/10.1038/s41586-024-08027-2 (2024)
Steinmetz, N. A. et al. Neuropixels 2.0: a miniaturized high-density probe for stable, long-term brain recordings. Science 372, eabf4588 (2021).
Peters, A. J., Fabre, J. M. J., Steinmetz, N. A., Harris, K. D. & Carandini, M. Striatal activity topographically reflects cortical activity. Nature 591, 420–425 (2021).
Lee, K. et al. Parvalbumin interneurons modulate striatal output and enhance performance during associative learning. Neuron 93, 1451–1463.e4 (2017).
Gunaydin, L. A. et al. Natural neural projection dynamics underlying social behavior. Cell 157, 1535–1551 (2014).
Ma, Y.-Y. et al. Bidirectional modulation of incubation of cocaine craving by silent synapse-based remodeling of prefrontal cortex to accumbens projections. Neuron 83, 1453–1467 (2014).
Oberto, V. J. et al. Distributed cell assemblies spanning the prefrontal cortex and striatum. Curr. Biol. 32, 1–13.e6 (2022).
Neumann, P. A. et al. Cocaine-induced synaptic alterations in thalamus to nucleus accumbens projection. Neuropsychopharmacol 41, 2399–2410 (2016).
Boxer, E. E., Kim, J., Dunn, B. & Aoto, J. Ventral subiculum inputs to nucleus accumbens medial shell preferentially innervate D2r medium spiny neurons and contain calcium-permeable AMPARs. J. Neurosci. 43, 1166–1177 (2023).
Stern, E. A., Jaeger, D. & Wilson, C. J. Membrane potential synchrony of simultaneously recorded striatal spiny neurons in vivo. Nature 394, 475–478 (1998).
Carter, A. G. & Sabatini, B. L. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron 44, 483–493 (2004).
Plotkin, J. L., Day, M. & Surmeier, D. J. Synaptically driven state transitions in distal dendrites of striatal spiny neurons. Nat. Neurosci. 14, 881–888 (2011).
Day, M. et al. GABAergic regulation of striatal spiny projection neurons depends upon their activity state. PLoS Biol. 22, e3002483 (2024).
Roitman, M. F., Wheeler, R. A. & Carelli, R. M. Nucleus accumbens neurons are innately tuned for rewarding and aversive taste stimuli, encode their predictors, and are linked to motor output. Neuron 45, 587–597 (2005).
Zachry, J. E. et al. D1 and D2 medium spiny neurons in the nucleus accumbens core have distinct and valence-independent roles in learning. Neuron 112, 835–849.e7 (2024).
Zalocusky, K. A. et al. Nucleus accumbens D2R cells signal prior outcomes and control risky decision-making. Nature 531, 642–646 (2016).
Nishioka, T. et al. Error-related signaling in nucleus accumbens D2 receptor-expressing neurons guides inhibition-based choice behavior in mice. Nat. Commun. 14, 2284 (2023).
Faust, T. W., Mohebi, A. & Berke, J. D. Reward expectation and receipt differentially modulate the spiking of accumbens D1+ and D2+ neurons. Curr. Biol. S0960982225001411 https://doi.org/10.1016/j.cub.2025.02.007 (2025).
O’Donnell, P., Greene, J., Pabello, N., Lewis, B. L. & Grace, A. A. Modulation of cell firing in the nucleus accumbens. Ann. N. Y. Acad. Sci. 877, 157–175 (1999).
Goto, Y. & O’Donnell, P. Timing-dependent limbic-motor synaptic integration in the nucleus accumbens. Proc. Natl. Acad. Sci. USA 99, 13189–13193 (2002).
Hadler, M. D., Tzilivaki, A., Schmitz, D., Alle, H. & Geiger, J. R. P. Gamma oscillation plasticity is mediated via parvalbumin interneurons. Sci. Adv. 10, eadj7427 (2024).
Sosa, M., Joo, H. R. & Frank, L. M. Dorsal and ventral hippocampal sharp-wave ripples activate distinct nucleus accumbens networks. Neuron 105, 725–741.e8 (2020).
van Daal, R. J. J. et al. Implantation of neuropixels probes for chronic recording of neuronal activity in freely behaving mice and rats. Nat. Protoc. 16, 3322–3347 (2021).
Zhou, P. et al. Efficient and accurate extraction of in vivo calcium signals from microendoscopic video data. Elife 7, e28728 (2018).
Rossant, C. et al. Spike sorting for large, dense electrode arrays. Nat. Neurosci. 19, 634–641 (2016).
Willett, J. A. et al. Nucleus accumbens core medium spiny neuron electrophysiological properties and partner preference behavior in the adult male prairie vole, Microtus ochrogaster. J. Neurophysiol. 119, 1576–1588 (2018).
Acknowledgements
We thank Daniel Denman, Juan Santiago, and Jordan Hickman for assistance with Neuropixels hardware; Jessica Abazaris and the Office of Animal Resources (OAR) staff for their excellent care of the voles; the Stem Cell Research and Technology Resource Center and Light Microscopy Core Facility at the University of Colorado Boulder for microscope use and advice; Vitor Lopes dos Santos for sharing code for PCA-ICA ensemble identification; Lyle Kingsbury, Genia Kozorovitskiy, and Kareem Zaghloul for feedback on the manuscript. National Institutes of Health F30 MH131300 (MEK). National Institutes of Health T32 HL149646 (JKK). National Institutes of Health F30 MH126607 (WMS). National Institutes of Health F31 MH132278 (LEB). National Institutes of Health R01 AG064465 (CAH). National Institutes of Health R01 AG083268 (CAH). National Institutes of Health R01 NS086933 (CAH). National Institutes of Health DP2 MH119427 (ZRD). National Institutes of Health R01 MH125423 (ZRD). National Institutes of Health UF1 NS122124 (ZRD). National Science Foundation CAREER IOS-2045348 (ZRD). Publication of this article was funded by the University of Colorado Boulder Libraries Open Access Fund.
Author information
Authors and Affiliations
Contributions
M.E.K. conceived and performed experiments, developed code, analyzed data, and wrote the manuscript. J.K.K. performed patch-clamp electrophysiology experiments. W.M.S. contributed to code development and manuscript writing. O.E.N. performed histology and tissue slicing, conducted viral injections, and carried out immunohistochemistry and cell counting. K.E.W. assisted with Neuropixels implants and electrophysiological data collection. L.E.B. collected sequencing data. M.A.K. assisted with behavioral video processing. C.A.H. provided resources for patch-clamp electrophysiology. Z.R.D. conceived experiments, provided all project resources, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Lisa Topolnik, Jessica Walsh, Wenxi Zhou and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
El-Kalliny, M.M., Kushner, J.K., Sheeran, W.M. et al. Accumbal calcium-permeable AMPA receptors orchestrate neuronal ensembles underlying social attachment. Nat Commun 17, 783 (2026). https://doi.org/10.1038/s41467-025-67459-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67459-0
