
Abstract
N6-methyladenosine (m6A) is the most prevalent RNA modification and plays diverse roles in mammalian cells. However, its significance in human trophoblast lineage commitment and placenta development remains poorly understood. Here we show that METTL3, the primary enzyme responsible for m6A depositions, specifically marks cytotrophoblasts in human placenta, and exhibits defective expression in cytotrophoblasts from preeclampsia patients. Through in-depth in vitro investigation, we identify critical roles of METTL3 in generating human trophoblast stem cells (hTSCs) from human expanded potential stem cells, and in maintaining hTSCs’ self-renewal and identity. In hTSCs, METTL3 loss induces pro-inflammatory responses and promotes premature senescence, leading to preeclampsia-associated transcriptomic signature. Mechanistically, METTL3-dependent m6A methylation directly regulates histone modifiers such as EP300 and EZH2 to maintain epigenetic homeostasis, as well as various transcription factors, including JUND. Targeted inhibition of EP300 in hTSCs successfully ameliorate preeclampsia signature caused by METTL3 deficiency. These findings reveal METTL3-dependent m6A methylation as a critical regulator of human trophoblast development and its potential involvement in pregnancy-related diseases.
Similar content being viewed by others


Introduction
The placenta is a transient organ that connects the developing embryo to the maternal systems during gestation. It supports fetal growth by facilitating nutrients exchange and oxygen supply while protecting the fetus from the maternal immune system. Defective placenta function and development can result in pregnancy-associated complications, such as preeclampsia (PE)1,2, fetal growth restriction3, and intrauterine lethality and miscarriage4. The functions of the human placenta are mediated by extensive proliferation and differentiation of trophoblast cells. Three main types of trophoblast cells in the placenta have been identified: cytotrophoblasts (CTBs), syncytiotrophoblasts (STBs), and extravillous trophoblasts (EVTs). CTBs are multipotent progenitors that can differentiate into both STBs and EVTs within the rapidly developing placenta5. STBs are multinuclear and are responsible for hormone secretion, while EVTs invade the maternal decidua and remodel the maternal vasculature5. Normal pregnancy involves senescence in the placenta as a physiological process6. For instance, EVTs acquire a senescence phenotype during placentation due to their intrinsic genome amplification feature7.
Recent advances have established human trophoblast stem cells (hTSCs) derived from human embryo and placenta tissues8, as well as human pluripotent stem cells (hPSCs)9,10,11,12 or reprogramming somatic cells13. hTSCs exhibit characteristics of in vivo CTBs and serve as an in vitro model for studying human placenta development and the pregnancy complications8. Notably, bona fide hTSCs can be derived from human expanded potential stem cells (hEPSCs)11, providing a valuable resource for studying human trophoblast lineage differentiation14,15 and clinical applications such as viral infection12, disease modelling16, and drug screening17.
The importance of RNA modifications in regulating chromatin status, gene transcription, and translation for diverse cellular activities has been well documented18,19,20. N6-methyladenosine (m6A) RNA methylation is primarily catalyzed by the methyltransferase like 3 (METTL3) and 14 (METTL14) methyltransferase writer complex18,21, playing a critical role in RNA stability22,23, chromatin state24,25,26,27,28,29, and translation efficiency23,30. m6A modification is critical for development such as maternal-to-zygotic transition31,32, stem cell differentiation33,34 and hematopoiesis35,36,37. Dysregulation of m6A modification is implicated in obesity, cancer, and other diseases38. METTL3 has also been reported to regulate aging and senescence process in human cells in an m6A dependent or independent manner39,40,41. However, its role in human trophoblast lineage development under physiological and pathological conditions remains poorly understood.
In this study, we systematically investigated the functions of METTL3 and RNA m6A methylation in the human trophoblast development from hEPSCs, hTSCs, and the human placenta. Single-nucleus RNA sequence data and immunostaining results revealed that CTBs in the human placenta are specifically marked by high METTL3 and METTL14 expressions. A doxycycline-inducible knockout (iKO) system demonstrated the essential roles of METTL3 in hTSC generation, self-renewal, and maintenance of cellular identity. Notably, METTL3 acts as a gatekeeper in hTSCs for cell survival and for preventing premature senescence. Its depletion in hTSCs leads to PE-like features, and inhibiting EP300 efficiently mitigates the observed METTL3 inactivation phenotypes. Mechanistically, METTL3-dependent RNA m6A methylation directly regulates histone modifiers such as EP300 and EZH2 to maintain epigenetic homeostasis, as well as various transcription factors, including JUND. Additionally, in vivo CTBs from PE patients show reduced METTL3 expression alongside elevated H3K27ac level. Collectively, these results highlight the crucial roles of METTL3 and RNA m6A methylation in human placentation and possibly in the pathogenesis of pregnancy-related disorders.
Results
METTL3 specifically marks cytotrophoblasts in human placenta
In order to investigate METTL3 expression during pregnancy, we re-analyzed the published single-nucleus RNA sequence (snRNA-seq) data of human placenta42. The reported uniform manifold approximation and projection (UMAP) was successfully reproduced for the early and late pregnancy with the previously defined 11 cell clusters42 (Fig. 1a and Supplementary Fig. 1a). These single-nucleus data captured trophoblast cells from CTBs to mature STBs, profiling the trophoblast syncytialization and placentation process (Fig. 1a and Supplementary Fig. 1a). The analysis revealed that METTL3 and METTL14 were exclusively expressed in CTBs and proliferation-associated CTBs (pCTBs), as identified by the CTB marker PEG1043 and the absence of the STB marker gene SDC18 (Fig. 1b, c and Supplementary Fig. 1b). Interestingly, both METTL3 and METTL14 expressions significantly declined when CTBs transitioned to the competence stage for syncytialization, corresponding to clusters 11 and 9 in early and late pregnancy, respectively (Fig. 1b and Supplementary Fig. 1b). Conversely, m6A demethylases FTO and ALKBH5 showed an increased expression during STB maturation, particularly in late pregnancy (Fig. 1b and Supplementary Fig. 1b). In vivo human placental villi at 8 weeks exhibited METTL3 and METTL14 proteins exclusively in CTBs, co-localized with PEG10 but not hCG (Fig. 1d, e). In late pregnancy, METTL3 remained exclusively expressed in CTBs alongside PEG10, even though the overall CTB population was markedly diminished (Supplementary Fig. 1c, d). These findings demonstrate that METTL3 specifically marks CTBs in human placentas, suggesting its potential roles in regulating CTBs cellular identity and their differentiation capacity.

a The uniform manifold approximation and projection (UMAP) of the published snRNA-seq data of human placenta at early pregnancy (8–10 week)42. b Dot plots showing trophoblast marker genes, m6A writers, erasers, and readers expression in the snRNA-seq data. Dot sizes indicate the percentage of cells in each cluster expressing the corresponding gene, and colors indicate scale average expression level. The yellow background indicates the CTB populations during early pregnancy, and the arrowhead indicates the CTB-fusion population (cluster 11) with substantially low METTL3 and METTL14 expression relative to other CTB sub-populations. c UMAP visualization of the expression of METTL3 and METTL14, CTB marker gene PEG10, and STB marker gene SDC1. The color intensity indicates the gene expression level. d Immunofluorescence of METTL3, METTL14, hCG, and PEG10 in human placental villi of 8-week pregnancy. The white dash lines indicate the layer of CTBs. Scale bar, 20 µm. e Quantification of METTL3 and METTL14 levels in CTBs and STBs of human placental villi (8-week pregnancy). The numbers of examined CTBs were 49 for METTL3 staining and 85 for METTL14 staining; of examined STBs were 67 for METTL3 staining and 130 for METTL14 staining. Data are presented as means ± SD, and p values are from two-tailed Student’s t-test.
METTL3 and m6A methylation decrease during hTSCs to STBs differentiation in vitro
hTSCs, as an in vitro model of CTBs, can differentiate into STBs and EVTs8,11,12. In line with the in vivo observation, immunofluorescence and western blotting analysis confirmed elevated METTL3 and METTL14 expression levels in hTSCs compared to the differentiated STBs (Fig. 2a–c). Re-analysis of the published RNA-seq data on hTSCs to STBs differentiation12 also revealed a decline in the expression of METTL3 and METTL14 during the process (Fig. 2d). Additionally, several m6A reader genes, including YTHDC2, YTHDF2/3, and IGF2BP1/3, and m6A eraser gene FTO showed decreased expression (Fig. 2d). As a result, m6A level markedly decreased during STBs differentiation (Fig. 2e). These findings suggest a downregulation of the whole m6A methylation pathway during STBs development from hTSCs.

a Bright field and immunofluorescence images of METTL3, METTL14, NANOG, SOX2, GATA3, and hCG in hEPSCs, hTSCs, and STBs on differentiation day 6 (3 biological replicates). Scale bar, 50 µm. b Western blotting of METTL3, METTL14, OCT4, and hCG in hEPSCs, hTSCs, and STBs on differentiation day 6 (3 biological replicates). β-ACTIN was used as loading control. c Relative expression levels of METTL3 and METTL14 as in (b). Data are presented as means ± SD from three biological replicates. p values are from two-tailed Student’s t-test. d Heatmap from RNA-seq data12 showing relative expression levels of m6A related genes and STB marker genes in hEPSCs, hTSCs, STBs on differentiation day 2, 4, and 6. Color intensity indicates gene expression level calculated by z-score. e Quantification of m6A level by ELISA in hTSCs and STBs on differentiation day 3 and 6. Data are presented as means ± SD from three biological replicates, and p values are from two-tailed Student’s t-test. f Distribution of m6A peaks in CT-hTSCs and differentiated day 6 STBs along the 5’UTR, CDS, and 3’UTR regions. Identified peaks were mapped to the known consensus m6A motif. Motif enrichment p values were from hypergeometric test. g Venn diagram showing identified hTSCs and STBs m6A peaks. The number of genes in each category is shown in parenthesis. h Volcano plot showing differential m6A peaks in hTSCs versus STBs. Horizontal dashed line represents the threshold of absolute log2 fold change greater than 1.0. Vertical dashed line represents the threshold of adjusted p value of 0.05. p values were calculated by Wald test. IGV tracks showing MeRIP-seq results of PEG10 and MKI67 (i), TFAP2C and GATA2 (j), TNFRSF10B, GADD45G and H3C4 (k) and CGA and CGB clusters (l) relative to the input in CT-hTSCs and differentiated day 6 STBs.
To further elucidate m6A dynamics during this process, we performed methylated RNA immunoprecipitation sequencing (MeRIP-seq) on hTSCs originally derived from in vivo placental villi44 (CT-hTSCs) and their differentiated day 6 STBs. The results demonstrated a substantial decrease in m6A signals on the 3’ UTRs of transcripts in STBs compared to hTSCs (Fig. 2f). Specifically, 15,725 m6A-modified peaks were identified in hTSCs, whereas 12,449 peaks were modified with m6A in STBs, with 5660 and 2384 peaks being specific to hTSCs and STBs, respectively (Fig. 2g and Supplementary Tables 3 and 4). De novo motif analysis of these peaks revealed the classical m6A “RRACH” motif (R denotes A or G, and H denotes A, C, or U) in both cells (Fig. 2f). Additionally, 4463 peaks lost m6A modification during hTSCs differentiation to STBs, while 511 peaks gained m6A (Fig. 2h and Supplementary Data 1). For example, marker genes for hTSCs, such as PEG3, PEG10, and MKI67, exhibited m6A modification in hTSCs but showed a substantial decrease in STBs (Fig. 2h, i). Several key trophoblast master regulator genes, including GATA2, TFAP2A, and TFAP2C, also displayed m6A methylation in hTSCs, with no significant m6A level changes observed upon differentiation (Fig. 2h, j). Although most transcripts in STBs lost m6A modification, a notable number of transcripts, such as TNFRSF10B, GADD45G, and H3C4, gained m6A modification (Fig. 2h, k). Interestingly, transcripts of CGA and CGB clusters, which encode the hormone hCG secreted by STBs, lacked m6A modification in both hTSCs and STBs (Fig. 2l), suggesting hCG regulation may not involve m6A methylation during pregnancy. These results demonstrate that, despite the distinct m6A methylation profiles observed in hTSCs and STBs, both METTL3 expression level and overall m6A methylation decline during the differentiation process.
METTL3 is required in hTSCs generation from hEPSCs
To explore the role of METTL3 in human trophoblast lineages development, we established an in vitro cellular model to generate hTSCs from hEPSCs, which possesses both embryonic and extraembryonic lineage potentials11,12,45. To this end, a doxycycline (DOX)-inducible METTL3 expression system was developed in METTL3-knockout (KO) hEPSCs (Supplementary Fig. 2a). Our previous study suggested that withdrawal of DOX in hEPSCs led to METTL3 transgene expression ceasing, resulting in cell proliferation defects and failure to undergo proper differentiation14. The defective trophoblast generation was also confirmed by immunofluorescence and western blotting, which revealed the decreased expression of trophoblast master regulators GATA2 and GATA3, while NANOG, a pluripotency factor, remained expressed (Supplementary Fig. 2b–d), consistent with the constitutive Nanog expression in Mettl3-KO mouse embryonic stem cells33,46. Additionally, hCG, secreted by mature trophoblasts, was significantly reduced (Supplementary Fig. 2e). These results underscore the necessity of METTL3 in hTSCs generation from hEPSCs and suggest that loss of METTL3 causes trophoblast differentiation defects, possibly resulting from the failure of pluripotency exit.
METTL3 loss leads to defective hTSCs
METTL3 expression is significantly higher in hTSCs than in the differentiated trophoblasts (Fig. 2a–d). We next investigated whether METTL3 is required in the established hTSCs. Multiple independent hTSC lines were successfully derived from the METTL3-iKO hEPSCs in the presence of DOX (Supplementary Fig. 3a). In contrast, no hTSC lines were derived from the iKO hEPSCs where DOX was removed (Supplementary Fig. 3a). The established hTSCs exhibited typical epithelial morphology when cultured in the TSC medium containing DOX (Fig. 3a). By titrating the DOX concentration, we confirmed that 5 ng/ml DOX maintained a comparable exogenous METTL3 protein expression as found in normal hTSCs (thereafter referred as P0(+DOX)) (Supplementary Fig. 3b). Furthermore, 50 ng/ml DOX induced METTL3 overexpression (Supplementary Fig. 3b). Upon DOX removal, METTL3 protein level decreased substantially, becoming barely detected at passage 1 and not detectable in passage 2 and passage 3 (thereafter referred to P1(-DOX), P2(-DOX) and P3(-DOX)) (Supplementary Fig. 3c). Notably, the protein level of another methyltransferase METTL14, which is stabilized by METTL347, was sharply decreased upon METTL3 loss, whereas YTHDC1, an m6A reader protein, was substantially upregulated (Supplementary Fig. 3c). This compensatory effect may reflect a response to m6A loss in hTSCs. Consistent with METTL3 loss, RNA m6A level decreased after DOX withdrawal (Supplementary Fig. 3d). Interestingly, METTL3 loss substantially decreased hTSC proliferation, whereas its overexpression promoted cell proliferation (Fig. 3a, b). Eventually, P3(-DOX) hTSCs became flattened, enlarged, non-proliferative and multinucleated (Fig. 3a), reminiscent of STB differentiation12, suggesting a tendency of cell fusion following METTL3 loss (Fig. 3c, d).

a Bright field of hTSCs cultured with 50 ng/ml, 5 ng/ml DOX, or without DOX for 3 passages. The zoom image shows multinucleated cells with yellow circle indicating the boundary of a single fused multinucleated cell. Scale bar, 100 μm. b Proliferation curve of hTSCs as in (a) during 3 passages. Data are presented as means ± SD of three independent experiments, and p values are from two-tailed Student’s t-test. c Immunofluorescence of ZO-1 in P0 and P3 hTSCs. The arrowhead indicates multinucleated cells. Scale bar, 20 μm. d Fusion index as in (c). Data are presented as means ± SD from three biological replicates, and p value is from two-tailed Student’s t-test. e Transmission electron microscopy showing subcellular changes upon METTL3 loss. Blue stars represent swollen mitochondria, black stars represent apoptotic bodies, and red stars represent lysosomes (3 biological replicates). Scale bar, 500 nm. f Western blotting of cell death-associated proteins in P0 and P3, control and STM2457-treated hTSCs (3 biological replicates). g SA-β-gal staining of P0 and P3 hTSCs. Scale bar, 100 μm. h Quantification of SA-β-gal positive cells as in (g). Data are presented as means ± SD from eight views (3 biological replicates), and p value is from two-tailed Student’s t-test. i Immunofluorescence of γH2AX and 53BP1 in P0 and P3 hTSCs (3 biological replicates). Scale bar, 20 μm. j Western blotting of γH2AX in P0, P2, and P3 hTSCs (3 biological replicates). Histone 3 was used as loading control. k Volcano plots showing DEGs comparing P2 or P3 to P0 hTSCs. p values were calculated by Walt test. l GSEA depicting the upregulation of indicated pathways in P2 versus P0 hTSCs. m Experimental design of STB differentiation from hTSCs with overexpressed, normal, and deficient METTL3 expression. n Western blotting of METTL3, METTL14, and hCG as in (m) (3 biological replicates). o Quantification of hCG level in day 5 STBs as in (n). Data are presented as means ± SD from three biological replicates, and p values are from two-tailed Student’s t-test.
Subsequently, we investigated the subcellular changes in METTL3-deficient hTSCs using transmission electron microscopy (TEM), which revealed subcellular alterations linked to cell death, such as apoptosis and necroptosis, as evidenced by the presence of apoptotic bodies and swollen mitochondria resembling necrosis in P3(-DOX) hTSCs (Fig. 3e). Consistently, cell death-associated proteins RIPK1, RIPK3, and MLKL were activated upon METTL3 loss (Fig. 3f), in line with the increased TUNEL signals in P3(-DOX) hTSCs (Supplementary Fig. 3e). Notably, a significant increase in the number of lysosomes was observed in P3(-DOX) hTSCs (Fig. 3e). Elevated lysosomal mass has been associated with senescence-associated β-galactosidase (SA-β-gal) activity, a well-known hallmark of senescence6,48. As expected, P3(-DOX) hTSCs exhibited enhanced β-gal signals (Fig. 3g, h). Additionally, we confirmed the presence of higher levels of DNA damage markers γH2AX and 53BP1 in P3(-DOX) hTSCs (Fig. 3i, j). Collectively, our findings indicate that loss of METTL3 in hTSCs leads to severe defects in self-renewal capacity and cell survival, resulting in cellular senescence.
METTL3 safeguards cellular identity and differentiation potential of hTSCs
To investigate the transcriptomic changes in hTSCs upon METTL3 loss, we performed bulk RNA-seq to analyze the global gene expression profiles in P0(+DOX), P2(-DOX), and P3(-DOX) of two independent hTSC lines. Functional enrichments of genes upregulated in P2(-DOX) and P3(-DOX) hTSCs revealed pathways related to hormone activity and inflammatory response, while downregulated genes were restricted in pathways associated with cell cycle checkpoint and DNA replication (Supplementary Fig. 3f). Specifically, a series of genes involved in mature trophoblast lineages, including STB marker genes such as ERVW-1, CGA, CGBs, and PSGs and EVT marker gene HLA-G, showed significant increased expression in P2(-DOX) and P3(-DOX) hTSCs (Fig. 3k and Supplementary Fig. 3g). In addition, gene set enrichment analysis (GSEA) revealed significant enrichment of pathways including the senescence-associated secretory phenotype (SASP), inflammatory response, TNFα signaling via NF-κB, and gene signature for STB43 in P2(-DOX) compared to P0(+DOX) hTSCs (Fig. 3l). We validated the increased expression of ERVW-1 and hCG, and decreased expression of Ki67 in P2(-DOX) and P3(-DOX) hTSCs at protein levels (Supplementary Fig. 3h–j). We further employed the trophoblast organoid (TO) model, which better mimics in vivo conditions than monolayer hTSCs49, and obtained consistent results: TOs derived from P2(-DOX) hTSCs exhibited impaired cellular proliferation (Supplementary Fig. 3k) and elevated expression of STB genes (Supplementary Fig. 3l). To examine whether the functions of METTL3 in hTSCs rely on m6A RNA methylation and to further reinforce our conclusions, we treated multiple wild-type hTSC lines, including two distinct hEPSC-derived hTSC lines and one hTSC line originally derived from in vivo placental villi44, with the specific METTL3 inhibitor STM2457, which inhibits the catalytic activity of METTL350. The results demonstrated that, similar to the genetic knockout of METTL3, STM2457 treatment in multiple hTSC lines consistently led to impaired proliferation, increased cell-cell fusion events, elevated DNA damage accumulation, and hormone secretion (Fig. 3f and Supplementary Fig. 4a–n). Notably, inhibition of METTL3 did not affect the expression of key trophoblast master regulators, including GATA2, GATA3, and TFAP2C (Supplementary Fig. 4f–j).
Cellular senescence is linked to inflammation, stem cell exhaustion, and tissue dysfunction51. Despite loss of METTL3 in hTSCs increased expression of genes of mature trophoblasts, particularly STBs (Fig. 3k, l), these genes were still expressed at much lower levels compared to those in normal mature STBs (see below), suggesting precocious differentiation of the mutant hTSCs rather than genuine STB development. To test this hypothesis, we examined gene expression changes in STBs derived from P0(+DOX) and P2(-DOX) hTSCs, as well as from hTSCs overexpressing METTL3 (hTSCs treated with 50 ng/ml DOX) (Fig. 3m). Key STB marker genes, including ERVW-1, SDC1, CGA, CGBs, and PSGs, were not fully activated in STBs derived from P2(-DOX) hTSCs compared to those from P0(+DOX) hTSCs (Supplementary Fig. 4o). Western blotting analysis further confirmed the inefficient expression of hCG hormone in STBs from P2(-DOX) hTSCs (Fig. 3n, o), supporting the idea that METTL3 loss impairs stem cell identity and promotes precocious differentiation toward STBs. These findings underscore the critical roles of METTL3 in maintaining hTSC stem cell identity and normal STB differentiation.
METTL3 regulates gene expression in hTSCs through both m6A-dependent and m6A-independent mechanisms
To explore the underlying mechanisms of how METTL3 maintains the stem cell identity of hTSCs, we performed MeRIP-seq to profile m6A modification in P0(+DOX) and P2(-DOX) hTSCs. In P0(+DOX) hTSCs, m6A modification was mainly found in 3’UTR and exon regions, while in P2(-DOX) cells, significant decreases were observed in the 3’UTR regions (Fig. 4a and Supplementary Fig. 5a). These modifications in P0(+DOX) hTSCs were primarily associated with the classical ‘GGACU’ consensus motif (Supplementary Fig. 5a). In P2(-DOX) cells, we identified 6,077 genes with decreased m6A methylation (Fig. 4b). We also observed a small fraction of transcripts showing increased m6A modification following METTL3 deletion (Fig. 4b), which is likely attributed to the presence of other m6A methyltransferases. We next focused on the regions with decreased m6A methylation after METTL3 loss, as they are likely METTL3-dependent. As expected, the regions with decreased m6A modification in P2(-DOX) cells were found around the 3’UTR regions and mapped to the classical ‘GGACU’ motif (Fig. 4c). Functional enrichment of these peaks revealed that the affected genes and pathways were relevant to protein and histone modification, utero embryonic and placenta development, and DNA replication and repair (Supplementary Fig. 5b). Notably, several trophoblast master regulator genes, such as GATA2/3, TFAP2C, TEAD1/4, displayed METTL3-dependent m6A methylation in hTSCs (Fig. 4b, d, e), but did not show significant differences in expression (Fig. 4d). Further analysis confirmed that loss of METTL3 did not affect their splicing isoforms or protein levels (Supplementary Fig. 5c–e). Interestingly, two imprinting genes PEG352 and PEG1053, essential in placenta development, displayed m6A modification and their expression drastically decreased in METTL3-deficient hTSCs (Fig. 4d, e and Supplementary Fig. 5f). Nevertheless, forced expressions of PEG3 and PEG10 in METTL3-deficient hTSCs did not rescue the proliferation defect or the premature expression of STB marker genes caused by METTL3 loss (Supplementary Fig. 5g–i).

a RNA m6A modification distribution profiles in P0 hTSCs along the 5’UTR, CDS, 3’UTR, and non-coding regions. b Volcano plot showing differentially methylated mRNAs comparing P2 to P0 hTSCs. Blue and red points represent significantly hypo- and hyper-methylated transcripts, respectively. Wald test of Poisson GLM with p_cutoff =1E-10 was performed. c Distribution of peaks with decreased m6A modification in P2 versus P0 hTSCs along the 5’UTR, CDS, and 3’UTR regions. These peaks were mapped to the known consensus m6A motif. Motif enrichment p value was from hypergeometric test. d Scatter plot showing differential analysis combining RNA-seq and MeRIP-seq results comparing P2 to P0 hTSCs. Red dots represent transcripts with m6A modification, and gray dots represent transcripts without m6A modification. e IGV tracks showing MeRIP-seq results of trophoblast master regulator genes GATA2 and TFAP2C, and key imprinting genes PEG3 and PEG10 relative to the input in P0 and P2 hTSCs. f Scatter plot showing differential analysis combining RNA-seq and MeRIP-seq results comparing P2 to P0 hTSCs within three indicated gene sets. Red dots represent transcripts with m6A modification, and gray dots represent transcripts without m6A modification. g Bar chart showing the number of transcripts with and without m6A modification within three gene sets. h IGV tracks showing MeRIP-seq results of STB marker genes ERVW-1 and CGA, SASP gene GDF15, and inflammation-related gene IL4R relative to the input in P0 and P2 hTSCs.
Next, we prioritized the analysis of three gene sets: gene signature for STB43, SASP54 and the hallmark of inflammatory response (MSigDB), due to their consistent upregulation upon METTL3 depletion, as observed in Fig. 3l. By integrating RNA-seq and MeRIP-seq data, the analysis revealed that only a minor proportion of upregulated genes within these three gene sets were modified with m6A (Fig. 4f). Specifically, only 10–20% of the total mRNA in these gene sets exhibited m6A modification (Fig. 4g and Supplementary Data 2). Additionally, upon METTL3 depletion, the fold changes in expression for m6A-modified transcripts were comparable to or even smaller than those observed for non-m6A-modified transcripts (Supplementary Fig. 5j), indicating that m6A methylation is unlikely to be a driving factor behind the upregulation of these genes. For example, the STB marker genes ERVW-1 and CGA were not modified with m6A but were upregulated upon METTL3 loss (Fig. 4d, h). On the other hand, several upregulated genes did carry m6A modification, such as the SASP gene GDF15 and the inflammation-related gene IL4R (Fig. 4h and Supplementary Data 2). These findings indicate that METTL3 modulates gene expression in hTSCs through both m6A-dependent and m6A-independent pathways. Given the relatively low proportion of m6A-modified mRNA, the regulatory effects of METTL3 may be predominantly mediated via indirect mechanisms rather than direct m6A modification of target transcripts.
METTL3 orchestrates trophoblast gene network through both epitranscriptomic and histone epigenetic mechanisms
RNA m6A methylations can influence gene expression by promoting or repressing transcript stability in mammalian cells, depending on the m6A readers that recognize these modifications23,55. To further elucidate the role of METTL3 in regulating gene expressions in hTSCs, we conducted an RNA stability assay. In this assay, we used Actinomycin D to transiently inhibit global transcription, followed by RNA-seq experiments at 0 h, 2 h, 4 h, and 6 h to assess transcription dynamics in P0(+DOX) and P2(-DOX) hTSCs (Supplementary Fig. 6a). The results demonstrated that, in general, transcripts in hTSCs tended to be more stable when METTL3 was absent (Supplementary Fig. 6b), suggesting that METTL3-dependent m6A RNA methylation may contribute to RNA decay in these cells. Regarding mRNA stability changes within three gene sets, the analysis indicated that their mRNAs tended to be more stable (Supplementary Fig. 6b). However, only the STB gene set exhibited statistically significant differences (Supplementary Fig. 6b), suggesting alternative mechanisms beyond m6A modification regulating their expressions. We further categorized all m6A modified transcripts into two clusters (cluster 1 and cluster 2) based on their half-lifetimes (Supplementary Fig. 6c). Genes categorized in cluster 1 exhibited longer half-lifetimes after METTL3 loss and were enriched in pathways related to in utero embryonic and placenta development, as well as histone modification and acetylation (Supplementary Fig. 6c). In contrast, genes categorized in cluster 2 exhibited shorter half-lifetimes after METTL3 loss and were enriched in pathways associated with DNA repair and replication, and cell cycle (Supplementary Fig. 6c).
To investigate the relationship between RNA stability with transcriptomic changes, we analyzed the overlap genes in cluster 1 with those showing upregulated expression, and the genes in cluster 2 with those exhibiting downregulated expression in P2(-DOX) hTSCs from bulk RNA-seq results (Fig. 5a). The identified differential expressed genes (DEGs) (168 up vs. 38 down) were considered to be directly regulated by m6A modification through RNA decay or stabilization (Fig. 5a, b and Supplementary Data 3). The upregulated gene list included genes encoding histone modifier EP300 for histone acetylation and several enhancer-binding transcription factors (TFs) like JUND, FOSL2, and CEBPA (Fig. 5b), which displayed METTL3-dependent m6A modification and exhibited higher RNA stability in P2(-DOX) cells (Fig. 5c, d). In contrast, the downregulated gene list included EZH2, which catalyzes H3K27me3, and its transcripts exhibited reduced stability in P2(-DOX) hTSCs (Fig. 5b and Supplementary Fig. 6d, e). Furthermore, employing a single-base elongation and ligation based PCR amplification method (SELECT)56, we confirmed EP300 and EZH2 transcripts were modified with m6A in control HUES8-hTSCs, with a reduction in m6A level following STM2457 treatment (Supplementary Fig. 6f). Western blotting confirmed elevated protein expression of EP300 as well as reduced protein expression of EZH2 in P2(-DOX) and P3(-DOX) hTSCs (Fig. 5e, f). These findings align with the observed changes in histone marks: METTL3-deficient hTSCs exhibited higher H3K27ac and lower H3K27me3 levels compared to P0(+DOX) cells (Fig. 5g, h), in contrast with the rapid decline of H3K27ac in normal hTSCs to STB differentiation (Supplementary Fig. 6g, h). Western blotting analysis further confirmed increased levels of EP300, c-JUN, JUND, and H3K27ac, as well as decreased levels of EZH2 and H3K27me3 upon STM2457 treatment in normal hTSCs (Supplementary Fig. 6i, j). These data suggest that dysregulated epigenetic dynamics may contribute to the defects in METTL3-deficient hTSCs.

a Venn diagrams showing the candidates’ analysis for directly m6A-regulated transcripts. Overlapping was performed among genes with m6A, higher expression and longer half lifetimes in P2 versus P0 hTSCs (upper), or among genes with m6A, lower expression and shorter half lifetimes in P2 versus P0 hTSCs (bottom). b Heatmap showing target genes from (a) (168 upregulation and 38 downregulation). c IGV tracks showing MeRIP-seq results of EP300, CEBPA, JUND, and FOSL2 in P0 and P2 hTSCs. d The mRNA half lifetimes of EP300, CEBPA, JUND, and FOSL2 in P0 and P2 hTSCs. Data are shown as mean ± SEM from RNA-seq data (2 biological replicates). e Western blotting of EP300 and EZH2 in P0, P1, P2, and P3 hTSCs (3 biological replicates). β-ACTIN was used as loading control. f Quantification of EP300 and EZH2 levels shown in (e). Data represent the means ± SD from three biological replicates, and p values are from two-tailed Student’s t-test. g Western blotting of METTL3, H3K27me3, and H3K27ac expression levels in P0, P2, and P3 hTSCs (5 biological replicates). Histone 3 was used as loading control. h Quantification of H3K27me3 and H3K27ac levels is shown in (g). Data represent the means ± SD from five biological replicates, and p values are from two-tailed Student’s t-test. i CUT&Tag profiles of H3K27ac, H3K27me3, EP300 and JUND in P0 and P3 hTSCs. Signals were plotted over the TSS ± 5 kb. j Binding profiles of H3K27ac, H3K27me3, EP300, and JUND for three indicated gene sets in P0 and P3 hTSCs. Signals were plotted over the TSS ± 5 kb. p values are from two-sided Wilcoxon rank-sum tests. All the p values regarding the enrichments of H3K27ac, H3K27me3, EP300, and JUND within the three gene sets comparing P3 to P0 hTSCs are less than 2.2E-16. k–m IGV tracks showing CUT&Tag results of H3K27ac, H3K27me3, EP300, and JUND in STB genes PSG clusters (k), inflammation-related gene TNFRSF1B (l), and SASP gene GDF15 (m) in P0 and P3 hTSCs.
We further analyzed the epigenetic landscape of P0(+DOX) and P3(-DOX) hTSCs by comparing H3K27ac and H3K27me3 profiles through CUT&Tag. Our findings revealed a significant elevation in H3K27ac level and a corresponding reduction in H3K27me3 level in P3(-DOX) cells relative to P0(+DOX) cells (Fig. 5i), which aligns with western blotting results (Fig. 5g, h). Additionally, an elevated number of binding sites for EP300 and JUND was observed in P3(-DOX) cells (Fig. 5i). Integrative analysis of RNA-seq and CUT&Tag datasets indicated that H3K27ac enrichment and EP300 occupancy exhibit a positive correlation with gene expression level, whereas H3K27me3 deposition demonstrates an inverse correlation with gene expression in P3(-DOX) compared to P0(+DOX) hTSCs (Supplementary Fig. 7a–f). As anticipated, H3K27ac, EP300, and JUND displayed increased enrichment across the STB, SASP, and inflammation gene sets upon METTL3 depletion, whereas H3K27me3 exhibited an inverse trend (Fig. 5j), indicating the pivotal roles of TFs and histone modifications in regulating gene expression in hTSCs through METTL3.
To further evaluate the direct regulatory role of m6A modification in hTSCs, we analyzed the overlap of genes within the STB, SASP, and inflammation gene sets that were co-modified by m6A, H3K27ac, and H3K27me3. The results demonstrated that nearly all m6A-modified genes in these gene sets also exhibited concurrent modifications by H3K27ac and/or H3K27me3 (Supplementary Fig. 7g). Notably, approximately two-thirds of the m6A-modified genes were simultaneously co-regulated by both H3K27ac and H3K27me3 (Supplementary Fig. 7g), suggesting a complex interplay of epitranscriptomic and histone epigenetic mechanisms in gene regulation within these pathways. For instance, STB genes within the PSG clusters and inflammation-associated genes such as TNFRSF1B did not exhibit m6A modification but were upregulated following METTL3 depletion (Supplementary Data 2). These genes showed increased enrichment of H3K27ac, EP300, and JUND, concomitant with decreased H3K27me3 deposition in P3(-DOX) hTSCs relative to P0(+DOX) hTSCs (Fig. 5k, l). A similar pattern was also observed in SASP-related genes, including SERPINE1 and VGF, as well as inflammation-associated genes such as IL6 (Supplementary Fig. 7h, i), indicating an indirect regulatory effect mediated through m6A methylation. Conversely, GDF15, a marker gene for STB and a component of SASP that was modified with m6A, exhibited increased mRNA stability upon METTL3 KO (Supplementary Data 2), along with elevated enrichment of H3K27ac, EP300, and JUND (Fig. 5m). A comparable trend was also observed in the inflammation-related gene RIPK2 (Supplementary Fig. 7j). Collectively, these results indicate that METTL3 orchestrates trophoblast gene regulation via both direct m6A-dependent mechanisms and indirect epigenetic pathways involving EP300-mediated H3K27ac and EZH2-mediated H3K27me3, with the histone epigenetic pathways playing a more prominent role.
Efficient rescue of METTL3-KO hTSC defects by chemical manipulation of histone modifications
We further asked whether correcting the dysregulated epigenetic modifications, specifically H3K27me3 and H3K27ac, could correct the defective gene expression in METTL3-KO hTSCs (Fig. 6a). As anticipated, treatment with the EP300 inhibitor A-48557 in P3(-DOX) hTSCs reduced H3K27ac level and significantly decreased the abnormal expression of STB proteins, including hCG and SDC1 (Fig. 6b–d). Previous studies have shown that PRC2 mediated H3K27me3 acts as a barrier to trophoblast lineage induction from human embryonic stem cells (hESCs)58,59, but the role of H3K27me3 in trophoblast lineage maintenance is yet understood. Initially, we treated normal hTSCs with the EZH2 inhibitors, UNC199959 or EPZ643860, for several passages, and the results were similar to those observed in hTSCs upon METTL3 loss (Supplementary Fig. 8). This included decreased proliferation, increased hCG hormone secretion, and enhanced cell-cell fusion (Supplementary Fig. 8a–e), indicating the essential roles of H3K27me3 for preserving cell identity of hTSCs. Since efficient EZH2 activators were unavailable, we used a KDM6B inhibitor, GSK-J161, to increase the H3K27me3 level in P3(-DOX) hTSCs (Fig. 6a). Immunostaining and western blotting analysis demonstrated that GSK-J1 treatment in P3(-DOX) hTSCs led to a slightly higher H3K27me3 level and significantly suppressed the expression of the STB proteins (Fig. 6b–d). These findings collectively demonstrate that chemical inhibition of H3K27ac or reinforcement of H3K27me3 could efficiently rescue the defective gene expression in METTL3-KO hTSCs.

a Schematic diagram showing the rescue experiments of treating P3 hTSCs with EP300 inhibitor A-485 (10 μM) or KDM6B inhibitor GSK-J1 (50 μM) for 48 h before analysis. b Immunofluorescence of SDC1 and hCG in DMSO, A-485 and GSK-J1-treated P3 hTSCs. Scale bar, 100 μm. c Percentage of hCG-positive cells as in (b). Data are presented as means ± SD from three biological replicates, and p values are from two-tailed Student’s t-test. d Western blotting of hCG, H3K27ac, and H3K27me3 in DMSO, GSK-J1, or A-485-treated P3 hTSCs (3 biological replicates). GAPDH and histone 3 were used as loading control. e Heatmap showing expression of severe PE marker genes64 in P0, P2, and P3 hTSCs, and in A-485-treated and control P3 hTSCs. Color intensity indicates the gene expression level calculated by z-score. f Enhancer activity prediction of transposable element (TE) families comparing P2 to P0 hTSCs. TE enhancer activity was predicted using a linear regression model67, and p value was established by null significance hypothesis and Benjamini-Hochberg procedure. g Profiles of ATAC-seq and CUT&Tag for H3K27ac and JUND on LTR10A loci from P0 and P3 hTSCs. h GSEA of PE signature comparing A-485-treated to DMSO-treated P3 hTSCs. Gene list for PE signature was obtained from the published study64. i IGV tracks showing ATAC-seq and CUT&Tag signals in PE-associated gene ATG9B, which contains a nearby LTR10A locus (red mark) as a putative enhancer with higher H3K27ac, EP300, and JUND deposition, and increased chromatin accessibility upon METTL3 loss. j RT-qPCR of LTR10A expression in control (Cas9 and non-target sgRNA) P3 hTSCs and P3 hTSCs with LTR10A excision (Cas9 and LTR10A-sgRNA). Data are presented as means ± SD from three biological replicates, and p values are from two-tailed Student’s t-test. k RT-qPCR of ATG9B and ENG expression in control P3 hTSCs and P3 hTSCs with LTR10A excision. Data are presented as means ± SD from three biological replicates, and p values are from two-tailed Student’s t-test.
METTL3-KO hTSCs exhibit preeclampsia molecular signatures, and inhibition of EP300 alleviates these features
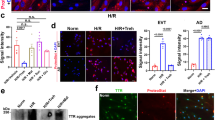
Premature trophoblasts and defective CTBs in the placenta are associated with preeclampsia (PE)62,63,64. Previous study has shown a widespread increase in H3K27ac occupancies in CTBs from patients with severe PE (sPE)65. Therefore, we tested whether METTL3 loss in hTSCs would induce PE-like signatures. Analysis of the published data64 revealed that genes highly expressed in the CTBs from sPE patients also showed higher expression levels in P3(-DOX) vs. P0(+DOX) hTSCs (Fig. 6e). Furthermore, following METTL3 KO in hTSCs, LTR10A, a transposable element (TE) associated with PE due to its hyperactivation66, exhibited increased enhancer activity indicated by the TE enhancer predicting linear regression model67 (Fig. 6f). Notably, LTR10A loci in P3(-DOX) hTSCs exhibited increased chromatin accessibility, elevated H3K27ac level, and increased JUND occupancies (Fig. 6g). Higher H3K27ac level and hyperactivation of LTR10A in both sPE CTBs and METTL3-deficient hTSCs prompted us to speculate that the inhibition of EP300-mediated H3K27ac might ameliorate the PE signature caused by METTL3 loss. Treatment with A-485 in P3(-DOX) cells effectively mitigated the PE signature, as evidenced by the reduced expression of genes that are enriched in CTBs from sPE (Fig. 6e, h). We further genetically deleted specific LTR10A loci in hTSCs and evaluated the nearby PE related gene expression. Genes such as ENG65,66 and ATG9B68, which contain the LTR10A element in their enhancer regions (Fig. 6i) and are associated with PE, were selected for analysis. Due to the challenges in clonally expanding hTSCs from single cells, we employed a population-wide approach and demonstrated over 50% deletion efficiency across various targets and experiments by examining LTR10A expression (Fig. 6j). Excising LTR10A loci significantly reduced the expression of ENG and ATG9B in P3(-DOX) hTSCs (Fig. 6k). In summary, our study indicates that loss of METTL3 in hTSCs induces PE transcriptomic signature, which can be effectively alleviated by inhibiting EP300-mediated H3K27ac, offering a potential therapeutic target for PE treatment.
The METTL3-EP300-H3K27ac axis is a key regulator of trophoblast development and is involved in the pathogenesis of PE during placentation
We finally investigated the correlations between METTL3 and histone modifications H3K27ac and H3K27me3 in human placentas in vivo. The results demonstrated that CTBs expressing METTL3 exhibited significantly lower level of H3K27ac compared to METTL3-negative trophoblasts (Fig. 7a, b). Notably, a subset of mature STBs lacked H3K27ac expression despite also being negative for METTL3 (Fig. 7a), indicating that H3K27ac may have stage-specific regulatory roles during trophoblast differentiation. Conversely, no significant correlation was observed between METTL3 expression and H3K27me3 level (Fig. 7a, b).

a Immunofluorescence of METTL3 with H3K27ac or H3K27me3 in human placental villi in 8-week pregnancy. White dash lines indicate the layer of CTBs. Yellow dash circle represents mature STBs with low H3K27ac expression. Scale bar, 20 µm. b Fluorescence intensity to area of H3K27ac and H3K27me3 in METTL3-positive and -negative trophoblasts as in (a). The numbers of quantified METTL3-positive cells were 122 (H3K27ac) and 116 (H3K27me3), and METTL3-negative cells were 144 (H3K27ac) and 147 (H3K27me3). Data are presented as means ± SD (two-tailed Student’s t-test). c Immunofluorescence of METTL3 and PEG10 in normal and PE human placental villi in term pregnancy. Yellow arrowheads indicate PEG10-positive CTBs in normal placentas co-localized with METTL3 expression. White arrowheads denote PEG10-positive CTBs in PE placentas with lower or undetectable METTL3 expression. Scale bar, 20 µm. d Percentage of METTL3-positive CTBs in normal and PE placental villi as in (c). Quantifications were from two independent experiments with 10 views for normal and 15 views for PE. Data are presented as means ± SD (two-tailed Student’s t-test). e Fluorescence intensity to area of METTL3 in PEG10-positive CTBs as in (c). 48 cells in normal group and 40 cells in PE group (first batch), and 48 cells in normal and 41 cells in PE group (second batch) were quantified. Data are presented as means ± SD (two-tailed Student’s t-test). f Immunofluorescence of H3K27ac and PEG10 in normal and PE human placental villi in term pregnancy. Yellow arrowheads indicate PEG10-positive CTBs in normal placentas, co-localized with lower H3K27ac expression. White arrowheads denote PEG10-positive CTBs in PE placentas with higher H3K27ac level. Scale bar, 20 µm. g Fluorescence intensity to area of H3K27ac in PEG10-positive CTBs as in (f). 52 cells in each group (first batch), and 53 cells in each group (second batch) were quantified. Data are presented as means ± SD (two-tailed Student’s t-test).
Our in vitro data demonstrated that METTL3-KO hTSCs acquired PE-associated signatures at the transcriptomic level. Given the significantly elevated H3K27ac level observed in CTBs of PE patients65 and the role of METTL3 in regulating EP300-mediated H3K27ac, we hypothesized that METTL3 deficiency may exist in PE CTBs. To investigate this, we analyzed two PE placentas from 35- and 37-week pregnancies and included two gestational age-matched normal placentas from 38-week as controls. The results demonstrated a significant reduction in the proportion of METTL3-positive trophoblasts in PE placentas compared to normal controls (Fig. 7c, d). More importantly, METTL3 expression was markedly decreased in PEG10-positive CTBs within PE placentas relative to those in normal placentas (Fig. 7c, e). Notably, a substantial subset of PEG10-positive CTBs in PE placentas lacked detectable METTL3 expression (Fig. 7c). Consistent with the previous study65, H3K27ac level was significantly elevated in PE CTBs (PEG10-positive cells) compared to normal controls (Fig. 7f, g). These findings suggest that the abnormal increase in H3K27ac level in PE CTBs may result from deficiencies in METTL3 expression, indicating a potential mechanistic link between METTL3 downregulation and epigenetic dysregulation in PE pathology.
Discussion
Normal human placentation and trophoblast development are not only important for the growth and development of the fetus but also crucial for the mother’s health69. This study utilized hEPSCs and hTSCs as in vitro models to investigate the roles of METTL3-mediated m6A RNA methylation in hTSC generation, identity maintenance, and differentiation. Our findings reveal that METTL3-mediated m6A RNA methylation is critical for deriving hTSCs from hEPSCs and preserving their cellular identity. Inactivation of METTL3 in the established hTSCs leads to loss of cell proliferation, increased cell death, and premature differentiation.
Previous studies have extensively explored m6A methylation in embryos and embryonic stem cells, highlighting its critical roles in pluripotency regulation and cell fate determination14,25,31,33,70,71. Using a METTL3-iKO system, we demonstrated here that METTL3 is required for deriving hTSCs from hEPSCs and for differentiating into trophoblast lineages. This finding aligns with the prior research showing that METTL3 knockdown in hESCs impairs differentiation potential33. A recent report suggested that DNA and RNA methylations could coordinately fine-tune gene expression during ESC differentiation72. It is worth investigating whether DNA methylation is also involved in the differentiation from hEPSCs to hTSCs, besides RNA methylation.
During human placentation, trophoblasts exhibit dynamic epigenomic regulation to ensure proper gene expression for cellular identity and functions60,65,73. Dysregulated epigenomes are implicated in pregnancy-associated disorders, such as excessive H3K27ac upregulation in CTBs, which is a hallmark of severe PE65. We found that METTL3-dependent m6A methylation safeguards normal epigenomes in hTSCs by directly regulating the turnover of transcripts for key histone modifiers, such as EP300 and EZH2, thereby shaping the hTSC epigenetic landscape. METTL3 depletion disrupts H3K27ac regulation in hTSCs, which is associated with a PE signature (Fig. 8). These epigenetic dysregulations lead to hyperactivation of inflammatory responses, SASP, and the emergence of PE signatures in vitro. We further discovered that A-485, a selective catalytic p300 inhibitor57, effectively mitigates PE signatures in METTL3-deficient hTSCs, offering novel insights into the pathology of pregnancy-associated disorders and suggesting potential therapeutic strategies for PE. Meanwhile, we do not exclude the potential involvement of additional histone modifications, such as H3K4me3 and H3K9me3, in the regulation of gene expression in hTSCs by METTL3. In particular, H3K9me3-mediated heterochromatin has been reported to be directly regulated by METTL327,28.

During normal placentation in vivo, CTBs exhibit elevated METTL3 expression (+++) and moderate level of H3K27ac (+), facilitating appropriate trophoblast proliferation and differentiation. In placentas affected by PE, CTBs display a reduction in METTL3 expression (+) concomitant with a marked increase in H3K27ac expression (+++). In vitro, hTSCs express METTL3, which catalyzes m6A modification on target transcripts such as EP300, JUND, and EZH2, ensuring their proper turnover. These transcripts are crucial for H3K27ac and H3K27me3 depositions, maintaining hTSCs identity and safeguarding their differentiation potential. Conversely, loss of METTL3 in hTSCs disrupts epigenomic homeostasis, potentially leading to premature senescence, activation of inflammatory pathways, and the emergence of an in vivo PE-like signature.
Programmed cellular senescence is a natural process during human placentation7,74, while premature placental aging is commonly observed in PE63,75. METTL3 has been shown to combat aging through both m6A-dependent and independent mechanisms in various cells and tissues39,40,41,76. Our results suggest that METTL3-dependent m6A modification directly regulates the stability of transcripts for AP-1 family members like FOSL2 and JUND. Dysfunctional enhancer landscapes and AP-1 depositions have been linked senescent cell fate77,78, which consistent with our observations of the close association between H3K27ac and JUND depositions and the SASP pathway in METTL3-deficient hTSCs. If RNA m6A dysfunction occurred in CTBs, it would be expected to lead to stem cell exhaustion in the entire organ, resulting in premature aging and pregnancy-associated disorders. As anticipated, our examination of PE placentas revealed a marked reduction in METTL3 expression in CTBs, which may play a contributory role in the underlying pathogenesis of PE. However, our in vitro results indicate that the inhibition of EP300 or AP-1 did not revert premature senescence phenotype upon METTL3 loss (data not shown), underscoring the EP300-independent role of METTL3 in resisting premature aging and maintaining self-renewal and cell identity in hTSCs. Notably, m6A RNA methylation has been demonstrated to regulate essential biological processes, including mitochondrial function in neuronal cells79 and glycolytic metabolism in cancer cells80. These pathways may contribute to the proliferation defects observed in hTSCs upon METTL3 depletion, indicating potential mechanistic links that merit further comprehensive investigation.
In summary, this study demonstrated that METTL3 plays a critical role in hTSCs generation from hEPSCs, maintaining their stem cell identity, and regulating their differentiation (Fig. 8). Our findings thus reveal m6A RNA methylation as a key regulator of human trophoblast development in vitro and in vivo and suggest its potential role in pregnancy-related diseases like PE. Further investigation utilizing mouse model of PE and METTL3 activator81 can be performed to examine the potential and safety of targeting METTL3 as a therapeutic treatment.
Methods
Ethical statement
The em1-hEPSC line was derived from human preimplantation embryos donated for research45. All the donors gave written informed consents for derivation of EPSC from their spare embryos after completed family through assisted reproduction. The derivation protocols were approved by the Institutional Review Boards of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (IRB number: UW 18-017) and the Council of Reproductive Technology, Hong Kong (research licence number: R5004). The human placental specimens used in this study of immunohistochemistry (IHC) were collected from two early pregnant women (8 weeks of pregnancy), two healthy women with normal-term pregnancies (38 weeks) and two pregnant women diagnosed with PE. For the PE patients, placental tissues were collected from term placenta of 35-week and 37-week respectively. PE was diagnosed according to the following criteria: new-onset hypertension (systolic blood pressure of 140 mm Hg or more or diastolic blood pressure of 90 mm Hg or more on two occasions at least 4 h apart after 20 weeks of gestation), and proteinuria: 300 mg or more per 24 h urine collection (or this amount extrapolated from a timed collection). All placental specimens were collected from Weifang People’s Hospital. Experiments using the placental specimens were reviewed and approved by the Ethics Committee of Weifang People’s Hospital under research license KYLL20230106-1 and KYLL20250826-4. Donors have signed the informed consent.
Culture of hEPSCs and hTSCs
Em1-hEPSCs were maintained on SNL 76/7 feeder layers and enzymatically passaged with TrypLE (Thermo Fisher Scientific, 12604013) at a four-day interval at a 1:4 to 1:8 ratio. The hEPSC medium (hEPSCM) was prepared based on a previous study with slight modification11. This medium is N2B27-based, supplemented with four inhibitors: 2.5 μM XAV939 (Tocris, 3748), 2.5 μM endo-IWR1 (Tocris, 3532), 1 μM CHIR99021 (Tocris, 4423), and 0.1 μM A419259 (Tocris, 3914). The N2B27 basal medium consists of a 1:1 mixture of DMEM/F12 (Thermo, 21331020) and Neurobasal medium (Thermo, 21103049), supplemented with 200 × N2 and 100 × B27, 1% ITS-X (Thermo Fisher Scientific, 5150056), 50 μM β-mercaptoethanol, 1% Penicillin-Streptomycin-Glutamine (Thermo Fisher Scientific, 15140163), 1% non-essential amino acid, 50 μg/ml Vitamin C (Sigma, A8960), and human leukaemia inhibitory factor (hLIF, 10 ng/ml). Primed state HUES8 hESCs were cultured in mTeSR1 (STEMCELL Technologies, 85850) medium on plates pre-coated with 1% Matrigel (Corning, 354230). To convert these cells into hEPSCs, primed HUES8 cells were passaged into hEPSCM and maintained for more than 10 passages, during which undifferentiated colonies were manually selected to establish stable hEPSC lines.
hTSCs derived from hEPSCs (em1-hTSCs and HUES8-hTSCs) or from in vivo placenta villi44 (CT-hTSCs) are maintained in hTSC medium (hTSCM) as described in the published study with slight modification8. hTSCM is made up of DMEM/F12 containing 0.5% penicillin-streptomycin, 1% foetal bovine serum, 1% ITS-X, 0.3% bovine albumin fraction V (Thermo Fisher Scientific, 15260037), supplemented with 50 μg/ml Vitamin C, 50 μg/ml human EGF (Peprotech, AF-100-15), 2 μM CHIR99021, 0.5 μM A-83-01 (Tocris, 2939), 1 μM SB431542 (Tocris, 1614), 10 μM valproic acid (Tocris, 2815), and 2.5 μM Y27632 (Selleckchem, S1049). Routine passage of hTSCs was conducted every four days by enzymatic digestion using TrypLE. Cells were resuspended and seeded in hTSCM on cell culture plate coated with 1% Matrigel at a split ratio of 1:3. hTSCM was refreshed every two days during culture. All cell cultures were maintained at 37 °C with 5% CO2.
Gene editing in hEPSCs and hTSCs
To generate METTL3-iKO hEPSCs, an exogenous METTL3 expression cassette was first constructed by inserting METTL3 coding sequence into Piggybac Tet-On plasmid. This plasmid was transfected into hEPSCs via electroporation using Neon transfection system (Thermo Fisher Scientific) with following parameters: 1100 V, 30 ms, and 1 pulse. Single colony formation was performed to obtain hEPSC lines with the exogenous METTL3 expression cassette. Subsequently, an sgRNA targeting exon 1 of METTL3 was cloned into the plasmid pKLV2-U6gRNA5(BbsI)-PGKpuro2ABFP-W (Addgene #67974) and co-transfected with the plasmid pKLV2-EF1a-Cas9Bsd-W (Addgene #68343), encoding Cas9, into hEPSCs using the same electroporation parameters. Transfected cells were cultured in hEPSCM with DOX unless otherwise specified. One day post-electroporation, cells were selected with 10 µg/ml blasticidin for 24 h, followed by selection with both 10 µg/ml blasticidin and 2 µg/ml puromycin for 48 h. Selected cells were then subjected to single colony formation to establish individual METTL3-iKO hEPSC lines. The loss of METTL3 in the selected iKO cell lines was verified by sanger sequencing of METTL3 alleles and by western blotting after culturing in hEPSCs without DOX. DOX was used at a concentration of 5 ng/ml unless otherwise specified. Primers used for sgRNAs are listed in Supplementary Table 1.
To overexpress PEG3 and PEG10 in METTL3-iKO hTSCs, cDNA of PEG3 and PEG10 was amplified through PCR and cloned into piggybac backbone and transfected into hTSCs via electroporation using Neon transfection system with following parameters: 1200 V, 40 ms, and 1 pulse. Expression of exogeneous PEG3 and PEG10 was driven by CAG promoter. The empty piggybac backbone was transfected as the control. At 24 h after transfection, drug selection was performed with 5 µg/ml blasticidin for two days. Selected cells were then used for subsequent experimental analysis.
To excise specific LTR10A loci in hTSCs, corresponding sgRNAs were cloned into the plasmid pKLV2-U6gRNA (BbsI)-PGKpuro2ABFP-W (Addgene #67974). Next, plasmids encoding the sgRNAs were co-transfected with the plasmid pKLV2-EF1a-Cas9Bsd-W (Addgene #68343) which encodes the Cas9 protein via electroporation using Neon transfection system (Thermo Fisher Scientific) with following parameters: 1200 V, 40 ms, and 1 pulse. Non-target sgRNA was transfected side-by-side as the control. At 24 h after transfection, cells were selected with 1 µg/ml puromycin and 5 µg/ml blasticidin for 2 days. DNA and RNA were extracted from the resultant cell populations and genotyped for the presence of excisions. Successful excision pools were further analyzed for the percent of excised alleles using RT-qPCR. Upon DOX removal for 3 passages, hTSCs with LTR10A excision were harvested for the expression analysis of genes nearby the targeted LTR10A using RT-qPCR. Primers of sgRNAs and primers used for genotyping and RT-qPCR are listed in Supplementary Table 1.
Differentiation of trophoblast lineage and derivation of stable hTSC lines from hEPSCs
For trophoblast lineage differentiation, METTL3-iKO hEPSCs with DOX or without DOX were harvested using TrypLE for 3 min and seeded in hTSCM on cell culture plate pre-coated with 1% Matrigel with a cell density of 10,000 cells per 6-well plate. Cells were collected at indicated timepoints for downstream experiments and analysis.
For the derivation of stable hTSC lines, 5 ng/ml DOX was continuously added to the hTSCM throughout trophoblast differentiation and establishment of hTSCs. By approximately day 8–10 of differentiation from hEPSCs, characteristic hTSC-like epithelial colonies were manually selected and transferred to individual wells of a 96-well plate pre-coated with 1% Matrigel, designated as passage 1 (P1). Some colonies were able to adhere to the plate and proliferate, while others did not. The proliferating hTSC colonies were dissociated into single cells using TrypLE and passaged at a ratio of 1:1–1:2 for early passages. The established hTSC lines were further characterized through gene expression analysis and assessment of their differentiation potential. DOX was used at a concentration of 5 ng/ml unless otherwise specified.
Differentiation of STBs from hTSCs
Differentiation of STBs from hTSCs was preformed according to the published study with minor modifications8. hTSCs were seeded in a 6-well plate pre-coated with 1% Matrigel at a density of 2 × 105 cells per well and cultured in 2 ml of STB medium: DMEM/F12 supplemented with 1% ITS-X, 0.5% penicillin-streptomycin, 0.3% bovine albumin fraction V, 0.1 mM β-mercaptoethanol, 4% KnockOut serum replacement, 2.5 μM Y27632, and 2 μM Forskolin (Tocris, 1099). The medium was refreshed every 2 days, and cells were analysed at indicated time points.
Cell proliferation assay
2 × 105 METTL3-iKO hTSCs (routinely cultured with 5 ng/ml DOX and referred as P0) were seeded in 6-well plates and cultured with 50 ng/ml DOX, 5 ng/ml DOX or without DOX. Cells were counted every 4 days and passaged into new 6-well plates with a ratio of 1:3. Number of cells was counted 3 times at passage 1, 2, and 3 (P1, P2, and P3) for each group.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, 15596026) following the manufacturer’s protocol and converted to cDNA using PrimeScript™ RT Master Mix (Takara, RR036A). Quantitative PCR (qPCR) was performed using iTaq™ Universal SYBR® Green Supermix (Biorad) with CFX Opus Real-Time PCR Systems (Biorad). The 2−∆Ct method with GAPDH as the endogenous control was used to calculate the relative gene expression. Primers of RT-qPCR are listed in Supplementary Table 1.
Immunofluorescence staining
Cells were fixed with freshly prepared 4% paraformaldehyde (Sigma, 158127) solution, permeabilized in 5% FBS/PBS supplemented with 0.1% Triton X-100 (Sigma, X100) for 30 min and blocked with 5% FBS/PBS for 2 h. Primary antibodies were diluted in blocking buffer according to the manufacturer’s protocols and incubated overnight at 4 °C. Signals of primary antibodies were detected using AlexaFluor-conjugated secondary antibodies, and cell nuclei were counterstained by DAPI. Images were acquired using a Zeiss LSM800 confocal laser scanning microscope. Antibodies used in this study are listed in Supplementary Table 2.
Immunohistochemistry (IHC)
The collected samples were immediately fixed in 4% paraformaldehyde for 24 hours at 4 °C. Subsequently, the samples were dehydrated through a graded ethanol series (70%, 80%, 90%, 100%), cleared in xylene, and embedded in paraffin through two cycles of immersion in molten wax (60 °C, 1 h each). 5 μm-thick sections were cut from the paraffin blocks, followed by deparaffinization in xylene and rehydration through a descending ethanol gradient (100%, 95%, 80%, 70%). Antigen retrieval was performed by heat-mediated epitope restoration in citrate buffer (pH 6.0, Beyotime Biotechnology, Cat# P0083) using a pressurized decloaking chamber (121 °C, 2 min). Sections then were blocked with 5% bovine serum albumin (BSA) and incubated with corresponding primary antibody at 4 °C overnight. After incubation, sections were washed three times (5 min each) with PBS, followed by incubation with fluorescence-conjugated secondary antibody for 1 h at room temperature, and cell nuclei were counterstained by DAPI. Images were acquired using a Zeiss LSM800 confocal laser scanning microscope. Antibodies used in this study are listed in Supplementary Table 2.
Western blot
For western blot analysis, one million cells were lysed using RIPA buffer (Thermo Fisher Scientific, 89900) supplemented with a protease inhibitor cocktail (Roche, 11697498001). SDS-PAGE was performed using 10% TGX polyacrylamide gels (Biorad, 1610173) with a thickness of 1.0 mm, followed by transfer onto Immun-Blot PVDF Membrane (Biorad, 1620177) using Trans-Blot Turbo Transfer System (Biorad). Membrane was blocked with either 5% non-fat dry milk (Cell Signalling Technology, #9999) or 5% BSA in TBST for 2-hour incubation at room temperature with gentle shaking. Transferred membranes were incubated with primary antibodies overnight at 4 °C and were detected using HRP-conjugated antibodies. Images were developed using SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) and captured using iBright™ FL1500 Imaging System (Thermo Fisher Scientific). Antibodies used in this study are listed in Supplementary Table 2. Unprocessed scans of all the blots were provided in the Source data.
Transmission electron microscopy (TEM)
Approximately 300,000 cells per ml of trypsinized P0(+DOX) and P3(-DOX) hTSCs were fixed using electron microscopy fixative (4% paraformaldehyde) at 4 °C overnight. The sample preparation was conducted in the Electron Microscopy Laboratory at Queen Mary Hospital. Following sample preparation, the specimens were examined using the Philips CM100 transmission electron microscope, and images at various magnifications were captured for subsequent analysis.
Senescence-associated-β-galactosidase (SA-β-gal) staining
SA-β-gal staining was performed according to manufacturer’s instruction (CST, #9860). Briefly, cultured cells were washed twice with PBS and fixed at room temperature for 5 min in 2% formaldehyde and 0.2% glutaraldehyde. Fixed cells were stained with SA-β-gal staining solution at 37 °C overnight, and images were captured using a microscope digital camera (Olympus).
RNA m6A methylation quantification
TRIzol reagent was used to extract the total RNA. m6A content was assayed using the m6A RNA methylation assay kit (EpigenTek, P-9005). Briefly, 80 μl of binding solution and 200 ng of sample RNA were added into each designated well and incubated at 37 °C for 90 min for RNA binding. 1 ng m6A-methylated RNA provided by the assay kit was used as positive control. Wells were washed three times with wash buffer, followed by the addition of 50 μl of diluted capture antibody and incubation at room temperature for 60 min. Subsequently, wells were incubated with detection antibody and enhancer solution at room temperature for 30 min. The reaction was developed with developer solution in the dark for 1–10 min at 25 °C, stopped with stop solution, and measured using a microplate reader at 450 nm within 2–10 min.
RNA stability assay
Assay for RNA stability analysis was performed as previously described in ref. 24 with some modifications. P0(+DOX) and P2(-DOX) hTSCs were cultured in six-well culture plates to ∼80% confluence. Actinomycin D (ActD) (Thermo, 11805017) was added at a final concentration of 5 μg/ml. Cells were collected at four time points (0, 2, 4, 6 h) after the addition of ActD and total RNA was extracted with TRIzol reagent, then performed RNA sequencing.
Single-base elongation- and ligation-based qPCR amplification (SELECT-qPCR)
SELECT was applied to detect m6A sites56. Briefly, 1.5 μg total RNA was mixed with 100 nM m6A or control primers and 5 μM dNTP with 1× CutSmart buffer in 17 μL reaction. The primers were annealed to RNA using following PCR steps: 90 °C for 1 min, 80 °C for 1 min, 70 °C for 1 min, 60 °C for 1 min, 50 °C for 1 min, 40 °C for 6 min, 30 °C for 1 min, 20 °C for 1 min, and 10 °C for 1 min. Then, a 3 μL mixture of 0.01 U Bst DNA polymerase, 0.5 U SplintR ligase, and 10 nM ATP was added to the RNA/primers sample and subsequently incubated at 40 °C for 20 min and 80 °C for 20 min. Next, 4 μL of mixture was used for RT-qPCR reaction in triplicate with SYBR Green Real-Time PCR Master Mix (Applied biosystems). Primers used in this study are listed in Supplementary Table 1.
RNA sequencing
Total RNA was extracted using TRIzol reagent. Preparation of the library for RNA sequencing was performed according to the method previously described82. mRNA was enriched from total RNA using magnetic beads with poly-A oligos attached. Divalent cations were used for fragmentation under elevated temperature with First-Strand Synthesis Reaction Buffer from New England Biolabs. First-strand cDNA was synthesized using M-MLV reverse transcriptase without RNase H in the presence of random hexamer primers. Second-strand cDNA synthesis was performed using DNA polymerase I and RNase H. Synthesized DNA fragments were ligated to Next Adaptors from New England Biolabs and amplified by PCR using Phusion high-fidelity DNA polymerase (Vazyme). Library quality was assessed using an Agilent Bioanalyzer 2100 system, and sequencing was performed on the Illumina NovaSeq 6000 System.
Cleavage under targets and tagmentation (CUT&Tag)
CUT&Tag was performed according to the manufacturer’s protocol (Vazyme, TD904). Briefly, 50,000 cells were harvested for nuclei isolation. Isolated nuclei were incubated with primary antibodies overnight at 4 °C, followed by capture using corresponding secondary antibodies. Unbound antibodies were removed using Dig-Wash buffer, and a pA-Tn5 adaptor complex was prepared and added to the nuclei with gentle vortexing. Nuclei were then subjected to tagmentation, and the resultant DNA was purified for library preparation and amplification. The spike-in DNA was added along with stop buffer to ensure normalization across samples. Sequencing was performed on the Illumina NovaSeq 6000 system.
Methylated RNA immunoprecipitation (MeRIP) sequencing
Total RNA from CT-hTSCs and their derived day 6 STBs, and P0(+DOX) and P2(-DOX) hTSCs was extracted using TRIzol. MeRIP-seq was performed by Shanghai Jiayin Biotechnology Company Limited. Briefly, total RNA was fragmented into ~100 nucleotide long fragments. Approximately 5% of the fragmented RNA was reserved as input RNA, while the remaining RNA was immunoprecipitated using an affinity-purified anti-m6A polyclonal antibody (Millipore, ABE572). The SMARTer Stranded Total RNA-Seq Kit-version 2 (Pico Input Mammalian, Takara/Clontech, 634413) was used to construct libraries for both the input and IP samples. Sequencing was performed using an Illumina NovaSeq 6000 platform.
Sequencing data analysis
General pre-processing of reads: the RNA-seq, MeRIP-seq, and CUT&Tag were performed using Illumina NovaSeq 6000 (Novogene). Raw reads were trimmed using Trim Galore (v0.6.10) (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), filtering out reads with a quality score lower than 20 and read lengths shorter than 20 base pairs. Alignment to the human genome (GRCh38 assembly) was performed using Hisat2 (v2.2.1) for RNA-seq and MeRIP-seq, and Bowtie2 (v2.5.3) for CUT&Tag. featureCounts (v2.0.6) was used to calculate the gene counts with gene annotation from Gencode (v45). The aligned reads were split into strand-specific BAM files and sorted using samtools (v1.10).
For RNA-seq, the batch effect between hTSC3# and hTSC4# cell lines was removed using ComBat-seq in sva R package (v2.48.0). The expression of transcripts was quantified as reads per kilobase of transcripts per million mapped reads (TPM). Differentially expressed genes (DEGs) were identified using DESeq2(v1.40.2) with a cutoff of |log₂(fold change)| > 1 and an adjusted p value < 0.05. For splicing isoform analysis, the assembly of mapped reads from each sample was performed utilizing StringTie (v2.0.4) as part of an approach that was based on reference data. Expression quantification was calculated as fragments per kilobase per million mapped reads (FPKM) for each annotated genomic region to evaluate transcript abundance and differential expression. Enhancer predictions for transposable elements were based on previous study67. Deviations in gene expression between P0(+DOX) and P2(-DOX) hTSCs for protein-coding genes were modelled as a linear combination of occurrences of nearby TE subfamily integrants (within 50 kb of the transcription start site).
For CUT&Tag, duplicated reads were removed using MarkDuplicates from Picard (v3.0.0). Read counts were normalized based on the spike-in chromatin to account for technical variation. Peaks calling was performed using SEACR (v3.1), with options: 0.05 non-stringent. Strand-specific bigwig files for visualization, heatmaps, and aggregation plots were generated using bamCompare from deepTools (v3.5.1). For differential peak analysis, a consensus set of peaks was created for each condition (P0 and P3). Read counts for these peaks were quantified from the normalized BAM files and analyzed using DESeq2.
For MeRIP-seq, samtools (v1.10) was used to separate the plus and minus stranded reads with “−d XS:+” and “−d XS:-”. For peaks calling and differential peaks analysis, exomePeak2 (v1.12.0)83 was used to determine the read-enriched regions from MeRIP-seq data. Based on the uniquely mapped reads, the p value for a given region was computed utilizing a dynamic Poisson distribution. When the p value was <1 × 10−10, the region was designated as a peak. Peaks were annotated to their associated genes based on genomic position and gene annotation data. In addition, the distribution of peaks on different functional regions, including the 3’UTR, 5’UTR, and CDS, was further assessed. Homer (v4.11) was used to detect the enriched motifs within m6A peaks with the parameter “-rna -len 5,6,7,8”. Identification of m6A-modified genes required both a detected m6A modification and reduced expression in the P2(-DOX) group relative to P0(+DOX).
Gene enrichment analysis
Curated gene sets of KEGG and Biological Processes (BP) were downloaded from the Molecular Signature Database (MSigDB, v7.5.1, http://software.broadinstitute.org/gsea/msigdb/index.jsp) and were used to calculate enrichment scores. Significantly enriched gene sets were identified with a Benjamini–Hochberg adjusted p ≤ 0.05. For the identified pathways, we selected DEGs and performed GO and KEGG enrichment analysis using the clusterProfiler (v4.8.3) package. Public gene sets were acquired from GSEA database. Gene sets were used to score each input cell with the Seurat function “AddModuleScore”. Changes in the scores across +DOX and -DOX group were calculated by ggpubr package (v 0.6.0) using Wilcoxon test.
Half lifetime RNA-seq analysis
Raw reads were trimmed with Trimmomatic (v0.39), then aligned to human genome and transcriptome (hg38) and external RNA Control Consortium (ERCC) RNA spike-in control (Thermo Fisher Scientific) using Hisat2 (v2.1.0). Reads on each GENCODE annotated gene were counted using featureCounts and then normalized to counts per million (CPM) using edgeR packages in R. CPM was converted to attomole by linear fitting of the RNA ERCC spike-in. Half lifetime of RNA was estimated using formula listed in previous study23. Specifically, as ActD treatment results in transcription stalling, the change of RNA concentration at a given time (dC/dt) is proportional to the constant of RNA decay (Kdecay) and the RNA concentration (C), leading to the following equation:
Thus, the RNA degradation rate Kdecay was estimated by:
To calculate the RNA half-life (t1/2), when 50% of the RNA is decayed (that is, C/C0 = 1/2), the equation was:
From where:
The final half-life was calculated by using the average value of 0 h, 2 h, 4 h, and 6 h.
Single nucleus RNA sequence (snRNA-seq) analysis
The published snRNA-seq data42 of human placenta tissue at early pregnancy (8–10 weeks) and late pregnancy (term) were downloaded (GSE247038), with the embedded UMAP and cell annotations. The cell clustering was reproduced, and the scaled average expression of genes of interest was visualized.
Statistical and reproducibility
No statistical methods were used to predetermine sample sizes, and sample sizes were chosen based on previously published studies in the field. All experiments, including transmission electron microscopy, RT-qPCR, western blotting, and immunofluorescence, for quantitative analysis and representative images were performed using at least three biological replicates, except for RNA-seq, ATAC-seq, MeRIP-seq, and CUT&Tag experiments, all of which had two biological replicates. The statistical analysis was conducted with Microsoft Excel or R (v 4.2.3). p values were calculated using unpaired Student’s t-test or Wilcoxon test. The threshold for statistical significance was p < 0.05. For the identification of differentially expressed genes, the Wald test was applied using the DESeq2 package in R. The number of measurements and independent experiments, and the statistical test used for each analysis performed were described in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw data generated in this study are available at the Gene Expression Omnibus (GEO) under the accession number GSE280039. There are no restrictions on data availability or use. The snRNA-seq data of human placenta from this study42 is available in the GEO database with the accession GSE247038 and the array data from this study64 is available in the GEO database under the accession code GSE40182. Source data are provided with this paper.
References
Magee, L. A., Nicolaides, K. H. & von Dadelszen, P. Preeclampsia. N. Engl. J. Med. 386, 1817–1832 (2022).
Redman, C. W. & Sargent, I. L. Latest advances in understanding preeclampsia. Science 308, 1592–1594 (2005).
Burton, G. J. & Jauniaux, E. Pathophysiology of placental-derived fetal growth restriction. Am. J. Obstet. Gynecol. 218, S745–S761 (2018).
Gunnarsdottir, J., Stephansson, O., Cnattingius, S., Akerud, H. & Wikstrom, A. K. Risk of placental dysfunction disorders after prior miscarriages: a population-based study. Am. J. Obstet. Gynecol. 211, 34 e31–34.e38 (2014).
Turco, M. Y. & Moffett, A. Development of the human placenta. Development 146, dev163428 (2019).
Suryadevara, V. et al. SenNet recommendations for detecting senescent cells in different tissues. Nat. Rev. Mol. Cell Biol. 25, 1001–1023 (2024).
Velicky, P. et al. Genome amplification and cellular senescence are hallmarks of human placenta development. PLoS Genet 14, e1007698 (2018).
Okae, H. et al. Derivation of human trophoblast stem cells. Cell Stem Cell 22, 50–63 e56 (2018).
Dong, C. et al. Derivation of trophoblast stem cells from naive human pluripotent stem cells. Elife 9, e52504 (2020).
Wei, Y. et al. Efficient derivation of human trophoblast stem cells from primed pluripotent stem cells. Sci. Adv. 7, eabf4416 (2021).
Gao, X. et al. Establishment of porcine and human expanded potential stem cells. Nat. Cell Biol. 21, 687–699 (2019).
Ruan, D. et al. Human early syncytiotrophoblasts are highly susceptible to SARS-CoV-2 infection. Cell Rep. Med. 3, 100849 (2022).
Tan, J. P., Liu, X. & Polo, J. M. Establishment of human induced trophoblast stem cells via reprogramming of fibroblasts. Nat. Protoc. 17, 2739–2759 (2022).
Zhang, W. et al. METTL3-dependent m6A RNA methylation regulates transposable elements and represses human naive pluripotency through transposable element-derived enhancers. Nucleic Acids Res. 53, gkaf349 (2025).
Huang, Y. et al. Tumor suppressor protein p53 governs human trophoblast lineage development. Cell Rep. 44, 116310 (2025).
Liu, X. et al. Recapitulating primary immunodeficiencies with expanded potential stem cells: Proof of concept with STAT1 gain of function. J. Allergy Clin. Immunol. 153, 1125–1139 (2024).
Wei, X. et al. A human organoid drug screen identifies alpha2-adrenergic receptor signaling as a therapeutic target for cartilage regeneration. Cell Stem Cell 31, 1813-1830.e8 (2024).
Wei, J. & He, C. Chromatin and transcriptional regulation by reversible RNA methylation. Curr. Opin. Cell Biol. 70, 109–115 (2021).
He, P. C. & He, C. m(6) A RNA methylation: from mechanisms to therapeutic potential. EMBO J. 40, e105977 (2021).
Shi, H., Wei, J. & He, C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 74, 640–650 (2019).
Yue, Y., Liu, J. & He, C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 29, 1343–1355 (2015).
Wang, X. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014).
Huang, H. et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20, 285–295 (2018).
Liu, J. et al. N (6)-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 367, 580–586 (2020).
Wei, J. et al. FTO mediates LINE1 m(6)A demethylation and chromatin regulation in mESCs and mouse development. Science 376, 968–973 (2022).
Deng, S. et al. RNA m(6)A regulates transcription via DNA demethylation and chromatin accessibility. Nat. Genet. 54, 1427–1437 (2022).
Chelmicki, T. et al. m(6)A RNA methylation regulates the fate of endogenous retroviruses. Nature 591, 312–316 (2021).
Xu, W. et al. METTL3 regulates heterochromatin in mouse embryonic stem cells. Nature 591, 317–321 (2021).
Wang, Y. et al. N(6)-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat. Neurosci. 21, 195–206 (2018).
Wang, X. et al. N(6)-methyladenosine modulates messenger RNA translation Efficiency. Cell 161, 1388–1399 (2015).
Wu, Y. et al. N(6)-methyladenosine regulates maternal RNA maintenance in oocytes and timely RNA decay during mouse maternal-to-zygotic transition. Nat. Cell Biol. 24, 917–927 (2022).
Zhu, W. et al. Reading and writing of mRNA m(6)A modification orchestrate maternal-to-zygotic transition in mice. Genome Biol. 24, 67 (2023).
Batista, P. J. et al. mA RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15, 707–719 (2014).
Wang, Y. et al. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 16, 191–198 (2014).
Yin, R. et al. Differential m(6)A RNA landscapes across hematopoiesis reveal a role for IGF2BP2 in preserving hematopoietic stem cell function. Cell Stem Cell 29, 149–159.e147 (2022).
Zhang, C. et al. m(6)A modulates haematopoietic stem and progenitor cell specification. Nature 549, 273–276 (2017).
Gao, L., Lee, H., Goodman, J. H. & Ding, L. Hematopoietic stem cell niche generation and maintenance are distinguishable by an epitranscriptomic program. Cell 187, 2801–2816.e2817 (2024).
Liu, Y. et al. N6-methyladenosine-mediated gene regulation and therapeutic implications. Trends Mol. Med. 29, 454–467 (2023).
Wu, Z. et al. METTL3 counteracts premature aging via m6A-dependent stabilization of MIS12 mRNA. Nucleic Acids Res. 48, 11083–11096 (2020).
Wu, Z. et al. m(6)A epitranscriptomic regulation of tissue homeostasis during primate aging. Nat. Aging 3, 705–721 (2023).
Liu, P. et al. m(6)A-independent genome-wide METTL3 and METTL14 redistribution drives the senescence-associated secretory phenotype. Nat. Cell Biol. 23, 355–365 (2021).
Wang, M. et al. Single-nucleus multi-omic profiling of human placental syncytiotrophoblasts identifies cellular trajectories during pregnancy. Nat. Genet. 56, 294–305 (2024).
Liu, Y. et al. Single-cell RNA-seq reveals the diversity of trophoblast subtypes and patterns of differentiation in the human placenta. Cell Res. 28, 819–832 (2018).
Lee, C. L. et al. Dysregulation of the CD147 complex confers defective placental development: A pathogenesis of early-onset preeclampsia. Clin. Transl. Med. 12, e826 (2022).
Chen, A. C. H. et al. Expanded potential stem cells from human embryos have an open chromatin configuration with enhanced trophoblast differentiation ability. Adv. Sci. 10, e2204797 (2023).
Geula, S. et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347, 1002–1006 (2015).
Zeng, Z. C. et al. METTL3 protects METTL14 from STUB1-mediated degradation to maintain m(6) A homeostasis. EMBO Rep. 24, e55762 (2023).
Tan, J. X. & Finkel, T. Lysosomes in senescence and aging. EMBO Rep. 24, e57265 (2023).
Sheridan, M. A. et al. Characterization of primary models of human trophoblast. Development 148, dev199749 (2021).
Yankova, E. et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 593, 597–601 (2021).
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Hiby, S. E. et al. Paternal monoallelic expression of PEG3 in the human placenta. Hum. Mol. Genet 10, 1093–1100 (2001).
Ono, R. et al. Deletion of Peg10, an imprinted gene acquired from a retrotransposon, causes early embryonic lethality. Nat. Genet. 38, 101–106 (2006).
Saul, D. et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat. Commun. 13, 4827 (2022).
Zaccara, S. & Jaffrey, S. R. A unified model for the function of YTHDF proteins in regulating m(6)A-modified mRNA. Cell 181, 1582–1595.e1518 (2020).
Xiao, Y. et al. An elongation- and ligation-based qPCR amplification method for the radiolabeling-free detection of locus-specific N(6)-methyladenosine modification. Angew. Chem. Int. Ed. Engl. 57, 15995–16000 (2018).
Lasko, L. M. et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 550, 128–132 (2017).
Zijlmans, D. W. et al. Integrated multi-omics reveal polycomb repressive complex 2 restricts human trophoblast induction. Nat. Cell Biol. 24, 858–871 (2022).
Kumar, B. et al. Polycomb repressive complex 2 shields naive human pluripotent cells from trophectoderm differentiation. Nat. Cell Biol. 24, 845–857 (2022).
Weigert, R. et al. Dynamic antagonism between key repressive pathways maintains the placental epigenome. Nat. Cell Biol. 25, 579–591 (2023).
Tang, L. et al. Dysregulated Gln-Glu-alpha-ketoglutarate axis impairs maternal decidualization and increases the risk of recurrent spontaneous miscarriage. Cell Rep. Med. 4, 101026 (2023).
Zhou, Y., Damsky, C. H. & Fisher, S. J. Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome?. J. Clin. Investig. 99, 2152–2164 (1997).
Wang, Y. et al. SIRT1 regulates trophoblast senescence in premature placental aging in preeclampsia. Placenta 122, 56–65 (2022).
Zhou, Y. et al. Reversal of gene dysregulation in cultured cytotrophoblasts reveals possible causes of preeclampsia. J. Clin. Investig. 123, 2862–2872 (2013).
Zhang, B. et al. Human placental cytotrophoblast epigenome dynamics over gestation and alterations in placental disease. Dev. Cell 56, 1238–1252.e1235 (2021).
Frost, J. M. et al. Regulation of human trophoblast gene expression by endogenous retroviruses. Nat. Struct. Mol. Biol. 30, 527–538 (2023).
Pontis, J. et al. Primate-specific transposable elements shape transcriptional networks during human development. Nat. Commun. 13, 7178 (2022).
Ermini, L. et al. Ceramide-induced lysosomal biogenesis and exocytosis in early-onset preeclampsia promotes exosomal release of SMPD1 causing endothelial dysfunction. Front. Cell Dev. Biol. 9, 652651 (2021).
Moffett, A. & Shreeve, N. Local immune recognition of trophoblast in early human pregnancy: controversies and questions. Nat. Rev. Immunol. 23, 222–235 (2023).
Wang, Y. et al. The RNA m(6)A landscape of mouse oocytes and preimplantation embryos. Nat. Struct. Mol. Biol. 30, 703–709 (2023).
Bayerl, J. et al. Principles of signaling pathway modulation for enhancing human naive pluripotency induction. Cell Stem Cell 28, 1549–1565.e1512 (2021).
Quarto, G. et al. Fine-tuning of gene expression through the Mettl3-Mettl14-Dnmt1 axis controls ESC differentiation. Cell 188, 998–1018.e26 (2025).
Yu, X. et al. Acetyl-CoA metabolism maintains histone acetylation for syncytialization of human placental trophoblast stem cells. Cell Stem Cell 31, 1280–1297.e1287 (2024).
Gal, H. et al. Molecular pathways of senescence regulate placental structure and function. EMBO J. 38, e100849 (2019).
Dimitriadis, E. et al. Pre-eclampsia. Nat. Rev. Dis. Prim. 9, 8 (2023).
Wang, Y. et al. METTL3-mediated m6A modification increases Hspa1a stability to inhibit osteoblast aging. Cell Death Discov. 10, 155 (2024).
Benayoun, B. A. et al. Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res. 29, 697–709 (2019).
Martinez-Zamudio, R. I. et al. AP-1 imprints a reversible transcriptional programme of senescent cells. Nat. Cell Biol. 22, 842–855 (2020).
Kahl, M. et al. m6A RNA methylation regulates mitochondrial function. Hum. Mol. Genet. 33, 969–980 (2024).
Shen, C. et al. m(6)A-dependent glycolysis enhances colorectal cancer progression. Mol. Cancer 19, 72 (2020).
Selberg, S. et al. Discovery of small molecules that activate RNA methylation through cooperative binding to the METTL3-14-WTAP complex active site. Cell Rep. 26, 3762–3771.e3765 (2019).
Fu, H. et al. Elevated retrotransposon activity and genomic instability in primed pluripotent stem cells. Genome Biol. 22, 201 (2021).
Meng, J. et al. A protocol for RNA methylation differential analysis with MeRIP-Seq data and exomePeak R/Bioconductor package. Methods 69, 274–281 (2014).
Acknowledgements
We thank our colleagues in P.L.’s lab for their critical comments and Shanghai Jiayin Biotechnology Limited for their MeRIP sequencing services. We acknowledge the following fundings and supports: Hong Kong Research Grant Council (GRF_17112422), NSFC/RGC Collaborative Research Scheme (CRS_HKU703/22), Projects of International Cooperation and Exchanges NSFC (32261160571), Tianjin Science and Technology Major Project (24ZXZSSS00490), Tianjin Natural Science Foundation (22JCZXJC00090), the InnoHK initiative of the Innovation and Technology Commission of the Hong Kong Special Administrative Region Government, and the S Y and H Y Cheng Professor endowment in Stem Cell Biology and Regenerative Medicine (to P.L.). Schematic diagram present in this manuscript (Fig. 8) was Created in BioRender (Liu, P. (2025) https://BioRender.com/61t1jq1). Instrument or analysis software was supported by the Imaging and Flow Cytometry Core of Centre for PanorOmic Sciences (CPOS) at the University of Hong Kong.
Author information
Authors and Affiliations
Contributions
P.L., B.S., and H.F. conceived the project and designed the experiments. The co-first authors, along with P.L. co-wrote the manuscript. H.F. conducted most of the experiments. C.C. and W.Z. conducted most of the bioinformatics analysis. Y.H. performed parts of multi-omics experiments and LTR10A excision experiment. L.X. conducted EP300 CUT&Tag. Z.F. conducted TEM. F.C. and C.W. conducted SELECT-qPCR experiment. X.O. and D.R. contributed to western blotting and IF experiments. D.H., S.L., and C.W. performed the staining of the human placenta specimen. L.X., K.Y., and J.A. contributed to the scRNA-seq bioinformatics analysis. Q.Z., L.M., Y.L.L., and W.S.B.Y. provided materials for the experiments. The final version of the manuscript has been approved by all the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fu, H., Chen, C., Huang, Y. et al. METTL3 safeguards cell identity and epigenome of human trophoblast stem cells. Nat Commun 17, 795 (2026). https://doi.org/10.1038/s41467-025-67484-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67484-z
