
Abstract
Imbalance in intestinal fluid homeostasis leads to nutrient malabsorption, intestinal tissue destruction, and systemic inflammation. Transient receptor potential melastatin 4 (TRPM4) is a calcium-activated, non-selective monovalent cation channel converting chemical signals (Ca2+) into electrical signals (membrane depolarization). Here, we show the TRPM4 channel as a direct target of bisacodyl (BIC), a widely used clinical drug for chronic constipation management, and its active metabolite, deacetyl bisacodyl (DAB). DAB-induced laxative effects are abolished in global and intestinal epithelium-specific TRPM4-knockout mice, establishing the essential role of TRPM4 in intestinal fluid regulation. Furthermore, our structural work reveals DAB bound to an uncharacterized pocket, marking it as a non-Ca2+ TRPM4 agonist and unveiling a noncanonical Ca2+-independent activation mechanism. Additionally, we delineate a signaling axis, TRPM4 → VGCC/NCX → ANO1, that governs ion homeostasis in the epithelium. Together, these findings establish TRPM4 as a key regulator of intestinal fluid balance and reveal its noncanonical calcium-independent activation as a therapeutic strategy for constipation.
Similar content being viewed by others


Introduction
Effective intestinal fluid homeostasis is essential for maintaining digestive health by balancing nutrient absorption with the elimination of waste. Disruptions in this balance manifest as constipation or diarrhea—conditions that pose significant health challenges. Although not lethal, chronic constipation arising from a variety of etiologies, including neurogenic bowel dysfunction, opioid-induced constipation, colorectal malignancy, and constipation-predominant irritable bowel syndrome, affects approximately one-third of the U.S. population, with a higher prevalence among the elderly and women. This condition not only significantly increases the health burden and impacts quality of life1 but can also lead to complications such as fecal impaction and hemorrhoids. Conversely, chronic diarrhea may result in dehydration, electrolyte imbalances, malabsorption, and systemic inflammation, often signaling more complex underlying disorders. Moreover, these conditions are not merely isolated ailments; they frequently serve as clinical indicators of broader molecular dysregulation, arising either from intrinsic gastrointestinal diseases or as adverse effects of pharmacological treatments. Despite the high prevalence and severe clinical implications of these disorders, the molecular mechanisms governing intestinal fluid dynamics remain poorly understood.
Among the therapeutic strategies for managing these conditions, Bisacodyl (BIC)—a stimulant laxative of the diphenylmethane family—has long been employed to treat chronic constipation2. Its active metabolite, deacetyl bisacodyl (DAB), is responsible for its efficacious laxative effect2. Clinical studies have consistently shown that BIC is more effective than alternatives such as prucalopride, lubiprostone, linaclotide, and tegaserod3. However, despite its popular clinical utility since the discovery in the 1950s, the precise molecular targets and intracellular signaling pathways underlying BIC’s action remain unknown. This critical gap in our understanding highlights the urgent need to decipher the molecular basis of intestinal fluid regulation, which is fundamental to developing more targeted and effective treatments for both constipation and diarrhea.
Recent advances in ion channel research have begun to shed light on the contribution of transient receptor potential (TRP) channels to gastrointestinal disorders. Among these, transient receptor potential melastatin 4 (TRPM4)—a calcium-activated, non-selective monovalent cation channel is widely expressed in tissues including the colon4. TRPM4 is responsible for transducing intracellular calcium (Ca2+) signals into membrane depolarization5. In excitable tissues such as the heart and brain, TRPM4 is well known for its roles in modulating cardiac conduction and neuronal excitability6,7. In the immune system, it is expressed in many immune cells, predominantly in T lymphocytes, where it regulates Ca2+ signaling critical for cell activation and migration8. While TRPM4 has been reported to be expressed in the colon tissues4, the pathophysiological role of TRPM4 in the gastrointestinal system remains largely unexplored.
Structurally, TRPM4 is organized as a homotetramer, with each subunit comprising six transmembrane helices and extensive intracellular N- and C-terminal domains that are essential for channel gating9,10. Recent cryo-electron microscopy studies have provided high-resolution insights into its architecture by delineating key binding pockets for agonist and modulators including Ca2+, ATP and decavanadate (DVT)9,10,11. Notably, emerging evidence highlights temperature-dependent structural dynamics that further modulate TRPM4 function, a feature that may be particularly relevant for understanding its physiological roles12.
Here, our in vitro and in vivo experiments demonstrate that anti-constipation drug BIC and its active metabolite DAB regulate ion transport by directly targeting TRPM4 in the intestinal epithelial cells, establishing a critical role of epithelial-specific TRPM4 as a regulator of intestinal fluid homeostasis and revealing noncanonical Ca2+-independent activation as a therapeutic strategy for chronic constipation. These findings underpin the laxative action of BIC in the treatment of chronic constipation and lay a molecular foundation for the rational design of targeted therapeutics for constipation arising from a variety of etiologies.
Results
BIC and DAB are potent TRPM4 activators
TRPM4 is widely expressed throughout the human body and has recently emerged as an attractive drug target due to its critical role in cancer, immune disorders and heart diseases13. However, there is lack of specific TRPM4 modulators, and Ca2+ remains the only known agonist. To address this gap, we conducted an unbiased high-throughput screen of 2154 structurally diverse compounds using a fluorescence-based Na⁺ mobilization assay in HEK-293 cells stably expressing human TRPM4 (HEK293-TRPM4) (Supplementary Fig. 1A). We discovered that BIC and its active metabolite, DAB activate TRPM4. Treatment with 10 µM BIC resulted in a gradual increase in Na⁺ influx, an effect that was absent in null HEK-293 cells (Supplementary Fig. 1B). DAB elicits a more rapid Na+ influx in HEK-293-TRPM4 cells, but not in null HEK-293 cells, suggesting a more potent effect on TRPM4 activation than its parent compound BIC (Supplementary Fig. 1E). This aligns with previous report that DAB is responsible for the efficacious laxative effect of BIC. Further concentration-response analyses confirmed that both compounds stimulate Na⁺ influx in a concentration-dependent manner, with EC50 values of 2.64 µM for BIC and 0.14 µM for DAB (Supplementary Fig. 1C, D, F, G), demonstrating that DAB is ~20-fold more potent than BIC.
Intestinal epithelial TRPM4 is essential for DAB-induced laxative effects
Given that TRPM4 is expressed in intestinal epithelial cells4 and functions as a monovalent ion channel contributing to sodium influx, we speculate that TRPM4 could be the effective molecular target of BIC and DAB, which alter electrolyte gradients and impact fluid movement in the intestine. To test our hypothesis, we performed in vivo animal experiments. Because DAB is the active metabolite of BIC and exhibits markedly higher potency, we focused on DAB in the following in vivo and in vitro experiments.
Immunofluorescence staining revealed strong TRPM4 expression in the colon tissues. The majority of TRPM4 fluorescence signals co-localized with CK-18, a well-established marker of intestinal epithelial cells, confirming TRPM4’s predominant expression in this cell type (Fig. 1A, upper panel). In contrast, TRPM4 knockout (Trpm4-/-) mice showed no detectable TRPM4 fluorescence in the colon (Fig. 1A, lower panel).

A Fluorescence images stained with Transient Receptor Potential Cation Channel Subfamily M Member 4 (TRPM4, green) and intestinal epithelial maker, Cytokeratin-18 (CK-18, orange) in colon sections derived from wild-type (WT) (upper panel) and Trpm4-/- (lower panel) mice. Hoechst staining (blue) was used to visualize the nuclei. Scale bar = 50 µm. B Procedure to investigate the laxative effect of DAB in mice. Fecal water content (C) and Bristol score (D) of the stool collected from WT and Trpm4-/- mice between 24 and 36 h after Veh or DAB exposure (n = 7 mice/group; four male and three female). Data are presented as Mean ± SEM. **, p < 0.0001, vs. WT-Veh; ##, p < 0.0001, vs. WT-DAB. Fecal water content (E) and Bristol score (F) of the stool collected from Trpm4flox/flox and TrpmΔIEC mice (n = 7 mice/group; four male and three female). **, p < 0.0001, vs. Trpm4flox/flox-Veh; ##, p < 0.0001, vs. Trpm4flox/flox-DAB. Two-way ANOVA followed by Bonferroni’s multiple comparisons test. Figure 1B was Created in BioRender. https://BioRender.com/8h3ia6w.
To functionally assess the role of TRPM4 in DAB-induced laxative effects, we administered DAB (20 mg kg-1) via oral gavage to both male and female wild-type (WT) and Trpm4-/- mice (Fig. 1B). In WT mice, DAB treatment led to a marked increase in fecal water content and an elevated Bristol stool score (a widely used measurement of stool consistency)14, indicative of an efficacious laxative effect (Fig. 1C, D; Supplementary Fig. 2A). These laxative effects were completely abolished in both male and female Trpm4-/- time-matched littermates, demonstrating that TRPM4 is indispensable for mediating DAB-induced intestinal fluid secretion and stool softening (Fig. 1C, D; Supplementary Fig. 2A).
To further confirm the role of epithelial-specific TRPM4 in this process, we generated intestinal epithelium-specific TRPM4 knockout mice (Trpm4ΔIEC) by cross-mating Trpm4flox/flox mice with Villin-Cre (Vil1-Cre) transgenic mice (Supplementary Fig. 3A). Immunofluorescent staining confirmed successful epithelium-specific deletion of TRPM4, as TRPM4 expression was robustly detected in the colon of Trpm4flox/flox mouse but completely absent in Trpm4ΔIEC mice, while the expression in the skin tissues remained unchanged (Supplementary Fig. 3B). We administrated DAB to both male and female Trpm4flox/flox and Trpm4ΔIEC littermates. In Trpm4flox/flox mice, DAB markedly increased fecal water content and Bristol stool scores, replicating effects observed in WT mice. However, such effect was not observed in DAB-treated Trpm4ΔIEC littermates (Fig. 1E, F; Supplementary Fig. 2B).
Biophysical characterization of DAB-induced activation on TRPM4
Our in vivo findings provide strong evidence that TRPM4 is the key mediator of DAB-induced epithelial fluid secretion and laxative action, we next looked into the biophysical characterization of DAB-induced activation on TRPM4 to find definitive evidence whether DAB directly activates TRPM4, and whether their effects are influenced by temperature or Ca2+ levels, two factors that critically impact TRPM4 activation12.
Using whole-cell patch-clamp recordings in HEK293-TRPM4 cells, we found that, at low concentrations (≤ 0.3 µM), DAB elicited outwardly rectified Na+ currents, whereas at higher concentrations, the I-V curves became linear (Fig. 2A–C), consistent with voltage-dependent gating of TRPM4 channels15. Importantly, introducing 1 µM free Ca2+ into the pipette solution did not alter the potency of DAB, suggesting that DAB-induced TRPM4 activation is independent of intracellular Ca2+ concentration (Fig. 2D–F; Supplementary Table 1). Furthermore, as Ca2+-dependent TRPM4 activation is temperature-dependent with substantial increase in current magnitude at higher temperature12, we explored the thermo-sensitivity of DAB-induced TRPM4 activation. Interestingly, DAB-induced TRPM4 activation showed no obvious difference between 25 °C and 37 °C (Fig. 2; Supplementary Table 1). Together, these findings suggest that DAB activates TRPM4 independent of both intracellular Ca2+ and temperature, implying an activation mechanism distinct from that of the endogenous agonist, Ca2+. In addition, DAB also activates mouse (m)TRPM4, but has no effect on hTRPM3, hTRPM5 and hTRPM8 (Supplementary Fig. 4A–D), demonstrating that DAB is a selective agonist of TRPM4.

Representative traces (A), I-V curves (B) and concentration-response relationship curves (C) of DAB-stimulated currents in HEK-293-TRPM4 cells. Currents were recorded in the pipette solution with 0 µM Ca2+ (with 5 mM EGTA) at 25 °C. Representative traces (D), I-V curves (E) and concentration-response relationship curves (F) of DAB-stimulated currents recorded in the pipette solution with 1 µM Ca2+ at 25 °C. Representative traces (G), I-V curves (H) and concentration-response relationship curves (I) of DAB-stimulated currents recorded in the pipette solution with 0 µM Ca2+ (with 5 mM EGTA) at 37 °C. Representative traces (J), I-V curves (K) and concentration-response relationship curves (L) of DAB-stimulated currents recorded in the pipette solution with 1 µM Ca2+ at 37 °C. Each data point represents Mean ± SEM in C (n = 8 cells), F (n = 10 cells), I (n = 7 cells) and J (n = 6 cells).
Structure of DAB—TRPM4 complex
Our in vivo and in vitro evidence establishes DAB as a non-Ca2+ agonist selective for TRPM4, highlighting the need to dive into molecular level to identify their binding site and underlying mechanism of activation.
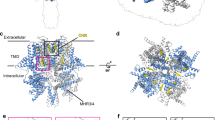
To this end, we determined the cryo-EM structures of human TRPM4 bound to DAB in the presence or absence of Ca²⁺ at physiological temperature, achieving overall resolutions of 3.1 and 3.3 Å, respectively (Fig. 3A–C; Supplementary Fig. 5; Supplementary Table 2). The overall structures resemble the previously published TRPM4 structures of a tetramer assembly with a transmembrane domain (TMD) and a large intracellular domain (ICD). In both conditions, we observed a well-defined density within the S1-S4 domain of each subunit, positioned above the CaTMD binding site (Fig. 3D, E). This density is tripod-shaped, with one pod smaller than the other two, closely resembling the shape of DAB. Notably, it seems this density has not been observed in any previously reported TRPM4 structures, suggesting a previously unrecognized ligand binding site in TRPM4.

A Cryo-EM map of TRPM4 bound to DAB and Ca2+, viewed parallel to the membrane. One subunit is highlighted in magenta. The transparent envelope outlines the detergent micelle and the disordered part of the C-terminal coiled-coil. B Atomic model of TRPM4 under the same condition, shown as a transparent surface with one subunit in cartoon representation. Gray ellipses mark the S1–S4 and S5–S6 domains. C Cryo-EM map of TRPM4 in the presence of DAB and EGTA. One subunit is highlighted in blue. D, E Close-up views of the S1–S4 binding site bound to DAB/Ca2+ (D) and DAB alone (E). CaTMD is shown as spheres; DAB and selected interacting residues are shown as sticks. Cryo-EM densities for the ligands are displayed as transparent surfaces. F Superposition of S1–S4 binding sites bound to DAB (blue) and Ca2+ (yellow). G Superposition of S1–S4 binding sites bound to DAB (blue) and DAB/Ca2+ (magenta). H Concentration–response curves of wild-type TRPM4 and mutants in response to DAB at +100 mV. WT (n = 6 cells), N786A (n = 10 cells), S789A (n = 11 cells), Y790A (n = 10 cells), F793A (n = 10 cells), L824A (n = 10 cells), D868A (n = 6 cells), F902A (n = 11 cells), R905A (n = 11 cells), R1072A (n = 11 cells) and F902A + R1072A (n = 10 cells). Each data point represents Mean ± SEM. Pore profiles of TRPM4 structures bound to DAB/Ca2+ (I) or DAB (J) viewed parallel to the membrane. The pore is shown as a surface, with two opposing subunits in cartoon representation. Residues forming the gate and selectivity filter are shown as sticks. K Pore radius plotted along the central axis, corresponding to the structures shown in (I, J). L Structural overlay of TRPM4 bound to Ca2+ (yellow), DAB/Ca2+ (magenta), and DVT/Ca2+ (salmon), showing the overall conformational trajectory of the TRP and S6 helices, as indicated by arrows.
The putative DAB binding site is primarily surrounded by both hydrophobic residues and hydrophilic residues (Fig. 3D). Alanine substitution of these key residues increased the EC50 values of DAB by 1–2 orders of magnitude (Fig. 3H; Supplementary Fig. 6; Supplementary Table 3), confirming this as the functional binding site. We further determined the structures of TRPM4 in the presence of BIC, the precursor of DAB. Despite attempting different concentrations up to 1 mM, we did not observe the density for BIC in the cryo-EM maps. We speculate that the two extra acetyl groups in BIC impede optimal engagement with the binding pocket, leading to reduced binding affinity, which may explain its weaker potency on TRPM4 compared to DAB.
DAB activates TRPM4 via a TMD-focused mechanism
Our structural data reveal that while DAB and Ca2+ bind to adjacent sites within the S1-S4 domain of TRPM4, they interact with entirely different sets of residues and activate TRPM4 independently (Fig. 3D). This distinction underscores the functional and pharmacological significance of the DAB binding site.
Previous studies have established that in non-excitable cells, Ca2+-induced activation of TRPM4 and its close homolog TRPM5 at physiologically relevant negative membrane potentials requires Ca2+ binding at two sites: the TMD agonist site and the ICD modulatory site12,16. While the CaTMD site functions as the orthosteric agonist site, the CaICD site is essential for triggering conformational changes—specifically, an upward movement of the ICD toward the TMD—that prime the channel for opening. This movement reflects an allosteric communication between the ICD and TMD, which is critical for activation at negative potentials. In the absence of CaICD binding, or more precisely, without the associated ICD conformational dynamics, TRPM4 requires membrane depolarization to open.
In contrast, our electrophysiological data show that DAB activates TRPM4 at negative membrane potentials independently of Ca2+, indicating a distinct mechanism (Fig. 2A–F). This DAB-induced activation appears confined to the TMD, where DAB binds, and does not rely on ICD movement triggered by Ca2+ binding at CaICD. Supporting this, TRPM4 adopts the same TMD conformation whether bound to DAB alone, Ca2+ alone, or both DAB and Ca2+, despite showing different ICD conformations depending on Ca2+ presence (Fig. 3F, G). Further supporting this distinction, DAB-induced activation is temperature independent (Fig. 2A–C, G–I), whereas Ca2+-induced activation requires both elevated temperature and Ca2+ binding to CaICD to stabilize the ICD in its active conformation12.
Together, these findings suggest that DAB activates TRPM4 exclusively through its action at the TMD, bypassing the TMD–ICD allosteric communication required for Ca2+-dependent activation. This may provide a valuable strategy for developing TRPM4-targeted therapeutics. Importantly, unlike Ca2+-mediated gating, which is tightly controlled by fluctuations in cytosolic Ca2+ levels and relies on ICD conformational dynamics, TMD-focused agonists like DAB and BIC operate independently of these regulatory mechanisms. As a result, they may offer more consistent pharmacological effects across diverse tissues and physiological conditions, particularly in settings like the intestine, where cytosolic Ca2+ levels can vary despite stable physiological temperature.
Although DAB activates TRPM4 and produces a near-linear current-voltage relationship at high concentrations (Fig. 2B, E, H, K), the DAB-bound TRPM4 structures exhibit a closed pore, regardless of Ca2+ presence (Fig. 3I–K). A plausible explanation is that additional factors, such as endogenous lipids, may be required to stabilize the open conformation which is missing in our cryo-EM study compared to the in vivo and in vitro study. To gain molecular insight into how DAB facilitates channel activation, we compared the DAB-bound structure with two reference states: a Ca2+-bound closed state and an open state induced by Ca2+ in combination with the positive modulator DVT (Fig. 3L). This comparison revealed that DAB triggers activation through a mechanism that partially overlaps with the Ca2+/DVT-induced opening pathway.
Compared to the Ca2+-bound closed state, DAB binding induces a conformational change in the S4 helix. This change propagates through the S4–S5 linker to the adjacent portion of the S5 helix and the TRP helix (Fig. 3L), causing an upward movement of the ICD toward the TMD. Notably, these DAB-induced conformational changes resemble those observed when DVT converts the Ca2+-bound closed state into an open state12, suggesting that DAB alone partially mimics the combined effect of Ca2+ and DVT. However, unlike in the Ca2+/DVT-bound structure, the structural rearrangement triggered by DAB does not extend to the distal portion of the S5 helix adjacent to S6, and therefore the S6 helices remain in their closed conformation, and the ion-conducting pore does not open (Fig. 3L). This difference likely arises because DAB binds exclusively within the S1–S4 domain and does not directly engage the S5–S6 pore domain, as DVT does12. Since the DAB-bound structure adopts a conformation that is intermediate between the Ca2+-bound closed state and the Ca2+/DVT-induced open state (Fig. 3L), and the channel did not show any desensitization during a prolonged patch-clamp electrophysiological recording, we speculate that it represents a pre-open, partially activated state of TRPM4.
TRPM4 → VGCC/NCX → ANO1 coupling in intestinal epithelium mediates the laxative effect of DAB
Given that the strong laxative effect of DAB was abolished in Trpm4ΔIEC mice, we next investigated the mechanism by which DAB disrupts ion homeostasis in intestinal epithelial cells. Na+ imaging revealed that DAB induced a robust increase in intracellular sodium concentration, an effect eliminated by pre-treatment with the TRPM4-selective inhibitor 9-Phe, indicating that TRPM4 is the primary molecular target mediating DAB-induced Na+ influx in HT-29 cells (Fig. 4A). Consistently, whole-cell patch-clamp recordings showed that DAB (10 μM) elicited a robust current in HT-29 cells, which was abolished by 9-Phe (Fig. 4B).

A Representative traces showing DAB-induced sodium influx in HT-29 cells that was abolished by the pretreatment of 9-Phe (30 μM) for 10 min. N = 229–236 cells. B Representative DAB (10 μM)-induced current in HT-29 cells that was blocked by the addition of 9-Phe (30 μM). Experiments were repeated in 6 independent cells. C–E Effects of Ani9 (10 μM), an ANO1 inhibitor on DAB (10 μM)-induced currents in HT-29 cells at the conditions of [Ca2+]e = 2 mM and [EGTA]p = 0 mM (C), [Ca2+]e = 2 mM and [EGTA]p = 5 mM (D), [Ca2+]e = 0 mM and [EGTA]p = 0 mM (E). [Ca2+]e: Extracellular cellular Ca2+ concentration; [EGTA]p: EGTA concentration in the pipette solution. (F) Quantification of Ani9 inhibition of DAB-induced currents in HT-29 cells at the conditions of (C–E). **, p < 0.0001, vs. DAB, n = 6 cells. G Procedure to determine the effect of CaCCinh-A01 (A01; 3 mg kg−1), an ANO1 inhibitor and CFTRinh-172 (172; 1 mg kg−1), a CFTR inhibitor on DAB-induced laxative effect (H, I). Fecal water content (H) and Bristol score (I) of the stool collected from WT mice after DAB exposure for 24–36 h pretreated with Veh, A01 or 172. **, p < 0.0001, vs. Veh; ##, p < 0.0001, vs. DAB. N = 6 mice/group, three male and three female. (J) Representative traces of DAB-induced Ca2+ influx and the effect of pretreatment of 9-Phe (30 μM). HT-29 cells were treated with 9-Phe for 10 min before addition of DAB. N = 56–71 cells. K Representative traces of 0 mM [Ca2+]e (without EGTA) on DAB-induced Ca2+ influx. N = 110–120 cells. L, M Representative traces (L) and quantification (M) of SEA0400 (1 μM), a Na+-Ca2+ exchanger inhibitor and nifedipine (1 μM), a voltage-gated Ca2+ channel inhibitor, on DAB-induced Ca2+ influx in HT-29 cells. **, p = 0.0061, ##, p < 0.0001, vs. DAB, n = 5 wells. Data in F, H–M are presented as Mean ± SEM, p values were calculated by one-way ANOVA followed by Bonferroni’s multiple comparisons test. Experiments in (A, J, K) were repeated four times in independent cultures with similar results. Figure 4A, B, G, J were Created in BioRender. (2025) https://BioRender.com/8h3ia6w.
In intestinal epithelial cells, passive water movement into the intestinal lumen is driven by Cl- efflux, primarily mediated by cystic fibrosis transmembrane conductance regulator (CFTR) and/or Ca2+-activated Cl- channels (CaCCs)17. To determine whether TRPM4-mediated Na⁺ influx couples to either or both Cl⁻ efflux systems, we conducted electrophysiological experiments. We found that DAB-induced currents were partially suppressed by Ani9, an inhibitor of Anoctamin1 (ANO1, a Ca2+-activated Cl- channel)18, but not by CFTRinh-172, a selective CFTR inhibitor19 (Fig. 4C, F; Supplementary Fig. 7). This suggests that DAB-induced current in HT-29 cells arises from Na⁺ influx through TRPM4 and Cl⁻ efflux through ANO1 (rather than CFTR). Consistent with this model, Ani9 no longer suppressed DAB-induced currents when either extracellular Ca2+ was removed (0 mM Ca2+ without EGTA) or intracellular Ca2+ was chelated with EGTA, whereas 9-Phe still completely abolished the response under both conditions (Fig. 4D–F). This suggests that DAB-induced Ca2+ influx is required for ANO1 activation and subsequent Cl- efflux. Together, these findings indicate that Na⁺ influx through TRPM4 functionally couples to Ca2+-dependent Cl⁻ efflux via ANO1.
To validate the TRPM4–ANO1 coupling in vivo, mice were pretreated with CaCCinh-A01 (A01), a specific ANO1 inhibitor with favorable pharmacokinetic properties20, prior to DAB administration. Pretreatment with A01—but not CFTR inhibitor CFTRinh-172—completely abolished DAB-induced laxative effect, as assessed by both Bristol score and fecal water content (Fig. 4G–I; Supplementary Fig. 8), confirming the functional relevance of the TRPM4–ANO1 axis in vivo.
We next explored how DAB might elevate intracellular Ca2+ to activate ANO1. In HT-29 cells, DAB triggered a rapid increase in cytosolic Ca2+ concentration, which was blocked by the pretreatment of 9-Phe, indicating the involvement of TRPM4 (Fig. 4J). Notably, this Ca2+ rise was completely abolished when extracellular Ca2+ was removed (0 mM Ca²⁺ without EGTA) (Fig. 4K), confirming that Ca2+ influx from the extracellular space is required for ANO1 activation.
To further dissect the route of Ca2+ entry, we found that DAB-induced Ca2+ influx was nearly eliminated by nifedipine, a blocker of voltage-gated Ca2+ channels (VGCCs), and modestly reduced by SEA0400, an inhibitor of the Na+-Ca2+ exchanger (NCX) (Fig. 4L, M). These findings support a model in which TRPM4-mediated Na⁺ promotes Ca²⁺ entry through two synergistic mechanisms: (1) membrane depolarization activates voltage-gated Ca2+ channels (VGCCs); and (2) the combination of elevated intracellular Na⁺ and membrane depolarization favors reverse-mode operation of NCX, leading to additional Ca2+ influx. The resulting rise in cytosolic Ca2+ activates ANO1, driving Cl⁻ efflux and contributing to the laxative effect of DAB.
Discussion
BIC is a widely used treatment for chronic constipation and bowel cleaning prior to medical procedures3. Its laxative effect has been attributed to its active metabolite, DAB, yet the underlying molecular mechanisms have remained elusive. In this study, we identify TRPM4 as the direct molecular target of DAB and establish a mechanism through which DAB regulates intestinal fluid secretion and homeostasis. Ex vivo studies have established that a high concentration of BIC (10 μg/mL, ~27 μM) is required to enhance contractility in human large intestine21. In contrast, DAB exerts its effects—including enhanced colon mucosal secretion and increased muscle tone in both the small bowel and colon—at much lower concentrations (0.5–5 µM)22. Here, we demonstrate that DAB and BIC activate TRPM4 with EC50 values of 0.14 and 2.64 μM, respectively, consistent with the observation that DAB is much more potent than BIC to stimulate muscle contraction and mucosal secretion21,22. The discrepancy between the low EC50 values for TRPM4 activation (BIC: 2.64 µM; DAB: 0.14 µM) and the higher concentrations required in ex vivo muscle contraction /mucosal secretion is likely due to reduced bioavailability and system complexity. In the ex vivo setting, penetration through dense tissue barriers lowers the effective concentration of both compounds at their target site. Furthermore, to trigger a measurable functional contraction, a threshold numbers of TRPM4 channels must be activated, a requirement that is absent in simplified in vitro systems.
We show that TRPM4 is highly expressed in intestinal epithelial cells, as evidenced by co-localization with the epithelial marker CK-18. Both global and intestinal epithelium-specific TRPM4 knockout mice fail to exhibit DAB-induced laxative effects, demonstrating that BIC-induced laxative effect is solely dependent on the activation of intestinal epithelial TRPM4 by its active metabolite, DAB. These data suggest that intestinal epithelial TRPM4 is an effective molecular target to cause diarrhea and an efficacious molecular target to treat chronic constipation.
Unlike endogenous TRPM4 activation, which is tightly coupled to intracellular Ca2+ and is subject to rapid desensitization, DAB activates TRPM4 independently of calcium and temperature23,24. DAB-induced activation results in a sustained Na+ current and a persistent [Na+]i increase in both HEK-293-TRPM4 and HT-29 cells. Such persistent and robust effect is likely required to effectively and consistently trigger a diarrhea effect, observed in the clinical settings. Our structural data reveal that DAB binds to a site in the S1-S4 domain. Interestingly, DAB bypasses the typical TMD-ICD allosteric coupling observed in calcium-dependent activation, providing a mechanism for TRPM4 gating that involves direct signal transduction within the TMD.
Mechanistically, intestinal fluid secretion is driven by transepithelial Cl– efflux into the intestinal lumen primary through the cystic fibrosis transmembrane conductance regulator (CFTR) and Ca2+-activated Cl- channels (CaCCs) expressed in the apical side of intestinal crypts17,25. ANO1 is one of the major CaCCs expressed in the intestinal epithelial cells, responsible for the Cl- secretion to the intestinal lumen26. Our data reveal that TRPM4 activation by DAB is functionally coupled to ANO1, forming a TRPM4/ANO1 signaling axis that promotes Cl- secretion. Inhibition of ANO1 abolishes DAB-induced laxative effect, confirming the physiological relevance of this coupling in vivo. Moreover, we show that DAB induces a rapid rise in intracellular Ca2+ in HT-29 cells, a response sensitive to inhibition of both VGCCs and NCX, implicating a TRPM4 → VGCC/NCX → ANO1 signaling cascade. This VGCC-mediated Ca2+ influx is consistent with the established finding that DAB-induced secretion in the human intestine is dependent on L-type Ca2+ channels22. This pathway coordinates sodium influx, Ca2+ mobilization, and chloride secretion to regulate epithelial fluid dynamics.
It is also worth noting that intestinal water absorption relies on electroneutral transport mechanisms mediated by sodium/glucose cotransporter, short-chain fat acid (SCFA)-activated Na+ channels, and epithelial Na+ channels (ENaCs)17,27,28,29. Since DAB activation of TRPM4 leads to a marked elevation of intracellular Na+, this may reduce the transmembrane Na+ gradient, potentially impairing Na⁺ uptake from the intestinal lumen and thereby hindering water reabsorption. This suggests that, in addition to promoting Cl⁻-driven fluid secretion, TRPM4 activation may also suppress Na⁺-driven fluid absorption, further enhancing the overall pro-secretory and laxative effect of DAB.
Although previous studies have identified TRPM4 modulators, including Ca2+, U73122, BTP2, necrocide 1 and DVT8,9,30,31,32,33, all of them lack specificity or exhibit limited cross-species efficacy. By contrast, we show that DAB is a potent and selective activator of TRPM4, making DAB a valuable pharmacological tool for dissecting TRPM4 pathophysiology. In summary, our findings establish a conceptual framework for understanding fluid homeostasis regulation and reveal the TRPM4 channel as the target for the treatment of constipation with distinct etiologies, including neurogenic bowel dysfunction, opioid-induced constipation, colorectal malignancy, and constipation-predominant irritable bowel syndrome (IBS-C).
Methods
All animal experiments were carried out in concordance with the National Institutes of Health Guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978) and approved by the Institutional Animal Care and Use Committee of China Pharmaceutical University (#SYXK 2021-0011, approved for Jan 25th, 2021 to Jan 24th, 2026).
Reagents and antibodies
Bisacodyl (BIS, #603-50-9), 9-Phenanthrol (9-Phe, #484-17-3), triphenylphosphine oxide (TPPO, #791-28-6), CIM0216 (#1031496-06-6), menthol (#2216-51-5), Ani9 (#356102-14-2), SEA0400 (#223104-29-8), nifedipine (#21829-25-4), CaCCinh-A01 (A01, #407587-33-1), CFTR(inh)-172 (172, #307510-92-5), Blasticidin S (#3513-03-9), Hygromycin B (#31282-04-9) and doxycycline (#564-25-0) were purchased from MedChemExpress (Monmouth Junction, NJ, USA). Deacetyl Bisacodyl (DAB, #603-41-8) was purchased from FishReag (Nanjing, Jiangsu, China). Poly-D-lysine (PDL, #27964-99-4), pluronic® F-127 (#9003-11-6), Leupeptin (#103476-89-7), Aprotinin (#9087-70-1), Pepstatin (#26305-03-3), PMSF (#329-98-6) and all the inorganic reagents were purchased from Sigma-Aldrich (St. Louis, MA, USA). Lipofectamine® 2000 reagent (#12566014), CoroNa™ Green/AM (#C36676), Fetal bovine serum (FBS, #A5670701), Penicillin and streptomycin (P/S, #15140122), Geneticin (G418, #108321-42-2), and Dulbecco minimum essential medium (DMEM, #12800017) were from Thermo Fisher Scientific (Waltham, NJ, USA). Fluo-4/AM (#20550) was purchased from AAT Bioquest (Sunnyvale, CA, USA). Primary antibodies against CK-18 (#bsm-33101M, RRID: AB_3678929) and TRPM4 (#ACC-044, RRID: AB_2040250) were from Bioss Antibodies (Beijing, China) and Alomone Labs (Jerusalem, IL, USA), respectively. Alexa Fluor 555 conjugated goat anti-mouse IgG (H + L) (#4409, RRID: AB_1500655) and Alexa Fluor 488-conjugated goat Anti-rabbit IgG (H + L) (#4412, AB_3067986) secondary antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). GDN (#GDN101 25 GM) was from Anatrace (Maumee, OH, USA). Strep-Tactin superflow high capacity resin (#2-1208-500) was obtained from IBA Lifesciences GmbH (Göttingen, Germany). Superose 6 Increase 10/300 GL (#29091596) was purchased from GE HealthCare Technologies Inc. (Chicago, Illinois, USA).
Animals
Both male and female C57BL/6 J mice (8 weeks, 20–22 g) were purchased from the Animal Core Facility of Nanjing Medical University (Nanjing, Jiangsu, China). Villin-Cre (Vil1-Cre) mice (Strain # 021504) were purchased from The Jackson Laboratory (Bar Harbor, MI, USA). Global TRPM4 knockout (Trpm4-/-, Strain # T014306) and Trpm4flox/flox (Strain # T013555) mice were obtained from GemPharmatech Co., Ltd. (Nanjing, Jiangsu, China). Mice were kept in individual ventilation cage in an SPF animal facility under a 12 /12 h light–dark cycle at 23 ± 2 °C with ad libitum access to food and water. Intestinal epithelial-specific TRPM4 knockout (Vil1+/--Trpm4flox/flox or Trpm4ΔIEC) mice were generated through cross-mating Vil1-Cre mice with Trpm4flox/flox mice. The genotypes of Trpm4-/-, Trpm4flox/flox and Trpm4ΔIEC mice were validated by RT-PCR and the expression levels of TRPM4 in colon tissues of these mice were accessed by immunofluorescence staining.
Cells, plasmid
HEK-293-TRPM8 cells were the same as previously described34. Human colorectal cancer epithelial cells (HT-29 cells) were purchased from the Chinese Academy of Medical Sciences (Beijing, China). The gene encoding full-length hTRPM3 (UniProtKB: Q9HCF6), hTRPM4 (UniProtKB: Q8TD43) and mTRPM4 (UniProtKB: Q7TN37), hTRPM5 (UniProtKB: Q9NZQ8) were synthesized by Bio Basic (Amherst, NY, USA).
Animal model
DAB laxative effect was evaluated in C57BL/6 J mice at 8 weeks old (both male and female, 20–22 g). After acclimation for 2 days, DAB (20 mg kg−1) was administrated by intragastric gavage (i.g.) at a volume of 20 ml kg−1 every 12 h. To access the role of ANO1 and CFTR on DAB-induced laxative effect, CaCCinh-A01 (3 mg kg−1), a specific ANO1 inhibitor and CFTRinh-172 (1 mg kg−1), a specific CFTR inhibitor, were intraperitoneal (i.p.) injected (10 ml kg−1) into the mouse 30 min prior to the administration of DAB, every 12 h. All the compounds were dissolved in 2% DMSO + 2% Tween-80 and 0.5% sodium carboxymethyl cellulose (CMC-Na) solution. Mice in control group were gavaged with vehicle (0.5% CMC-Na + 2% DMSO + 2% Tween-80). After 24 h, the stool of each mouse was collected, pictured, and the Bristol score was used to assess the stool consistency based on cohesion and surface characteristics categorized on a 7-point scale14. The freshly expelled fecal pellets were weighed and dried at 75 °C for 24 h. Water content of the feces was calculated by the following formula: Water content % = [wet weight of the feces (mg) − dried weight of the feces (mg)]/wet weight of the feces (mg) × 100.
To access the role of TRPM4 on DAB laxative effects, time-matched C57BL/6 J wild type (WT) and Trpm4-/- littermates as well as Trpm4flox/flox and Trpm4ΔIEC littermates at 8 weeks were used.
Cell cultures
HEK-293 cells and HT-29 cells were cultured in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin (P/S) and 1% HEPES. HEK-293-TRPM8 cells were cultured in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin (P/S), 1% HEPES, Blasticidin (5 μg ml−1) and Hygromycin B (50 μg ml−1). TRPM8 expression was induced with 1 µg ml−1 of doxycycline for 24 h before recording. HEK-293-hTRPM4 cells were cultured in DMEM supplemented with 10% FBS, 1% P/S and 500 μg ml-1 G418. All cells were cultured in a humidified incubator at 37 °C with 5% CO2.
Transfection and site-directed mutagenesis
HEK-293 cells were transfected with cDNA constructs using Lipofectamine® 2000 reagent according to the manufacturer’s protocol. To facilitate the identification of hTRPM3, hTRPM4, mTRPM4 or hTRPM5 transfected cells, a green fluorescence protein (GFP) was genetically linked to the C terminus of these genes. Patch-clamp recordings were performed after 24–48 h transfection. Mutants of hTRPM4 were generated using KOD plus mutagenesis kit (# KOD-401, Toyobo, Osaka, Japan) as described previously35. All mutants were confirmed by DNA sequencing. Transiently transfected cells were cultured in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin (P/S) and 1% HEPES.
Sodium influx assay
To discover TRPM4 ligands, changes in intracellular Na+ levels were monitored using Na+ sensitive dye, CoroNa™ Green/AM in HEK-293-TRPM4 cells36. Briefly, HEK-293-TRPM4 cultured in black-walled clear bottom 96-well plates (#3603, Corning, New York, USA) were incubated with 5 μM of CoroNa™ Green/AM (dissolved in Lock’s buffer containing 0.2 mg ml-1 Pluronic® F-127) for 60 min at 25 °C. After washing, plates were loaded onto the compound plate position of FLIPRTETRA® (Molecular Devices, Sunnyvale, CA). Emitted fluorescence excited at 488 nm wavelength was recorded at 510–570 nm. Compounds were automatically added to the cell plate by a programmable and automatic pipetting system after 60 s baseline fluorescence recording and the fluorescence signals were continuously recorded for an additional 240 s. Data are presented as F/F0 where F is the fluorescence signal at any time point whereas F0 is the fluorescence signal averaged from initial 5 data points. To quantify the concentration-response relationships of BIS and DAB activation of TRPM4, the area under the curve (AUC) calculated from a 240 s duration right after the addition in each well was used.
To investigate the changes of [Na+]i in HT-29 cells, HT-29 cells were loaded with 5 µM CoroNa Green/AM for 1 h, and the fluorescence was digitalized using a Flash4.0 V2 digital CMOS camera attached to a Leica DMi8 microscope at a sampling rate of 1 Hz. The excitation wavelength was filtered through a 460–495 nm excitation filter, and the emitted fluorescence was collected through a 510–550 nm filter. The arbitrary fluorescence unit of each region of interest (ROI) was quantified. Drugs were applied by bulk perfusion after a baseline fluorescence recording. Fluorescent signal at any time point was denoted as F and the mean of the first 5 data points was denoted as F0. Data are expressed as F/F0. Inhibitors were preincubated for 10 min before fluorescence recording.
Electrophysiology
Electrophysiological recordings were performed using an EPC-10 amplifier controlled by the PatchMaster software at 25 °C unless mentioned. Recording pipettes were pulled from borosilicate glass using P-1000 Micropipette Puller to 4-6 MΩ. For recording of hTRPM4, mTRPM4 and hTRPM5 currents, the pipette and extracellular solution contains (in mM) 130 NaCl, 3 HEPES, 5 EGTA. To investigate the influence of Ca2+ on DAB response on TRPM4. The pipette solution contains a free Ca2+ concentration of 1 µM obtained by a combination of CaCl2 (3.11 mM) and EGTA (5 mM) according to the following website: https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/CaEGTA-TS.htm. To recording the hTRPM8 currents in HEK-293 cells, the pipette solution contains (in mM) 150 NaCl, 5 MgCl2, 5 EGTA and 10 HEPES, pH 7.4 with NaOH, while the extracellular solution is (in mM) 150 NaCl, 1 MgCl2 and 10 HEPES, pH 7.4 with NaOH. The pipette solution for recording of hTRPM3 contains (mM) 110 CsCl, 20 tetraethylammonium chloride, 3.5 MgCl2, 2.5 Na2ATP, 2.41 CaCl2, 5 HEPES, 5 EGTA, pH 7.3 (with CsOH) while the extracellular solution is (mM) 144 NaCl, 5.8 KCl, 1.3 CaCl2, 0.9 MgCl2, 0.7 NaH2PO4, 10 HEPES and 5.6 D-glucose, pH 7.4 (with NaOH). When recording the DAB induced currents in HT-29 cells, the extracellular solution contains (in mM) 150 NaCl, 2 CaCl2 (or 0, without EGTA), 10 HEPES, PH 7.3, and the pipette solution comprises (mM) 140 KCl, 10 NaCl, 2 MgCl2, 10 HEPES, 0 Ca2+ (without EGTA or with 5 EGTA), PH 7.3. Cells were held at 0 mV and the currents, elicited by a voltage ramp from -100 mV to +100 mV in 400 ms, were recorded at 5 kHz. The ramp was applied every 1 s. Compounds were applied through a press-driven multichannel system (ALA Scientific Instruments, Farmingdale, NY, USA) with the outlet placed approximately 50 µm away from the cell being recorded.
Immunofluorescence
Immunofluorescence staining was conducted as described previously37. Briefly, after deparaffinization and antigen recovery, sections were blocked with 10% goat serum for 30 min and incubated with primary antibodies overnight at 4 °C. The primary antibodies were rabbit anti-TRPM4 (1:100) and mouse anti-CK-18 (1:200). After washing, sections were incubated with Alexa Fluor 555 goat anti-mouse (1:1,000) or Alexa Fluor 488 goat anti-rabbit (1:1,000) secondary antibodies for 1 h. Hoechst 33342 (10 μg ml−1) was added and incubated for 10 min to stain the nuclei of cells. Fluorescent images were taken using a confocal laser scanning microscope.
Intracellular Ca2+ levels determination
To investigate the DAB effects on intracellular Ca2+ levels, HT-29 cells were loaded with 5 µM Fluo-4/AM (dissolved in Lock’s buffer containing 5 mg ml−1 BSA) for 2 h, and the fluorescence was digitalized using a Flash4.0 V2 digital CMOS camera attached to a Leica DMi8 microscope at a sampling rate of 0.5 or 1 Hz. The excitation wavelength was set at 460–495 nm, and the emitted fluorescence was recorded at 510–550 nm. The arbitrary fluorescence unit of each region of interest (ROI) was quantified. DAB was applied by bulk perfusion after a baseline fluorescence recording. Fluorescent signal at any time point was denoted as F and the mean of the first 5 data points was denoted as F0. Data are expressed as F/F0. Inhibitors were preincubated for 10 min before fluorescence recording.
Human TRPM4 protein expression and purification
The gene encoding human full-length TRPM4 (UniProtKB: Q8TD43) was subcloned into pEG BacMam vector with a 2×Strep tag, GFP and a thrombin-cleavage site at the N terminus38. Bacmid and baculovirus of TRPM4 in a BacMam vector were generated, and P2 viruses were used to infect a suspension of tsA cells. Cells were incubated at 37 °C for 8 h and 10 mM sodium butyrate was added to the culture and the temperature was changed to 30 °C. The cells were collected 72 h after infection and resuspended in a buffer containing 100 mM Tris pH 8.0 and 150 mM NaCl (TBS buffer) in the presence of 1 mM phenylmethylsulphonyl fluoride, 0.8 μM aprotinin, 2 μg ml−1 leupeptin and 2 mM pepstatin A. The cells were lysed by sonication and the membrane fraction was collected by centrifugation at 186,000 g using a 45 Ti rotor (Beckman Coulter) for 1 h at 4 °C. The membrane then homogenized with a Dounce homogenizer in TBS buffer supplemented with protease inhibitors. The protein was extracted from the membrane with TBS buffer supplemented with 1% GDN and protease inhibitors for 3 h at 4 °C. The solubilized proteins were loaded to Strep-Tactin resin. After washing with TBS buffer supplemented with 0.02% GDN, TRPM4 was eluted with the same buffer, supplemented with 10 mM desthiobiotin. The GFP tag was cleaved, and proteins were concentrated and further purified by size-exclusion chromatography (Superose 6). The peak fractions containing the TRPM4 were pooled and concentrated to 8 mg ml−1.
EM sample preparation and data acquisition
Purified TRPM4 was incubated with 5 mM calcium chloride or 5 mM EGTA, as required for each experiment, for 30 s at 37 °C. Then 0.1 mM DAB was added and further incubation for 2 min. 2.5 μl sample was applied to a glow-discharged Quantifoil holey carbon grid (gold, 2/1 μm size/hole space, 300 mesh). The grids were blotted for 1.5 s in the Vitrobot Mark III set to 100% humidity and 37 °C with a 15 s wait time before being plunge-frozen into liquid ethane cooled by liquid nitrogen.
For the Ca2+ and DAB sample, images were recorded using the FEI Titan Krios electron microscope at 300 kV and a nominal magnification of 105,000×. Data were collected on the Gatan K3 Summit direct electron detector in super-resolution mode, resulting in a binned pixel size of 0.828 Å. SerialEM39 was used for automated acquisition. Nominal defocus ranged from −1.2 to −1.9 μm.
For the DAB and EGTA sample, images were recorded on an FEI Glacios electron microscope at 200 kV and a nominal magnification of ×130,000. A Falcon 4i direct electron detector with Selectris energy filter was used resulting a pixel size of 0.91 Å. EPU was used for automated acquisition. Nominal defocus ranged from −0.5 to −1.4 μm.
Cryo-electron microscopy data analysis procedure
The detailed workflow for the data-processing procedure is summarized in Supplementary Fig. 5. In general, the raw movies for each dataset were motion-corrected and 2× binned (for K3 data) using MotionCor2 (v.1.1.0)40. The per-micrograph defocus values were estimated using ctffind (v.4.1.10)41. Particle picking was performed using relion Laplacian-of-Gaussian blob42 and topaz (v.0.2.4)43. Junk particles were removed by rounds of 3D heterogeneous refinement using CryoSPARC44. Good particles were selected for non-uniformed refinement with C4 symmetry in CryoSPARC to generate a 3D map. Multiple rounds of CTF refinement and Bayesian polishing were performed in RELION to further improve the map resolution.
For Ca2+ and DAB dataset, symmetry expansion at the single subunit level was done from the map refined with C4 symmetry, followed by monomer subtraction. The subtracted images of the monomer underwent a round of local refinement with C1 symmetry followed by 3D classification without image alignment in RELION to improve the local resolution of MHR1/4 domain. Similar procedures were performed at the TMD to further increase the local resolution. Map resolution estimates were based on the gold standard Fourier shell correlation 0.143 criterion for all datasets.
Model building
Atomic models were generated by rigid-body fitting of the TMD, MHR1/2 and MHR3/4 and C-terminal domains from a published human TRPM4 model (PDB: 5WP6) into the final cryo-EM maps. Ligands were fitted into the density through real-space refinement using COOT45. The CIF file of DAB was generated using grade web server (Global Phasing Ltd.). The models were then manually adjusted in COOT and subjected to phenix.real_space_refine46 to improve the model metrics. The final models were validated using phenix.molprobity47. Figures were generated using UCSF ChimeraX48.
Statistics and reproducibility
Data were analyzed and plotted using GraphPad Prism software (ver 8.04, GraphPad Software, Inc., San Diego, CA, USA). Concentration-response curves were fitted by a non-linear regression analysis. All values are expressed as Mean ± SEM. One-way or two-way ANOVA followed by post hoc Bonferroni tests were used to examine statistical significance. A value of P < 0.05 was considered statistically significant. Each experiment was repeated at least three times. No statistical method was used to predetermine sample size, and no datapoints were excluded. The experimental procedures were randomized, and the investigators were blinded during experimentation or outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Cryo-EM density maps have been deposited at the EMDB (Electron Microscopy Data Bank) under accession code EMD-72405 (DAB/Ca2+ consensus), EMD-72407 (DAB/Ca2+ subunit), EMD-72406 (DAB/EGTA consensus) and the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCS-PDB) under accession code 9Y2A (DAB/Ca2+ consensus), 9Y2C (DAB/Ca2+ subunit), 9Y2B (DAB/EGTA consensus). This study did not generate new unique reagents. All the other data generated in this study are provided in the Supplementary Information/Source Data file. Source data are provided with this paper.
References
Lucak, S., Lunsford, T. N. & Harris, L. A. Evaluation and treatment of constipation in the geriatric population. Clin. Geriatr. Med. 37, 85–102 (2021).
Corsetti, M., Landes, S. & Lange, R. Bisacodyl: A review of pharmacology and clinical evidence to guide use in clinical practice in patients with constipation. Neurogastroenterol. Motil. 33, 14123 (2021).
Rao, S. S. C. & Brenner, D. M. Efficacy and safety of over-the-counter therapies for chronic constipation: an updated systematic review. Am. J. Gastroenterol. 116, 1156–1181 (2021).
Cantero-Recasens, G. et al. Sodium channel TRPM4 and sodium/calcium exchangers (NCX) cooperate in the control of Ca2+-induced mucin secretion from goblet cells. J. Biol. Chem. 294, 816–826 (2019).
Launay, P. et al. TRPM4 Is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 109, 397–407 (2002).
Amarouch, M.-Y. & El Hilaly, J. Inherited cardiac arrhythmia syndromes: focus on molecular mechanisms underlying TRPM4 Channelopathies. Cardiovasc. Ther. 2020, 6615038 (2020).
Li, K. et al. TRPM4 mediates a subthreshold membrane potential oscillation in respiratory chemoreceptor neurons that drives pacemaker firing and breathing. Cell. Rep. 34, 108714 (2021).
Yu, F. et al. Loss of the TRPM4 channel in humans causes immune dysregulation with defective monocyte migration. J. Allergy Clin. Immunol. 154, 792–806 (2024).
Winkler, P. A., Huang, Y., Sun, W., Du, J. & Lu, W. Electron cryo-microscopy structure of a human TRPM4 channel. Nature 552, 200–204 (2017).
Autzen, H. E. et al. Structure of the human TRPM4 ion channel in a lipid nanodisc. Science 359, 228–232 (2018).
Ekundayo, B. et al. Identification of a binding site for small molecule inhibitors targeting human TRPM4. Nat. Commun. 16, 833 (2025).
Hu, J. et al. Physiological temperature drives TRPM4 ligand recognition and gating. Nature 630, 509–515 (2024).
Tian, Y. & Zheng, J. The TRP channels serving as chemical-to-electrical signal converter. Physiol. Rev. 105, 1033–1074 (2025).
Koh, H., Lee, M. J., Kim, M. J., Shin, J. I. & Chung, K. S. Simple diagnostic approach to childhood fecal retention using the Leech score and Bristol stool form scale in medical practice. J. Gastroenterol. Hepatol. 25, 334–338 (2010).
Nilius, B. et al. Voltage dependence of the Ca2+-activated cation channel TRPM4. J. Biol. Chem. 278, 30813–30820 (2003).
Ruan, Z. et al. Structures of the TRPM5 channel elucidate mechanisms of activation and inhibition. Nat. Struct. Mol. Biol. 28, 604–613 (2021).
Thiagarajah, J. R., Donowitz, M. & Verkman, A. S. Secretory diarrhoea: mechanisms and emerging therapies. Nat. Rev. Gastroenterol. Hepatol. 12, 446–457 (2015).
Seo, Y. et al. Ani9, A Novel Potent Small-Molecule ANO1 Inhibitor with Negligible Effect on ANO2. PLoS. One 11, 0155771 (2016).
Young, P. G., Levring, J., Fiedorczuk, K., Blanchard, S. C. & Chen, J. Structural basis for CFTR inhibition by CFTRinh-172. Proc. Natl. Acad. Sci. USA 121, 2316675121 (2024).
Shi, S. et al. Molecular mechanism of CaCCinh-A01 inhibiting TMEM16A channel. Arch. Biochem. Biophys. 695, 108650 (2020).
Mitznegg, P. et al. Mode of action of bisacodyl (dulcolax) on isolated muscles of human colon (author’s transl). Klin. Wochenschr. 53, 493–495 (1975).
Krueger, D., Demir, I. E., Ceyhan, G. O., Zeller, F. & Schemann, M. bis-(p-hydroxyphenyl)-pyridyl-2-methane (BHPM)—the active metabolite of the laxatives bisacodyl and sodium picosulfate—enhances contractility and secretion in human intestine in vitro. Neurogastroenterol. Motil. 30, 13311 (2018).
Nilius, B. et al. Regulation of the Ca2+ sensitivity of the nonselective cation channel TRPM4. J. Biol. Chem. 280, 6423–6433 (2005).
Nilius, B. et al. The Ca2+-activated cation channel TRPM4 is regulated by phosphatidylinositol 4,5-biphosphate. Embo. J. 25, 467–478 (2006).
Barrett, K. E. & Keely, S. J. Chloride secretion by the intestinal epithelium: Molecular basis and regulatory aspects. Annu. Rev. Physiol. 62, 535–572 (2000).
Lee, B. et al. Anoctamin 1/TMEM16A controls intestinal Cl− secretion induced by carbachol and cholera toxin. Exp. Mol. Med. 51, 1–14 (2019).
Xia, W. et al. The distinct roles of anion transporters Slc26a3 (DRA) and Slc26a6 (PAT-1) in fluid and electrolyte absorption in the murine small intestine. Pflug. Arch. 466, 1541–1556 (2014).
Seidler, U. E. Gastrointestinal HCO3- transport and epithelial protection in the gut: new techniques, transport pathways and regulatory pathways. Curr. Opin. Pharmacol. 13, 900–908 (2013).
Krishnan, S., Ramakrishna, B. S. & Binder, H. J. Stimulation of sodium chloride absorption from secreting rat colon by short-chain fatty acids. Dig. Dis. Sci. 44, 1924–1930 (1999).
Leitner, M. G. et al. Direct modulation of TRPM4 and TRPM3 channels by the phospholipase C inhibitor U73122. Br. J. Pharmacol. 173, 2555–2569 (2016).
Takezawa, R. et al. A pyrazole derivative potently inhibits lymphocyte Ca2+ influx and cytokine production by facilitating transient receptor potential melastatin 4 channel activity. Mol. Pharmacol. 69, 1413–1420 (2006).
Fu, W. et al. Persistent activation of TRPM4 triggers necrotic cell death characterized by sodium overload. Nat. Chem. Biol. 21, 1238–1249 (2025).
Nilius, B., Prenen, J., Janssens, A., Voets, T. & Droogmans, G. Decavanadate modulates gating of TRPM4 cation channels. J. Physiol. 560, 753–765 (2004).
Zhao, F. et al. Surfactant cocamide monoethanolamide causes eye irritation by activating nociceptor TRPV1 channels. Br. J. Pharmacol. 178, 3448–3462 (2021).
Ding, M. et al. Plumbagin, a novel TRPV2 inhibitor, ameliorates microglia activation and brain injury in a middle cerebral artery occlusion/reperfusion mouse model. Br. J. Pharmacol. 182, 87–103 (2025).
Ozhathil, L. C. et al. Identification of potent and selective small molecule inhibitors of the cation channel TRPM4. Br. J. Pharmacol. 175, 2504–2519 (2018).
Li, S. et al. Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2b) mediates oxidation-induced endoplasmic reticulum stress to regulate neuropathic pain. Br. J. Pharmacol. 179, 2016–2036 (2022).
Goehring, A. et al. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat. Protoc. 9, 2574–2585 (2014).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Scheres, S. H. W. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Bepler, T., Kelley, K., Noble, A. J. & Berger, B. Topaz-Denoise: general deep denoising models for cryoEM and cryoET. Nat. Commun. 11, 5208 (2020).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D. Struct. Biol. 74, 531–544 (2018).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 66, 12–21 (2010).
Pettersen, E. F. et al. UCSF chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Acknowledgements
We thank for the support with data collection at the David Van Andel Advanced Cryo-Electron Microscopy Suite and Structural Biology Facility at Northwestern University. We appreciate the high-performance computing team of VAI, structural biology facility and Quest High-Performance Computing Cluster at Northwestern University for computational support. This project is jointly supported by National Natural Science Foundation of China (82373929, 81972960, Z.C.), Major Program of Jiangsu Provincial Administration for Market Regulation (KJ2024014, Z.C.), Double First-Class University project (CPU2025CXY02, Z.C.), The Natural Science Foundation of Jiangsu Province of China (BK20241599, C.X.), China Postdoctoral Science Foundation (2024M763667, C.X.), and the Fundamental Research Funds for the Central Universities (2632024PY10, C.X.), a McKnight Scholar Award (J.D.), a Klingenstein-Simon Scholar Award (J.D.), a Pew Scholar in the Biomedical Sciences Award (J.D.), a Sloan Research Fellowship in neuroscience (J.D.), Van Andel Institute and Northwestern University startup funds (J.D. and W.L.), and American Heart Association Postdoctoral Fellowship (24POST1196982, J.H.). Figures were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
Conceptualization: W.L., J.D., Z.C.; Data curation: Y.L., H.J., C.X., W.H., S.I., Z.C.; Formal analysis: Y.L., H.J., C.X., W.H., S.I., Z.C.; Investigation: Y.L., H.J., C.X., W.H., S.I.; Funding acquisition: C.X., W.L., J.D., Z.C.; Project administration: W.L., J.D., Z.C.; Resources: W.L., J.D., Z.C.; Supervision: W.L., J.D., Z.C.; Validation: C.X., W.L., J.D., Z.C.; Writing–original draft: Y.L., H.J., C.X., W.H.; Writing–review & editing: W.L., J.D., Z.C.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Y., Hu, J., Xue, C. et al. Noncanonical calcium-independent TRPM4 activation governs intestinal fluid homeostasis. Nat Commun 17, 1253 (2026). https://doi.org/10.1038/s41467-025-68014-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68014-7
