
Abstract
Susceptibility transcription factors (TF) whose DNA bindings are altered by genetic variants regulating colorectal cancer (CRC) risk genes remain poorly defined. Using generalized linear mixed models, we analyze 218 TF ChIP-Seq datasets alongside GWAS data from 100,204 CRC cases and 154,587 controls of East Asian and European ancestries. We identify 51 TFs and TF-cofactor interactions, including VDR-cofactors, as key regulators of CRC risk. Integrating these TF insights with transcriptome-wide association studies (TWAS), we further evaluate associations between genetically predicted gene expression, alternative splicing, and alternative polyadenylation with CRC risk, using RNA-seq data from 364 Asian-ancestry and 707 European-ancestry individuals. Multi-ancestry TWAS identify 222 risk genes, including 95 novel genes and 48 potentially druggable targets. Single-cell analysis provides additional functional evidence supporting ~45% of these genes, and experimental validation confirms oncogenic roles for RHPN2, IRS2, and TXN. Our findings elucidate key TF–gene regulatory networks and uncover novel CRC risk genes.
Similar content being viewed by others

Introduction
Transcription factors (TFs) play a pivotal role in mediating the effects of genetic risk variants by regulating gene expression in a context-specific manner, thereby influencing disease risk1,2. A better understanding of the regulatory mechanisms by which TFs mediate these genetic variants is critical for elucidating the molecular basis of complex traits, including cancer. Previous studies have identified susceptible TFs through enrichment analyses of genome-wide association studies (GWAS)-identified risk variants in TF binding sites through chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq)3,4,5,6. However, these approaches often suffer from suboptimal statistical power, as they focus primarily on a narrow range of GWAS-identified risk variants. Recently, we developed a statistical method using a generalized linear mixed model (GLMM) to enhance power by analyzing TF ChIP-seq data in conjunction with GWAS summary statistics of variants across the genome7. This method evaluated associations between variants’ risk (e.g., Chi-squared values from GWAS) and their binding status by a TF, and has identified known susceptible TFs, such as FOXA1, ESR1 and GATA3 for breast cancer, by showing significantly higher risk for variants occupied by these TFs compared to genome-wide controls7. However, the specific TFs involved in colorectal cancer (CRC) susceptibility remain unclear. Thus, identifying TFs that interact with CRC risk variants could provide insights into TF-mediated gene regulation and enhance our understanding of CRC susceptibility.
GWASs have successfully identified more than 250 risk loci for CRC1,2,8, and integrative analyses of expression quantitative trait loci (eQTL) and these GWAS loci have revealed numerous candidate susceptibility genes9,10,11,12. Further transcriptome-wide association studies (TWAS) have linked predicted gene expression levels with disease risk, offering higher statistical power and potentially revealing novel associations overlooked in GWAS13,14. Prior research, including our own, has conducted TWAS in CRC and identified more than 200 putative susceptibility genes8,15,16. However, conventional TWAS methods are prone to false positives due to the inclusion of non-regulatory variants17,18,19. To address this, we developed the sTF-TWAS approach, which refines gene expression prediction by focusing only on regulatory variants within TF-occupied cis-regulatory elements (STFCREs)7,20. This approach significantly enhances disease gene detection, outperforming conventional TWAS methods20. In addition, our recent alternative polyadenylation (APA)-WAS framework has uncovered new cancer risk genes21, while its application to CRC has yet to be explored. Moreover, while TWAS has primarily been conducted in European populations, CRC studies in East Asian populations are limited due to a lack of relevant RNA-seq data from colon tissue.
In this study, we integrate 218 ChIP-seq datasets from CRC-related cells with GWAS data from 100,204 CRC cases and 154,587 controls of both European and East Asian ancestries to identify susceptible TFs for CRC. We further utilize RNA-seq data from 364 Asian-ancestry and 707 European-ancestry individuals to build ancestry-specific models for gene expression, alternative splicing (AS), and APA) under the sTF-TWAS framework. Our goal is to identify novel CRC susceptibility genes through multi-ancestry TWAS analyses, providing a deeper understanding of the genetic basis of CRC.
Results
Identification of susceptible CRC-associated TFs and key TF-cofactor interactions
We integrated 218 ChIP-seq data for 84 TFs (Supplementary Data 1) with CRC GWAS summary statistics from European and East Asian ancestry to identify CRC susceptible TFs (Fig. 1A). At Bonferroni-corrected P < 0.05, we identified 51 TFs associated with CRC susceptibility. Among the top TFs were VDR, MYC, JUN, CDX2, NIPBL, MED12, and ETV5 (Fig. 1B and Supplementary Data 2, “Methods”). We conducted motif analysis for each TF-binding region derived from ChIP-seq data and observed significant enrichment for host motifs, including those for VDR (24.2%), JUN (11.1%), KLF5 (49.7%), and HNF4A (36.4%) (Fig. 1C). We further explored the impact of genetic variations in co-occupied cis-regulatory regions of these 51 TFs and observed a substantial proportion of genetic variants located in co-occupied binding sites (Fig. 1D and Supplementary Data 3). Pair-wise analyses identified 154 significant interactions at Bonferroni-corrected P < 0.05, showing that genetic variants in regions co-occupied by multiple TFs exhibited stronger associations with CRC risk compared to variants bound by a single TF (Fig. 1D and Supplementary Data 3). Notably, VDR emerged as a key player in these interactions, highlighting its central role in CRC susceptibility (Fig. 1D and Supplementary Data 3). In addition, we applied stratified LD Score Regression (S-LDSC)22 as an alternative approach and found that 40 of the 51 risk TFs identified by our method were also detected by S-LDSC at a Bonferroni-corrected P < 0.05. Most of the remaining 11 TFs showed nominal significance (P < 0.05) (Supplementary Fig. 1A), providing further support for the validity of our findings. We also conducted simulation analyses to compare the statistical power of our GMM with S-LDSC. Our results demonstrate that GLMM consistently outperformed S-LDSC in detecting causal TFs across varying levels of heritability (Supplementary Fig. 1B). Quantile-quantile (Q-Q) plots demonstrate that Type I error rates are under control for both approaches, with overall comparable performance (Supplementary Fig. 1C–F). These simulation results are consistent with our observations in real data, supporting GLMM as a robust approach for identifying risk TFs in colorectal cancer.

A A flow chart to illustrate the integrative analysis of ChIP-seq data (n = 218) for 84 TFs and CRC GWAS summary statistics from 100,204 cases and 154,587 controls of European and East Asian ancestry. B A total of 51 identified TFs with genetic variation of TF-DNA bindings significantly associated with CRC risk. P-values were determined by a two-sided Wald Z test. The dashed line represents a Bonferroni-corrected P < 0.05. C The host motifs of identified TFs were enriched in their ChIP-seq peaks. D Analysis of co-occupied binding regions of the top 10 CRC risk-associated TFs. Venn diagrams in the upper-right triangle show the number of genetic variants (multiplied by 1000) that are occupied by specific TFs or co-occupied by two TFs in each TF pair. Bar plots in the lower-left triangle show the association strengths (regression coefficients) for the genetic variants occupied by two TFs (only the first TF and only the second TF, respectively) as indicated from left to right. Two TFs with significant interactions at the Bonferroni-threshold of P < 3.92 × 10−5 (0.05/1,275 TF pairs from 51 TFs) are highlighted in red. P-values were determined by a two-sided Wald Z test.
Associations of genetically predicted gene expression with CRC risk
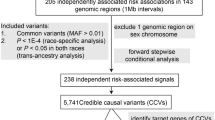
For the multi-ancestry TWAS under the sTF-TWAS framework, we built gene expression prediction models using RNA-seq data from 364 participants of East Asian ancestry (from the ACCC) and 707 participants of European ancestry (N = 423 from the BarcUVa-seq and N = 284 from the GTEx projects) (Methods)16. A total of 7096 (for ACCC), 14419 (for BarcUVa-seq), 11361 (for GTEx) gene expression prediction models were built with a coefficient of determination R2 > 0.01 (10% correlation) using the elastic net approach (Supplementary Data 4). We then applied these ancestry-specific prediction models to summary statistics of CRC GWAS from 80,774 cases and 105,298 controls of European ancestry and from 23,572 cases and 48,700 control of East Asian ancestry. At a Bonferroni-corrected P < 0.05, we identified 138 genes whose genetically predicted expression was associated with CRC risk after applying a meta-analysis of TWAS results based on models built in both European and East Asian populations (Supplementary Fig. 2, “Methods”). In addition, we evaluated the associations separately in European and Asian populations. At the Bonferroni-corrected threshold, we identified two additional genes (SENP8 and PCNT) from the European population, while no additional significance was identified in the East Asian population (Supplementary Data 5).
Together, 140 genes showed an association between their genetically predicted expression and CRC risk (Fig. 2A). Of them, 41 (29% of 140) genes were not previously reported to associate with CRC risk1,2,8,10,15,16 (Table 1). Of note, seven putative susceptibility genes (PPAN, DERL3, PROX1, TMEM87B, SV2A, FBXL20, and MED1) are located at loci, with > 2 Mb away from the closest GWAS-identified risk variants (Table 1). To assess whether the genes identified were independent of the established GWAS association signals, we conducted conditional analyses for their associations with CRC risk, adjusting for the associations with the closest lead SNP for each locus (“Methods”). Of the 99 previously reported genes located in GWAS-identified risk loci, conditional analysis showed that 39 genes remained statistically significant at a nominal P < 0.05 (Supplementary Data 6).

A The meta-analysis result from the associations among European ancestry and East Asian ancestry participants. P-values were calculated from a two-sided Wald Z test conducted by sTF-TWAS. The blue line indicates a Bonferroni-threshold of nominal P < 5.8 × 10−6 for TWAS (A), and P < 1.95 × 10−6 for AS-WAS (B), and P < 8.25 × 10−6 for APA-WAS (C), respectively. The novel identified risk genes from this study are highlighted in green. Four genes showed association signals with P-values exceeding the y-axis scale: POU5F1B (P = 1.09 × 10⁻⁶⁴) in (A); COLCA2 (P = 5.49 × 10⁻⁴⁵) in (B); LAMC1 (P = 9.57 × 10⁻²⁴) and DIP2B (P = 1.25 × 10⁻²²) in (C).
Additional risk genes identified by AS-WAS and APA-WAS
We built prediction models for AS and APA using the aforementioned RNA-seq data in colon tissues, generating 9680 AS models for ACCC, 10,025 for BarcUVa-seq, and 20,240 for GTEx, at prediction performance R2 > 0.01 (Methods, Supplementary Data 7). A meta-analysis of AS-WAS results from these models, we identified 82 risk genes at a Bonferroni-corrected threshold P < 0.05 (Fig. 2B). We identified three additional genes, POLD3, PARP11, and CEP89, specific in European populations (Supplementary Data 8). In total, we identified 85 genes after combining the results from the meta-analysis and population-specific analysis. For APA, a total of 2569 (for BarcUVa-seq), 4399 (for GTEx), and 2744 (for ACCC) prediction models were built successfully with R2 > 0.01 (Supplementary Data 9). After meta-analysis of APA-WAS results from these models, we identified 29 risk genes, at a Bonferroni-corrected P < 0.05 (Fig. 2C). Similarly, we identified four additional genes (ARPC5, PHLDB2, COA4, NEIL1) specifically in the European population (Supplementary Data 10). In total, we identified 33 genes after combining the results from the meta-analysis and population-specific analysis.
In total, 112 genes were identified for CRC risk combining results of SP-TWAS and APA-WAS. Of them, 58 were not previously reported for CRC risk (Table 2, 3). Among the remaining 54 previously reported genes, conditional analysis showed that 24 genes remained statistically significant at a nominal P < 0.05 (Supplementary Data 11).
Risk genes identified by combining TWAS, AS-WAS, and APA-WAS analyses
By integrating results from TWAS, AS-WAS, and APA-WAS, we identified 222 unique CRC risk genes, 95 of which had not been previously associated with CRC (Fig. 2). Among them, four genes (COLCA2, METRNL, ARPC5, COX14) were commonly identified by all three TWAS analyses and 28 genes commonly detected by any two of the TWAS analyses (Supplementary Fig. 3). By comparing genes reported in previous CRC TWAS and other genetic studies1,2,8,10,15,16, 95 of them were not previously identified as CRC susceptibility genes, particularly including 13 genes reported in new loci (Tables 1–3).
Using scRNA-seq data to explore risk genes potentially contributing to CRC development
We next analyzed scRNA-seq datasets spanning CRC developmental stages, including colon normal, serrated polyps, conventional adenomas, and carcinomas tissues from the COLON MAP23. Our scRNA-seq data analysis identified several major cell types (e.g., ABS, GOB, STM) in normal tissues, SSC in serrated polyps, and ASC in conventional adenomas, consistent with previous studies23 (Supplementary Fig. 4). Differential analysis of these major cell types was performed using DESeq224 to evaluate their contribution to two CRC etiology pathways: the normal-serrated polyp-carcinoma pathway and the normal-adenoma-carcinoma pathway (“Methods”). At an FDR-adjusted P < 0.05, 48 genes exhibited significantly differential expression with consistent associations from TWAS analysis across one of these major cell types in the normal-serrated polyp-carcinoma pathway (Supplementary Data 12), while 92 genes showed significant differential expression in the normal-adenoma-carcinoma pathway (Supplementary Data 13). Combined with these results, we provide additional evidence that 99 genes (44.6% of the identified 222 risk genes) are potentially involved in CRC development.
Druggable genes for CRC prevention and intervention
We comprehensively annotated 222 identified risk genes as therapeutic targets for existing drugs, including those either approved or in clinical development (“Methods). Our analysis identified nine CRC susceptibility genes that are druggable by 125 drugs approved or in clinical trials for CRC treatment (Fig. 3 and Supplementary Data 14). In addition, we identified 26 genes targeted by 519 drugs approved or in clinical trials for the treatment of other cancers (Supplementary Data 15). Furthermore, 40 genes were identified as targets of 585 drugs approved or in clinical trials for non-cancer indications (Supplementary Data 16). In total, 48 of the 222 CRC susceptibility genes (21.6%) were found to be druggable, with potential links to 1216 drugs, either approved or under investigation.

The innermost to outermost layers represent CRC, the identified druggable risk genes, and therapeutic drugs - approved (green) or currently under clinical trials (Phase 3: yellow, Phase 2: red).
Functional assays for three putative oncogenes, RHPN2, IRS2, and TXN
To functionally examine the CRC susceptibility genes, we performed CRISPR/Cas9-mediated gene knockout. Three risk genes, including one previously reported gene, RHPN2, and two newly identified risk genes, IRS2 and TXN, were selected for downstream experiments based on evidence of their potential oncogenic functions and literature review (Methods). This was supported by their predicted elevated expressions being associated with an increased risk of CRC, as well as differential analysis of single-cell data highlighting their oncogenic roles. For each gene (RHPN2, IRS2, or TXN), we employed two independent sgRNAs for protein coding region targeting (Supplementary Data 17). We used western blotting for confirming target protein reduction and CCK-8 proliferation and colony formation (Fig. 4) assays for gene’s functions. TXN proteins displayed 65-98% reductions between knockout cells and control cells (sgRNA targeting LacZ) in the CRC cell lines (RKO, HCT116, and SW480) (Supplementary Fig. 5). We showed that TXN knockout significantly inhibited cell proliferation and colony formation in all CRC cell lines (Fig. 4A–C). IRS2 proteins had 66–86% reductions between knockout cells and control cells in three CRC cell lines (Supplementary Fig. 5). There was only significant inhibition of colony formation (Fig. 4E, F), indicating a long-term effect of knocking out the IRS2 protein. RHPN2 proteins had 34–61% reductions between knockout cells and control cells (Supplementary Fig. 5). However, the data showed a significant inhibition of cell proliferation and colony formation only in RKO cells, together with a trend of inhibition in HCT116 cells (Fig. 4G–I). There was a minimal effect in SW480 cells by knocking out RHPN2. The variable extents of RHPN2 dependency in cell proliferation and growth were matched to the consistent protein expression levels in these cells. Our experimental verification demonstrated the potential oncogenic functions of TXN, IRS2 and RHPN2, aligning with our TWAS analysis.

CRC cell lines with knocking down by two sgRNAs for each gene were compared to CRC cell lines with a non-targeting sgRNA control. Cell viability was detected with CK8 assay for TXN (A), IRS2 (D), and RRHPN2 (G). Colony formation assays were performed with cell stained by crystal violet and were further quantified for TXN (B and C), IRS2 (E and F), and RRHPN2 (H and I). The independent experiments were performed in triplicate. P-values were determined by two-sided t test from the comparison of knockdown and control cells. “*”, P < 0.05; “**”, P < 0.01; “***”, P < 0.001. The height of each bar represents the mean value across replicates, and the error bar indicates the standard error of the mean.
Discussion
Key TFs transcribed in a cell type-specific manner and co-occupy cis-regulatory elements that control gene expression programs to establish and maintain cell identity25,26,27,28,29. Identifying these TFs, whose DNA-binding activities are altered by risk-associated genetic variations, and understanding the genes they regulate, is crucial for advancing our knowledge of transcriptional dysregulation in human cancers30,31,32,33. In this study, we identified 51 TFs associated with CRC susceptibility, advancing our understanding of key TF–gene regulatory networks underlying CRC etiology. By incorporating prior knowledge on TF-binding sites for CRC into the sTF-TWAS framework, we demonstrated that our approach significantly improves predictive model construction and facilitates the discovery of risk genes. Compared to traditional TWAS analysis in our previous study16, the sTF-TWAS method improved the prediction of gene expression and AS, increasing the number of predicted genes by approximately 1500 and 2000, respectively. Moreover, sTF-TWAS identified 94 additional genes beyond those detected by the traditional TWAS and AS-WAS. For the APA analysis, we integrated RNA-seq data from ACCC and BarcUVa-seq, compared to our previous study relying on GTEx data, identifying 30 novel risk genes21. Notably, the sTF-TWAS analysis, using these diverse prediction models, complements and strengthens risk gene discovery. Among the 222 identified risk genes, only four genes (COLCA2, METRNL, ARPC5, COX14) were detected by TWAS, AS-WAS, and APA-WAS. This observation aligns with prior research showing that different molecular traits within the same gene can represent largely independent signals21,34. For these four commonly detected genes, we found that very few shared variants were observed among them, indicating genetic heritability may independently contribute to each molecular trait, while a small proportion of shared risk variants may influence expression, splicing, and APA through interconnected mechanisms under multi-layered regulatory control.
Our sTF-TWAS analysis highlighted risk TF-based transcriptional networks underlying genetic susceptibility to CRC. By examining TF occupancy information for the genetic variants included in the prediction model, we found that 117 of the 222 putative susceptibility genes (52.7%) are commonly regulated by the top five TFs (RAD21, CBX3, MAX, SP1, and POLR2A). In addition, we conducted a new TF enrichment analysis on these 222 genes using publicly available TF ChIP-seq data from the Enrichr35,36. This analysis identified four CRC-associated TFs (VDR, EP300, MYC, and SOX2) significantly enriched at FDR < 0.05, and an additional set of 17 TFs (Supplementary Data 18) enriched at nominal P < 0.05. Of note, among the 222 risk genes, several TFs themselves, such as MYC, PROX1, SOX9, KLF5, and RAD21, were identified as significant genes. It is worth noting that most CRC-associated TFs were not detected by TWAS, likely because TWAS captures genetic heritability primarily from cis-variants. However, our GMM analysis using genome-wide variants provided evidence of their susceptibility. Taken together, these findings underscore the role of TFs and TF-based transcriptional networks in CRC genetic susceptibility. Moving forward, in our prediction models, we identified 144 lead variants—representing the strongest associations with CRC risk for each model, at P < 5 × 10⁻⁸. These variants are predicted to influence the binding affinity of CRC-associated TFs and are linked to 162 risk genes (Supplementary Data 19). Taken together, these findings offer valuable insights for prioritizing functional studies aimed at fine-mapping causal variants and genes for CRC through in vitro and in vivo assays. Our integration of scRNA-seq data analyses enhances causal gene discovery and improves our understanding of the cellular processes driving CRC. We acknowledge that some genes identified through differential expression analysis across developmental stages may not reflect true causal associations, particularly given the high correlation among many gene expression levels. In the trans-ancestry TWAS analysis, meta-analysis improved statistical power, enabling robust gene prediction in both East Asian and European populations. Notably, 37 CRC risk genes identified through meta-analysis exhibited consistent associations with CRC risk (P < 0.05) across both populations, providing strong evidence for these genes as potential causal factors for CRC (Supplementary Data 5, 8, 10).
Our identification of risk genes has significantly advanced our understanding of CRC biology and etiology. Among the 222 identified genes, nine are known CRC driver genes, including CDH1, ERBB2, KLF5, SOX9, SRSF6, TBX3, TET2, TRPS1, and ZNRF3, highlighting their genetic contributions to CRC development37,38,39,40. We also uncovered seven previously unreported CRC risk genes that map to six established pathways crucial for CRC development41,42,43,44. Specifically, PPP2R1B regulates both the Wnt/β-catenin and TGF-β signaling pathways, while RICTOR modulates the PI3K-AKT pathway, a key driver of cell proliferation and survival. RAD21 is involved in both the Cell Cycle and Mismatch Repair pathways, reinforcing its role in genomic maintenance. NFATC2 supports MAPK signaling and DNA mismatch repair, while ARRB1 and MAPK13 further emphasize the significance of MAPK signaling. RTEL1 strengthens the Mismatch Repair pathway. In addition to these established oncogenic pathways, we identified three novel oncogenes that enhance our understanding of CRC susceptibility. In addition to the established CRC oncogenic pathways, we provided strong evidence of three oncogenes to advance the understanding of CRC susceptibility. RHPN2, a Rho GTPase-binding protein that regulates the cell cytoskeleton, may contribute to cancer cell malignant growth45. IRS2 has been shown to activate the oncogenic PI3 kinase pathway and increase CRC cell lines adhesion (signal of invasive cells)46. Moreover, over-expression of IRS2 is also observed in many other cancers47,48 and its amplification has been proposed as a biomarker for inhibitor-based cancer therapy49. TXN, a redox protein, may regulate antioxidant reactions that promote cancer cell metabolism and survival50,51. In our prior work, we have validated the functionality of another five reported genes, DIP2B, SFMBT1, TRPS1, METRNL, and C14orf166, using siRNA/shRNA-mediated gene knockdown cell lines in vitro10,16. Furthermore, public CRISPR-Cas9 data from the Cancer Dependency Map (DepMap) provided additional evidence for the essential roles of 21 risk genes in colorectal adenocarcinoma cell proliferation (Supplementary Data 20). Together, these discoveries expand the repertoire of CRC susceptibility genes and reveal previously unexplored molecular mechanisms underlying CRC development.
Through the analysis of drug-protein interaction databases52,53,54,55, we identified nine druggable proteins that are either approved or in clinical trials for CRC treatment. Notably, six of these genes—DDR1, SIK2, EHMT2, MAPK13, PKM and PARP11—are novel CRC risk genes reported in this study. Among these six novel genes, PKM2 from PKM family, a member of the PKM family, has been reported to mediate VEGF-induced angiogenesis in human endothelial cells. Resveratrol inhibits this process by suppressing aerobic glycolysis, potentially through modulation of PKM2 nuclear translocation56. PARP11, implicated in DNA repair, can be inhibited by approved PARP inhibitors, including Olaparib, Niraparib, and Talazoparib57. However, these results are observations are exploratory and require further validation. Overall, our protein–drug mapping expands the catalog of druggable genes, candidate therapeutics and supports the development of precision medicine strategies for CRC prevention and intervention.
Methods
Study populations
This study utilized a meta-analysis summary statistics dataset of 31 GWAS comprised of 186,072 individuals of European ancestry (80,774 CRC cases and 105,298 controls) and 72,272 individuals of Asian ancestry (23,572 CRC cases and 48,700 controls). The meta-analysis was conducted using the fixed-effects inverse variance weighted model implemented in METAL58. Details on sample selection and matching, sample numbers, and demographic characteristics of study participants have been described previously59,60,61,62. All participants provided written informed consent, and all study protocols were approved by the relevant research ethics committee or institutional review board.
We included RNA-seq data from tumor-adjacent normal colon tissues and genotyping data of 364 individuals of East Asian ancestry who participated in the Asia Colorectal Cancer Consortium (ACCC). We also included data from individuals of European ancestry, including RNA-seq in normal colon tissues and genotyping data of 423 individuals in the BarcUVa-Seq project and 284 individuals in the GTEx project. The sample collection for these data has been described in detail in previous studies1,63,64.
Generalized linear mixed models
We systematically analyzed TF ChIP-seq data generated in normal colorectal epithelium and CRC cell lines from the Cistrome database65. Only ChIP-seq datasets meeting Cistrome’s recommended quality control standards were included. After evaluation, we curated 218 ChIP-seq datasets corresponding to 84 TFs. To ensure robust analysis, we focused on datasets with more than 1000 peaks and a fold change greater than 10. For TFs with multiple ChIP-seq datasets, only the dataset with the highest number of peaks was retained. This process resulted in a high-quality set of ChIP-seq data for 69 TFs, which were used in downstream analyses.
We used our recently developed generalized linear mixed model approach7 to investigate associations of CRC risk with variations of TF-DNA binding by a single TF (Eq. 1). In brief, a NxM matrix for all genetic variants from the GWAS summary statistics and the annotation from all available TF-DNA binding regions (“N” refers to the number of genetic variants and “M” refers to the number of TF ChIP-seq datasets; the variant in ChIP-seq peak is coded as “1” and the one not in ChIP-seq peak is coded as “0”). We used the Chi-squared value for each genetic variant reported in the GWAS summary data to measure its association with CRC risk. We then used generalized linear mixed models to estimate the associations between the Chi-squared values (Y) and TF binding status of genetic variants located in binding sites of each TF, given LD blocks (non-overlapping segments of 100 kb) of genetic variants to handle the dependence between genetic variants (Eq. (1)).
In Eq. (1), \({Y}_{{ij}}\) is the Chi-squared value for the j-th variant in the i-th LD block; β0 is the fixed intercept, and β1 is the fixed slope, which measure the mean difference of the Chi-Squared values (\(\Delta {\bar{X}}^{2}\)) between TF status; \({{TF}}_{{ij}}\) is the j-th TF value (i.e., 1 for a variant located in a TF binding site, 0 otherwise) in the i-th LD block; \({V}_{i}\) is the random intercept for the i-th LD block; and εij is the error term. Based on this statistical model, we identified cis-regulatory elements occupied by TFs whose genetic variations of TF-DNA bindings are associated with target cancer risk at Bonferroni-corrected P < 0.05. We additionally used generalized linear mixed models to estimate the associations of the Y values of variants with the TF-pair occupancy if they showed a significant interaction (Eq. (2)). Of note, we used genetic variants non-occupied by any of the TFs investigated as the referenced control group for all the analyses.
Simulation studies
To benchmark the statistical power of GLMM against S-LDSC, we conducted simulations under a polygenic causality scenario where genotypes influence phenotypes through TF binding sites. Specifically, we assumed that only SNPs located within TF binding sites contribute to disease risk, while SNPs outside these regions have no effect.
For each replicate, we randomly selected K causal variants (K = 500, 1000, 5000, or 10,000) from a pool of 600,000 SNPs annotated to 84 TFs in CRC cell lines (Supplementary Data 1). Under an additive architecture, phenotypes were simulated using Eq. 3:
where, \({{{\rm{X}}}}_{{{\rm{ij}}}}^{{{\rm{TF}}}}\) is the genotype (0,1,2) of j-th variant for subject i use data from 489 European ancestry from1000G project phase 366. β1 was set to 1 and \({{{\rm{\varepsilon }}}}_{{{\rm{j}}}}\) is sampling from a normal distribution \({{\rm{N}}}\left(0,\frac{1-{{{\rm{h}}}}^{2}}{{{{\rm{h}}}}^{2}}{\mathrm{var}}\left({\sum }_{{{\rm{j}}}=1}^{{{\rm{K}}}}{{{\rm{\beta }}}}_{1}\times {{{\rm{X}}}}_{{{\rm{ij}}}}^{{{\rm{TF}}}}\right)\right)\), with heritability \({{{\rm{h}}}}^{2}\,\)= 0.5, 0.25, 0.1, 0.05 and \({\mathrm{var}}\left(\mathop{\sum }_{{{\rm{j}}}=1}^{{{\rm{K}}}}{{{\rm{\beta }}}}_{1}\times {{{\rm{X}}}}_{{{\rm{ij}}}}^{{{\rm{TF}}}}\right)\) is the variance from the genotype part.
To facilitate downstream GWAS, we binarized the continuous trait into a case–control phenotype \({{{\rm{C}}}}_{{{\rm{i}}}}^{*}\) based on the mean value of \({{{\rm{Y}}}}_{{{\rm{i}}}\,}^{*}\) across individuals using Eq. (4).
\({{\rm{We}}}\; {{\rm{performed}}}\; {{\rm{GWAS}}}\) using PLINK 2.067 and applied both GMM and S-LDSC to the resulting summary statistics to estimate TF–disease associations. For each K, we conducted 50 replicates, each with a different random subset of causal SNPs, ensuring reproducibility while keeping K fixed. Results were summarized as the number of true-positive TFs detected at a nominal threshold (P < 0.05) by each method.
To evaluate the type I error rate of GLMM, we conducted simulation analyses under the null hypothesis of no association between genotype and phenotype. Specifically, we randomly assigned binary phenotype values (0 or 1) to 489 individuals of European ancestry and performed logistic regression to generate GWAS summary statistics under the null. We then applied GLMM and S-LDSC using different numbers of TF binding variants (500, 1000, 5000, and 10000) and repeated the entire procedure 1000 times. Finally, we compared the Type I error rates of GLMM and S-LDSC using quantile-quantile (QQ) plots of p-values across the 1000 replicates for each binding variant set size.
Gene expression profiling and data processing
The RNA-seq library preparation, sequencing, data processing, and quality control for the ACCC1 and the BarcUVa-Seq project63 have been described previously. In brief, paired-end sequencing was conducted for mRNA obtained from the BarcUVa-Seq project using either an Illumina HiSeq 2500 or a NovaSeq 6000 instrument and for mRNA obtained from the ACCC project using a HiSeq 3000 Sequencer. The raw RNA-seq data were processed following the GTEx Consortium pipeline64. BAM files containing aligned reads to the reference genome (GRCh37 for ACCC and GRCh38 for BarcUVa-Seq and GTEx project), were generated using STAR68 (v2.5.4). These BAM files, along with GENCODE release 19 (for ACCC) and 26 (for BarcUVa-Seq and GTEx) were used for quantification of gene expression, AS and APA.
Building on our previous work in processing genet expression and AS16, gene expression quantification was performed using RNA-SeQC69. Read counts and Transcripts Per Kilobase Million (TPM) values were calculated from aligned reads based on the following criteria: (1) reads were uniquely mapped; (2) aligned reads were properly paired; (3) the read alignment distance was < 6. Genes with expression thresholds of ≥ 0.1 TPM in ≥ 20% of samples and ≥ 6 reads (unnormalized) in ≥ 20% of samples were selected. Quantile normalization was applied to standardize the gene expression data within each sample to the same distribution, followed by rank-based inverse normal transformation across all samples.
AS was quantified using LeafCutter70 and RegTools71. In brief, the intron usage was quantified from aligned BAM files using the junction extract from the RegTools with the default options. Next, the introns were clustered together using the leafcutter_cluster_regtools.py script from LeafCutter, with the following options: -m 50 -l 500000. The clusters were mapped to each gene using the map_clusters_to_genes.R script from the cluster_prepare_fastqtl.py provided by the GTEx. Introns with low counts or low complexity were filtered based on the criteria in the in the GTEx pipeline (e.g., cluster_prepare_fastqtl.py). The filtered counts were normalized using the prepare_phenotype_table.py script from LeafCutter.
For APA analysis, following our recent APA-WAS study21, APA was qualified using the percentage of PDUI estimated from DaPars v2.021. We performed quantile normalization to transform the quantified PDUI values of APA for each sample to the same distribution. We used 60 probabilistic estimation of expression residuals (PEER) factors72 to correct batch effects and experimental confounders in downstream prediction model building for gene expression, AS and APA.
Genotyping data processing
The generation and processing of genotype data have been described in our previous work1,16. Briefly, CRC patients of East Asian ancestry from the ACCC were genotyped using the Illumina OncoArray and the expanded Illumina MEGA Array. For genotype quality control, genetic variants were excluded based on the following criteria: (1) genotype call rate < 95%; (2) ambiguous variants; (3) duplicated variants; (4) P for Hardy-Weinberg equilibrium (HWE) < 1.0 × 10−6. For quality control of samples, we excluded those with: (1) genotype call rate < 95%; (2) genetically identical or duplicated samples; (3) first- or second-degree relatives; (4) racial outliers. To estimate the genetic relatives among samples, the quality-controlled common SNPs (minor allele frequency [MAF] > 0.01) were first pruned based on the window size = 1500 SNPs and pairwise r2 > 0.2. Identity-by-descent (IBD) was calculated using the pruned genotype data. Quality-controlled genotypes were imputed using the 1000 Genomes Project Phase 3 mixed reference haplotypes with the Michigan Imputation Server (Minimac4 for imputation and Eagle v2.4 for phasing). Variants with MAF < 0.01 were excluded before imputation. Approximately 4.5 million variants with MAF > 0.05 and R2 > 0.3 were included for prediction model building.
For the BarcUVa-Seq project, approximately 400,000 SNPs were genotyped from DNA isolated from peripheral blood samples, using the OncoArray BeadChip. Quality controls and genotyping data processing details were provided in a previous study63. Genotypes were aligned to the reference genome GRCh38 and imputed using the TOPMed (Version R2) reference panel63. Genetic variants with an imputation quality of R2 < 0.3 were excluded.
In the GTEx project, whole genome sequencing (WGS) was performed on DNA samples from 284 individuals of European ancestry. WGS-based genotype data for these individuals, in VCF format, were accessed from the dbGap (phs000424.v8.p2). Detailed information on WGS variant quality control is provided in GTEx documentation64.
Conditional analyses based on adjusted GWAS association signals
To investigate whether the identified associations between genetically predicted gene expression (or AS, APA) and CRC risk are independent of the association signals identified in previous GWAS, we performed conditional analyses by adjusting for the lead variant using the GCTA-COJO method73. The lead variant in each risk locus was determined based on the strongest association from GWAS summary statistics derived from the meta-analysis of European and East Asian populations or from Europeans alone. Genotype data from either European or East Asian populations in the 1000 Genomes Project Phase 3 were used to estimate linkage disequilibrium (LD). We then conducted TWAS analyses (or AS, APA) using the adjusted GWAS summary statistics to explore the association between genetically predicted gene expression and CRC risk. In addition, joint analyses were performed to combine p-values for the European population, as well as datasets from both European and East Asian populations, for gene expression (or AS, APA).
Building genetically predicted models of gene expression, AS, and APA
To construct genetically predicted models for gene expression, AS, and APA, based on our sTF-TWAS framework20, we selected only genetic variants that located in the binding regions of any susceptible TFs for variants with MAF > 0.05, and HWE P < 10−6. We also excluded variants missing in CRC GWAS summary statistics from either European or Asian populations, which resulted in approximately 378,000 genetic variants. Prediction models were trained using the elastic net approach based on their flanking variants within a ± 1 Mb region, accounting for potential confounding factors, such as the top five principal components (PCs), gender, potential batch effects, and other factors derived from the PEERs method. A tenfold cross-validation approach was used to fine-tune the model parameters. The performance of the prediction models was evaluated by calculating the squared correlation (R2) between the predicted values and the observed levels for gene expression, AS and APA, respectively.
Association analyses of genetically predicted gene expression, AS, and APA with CRC risk
Using the weight matrix and summary statistics from CRC GWAS datasets consisting of 186,072 individuals of European ancestry and 72,272 individuals of East Asian ancestry, we evaluated the association between gene expression (or AS, APA) and CRC risk under the sTF-TWAS framework20. The statistical method was outlined in Eq. (3)
Briefly, the Z-score was used to estimate the association between predicted gene expression (or AS, APA) and CRC risk, \({w}_{{\mathrm{lg}}}\) is the weight of variant \(l\) for predicting the expression of gene \(g\) (or AS, APA), \({\hat{\beta }}_{l}\) and \({{se}}(\hat{\beta }_{l})\) are the association regression coefficient and its standard error, respectively, for variant \(l\) in GWAS, and \({\hat{\sigma }}_{l}\) and\(\,{\hat{\sigma }}_{g}\) are the estimated variances of variant \(l\) and the predicted expression of gene \(g\) (or AS, APA). We further conducted a joint analysis to combine TWAS p-values for each gene from the BarcUVa-Seq and GTEx datasets for the European population, as well as from datasets across European and East Asian populations (ACCC), using the aggregated Cauchy association test (ACAT) method74. Similarly, joint analyses were also performed to combine p-values for AS and APA, respectively. Statistically significant genes were determined based on Bonferroni corrected P < 0.05, considering the total number of tests for the models of gene expression, AS, and APA, respectively.
Differential gene expression analysis from single cell data
We downloaded and analyzed single-cell RNA sequencing (scRNA-seq) data spanning CRC developmental stages, including colon normal tissues (n = 31), serrated polyps (n = 19), conventional adenomas (n = 29), and carcinomas (microsatellite stability [MSS], n = 17; and microsatellite instability-high [MSI-H], n = 15) from the Colorectal Molecular Atlas Project (COLON MAP)23. We filtered the cell count matrix for each sample by removing low-expressed genes (expressed in ≤3 cells) and excluded cells with high mitochondrial activity ( ≥ 20% of transcripts attributed to mitochondrial genes) using Seurat (version 4.2.1)75,76. We next identified the top 2000 highly variable genes for principal component analysis (PCA) and clustered cells based on the first 30 principal components using the Shared-Nearest-Neighbors (SNN) graph and the Louvain community detection algorithm. These clusters were visualized using the dimensionality reduction technique Manifold Approximation and Projection (UMAP).
For differential expression analysis across CRC developmental stages, we examined risk genes associated with the normal-serrated polyp-carcinoma pathway and the normal-adenoma-carcinoma pathway. We performed integrative analysis of the cell count matrix across stages, generating a pseudobulk gene count matrix for each cell type within each tissue sample. Using these pseudobulk count data, we applied DESeq224 to identify differentially expressed genes between developmental stages. DESeq2 accounts for heterogeneous variability (heteroscedasticity) in gene expression, where variance depends on mean values, by modeling this relationship. This approach effectively adjusts for differences in variability between low- and high-expressing genes, leading to more accurate differential expression analysis. Specifically, we focused on potential CRC cell origins in major cell types, including absorptive cells (ABS), goblet cells (GOB), and stem cells (STM), comparing normal tissues with serrated polyps and normal tissues with conventional adenomas. Furthermore, we analyzed differential gene expressions between adenoma-specific cells (ASC) and MSS, as well as ASC and MSI-H. Similarly, differential gene expression analyses were performed between serrated-specific cells (SSC) and MSS, and SSC and MSI-H. Statistically significant genes were considered as a False Discovery Rate (FDR)-adjusted p-value < 0.05.
Identifying druggable targets for the identified risk genes
We identified potentially druggable risk genes and associated drugs by integrating drug-protein interaction data from four well-established drug databases: DrugBank52, ChEMBL53, the Therapeutic Target Database54 (TTD) and OpenTargets55. Specifically, we extracted key information for each drug, including commercial names, maximum clinical trial phases, and indications. Using this data, we annotated the identified risk genes as druggable targets and linked their corresponding drugs that are either approved or in clinical trials. In addition, we categorized therapeutic drugs based on their indications, grouping them into those targeting CRC, other types of cancer, and non-cancer diseases.
Cell culture and cell proliferation and colony formation assays
Human colorectal cancer cell lines (HCT116, RKO, and SW480) from ATCC were cultured in RPMI 1640 supplemented with 10% FBS and penicillin/streptomycin. Cells were kept in a humidified incubator at 37˚ with 5% CO2. CCK-8 cell proliferation was performed for four days. In detail, 5 × 103 cells were seeded into a 96-well plate and cultured for four days. The CCK-8 reagent (APExBIO) was then added into each well, and the plate was read at OD 450 nm using a microplate reader (BioTek). Relative cell growth was calculated by normalizing the OD value of knockout cells to the control cells. For long-term effect, colony formation assay was performed for 10–15 days. In detail, 200 cells (HCT116 and RKO) or 500 cells (SW480) were seeded into a 12-well plate. Cell colonies were visualized by crystal violet staining. The colonies were photographed, and numbers were counted.
CRISPR/Cas9-mediated gene knockout
For gene knockout, the lentiviral vector LentiCRISPR v2 (Addgene) was used to clone sgRNA of the individual target gene. Lentiviruses were amplified in HEK 293 T cells. Briefly, lentiviral vector and packaging vectors, psPAX2 and pVSV-G, were transfected into 293 T cells with lipofectamine 2000 (Thermo Fisher Scientific). Viral supernatants were collected by centrifugation at 48 and 96 hours after transfection. CRC lines were infected with the corresponding lentiviruses for 24 h. Cells were changed to new growth medium for another 24 h and then selected with puromycin (2 μg/ml, Sigma-Aldrich) for 5 days. The CRISPR/Cas9 edited cells were verified by western blotting of the target protein before using for the functional assays.
Western blotting
Total proteins were extracted from the corresponding cells lysed with RIPA buffer plus protease and phosphatase inhibitors (Roche). Twenty ug of total proteins were separated by SDS-PAGE, transferred to PVDF membrane and probed with the indicated primary antibodies. Anti-rabbit or anti-mouse secondary antibody was subsequently applied, and western blot signals were detected using enhanced chemiluminescence substrate (West Femto, Thermo Fisher Scientific). β-Tubulin was used as the loading control. Band intensity was calculated with NIH ImageJ software.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The GWAS summary statistics are used in this study are publicly available from the GWAS Catalog under accession GCST90129505. The RNA-seq data and genotype data of subjects of East Asian ancestry from the ACCC were generated as part of this study and have been deposited to the NIH Genotypes and Phenotypes (dbGaP) under accession number phs002813.v1.p1. Access to these data is controlled to protect participant privacy in accordance with NIH policies and is freely available to qualified academic researchers through dbGaP. Data access requests are reviewed by the appropriate Data Access Committee, with an expected response timeframe of approximately two months, based on estimates provided by dbGaP. The data from the GTEx (version 8) used in this study are available through dbGaP under accession phs000424.v8.p2. Transcriptome and genotype data, along with sample covariates, from the BarcUVa-Seq project are available through dbGaP under accession phs003338.v1.p1. Single-cell RNA-sequencing datasets were obtained from the Colorectal Molecular Atlas Project (COLON MAP) through the National Cancer Institute-funded Human Tumor Atlas Network (HTAN) Data Portal. The CHIP-seq data for 218 TFs were obtained from the Cistrome database. Drugs and compounds data was retrieved from publicly available databases, including ChEMBL: https://www.ebi.ac.uk/chembl/; Therapeutic Target Database: https://db.idrblab.net/ttd/; Open Targets: https://www.opentargets.org/; DrugBank: https://go.drugbank.com/. Summary-level results generated in this study, including all TWAS, AS-WAS, and APA-WAS findings, are provided in Supplementary Data 5, 8 and 10. Source data are provided in this paper.
Code availability
All analysis code used in this study is publicly available in the GitHub repository (https://github.com/XingyiGuo/sTF-TWAS-CRC) or a Code Ocean capsule (https://doi.org/10.24433/CO.1946831.v1).
References
Chen, Z. et al. Fine-mapping analysis including over 254,000 East Asian and European descendants identifies 136 putative colorectal cancer susceptibility genes. Nat. Commun. 15, 3557 (2024).
Law, P. J. et al. Systematic prioritization of functional variants and effector genes underlying colorectal cancer risk. Nat. Genet. 56, 2104–2111 (2024).
Cowper-Sal lari, R. et al. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat. Genet. 44, 1191–1198 (2012).
Li, Q. et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell 152, 633–641 (2013).
Castro, M. A. et al. Regulators of genetic risk of breast cancer identified by integrative network analysis. Nat. Genet. 48, 12–21 (2016).
Iotchkova, V. et al. GARFIELD classifies disease-relevant genomic features through integration of functional annotations with association signals. Nat. Genet. 51, 343–353 (2019).
Wen, W. et al. Genetic variations of DNA bindings of FOXA1 and co-factors in breast cancer susceptibility. Nat. Commun. 12, 5318 (2021).
Fernandez-Rozadilla, C. et al. Deciphering colorectal cancer genetics through multi-omic analysis of 100,204 cases and 154,587 controls of European and east Asian ancestries. Nat. Genet. 55, 89–99 (2023).
Chen, Z. et al. Identifying Putative Susceptibility Genes and Evaluating Their Associations with Somatic Mutations in Human Cancers. Am. J. Hum. Genet. 105, 477–492 (2019).
Yuan, Y. et al. Multi-omics analysis to identify susceptibility genes for colorectal cancer. Hum. Mol. Genet. 30, 321–330 (2021).
Yao, L., Tak, Y. G., Berman, B. P. & Farnham, P. J. Functional annotation of colon cancer risk SNPs. Nat. Commun. 5, 5114 (2014).
Moreno, V. et al. Colon-specific eQTL analysis to inform on functional SNPs. Br. J. Cancer 119, 971–977 (2018).
Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47, 1091–1098 (2015).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
Guo, X. et al. Identifying novel susceptibility genes for colorectal cancer risk from a transcriptome-wide association study of 125,478 subjects. Gastroenterology 160, 1164–1178.e1166 (2021).
Chen, Z. et al. Novel insights into genetic susceptibility for colorectal cancer from transcriptome-wide association and functional investigation. J. Natl. Cancer Inst. 116, 127–137 (2024).
Mancuso, N. et al. Probabilistic fine-mapping of transcriptome-wide association studies. Nat. Genet. 51, 675–682 (2019).
Wainberg, M. et al. Opportunities and challenges for transcriptome-wide association studies. Nat. Genet. 51, 592–599 (2019).
Amariuta, T. et al. IMPACT: Genomic annotation of cell-state-specific regulatory elements inferred from the epigenome of bound transcription factors. Am. J. Hum. Genet. 104, 879–895 (2019).
He, J. et al. Integrating transcription factor occupancy with transcriptome-wide association analysis identifies susceptibility genes in human cancers. Nat. Commun. 13, 7118 (2022).
Guo, X. et al. Large-scale alternative polyadenylation-wide association studies to identify putative cancer susceptibility genes. Cancer Res. 84, 2707–2719 (2024).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Chen, B. et al. Differential pre-malignant programs and microenvironment chart distinct paths to malignancy in human colorectal polyps. Cell 184, 6262 (2021).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Wontakal, S. N. et al. A large gene network in immature erythroid cells is controlled by the myeloid and B cell transcriptional regulator PU.1. PLoS Genet. 7, e1001392 (2011).
Lee, T. I. & Young, R. A. Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251 (2013).
Saint-Andre, V. et al. Models of human core transcriptional regulatory circuitries. Genome Res. 26, 385–396 (2016).
Vaquerizas, J. M., Kummerfeld, S. K., Teichmann, S. A. & Luscombe, N. M. A census of human transcription factors: function, expression and evolution. Nat. Rev. Genet. 10, 252–263 (2009).
Wontakal, S. N. et al. A core erythroid transcriptional network is repressed by a master regulator of myelo-lymphoid differentiation. Proc. Natl. Acad. Sci. USA 109, 3832–3837 (2012).
Deplancke, B., Alpern, D. & Gardeux, V. The genetics of transcription factor DNA binding variation. Cell 166, 538–554 (2016).
Tehranchi, A. K. et al. Pooled chIP-seq links variation in transcription factor binding to complex disease risk. Cell 165, 730–741 (2016).
Yan, J. et al. Systematic analysis of binding of transcription factors to noncoding variants. Nature 591, 147–151 (2021).
Choudhuri, A. et al. Common variants in signaling transcription-factor-binding sites drive phenotypic variability in red blood cell traits. Nat. Genet. 52, 1333–1345 (2020).
Wu, Y. et al. Joint analysis of GWAS and multi-omics QTL summary statistics reveals a large fraction of GWAS signals shared with molecular phenotypes. Cell Genom. 3, 100344 (2023).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 14, 128 (2013).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Bailey, M.H. et al. Comprehensive characterization of cancer driver genes and mutations. Cell 173, 371–385 (2018).
Dietlein, F. et al. Identification of cancer driver genes based on nucleotide context. Nat. Genet. 52, 208 (2020).
Nunes, L. et al. Prognostic genome and transcriptome signatures in colorectal cancers. Nature 633, 137–146 (2024).
Cornish, A. J. et al. The genomic landscape of 2,023 colorectal cancers. Nature 633, 127–136 (2024).
Ahmad, R. et al. Emerging trends in colorectal cancer: Dysregulated signaling pathways (Review). Int. J. Mol. Med. 47, https://doi.org/10.3892/ijmm.2021.4847 (2021).
Koveitypour, Z. et al. Signaling pathways involved in colorectal cancer progression. Cell Biosci. 9, 97 (2019).
Patel, M., Horgan, P. G., McMillan, D. C. & Edwards, J. NF-kappaB pathways in the development and progression of colorectal cancer. Transl. Res. 197, 43–56 (2018).
DeDecker, L., Coppedge, B., Avelar-Barragan, J., Karnes, W. & Whiteson, K. Microbiome distinctions between the CRC carcinogenic pathways. Gut Microbes 13, 1854641 (2021).
Peck, J. W., Oberst, M., Bouker, K. B., Bowden, E. & Burbelo, P. D. The RhoA-binding protein, rhophilin-2, regulates actin cytoskeleton organization. J. Biol. Chem. 277, 43924–43932 (2002).
Day, E. et al. IRS2 is a candidate driver oncogene on 13q34 in colorectal cancer. Int. J. Exp. Pathol. 94, 203–211 (2013).
Gibson, S. L., Ma, Z. & Shaw, L. M. Divergent roles for IRS-1 and IRS-2 in breast cancer metastasis. Cell Cycle 6, 631–637 (2007).
Porter, H. A., Perry, A., Kingsley, C., Tran, N. L. & Keegan, A. D. IRS1 is highly expressed in localized breast tumors and regulates the sensitivity of breast cancer cells to chemotherapy, while IRS2 is highly expressed in invasive breast tumors. Cancer Lett. 338, 239–248 (2013).
Lee, M. S. et al. IRS2 Amplification as a predictive biomarker in response to ceritinib in small cell lung cancer. Mol. Ther. Oncolytics 16, 188–196 (2020).
Liu, W. et al. Thioredoxin-1 ameliorates myosin-induced autoimmune myocarditis by suppressing chemokine expressions and leukocyte chemotaxis in mice. Circulation 110, 1276–1283 (2004).
Cortes, R. et al. Enhanced reduction in oxidative stress and altered glutathione and thioredoxin system response to unsaturated fatty acid load in familial hypercholesterolemia. Clin. Biochem. 47, 291–297 (2014).
Knox, C. et al. DrugBank 6.0: the DrugBank Knowledgebase for 2024. Nucleic Acids Res. 52, D1265–D1275 (2024).
Gaulton, A. et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40, D1100–D1107 (2012).
Zhou, Y. et al. TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 52, D1465–D1477 (2024).
Ochoa, D. et al. The next-generation Open Targets Platform: reimagined, redesigned, rebuilt. Nucleic Acids Res. 51, D1353–D1359 (2023).
Wu, H. et al. Resveratrol inhibits VEGF-induced angiogenesis in human endothelial cells associated with suppression of aerobic glycolysis via modulation of PKM2 nuclear translocation. Clin. Exp. Pharmacol. Physiol. 45, 1265–1273 (2018).
Kim, D. S., Camacho, C. V. & Kraus, W. L. Alternate therapeutic pathways for PARP inhibitors and potential mechanisms of resistance. Exp. Mol. Med. 53, 42–51 (2021).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Huyghe, J. R. et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat. Genet. 51, 76–87 (2019).
Lu, Y. et al. Large-Scale Genome-Wide Association Study of East Asians Identifies Loci Associated With Risk for Colorectal Cancer. Gastroenterology 156, 1455–1466 (2019).
Law, P. J. et al. Association analyses identify 31 new risk loci for colorectal cancer susceptibility. Nat. Commun. 10, 2154 (2019).
Lu, Y. et al. Identification of Novel Loci and New Risk Variant in Known Loci for Colorectal Cancer Risk in East Asians. Cancer Epidemiol. Biomarkers Prev. 29, 477–486 (2020).
Díez-Obrero, V. et al. Genetic effects on transcriptome profiles in colon epithelium provide functional insights for genetic risk loci. Cell. Mol. Gastroenterol. Hepatol. 12, 181–197 (2021).
GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Zheng, R. et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 47, D729–D735 (2019).
Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
DeLuca, D. S. et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 28, 1530–1532 (2012).
Li, Y. I. et al. Annotation-free quantification of RNA splicing using LeafCutter. Nat. Genet. 50, 151–158 (2018).
Cotto, K. C. et al. Integrated analysis of genomic and transcriptomic data for the discovery of splice-associated variants in cancer. Nat. Commun. 14, 1589 (2023).
Stegle, O., Parts, L., Piipari, M., Winn, J. & Durbin, R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc. 7, 500–507 (2012).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Acknowledgements
This research was primarily supported by US National Institutes of Health (NIH) grant R37CA227130, R01CA269589 and R01CA297582 (to X.G.). This study has been supported by: Spanish Association Against Cancer (AECC) Scientific Foundation grant GCTRA18022MORE (to V.M.). Consortium for Biomedical Research in Epidemiology and Public Health (CIBERESP), action Genrisk (to V.M.); Instituto de Salud Carlos III (ISCIII), “Programa FORTALECE del Ministerio de Ciencia e Innovación” (FORT23/00032) (to V.M.). We acknowledge Dr. Chong Wu from MD Anderson for the valuable discussions regarding simulation analysis.
Author information
Authors and Affiliations
Contributions
X.G. conceived and designed the study. Z.C. and Q.L. conducted the main genetic and statistical analyses, and W.S. performed the experimental validation. Q.L., C. L., K.S.L., and X.G. carried out the single-cell RNA-seq analysis and interpretation. Z.C., W.S., Q.L., and X.G. wrote the manuscript with feedback from all other authors, including C.L., W.W., J.R.H., P.J.L., C.F.R., M.N.T., M.T., S.L.S., V.M., M.D., C.D., F.M.-N., Q.C., J.W., J.S., S.K., C.T., W.H.J., X.S., J.L., J.G., J.K., A.S., K.M., S.H.J., K.J.J., N.W., D.H.K., J.P., G.Y., M.H.S., Z.R., J.H.O., I.O., Y.A., Y.T.G., Z.Z.P., Y.K., L.V.K., L.W., B.L., K.M., X.O.S., L.H., M.G.D., S.B.G., R.H., I.T., L.L., K.S.L., V.M., G.C., U.P., and W.Z. All authors reviewed and approved the final manuscript. X.G. supervised the study and acquired funding.
Corresponding author
Ethics declarations
Competing interests
Luc Van Kaer is a member of the scientific advisory board of Isu Abxis Co., Ltd. (South Korea). Stephen B. Gruber is co-founder of Brogent International LLC and reports research grant support from AbbVie, AstraZeneca, Eisai, HaloDx, Invitae, and Johnson & Johnson (all unrelated to this work). The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, Z., Song, W., Li, Q. et al. Mixed-model and transcriptome-wide association analyses identify transcription factors and genes associated with colorectal cancer susceptibility. Nat Commun 17, 1377 (2026). https://doi.org/10.1038/s41467-025-68127-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68127-z
