
Abstract
Polygenic risk scores (PRS) have demonstrated predictive validity across a range of cohorts and diseases, but quantifying their clinical utility remains a challenge. As PRS can be derived from a single biological sample and remains stable throughout life, we explore the potential of PRS to optimize existing screening programs. Via an integrated modelling approach, we quantify the potential clinical benefits arising from a knowledge of PRS across seven diseases with existing screening programs (abdominal aortic aneurysm, breast cancer, colorectal cancer, coronary artery disease, hypertension, prostate cancer, and type 2 diabetes). We identify individuals at high genetic risk (PRS OR>2) and very high genetic risk (PRS OR>3) and estimate the optimal screening ages for these genetically high-risk individuals, based on the equivalent risk to population-level risk at recommended screening ages. We then leverage published data on differential mortality and other outcomes, with and without screening-based interventions, to assess the potential benefits of tailoring screening age based on genetic risk. Very high risk individuals reach the risk level associated with usual starting screening age on average 10.8 years earlier, high risk individuals 8.9 years earlier and reduced risk individuals (OR<0.5) 16.8 years later. During this time, case enrichment (the ratio of the percentage of cases in the high PRS risk group and in the total population) in the high risk group is between 1.7 and 3.0, depending on the disease. Across all seven diseases, appropriate interventions following PRS-guided screening could reduce premature deaths in high-risk individuals by 23.3%. Knowledge of genetic risk, measured using PRS, has the potential to deliver substantial public health benefits when aggregated across conditions, and could reduce premature mortality by tailoring existing screening programs.
Similar content being viewed by others

Introduction
Common, chronic diseases account for over 90% of the $4.5 trillion annual healthcare spend in the US1. There is a range of public health screening programs aimed at reducing the burden of these diseases, either via early detection (e.g., screening for asymptomatic cancers) or prevention (e.g., screening for cardiovascular disease risk). Most programs use simple risk factors for determining initial access to screening, notably age and sometimes sex. This approach leads to overtreatment in some2, and undertreatment in others3.
Despite its widespread use, fixed age cutoffs fail to account for the considerable variability in individual risk, often missing high-risk individuals who are younger than the minimum age threshold, whilst including lower-risk individuals just because they are within the target age-group4. Data on the effectiveness of age-based criteria in capturing true high-risk populations are limited, and the current approach may inadvertently reduce the efficiency and equity of screening programs. Understanding the spectrum of risk across the population may allow better targeting of interventions to those most likely to benefit, and least likely to be harmed.
One promising avenue for improving risk stratification lies in genetic factors. Polygenic risk scores (PRS), which aggregate the effects of common genetic variants associated with a disease into a single score, have emerged as novel biomarkers that are stable throughout life and available for a broad spectrum of diseases5,6. For many diseases, the strongest measurable risk factor is a PRS7. Unlike other biomarkers, a single genetic test allows the construction of multiple PRSs that can be used to predict the risks of as many diseases as required.
In this study, we estimate the potential clinical benefits of tailoring current screening programs to genetic risk. Specifically, we calculate the proportion of the population who are at sufficient risk of disease to warrant earlier initiation, identify the age at which this earlier screening should occur, and quantify the mortality benefits of earlier, PRS-guided screening.
Results
Individuals at risk for multiple diseases
PRS effect size estimates (OR per SD) were determined in the UKB training set (Supplementary Data 1) and used to define the disease-specific high-risk and very high-risk individuals in the UKB testing set. Of the 332,664 individuals in the UKB testing set, there were 83,950 individuals at high risk (PRS OR>2) and 18,956 individuals at very high risk (PRS OR>3) for at least one of the seven diseases for which current screening programs exist (Table 1), corresponding to approximately 25% and 6% of the cohort, respectively.
High PRS individuals are predicted to reach equivalent risk earlier - Risk Advancement Period (RAP)
For high PRS risk groups (PRS OR>2), we predict that high-risk individuals would reach the target incidence rate between 4 and 22 years (mean 8.9 years) before the conventional starting age for screening, depending on the disease (Fig. 1, Supplementary Data 2). The disease with the largest RAP for high-risk individuals was hypertension (Fig. 1, Supplementary Data 2). A similar pattern is seen for the very high risk groups (OR>3), with RAP ranging from 5 to 22 years (Supplementary Data 2) (mean 10.8 years). Individuals with a reduced PRS risk (OR <0.5) are predicted to reach population level risk 6 to 40 years later (mean 16.8 years) than the conventional starting age for screening; for prostate cancer (based on a screening start age 55) and hypertension, we predict that the reduced risk groups will never reach the target incidence (Fig. 1, Supplementary Data 2).

Incidence data are based on US White Non-Hispanic self-reported race/ethnicity, and PRS effect sizes are based on European genetic ancestry (see Supplementary Information for figures based on alternative sources). Incidences are sex-averaged, apart from prostate cancer (male), breast cancer (female), and abdominal aortic aneurysm (male). Vertical dotted lines indicate the conventional screening start age. Horizontal dotted lines indicate the RAPs for different PRS-based risk groups. AAA abdominal aortic aneurysm, BC female breast cancer, CRC colorectal cancer, CAD coronary artery disease, HT hypertension, PC prostate cancer, T2D type 2 diabetes.
Alternative conventional starting ages for breast cancer, colorectal cancer and coronary artery disease were explored for sensitivity analyses, with results summarized in Supplementary Fig. 1 and Supplementary Data 2. The analyses were also repeated, replacing the population incidence rates with those for Black Non-Hispanic self-reported race/ethnicity and using a PRS effect size estimated in the genetically-inferred African-ancestry UKB training set (Supplementary Figs. 2 and 3, Supplementary Data 1). Here, the PRS effect sizes are smaller, meaning that fewer individuals have a PRS OR high enough to be in a high-risk group. However, the slightly lower average OR within each high-risk group leads to only a relatively small reduction in the length of the RAP for these diseases. For some diseases (breast cancer, colorectal cancer, prostate cancer, and type 2 diabetes), we also repeated RAP calculations based on UK-based conventional starting ages, UK self-reported White race/ethnicity incidence rates, and European ancestry PRS effect sizes, which yield similar RAP durations (Supplementary Fig. 4).
The clinical benefits of PRS-guided screening and treatment
For the seven diseases with existing screening programs, we determined the number of disease-specific deaths that could be prevented by offering earlier PRS-guided screening or intervention to individuals who are at risk due to their PRS. For each disease, we identified the probability of disease-associated death once diagnosed as a case within control groups of relevant randomized controlled trials; the appropriate screening or screening-based intervention based on guidelines; and the corresponding screening or intervention efficacy (relative reduction in relevant outcome) based on randomized controlled trials (Table 2). For breast cancer, we identified efficacies for both screening (mammography) and for risk-lowering preventive intervention (tamoxifen).
Based on various modelling assumptions (for example, that trial-derived values reflect current real world practice, and that PRS effect sizes estimated in UK Biobank would apply to US settings, see Discussion for further considerations) we proceeded to estimate the early detection and early prevention benefits of PRS-guided screening, using a PRS threshold of OR>2 to define disease-specific high risk groups (Tables 3 and 4). Across the seven diseases, between 2.9% (colorectal cancer) and 10.0% (Type 2 diabetes) of the population are identified as being at high risk due to their PRS. Within these high risk groups, there is a case enrichment within the RAP of between 1.7 (hypertension in men) and 3.0 (Type 2 diabetes in men) compared to the general population, resulting from their increased genetic risk (Fig. 2). If relevant screening and screening-based interventions were applied within these high risk groups, we estimate that between 12% (breast cancer) and 35% (abdominal aortic aneurysm) of disease-specific deaths within these high risk groups could be prevented via early detection (Table 3), and between 23% (coronary artery disease) and 31% (breast cancer) could be prevented via early preventive intervention (Table 4). Summing across both types of benefit (but counting only the early detection benefit for breast cancer to avoid double-counting), we estimate that, in a population of 100,000 individuals of each sex, 6387 disease cases could potentially occur within the RAP in individuals with a high PRS risk for the relevant disease. These cases, which fall outside of current screening ages, could be expected to lead to 256 deaths. If a PRS-guided early detection and/or early prevention strategy were adopted, 60 of these deaths (23.3% of disease-specific deaths in the respective high-risk groups) could be saved.

Case enrichment (ratio of the percentage of cases in high-risk (PRS OR>2) individuals compared to that in the whole population), in men and women, for the seven screening diseases.
For example, in a cohort of 100,000 women who were without breast cancer at the start of the RAP, the 7200 women in the high PRS risk group for breast cancer (7.2% of the cohort) could begin screening for early detection at age 34, instead of at the conventional age of 40 (Table 3). Among these 7200 high-risk 34-year-olds, we could expect to see 75 breast cancer diagnoses between the ages of 34 and 40. Assuming that 29% of cases would lead to breast cancer-related death (Table 2)8, these cases could result in 22 deaths. The UK Age randomized control trial reported a 12% reduction in breast cancer mortality associated with mammographic screening between ages 40 and 49 (as opposed to the UK standard screening age of 50 years)8. We hence estimate that 2.6 deaths could be prevented by PRS-guided screening in a cohort of this size.
We performed various sensitivity analyses to explore the impact of applying alternative modelling scenarios. Altering the conventional starting age for screening tended to increase PRS-guided screening benefit if the age was later, and decrease it if the age was earlier (Supplementary Data 3). Restricting interventions to individuals with a very high risk PRS (OR>3) resulted in longer risk advancement periods, but a more modest overall impact, because the earlier interventions would be offered to fewer people (e.g., 1.6% of women at very high risk of breast cancer, as opposed to 7.2% at high risk). Over all seven diseases, we estimate that 28 disease-specific deaths in the very high-risk groups could be prevented in a population of 100,000 individuals of each sex, combining early detection and early prevention benefits (Supplementary Data 4 and 5). Using UK-based disease incidences, either using empirical case data from the UK Biobank testing set (Supplementary Data 6 and 7) or using national data sources (Supplementary Data 8), generated only modest differences in results.
Discussion
Healthcare systems continue to invest large amounts of resources into screening for many common diseases. In order to optimize cost-effectiveness and minimize harms from overtreatment, access to screening for early detection or prevention is targeted at population groups for whom the benefits are deemed to be greatest. This targeting is achieved using a very minimal number of predictors, typically age and sex. As these are relatively crude predictors of risk, a reliance upon these factors can lead to overtreatment in some groups and undertreatment in others. Here, we consider using an additional immutable risk factor, PRS, to support the stratification of screening and screening-based intervention opportunities. Specifically, we focus upon the potential clinical benefit of earlier intervention based upon genetic stratification across a range of important and common diseases.
Our paper shows three main results: 1) a single biological sample, obtained once, can predict the risk of many important common diseases, and can identify a substantial proportion of the population at high risk; 2) PRS can be used to propose rational starting ages for early screening in high risk people; and 3) if PRS-guided interventions were implemented, premature mortality could be substantially reduced.
First, we show that PRS can predict the risk of seven common diseases (abdominal aortic aneurysm, female breast cancer, colorectal cancer, coronary artery disease, hypertension, prostate cancer, and type 2 diabetes), and that a quarter of the population is at high risk of at least one of these diseases. PRS has the advantageous property that data from a single test can be obtained once at an early age, in principle from birth, and can capture risk simultaneously across many diseases.
Second, regarding screening for these seven diseases, we show how PRS information could be used to propose rational earlier screening start times for people at high risk. Compared to a strategy offering earlier invitations to individuals with a high PRS, we show that current screening misses opportunities to intervene in 6387 disease-specific cases per 100,000 individuals of each sex. Under current practice, notwithstanding infrequent familial cases detected by cascade screening9,10, most of these cases would either be detected at later symptomatic stages, where therapeutic interventions typically have poorer outcomes, or would manifest without the presence of preventive interventions that could have reduced their risk of occurring.
Compared to when conventional screening begins, we predict that high-risk individuals (PRS OR>2) would reach the equivalent population disease incidence rate around a decade earlier (ranging from 4 to 22 years across diseases). Hypertension (using a conventional starting age of 40) and breast cancer (using a conventional starting age of 50) were the diseases with the longest risk advancement periods. Conversely, reduced-risk individuals (OR < 0.5) reached the equivalent population disease incidence rate on average more than a decade after conventional screening began. Together, this demonstrates that using age alone is a relatively crude way to stratify population health programs.
Third, we show that premature disease-specific deaths that occur before conventional screening occurs are potentially preventable with PRS-guided interventions. Across our included diseases, our results suggest that 256 disease-specific deaths (per 100,000 individuals of each sex) would occur in the corresponding high-risk groups before conventional screening occurs, and 23% of the disease-specific deaths within these high-risk groups could be prevented with PRS-guided interventions. We estimate that the highest proportions of preventable deaths during the risk advancement period are for abdominal aortic aneurysm, breast cancer, and type 2 diabetes, with respectively an estimated 35%, 31% and 27% of disease-specific deaths preventable with PRS-guided interventions. In absolute terms, we found that type 2 diabetes, hypertension, and breast cancer had the potential for the largest number of deaths saved by this approach.
Our estimate of the absolute number of deaths prevented is small (60 per 100,000 individuals of each sex), but identifying and preventing mortality in younger adults is likely to lead to large improvements in quality-adjusted life years. More broadly, earlier intervention would be associated with other benefits, beyond mortality reduction, including less aggressive (and less expensive) treatments, likely resulting in fewer side-effects. Identifying high-risk individuals or those with diseases earlier also has the potential to reduce events such as non-fatal myocardial infarction and stroke. Indeed, in absolute terms, the reduction in non-fatal events is likely to be greater.
An alternative to PRS-informed intervention programs would be to lower the screening invitation age for the whole population. However, we note this would involve considerably more resources. For example, the RAP period for breast cancer (using a conventional starting age of 40) is 6 years, with 7.2% of females in the high PRS risk group. Biennial screening for this high-risk group (i.e., 3 extra screens) would be equivalent to 0.21 extra screens for the whole female population. Adding even just one additional screen for the whole population would result in nearly 5 times as many total screens as targeting 3 extra screens to the high PRS risk group.
The existing literature regarding PRS largely examines the prediction of lifetime disease susceptibility11,12, and integration of PRS into existing clinical risk scores11,13,14. However, there are only a few studies concerning the identification of earlier conventional screening ages with PRS12,15,16, or predicting cumulative disease burden across a number of diseases using PRS17. Our 8-year advancement period for colorectal cancer is broadly in line with the 5- and 11-year risk advancement periods estimated for the 80-99% and >99% PRS groups modelled in the Finnish population16. Our results are also congruent for men compared to the study of Chen et al15, who estimated personalized screening ages for men and women in the UK Biobank in the highest PRS decile for colorectal cancer, but not for women (we predicted women would reach the same disease incidence rate at 34, whereas they predicted 48). Reasons for this difference may include the use of different conventional screening ages (50 versus 55 years), and also our use of national sources of incidence data to support our estimates. Jermy et al examined the ability of PRS to estimate PRS-guided screening ages for breast cancer and type 2 diabetes12. This paper used a different methodology, comparing PRS percentiles, and used the old USPSTF breast cancer screening age (50), and found that women at the top 5% PRS risk would be eligible for breast cancer screening at 43 years. In contrast, we used the new USPSTF breast cancer screening age of 40 and found a PRS-guided screening age of 34 years. For type 2 diabetes, Jermy et al found that people in the top 5% PRS risk for type 2 diabetes would be eligible for screening at 29 years, whereas our results suggest 25 years of age. However, Jermy et al did not use USPSTF guidelines to define conventional screening ages and did not use national US incidence data to support estimates. Widen et al examined the ability of PRS to identify people at high risk across a number of diseases17. These authors constructed polygenic health indexes and estimated the ability of PRS to predict lifetime disease burden when combining PRS across a number of diseases. Our study quantifies the number of people at high and very high risk for a number of diseases, and then estimates the associated mortality estimates.
Assumptions and limitations
The findings of this study are based on an integrated modelling approach, combining model parameters from a wide variety of different sources. This approach introduces a number of assumptions at various stages of the modelling process, which we consider here.
The estimates of PRS effect size (the odds ratio per standard deviation of PRS) were obtained from UK Biobank, a UK-based cohort, and these estimates were then applied to modelling primarily using US guidelines and US estimates of disease incidence to assess PRS-guided screening benefits. This assumes that ancestry-specific PRS effect sizes are transferable from the UK to the US, and are robust to known health biases in UK Biobank, to differences in disease prevalence between the UK and US, and to potential differences in the way diseases are recorded or defined between the UK Biobank and the target US setting (for example, we assume the PRS has the same relative effect on rates of screen-detected and symptomatically-detected abdominal aortic aneurysm). Studies have noted generally consistent PRS effect size between target populations of the same ancestry18,19, and that health biases in the UK Biobank do not necessarily impact the valid assessment of exposure-disease relationships20. We used ICD10 codes wherever possible to rely on international coding standards, but accept that application of these codes may vary between the UK and the US. Some diseases, such as type 2 diabetes, are not well captured by ICD10 codes in a hospital setting, because they are typically diagnosed and treated within a primary healthcare setting. For these diseases, we used primary healthcare data from UK Biobank to define the disease status when carrying out the PRS effect size estimation (see Supplementary Information)20.
We also assumed that PRS effect sizes are constant with age. Studies have noted a small but consistent tendency towards larger PRS effect sizes in younger age groups6,21. To the extent that this effect holds, our modelling will underestimate the effect of PRS-guided screening on deaths prevented in younger age groups. We also assumed that a PRS effect size calculated as an odds ratio can be applied as a hazard ratio in subsequent modelling. We have shown elsewhere that, in the context of UK Biobank, these two ways of measuring effect size are largely equivalent6.
US and UK national disease incidence and mortality data were obtained from a variety of recognised sources (see Supplementary Information), but nevertheless these can be subject to potential uncertainties associated with their sampling and recording methods. For diseases such as abdominal aortic aneurysm, where the purpose of screening is for early detection, these sources will underestimate the expected disease detection rate under screening, because they do not capture disease events that would not have otherwise manifested during the RAP time interval, thus deflating the modelled estimates of screening benefit.
Estimates of the conditional probability of disease-specific death under no screening, given that a person is a diagnosed case for that disease, were obtained from the control arms of relevant randomised controlled trials. Estimates of screening or intervention efficacy were typically obtained from the same randomised controlled trials. Differences in screening or subsequent intervention procedures between these trials and current real-world practice could influence those estimates. These trials were conducted in a variety of settings, both in and outside the USA, at various timepoints, and with a range of trial conditions and follow-up times. Being relative or conditional metrics, our metrics do not depend directly on trial-specific disease incidence, but they do depend on a proportional hazards assumption for situations where incidence may vary between the trial and our proposed application (for example, between the general population and smokers for abdominal aortic aneurysm). We note that, since many of these trials were trials of population-based screening, the control arms often reflected real-world standard of care, enhancing their utility for our purposes. Our modelling assumes that follow-up times are sufficiently long to capture pertinent disease-specific death events (we note that our modelling assumes that diagnosed case events must occur with the RAP periods, but disease-specific deaths caused by those case events can occur afterwards); and that both the conditional probability of death and efficacy benefit of screening is the same in people with high PRS risk as it is in the general population. We note this last point may sometimes be a conservative assumption - for example, there is evidence of an increased efficacy of statins in people with high PRS risk22,23. The randomised controlled trials were typically conducted on older individuals: we assume that both the conditional probability of death and the screening and intervention efficacies and modalities are the same for events diagnosed in younger people in the RAP periods. We note that optimal screening modalities and frequencies in younger people may differ - for example, ultrasound may be more efficient for screening for breast cancer in younger women, which carries its own costs and characteristics, and screening for abdominal aortic aneurysm in younger individuals may imply repeated screening (rather than a single screen at an older age).
We used conditional probabilities and relative screening and intervention efficacies from randomised controlled trials to estimate PRS-guided screening benefits. More complex approaches could consider the impact of individual disease trajectories, for example, using models that predict cancer stage progression over time. We performed our modelling separately for each disease, and then summed the impact on deaths as a last step. This approach requires a low per-disease death rate (to avoid issues relating to double counting), a requirement which was met by the relatively young age group in which our modelling was performed, and assumes independence between diseases. The confidence intervals we provide on deaths prevented per disease only take uncertainty on PRS effect size into account, and so do not reflect all sources of uncertainty.
Our primary analyses present results for individuals of European ancestry and White race/ethnicity, using guidelines and modelling tailored to the USA. However, we present various sensitivity analyses to indicate that PRS-guided screening could also be beneficial to individuals of non-European ancestry and race/ethnicity, in the UK as well as the US, and using alternative starting ages for conventional screening and alternative thresholds for defining the at-risk PRS group. Our analyses consider one of the simplest possible alternatives to a single conventional starting age for screening, which is to propose a single alternative starting age for individuals declared to be at risk due to their PRS. We note that a PRS affords a quantitative estimate of a person’s disease risk, and that therefore alternative schemes could be devised in which the screening starting age is individually tailored to a person’s PRS score, perhaps combined with other risk factors such as their sex (although we note there could be administrative complexities associated with such schemes). PRS-guided screening could also be applied to the consideration of delayed starting ages for individuals with low PRS, to the consideration of tailored screening stopping ages, or to the consideration of tailored frequency of screening once screening starts. These alternative schemes are fruitful areas for future research. We also note that our simple PRS-only approach assumes that other risk factors, such as rare pathogenic variants for breast cancer, or the use of clinical risk tools for screening for high risk in coronary artery disease and type 2 diabetes, are dealt with via other pathways. We did not carry out a formal cost-benefits analysis of our PRS-guided scheme, but note this would be a priority for future work.
In summary, our modelling approach applies a number of assumptions and simplifications, which influence the accuracy of our calculations. We carried out various sensitivity analyses, all of which demonstrated a benefit to PRS-guided screening, but inevitably, these analyses could not cover all possible eventualities.
Concluding remarks
Collectively, our results show the power of PRS across multi-disease risk prediction and precision-guided screening, with the potential to save premature, preventable deaths in younger adults. When considered in combination with prior work showing the feasibility of integrating PRS into primary care24, our results show a promising clinical use case for PRS. Our results have implications for patients, clinicians, researchers and policy makers. For clinicians, PRS are increasingly available both via direct-to-consumer tests and within healthcare systems themselves25. We anticipate our data may provide some guidance by providing data to support conversations regarding risk and screening for patients presenting to clinics with PRS results. For patients, our results show that PRS should be considered a risk factor that may help guide health behaviours. For policymakers, our results highlight the use of PRS as a risk factor that can improve screening. PRS can enhance accuracy and potentially identify people who are being missed by conventional screening programs.
Methods
Screening diseases
We identified a list of seven common diseases for which age-related public health screening programs exist: abdominal aortic aneurysm, female breast cancer, colorectal cancer, coronary artery disease (which we define as a combination of either fatal coronary heart disease and/or myocardial infarction), hypertension, prostate cancer, and type 2 diabetes (see Supplementary Information for more information). ‘Screening’ refers here either to screening for an existing condition to facilitate earlier therapeutic intervention, or to screening for risk to facilitate preventive intervention. For some diseases (abdominal aortic aneurysm, prostate cancer, type 2 diabetes), screening is only recommended in certain high-risk groups, or following a patient-clinician discussion of benefits and harms based on the patient’s risk. We assume that possession of a high-risk PRS value constitutes sufficient motivation for screening in this group (see Supplementary Information).
PRS and PRS effect sizes
We previously constructed and released the UK Biobank PRS Release v2 as a freely-available resource for UK Biobank researchers6. The UK Biobank (UKB) is a UK-based prospective cohort of ~500,000 individuals, aged 40–69 when recruited between 2006 and 201026,27. Our research project (project application number 9659) was approved by the UK Biobank according to their established access procedures, and legal and ethical approval is covered by the Research Tissue Bank approval obtained from the UK Biobank’s governing Research Ethics Committee (REC 16/NW/0274), as recommended by the National Research Ethics Service. We used the Standard PRS set, which was constructed from non-UKB training data only, as our source of high-performance PRS, along with their corresponding trait definitions. Briefly, the PRS were generated using meta-analyzed external genome-wide association study summary statistics. Per-individual PRS values were calculated as the genome-wide sum of the per-variant posterior effect size multiplied by allele dosage. All PRS were centred and standardized based on external reference data (1000 Genomes Project) to ensure means of approximately zero and standard deviations of approximately one within major genetically inferred ancestry groups. The same pipeline was used to construct a new PRS for abdominal aortic aneurysm. Disease phenotypes are defined using a combination of linked hospital episode statistics, cancer registry data and primary care data (see Supplementary Information).
The UKB cohort was split into study training and test sets. Genetic ancestry was inferred using scores derived from principal components analysis of genetic data6. The UKB training set was designed to include all participants with available PRS and non-European genetic ancestry (2867 with inferred East Asian (EAS) ancestry, 9543 with South Asian (SAS), 9486 with Sub-Saharan African (AFR), and 389 with Native/Indigenous American (AMR) ancestry), along with a random sample of 100,000 participants with European (EUR) ancestry. This UKB training set (122,285 individuals) was used to estimate the odds ratio per standard deviation (OR per SD) of each PRS in each ancestry group, using logistic regression.
Following the removal of 31,300 UKB individuals who were related (up to third degree) to individuals in the training set, the remaining 332,664 individuals with available PRS were designated as an independent UKB testing set. The testing set consisted solely of individuals with inferred European ancestries, as the numbers with other ancestries were not large enough to allow useful training/testing splits. The UKB testing set was only used for two purposes: to calculate percentages of individuals at high genetic risk for multiple diseases, and to perform a sensitivity analysis to compare the primary results based on national incidence data with results based on observed case numbers.
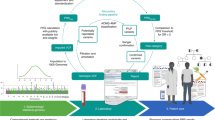
Following previous practice7,11,28,29, we defined individuals to be at ‘high risk’ of a disease if their PRS value was associated with an odds ratio (OR) >2 compared to the population mean risk. For ‘very high risk’, we used a PRS value associated with OR>3, and for ‘reduced risk’, we used a PRS value associated with OR<0.5 (Fig. 3A). A detailed description of the calculation of risk groups is available in the Supplementary Information.

For each of seven screening diseases, we estimated the proportion of the population that could be considered to be at high genetic risk on the basis of their PRS (A). We then identified the age at which PRS-defined high risk individuals would reach the same disease incidence rate as that of the average population at the conventional starting age for screening, which defines the start of the risk advancement period (B), and modelled the number of cases which would be diagnosed in these high-risk individuals between this earlier screening age and the current screening age (see “Estimation of events and deaths without PRS-guided screening”). From this, we estimated the number of cause-specific deaths that would result from cases diagnosed within this period, and hence the potential reduction in mortality that could be achieved by earlier intervention in this group (C).
Risk advancement periods (RAP)
For each of the seven diseases, we extracted recommendations on screening and screening start age, predominantly relying on US-based recommendations from the United States Preventive Services Task Force (USPSTF) (see Supplementary Information). Following USPSTF recommendations, we only considered screening for abdominal aortic aneurysm in men.
For each disease, we then used published incidence data for the US population, stratified by age, sex, and self-reported race/ethnicity, to calculate a set of baseline one-year incidence rates from age 0 to 80 years (see Supplementary Information). Disease incidence data were obtained from a number of sources and reflect the real-world diagnosis rate at each age under the current standard of care. We were unable to identify suitable US population incidence data for abdominal aortic aneurysm, so we instead used male incidence data derived from the UK Biobank. To estimate the incidence rate in each disease-specific high risk group, baseline incidence rates were multiplied by the mean relative risk within the high risk group compared to baseline, which was derived theoretically from the mean OR per SD in the relevant upper portion of the PRS distribution (using the equivalence between relative risk and OR that applies when baseline incidences are low, adjusting for bias in the transformation from PRS to OR (see Supplementary Information), and assuming a constant relationship between PRS effect size and age)30,31,32.
Following standard methodology for the construction of the risk advancement period33, we then compared the incidence-by-age curves in order to find the age at which individuals in the high-risk group reached the same level of risk as that found in the whole population at the conventional starting age for screening. We refer to this starting age as the PRS-guided screening age. The difference between the conventional screening age and the PRS-guided screening age defines the risk advancement period (RAP) (Fig. 3B)33.
Analyses were repeated for disease-specific very high-risk groups (OR > 3) and for reduced-risk groups (OR<0.5). For reduced risk groups, the PRS-adjusted screening ages were later than the conventional screening ages, reflecting a negative RAP (i.e. a risk delay period). For some diseases, the reduced risk group does not reach, at any age, the population risk at the conventional screening age. In this situation, calculations were based on taking the PRS-adjusted screening age to be 80 years, although we note that this does not reflect a realistic use-case.
The primary analyses used US incidence rates corresponding to White Non-Hispanic self-reported race/ethnicity, PRS effect size estimates corresponding to genetically inferred European ancestry, and PRS-adjusted screening ages that were calculated using baseline rates averaged over males and females, where applicable (sex-specific ages are reported in the Supplementary Information). Sensitivity analyses explored the use of alternative conventional starting ages for screening, alternative US-based race/ethnicity for incidence rates and genetic ancestry for PRS effect size estimates, and alternative UK-based incidence rates derived either from external data sources or empirically from UK Biobank data.
Estimation of events and deaths without PRS-guided screening
We used an integrated modelling approach to estimate events and deaths without PRS-guided screening. Population disease incidence data, stratified by age, sex and self-reported race/ethnicity and obtained from various national sources (see previous section and Supplementary Information), were combined with estimates of mortality from other causes and with estimates of PRS effect size into a competing mortality survival model34. The cumulative incidence function of this model was used to estimate the ‘without screening’ events, which is the number of diagnosed disease events, per 100,000 males and 100,000 females, that are predicted to occur without the imposition of a formal PRS-guided screening program, and without the additional interventions that would flow from such a program, during the RAP time interval between the earlier PRS-guided starting age for screening and the conventional starting age for screening. We note that the ‘without screening’ events are estimated from real-world data, and therefore include any opportunistic screening, special screening and standard-of-care interventions that may occur during the RAP time interval. The cohort of 100,000 males and 100,000 females is modelled separately for each disease, and is assumed to be free of that disease at the start of the RAP period.
The without-screening events in the high PRS risk groups were found by rerunning the same competing mortality survival model, including a subdistribution hazard rate multiplier to represent the PRS effect size, which is sampled from the appropriate top truncated portion of the PRS distribution. These without-screening disease events represent opportunities for early detection or preventive intervention (Fig. 3C). As with the RAP calculation, we use the equivalence between relative risk and OR that applies when baseline incidences are low, we adjust for bias in the transformation from PRS to OR (see Supplementary Information), and we assume a constant relationship between PRS effect size and age30,31.
We note that the without-screening events in the RAP (both overall and in the high PRS risk groups) are expected to be an underestimate of the true number of disease events for diseases where the purpose of screening is for early detection, because they are based on reported population incidence rates under current standard of care, and so do not include early-stage cases that might additionally be detected through earlier screening. Our modelling results may therefore be considered conservative in this regard.
The without-screening events in the high PRS risk groups were calculated separately for males and females, and the confidence intervals (CI) were derived from the 95% CI of the log(OR) per SD of the PRS, assuming that the PRS-guided screening age and the proportion of the population in the high risk groups are known without error, and ignoring uncertainty in the population baseline rates.
We estimated the ‘case enrichment ratio’ as the ratio of the expected case incidence in each high PRS risk group to the expected case incidence in the total population (equivalently, by rearrangement, it is the ratio of the percentage of high PRS risk group membership within cases to the percentage of high PRS risk group membership in the total population). For example, for breast cancer, 7.08% of all cases occurring during the RAP are in the high-risk group, and 2.98% of the female population is in the high-risk group; thus, the case enrichment ratio is 2.37.
To estimate without-screening deaths, we conducted a literature search to identify large randomized controlled trials studying the efficacy of screening and screening-based interventions, and for which information on the number of disease-related events and deaths occurring in the control arms of the trial was available (see Supplementary Information). From these data, we derived the conditional probability of disease-specific death, conditional on experiencing a disease event within the control arm. The without-screening events were multiplied by this conditional probability to give the estimated number of without-screening deaths per 100,000 males and 100,000 females, arising from events occurring within the RAP. Note that while these deaths are conditional on disease events that are diagnosed within the RAP, the deaths themselves can occur after the RAP time period (see next section).
Quantification of clinical benefits of PRS-guided screening
We consider two kinds of clinical benefit: an ‘early detection’ benefit that arises from earlier screening, and thus, earlier therapeutic intervention for current disease, and an ‘early prevention’ benefit that arises from earlier preventive intervention that is administered to individuals at high risk due to their PRS. A literature search was conducted to identify randomized controlled trials on the effects of screening and screening-based interventions on relevant outcomes over sufficiently long follow-up time periods (see Supplementary Information). From these data, we extracted relative risks describing the efficacy of screening and screening-based intervention, assuming the same level of compliance to screening and intervention as seen in the randomized controlled trials. For diseases where screening is for early detection, the relative risk is the relative mortality seen in the intervention versus control arms. For diseases where screening is for early prevention, the relative risk describes either the relative event incidence or relative mortality in the intervention versus control arms. Under both scenarios, the estimate of deaths expected under screening and screening-based intervention is obtained by multiplying the without-screening deaths by the relevant relative risk. The screening benefit arises either from improved survival outcomes following earlier detection (for diseases where screening is for early detection), or from reducing the disease event rate within the RAP time period (for diseases where screening is for early prevention).
When quantifying the combined benefit over all diseases, to avoid double-counting, we only included the benefit of ‘early detection’ and excluded the benefit of ‘early prevention’ for breast cancer. We did not estimate the clinical benefits of delaying screening in the reduced risk groups, as there are no randomized controlled trials or efficacy estimates for delayed screening.
We conducted a series of sensitivity analyses to explore the impact of altering various inputs on our modelling. These included: (1) using very high PRS risk (OR>3) to define the at-risk PRS groups; (2) using alternative conventional starting ages for screening; (3) using observed cases in the UK Biobank testing set during the RAP period, rather than modelling from separate sources of incidence and mortality data; and (4) modelling UK based incidence and mortality rates for a subset of diseases (see Supplementary Information).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data used in this study are available via the usual research application procedures to UK Biobank, or via application to the other external data sources listed in Supplementary Information, or are available from previous publications (all the clinical trials data used in this study fall into this latter category). The UK Biobank PRS Release is available via application to the UK Biobank’s Research Access Platform (Category 300, https://biobank.ndph.ox.ac.uk/ukb/label.cgi?id=300). All other data supporting the findings of this study are available in the article and its Supplementary Information files.
Code availability
Custom code used in the RAP calculations is available at https://github.com/genomics-dev/clinical_benefits35.
References
Centers for Disease Control and Prevention. Fast Facts: Health and Economic Costs of Chronic Conditions. https://www.cdc.gov/chronic-disease/data-research/facts-stats/index.html (accessed 15 Aug 2024).
Welch, H. G., Prorok, P. C., O’Malley, A. J. & Kramer, B. S. Breast-Cancer Tumor Size, Overdiagnosis, and Mammography Screening Effectiveness. N. Engl. J. Med. 375, 1438–1447 (2016).
Saw, S. T., Leong, B. D. K. & Aziz, D. A. A. Early Detection of Undiagnosed Abdominal Aortic Aneurysm and Sub-Aneurysmal Aortic Dilatations in Patients with High-Risk Coronary Artery Disease: The Value of Targetted Screening Programme. Vasc. Health Risk Manag. 16, 215–229 (2020).
Mukama, T. et al. Risk-Adapted Starting Age of Screening for Relatives of Patients With Breast Cancer. JAMA Oncol. 6, 68–74 (2020).
Lambert, S. A. et al. The Polygenic Score Catalog as an open database for reproducibility and systematic evaluation. Nat. Genet. 53, 420–425 (2021).
Thompson, D. J. et al. A systematic evaluation of the performance and properties of the UK Biobank Polygenic Risk Score (PRS) Release. PLOS ONE 19, e0307270 (2024).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018).
Duffy, S. W. et al. Effect of mammographic screening from age 40 years on breast cancer mortality (UK Age trial): final results of a randomised, controlled trial. Lancet Oncol. 21, 1165–1172 (2020).
Maxwell, K. N., Domchek, S. M., Nathanson, K. L. & Robson, M. E. Population Frequency of Germline BRCA1/2 Mutations. J. Clin. Oncol. 34, 4183–4185 (2016).
Beheshti, S. O., Madsen, C. M., Varbo, A. & Nordestgaard, B. G. Worldwide Prevalence of Familial Hypercholesterolemia. JACC 75, 2553–2566 (2020).
Mars, N. et al. Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat. Med. 26, 549–557 (2020).
Jermy, B. et al. A unified framework for estimating country-specific cumulative incidence for 18 diseases stratified by polygenic risk. Nat. Commun. 15, 5007 (2024).
Weale, M. E. et al. Validation of an Integrated Risk Tool, Including Polygenic Risk Score, for Atherosclerotic Cardiovascular Disease in Multiple Ethnicities and Ancestries. Am. J. Cardiol. 148, 157–164 (2021).
Riveros-Mckay, F. et al. Integrated Polygenic Tool Substantially Enhances Coronary Artery Disease Prediction. Circ. Genom. Precis. Med. 14, e003304 (2021).
Chen, X., Heisser, T., Cardoso, R., Hoffmeister, M. & Brenner, H. Personalized Initial Screening Age for Colorectal Cancer in Individuals at Average Risk. JAMA Netw. Open 6, e2339670 (2023).
Tamlander, M. et al. Genome-wide polygenic risk scores for colorectal cancer have implications for risk-based screening. Br. J. Cancer 130, 651–659 (2024).
Widen, E., Lello, L., Raben, T. G., Tellier, L. C. A. M. & Hsu, S. D. H. Polygenic Health Index, General Health, and Pleiotropy: Sibling Analysis and Disease Risk Reduction. Sci. Rep. 12, 18173 (2022).
Mars, N. et al. Genome-wide risk prediction of common diseases across ancestries in one million people. Cell Genomics 2, 100118 (2022).
Wang, Y. et al. Global Biobank analyses provide lessons for developing polygenic risk scores across diverse cohorts. Cell Genomics 3, 100241 (2023).
Fry, A. et al. Comparison of Sociodemographic and Health-Related Characteristics of UK Biobank Participants With Those of the General Population. Am. J. Epidemiol. 186, 1026–1034 (2017).
Jiang, X., Holmes, C. & McVean, G. The impact of age on genetic risk for common diseases. PLOS Genet 17, e1009723 (2021).
Oni-Orisan, A. et al. Polygenic Risk Score and Statin Relative Risk Reduction for Primary Prevention of Myocardial Infarction in a Real-World Population. Clin. Pharmacol. Ther. 112, 1070–1078 (2022).
Schunkert, H. et al. Clinical utility and implementation of polygenic risk scores for predicting cardiovascular disease: A clinical consensus statement of the ESC Council on Cardiovascular Genomics, the ESC Cardiovascular Risk Collaboration, and the European Association of Preventive Cardiology. Eur. Heart J. 46, 1372–1383 (2025).
Fuat, A. et al. A polygenic risk score added to a QRISK®2 cardiovascular disease risk calculator demonstrated robust clinical acceptance and clinical utility in the primary care setting. Eur. J. Prev. Cardiol. 31, 716–722 (2024).
Park, J. K. & Lu, C. Y. Polygenic Scores in the Direct-to-Consumer Setting: Challenges and Opportunities for a New Era in Consumer Genetic Testing. J. Pers. Med. 13, 573 (2023).
Sudlow, C. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Hao, L. et al. Development of a clinical polygenic risk score assay and reporting workflow. Nat. Med. 28, 1006–1013 (2022).
Fahed, A. C. et al. Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat. Commun. 11, 3635 (2020).
Zhang, J. & Yu, K. F. What’s the Relative Risk? A Method of Correcting the Odds Ratio in Cohort Studies of Common Outcomes. JAMA 280, 1690–1691 (1998).
Pharoah, P. D. P. et al. Polygenic susceptibility to breast cancer and implications for prevention. Nat. Genet. 31, 33–36 (2002).
Thompson, H. R. Truncated lognormal distributions: I. Solution by moments. Biometrika 38, 414–422 (1951).
Brenner, H., Gefeller, O. & Greenland, S. Risk and rate advancement periods as measures of exposure impact on the occurrence of chronic diseases. Epidemiology 4, 229 (1993).
Gail, M. H. et al. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J. Natl. Cancer Inst. 81, 1879–1886 (1989).
Thompson, D. & Chuong, M. genomics-dev/clinical_benefits: 1.0.0 software for ‘Preventing premature deaths through polygenic risk scores’. https://doi.org/10.5281/zenodo.17877984 (accessed 11 Dec 2025).
Acknowledgements
We thank the participants for agreeing to take part in the UK Biobank and other studies that make this work possible. In addition to the named Genomics authors, the study was made possible by contributions from many others at Genomics, including but not limited to the Engineering, Data Services, Science and Medical teams.
Author information
Authors and Affiliations
Contributions
Conceptualization: V.P., G.M., E.A.A., P.D., S.H., J.W.O’S.; Methodology: M.E.W., V.P., S.H., J.W.O’S.; Data Curation, Software, Validation, Investigation and Formal Analysis: M.C., D.T., F.R-M., D.W.; Validation: M.C., D.T., F.R-M., D.W.; Resources: V.P., G.M., P.D.; Writing - Original Draft: M.C., D.T., M.E.W., J.W.O’S.; Writing - Review & Editing: N.J.S., V.P., S.H.; Visualization: M.C., D.T.; Supervision: M.E.W., V.P., G.M., P.D, S.H, J.WO’S.
Corresponding author
Ethics declarations
Competing interests
M.C., D.T., M.E.W., F.R-M., D.W., V.P., G.M., P.D., and S.H. are current or former employees of Genomics Ltd, and own or have owned shares or stock options in the company. G.M. and P.D. are also partners in Peptide Groove LLP. E.A.A. reports advisory board fees from SequenceBio, Foresite Labs, Pacific Biosciences, and Versant Ventures. E.A.A. has ownership interest in Personalis, Deepcell, Svexa, Candela, Parameter Health, Saturnus Bio, outside the submitted work. E.A.A. is a non-executive director of AstraZeneca and Svexa. E.A.A. receives collaborative research support from Illumina, Pacific Biosciences, Oxford Nanopore, Cache, Cellsonics, outside the submitted work. JWO’S is supported by the AHA Postdoctoral fellowship and ACC postdoctoral fellowship and has had consultancy relationships with Google AI, Google Deepmind and Foresite Labs (outside the current work).
Peer review
Peer review information
Nature Communications thanks Nora Pashayan and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chuong, M., Thompson, D., Weale, M.E. et al. Preventing premature deaths through polygenic risk scores. Nat Commun 17, 1379 (2026). https://doi.org/10.1038/s41467-025-68129-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68129-x
