
Abstract
Sigma Non-Opioid Intracellular Receptor 1 (SigmaR1) is a member of the sigma family of receptors that interacts with a variety of psychotomimetic drugs and is involved in a wide range of cellular and physiological functions. Despite its increasing importance in human physiology and disease, the subcellular localization of SigmaR1 and its molecular function remain poorly defined. Using endogenous tagging and cell fractionation, we show that SigmaR1 is a type II integral ER membrane protein that is specifically enriched at ER sheets. A short region at the N-terminus of SigmaR1 promotes its ER-sheet localization. Importantly, our biochemical studies demonstrate that SigmaR1 directly interacts with components of the translocon complex including TRAPα and Nicalin. In addition, we found that a β-barrel at the C-terminal of SigmaR1 binds phosphatidylcholine (PC), and the binding of PC strengthens the association of SigmaR1 with the translocon complex. SigmaR1 knockout systematically impaired cellular protein and lipid homeostasis, resulting in accumulation of lipid droplets in hepatocytes. Collectively, we propose that SigmaR1 is an auxiliary translocon factor that binds lipids to regulate protein and lipid droplet homeostasis, which may underlie the broad and vital roles of SigmaR1 in physiology and disease.
Similar content being viewed by others


Introduction
Despite the continuity of the endoplasmic reticulum (ER) membrane system, the ER has a distinct morphological domain consisting of sheets and tubules that differ in their characteristic membrane curvature1,2,3, which can be clearly observed in electron microscopy (EM) studies2,4,5,6. Critical proteins drive the formation of these structural morphologies, which in turn generate rough and smooth functional domains of the ER7,8,9,10. ER sheets, which have a higher density of ribosomes and associated protein translocation machinery than ER tubules, are also referred to as rough ER1. Accordingly, the flat and stacked membrane of the rough ER can also better accommodate the bulky membrane-bound polyribosomes that support protein synthesis1,8,11,12,13.
In mammalian cells, the import of proteins into the ER begins before the polypeptide chain is completely synthesized, i.e., the import is a co-translational process. During co-translational translocation, the signal recognition particles interact with their cognate receptor to deliver the ribosome nascent chain complex into the rough ER and bind to the translocon complex14,15. The translocon complex is a large multi-subunit channel that transports the nascent peptides across ER membrane and undergo post-translational modifications and folding16,17,18,19,20,21. The eukaryotic translocon is composed of Sec61 core channels associated with multiple co-factors, including the translocon -associated protein (TRAP) complex22, translocation-associated membrane protein (TRAM)23,24, Sec62/6325, oligosaccharyl transferase (OST)21, and signal peptidase complex26. A ribosome-associated complex consisting of the core Sec61 channel and five accessory factors—TMCO1, CCDC47, Nicalin, TMEM147, and NOMO-has recently been characterized and plays a specific role in the biogenesis of multipass membrane proteins27,28,29,30. Nevertheless, the molecular composition of the translocon complex remains incompletely clear.
Sigma Non-Opioid Intracellular Receptor 1 (SigmaR1) is a member of the Sigma family of receptors that interact with a wide range of psychotomimetic drugs. SigmaR1 has been shown to play an important role in the cellular functions of various tissues associated with the endocrine, immune, and nervous system. Mutations of SigmaR1 have been associated with distal hereditary motor neuropathy, type II and juvenile amyotrophic lateral sclerosis. In addition, SigmaR1 regulates protein stability by modulating the unfolded protein response31,32. SigmaR1 is also reported to play a role in calcium signaling by modulating of the IP3R3-dependent calcium efflux at the ER33. Although the cellular and physiological importance of SigmaR1 has been increasingly appreciated, the subcellular localization and molecular function of SigmaR1 have not yet been fully elucidated.
In agreement with a recent study34, we demonstrate that SigmaR1 is specifically enriched in ER sheets. This localization is facilitated by a short N-terminal region, which we found mediates direct interactions with the translocon components TRAPα and Nicalin. In addition, the C-terminal β-barrel structure of SigmaR1 binds phospholipids, including phosphatidylcholine (PC), and the binding strengthens the association of SigmaR1 with the translocon complex. SigmaR1 knockout (KO) systematically impaired cellular protein and lipid homeostasis, leading to dramatic accumulation of lipid droplets (LDs) in hepatocytes. Together, these results elucidate the molecular mechanisms underlying the diverse roles of SigmaR1 in physiology and disease.
Results
SigmaR1 is an ER sheet protein
Using high-resolution confocal microscopy, we investigated the subcellular localization of SigmaR1. SigmaR1-GFP was specifically enriched in ER sheets, as demonstrated by its strong co-localization with the ER-sheet marker Halo-Sec61β35 (Fig. 1a). This enrichment was significantly greater than its co-localization with the ER-tubule markers Reticulon-47, Reep57, and Tex236 (Fig. 1b–e). Furthermore, in accord with the recent study34, overexpression of SigmaR1 appears to increase the abundance of ER sheets, evident from their expansion into the cell periphery (Fig. 1a).

a–d Representative confocal images of live COS7 cells expressing SigmaR1-GFP (green) and ER markers including Halo-Sec61β (magenta, an ER sheet marker, a), Reticulon4-Halo (b), Reep5-Halo (c) or Halo-Tex2 (d) (magenta, ER tubule markers) with insets. e Pearson’s correlation coefficient of SigmaR1-GFP vs Halo-Sec61β (32 cells), Reticulon4-Halo (25 cells), Reep5-Halo (22 cells) and Halo-Tex2 (19 cells) as in (a–d) from more than 3 independent experiments. Ordinary one-way ANOVA with Tukey’s multiple comparisons test. Mean ± SD. f Representative images of a live sfGFP-SigmaR1 KI (green) HeLa cell expressing Halo-Sec61β (magenta) with insets. g Immunofluorescence of a fixed sfGFP-SigmaR1 KI (green) HeLa cell stained with Sec61β antibody (magenta) with insets. h Representative images of a live sfGFP-SigmaR1 KI (green) HeLa cell expressing Reticulon4-Halo (magenta) with insets. i Pearson’s correlation coefficient of endogenous SigmaR1-GFP and Halo-Sec61β (32 cells), anti-Sec61β (45 cells) or Reticulon4-Halo (45 cells) as in (f–h) from 3 independent experiments. Two-tailed unpaired Student’s t-test. Mean ± SD. j Membrane fractionations isolated from rat liver lysate showing the distribution of endogenous SigmaR1 relative to other proteins. SigmaR1 was enriched in fractions 3, 4, and 5 similar as known ER sheet proteins (anti-Sec61β and anti-Calnexin). The ER fractions with different density were labelled from fraction 0 (heavy) to 11 (light). k The distribution of SigmaR1 relative to other known ER proteins as in (j). A red arrow denotes the ER-sheet membrane fractions from 3 independent experiments. Mean ± SD.l Representative images of live ER-GFP-expressing COS7 cells treated with either scrambled or SigmaR1 siRNAs under normal or starvation (EBSS, 4 h) with insets. m Quantification of ER sheet abundance under normal [Scrambled (17 cells) or SigmaR1 siRNAs (20 cells)] or starvation (EBSS, 4 h) [Scrambled (16 cells) or SigmaR1 siRNAs (19 cells)] as in (l) from 3 independent experiments. Ordinary one-way ANOVA with Tukey’s multiple comparisons test. Mean ± SD. Scale bars, 10 μm in the whole cell images and 2 μm in the insets (a–d, f–h, l).
To avoid potential overexpression artifacts, we endogenously tagged the N-terminus of SigmaR1 with monomeric super folder GFP (sfGFP A206K) in HeLa cells using CRISPR-Cas9, generating an sfGFP-SigmaR1 knock-in (KI) line (Supplementary Fig. 1a). The functionality of the construct was confirmed later in this manuscript (Fig. 6n). We chose HeLa cells due to the expression level of SigmaR1 was significantly higher compared to other cell lines commonly used in our laboratory (Supplementary Fig. 1b). The KI line (clone-2, used throughout this study) was validated by immunoblotting (Supplementary Fig. 1c). Live-cell and immunofluorescence imaging revealed that endogenous sfGFP-SigmaR1 was preferentially enriched in ER sheets, as evidenced by strong colocalization with the sheet marker Sec61β but minimal overlap with the tubule marker Reticulon-4 (Fig. 1f–i). Together, these results support that SigmaR1 is an ER sheet-resident protein.
To corroborate these findings, we investigated the distribution of endogenous, untagged SigmaR1 by cell fractionation. We selected rat liver tissue for this assay due to its high endogenous SigmaR1 expression (Supplementary Fig. 1d) and high abundance of rough ER37,38,39,40. Consistent with our imaging results, SigmaR1 was predominantly enriched in the rough ER fractions (3, 4 and 5), which were marked with the ER-sheet proteins Sec61β and Calnexin, but absent from fractions containing the tubular markers Reticulon-4 and Reep5 (Fig. 1j, k; Supplementary Fig. 1e). These results provide independent biochemical evidence that SigmaR1 is specifically localized to ER sheets.
The rough ER is characterized by the presence of membrane-bound ribosomes in the form of a thread-like structure, the polysome. Polysomes not only promote the formation of ER sheet but also facilitate the localization of certain membrane proteins to ER sheets8. We therefore investigated whether the polysome is required for the ER-sheet localization of SigmaR1. Live-cell imaging showed that localization of SigmaR1 was not significantly affected by treatment of puromycin (Puro) (Supplementary Fig. 1f, g), which releases nascent polypeptide chains from the ribosome and disassembles polysomes41. Similarly, cycloheximide (CHX), which halts polypeptide chain elongation but leaves the polysomes intact42,43, also had no substantial effect (Supplementary Fig. 1f, g). These results indicate that the localization of SigmaR1 to ER sheet is independent of polysome.
We next investigate whether the role of SigmaR1 in maintaining ER sheet-to-tubule ratio is responsive to cellular stress. Starvation triggers the expansion of ER sheet44—a phenomenon we could recapitulate in our system (Fig. 1l, m). Importantly, the starvation-induced expansion of ER sheet was significantly inhibited upon SigmaR1 depletion (Fig. 1l, m), indicating that starvation-induced ER sheet formation requires SigmaR1.
A short region at the N-terminus promotes the ER-sheet localization of SigmaR1
To determine the mechanism underlying the specific targeting of SigmaR1 to ER sheets, we investigated its membrane topology. SigmaR1 has been reported as a type II integral membrane protein45,46. We therefore sought to confirm this topology in our cellular system (Supplementary Fig. 2a). We utilized a live-cell assay based on a high-affinity interaction between a Flag tag and a genetically encoded cytosol-localized Halo-Flag frankenbody (a single-chain variable fragment)47. When co-expressed, Flag-SigmaR1, but not SigmaR1-Flag, recruited the frankenbody (Supplementary Fig. 2b, c), confirming that the N-terminus is cytosolic and the C-terminus is luminal. These results validate the canonical type II topology of SigmaR1 in the ER.
To identify the ER-sheet targeting determinant, we dissected the SigmaR1 protein. We found that a conserved N-terminal region containing the transmembrane domain (TM) (residues 1-30; SigmaR1-NT) was sufficient for ER-sheet localization, albeit to a slightly lesser extent than the full-length protein (Fig. 2a, b, e & f; Supplementary Fig. 2d). In contrast, a construct containing only the TM domain (residues 10-30; SigmaR1-TM) failed to target sheets and instead colocalized with the tubule marker Reticulon-4, regardless of the position of the GFP tag (Fig. 2c, e, f; Supplementary Fig. 2e). This implicated the upstream residues 2-8 as critical. Consistent with this, their deletion (SigmaR1Δ2-8) abolished specific ER-sheet targeting (Fig. 2d–f), demonstrating that this short N-terminal segment is important for ER sheet localization.

a–d Representative images of live COS7 cells expressing full-length (a, green) or SigmaR1 truncated mutants including SigmaR1-1-30 (b, green), SigmaR1-10-30 (c, green), SigmaR1∆2-8 (d, green) along with Reticulon4-Halo (top panel; magenta) or Halo-Sec61β (bottom panel; magenta) with insets. e, f Pearson’s correlation coefficient of SigmaR1-GFP (32 cells), SigmaR1∆2-8 (26 cells), SigmaR1-1-30 (30 cells) and SigmaR1-10-30 (32 cells) vs Reticulon4-Halo (e) or Halo-Sec61β [SigmaR1-GFP (28 cells), SigmaR1∆2-8 (20 cells), SigmaR1-1-30 (26 cells) and SigmaR1-10-30 (26 cells)] (f) in more than three independent experiments. Ordinary one-way ANOVA with Tukey’s multiple comparisons test. Mean ± SD. g, h Representative images of live COS7 cells expressing SigmaR1(1-30)-Tex2(∆1-517)-GFP (g, green) or SigmaR1(2-8)-Tex2(∆1-473)-GFP (h, green) along with Halo-Tex2 (magenta) and mCh-Sec61β (blue) with insets. i Pearson’s correlation coefficient of two chimeric proteins and Halo-Sec61β [SigmaR1(1-30)-Tex2(∆1-517)-GFP (29 cells) or SigmaR1(2-8)-Tex2(∆1-473)-GFP (24 cells)] as in (g, h) from 3 independent experiments. Two-tailed unpaired Student’s t-test. Mean ± SD. Scale bars, 10 μm in the whole cell images and 2 μm in the insets in (a–d, g, h).
We next investigated whether the SigmaR1-NT is able to redirect other proteins to ER sheet. To test this, we generated a chimera in which SigmaR1-NT was fused to a truncated version of the tubular ER protein Tex2 [Tex2Δ(NT-TM); Supplementary Fig. 2f, g]. This chimeric protein [SigmaR1-NT-Tex2Δ(NT-TM)-GFP] was highly enriched on ER sheets (Fig. 2g). In contrast, the short N-terminal segment (residues 2-8) alone was insufficient to redirect Tex2 to sheets (Fig. 2h, i). These results demonstrate that both the short N-terminal sequence and the TM domain are required for efficient ER-sheet targeting.
SigmaR1 directly interacts with the translocon complex
To elucidate the functional significance of the ER sheet localization of SigmaR1, we sought to identify its interacting partners. We performed coimmunoprecipitation (CoIP) using SigmaR1-GFP as bait, followed by mass spectrometry (MS) analysis. Consistent with its ER localization, functional enrichment analysis revealed that the identified SigmaR1-interacting proteins were predominantly associated with ER-related cellular components in the GO database (Supplementary Fig. 3a) and metabolic pathways in the KEGG database (Supplementary Fig. 3b). Interestingly, our analysis identified numerous translocon components, including TRAPα, Nicalin, SEC11A, CCDC47, TMEM147, NOMO1, RRBP1, TRAM1, and TMCO1, as potential SigmaR1-interacting partners (Supplementary Fig. 3c). Consistent with this finding, all of these proteins exhibited strong colocalization with SigmaR1 on ER sheets, with the exception of TRAM1. TRAM1 was mainly found in distinct punctate structures in addition to its ER sheet localization (Fig. 3a, Supplementary Fig. 3d-h).

a Representative confocal images of live COS7 cells expressing SigmaR1-GFP (green) and translocon proteins including Nicalin-Halo, Halo-TRAPα, CCDC47-Halo, Halo-SEC11A (magenta) with insets from at least 3 independent experiments. b GFP-Trap assays in sfGFP-SigmaR1 KI HeLa cells from 3 independent experiments demonstrated endogenous interactions between SigmaR1 and the translocon components such as Nicalin, TRAPα, SEC11A, and CCDC47. c GFP-Trap assays from 3 independent experiments demonstrated interactions between SigmaR1-GFP or SigmaR1-GFP∆2-8 and endogenous Nicalin. d In vitro pull-down assays from 3 independent experiments demonstrated direct interactions between purified His-Flag-SigmaR1 or His-Flag-SigmaR1∆2-8 and GFP-Nicalin or GFP-TRAPα. Asterisks denote the His-Flag-SigmaR1 (blue) or His-Flag-SigmaR1∆2-8 (red) band in Coomassie blue staining. e ER Membrane fractionations isolated from the sfGFP-SigmaR1 KI cells showing the distribution of endogenous SigmaR1 relative to other proteins. SigmaR1 was enriched in rough ER fractions containing known ER translocon components (Sec61β, Nicalin and TRAPα) and ribosomal protein (RPL10) from 3 independent experiments. The ER fractions with different density were labelled from fraction 1 (light) to 14 (heavy). The fractions (12-14) enriched with rough ER proteins were highlighted in blue. f GFP-Trap assays performed in smooth or rough ER fractions from 3 independent experiments demonstrated interactions between SigmaR1 and Nicalin or TRAPα specifically occurred in rough ER fractions. g FLIM assays illustrate a direct and specific interaction between SigmaR1-mClover2 (green) and mRuby2-Nicalin (magenta), but not Reep5-mRuby2 (magenta) or Sec61β-mRuby2 (magenta). h Mean Clover2 fluorescence lifetime [SigmaR1-mClover2 (23 cells), SigmaR1-mClover2 and mRuby2-Nicalin (14 cells), Reep5-mRuby2 (8 cells) or Sec61β-mRuby2 (14 cells)] as in (g) from 3 independent experiments. Ordinary one-way ANOVA with Tukey’s multiple comparisons test. Mean ± SD. Scale bars, 10 μm in the whole cell images and 2 μm in the insets (a) and 10 μm in (g).
To validate these interactions, we performed GFP-Trap assays. Consistent with our imaging data, SigmaR1 interacted with Halo-tagged Nicalin, TRAPα, SEC11A, TMEM147, NOMO1, and TMCO1, as well as GFP-tagged RRBP1 and TRAM1 (Supplementary Fig. 4a-h). Importantly, immunoprecipitation using our sfGFP-SigmaR1 KI cell line revealed specific interactions with Nicalin, TRAPα, SEC11A, and CCDC47 at endogenous levels, but not with NOMO1, RRBP1, SRP54, or TRAM1; a weak interaction was detected with TMEM147 (Fig. 3b, Supplementary Fig. 4i). Consistent with the crucial role of residues 2-8 in targeting SigmaR1 to ER sheets, GFP-Trap assays demonstrated that deletion of this short sequence significantly impaired its interaction with endogenous Nicalin (Fig. 3c).
We next employed in vitro GST pull-down assays to determine if these interactions were direct. Recombinant GST tagged SigmaR1 (GST-SigmaR1) bound directly to His-Nicalin and His-TRAPα (Supplementary Fig. 4j, k), but not to His-SEC11A or His-CCDC47 (Supplementary Fig. 4l, m). To further validate these direct interactions, we purified His-Flag-SigmaR1 alongside GFP-TRAPα and GFP-Nicalin from HEK293F cells, and found that purified SigmaR1 bound directly to both GFP-TRAPα and GFP-Nicalin, but not to GFP alone (Fig. 3d). Importantly, the deletion of the residues 2-8 in SigmaR1 significantly reduced its binding to these translocon components (Fig. 3d). Collectively, these results demonstrate that SigmaR1 directly interacts with specific components of the translocon complex, and indicate an essential role of the NT sequence (residues 2-8) of SigmaR1 in its binding to the translocon complex.
To validate the specificity of the association of SigmaR1 with the translocon, we employed sucrose gradient centrifugation. Using solubilized ER microsomes isolated from the sfGFP-SigmaR1 KI cell line, we compared SimgaR1 distribution in ribosome-bound and ribosome-free fractions. Endogenous SigmaR1 was specifically co-enriched with the translocon components Nicalin and TRAPα in the ribosome-bound ER sheet fractions (Fig. 3e). GFP-Trap assays further confirmed that the interactions between SigmaR1 and these two proteins occurred specifically within these ER sheet fractions (Fig. 3f).
It should be noted that no interaction was detected between SigmaR1 and the core channel of the translocon complex, Sec61β, in the ribosome-bound ER fraction (Supplementary Fig. 4n). This finding was further corroborated by in vitro pulldown assays, which showed no binding between His‑Flag‑SigmaR1 and GFP‑Sec61β (Supplementary Fig. 4o). Together, these results suggest that SigmaR1 is not a structural subunit of the core translocon channel.
Furthermore, we performed fluorescence Lifetime Imaging Microscopy (FLIM) to validate a specific interaction between SigmaR1 and Nicalin at ER sheets. The FLIM results demonstrated that mClover2 fluorescence lifetime was significantly reduced in a cell co-expressing SigmaR1-mClover2 and mRuby2-Nicalin (yellow asterisk) compared to that in cells expressing SigmaR1-mClover2 alone (white asterisk) in the same imaging field (Fig. 3g, top panel; 3 h). In contrast, the mClover2 fluorescence lifetime was not significantly impacted in cells co-expressing SigmaR1-mClover2 and Reep5-mRuby2 (Fig. 3g, middle panel; 3 h), indicating no direct interaction between these two proteins. Consistent with our co-IP and pulldown data, a slight reduction in lifetime was observed with the translocon channel Sec61β-mRuby2, suggesting these proteins are co-enriched in the translocon complex but do not directly interact (Fig. 3g, bottom panel; 3 h). Taken together, these findings support that SigmaR1 is an auxiliary component that physically engages with the translocon machinery.
SigmaR1 binds to PC via a C-terminal β-barrel structure
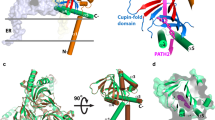
SigmaR1 harbored a V-shaped double helix bundle and a cupin-like β-barrel structure in the C-terminal region48,49,50. Notably, the β-barrel exhibits a wide, flat, and hydrophobic membrane-proximal surface that forms a large binding cavity, suggesting a potential site for hydrophobic lipid interactions (Fig. 4a). To test this hypothesis, we analyzed lipids bound to endogenous SigmaR1 in the SigmaR1 KI cell line using non-targeted lipidomics (LC-MS/MS). This revealed associations with various phospholipids, including phosphatidylcholine (PC), phosphatidylethanolamine (PE), sphingolipids/ceramides, and cholesterol (Fig. 4b), with a marked preference for PC (Fig. 4b). Molecular docking simulations further supported these findings, demonstrating a favorable binding mode between the β-barrel of trimeric SigmaR1 and PC (Fig. 4c; Supplementary Fig. 5a, b).

a Protein structure of SigmaR1 relative to ER membrane. The N-terminal (residues 1-30) portion is in red, and the C-terminal region in ER lumen is in green with the β barrel (residues 80-168) is colored in purple with insets. b Volcano plot of lipids bound to endogenous Sigmar1 from the Sigmar1 KI line. Lipids that were considered significant (−log [P value] > 1.3; P < 0.05; Two-sided t-test with Benjamini-Hochberg FDR adjustment). PC species and cholesterol were highlighted by different colors (PC in blue and cholesterol in magenta). c. Molecular docking analysis showing the putative binding of PC (grey) in the β-barrel structure of trimeric SigmaR1 (blue) with interfaces highlighted in yellow. Note that SigmaR1 monomer was shown. d, e. Left: crystal structures of SigmaR1∆TM (d) and SigmaR1∆2-30 (e); Right: In vitro protein-lipid migrating assays for SigmaR1∆TM (d) and SigmaR1∆2-30 (e). Purified GST-SigmaR1 and mutants were incubated with NBD-tagged lipids and examined by native PAGE. Phospholipids, visualized by their fluorescence, comigrated with protein, visualized by Coomassie blue staining. Experiments were repeated three times independently with consistent results. f Conservative analysis of amino acids of the β-barrel structure in different species with hydrophobic amino acids highlighted in yellow, among which evolutionarily conserved amino acids were indicated by red asterisks. g Schematic diagram of liposome (70% DOPC, 30% PE and 0.75 M sucrose) pelleting assays using purified GST-SigmaR1∆TM proteins. h The crystal structures of SigmaR1∆TM-Mut2 (104 V, 105 L, 106 L, 107F-W). i. Liposome pelleting assays showing the distribution of GST-SigmaR1∆TM (0.1 μg/uL) or GST-SigmaR1∆TM-Mut2 (104 V, 105 L, 106 L, 107F-W) (0.1 μg/uL) in supernatant and pellet. Multiple amounts of proteins (0, 5, 10, 20 μL) were used in these assays. Immunoblots were performed using GST antibody from 3 independent experiments. j In vitro pull-down assays from 3 independent experiments demonstrated a stronger binding between purified His-Flag-SigmaR1 and GFP-Nicalin or GFP-TRAPα in the presence of PC. A red asterisk denotes the His-Flag-SigmaR1 band in Coomassie blue staining.
To determine whether SigmaR1 binds glycerophospholipids directly, we performed in vitro protein-lipid co-migration assays using purified GST tagged SigmaR1 with a deletion of the TM (GST-SigmaR1∆TM). Using native gel electrophoresis, we found that GST-SigmaR1-∆TM co-migrated with nitrobenzoxazole (NBD)-labeled PC, phosphatidic acid (PA), and phosphatidylserine (PS), demonstrating direct binding to these lipids. In contrast, it did not bind to cholesterol (Fig. 4d). The lack of cholesterol binding is consistent with previous reports that cholesterol association is mediated by the TM domain of SigmaR151, which was absent in this construct. Notably, the purified SigmaR1 also failed to bind PE and ceramide in vitro (Fig. 4d), suggesting that its cellular associations with these lipids are likely indirect.
To determine whether the β-barrel domain of SigmaR1 mediates lipid binding, we first purified the GST-SigmaR1∆2-30 mutant, which is deficient in ER-sheet targeting. This mutant retained the ability to bind lipids, though with reduced affinity compared to the wild-type protein (Fig. 4e). In addition, a truncation mutant containing only the β-barrel domain (GST-SigmaR1-80-168) could bind these lipids (Supplementary Fig. 5c), suggesting that this structural region may mediate lipid interactions. We further generated four space-filling mutants (Mut1, 2, 3 and 4) of SigmaR1 in which several conserved residues with small hydrophobic side chains of the β-barrel structure were substituted with bulky tryptophan residues (Fig. 4f), based on a previous study52. We purified these mutants and found that all the four mutants reduced the lipid binding affinity to varying degrees, with the Mut2 being most efficient (Supplementary Fig. 5d-g).
To further validate these results, we performed in vitro liposome pelleting assays (Fig. 4g). The GST-SigmaR1, but not GST-SigmaR1-Mut2 (104 V, 105 L, 106 L, 107F-W), was efficiently pelleted by sucrose-loaded liposomes (Fig.4h, i), confirming that lipid interaction is mediated specifically by the β-barrel domain of SigmaR1.
We next investigated the functional significance of lipid binding of SigmaR1. In vitro pulldown assays showed that PC binding enhances the binding of SigmaR1 to these two translocon components Nicalin and TRAPα (Fig. 4j). This regulatory relationship supports that SigmaR1 functions not as a static structural element, but as a dynamic auxiliary factor whose interaction with the translocon complex is modulated by lipid binding.
SigmaR1 KO impairs cellular protein homeostasis
Given the direct interaction of SigmaR1 with the translocon complex, we next investigated the function of SigmaR1 relative to protein homeostasis in SigmaR1 KO HeLa cells generated by CRISPR-Cas9 (Supplementary Fig. 6a, b). Of note, SigmaR1 KO did not affect the levels of other translocon components (Supplementary Fig. 6c), suggesting that SigmaR1 is not directly involved in the stability of the translocon complex.
Importantly, we observed a marked increase in phosphorylated eIF2α—a key integrator of the stress response—in SigmaR1 KO cells, despite unchanged total eIF2α levels (Fig. 5a). As eIF2α phosphorylation globally represses translation in response to cell stress, this indicates that SigmaR1 is critical for maintaining protein homeostasis. Notably, this effect is not indirectly caused by ER stress, as SigmaR1 deficiency neither induced nor potentiated thapsigargin-triggered ER stress. This was evidenced by unaltered levels of the chaperone BiP (Fig. 5a) and ER whorls (marked by Sec61β) (Supplementary Fig. 6d, e), both established markers of ER stress53.

a Immunoblots showing the levels of p-eIF2α, eIF2α and BiP in control or SigmaR1 KO HeLa cells upon Thapsigargin treatments (1 μM) at multiple timepoints (0, 3, 6, 12, 24 h) from 3 independent experiments. b GO enrichment analysis by comparing candidate proteins in SigmaR1 KO#3 HeLa cells with those in the control group, with cellular component (left; green), molecular function (middle; orange) and biological process (right; blue) being shown individually. Significance was assessed using a two-sided hypergeometric test with Benjamini-Hochberg FDR adjustment (FDR < 0.05). c The workflow of rescue experiments in SigmaR1 KO HeLa cells. d–f: as in (b), the GO enrichment analysis by comparing levels of candidate proteins in SigmaR1 KO#3 HeLa cells supplemented with WT SigmaR1-GFP (d), the ER sheet localization mutant (SigmaR1∆2-8-GFP; e), or the lipid binding mut (f), with those in SigmaR1 KO cells. Significance was assessed using a two-sided hypergeometric test with FDR adjustment (FDR < 0.05).
To systematically identify pathways affected by SigmaR1 loss, we performed quantitative MS. SigmaR1 deficiency resulted in a substantial downregulation of ribosome-related proteins, as highlighted by enrichment analyses using the Gene Ontology (GO) and KEGG databases (Fig. 5b, Supplementary Fig. 6f). This is consistent with the role of SigmaR1 as an auxiliary factor of the translocon on ER sheets. KEGG analysis further implicated SigmaR1 in a broad range of physiological, pathological, and disease contexts, including infectious diseases (e.g., COVID-19), neurodegenerative disorders, cell cycle regulation, and endocytosis (Supplementary Fig. 6f). In addition, proteins involved in lipid metabolism were also affected (Supplementary Fig. 6f), suggesting a role for SigmaR1 in lipid regulation.
To investigate the mechanistic link between molecular function of SigmaR1 and protein homeostasis dysregulation, we performed rescue experiment in SigmaR1 KO HeLa cells by reconstituting expression of SigmaR1-GFP, an N-terminal deletion mutant (defective in ER sheet targeting), or the lipid-binding variant. Cells expressing these constructs were isolated by flow cytometry for subsequent proteomic analysis (Fig. 5c). Introduction of SigmaR1-GFP efficiently restored global protein homeostasis, particularly rescuing these ribosomal pathways (Fig. 5d; Supplementary Fig. 6g). In contrast, the NT-deletion mutant failed to fully restore the proteome (Fig. 5e; Supplementary Fig. 6h), underscoring the importance of ER sheet localization and translocon interaction for SigmaR1 function. Similarly, the lipid-binding mutant was also insufficient for complete rescue (Fig. 5f; Supplementary Fig. 6i), indicating that the ability of SigmaR1 to bind lipids is also involved in maintaining protein homeostasis.
SigmaR1 depletion resulted in an accumulation of lipid droplets in hepatocytes
Unexpectedly, small interfering RNA (siRNA)-mediated depletion of SigmaR1 triggered a dramatic accumulation of LDs in the hepatic cell line HepG2 cells following oleic acid (OA) stimulation (Fig. 6a; Supplementary Fig. 7a). To confirm the phenotype, we measured intracellular levels of triglycerides (TG) and cholesterol. In agreement with our imaging results, both TG and cholesterol were significantly increased in SigmaR1-depleted cells compared to controls (Fig. 6b, c).

a Representative images of live HepG2 cells stained by BODIPY 558/568 (gray) treated with scrambled or two different SigmaR1 siRNAs (#1, #3) with insets under treatment of OA (200 μM) for 16 h. b, c The levels of TG (b) and cholesterol (c) in HepG2 cells as in (a) in four independent experiments. Two-tailed unpaired Student’s t test. Mean ± SD. d Representative images of BODIPY 558/568 (gray)-stained mouse primary liver cells treated with scrambled (left) or SigmaR1 siRNA (right) with insets under treatment of OA (200 μM) for 16 h. e Western blots showing the efficiency of siRNAs-mediated SigmaR1 depletion in primary hepatocytes from 3 independent experiments. f Quantification of LDs size in mouse primary liver cells as in (d) treated with scrambled (139 cells) or SigmaR1 siRNA (99 cells) in at least three independent experiments. Two-tailed unpaired Student’s t test. Mean ± SD. g, h. The levels of TG (g) and cholesterol (h) in mouse primary liver cells as in (d) from three independent experiments. Two-tailed unpaired Student’s t test. Mean ± SD. i, j Representative TEM images of control (i) or SigmaR1-depleted (j) HepG2 cells under normal culture condition with insets. k, l The LD diameter [scrambled (11cells) or SigmaR1 siRNA (13 cells)] (k) and LD number [scrambled (11 cells) or SigmaR1 siRNA (11 cells)] per cell (l) as shown in (i, j). Two-tailed unpaired student’s t-test. Mean ± SD. m–q BODIPY 558/568 (magenta)-stained HepG2 cells treated with SigmaR1 siRNA expressing siRNA-resistant SigmaR1-GFP (m, green), sfGFP-SigmaR1 (n), SigmaR1-GFP∆81-160 (o), SigmaR1-GFP lipid-binding Mut2 (p), or SigmaR1-GFPΔ2-8 (q) with insets. r Quantification of LD size in HepG2 cells [scrambled (39 cells) or SigmaR1 siRNA#1 (42 cells), SigmaR1 siRNA#3 (43 cells), SigmaR1 siRNA expressing siRNA-resistant SigmaR1-GFP (83 cells), sfGFP-SigmaR1 (24 cells), SigmaR1-GFPΔ2-8 (92 cells), SigmaR1-GFP∆81-160 (41 cells) or SigmaR1-GFP lipid-binding Mut2 (68 cells)] as in (a, m–q) from three independent experiments. Ordinary one-way ANOVA with Tukey’s multiple comparisons test. Mean ± SD. Scale bars, 10 μm in the whole cell images and 2 μm in the insets (a, d, m–q) and 2 μm in (i, j).
We next confirmed that the ER sheet localization of SigmaR1 and the aberrant accumulation of LDs upon its depletion are conserved across other hepatic-related cells, including the Huh7 cell line (Supplementary Fig. 7b-d) and mouse primary liver cells (Fig. 6d–f; Supplementary Fig. 7e). In primary hepatocytes, siRNA-mediated depletion of SigmaR1 not only resulted in a striking increase in LD size but also significantly elevated levels of TG and cholesterol/esters levels following OA treatment (Fig. 6g, h).
Notably, this lipid accumulation phenotype exhibited cell-type specificity. Although proteomic data from SigmaR1-KO HeLa cells indicated a predisposition toward dysregulated lipid metabolism, these cells exhibited only moderate, statistically insignificant LD accumulation (Supplementary Fig. 7f–i). Similarly, SigmaR1 depletion in non-hepatic cell lines, including pancreatic PaCa2 cells (Supplementary Fig. 7j–l) and mouse neuroblastoma Neuro2a cells (Supplementary Fig. 7m–o), resulted in a moderate LD phenotype, despite robust conservation of its ER sheet localization. We attribute this cell-type-dependent effect to the specialized role of hepatocytes in lipid synthesis, storage, metabolism and secretion, which appears to amplify the phenotypic severity of SigmaR1 loss.
Next, we performed transmission electron microscopy (TEM) to examine the ultrastructural details of LDs in SigmaR1-depleted HepG2 cells. TEM micrographs revealed a strong increase in LD size without a significant change in their number (Fig. 6i–l).
Importantly, the strong accumulation of LDs was specific to SigmaR1, as the introduction of SigmaR1-GFP substantially rescued the phenotype compared to an untransfected cell in the same imaging field (Fig. 6m). The sfGFP-tagged version of SigmaR1 (sfGFP-SigmaR1) also effectively reversed the LD accumulation in SigmaR1-depleted HepG2 cells (Fig. 6n), confirming its functionality.
Next, we investigated whether and to what extent LD accumulation was dependent on lipid-binding activity or ER sheet-localization of SigmaR1. Importantly, introduction of SigmaR1∆81-160 (Fig. 6o) or lipid binding deficient mutant (GST-SigmaR1-Mut2; Fig. 6p, r) could not completely restore the LD defect. Similarly, the LD accumulation phenotype was not fully rescued by expression of SigmaR1∆2-8, the mutant defective in ER sheet targeting and translocon association (Fig. 6q, r). These results suggest that both the ligand-binding and ER-sheet localization are required for the role of SigmaR1 in LD dynamics.
In addition, depletion of SigmaR1, but not other known ER sheet factors p180/RRBP1 and Climp63, significantly impacted LD dynamics. Notably, depletion of Kinectin, the kinesin adaptor associating with ER sheet, also caused accumulated LDs, but to a lesser extent compared to SigmaR1 depletion (Supplementary Fig. 8a-c). This suggests that the LD defect in SigmaR1-depleted cells is not merely a secondary consequence of an altered ER sheet-to-tubule ratio but rather points to a more direct, unique role for SigmaR1 in lipid storage.
Discussion
SigmaR1 is involved in a broad spectrum of cellular and physiological functions in various tissues including the endocrine, immune and nervous systems. Despite its importance in human physiology and disease, the subcellular localization and related functional significance of SigmaR1 are not yet fully understood. In this study, we demonstrate that SigmaR1 is a type II integral membrane protein specifically enriched in ER sheets. This localization is promoted by a short N-terminal region and facilitates direct interaction with specific translocon components, including TRAPα and Nicalin. In addition, the β-barrel structure at the C-terminal of SigmaR1 binds to phospholipids including PC, and the binding of PC promotes the association of SigmaR1 with the translocon complex. SigmaR1 KO impairs protein and lipid homeostasis, resulting in accumulation of LDs. Based on these findings, we propose a working model in which SigmaR1, enriched within ER sheets, functions as a dynamic auxiliary factor for the translocon complex. In this model, the binding of SigmaR1 to PC enhances its association with specific translocon components (Nicalin and TRAPα), thereby potentially modulating translocon activity by promoting ribosome binding to the rough ER, consequently contributing to the maintenance of protein and lipid homeostasis. It should be noted that the evidence remains insufficient to establish a definitive causal link between PC binding, enhanced translocon engagement, and functional regulation in a physiological context at this stage. The model therefore represents a plausible framework that requires further validation (Supplementary Fig. 8f).
During the preparation of this manuscript, a study was published showing that SigmaR1 is an ER-sheet protein by a proteomics screen using ER sheet-localized RNA-binding proteins as bait34. They also show that SigmaR1 oligomers bind to and flatten the luminal leaflet of the ER membrane to counteract the membrane curvature, thereby promoting the amount of ER sheets in cells via their extended arrays of amphipathic helices34. In this study, we have provided more evidence to demonstrate the localization of SigmaR1 and further investigate its functional significance. We made a knock-in cell line to tag endogenous SigmaR1 with sfGFP using CRISPR-Cas9, and further confirmed the ER-sheet localization of endogenous SigmaR1. In addition, our cell fractionation results confirmed that endogenous SigmaR1 was enriched in the rough ER fraction. Furthermore, we dissected the mechanism through which SigmaR1 specifically targets to ER sheet by identifying a short region at the N-terminus of SigmaR1, which promote its ER-sheet localization. Therefore, the role of SigmaR1 in ER morphology is mediated by distinct structural elements: α-helices 4&5 for membrane flattening as reported34 and the N-terminal α-helix 1 for specific targeting identified in this study (Supplementary Fig. 8d, e).
The flat, stacked architecture of ER sheets is ideally suited to accommodate the translocon complex and bound polysomes for efficient protein synthesis1,8,11,12. While the recent study has shown that SigmaR1 overexpression caused a dramatic expansion of the ER sheet without altering the density of ribosomes on ER membrane34, implying that SigmaR1 overexpression somehow increased the number of ribosomes on ER sheet, which in turn may lead to an increase in the amount of the translocon complex. Our proteomic and biochemical studies showed that SigmaR1 directly interacts with the translocon complex, and the enrichment analysis of GO function and KEGG pathway enrichment in proteomics shows that ribosome-related processes were substantially down-regulated in SigmaR1 KO cells, suggesting that SigmaR1 may facilitate the association of ribosomes and the translocon with ER sheet. Our results link the role of SigmaR1 in ER-sheet formation to co-translational protein translocation, the major function of ER sheet. Therefore, our findings not only validate the ER-sheet localization of SigmaR1 but demonstrate the functional significance of SigmaR1 at ER sheets.
Sigma receptors including SigmaR1 have no known homology with other receptor systems and have no definitive endogenous natural ligand54. Currently, the neurosteroids, sphingolipids, N, N-dimethyltryptamine, myristic acid, and choline have been shown to bind to SigmaR1 and are considered as candidates for endogenous ligands48,55,56,57,58,59. In this study, we found that SigmaR1 was able to bind to PC via the β-barrel structure, nominating PC as a potential ligand for SigmaR1. Although lipid-binding proteins are remarkably diverse at the sequence level, the characteristic β-barrel folding architecture remains the most common structural feature60,61. In fact, the interior surface of the β-barrel structure of most lipid transfer proteins forms a cavity or pit that accommodates hydrophobic ligands of various sizes and shapes, including lipids and hydrophobic hormones as well as secondary metabolites, such as vitamins, cofactors, or odorants, thereby achieving various biological functions60,62,63,64,65,66. Consistently, a structural analysis reveals a trimeric structure of SigmaR1 exhibits a broad, hydrophobic surface adjacent to membranes via the C-terminal region, suggesting a close interaction with the luminal side of ER membranes. This domain features a cupin-like β-barrel with a central ligand-binding site with remarkable flexibility in recognizing and binding ligands48,49,50,67. Taken together, our results suggest that SigmaR1 may be a PC-binding protein with the β-barrel structure at the CT as the putative lipid-binding sites. Noteworthy, in view of the fact that the ER is the major site for cellular lipid synthesis68,69,70, it is plausible that the lipid-binding activity of SigmaR1 is functionally coupled to lipid homeostasis and regulates the organization of ER microdomains, which has been proposed previously71,72,73,74. An alternative or complementary mechanism is revealed by our in vitro pull-down assays, which demonstrate that PC binding enhances the association of SigmaR1 with the translocon complex (Fig. 4j), potentially enabling modulation of translocon function. To fully elucidate how the lipid-binding capacity of SigmaR1 regulates both its translocon engagement and subsequent fine-tuning of translocon function, future studies employing integrated structural biology and functional reconstitution approaches will be essential.
The mechanism underlying the accumulation of LDs in SigmaR1-depleted hepatic cells is likely multifactorial. Our rescue experiments implicate both ER sheet localization, as the mutant deficient in ER sheet (SigmaR1∆2-8) fail to fully rescue the phenotype. This result suggests that ER morphology or the translocon-associated function plays a key role in LD dynamics. How is the ER-sheet shaping process linked to LD dynamics? In view of the specific role of ER tubules and associated proteins in LD formation75, we speculate that SigmaR1 deficiency may disturb the ratio of ER sheet to tubule, resulting in more tubules than sheets thereby increasing LD biogenesis, an event that specifically originates from ER tubules. In addition, since the assembly of translocon complex is coupled to ER sheet formation and a wide-ranging effect in protein homeostasis induced by SigmaR1 depletion, the loss of SigmaR1 could also directly or indirectly impair LD formation and turnover and consequently lead to an accumulation of LDs inside cells. Notably, the lipid binding mutant Mut2 that was capable in targeting ER sheet was also insufficient in rescuing the LD defect. Furthermore, silencing other ER sheet factors such as Climp63, p180 and Kinectin did not caused accumulated LDs as pronounced as SigmaR1 silencing. These results suggest that ER sheet/translocon function does not solely account for LD defect in SigmaR1-depleted cells. Given that SigmaR1 has been involved in regulating ER microdomains and the trafficking of neutral lipids71,72, it is also possible that SigmaR1 may regulate LD dynamics via the remodeling of ER microdomain by binding to PC at the luminal face of ER membranes. Therefore, we propose that the role for SigmaR1 in LD dynamics depends on an orchestrated interplay between ligand binding and ER sheet formation.
Interestingly, the regulatory role of SigmaR1 in LD abundance was most pronounced in hepatocytes. This cell-type specificity aligns with the high endogenous expression of SigmaR1 in the liver and the central role of hepatocytes in lipid synthesis, storage, and secretion. We therefore speculate that fine-tuning of ER morphology or ligand-induced microdomain organization by SigmaR1 is critical for the maintenance of lipid homeostasis in liver cells. Finally, given the vital role of the translocon complex in protein homeostasis, the association of SigmaR1 with the translocon may underlie its broad physiological and pathological implications (Model; Supplementary Fig. 8f). Importantly, in light of the hepatocyte-predominant role of SigmaR1 in LD dynamics, our results also position SigmaR1 as a potential therapeutic target for metabolic liver disorders, such as non-alcoholic fatty liver disease.
Methods
Cell cultures, transfection and RNAi
African green monkey kidney fibroblast-like COS7 cell line (ATCC), human embryonic kidney 293 T (ATCC), the derivative of the commonly used HEK293 cell line AD293 and HEK293F (ATCC), the mouse hippocampal neuroblast line Neuro-2a (ATCC),the human pancreatic cancer cell line PaCa2 (ATCC), the human cervical cancer HeLa (ATCC), and the human hepatocellular carcinoma cell lines [HepG2 (ATCC) and Huh-7 (ATCC)] were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (VivaCell) and 1% penicillin/streptomycin at 37 °C with 5% CO2. Mouse hepatocytes were isolated from male C57 mouse by collagenase perfusion and were cultured in DMEM with 10% fetal bovine serum (PAN Seratech) at 37 °C under 5% CO2 as described by previously76. All cells in this study were tested and confirmed to be mycoplasma-free. Transfection of plasmids and RNAi oligos was carried out with Lipofectamine 2000 and RNAi MAX, respectively. For transfection, cells were seeded at 4 × 105 cells per well in a six-well dishes approximately 16 h prior to transfection. Plasmid transfections were performed in OPTI-MEM (Gibco) with 2 μL Lipofectamine 2000 (Invitrogen) per well for 6 h, followed by trypsin (Gibco) digestion and re-plated onto glass-bottom confocal dishes at ~3.5 × 105 cells per well. Cells were imaged in live-cell medium (DMEM with 10% FBS and 20 mM Hepes without penicillin or streptomycin) approximately 16-24 h after transfection (individually or combined for co-transfection). For all transfection experiments in this study, 1000 ng DNA was used per 3.5 cm well. For siRNA transfections, cells were plated on 3.5 cm dishes at 30-40% density, and 2 μL Lipofectamine RNAimax (Invitrogen) and 50 ng siRNA were used per well. At 48 h after transfection, a second round of transfection was performed with 50 ng siRNAs. Cells were harvested and analyzed 24 h after the second transfection for suppression. All siRNAs used in all experiments were synthesized by RiboBio. siRNA targeting sequences for human SigmaR1#1: TGCAGTGGGTGTTCGTGAA, Sigmar1#2: TCACCCTCTTCTATACTCT, SigmaR1#3: GCGAAGAGATAGCGCAGTT. siRNA targeting sequences for mouse SigmaR1#1: CCCTGATTCTGACTATTAT, SigmaR1#2: GCTGGAACTCATATTCAGA, SigmaR1#3: CTCACCATATGCCTTATCT. siRNA targeting sequences for human Climp63: GGTGCAGTCTTTGCAAGCCACATTT. siRNA targeting sequences for human p180: GGCAGCAGTTGAGTGAAAT. siRNA targeting sequences for human Kinectin: GCAGTTCCATTCCCAGATA.
OA induced LD accumulation
To examine the colocalization of LD with different proteins or measure cellular triglyceride (TG) and cholesterol (CHO), cells were treated with 200 μM OA (O1008, Sigma-Aldrich) for 16 h.
Plasmid construction
SigmaR1 (NM_005866.4), TRAPα (NM_003144.5), SEC11A (NM_014300.4), Nicalin (NM_020170.4), CCDC47 (NM_020198.3), TMEM147 (NM_032635.4), TMCO1 (NM_019026.6), RRBP1 (NM_001042576.2), TRAM1 (NM_014294.6), NOMO1 (NM_014287.4) were cloned from HeLa or 293 T cDNA. SigmaR1, CCDC47, TMEM147, TMCO1, RRBP1, TRAM1, NOMO1 were cloned into mEGFP-N1 or Halo-N1 between the HindIII and BamHI. GFP/Halo-SigmaR1, Halo-TRAPα, Halo-SEC11A, Halo-Nicalin were generated by cloning respective ORF into mEGFP-C1 or Halo-C1 between the SacI and BamHI/ The ORFs of SigmaR1, TRAPα, SEC11A, Nicalin were cloned into mEGFP-C1 or Halo-C1 between the SacI and BamHI. GST-SigmaR1∆TM, GST-SigmaR1∆2-30, GST-SigmaR1-β barrel and GST-SigmaR1-β barrel mut1-4 were cloned into PGEX-2T vector between the BamHI and EcoRI. 14xHis-NEDD8-TRAPα, SEC11A, Nicalin and CCDC47 were cloned into 14xHis-NEDD8 vector between the BamHI and HindIII. Reticulon 4-Halo, Reep5-Halo, Halo-Tex2, Halo-Sec61β and ER-eBFP2 were previously described77. All mutant constructs in this study were generated by PCR mediated site directed mutagenesis on the basis of their respective wild-type constructs. All constructs were confirmed by DNA sequencing. A list of plasmids and primers for DNA constructs were provided in Supplementary Table 1 and 2, respectively.
Generation of KO/KI cell line using CRISPR/Cas9
To make sfGFP-SigmaR1-KI HeLa cell line, two gRNAs (5’-CGCCGTGCCAGGCCGCCCGCCGG-3’ and 5’ -AGTGGGCCGTGGGCCGGCGGTGG-3’) were used to target the N-terminus of SigmaR1 gene. The sgRNA was annealed and ligated into the BsmBI-cleaved/cut pLenti-CRISPR-V2 vector (#52961, Addgene). Then the plasmid encoding sgRNA and the donor construct containing super folded GFP (sfGFP) and two homology arms were transfected into HeLa cells with Lipofectamine 2000, and single clones were sorted 48 h after transfection. Four positive clones were verified by imaging and Western blots.
To generate the SigmaR1 KO HeLa cell line, 630 bp bases between exon 1 and exon 3 of the SigmaR1 gene were deleted using two gRNAs (5’-CGTGGGCCGGCGGTGGGCGTGGG-3’ and 5’-TCTCTGGCACCTTCCACCAGTGG-3’). Complementary gRNAs were annealed and subcloned into the pSpCas9 (BB) -2A-GFP (pX-458) vector (#48138, Addgene) between restriction sites of the BbsI endonuclease. After 48 h transfection, single-cell was sorted by fluorescence-based flow cytometry. Two independent clones were verified by PCR validation and Western blot. All single-guide RNAs (sgRNAs) were designed using CCTop (https://cctop.cos.uni-heidelberg.de:8043/index.html).
Antibodies and reagents
Anti-SigmaR1 (B-5) (sc-137075, Santa Cruz), anti-Sec61β (E-6) (sc-393633, Santa Cruz), anti-Tom20 (sc17764, Santa Cruz), anti-NOMO (B-10) (sc-390565, Santa Cruz) and anti- Nicalin (P09771, promab) were used at 1:500 for western blots. Anti-LAMP1 (H4A3) (sc-20011, Santa Cruz), anti-REEP5 (H-10) (sc-393508, Santa Cruz), anti-RRBP1 (A12239, ABclonal), anti-SEC11A (P02842, promab), anti-TRAPα (201011-T32, Sino Biological Inc), anti-CCDC47 (201332-T44, Sino Biological Inc), anti-TMEM147 (K113174P, Solarbio), anti-TRAM1 (K113836P, Solarbio) anti-Perilipin-2 (A6276, Abclonal), anti-BiP (A11366, Abclonal), eIF2α (9722, Cell Signaling Technology) and Phospho-eIF2α (Ser51) (9721, Cell Signaling Technology) were used at 1:1000 for western blots. Anti-Flag (F1804, Sigma-Aldrich) and anti-RPL10 (K009842P, Solarbio) were used at 1:1500 for western blots. Anti-Calnexin (ab22595, Abcam), anti-PMP70 (ab3421, Abcam) and anti-GM130 mAb (M179-3, MBL) were used at 1:2000 for western blots. Anti GFP-Tag pAb (AE011, ABclonal), anti-Halo Tag pAB (G928A, Promega), anti-GAPDH (AB9485, Abcam), anti-tubulin (100109-MM05T, Sino Biological Inc), anti GST-Tag mAb (AE001, Abclonal) and anti-His (66005-1-Ig, Proteintech) were used at 1:5000 for western blots. Anti-Sec61β(15087-1-AP, Proteintech) were used at 1:200 for Immunofluorescence (IF). Donkey anti-Rabbit IgG (H + L) Secondary Antibody, Alexa Fluor 546 (A10040, Invitrogen) was performed for IF. For immunoblotting, HRP-conjugated goat anti-mouse IgG (H + L) (AS003, Abclonal), HRP-conjugated goat anti-rabbit IgG (H + L) (AS014, Abclonal) were used as secondary antibodies. For live cell imaging, Janilia Fluo® 646 HaloTag® Ligand (GA1120, Promega) and BODIPY-558/568 C12 (D3835, ThermoFisher Scientific) were added to added to OPTI-MEM (Invitrogen) medium in concentration of 20 nM, followed by incubation for 20 min. Cells were washed three times with PBS before imaging. For translation inhibition experiments, cells were treated with 200 μM cycloheximide (C112766, Aladdin) and 200 μM puromycin (P8230, Solarbio) in complete media for 20 min. For the ER stress test, cells were treated with EBSS buffer (G4213, Servicebio) for 4 h. Cells were treated with Thapsigargin (TG) (SC0389, Beyotime) for different durations before imaging. For in vitro lipid binding assay, the following lipids were purchased from Avanti Polar Lipids: NBD-PC (810133), NBD-PE (810144), Rh-PE (810150), NBD-PS (810198), NBD-PA (810138), NBD-ceramide (810211) and NBD-Cholesterol (810252).
Live imaging by confocal microscopy
Cells were grown on 35 mm glass-bottom confocal dishes (801001, NEST). Confocal dishes were loaded to a spinning disk confocal microscope (Dragonfly CR-DFLY-201-40, Andor) equipped with multiple excitation lasers (405-nm, 488-nm, 561-nm, 637-nm). Cells were imaged with the 60×/1.4 NA iPlan-Apochromat 60× oil objective using the 405-nm laser for BFP, 488-nm for GFP, 561-nm for mStrawberry, OFP, tagRFP or mCherry and 637-nm for Janilia Fluo® 646 HaloTag® Ligand. All analyses/quantifications were done on raw 16-bit images using Image J.
Immunofluorescence staining
Cells were washed with PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. After three PBS washes, cells were permeabilized with 0.1% Triton X-100 in PBS for 12 min on ice (4 °C). Following another three PBS washes, cells were blocked with 1% BSA for 1 h at room temperature, then incubated with specific primary antibodies overnight at 4 °C. After thorough PBS washing, cells were incubated with Donkey anti-Rabbit IgG (H + L) Secondary Antibody, Alexa Fluor 546 (Invitrogen, A10040) for 1 h at room temperature protected from light. Finally, cells were washed three times with PBS, mounted with antifade reagent, and imaged.
Fluorescence lifetime imaging microscopy (FLIM)
Cos7 cells were transiently co-transfected with plasmids encoding SigmaR1-mClover2 and mRuby2-Nicalin (or Reep5-mRuby2/mRuby2-Sec61β) for 24 h. FLIM assays were performed using a Leica Stellaris STED laser confocal microscope. Data were analyzed using the TauSense FLIM module.
Quantitative RT-PCR
For quantification of SigmaR1 mRNA levels in different cells, total RNA from cells were extracted with Trizol reagent (15596026, Invitrogen). According to the reagent instructions, cDNA (1000 ng) was synthesized using gDNA Clear cDNA Synthesis Kit/ HiScript II 1st Strand cDNA Synthesis Kit (+gDNA wiper) (R212, Vazyme), and qPCR experiments were performed using ChamQ universal SYBR Green SYBR qPCR Master Mix (Q711, Vazyme) via CFX connect™ fluorescent quantitative PCR detection system (BIO-RAD). Primers used for mRNA quantification were:
h-GAPDH: 5’- GACAAGCTTCCCGTTCTCAG - 3’, 5’- GAGTCAACGGATTTGGTCGT - 3’. h-SigmaR1: 5’- GGGCCAAACACATGGATG - 3’, 5’- GCATAGGAGCGAAGAGTATAGAAG - 3’.
h-Climp63: 5’- GCTCACCAAATCCATCAACG - 3’, 5’- TGCTTGTTCCCCTCAGATTC - 3’. h- p180: 5’- TTGAAAGTTCGGACCAGGTG-3’, 5’- CGAGCTGAGATTGAGATTCTAGG - 3’. h- Kinectin: 5’- TGGAATCAGAGCAGAAAAGGG - 3’, 5’-GAACTGAAGCGGAGGTCTG - 3’.
GST pull-down assay
GST or His-tagged proteins were constructed into pGEX-2T vector or 14×His-NEDD8 vector. E. coli Rosetta (DE3) or BL21 (DE3) transformed with GST-SigmaR1, 14×His- TRAPα, 14×His-SEC11A, 14×His-Nicalin, 14×His-CCDC47 plasmids were inoculated into 5 mL LB medium and grown to OD600 of 0.6-0.8 at 37 °C, and then E. coli transformed with different plasmids was induced with the corresponding optimal induction concentration of IPTG for 16 h at 16 °C. Bacteria were harvested by centrifugation and resuspended in lysis buffer (20 mM Tris, pH 7.4, 300 mM NaCl, 0.1% NP-40, 1% Protease Inhibitor Cocktail (C0001, TargetMol), 1 mM DTT)and then lysed by sonication on ice. Lysates were centrifuged at 12,000 × g for 30 min and supernatants were collected. After washing the GST agarose beads (C600031-0025, Sangon) three times with lysis buffer, the supernatant was incubated with the beads overnight at 4 °C. Then GST agarose beads were washed three times with lysis buffer, collected, and resuspended in 50 μL of 2× SDS loading buffer. Samples were analyzed by Western blot.
GST-tagged protein purification
The constructed plasmids GST-SigmaR1, GST-SigmaR1∆2-30, GST-β barrel and GST-β barrel Mut1 (83F-W, 95 L, 96L-W), Mut2 (104 V, 105 L, 106 L, 107F-W), Mut3 (128I, 129I-W) and Mut4 (152 V, 153V-W) were transformed into E. coli Rossetta (DE3) cells and coated onto LB plates overnight. The following day, healthy monoclonal bacteria were picked and inoculated into a small amount of LB culture medium, and then fresh bacterial solution was inoculated into 3 L of fresh LB medium at a ratio of 1:100 and expanded the culture at 37 °C. When the optical density at 600 nm (OD600) reached 0.6–0.8, protein expression was induced with 0.5 mM IPTG for 20 h at 16 °C. The supernatant containing these proteins was incubated overnight with GST agarose beads after sonication and high-speed centrifugation. Finally, the proteins were eluted with washing buffer containing 20 mM GSH and stored at −80 °C. Protein expression and size were confirmed by Coomassie staining and protein concentration was measured by Nanodrop.
In vitro protein-lipid co-migrating assays
The in vitro lipid binding assays were performed according to a previous study with modifications77. Briefly, 19 μL purified GST-SigmaR1, purified GST-SigmaR1∆2-30, purified GST-β and GST-β Mut1 (83F-W, 95 L, 96L-W), Mut2 (104 V, 105 L, 106 L, 107F-W), Mut3 (128I, 129I-W) and Mut4 (152 V, 153V-W) (1 mg/mL) were mixed with 1 μL of methanol and NBD-labeled ceramide, PE, PS, PA, PC, cholesterol (1 mg/ml in methanol) in 20 μL total reaction volumes and incubated at 4 °C for 3 h. Samples mixed with 5 μL of non-SDS 5xloading buffer were loaded onto 10% non-denaturing native PAGE gel for 4 h at a low current (80-100 V), during which low temperature and dark conditions were maintained. Finally, the NBD fluorescence was visualized using Bio-Rad ChemiDoc XRS + 170-8265 and migration of total protein was observed by Coomassie staining.
Liposome pelleting assay
The purified protein was pre-cleared by ultracentrifugation at 20,000 g for 1 h. Various amounts of the pre-cleared protein were incubated with liposomes (250 μM,70% DOPC, 30% PE, 0.75 M sucrose) in a shaking incubator at 30 °C for 1 h. The mixtures were then centrifuged at 16,000 g for 15 min. Subsequently, both the supernatant and the pellet fractions were carefully collected and analyzed by Western blotting.
GFP-Trap assays
GFP-Trap was used to detect protein-protein interactions and GFP-Trap assays were performed according to the manufacturer’s protocol. For exogenous protein-protein interaction assays, 293 T cells transfected with the indicated constructs were collected and resuspended in lysis buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% NP-40, 0.5 mM EDTA) supplemented with 1% Protease Inhibitor Cocktail for 30 min on ice. 10% supernatant after high-speed centrifugation was saved for input sample of GFP-Trap, and the remaining supernatant samples were incubated with GFP-Nanoab-agarose beads (GNA-50-1000, LABLEAD) for 4 h at 4 °C. The beads were washed three times with lysis buffer and collected. For endogenous protein-protein interaction assays, two equal amounts of cells were cultured and collected, one was HeLa cells transfected with mEGFP-C1 empty vector as the control group and the other was sfGFP-SigmaR1 KI#2 HeLa cells in the experimental group. Subsequently, the experimental samples were processed and collected according to the GFP-Trap experimental manufacturer ‘s protocol. Western blot analysis of protein co-IP.
In vitro pull-down assay of his-flag-SigmaR1 and GFP-TRAPα/GFP-nicalin
His-Flag-SigmaR1 was purified from suspension HEK293F cells transiently transfected with a corresponding plasmid. Cells were harvested 72 h post-transfection and lysed via sonication in lysis buffer containing 150 mM NaCl, 25 mM HEPES, 10% glycerol, and 1 mM PMSF. The lysate was supplemented with detergents to final concentrations of 1% (w/v) n-Dodecyl-β-D-maltopyranoside (DDM) and 0.1% (w/v) cholesteryl hemisuccinate tris salt (CHS), followed by membrane solubilization for 2 h. The mixture was then centrifuged at 20,000 × g for 1 h at 4 °C. The supernatant was incubated with anti-Flag Magnetic Beads (P2115, Beyptime) for 3 h. Bound proteins were eluted using 3×FLAG peptide. All purification steps were carried out on ice or at 4 °C to ensure protein stability.
HEK293F cells transiently transfected with GFP-TRAPα or GFP-Nicalin were lysed in a high-salt lysis buffer. GFP-Trap beads were used to capture the respective GFP-fusion proteins from the cell lysates. The beads were subsequently washed ten times with the high-salt lysis buffer to eliminate any potential interacting molecules.
For the pull-down assay, the purified His-Flag-SigmaR1 protein was incubated with immobilized GFP-TRAPα or GFP-Nicalin complexes for 4 h at 4 °C. The beads were washed extensively with HNM buffer (20 mM HEPES pH 7.4, 100 mM NaCl, 5 mM MgCl₂, 1 mM DTT, 0.01% DDM). Finally, the beads were resuspended in 100 μL of 2× SDS sample buffer and boiled at 95 °C for 10 min to elute bound proteins.
Transmission electron microscopy
HepG2 cells treated with either scrambled or SigmaR1 siRNA were fixed with 2.5% glutaraldehyde in 0.1 M Phosphate buffer, pH7.4 for 2 h at room temperature. After washing three times with 0.1 M Phosphate buffer, cells were scraped and collected with 0.1 M phosphate buffer followed by centrifugation at 956 × g. The pellet was resuspended in PBS (0.1 M), and centrifuged at 956 ×g for 10 min. This step was repeated three times. The samples were post-fixed with pre-cold 1% OsO4 in 0.1 M Phosphate buffer for 2–3 h at 4 °C, followed by rinsing with PBS 3 times (3 × 20 min). The samples were dehydrated in graded ethanol (50%, 70%, 85%, 90%, 95%, 2 × 100%) with 15 min for each condition. The penetrations were performed in an order of acetone-epoxy (2:1); acetone-epoxy (1:1); epoxy. Each round of penetration was performed at 37 °C for 12 h. The samples were embedded in epoxy resin using standard protocols78. Sections parallel to the cellular monolayer were obtained using a Leica EM UC7 with the thickness of 60-100 nm and examined under Tecnai G2 20 TWIN (FEI) with accelerating voltage 200 kv. LDs and nucleus were identified based on their respective morphology. The number and size (diameter) of LDs were measured using the “measure” tool in ImageJ (NIH).
Mass spectrometry for identification of SigmaR1-GFP interacting proteins
Identification of Sigmar1-GFP interacting proteins by MS was described in our previous study77. For processing of samples before loading, bound proteins were first extracted from GFP-Trap agarose beads using SDT lysis buffer (4% SDS, 100 mM DTT, 100 mM Tris-HCl, pH 8.0), and then samples were boiled for 3 min and further sonicated. Undissolved beads were removed by centrifugation at 16,000 ×g for 15 min. Collect the supernatant containing protein. Subsequently, the FASP method was used for protein digestion: detergent, DTT, and IAA in UA buffer were added to blocked reduced cysteines. The proteins were digested with 2 µg trypsin (Promega) for overnight at 37 °C. Peptides were collected by centrifugation at 16,000 × g for 15 min. Finally, peptides were desalted with C18 StageTip for further LC-MS analysis. LC-MS/MS experiments were performed on a Q Exactive Plus mass spectrometer coupled to an Easy nLC (Thermo Fisher Scientific). Firstly, three independent sample were loaded onto the capture column (100 µm × 20 mm, 5 µm, C18, Dr. Maisch GmbH) in buffer A (0.1% formic acid in water). Reversed-phase high-performance liquid chromatography (RP-HPLC) separation was performed using a self-packed column (75 µm × 150 mm; 3 µm ReproSil Pur C18 beads, 120 Å; Dr. Maisch GmbH, Ammerbuch) at a flow rate of 300 nl/min. Specifically, the RP-HPLC mobile phase (A was 0.1% formic acid in water and B was 0.1% formic acid in 95% acetonitrile) eluted the sample from 2-4% buffer B gradient to 90% buffer B within 60 min. MS data were acquired using a data-dependent top20 method dynamically choosing the most abundant precursor ions from the survey scan (350–1800 m/z) for HCD fragmentation. A locked mass of 445.120025 Da was used as the internal standard for mass calibration. Full MS scans were acquired at resolutions of 70,000 at m/z 200 and 17,500 at m/z 200. The maximum injection time was set to 50 ms for MS and MS/MS, respectively. Normalized collision energy was 27 and the isolation window was set to 1.6 Th. Dynamic exclusion duration was 60 s. MS data were analyzed using MaxQuant software version 1.6.1.0. MS data were searched against the UniProtKB human database (36,080 entries in total, downloaded on 25 June 2019). Trypsin was chosen as the digestion enzyme. A maximum of two missed cleavage sites and a mass tolerance of 4.5 ppm for precursor ions and 20 ppm for fragment ions were defined for database search. Carbamidomethylation of cysteine was defined as a fixed modification, whereas acetylation of the protein N-terminus and the oxidation of methionine were set as variable modifications for database search. Database search results were filtered and exported at <1% false discovery rate (FDR) at the peptide spectrum matching level and protein level, respectively.
Non-targeted lipidomics using LC-MS/MS
The identification of sfGFP-SigmaR1-associated lipids in the KI cells was described in our previous study77. In particular, to extract lipids, 1 mL methyl tert-butyl ether (MTBE) was added to GFP-Trap agarose beads (Chromoteck) and the samples were shaken for 1 h at room temperature. Next, phase separation was induced by adding 250 μl water, letting it sit for 10 min at room temperature, and centrifuging for 15 min at 14,000 × g, 4 °C. Due to the low density and high hydrophobicity of MTBE, lipids and lipophilic metabolites were mainly extracted to the upper MTBE-rich phase. The lipid was transferred to fresh tubes and dried with nitrogen.
In addition, to ensure data quality for metabolic profiling, quality control (QC) samples were prepared by pooling aliquots from representative samples for all of the analysis samples, and were used for data normalization. QC samples were prepared and analyzed with the same procedure as that for the experiment samples in each batch. Dried extracts were then dissolved in 50% acetonitrile. Each sample was filtered with a disposable 0.22 µm cellulose acetate and transferred into 2 mL HPLC vials and stored at −80 °C until analysis. For UHPLC-MS/MS analysis, lipid analysis was performed on Q Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific) coupled to a UHPLC system Ultimate 3000 (Thermo Fisher Scientific). Samples were separated using a Hypersil GOLD C18 column (100 × 2.1 mm, 1.9 μm) (Thermo Fisher Scientific). Mobile phase A was prepared by dissolving 0.77 g of ammonium acetate to 400 mL of HPLC-grade water, followed by adding 600 ml of HPLC-grade acetonitrile. Mobile phase B was prepared by mixing 100 ml of acetonitrile with 900 ml isopropanol. The flow rate was set as 0.3 ml/min. The gradient was 30% B for 0.5 min and was linearly increased to 100% in 10.5 min, and then maintained at 100% in 2 min, and then reduced to 30% in 0.1 min, with 4.5 min re-equilibration period employed. Both electrospray ionization (ESI) positive-mode and negative-mode were applied for MS data acquisition. The positive mode of spray voltage was 3.0 kV and the negative mode 2.5 kV. The ESI source conditions were set as follows: heater temperature of 300 °C, Sheath Gas Flow rate, 45arb, Aux Gas Flow Rate, 15 arb, Sweep Gas Flow Rate, 1 arb, Capillary Temp, 350 °C, and S-Lens RF Level, 50%. The full MS scans were acquired at a resolution of 70,000 at m/z 200, and 17,500 at m/z 200 for MS/MS scans. The maximum injection time was set to for 50 ms for MS and 50 ms for MS/MS. MS data was acquired using a data-dependent Top10 method dynamically choosing the most abundant precursor ions from the survey scan (200–1500 m/z) for HCD fragmentation. Stepped normalized collision energy was set as 15, 25, 35 and the isolation window was set to 1.6 Th. QC samples were prepared by pooling aliquots that were representative of all samples under analysis, and used for data normalization. Blank samples (75% acetonitrile in water) and QC samples were injected every six samples during acquisition.
For data preprocessing and filtering, lipids were identified and quantified using LipidSearch 4.1.30 (Thermo, CA). Mass tolerance of 5ppm and 10ppm were applied for precursor and product ions. Retention time shift of 0.25 min was performed in ‘alignment’. M-score and chromatographic areas were used to reduce false positives. The lipids with less than 30% relative standard deviation (RSD) of MS peak area in the QC samples were kept for further data analysis. SIMCAP software (Version 14.0, Umetrics, Sweden) was used for all multivariate data analyses and modeling. Data were mean-centered using Pareto scaling. Models were built on principal component analysis (PCA), orthogonal partial least-square discriminant analysis (PLS-DA) and partial least-square discriminant analysis (OPLS-DA). All the models evaluated were tested for overfitting with methods of permutation tests. The descriptive performance of the models was determined by R2X (cumulative) [perfect model: R2X (cum)=1] and R2Y (cumulative) [perfect model: R2Y (cum)=1] values while their prediction performance was measured by Q2 (cumulative) [perfect model: Q2 (cum)=1] and a permutation test (n = 200). The permuted model should not be able to predict classes -R2 and Q2 values at the Y-axis intercept must be lower than those of Q2 and the R2 of the non-permuted model. OPLS-DA allowed the determination of discriminating metabolites using the variable importance on projection (VIP). The VIP score value indicates the contribution of a variable to the discrimination between all the classes of samples. Mathematically, these scores are calculated for each variable as a weighted sum of squares of PLS weights. The mean VIP value is 1, and usually VIP values over 1 are considered as significant. A high score is in agreement with a strong discriminatory ability and thus constitutes a criterion for the selection of biomarkers. The discriminating metabolites were obtained using a statistically significant threshold of variable influence on projection (VIP) values obtained from the OPLS-DA model and two-tailed Student’s t-test (P-value) on the normalized raw data at a univariate analysis level. The P-value was calculated by one-way analysis of variance (ANOVA) for multiple groups analysis. Metabolites with VIP values greater than 1.0 and P-value less than 0.05 were considered to be statistically significant metabolites. Fold change was calculated as the logarithm of the average mass response (area) ratio between two arbitrary classes. On the other side, the identified differential metabolites were used to perform cluster analyses with R package.
Preparation of rough ER fraction from rat liver and sfGFP-Sigmar1-KI cell
Based on the protocol used in the preparation of crude microsomes from rat livers79, 5 female Balb/c rats (age, 6 weeks; weight, 80–100 g) were euthanized and the livers were collected quickly and washed with cold homogeneous medium (HM) (0.5 M sucrose, 1 mM dithiothreitol (DTT)). Firstly, Liver tissue was ground with a tissue grinder until a paste-like consistency form. The homogenate volume was diluted to 5 times the liver weight (i.e., 20% [w/v]) with HM and centrifuged at 1400 × g for 10 min mainly to remove nuclei and cellular debris, and the supernatant was centrifuged at 13,800 × g for 10 min at 4 °C to remove mitochondria. After transferring the supernatant enriched in cytosol and ER into a 50 ml tube on ice, the pellet was resuspended with a 5 × volume of HM, ground 5 times, and then centrifuged at 13,800 × g for 10 min at 4 °C. The above step was repeated for once. Subsequently, the supernatants (post-mitochondrial supernatant, PMS) were combined and kept on ice. Next, four sucrose gradients solutions were prepared using PMS and 2.5 M sucrose: PMS containing 1 M sucrose, PMS containing 1.35 M sucrose, PMS containing 1.55 M sucrose, and PMS containing 1.8 M sucrose. Finally, different sucrose gradients solutions (Layer the solutions from the bottom to the top starting with 0.67 mL of 1.8 M sucrose-PMS, 0.4 mL of 1.55 M sucrose-PMS, 1.5–2.5 mL of 1.35 M sucrose-PMS, and finally 0.4 mL of 1 M sucrose-PMS) were transferred in ultracentrifuge tubes (Ultra-Clear Beckman, USA) with a Ti60 rotor and centrifuged for at least 6 h at maximum 240,000 × g at 4 °C. A total of 11 samples within and between each layer were collected for Western blotting.
For the preparation of crude rough microsomes from culture cells80, we added cycloheximide to the culture medium to a final concentration of 10 µM, and incubated the culture at 37˚C for 10 min, and washed the cells three times with cold PBS containing a concentration of 10 µM cycloheximide. Finally, the collected cells were added 0.7 mL of HM (10 mM HEPES-KOH buffer (pH 7.5), 10 mM KCl, 1 mM MgCl2, 1 mM DTT, 0.5 mM PMSF) containing 3 units/mL RNase inhibitor and ground with the Dounce homogenizer until the cell homogenate was in a milky white state. After cell homogenization, the cell homogenate was centrifuged at 1400 × g for 10 min mainly to remove nuclei and cellular debris, and the supernatant was centrifuged at 13,800 ×g for 10 min at 4 °C to remove mitochondria. After transferring the supernatant enriched in cytosol and ER into a tube, 2.2 times the volume of 2.5 M sucrose was added to achieve a sucrose concentration of 1.8 M in the sucrose PMS mixture. Finally, different sucrose gradient solutions (layered from bottom to top, starting from 1.67 mL of 1.8 M sucrose-PMS mixture, 0.5 mL of HM containing 1.5 M sucrose, 0.5 mL of HM containing 1.3 M sucrose, 0.5 mL of HM containing 1 M sucrose, 0.5 mL of HM containing sucrose, and finally 0.33 mL of HM containing 0.25 M sucrose) were transferred to ultracentrifuge tubes (Ultra Clean Beckman, USA) with Ti60 rotors and centrifuged at 263,000 × g for at least 6 h at 4 °C. A total of 14 samples within and between each layer were collected for Western blotting.
Fluorescence-activated cell sorting (FACS) analysis
SigmaR1 KO#3 HeLa cells were transfected with either SigmaR1-GFP, SigmaR1-GFPΔ2-8, or SigmaR1-GFP lipid-binding mutant plasmids and cultured for 36 h. Cells (40 10 cm dishes with ~100% confluency for each condition) were collected, washed with PBS, and resuspended as single-cell suspensions. The suspensions were then filtered through a cell strainer to remove clumps and debris. GFP-positive cells were isolated using a BD FACSAria II cell sorter equipped with a FITC fluorescence channel, and collected for subsequent experiments.
Total triglyceride and cholesterol/cholesterol esters measurements
Cells were treated with 200 µM OA for 16 h, and cellular triglyceride and cholesterol levels were measured using total triglyceride (TG) assay kit (A110-1-1, NanJing JianCheng Bioengineering Institute) and total cholesterol assay kit (A111-1-1, NanJing JianCheng Bioengineering Institute), respectively. Briefly, cells were collected by centrifugation at 1000 × g for 10 min at 4 °C, then cells were resuspended in 500 μL of cold diluted PBS buffer and homogenized on ice by ultrasonic. For triglyceride and cholesterol assay, 10 μL standard or cell suspension was added to 96 plate and the reaction was initiated by adding 200 μL diluted Enzyme Mixture solution to each well. The plate was incubated for 30 min at 37 °C, the absorbance at 500 nm was measured.
Image analysis
All image analysis and processing were performed using ImageJ (2.1.0/1.53c; National Institutes of Health). The colocalization analysis of Pearson’s correlation coefficient was performed using a plugin named JACoP in ImageJ.
Proteomics analysis
For functional analysis of mass spectrometry data, the corresponding IDs of the screened proteins (any two groups of unique peptides enriched in GFP-C1 control group =0, and any two groups of unique peptides enriched in Sigmar1-GFP experimental group≠0) were converted to gene IDs by the DAVID knowledgebase tool81,82, and then enrichment analysis and mapping of GO function and KEGG pathway database of the converted genes were performed using the OmicShare tools (www.omicshare.com/tools).
Prediction of the conservation and the secondary structure of SigmaR1
In order to analyze the secondary structure and sequence conservation of SigmaR1, the sequence similarities and secondary structure of SigmaR1 were performed ESPript 3.x83.
Molecular docking
To analyze the binding affinities and modes of interaction between SigmaR1(PDB:6DJZ) and PC (CID: 10425706), CB-Dock2, the silico protein-ligand docking blind softwares were employed84,85.
Statistical analysis
All statistical analyses and p-value determinations were performed in GraphPad Prism6. All the error bars represent Mean ± SD. To determine p-values, ordinary one-way ANOVA with Tukey’s multiple comparisons test was performed among multiple groups and a two-tailed unpaired student t-test was performed between two groups.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the iProX partner repository with the dataset identifier PXD058640. All the data and relevant materials, including reagents and primers, that supports the findings of this study are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
West, M., Zurek, N., Hoenger, A. & Voeltz, G. K. A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J. Cell Biol. 193, 333–346 (2011).
Shibata, Y., Hu, J. J., Kozlov, M. M. & Rapoport, T. A. Mechanisms shaping the membranes of cellular organelles. Annu. Rev. Cell Dev. Bi 25, 329–354 (2009).
Hu, J. J. et al. Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science 319, 1247–1250 (2008).
Voeltz, G. K., Rolls, M. M. & Rapoport, T. A. Structural organization of the endoplasmic reticulum. Embo Rep. 3, 944–950 (2002).
Shibata, Y., Voeltz, G. K. & Rapoport, T. A. Rough sheets and smooth tubules. Cell 126, 435–439 (2006).
Terasaki, M. et al. Stacked endoplasmic reticulum sheets are connected by helicoidal membrane motifs. Cell 154, 285–296 (2013).
Voeltz, G. K., Prinz, W. A., Shibata, Y., Rist, J. M. & Rapoport, T. A. A class of membrane proteins shaping the tubular endoplasmic reticulum (vol 124, pg 573, 2006). Cell 130, 754–754 (2007).
Shibata, Y. et al. Mechanisms determining the morphology of the peripheral ER. Cell 143, 774–788 (2010).
Gao, G., Zhu, C. J., Lie, E. M. & Nabi, I. R. Reticulon and CLIMP-63 regulate nanodomain organization of peripheral ER tubules. Plos Biol. 17, e3000355 (2019).
Xu, L., Xiang, Y. & Hu, J. J. Molecular basis of Climp63-mediated ER lumen spacing. J. Cell Sci. 136, jcs260976 (2023).
Rolls, M. M., Hall, D. H., Victor, M., Stelzer, E. H. & Rapoport, T. A. Targeting of rough endoplasmic reticulum membrane proteins and ribosomes in invertebrate neurons. Mol. Biol. Cell 13, 1778–1791 (2002).
Puhka, M., Vihinen, H., Joensuu, M. & Jokitalo, E. Endoplasmic reticulum remains continuous and undergoes sheet-to-tubule transformation during cell division in mammalian cells. J. Cell Biol. 179, 895–909 (2007).
Westrate, L. M., Lee, J. E., Prinz, W. A. & Voeltz, G. K. Form follows function: the importance of endoplasmic reticulum shape. Annu Rev. Biochem. 84, 791–811 (2015).
Egea, P. F., Stroud, R. M. & Walter, P. Targeting proteins to membranes: structure of the signal recognition particle. Curr. Opin. Struct. Biol. 15, 213–220 (2005).
Evans, E. A., Gilmore, R. & Blobel, G. Purification of microsomal signal peptidase as a complex. Proc. Natl. Acad. Sci. USA 83, 581–585 (1986).
Gemmer, M. & Förster, F. A clearer picture of the ER translocon complex. J. Cell Sci. 133, jcs231340 (2020).
Nyathi, Y., Wilkinson, B. M. & Pool, M. R. Co-translational targeting and translocation of proteins to the endoplasmic reticulum. Biochim. Biophys. Acta 1833, 2392–2402 (2013).
Voorhees, R. M. & Hegde, R. S. Toward a structural understanding of co-translational protein translocation. Curr. Opin. Cell Biol. 41, 91–99 (2016).
Braunger, K. et al. Structural basis for coupling protein transport and N-glycosylation at the mammalian endoplasmic reticulum. Science 360, 215–219 (2018).
Pfeffer, S. et al. Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat. Commun. 5, 3072 (2014).
Chavan, M., Yan, A. & Lennarz, W. J. Subunits of the translocon interact with components of the oligosaccharyl transferase complex. J. Biol. Chem. 280, 22917–22924 (2005).
Fons, R. D., Bogert, B. A. & Hegde, R. S. Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J. Cell Biol. 160, 529–539 (2003).
Görlich, D. & Rapoport, T. A. Protein translocation into proteoliposomes reconstituted from purified components of the endoplasmic reticulum membrane. Cell 75, 615–630 (1993).
Voigt, S., Jungnickel, B., Hartmann, E. & Rapoport, T. A. Signal sequence-dependent function of the TRAM protein during early phases of protein transport across the endoplasmic reticulum membrane. J. Cell Biol. 134, 25–35 (1996).
Conti, B. J., Devaraneni, P. K., Yang, Z., David, L. L. & Skach, W. R. Cotranslational stabilization of Sec62/63 within the ER Sec61 translocon is controlled by distinct substrate-driven translocation events. Mol. Cell 58, 269–283 (2015).
Kalies, K. U., Rapoport, T. A. & Hartmann, E. The beta subunit of the Sec61 complex facilitates cotranslational protein transport and interacts with the signal peptidase during translocation. J. Cell Biol. 141, 887–894 (1998).
Haffner, C., Dettmer, U., Weiler, T. & Haass, C. The Nicastrin-like protein Nicalin regulates assembly and stability of the Nicalin-nodal modulator (NOMO) membrane protein complex. J. Biol. Chem. 282, 10632–10638 (2007).
Dettmer, U. et al. Transmembrane protein 147 (TMEM147) is a novel component of the Nicalin-NOMO protein complex. J. Biol. Chem. 285, 26174–26181 (2010).
Anghel, S. A., McGilvray, P. T., Hegde, R. S. & Keenan, R. J. Identification of Oxa1 homologs operating in the eukaryotic endoplasmic reticulum. Cell Rep. 21, 3708–3716 (2017).
McGilvray, P. T. et al. An ER translocon for multi-pass membrane protein biogenesis. Elife 9, e56889 (2020).
Mori, T., Hayashi, T., Hayashi, E. & Su, T. P. Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS One 8, e76941 (2013).
Rosen, D. A. et al. Modulation of the sigma-1 receptor-IRE1 pathway is beneficial in preclinical models of inflammation and sepsis. Sci. Transl. Med. 11, eaau5266 (2019).
Hayashi, T. & Su, T. P. Sigma-1 receptor chaperones at the ER-Mitochondrion interface regulate Ca signaling and cell survival. Cell 131, 596–610 (2007).
Sawyer, E. M. et al. SigmaR1 shapes rough endoplasmic reticulum membrane sheets. Dev. Cell (2024).
Shibata, Y. et al. The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. J. Biol. Chem. 283, 18892–18904 (2008).
Du, Y. J., Chang, W. P., Gao, L., Deng, L. & Ji, W. K. Tex2 is required for lysosomal functions at TMEM55-dependent ER membrane contact sites. J. Cell Biol. 222, e202205133 (2023).
Mei, J. & Pasternak, G. W. Molecular cloning and pharmacological characterization of the rat sigma1 receptor. Biochem. Pharm. 62, 349–355 (2001).
Pan, L. et al. Isolation and characterization of alternatively spliced variants of the mouse sigma1 receptor gene, Sigmar1. PLoS One 12, e0174694 (2017).
Franko, A. et al. Identification of the secreted proteins originated from primary human hepatocytes and HepG2 cells. Nutrients 11, 1795 (2019).
Kim, K. E. et al. Dynamic tracking and identification of tissue-specific secretory proteins in the circulation of live mice. Nat. Commun. 12, 5204 (2021).
Aviner, R. The science of puromycin: From studies of ribosome function to applications in biotechnology. Comput Struct. Biotec 18, 1074–1083 (2020).
Obrig, T. G., Culp, W. J., McKeehan, W. L. & Hardesty, B. The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. J. Biol. Chem. 246, 174–181 (1971).
Schneider-Poetsch, T. et al. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol. 6, 209–217 (2010).
Jang, W. et al. Endosomal lipid signaling reshapes the endoplasmic reticulum to control mitochondrial function. Science 378, eabq5209 (2022).
Sharma, N. et al. The Sigma-1 receptor is an ER-localized type II membrane protein. J. Biol. Chem. 297, 101299 (2021).
Schmidt, H. R. et al. Crystal structure of the human sigma1 receptor. Nature 532, 527–530 (2016).
Liu, Y. et al. Visualizing looping of two endogenous genomic loci using synthetic zinc-finger proteins with anti-FLAG and anti-HA frankenbodies in living cells. Genes Cells 26, 905–926 (2021).
Fu, C. T., Xiao, Y., Zhou, X. M. & Sun, Z. Y. Insight into binding of endogenous neurosteroid ligands to the sigma-1 receptor. Nat. Commun. 15, 5619 (2024).
Schmidt, H. R., Betz, R. M., Dror, R. O. & Kruse, A. C. Structural basis for σ1 receptor ligand recognition. Nat. Struct. Mol. Biol. 25, 981–987 (2018).
Schmidt, H. R. et al. Crystal structure of the human σ1 receptor. Nature 532, 527 (2016).
Zhemkov, V. et al. The role of sigma 1 receptor in organization of endoplasmic reticulum signaling microdomains. Elife 10, e65192 (2021).
Saheki, Y. et al. Control of plasma membrane lipid homeostasis by the extended synaptotagmins. Nat. Cell Biol. 18, 504 (2016).
Becuwe, M. et al. FIT2 is an acyl-coenzyme A diphosphatase crucial for endoplasmic reticulum homeostasis. J. Cell Biol. 219, e202006111 (2020).
Schmidt, H. R. & Kruse, A. C. The Molecular Function of σ Receptors: Past, Present, and Future. Trends Pharm. Sci. 40, 636–654 (2019).
Su, T. P., London, E. D. & Jaffe, J. H. Steroid binding at sigma receptors suggests a link between endocrine, nervous, and immune systems. Science 240, 219–221 (1988).
Ramachandran, S. et al. The sigma1 receptor interacts with N-alkyl amines and endogenous sphingolipids. Eur. J. Pharm. 609, 19–26 (2009).
Fontanilla, D. et al. The hallucinogen,-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science 323, 934–937 (2009).
Tsai, S. Y. A., Pokrass, M. J., Klauer, N. R., Nohara, H. & Su, T. P. Sigma-1 receptor regulates Tau phosphorylation and axon extension by shaping p35 turnover via myristic acid. Proc. Natl. Acad. Sci. USA 112, 6742–6747 (2015).
Brailoiu, E. et al. Choline is an intracellular messenger linking extracellular stimuli to IP-evoked Ca signals through Sigma-1 receptors. Cell Rep. 26, 330 (2019).
Curto, L. M., Angelani, C. R. & Delfino, J. M. Intervening in the β-barrel structure of lipid binding proteins: consequences on folding, ligand-binding and aggregation propensity. Prostaglandins Leukot. Ess. Fat. Acids 93, 37–43 (2015).
Schiefner, A. & Skerra, A. The menagerie of human lipocalins: a natural protein scaffold for molecular recognition of physiological compounds. Acc. Chem. Res. 48, 976–985 (2015).
Flower, D. R. Multiple molecular recognition properties of the lipocalin protein family. J. Mol. Recognit. 8, 185–195 (1995).
Flower, D. R., North, A. C. & Sansom, C. E. The lipocalin protein family: structural and sequence overview. Biochim. Biophys. Acta 1482, 9–24 (2000).
Reese, A. J. & Banaszak, L. J. Specificity determinants for lipids bound to beta-barrel proteins. J. Lipid Res. 45, 232–243 (2004).
He, Y. et al. Solution-state molecular structure of apo and oleate-liganded liver fatty acid-binding protein. Biochemistry 46, 12543–12556 (2007).
Kielkopf, C. S., Whitten, A. E., Garner, B. & Brown, S. H. J. Small angle X-ray scattering analysis of ligand-bound forms of tetrameric apolipoprotein-D. Biosci. Rep. 41, BSR20201423 (2021).
Meng, F. H., Xiao, Y., Ji, Y. J., Sun, Z. Y. & Zhou, X. M. An open-like conformation of the sigma-1 receptor reveals its ligand entry pathway. Nat. Commun. 13, 1267 (2022).
Vance, J. E. Phospholipid synthesis and transport in mammalian cells. Traffic 16, 1–18 (2015).
Balla, T., Sengupta, N. & Kim, Y. J. Lipid synthesis and transport are coupled to regulate membrane lipid dynamics in the endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1865, 158461 (2020).
Nishimura, T. & Stefan, C. J. Specialized ER membrane domains for lipid metabolism and transport. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1865, 158492 (2020).
Hayashi, T. & Su, T. P. σ-1 receptors (σ binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum:: Roles in endoplasmic reticulum lipid compartmentalization and export. J. Pharmacol. Exp. Therapeutics 306, 718–725 (2003).
Hayashi, T. & Su, T. P. Sigma-1 receptors at galactosylceramide-enriched lipid microdomains regulate oligodendrocyte differentiation. Proc. Natl. Acad. Sci. USA 101, 14949–14954 (2004).
Hayashi, T. & Su, T. P. The potential role of sigma-1 receptors in lipid transport and lipid raft reconstitution in the brain: implication for drug abuse. Life Sci. 77, 1612–1624 (2005).
Takebayashi, M., Hayashi, T. & Su, T. P. σ-1 receptors potentiate epidermal growth factor signaling towards neuritogenesis in PC12 cells:: Potential relation to lipid raft reconstitution. Synapse 53, 90–103 (2004).
Chen, F. et al. FIT2 organizes lipid droplet biogenesis with ER tubule-forming proteins and septins. J. Cell Biol. 220, e201907183 (2021).
Cabral, F. et al. Purification of hepatocytes and sinusoidal endothelial cells from mouse liver perfusion. Jove-J. Vis. Exp. (2018).
Gao, Y., Xiong, J., Chu, Q. Z. & Ji, W. K. PDZD8-mediated lipid transfer at contacts between the ER and late endosomes/lysosomes is required for neurite outgrowth. J. Cell Sci. 135, jcs255026 (2022).
Kumar, S., Ciraolo, G., Hinge, A. & Filippi, M. D. An efficient and reproducible process for transmission electron microscopy (TEM) of rare cell populations. J. Immunol. Methods 404, 87–90 (2014).
Sabatini, D. D. Preparation of rough microsomes from rat liver. Cold Spring Harb. Protoc. 2014, 845–851 (2014).
Sabatini, D. D. Preparation of crude rough microsomes from tissue culture cells. Cold Spring Harb. Protoc. 2014, 980–987 (2014).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Sherman, B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221 (2022).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Morris, G. M., Huey, R. & Olson, A. J. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinforma. Chapter 8, 14 (2008). Unit 8.
Wang, Y. et al. PubChem BioAssay: 2017 update. Nucleic Acids Res. 45, D955–d963 (2017).
Acknowledgements
We thank the School of Basic Medicine Innovation Research Center, Huazhong University of Science and Technology, for providing technical support in imaging, FACS and Ultracentrifugation. We thank the Mass Spectrometry Core Facility of Shenzhen Bay Laboratory for assistance in proteomic experiments. W. Ji was supported by National Natural Science Foundation of China (32371343; 32570822; 32541022).
Author information
Authors and Affiliations
Contributions
X. Hu and W. Ji conceived the project and designed the experiments. X. Hu, T. Zhou T. Cui, Y. Du, C. Song performed the experiments. X. Hu, Y. Du, W. Chang, L Deng, J. Qin and J. Xiong analyzed and interpreted the data. W. Ji prepared the manuscript with inputs and approval from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, X., Zhou, T., Cui, T. et al. SigmaR1 is an auxiliary translocon factor with lipid-binding activity that regulates protein and lipid droplet homeostasis. Nat Commun 17, 1415 (2026). https://doi.org/10.1038/s41467-025-68157-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68157-7
