
Abstract
Conventional chromatin profiling techniques are often limited by antibody availability and performance. Here, we introduce Af-CUT&Tag, a target antibody-free method that overcomes these limitations by using CRISPR-integrated peptide tags (HiBiT/ALFA-tag) recognized by engineered binders (LgBiT/NbALFA) fused to a Tn5 transposase. Af-CUT&Tag eliminates dependence on traditional target antibodies, achieving robust specificity and sensitivity with as few as 500 cells. It provides high-quality chromatin profiles, with improved signal-to-noise ratios and library quality compared with conventional antibody-based counterparts, while also enabling single-cell resolution (scAf-CUT&Tag). Applying Af-CUT&Tag to Hippo effectors (YAP1/TAZ) during liver regeneration reveals dynamic chromatin remodeling, including YAP1/TAZ-mediated control of lipid metabolism (e.g., Lpin1, Fasn) and heme clearance (Hpx, Trf). We further identify miR-122 as a critical regulator of these processes, impacting liver regeneration. The versatility of Af-CUT&Tag in cell lines, bulk tissues, and single nuclei establishes it as a powerful tool for studying gene regulation in development, disease, and regeneration.
Similar content being viewed by others
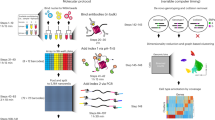


Introduction
Chromatin landscape analysis has revolutionized our understanding of gene regulation by enabling precise mapping of transcription factor binding sites, DNA-binding proteins, and nucleosome features1. This advanced resolution allows systematic profiling of histone modifications, transcription factor positioning, and chromatin structure, facilitating deeper insights into epigenetic regulation2. Among these, transcription factors play a central role in regulating gene expression by binding to specific DNA sequences, recruiting co-regulators, and forming transcriptional complexes3,4. Understanding these regulatory mechanisms is essential for elucidating the underlying processes that govern cellular function, differentiation, and responses to environmental stimuli5.
Current methods for transcription factor (TF) profiling such as CUT&Tag6 have emerged as powerful tools in transcriptional regulation and epigenetics, offering superior efficiency, reduced starting material, and high sensitivity compared to techniques like ChIP-seq1, ChEC-seq7, and CUT&RUN8,9. By utilizing a transposase enzyme to insert sequencing adapters into chromatin fragments, CUT&Tag yields high-resolution fragment libraries with low background noise, making it ideal for chromatin profiling10.
Recent advancements have focused on enhancing precision and accuracy through the fusion of nanobodies and Tn5 transposase, such as Nano-CUT&Tag11 and NTT-seq12. These developments have significantly improved the versatility of CUT&Tag, enabling the simultaneous analysis of multiple epigenetic modifications within single cells13,14,15. Nanobodies, or single-domain antibodies (VHH), are derived from the variable regions of camelid heavy-chain antibodies and are smaller and more stable than conventional antibodies16. Their integration enhances the targeting specificity for particular chromatin regions, which is critical for gaining deeper insights into gene regulation and cellular differentiation.
However, despite the advantages of CUT&Tag or Nano-CUT&Tag, their reliance on conventional target antibodies for chromatin targeting poses significant challenges. Target antibody-based methods are limited by the quality of antibodies, their high molecular weight, and low cell penetration, which can lead to potential targeting errors17,18. Additionally, protein post-translational modifications—such as phosphorylation19,20, methylation, and acetylation—can disrupt antibody binding21, further complicating the accurate profiling of epigenetic features. These limitations highlight the need for alternative approaches that bypass dependence on target antibodies, providing more robust and flexible solutions for epigenetic studies.
In this study, we develop a target antibody-free chromatin profiling method: Af-CUT&Tag. This approach leverages high-affinity small peptide tags, such as HiBiT22,23,24 and ALFA-tag25,26, and their corresponding binders or nanobodies, coupled with the Tn5 transposase. These peptide-based systems offer enhanced stability27, sub-nanomolar-level affinity28, and resistance to interference from protein modifications. By tagging proteins of interest (POIs) with small peptides and using CRISPR for knock-in of these tags29, Af-CUT&Tag allows precise and efficient chromatin profiling across different cell types, tissues, and proteins, as exemplified by uncovering YAP/TAZ-mediated Hippo pathway dynamics during liver regeneration. Importantly, this method is unaffected by protein modifications and provides a universal platform for chromatin landscape analysis, paving the way for more comprehensive and accurate studies of gene regulation and chromatin dynamics.
Results
Development and validation of the HiBiT/LgBiT-Tn5-based Af-CUT&Tag system
To create a more direct and efficient method for studying protein-DNA interactions, we developed a target antibody-free CUT&Tag system using genetically encoded tags. Our approach relies on fusing the Tn5 transposase to high-affinity protein binders. We selected two short peptide tags, HiBiT23 and ALFA-tag25, which exhibit nanomolar-level affinities and high resistance to disruption due to their tight binding configuration (Fig. 1a, b).

a, b. Schematic diagram of the protein-protein interactions between HiBiT (yellow) binding with LgBiT (dark blue) and ALFA-tag (light blue) binding with NbALFA (green). c. The binding kinetics and affinity between the recombinant protein ExoⅢ-HiBiT and LgBiT (left) or LgBiT-Tn5 (right) were determined using Bio-Layer Interferometry (BLI). The ExoⅢ-HiBiT protein was immobilized on the sensor surface, and the sensors were dipped into wells containing different concentrations of LgBiT (1-25 nM) or LgBiT-Tn5 (1-20 nM). d. The binding kinetics and affinity between the recombinant protein GFP-ALFA-tag and NbALFA (left) or NbALFA-Tn5 (right) were determined using Bio-Layer Interferometry (BLI). The GFP-ALFA-tag protein was immobilized on the sensor surface, and the sensors were dipped into wells containing different concentrations of NbALFA (1-8 nM) or NbALFA-Tn5 (1-8 nM). e. Schematic representation of the assembly of the fusion protein LgBiT/NbALFA-Tn5 with the transposon adapter. f. Construction of Nano-Luciferase reporter gene knock in system based on CRISPR/Cas9 (Left). 100,000 SW480 RPB1-HiBiT and SW480 CTCF-HiBiT gene knock-in cells were separately lysed using Nano-Glo® Lytic Buffer (Promega). Excess substrate furimazine and 1 μL (26 μM) of LgBiT-Tn5 were added for chemiluminescent detection (Right). The detection parameters were set as follows: LUM-EndPoint, Single label, 700 nm Blocker, Measurement direction: top, Measurement time: 1,000 ms, Default Z focus: 8.5 mm. Each sample was measured in three technical replicates, and data are presented as mean ± SD (unpaired t-test, ****p < 0.0001). g. The assembled transposome LgBiT-Tn5 (0.5 μM) and NbALFA-Tn5 (0.5 μM) were incubated with 200 ng gDNA at 55 °C for 10 min. The cleavage of gDNA was analyzed using agarose gel electrophoresis. The gel image shown is representative of results obtained from multiple independent experiments. h. Comparison of strategies between antibody-dependent CUT&Tag, nano-CUT&Tag and Af-CUT&Tag. Adding LgBiT-Tn5 can directly target HiBiT/ALFA-tag at the C-terminus of the target transcription factor and bind with high affinity. After multiple rounds of washing to remove excess transposons, add Mg²⁺ to activate transposons, insert adapters into transcription factor binding sites, and construct DNA sequencing libraries. In the CUT&Tag strategy, primary antibody against the target protein, secondary antibody, and pA/G-Tn5 are required, with washing steps after each incubation. In nano-CUT&Tag, primary antibody and nanobody-Tn5 are added, followed by washing. The library construction steps are consistent across all strategies.
In vitro binding assays confirmed that both systems exhibited nanomolar-level affinities, with the dissociation constants (KD) for the HiBiT/LgBiT-Tn5 and ALFA-tag/NbALFA-Tn5 interactions measured at 0.58 nM and 0.202 nM, respectively (Fig. 1c, d). The affinity of the HiBiT/LgBiT-Tn5 system was comparable to that of the native interaction, whereas the affinity of NbALFA-Tn5 was reduced by approximately sevenfold compared with NbALFA alone, although it remained very high (Supplementary Fig. 1a). Despite the higher affinity of the NbALFA-Tn5 system, its poor solubility resulted in reduced protein purity (Supplementary Fig. 1c). In contrast, fusion with LgBiT significantly improved the solubility, yield, and overall quality of the LgBiT-Tn5 protein, making it more robust and efficient to purify (Supplementary Fig. 1b). Given these advantages, we selected the HiBiT/LgBiT-Tn5 system as the focus of our study.
To validate this target antibody-free protein-DNA interaction system, we co-transfected SW480 or DLD1 colorectal cancer cells with RNP complex-targeting transcription factor plasmids and donor template plasmids. Gene-edited cells with HiBiT and ALFA-tag knock-ins were efficiently generated in 96-well plates using dual fluorescence labeling and flow cytometry sorting. HiBiT and ALFA-tags were inserted into the C-terminus of RNA polymerase II’s largest subunit, RPB1, which regulates transcription of protein-coding and non-coding genes. A similar approach was applied to the transcription factor CTCF (Supplementary Fig. 1d).
Subsequently, LgBiT-Tn5 was assembled with the transposon adapter, and its activity was validated in HiBiT knock-in cell lines by detecting chemiluminescence from furimazine, the NanoLuc substrate (Fig. 1e, f and Supplementary Fig. 1e). The catalytic stability of LgBiT-Tn5 at varying concentrations in different buffers showed minimal deviation from the Nano-Glo® Lytic Buffer standard (Supplementary Fig. 1f). Additionally, LgBiT-Tn5 and NbALFA-Tn5 tagged with 3xFlag proteins were purified and used with Flag nanobody magnetic beads to enrich HiBiT and ALFA-tagged endogenous target protein from cell lysates, confirming stable interactions between the fusion proteins and their tagged targets (Supplementary Fig. 2a).
The DNA cleavage activity of LgBiT-Tn5 and NbALFA-Tn5 was confirmed, with Tn5-tagmented DNA fragments predominantly ranging from 200 to 300 base pairs (Fig. 1g and Supplementary Fig. 2b). Both fusion proteins exhibited equivalent transposase activity with control Tn5. To assess system versatility, Tn5 was fused with high-affinity binders, including G4PID30 and SG4 nanobody31 targeting DNA G-quadruplexes, avidin for biotin, and affibodies for EGFR and HER232. All constructs demonstrated robust DNA cleavage activity in vitro (Supplementary Fig. 2c). These results indicate that binder-fused Tn5 proteins can specifically bind targets and insert sequencing adapters near TF-binding sites, offering a specific, and streamlined protein-DNA interaction system suitable for high-throughput sequencing (Fig. 1h).
Af-CUT&Tag Provides High-Resolution Mapping of RNA Polymerase Ⅱ
After incubation with Concanavalin A-coated paramagnetic beads to immobilize the cells, the cells were permeabilized, and the fusion protein LgBiT-Tn5 or NbALFA-Tn5, pre-assembled with adapters, was introduced. Owing to its lower molecular weight (70 kDa) compared to antibodies (150 kDa), the fusion protein exhibited increased permeability through both cellular and nuclear membranes. Thus, nuclear extraction for subsequent membrane penetration was unnecessary. LgBiT-Tn5 or NbALFA-Tn5 specifically binds to the HiBiT or ALFA-tag at the protein of interest’s C-terminus, forming a stable protein complex. After multiple washes to remove unbound fusion protein, Mg2+ was added to activate the Tn5 transposase, generating high-quality libraries for investigating TF-DNA interactions (Fig. 1h).
To evaluate system specificity, library sequencing was performed using LgBiT-Tn5 in SW480 RPB1-HiBiT cells and SW480 WT cells lacking HiBiT integration. Two parallel replicates were included in the experimental group. To minimize nonspecific genomic interactions of LgBiT, purified full-length Nanoluciferase was used as a blocking agent, as it does not bind the HiBiT subunit. This effectively reduced LgBiT’s nonspecific binding. The results showed no prominent peaks in SW480 WT cells following LgBiT-Tn5 cleavage, confirming the specificity of the system (Fig. 2a).

a. Representative chromatin landscapes across a 419 kb segment of the human genome generated by Af-CUT&Tag, CUT&Tag, Nano-CUT&Tag, and ATAC-seq for RNA polymerase II. SW480 WT (with equal LgBiT-Tn5 input, processed in parallel), SW480 WT (CUT&Tag), and IgG control are included for comparison. For RNA PolⅡ, CUT&Tag, Nano-CUT&Tag, and ATAC-seq datasets were downsampled to the same total mapped read counts as Af-CUT&Tag for direct comparison. The RefSeq gene annotations are displayed at the bottom. b. The enrichment heatmap of RNA PolⅡ around 3 kb upstream and downstream of the TSS generated using Af-CUT&Tag, CUT&Tag, and Nano-CUT&Tag. For all three methods, reads were mapped to the genome and downsampled to 8 million for comparison. c. Overall TSS enrichment profile of RNA PolⅡ signal in Af-CUT&Tag, CUT&Tag, and Nano-CUT&Tag in SW480 RPB1-HiBiT cells. d. Comparison of the proportion of reads mapping to the promoter region for RNA polymerase II signals in Af-CUT&Tag, CUT&Tag, and Nano-CUT&Tag in SW480 RPB1-HiBiT cells. The numerical values represent the proportion of reads in the promoter region, and statistical significance between groups was assessed using a unpaired t-test. e. Library complexity analysis for Af-CUT&Tag, CUT&Tag, and Nano-CUT&Tag, plotted using three ENCODE standard metrics: NRF, PBC1, and PBC2. The NRF values are labeled in the figure, with the size of the points representing PBC1 and the color representing PBC2. f. Hierarchically clustered correlation matrix of Af-CUT&Tag replicates and with CUT&Tag and Nano-CUT&Tag profiling for the RNA PolⅡ. The same antibody was used in all experiments. Spearman correlations were calculated using values of read counts split into 10 kb bins across the genome.
To evaluate the signal-to-noise ratio of Af-CUT&Tag relative to other methods, we conducted experiments using CUT&Tag and nano-CUT&Tag with the same RPB1 antibody in 100,000 SW480 RPB1-HiBiT cells. Each method included two replicates and was benchmarked against ATAC-seq data generated with in-house LgBiT-Tn5. CUT&Tag and nano-CUT&Tag systems required antibody incubation, whereas Af-CUT&Tag did not (Fig. 1h). Downsampling of ~8 million reads aligned to the human genome revealed similar chromatin landscape patterns among the methods, with Af-CUT&Tag achieving high-quality data in promoter regions within the 100,000-cell dataset (Fig. 2a). Due to the fact that the RNAPII-S2P antibody labels elongating RNA polymerase II, CUT&Tag and nano-CUT&Tag datasets exhibited stronger signals across gene bodies compared to Af-CUT&Tag (Supplementary Fig. 3a). We also included WT cells, which had not undergone gene editing, as a positive control for RNA Pol II profiling and performed a quantitative comparison of genome-wide enrichment, further confirming that HiBiT knock-in does not appear to markedly affect the binding of the target protein to DNA (Fig. 2a and Supplementary Fig. 3b). Remarkably, this approach enabled profiling from as few as ~500 cells, yielding signal levels comparable to ~50,000-cell datasets, with minimal background noise (Supplementary Fig. 3c, d). Crucially, our Af-CUT&Tag approach profiles total RPB1, whereas the antibody-based CUT&Tag and nano-CUT&Tag targeted the elongating form (RNAPII-S2P). This distinction accounts for the observed enrichment of Af-CUT&Tag signals at promoter regions compared to the gene body enrichment seen with the antibody-based methods.
For a quantitative comparison, we downsampled all samples to 8 million mapped reads and used deepTools to generate heatmaps of transcription start site (TSS) enrichment across all genes. The results revealed that Af-CUT&Tag produced moderately higher signals than CUT&Tag and nano-CUT&Tag across most TSS sites, despite exhibiting a narrower dynamic range (~4/5 of CUT&Tag’s). This may be attributed to the closer spatial proximity of the transposase to the target protein, and to the fact that Af-CUT&Tag captures both phosphorylated and unphosphorylated forms of RPB1 (Fig. 2b).
To further assess method performance, the TSS profile plots were calculated and plotted for RPB1 Af-CUT&Tag, CUT&Tag and nano-CUT&Tag datasets (Fig. 2c). Af-CUT&Tag demonstrated significantly higher signal accumulation at these sites, highlighting its efficiency in distinguishing POI regulatory states using fewer reads. Library quality analysis indicated enhanced performance for Af-CUT&Tag in terms of promoter region coverage and complexity (Fig. 2d, e). Replicates for Af-CUT&Tag RPB1 profiling showed high reproducibility (Fig. 2f). Comparison of RNAPⅡ read counts against accessible sites (identified through ATAC-seq) further revealed that Af-CUT&Tag exhibited fewer nonspecific accessible sites, distinguishing RNAPⅡ TSS-binding sites more effectively (Supplementary Fig. 3e).
To further validate the system, homemade NbALFA-Tn5 was tested in DLD1 cells with endogenous ALFA-tagged RPB1 knock-in alleles. Comparisons were made between two parallel replicates and DLD1 WT cells lacking ALFA-tag in heatmaps, and profile plots to Nano-CUT&Tag (Supplementary Fig. 3f–h). These findings highlight the versatility and reliability of Af-CUT&Tag.
Af-CUT&Tag enables profiling of CTCF binding across genome in bulk and single-cell levels
To assess the applicability of Af-CUT&Tag for profiling abundant transcription factor binding sites, we profiled the CCCTC-binding factor (CTCF) DNA-binding protein. Equal concentrations of LgBiT-Tn5 and NbALFA-Tn5 were introduced into SW480 cells with HiBiT or ALFA-tag knocked-in alleles, respectively. Following identical experimental protocols, CTCF antibody was used concurrently as a control. Each sample contained 50,000 cells, and the read depth was uniformly maintained across all datasets. We selected a subset of approximately 8 million mapped reads for downstream analysis, revealing distinct patterns of chromatin domains regulated by CTCF. The chromatin landscapes generated by these three methods exhibited high similarity (Fig. 3a). A genome-wide enrichment comparison using CTCF antibody targeting WT control and HiBiT knock-in cells further indicated that the introduction of tagged proteins did not appear to significantly affect CTCF binding to DNA (Fig. 3a and Supplementary Fig. 4a). The heatmaps and profile plots for the CTCF dataset were generated for Af-CUT&Tag (HiBiT and ALFA-tag) and Nano-CUT&Tag datasets (Fig. 3b and Supplementary Fig. 4b), all of which exhibited significant enrichment near the transcriptional start site.

a. Representative chromatin landscapes across a 203 kb segment of the human genome generated by the indicated method. For CTCF, we downsampled Nano-CUT&Tag (anti-CTCF and anti-HiBiT) datasets to the same total mapped read counts as Af-CUT&Tag (HiBiT and ALFA-tag) for direct comparison. SW480 WT (CUT&Tag), IgG control, and SW480 CTCF-HiBiT (ATAC-seq) are included for reference. b. Enrichment heatmap of CTCF around 3 kb upstream and downstream of the TSS generated using Af-CUT&Tag (HiBiT and ALFA-tag) and Nano-CUT&Tag profiling. For all methods, reads were mapped to the genome and downsampled to 8 million for comparison. c. The distribution of read counts for all CTCF peaks in Af-CUT&Tag (LgBiT-Tn5 and NbALFA-Tn5) and Nano-CUT&Tag (anti-CTCF), as well as the distribution of read counts for ATAC-seq peaks, was analyzed. d. The schematic workflow of single-cell Af-CUT&Tag (scAf-CUT&Tag). Permeabilized HiBiT knock-in cells are distributed into 96 wells and incubated with LgBiT-Tn5 complexes carrying unique 5’ and 3’ barcodes. After the removal of unbound complexes, the cells are sorted into a 96-well plate. Library construction and amplification are performed in each well using primers with distinct index barcodes. The final products are purified and sequenced. e. Genome browser image of CTCF peaks on chromosome 1 (chr1) from bulk Af-CUT&Tag (blue) and aggregated scAf-CUT&Tag (red). The mapped reads from all 128 individual cells are plotted below the aggregate peaks. Each row represents a single cell. f. Profile plot of CTCF enrichment at the TSS region of genes from the hg19 genome for all single cells. The red line indicates average enrichment for 128 single cells from the scAf-CUT&Tag data set. The blue lines represent bulk Af-CUT&Tag enrichment. g. Fraction of reads in single SW480 CTCF-HiBiT cells falling within the designated CTCF binding peaks using stringent criteria. Narrow peaks were called using MACS2 on bulk profiling data, and reads from scAf-CUT&Tag were assigned to those peaks. The purple dashed line represents the fraction of bulk Af-CUT&Tag reads that fall within the designated CTCF binding peak. Violin plots show the distribution of data, with the middle line representing the median, the box bounds representing the 25th and 75th percentiles, and the upper and lower edges of the violin indicating the maximum and minimum values.
To confirm that Af-CUT&Tag captures specific protein-DNA interactions rather than general chromatin accessibility, we first performed motif analysis on CTCF peaks. As expected, the canonical CTCF binding motif was the most significantly enriched motif (Supplementary Fig. 4c). We then compared our data to ATAC-seq, which further supported the system specificity of Af-CUT&Tag in effectively distinguishing CTCF binding sites (Fig. 3c). Replicates for CTCF profiling using Af-CUT&Tag (HiBiT and ALFA-tag) exhibited high similarity, reaffirming the method’s reproducibility (Supplementary Fig. 4d).
To evaluate the effectiveness of epigenetic regulation validation targeting HiBiT protein, multiple indicators were compared, including peak occurrence in promoters, peak occurrence in introns, PCR Bottlenecking Coefficient 1 (PBC1), Non-Redundant Fraction (NRF), and duplication ratio, using CTCF and HiBiT antibodies for CTCF CUT&Tag and Af-CUT&Tag (HiBiT) libraries (Supplementary Fig. 4e). As expected, sequencing library quality derived from HiBiT was enhanced, irrespective of antibody-free or antibody-dependent methods. Additionally, TSS profile plots were calculated and generated by HiBiT based analysis (Supplementary Fig. 4f). Consistent with expectations, the use of either LgBiT-Tn5 or HiBiT-targeting antibody based classical CUT&Tag produced similar genomic landscapes. The overarching patterns closely resembled those obtained via CTCF antibody-based approaches, all of which displayed notably low background noise (Supplementary Fig. 4g).
Next, we evaluated the application of the HiBiT in Af-CUT&Tag single-cell assays. Since Af-CUT&Tag does not require antibody binding and the entire reaction, from the addition of fusion transposase to adapter integration, occurs within digitonin-treated, semi-permeabilized cells, fragmented DNA remains within each cell nucleus. Consequently, we developed a straightforward strategy to generate chromatin profiles from individual cells, which we have termed single-cell Af-CUT&Tag (scAf-CUT&Tag) (Fig. 3d). We employed a barcoding multiplexing strategy analogous to previously published methods33,34, incorporating barcode sequences from ACT-seq35. SW480 CTCF-HiBiT cells were divided into 96 wells by volume, with a density of 10,000 cells per well. Each well was treated with a separate LgBiT-Tn5 complex, carrying unique 5’ and 3’ sequence barcode combinations. Following the removal of any unbound complexes, the cells were pooled and redistributed into a second 96-well plate using FACS, with 200 cells per well (Fig. 3d). The transposition reaction was initiated by adding tagmentation buffer containing Mg2+ and terminated by the addition of EDTA and protease. Library construction and amplification were performed in each well using a second set of unique index barcodes. After sequencing, the samples were demultiplexed to obtain single-cell data.
The aggregate of single-cell CTCF enrichment peaks aligns closely with the profiles generated in bulk samples (Fig. 3e), exhibiting a high correlation (Pearson’s r = 0.88) (Supplementary Fig. 4h). Cells were ranked by their genome-wide read counts, and unique fragments for each cell are displayed in individual tracks. The majority of reads from single cells fall within the CTCF binding peaks defined by bulk analysis, indicating a high recovery rate for single-cell chromatin profiling and effective enrichment of CTCF binding motifs (Supplementary Fig. 4i). Additionally, the average enrichment pattern of annotated gene TSS regions demonstrates high reproducibility in single cells (Fig. 3f). Fraction of reads in single SW480 CTCF-HiBiT cells falling within the designated CTCF binding peaks reaches an optimal value (Median = 0.28) (Fig. 3g). Thus, Af-CUT&Tag can be used to profile POIs in single cells and can be further extended to multiplex chromatin profiling through the introduction of diverse tags.
Af-CUT&Tag efficiently profiles chromatin landscapes bound by the POI in vitro and in vivo through combination with CRISPR-based tag knockin
Injury, disease, and aging often result in tissue damage, which triggers intrinsic cellular repair mechanisms to facilitate regenerative processes and restore organ function36. The Hippo pathway is foundational to the orchestration of organ growth and regeneration, as extensively established in prior research37. Within this pathway, the downstream effectors YAP1 and TAZ play critical roles in coordinating reparative functions across diverse tissue types38. However, many aspects of how these effectors modulate downstream gene expression to drive tissue repair remain poorly understood, particularly in the context of regeneration and repair.
The HiBiT-tagged protein exhibits diverse functionalities39,40, serving not only as a nanoluciferase reporter gene system but also as a tool in the Af-CUT&Tag system to investigate transcriptional regulatory mechanisms of YAP1 and TAZ. We validated this approach using YAP1- or TAZ-HiBiT knock-in cell models. This system allowed real-time monitoring of changes in YAP1 or TAZ expression levels via chemiluminescence detection, with signal stability persisting for over 40 min without notable attenuation (Supplementary Fig. 5a). Additionally, it effectively captured changes in YAP1 and TAZ expression levels following treatment with MG-132 (a proteasome inhibitor) and CHX (cycloheximide, an inhibitor of eukaryotic protein synthesis), aligning with results from Western blot experiments (Supplementary Fig. 5b, c).
The system also proved effective for characterizing the regulatory states of YAP1 and TAZ. We generated heatmaps to examine genomic loci enriched by these proteins and compared the TSS profiles between the Af-CUT&Tag and CUT&Tag datasets (Supplementary Fig. 5d, e). Genome browser visualizations further revealed enriched signals of YAP1 and TAZ at the CCN1 gene promoter region in a subset of samples (Supplementary Fig. 5f). These result was confirmed in DLD1 TAZ-ALFA-tag knock-in cells (Supplementary Fig. 5g).
To investigate liver regeneration mechanisms, we constructed recombinant rAAV2/8 vectors encoding TBG-Cre, U6 promoter-sgRNA, and Yap1/Taz-HiBiT knock-in templates. By administering AAV injection, Cas9 expression was induced in Cas9 knock-in mouse liver cells, enabling precise HiBiT tag insertion at the C-terminus of Yap1 or Taz (Fig. 4a). Subsequently, partial hepatectomy (PHx) was performed, revealing significantly higher HiBiT protein expression in knock-in mice compared to controls, while YAP1/TAZ antibody staining showed no overall protein level changes (Fig. 4b). Liver tissues were collected 24 h post-surgery for further analysis, and HiBiT protein expression was confirmed by immunohistochemical staining (Supplementary Fig. 6a).

a. Schematic diagram of Rosa26-CAG-LSL-Cas9-tdTomato mice injected with rAAV2/8-type virus carrying Yap1/Taz-HiBiT knock-in template for liver cell-specific gene knock-in. b. Western blot detected the expression levels of HiBiT and YAP1 in Yap1-HiBiT liver tissue protein (top), and detected the expression levels of HiBiT and TAZ in Taz1-HiBiT liver tissue protein (bottom). The blots shown are representative of results obtained from multiple independent experiments. c. Comparison of the Fraction of Reads in Peaks (FRiP) for Yap1/Taz signals in Af-CUT&Tag and Nano-CUT&Tag profiles in liver tissue cells. The bar plot represents the mean FRiP value for each condition, with pre- and post-hepatectomy (PHx-pre and PHx-post) samples indicated by distinct colored points. Statistical significance was assessed using a unpaired t-test to compare the mean values between the two groups. d. Peak in promoter, reads in promoter, PCR Bottlenecking Coefficient 1 (PBC1), Non-Redundant Fraction (NRF), and duplication ratio comparison of Yap1/Taz signal in Af-CUT&Tag and Nano-CUT&Tag in liver tissue cells. e. Comparison of unique reads per cell detected by the scAf-CUT&Tag method targeting Yap1 between PHx-pre and PHx-post samples without downsampling the data. The mean number of unique reads per cell is 1153 for PHx-pre and 5844 for PHx-post. Data are shown on a log10 scale. Violin plots illustrate the distribution of unique reads per cell, with the central line representing the median, the box edges corresponding to the 25th and 75th percentiles, and the upper and lower edges of the violin indicating the maximum and minimum values. f. Uniform manifold approximation and projection (UMAP) plots of Yap1 signals for PHx-pre (left) and PHx-post (right) (PHx-pre: n = 12,775; PHx-post: n = 12,812). g. Chromatin landscapes showing comparing bulk Af-CUT&Tag maps with scAf-CUT&Tag maps (PHx-pre Yap1 cluster3 and PHx-post Yap1 cluster 2), both in aggregate over all single cells and individual cells, at regions of enrichment of Yap1. Cells were ordered by read coverage within the regions depicted.
Resected and residual liver tissues were categorized as pre- and post-regeneration samples and subjected to nuclear extraction. Following DAPI staining, 100,000 nuclei per sample were isolated. Af-CUT&Tag (LgBiT-Tn5) and Nano-CUT&Tag (antibody-based) were employed to elucidate Yap1/Taz-mediated regulatory mechanisms in early liver repair and regeneration. Six parallel experiments were performed to ensure reproducibility and reliability.
Graphical analyses of average read counts in the Af-CUT&Tag and Nano-CUT&Tag datasets revealed significantly enhanced signal accumulation at TSS regions in Af-CUT&Tag profiles. Heatmap analyses corroborated these trends (Supplementary Fig. 6b). Replicates demonstrated substantial reproducibility in Yap1 and Taz profiling using Af-CUT&Tag (Supplementary Fig. 6c). Comparative analyses of FRiP, promoter peaks, reads in promoters, and library quality metrics (e.g., PBC1, NRF, duplication ratio) further underscored the superior performance of Af-CUT&Tag in complex hepatocellular tissue (Fig. 4c, d). Motif enrichment analysis of pre-regeneration liver samples from Yap1-HiBiT and Taz-HiBiT knock-in mice revealed significant enrichment of canonical TEAD4 and AP-1 binding motifs across all profiling strategies, including LgBiT-Tn5, anti-HiBiT, and anti-Yap1/Taz-based Af-CUT&Tag and Nano-CUT&Tag approaches (Supplementary Fig. 6d).
scAf-CUT&Tag enables single-cell applications in liver regenerative tissues
We further applied Af-CUT&Tag to the single-cell analysis of regenerating liver tissues. To enhance HiBiT-tagged protein expression, we constructed a recombinant AAV vector encoding TBG-Yap1-HiBiT, packaged the virus, and administered it to mice for liver-specific overexpression of Yap1-HiBiT. Liver tissues were collected pre- and 24 h post-partial hepatectomy, and nuclei were extracted and fixed. Using barcoding multiplexing strategy, sequencing data were demultiplexed to obtain single-cell datasets. A total of 17,416 barcodes were identified in the PHx-pre samples, while 17,855 barcodes were identified in the PHx-post samples. Similar to other NGS-based approaches41,42,43, scAf-CUT&Tag generated unique reads for each nucleus. To ensure data quality, libraries with excessively high or low unique read distributions were excluded from the analysis. After filtering, the mean number of unique reads per nucleus was 1153 for PHx-pre and 5844 for PHx-post (Fig. 4e). Subsequently, clustering analysis was performed on 12,775 nuclei from the PHx-pre sample and 12,812 nuclei from the PHx-post sample, yielding eight distinct single-cell datasets for each condition (Fig. 4f).
To ensure that single-cell isolation and library preparation retained the high specificity observed in bulk Af-CUT&Tag, single-cell datasets from pre- and post- PHx were correlated with bulk samples (Supplementary Fig. 7a, b). Single-cell datasets with the highest correlation to bulk samples were selected, and nuclei from pre- and post-PHx were ranked by genome-wide read counts, with unique fragments from each nucleus displayed on individual tracks. The aggregated single-cell profiles closely resembled Yap1 bulk Af-CUT&Tag profiles across various genomic loci, exhibiting similarly high signal-to-noise ratios (Fig. 4g). As a proof of concept, although AAV-mediated expression of Yap1-HiBiT in liver tissues was heterogeneous, the exceptional sensitivity of Af-CUT&Tag rendered it suitable for single-cell transcriptional regulatory analysis in tissues.
Investigating YAP1/TAZ-mediated early liver regeneration mechanisms via Af-CUT&Tag
To further investigate the genes potentially regulated by YAP1/TAZ-associated chromatin binding events after liver resection, we identified the differential binding peaks (|log2FC | > 1, p-value < 0.05) from bulk data using the edgeR44 package. These peaks represent genomic regions with altered YAP1/TAZ occupancy, and were annotated to the corresponding genes using the ChIPseeker45 package. The volcano plot illustrates the distribution of differential binding peaks across the different groups, with peaks associated with genes involved in fatty acid metabolism highlighted (Fig. 5a, b). Gene Ontology (GO) enrichment analysis on the genes corresponding to the differential binding peaks at 24 h post-regeneration revealed that upregulated genes were linked to regeneration, immune response, inflammation, hemostasis, and iron ion transport, while downregulated genes were associated with fatty acid metabolism, acyl-CoA processes, and organic acid biosynthesis (Fig. 5c). These pathways were not significantly enriched in the Nano-CUT&Tag antibody-treated group (Supplementary Fig. 8a). Heatmaps of peak enrichment values further highlighted the superior sensitivity of Af-CUT&Tag (Fig. 5d).

a. Schematic overview of Af-CUT&Tag for investigating the YAP1/TAZ regulatory dynamics during liver regeneration. Recombinant AAV2/8 vectors encoding Yap1-HiBiT or Taz-HiBiT knock-in templates were packaged into AAV particles and delivered via intravenous injection to achieve liver-specific expression of HiBiT-tagged proteins. Mice were subjected to PHx to induce liver regeneration. Nuclei were isolated from liver tissues before (PHx-pre) and after (PHx-post) surgery, followed by preparation of single-cell suspensions. Af-CUT&Tag was then performed using LgBiT-Tn5, which directly targets the HiBiT-tagged YAP1 or TAZ proteins, allowing for the mapping of chromatin-bound regions and regulatory elements involved in the regenerative response. Created in BioRender. hualei, Z. (https://BioRender.com/xvrbur2). b. To generate a multi-group differential peak beehive plot, we calculated the average coverage across all peak intervals using bigWig files and employed the edgeR package to identify peaks with differential binding of YAP1 and TAZ before and after PHx. In the plot, points represent differential peaks (coverage ≥ 10, p-value ≤ 0.01, FDR ≤ 0.05, |log2FC | > 1), with upregulated peaks colored in red and downregulated peaks in blue. Peaks corresponding to genes associated with fatty acid metabolism are annotated in the figure. c. Annotate the up- and down-regulated peaks in the Af-CUT&Tag (LgBiT-Tn5) dataset to genes, and use the annotated genes to perform GO enrichment analysis. The top 10 enriched GO pathways for both upregulated and downregulated gene sets were identified based on their adjusted p-values (FDR ≤ 0.05, right for upregulated, left for downregulated). d. Use bamCoverage of deepTools to calculate the coverage ratio (RPGC) within the gene interval. Heatmap showing coverage of 40 selected genes from the enrichment analysis results, with upregulated genes focusing on pathways related to liver regeneration, acute inflammatory response, hemostasis, iron ion homeostasis and iron ion transport pathways. The down-regulated genes primarily clustered in fatty acid metabolic processes, very long-chain fatty acid metabolism, and acyl-CoA metabolic pathways. e. Evaluate the enrichment levels of transcription factors Yap1 and Taz at the Lpin1, Acox1, Fasn and Scd1 loci, indicating the changes pre and post partial hepatectomy. Positive values indicate an increase, while negative values indicate a decrease. f. Spatial visualization of Lpin1, Acox1, Fasn and Scd1 gene expression levels at various time points (0, 8, 24, 48, 72, 168 h) following PHx, with data sourced from https://db.cngb.org/stomics/lista57. g. Representative Oil Red O staining images from the same mouse at 24 h before and after PHx. The images shown are representative of results obtained from multiple independent experiments. Magnified views (200×) of the regions highlighted at 100× by red squares are shown in the right column. Scale bar, 100 μm. h. Using liver RNA-seq data from the GSE180012 dataset, which includes samples from PHx and sham surgery groups at various time points, we analyzed the differential gene expression of Plin2 and Cidec between the PHx and sham surgery groups across these time points (unpaired t-test, *p < 0.05). Each group comprised three independent biological replicates. Box plots illustrate the distribution of the data, with the central line representing the median, the box edges corresponding to the 25th and 75th percentiles, and the whiskers extending to the minimum and maximum values within 1.5 times the interquartile range from the lower and upper quartiles, respectively. Values beyond this range are considered outliers and plotted individually. i. Spatial visualization of Plin2 and Cidec gene expression levels at various time points (0, 8, 24, 48, 72, 168 h) following PHx.
After partial hepatectomy, Yap1/Taz promoted the expression of Hemopexin (Hpx) and Serotransferrin (Trf) to clear free heme released during erythrocyte rupture, providing antioxidant and immunomodulatory effects crucial for initiating liver regeneration (Supplementary Fig. 9a). Spatial transcriptomic analysis revealed that Hpx and Trf were significantly upregulated at 24 h and returned to baseline by 72 h (Supplementary Fig. 9b).
Lipid metabolic homeostasis is vital for liver regeneration46, with acute lipid accumulation playing a critical early role. At 24 h post-hepatectomy, Yap1/Taz enrichment in the promoters of fatty acid metabolic enzymes and synthases (e.g., Acox1, Lpin1, Fasn, Scd1) was markedly reduced, especially in the Af-CUT&Tag system (Fig. 5e). Notably, Lpin1, essential for lipid metabolism, causes metabolic dysfunction and impairs regeneration when deficient47,48. Reduced chromatin accessibility and decreased RNA PolⅡ enrichment in these genes evidenced chromatin remodeling (Supplementary Fig. 8b). Except for transient Acox1 upregulation, these genes were significantly downregulated 24 h post-regeneration, exhibiting spatiotemporal expression differences (Fig. 5f).
Additionally, genes associated with fatty acid metabolism, including Cyp4a32, Acsl1, and Thrsp, showed decreased peak enrichment values (Fig. 5d). Analysis of the GSE180012 dataset confirmed that genes regulated by Yap1/Taz, such as Lpin1, Scd1, Thrsp, and Hpx, exhibited similar expression trends 24 h post-hepatectomy (Supplementary Fig. 9c). Differential gene expression analysis during liver regeneration demonstrated that the expression levels of several other fatty acid synthases were significantly downregulated within the first 24 h, recovering only thereafter (Supplementary Fig. 9e).
Subsequently, we assessed lipid accumulation 24 h after partial hepatectomy, revealing a marked increase (Fig. 5g). This raised the question of the mechanisms underlying the origin of these accumulated lipids. Transcriptomic analysis at 24 h post-hepatectomy showed that most genes involved in fatty acid metabolism or synthesis were significantly downregulated during this period (Supplementary Fig. 9d). In contrast, the additional accumulation of hepatic lipids can also arise from two sources: lipid droplet enlargement via fusion and enhanced fatty acid uptake. Examination of genes associated with lipid droplet fusion and storage, such as Plin2 and Cidec, revealed early upregulation prior to 24 h, followed by a return to baseline expression levels (Fig. 5h, i). Similarly, fatty acid transport genes like Cd36 and Fabp449 were upregulated at 24 h, contributing to rapid lipid droplet accumulation (Supplementary Fig. 9f, g).
Notably, although Plin2 and Cidec are regulated by Yap1/Taz, the enrichment of Yap1/Taz at the promoters of these two genes had largely returned to pre-hepatectomy levels by the 24-h mark, with a slight decrease even observed (Supplementary Fig. 9h). Fatty acid transport genes, primarily regulated by the PPAR signaling pathway50, also reached peak upregulation within the first 24 h, followed by a gradual decline in expression levels thereafter (Supplementary Fig. 9f, g).
These findings highlight the dynamic nature of lipid accumulation during liver regeneration. In the early stages, Yap1/Taz-driven lipid droplet fusion and PPAR-enhanced fatty acid transport promote acute lipid accumulation. Subsequently, Yap1/Taz mediates the inhibition of lipid metabolism and synthesis, reducing inflammation and oxidative stress from metabolic byproducts and providing energy and a favorable environment for regeneration.
miR-122 identified by Af-CUT&Tag plays a crucial role in early liver regeneration
An intriguing aspect pertains to the notable increase in the enrichment of Yap1/Taz on Mir122 (Fig. 6a), which is notably abundant in the liver, comprising an impressive 70% of all microRNAs, and exhibits multifaceted robust functionalities by regulating the expression of a series of downstream genes51. We also observed a significant increase in both the chromatin accessibility and H3K27 acetylation of Mir122 (Fig. 6b). The actual levels of miR-122 in the liver were validated by microRNA-specific RT-qPCR, concomitant with the acute inflammatory response induced by liver injury, inflammatory factors such as Tnf-α, Il-6 and Cxcl-1 show an upregulation (Fig. 6c and Supplementary Fig. 10a).

a. Evaluate the enrichment levels of transcription factors Yap1 and Taz at the Mir122 loci, indicating the changes pre and post partial hepatectomy. Positive values indicate an increase, while negative values indicate a decrease. b. Integrative Genomics Viewer snapshot show the distribution of normalized reads of H3K27Ac and ATAC-seq at gene loci of miR-122 in liver cells pre- and post-partial hepatectomy. c. RT-qPCR detects the expression changes of miR-122 at 24 h pre and post partial hepatectomy (the left shows the Yap1-HiBiT group mice, and the right shows the Taz-HiBiT group mice). RT-qPCR data are shown as the mean ± SD from three independent replicates (unpaired t-test. *p < 0.05,**p < 0.01, ***p < 0.001). d. Schematic diagram of Rosa26-CAG-LSL-Cas9-tdTomato mice injected with rAAV2/8-type virus carrying sgRNA targeting the Mir122 locus for liver cell-specific knockout of the Mir122 gene. e. Representative Oil Red O staining images from the control and miR-122#KO at 24 h after PHx. Magnified view at 200× of the area highlighted at 100× by the red squares is depicted in the right column. Scale bar, 100 μm. RT-qPCR detects the expression levels of miR-122 in the control and miR-122#KO liver sample (n = 3). RT-qPCR data are shown as the mean ± SD from three independent replicates (unpaired t-test. ****p < 0.0001). f. Representative liver samples that were stained with H&E in the control and miR-122#KO at 24 hous after PHx. All fields are shown at magnification 200×. Arrows are used to designate areas of inflammation in the miR-122#KO at 24 h post PHx. Scale bar, 100 μm. g, h. Liver RNA-seq data from the GSE111805 dataset58 were utilized, comparing miR-122 knockout and control conditions. Limma (3.62.1) was employed to identify differentially expressed genes (FDR ≤ 0.05, |log2FC| > 1), followed by GSEA for functional enrichment analysis. The results indicate that specific knockout of miR-122 in the liver significantly upregulates (I) or downregulates (J) certain signaling pathways compared to the control group. i, j. Representative liver samples that were stained for Ki-67 and PCNA in the control (n = 6) and miR-122#KO (n = 6) at 24 h after partial hepatectomy. Magnified view at 400× of the area highlighted at 200× by the red squares is depicted in the right column. Scale bar, 100 μm. Data are presented as mean ± SD., and **p < 0.01, ***p < 0.001 compared to control group, based on unpaired t-test.
To ascertain whether the upregulation of miR-122 is one of the significant determinants in liver regeneration, the Cas9 knockin animal was adopted for specific in vivo miR-122 depletion in hepatocytes via AAV-TBG-Cre vectors carrying Mir122-sgRNA, with vectors expressing Cre recombinase were used as control (Fig. 6d). Subsequently, partial hepatectomy was performed and miR-122 expression levels were assessed, and the regenerated liver tissue was examined 24 h post-surgery. The results showed that, compared to the control group, the liver-specific miR-122 knockout group exhibited increased lipid accumulation (Fig. 6e), and H&E staining revealed a certain degree of inflammation (Fig. 6f). Transcriptomic analysis following miR-122 depletion supported these findings, showing upregulation of inflammation-related pathways and a marked suppression of lipid metabolism pathways (Fig. 6g, h). We then assessed liver regeneration following miR-122 inhibition, observing a reduction in the proportion of Ki-67+ hepatocytes (Fig. 6i) and a significant decrease in the percentage of PCNA-positive hepatocytes (Fig. 6j). These results confirm that excessive lipid accumulation and inflammatory responses are critical factors affecting the early stages of liver regeneration.
Correspondingly, the downstream target genes of miR-122 involved in iron ion metabolism and fatty acid metabolism, such as Hfe, Tfr2, Ndrg3 (downregulation of these three genes maintains iron ion homeostasis), and E2f1 (regulates fatty acid metabolism), demonstrate a favorable declining trend during liver regeneration as miR-122 expression increases. However, the loss of miR-122 does not reverse this phenomenon; it merely decelerates the trend (Supplementary Fig. 10b). Interestingly, miR-122 expression initially increases and then decreases during liver regeneration. Conversely, E2F1, a transcription factor regulating lipid metabolism, displays an expression pattern inversely correlated with miR-12252 (Supplementary Fig. 10c), aligning with the lipid metabolism dynamics observed during liver regeneration.
Thus, Yap1/Taz likely facilitates liver tissue regeneration and repair by regulating the expression of genes such as Mir122. This phenomenon was maximally captured utilizing Af-CUT&Tag, with DNA peaks highly enriched in the regulatory regions of target genes, demonstrating the system’s exceptional sensitivity in complex tissues. In contrast, antibody-dependent Nano-CUT&Tag signals considerably attenuate in comparison.
Discussion
Af-CUT&Tag advances chromatin profiling by providing a highly sensitive, target antibody-free method for epigenetic analysis across diverse biological systems. This technique overcomes the limitations of traditional methods like CUT&Tag and Nano-CUT&Tag, enabling precise, cost-effective profiling of chromatin features in multiple cell lines, tissues, and even at single-cell resolution. Our results show that Af-CUT&Tag generates high-quality chromatin maps from minimal input material and can be applied to both in vitro and in vivo systems. Notably, this method reveals the critical role of Yap1/Taz in liver regeneration through their control of chromatin accessibility and the regulation of key genes involved in metabolic processes, including miR-122 and lipid metabolism. In particular, the HiBiT-tagged protein serves as a versatile tool for studying the transcriptional regulation of YAP1 and TAZ, enabling real-time monitoring of their expression levels and providing insights into their genomic binding sites through the Af-CUT&Tag system.
Af-CUT&Tag also enables precise and quantitative comparisons of proteins across different conditions through uniform peptide tagging, making it a standardized and broadly applicable tool for diverse proteins, cell types, and tissues. It also bypasses interference from common post-translational modifications such as phosphorylation, methylation, and acetylation. While the generation of knock-in tags may present technical challenges and this strategy cannot distinguish between different modification states of the target protein, and may even introduce unknown knock-in effects, it nonetheless improves the reliability of chromatin profiling, particularly for targets whose chromatin binding is unaffected by such modifications. In addition, applying this approach to epigenetic markers such as histone modifications would require the development of highly specific binders or nanobodies, especially for modifications that are transient or of low abundance.
Beyond the tag-binder design, a key strength of Af-CUT&Tag lies in its modular use of high-affinity binders recognizing genetically encoded tags. This binder-centered framework allows flexibility across different binder types, including nanobodies, engineered scaffolds, and ligand-based binders, without relying on traditional antibody architectures. At the same time, we acknowledge that producing binder-Tn5 fusion proteins as recombinant constructs can present practical challenges, such as reduced solubility or lower expression yield. Fusion to a large enzyme like Tn5 can influence the folding efficiency or stability of the binder domain, and certain binders may still benefit from antigen-mediated stabilization53. These production challenges represent a genuine but manageable limitation, underscoring the importance of rational binder engineering and expression optimization for future applications.
Looking ahead, the modularity of Af-CUT&Tag establishes a versatile foundation for chromatin profiling across a wide spectrum of biological and experimental contexts. By enabling the rational design of binders optimized to target endogenous proteins in specific tissues, Af-CUT&Tag can be readily extended to diverse applications, such as endogenous protein tracking, cell-type-specific regulatory mapping, and profiling of primary tissues or limited clinical samples where conventional antibody-based methods may be limited by insufficient affinity. Continued advances in computational protein engineering will further enhance this potential. Recent AI-driven frameworks for binder and interface design, such as RFdiffusion54 and AlphaFold55, provide powerful tools to refine structural complementarity, improve solubility, and enhance fusion stability, thereby facilitating the generation of next-generation binders suited for Af-CUT&Tag. Together with ongoing innovations in CRISPR-based genome editing and single-cell epigenomics, these developments will expand Af-CUT&Tag into a broadly applicable and scalable platform for dissecting transcriptional regulation and chromatin dynamics in development, disease, and regeneration.
Methods
Ethical Statement
All animal experiments in this study were conducted in compliance with relevant ethical regulations and were approved by the Institutional Animal Care and Use Committee of Xiamen University Laboratory Animal Center (ethics approval number: XMULAC20230071). The study protocol was reviewed and approved by the appropriate ethics review board at Xiamen University. All procedures involving animals were performed in accordance with institutional guidelines to ensure humane treatment.
Biological materials
Human RKO (Cat#TCHu116), DLD1 (Cat#CC0507), and SW480 (Cat#CCL-228) cell lines were obtained from the Shanghai Institute of Biochemistry and Cell Biology, Guangzhou Cellcook Biotech Company, and the American Type Culture Collection (ATCC), respectively, and cultured according to the suppliers’ instructions. We used the following antibodies: Rabbit Control IgG (Abclonal, AC005), Phospho-Rpb1 CTD (Ser2) (Cell Signaling Technology, 13499), CTCF (Cell Signaling Technology, 3418), YAP1 (Cell Signaling Technology, 14074), TAZ (Cell Signaling Technology, 72804), HiBiT (Promega, N7200), Histone H3 (acetyl K27) (Abcam, ab4729), Ki-67 (Abcam, ab279653), PCNA (Santa Cruz, sc-56), Goat anti-rabbit secondary antibody (Thermo Fisher Scientific, 31210), and Goat anti-mouse secondary antibody (Thermo Fisher Scientific, 31160). C57BL/6JGpt-Rosa26tm1(CAG-LSL-Cas9-tdTomato)/Gpt mice were purchased from GemPharmatech (T002249). Mice were maintained under standard laboratory conditions with a normal light/dark cycle, ambient room temperature, and standard humidity.
Generation of isogenic HiBiT/ALFA-tagged cell lines
To generate endogenous HiBiT/ALFA-tagged YAP1/TAZ cell lines, sgRNA targeting the stop codon regions were co-transfected with donor vector contained the following elements: left and right homology arms, as well as linker and HiBiT or ALFA-tag using pSpCas9(BB)-2A-GFP(PX458) and pAAV-mCherry-donor. The transfected cells were sorted 48 h later, and GFP/mCherry-double-positive cells were selected and plated into individual well of 96 well plate, Homozygous or heterozygous clones were selected for further analysis. All homozygous/heterozygous knock-ins were confirmed by Sanger sequencing and western blotting. A list of the oligos and the sequences of the sgRNAs is provided in Supplementary Data 1.
Flow cytometry
Prior to flow cytometry analysis, approximately 1 × 106 cells expressing pSpCas9(BB)-2A-GFP and pAAV-mCherry-donor were treated with trypsin and then washed with PBS. The cells were subsequently sorted and examined using a BD FACSAria FUSION cell sorter, with data analyzed using BD FACSDiva™ Software. The instrument employed 488- and 561-nm excitation lasers, and emission signals were detected using filters at 525/20 nm (wavelength/bandwidth) for GFP and either 610/20 nm or 615/20 nm for mCherry. The sorted cells, both bulk populations and single cells, were placed into either 12 well plates or 96 well plates containing DMEM, RPMI-1640, and L-15 with 10% FBS and 1% penicillin-streptomycin.
LgBiT/NbALFA-Tn5 construction, purification and activity test
Using the pETM11-His-SUMO3-LgBiT-Tn5 expression vector, sequences of LgBiT were PCR amplified from the LgBiT vector. The C terminus of LgBiT was separated from the transposase by a 7-residue flexible linker peptide composed of GSSGGGS. The pTXB1-Tn5 plasmid was a gift from Rickard Sandberg (Addgene plasmid#60240). Previously published sequences coding for NbALFA were synthesized as a gene fragment (Tsingke), with purification steps for NbALFA-Tn5 followed as LgBiT-Tn5. Sequences of other binder-Tn5 fusion proteins were designed according to Bartosovic and Castelo-Branco11. A His-3×FLAG tag was added to the N terminus of each binder, separated by a GGGGS linker to eliminate the need for tag-cleavage steps. Protein purification was performed following the same column-based procedure as described for LgBiT-Tn5. The amino acid sequences of the proteins are provided in Supplementary Data 3.
The pETM11-His-SUMO3-LgBiT-Tn5 plasmid was transformed into E. coli ER2566 Star following the manufacturer’s protocol. Each colony tested was inoculated into 10 mL LB medium and growth was continued at 37 °C for 12 h. The overnight culture was used to inoculate 1.5 L of LB medium supplemented with 50 µg/mL kanamycin and incubated at 37 °C with shaking. When the culture reached an optical density (OD₆₀₀) of ~1.0, the temperature was reduced to 20 °C, and protein expression was induced with 0.25 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). The culture was then incubated overnight at 20 °C before harvesting. Cells were harvested by centrifugation (3900×g, 10 min). The bacterial pellet was frozen and stored at -80 °C. A frozen pellet was resuspended in 50 mL Binding Buffer (20 mM HEPES-KOH at pH 7.2, 0.8 M NaCl, 1 mM EDTA, 10% glycerol, 0.2% Triton X-100) including 1×Roche Complete EDTA-free protease inhibitor tablets. The lysate was sonicated for a total of 15 min using a 2 s on / 3 s off pulse cycle at an output level of 6 (60% amplitude), while keeping the sample chilled on ice throughout the process. The lysate was centrifuged at 12,000×g for 30 min at 4 °C, and the supernatant was loaded onto a HisTrap HP column (Cytiva). The column was washed with 10×column volume of Wash buffer 1 (20 mM HEPES pH 7.2, 800 mM NaCl, 20 mM imidazole, 10% Glycerol and 1 mM EDTA) and 20×column volume of Wash buffer 2 (20 mM HEPES pH 7.2, 1.5 M NaCl, 20 mM imidazole, 10% Glycerol and 1 mM EDTA) sequentially. The recombinant His-SUMO3-LgBiT Tn5 was eluted by 5×column volume of Elution buffer (25 mM HEPES pH 7.2, 800 mM NaCl, 300 mM imidazole, 10% Glycerol and 1 mM EDTA). Each fraction was analyzed by SDS-PAGE, and the one containing a high concentration of recombinant Tn5 was combined and concentrated to 2 mL using Millipore Amicon ultra-15 centrifugal filter unit (30 kDa) to remove the imidazole. The eluent was stored in dialysis buffer (25 mM HEPES pH 7.2, 800 mM NaCl, 0.1 mM EDTA, 10% Glycerol and 1 mM DTT). SENP2 protease was added at a 1:100 ratio to remove the SUMO3 tag from SUMO3-LgBiT-Tn5, followed by incubation with His magnetic beads to eliminate the His-tagged SUMO3, yielding purified LgBiT-Tn5. The concentrated LgBiT-Tn5 was then loaded onto Superdex™ 75 Increase 10/300 GL column with Buffer A. The first peak (peak at 8 mL) was collected and combined, which was the dimer form of recombinant LgBiT-Tn5. The LgBiT-Tn5 dimer fractions were concentrated using Millipore Amicon ultra-15 centrifugal filter unit (30 kDa), and mixed with 100% glycerol to make the glycerol up 50%. The final recombinant LgBiT-Tn5 was stored at -20 °C.
To generate the LgBiT-Tn5 adapter transposome, The lyophilized DNA oligos were dissolved in annealing buffer (40 mM Tris pH 8, 50 mM NaCl) to get 100 μM. The oligo sequences are as follows: Tn5MErev, 5′-CTGTCTCTTATACACATCT-3’; Tn5ME-A,5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3’; Tn5ME-B, 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3’. One volume of Tn5MErev was mixed with one volume of Tn5ME-A or Tn5ME-B to get 50 μM Adaptor A/B. The adaptor A/B was annealed in a thermocycler with the program: 75 °C, 15 min; 60 °C, 10 min; 50 °C, 10 min; 40 °C, 10 min; 25 °C, 30 min; 4 °C, hold. The equal volume of adaptor A and adaptor B were mixed to get 50 μM adaptor mix. Assembly of LgBiT-Tn5 transposome was performed by mixing 2 μL LgBiT-Tn5 (26 μM), 2.4 μL adapter mix (50 μM) and 10.6 μL dialysis buffer. The mixture was incubated at room temperature for 60 min and then stored at 4 °C for several days, then mixed with 100% glycerol to make the glycerol up 50%, The final LgBiT-Tn5 adapter transposome was stored at -20 °C.
Liver tissue nuclei extraction
Mice were sacrificed. Subsequently, 0.1-0.2 g of liver tissue was excised and washed with 2 mL of swelling buffer (10 mM Tris-HCl pH 7.5, 2 mM MgCl2, 3 mM CaCl2, 1×Protease inhibitor cocktail). The liver was then dounced in 1 ml of swelling buffer (dounced 10 times, piston A) and incubated at 4 °C for 20 min. Afterward, the liver was dounced again 20 times (piston B), and 1 mL of swelling buffer was added. The cells were filtered using a 100 μm cell strainer and spun at 400×g at 4 °C for 10 min. The supernatant was removed, and the cells were resuspended in 0.5 ml of swelling buffer + 10% glycerol. Slowly vortex while adding drop by drop 0.5 mL of lysis buffer (Swelling buffer + 10% glycerol + 1% NP-40). Incubate for 5 min at 4 °C and add 1.5 mL of lysis buffer. Spin at 600×g at 4 °C for 5 min, remove the supernatant, and wash the nuclei again with 1.5 mL of lysis buffer. Spin at 600×g at 4 °C for 5 min, remove the supernatant, and resuspend the nuclei in 1 mL of freezing buffer (50 mM Tris-HCl pH 7.5, 5 mM MgCl₂, 0.1 mM EDTA, 40% glycerol, and 1× Protease inhibitor cocktail). Finally, the nuclei were stained with DAPI and counted.
Af-CUT&Tag bulk
Nuclei or permeabilized cells were counted and centrifuged for 3 min at 600×g at room temperature. Aliquots of nuclei or cells (100–50,000 nuclei or cells), were washed twice in 0.5 mL Wash Buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM Spermidine, 1×Protease inhibitor cocktail). Concanavalin A coated magnetic beads (Vazyme#N515) were prepared and 10 µL of activated beads were added per sample and incubated at RT for 10-15 min. And then nuclei or permeabilized cells were directly suspended with 100 µL antibody buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 2 mM EDTA, 0.5 mM spermidine, 0.05% Digitonin, 1% BSA, 1×protease inhibitors) with a cocktail of blocking protein (Nanoluciferase composed of LgBiT and SmBiT, final concentration: 2 μM) and LgBiT-Tn5 or NbALFA-Tn5 (final concentration: 0.04 μM), and then incubated on a rotator at room temperature for 30 min. Subsequently, cells were washed three times with 200 μL high salt wash buffer (20 mM HEPES pH 7.5, 300 mM NaCl, 0.5 mM spermidine, 0.01% Digitonin, 1×protease inhibitors). The nuclei or cells were then resuspended in 50 μL tagmentation buffer (20 mM HEPES pH 7.5, 300 mM NaCl, 0.5 mM spermidine, 10 mM MgCl2, 0.01% Digitonin, 1×protease inhibitors). The samples were incubated for 1 h at 37 °C. Tagmentation steps were performed in 0.2 mL tubes to minimize cell loss. To stop tagmentation, 1 µL of 0.5 M EDTA, 1 µL of 10% SDS and 0.5 µL of 20 mg/mL proteinase K was added to 50 µL of sample, which was incubated at 55 °C for 10 min. To extract the DNA, 20 µL VAHTS Particles G beads (Vazyme#N903) were added to each tube with vortexing, incubated for 5 min at room temperature. Tubes were placed on a magnet stand to clear, then the liquid was carefully withdrawn. Without disturbing the beads, beads were washed twice in 200 µL 80% ethanol. After allowing to dry ~5 min, 35 µL of 10 mM Tris pH 8 was added, the tubes were vortexed, quickly spun and allowed to sit for 5 min. Tubes were placed on a magnet stand and the liquid was withdrawn to a fresh tube.
To amplify libraries, 35 µL of DNA was mixed with 2 µL of a universal i5 and a uniquely barcoded i7 primer, using a different barcode for each sample. A volume of 1 µL of TAE and 5×TAB (Vazyme#TD601) were added and mixed. The sample was placed in a thermocycler with a heated lid using the following cycling conditions: 72 °C for 3 min (gap filling); 98 °C for 30 s; 15 cycles of 98 °C for 15 s, 60 °C for 30 s and 72 °C for 30 s; final extension at 72 °C for 5 min and hold at 4 °C. Post-PCR clean-up was performed by adding 1.5×volume of VAHTS DNA Clean Beads (Vazyme#N411), and libraries were incubated with beads for 5 min at RT, washed twice gently in 80% ethanol, and eluted in 22 µL 10 mM Tris pH 8.0.
Single-cell Af-CUT&Tag
The preparation of the LgBiT-Tn5 complex with various barcodes was conducted in a total of 192-well PCR strip tubes, each containing a unique combination of 5′ and 3′ complex barcodes, with sequences referenced from ACT-seq35. The following components were mixed in each well: 1.5 μL of 26 μM recombinant LgBiT-Tn5 protein, 0.75 μL of 50 μM 5′ composite barcode, 0.75 μL of 50 μM 3′ composite barcode, and 4 μL of dialysis buffer. The strip tubes were incubated at 25 °C for 60 min to form the complex.
Wash and permeabilize 2 million HiBiT-tagged cells or nuclei as described above. Resuspend all permeabilized cells in antibody buffer and add NanoLuciferase. Distribute the sample into 96 wells at a density of 10,000 cells or nuclei per 50 μL. Add 1 μL of the LgBiT-Tn5 complex to each well and incubate at room temperature for 30 min. Centrifuge the PCR strip tubes at 300×g for 3 min, retaining ~10 μL of solution to remove unbound complexes. Add 100 μL of Dig-300 wash buffer to each well and repeat the centrifugation and wash steps three times. Resuspend the cell or nuclear solution and pool it into a 15 mL centrifuge tube. Centrifuge the tube containing the pooled cells at 600×g for 3 min and carefully remove all but ~20 μL of the solution. Resuspend the cells in 400 μL of Dig-300 buffer and filter using a cell strainer test tube. Using a FACS instrument, distribute the cells or nuclei into 96 clean wells, with each well containing 200 cells or nuclei. Initiate the tagmentation reaction by adding 25 μL of tagmentation buffer to each well and incubate at 37 °C for 60 min. Subsequently, terminate the reactions and extract DNA as described above.
For library preparation, 1.25 μL of 10 µM 5′ library index barcode was added to each of the 96 wells, with each well receiving a 3′ barcode with a unique sequence. Each well then received 18 μL of DNA, 1.25 μL of 10 μM universal 5’ library index barcode, 0.5 μL of TAE, and 5 μL of 5×TAB. Twenty cycles of PCR amplification were performed using the program described above. DNA purification was consistent with the aforementioned bulk protocol. All barcode sequences and unique library primer sequences are provided in Supplementary Data 2.
ATAC-seq
Harvest cells by centrifuging at 600×g for 5 min, discard the supernatant, and resuspend in PBS. Centrifuge a single-cell suspension containing the desired number of target cells at 600×g for 5 min, discard the supernatant, and resuspend in 50 µL of pre-cooled lysis buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40, 0.02% digitonin, 0.1% Tween-20). Allow the cells to lyse by placing them on ice for 5 min. Centrifuge at 500×g for 10 min at 4 °C to collect the cell nuclei and immediately continue to transposition reaction. To make the transposition reaction mix, combine the following: 10 μL 5×TD Buffer (50 mM Tris pH 7.5, 25 mM MgCl2, and 50% dimethylformamide), 16.5 µL PBS, 0.5 µL 10% Tween-20, 0.5 µL 1% digitonin, 18.5 ddH2O and 4 µL LgBiT/NbALFA-Tn5 Transposase (~1 µM). Resuspend nuclei in the transposition mix and incubate at 37 °C for 30 min. PCR mix (Vazyme#TD601) and adapters (VazymeTD#202) were used for library amplification, the total volume is 50 µL. The fragmented DNA library was initially incubated with a 0.55× (27.5 µL) volume of DNA purification magnetic beads (Vazyme#N411). After discarding the magnetic beads, the supernatant was retained, and a 1.2× (60 µL) volume of DNA magnetic beads was added to further purify the DNA fragment library.
CUT&Tag and Nano-CUT&Tag bulk
CUT&Tag was performed as described in Henikoff6, and all reagents and transposase used in this assay were obtained from the Hyperactive Universal CUT&Tag Assay Kit for Illumina (Vazyme Biotech Co., Ltd). Nano-CUT&Tag was performed as described in Bartosovic and Castelo-Branco11, with NanoRabbit/NanoMouse-Tn5 expressed and purified according to the sequences provided by the authors, and all salt concentrations strictly matched those specified in the original protocol. The sequencing library was prepared consistent with Af-CUT&Tag.
DNA sequencing
The size distribution of libraries were determined by Agilent 2100 TapeStation analysis, and concentration of the libraries were determined by Qubit 3.0. Paired-end Illumina sequencing was performed on the barcoded libraries following the manufacturer’s instructions.
Bulk Af-CUT&Tag data processing
The raw reads were first filtered to remove the adapter sequences and low-quality sequences using fastp (version 0.20) with options: -g -q 30 -u 50 -n 15 -l 36 -w 4, clean reads were mapped to the mouse mm10 or human hg19 reference genome using Bowtie2 (version 2.4) with options: --end-to-end --very-sensitive --no-mixed --no-discordant --phred33 -I 10 -X 700. Duplicate reads were removed with Picard MarkDuplicates (version 2.27) with default parameters, mitochondrial reads, reads from unmapped and reads with a mapping quality<30 were removed using SAMtools (version 1.15). BigWig files were generated with deepTools bamCoverage (version 3.3.1) tools from reads with --binSize 10 --normalizeUsing RPGC. The results were visualized in the Integrative genomics viewer IGV (version 2.16.2). TSS heatmap and profile plot are plotted using the plotHeatmap and plotProfile functions respectively. Peak calling was performed on each individual sample by MACS2 callpeak (version 2.2) with options: -c control_sample --bdg --SPMR --gsize hs --keep-dup all --qvalue 0.05 --shift -75 --extsize 150 --nomodel --call-summits --nolambda. Use the ChIPseeker R package to annotate the genes corresponding to the peaks. Homemade Python scripts were used to calculate the quality control indicators of the sample. For more details see https://github.com/xmuzhanglab/Workflow.
Single-cell Af-CUT&Tag data processing
The single-cell raw data were split into individual samples at the single-cell level using custom Python scripts. Sequencing adapters were removed, and quality control was performed using methods commonly employed in bulk sequencing analysis. The sequences were then aligned to the reference genome. To ensure data quality, reads that could not be aligned were filtered out, along with cells that had an insufficient number of reads. Cells with an excessively high number of reads, which could indicate doublets, were also excluded. Subsequently, the genome was divided into bins of 10,000 bases using bamCoverage (version 3.3.1), and the read coverage for each bin was calculated. The results were converted into bedGraph files using the map tool in BEDtools (version 2.30). Dimensionality reduction was performed using scikit-learn for PCA and UMAP, followed by k-means clustering analysis to identify distinct cell populations.
Differential binding peak analysis
To compare the accessibility differences of YAP1/TAZ pre and post PHx, we first utilized UCSC tools BigWigAverageOverBed download from GitHub (https://github.com/kevlim83/ucsctools) to calculate the average score of BigWig over each peak and filtered out peaks with scores below 10 in both pre- and post-resection samples. Subsequently, we employed edgeR (version 3.36.0) R package to identify differential peaks between the samples with cutoff p value < 0.01, FDR < 0.05 and |log2FC | > 1.
GO enrichment
To investigate the functional impact of differentially accessible peaks, we utilized ChIPseeker (version 1.30.3) to annotate the upregulated and downregulated peaks to their nearest genes. We then performed functional enrichment analysis separately for these genes using the clusterProfiler (version 4.2.2) R package.
Bio-layer interferometry (BLI)
The determination of HiBiT/LgBiT-Tn5 and ALFA-tag/NbALFA-Tn5 binding affinities was conducted using the Octet K2 Protein Analysis System and Streptavidin (SA) biosensors. Specifically, biotinylated ExoIII-HiBiT and GFP-ALFA-tag proteins were immobilized onto the SA biosensors. Serial dilutions of LgBiT/LgBiT-Tn5 and NbALFA/NbALFA-Tn5 proteins, prepared in assay buffer (1×PBS, pH 7.4, 0.05% Tween-20), were employed to investigate their binding interactions with the immobilized peptides. To mitigate any potential interference from the assay buffer, an equivalent volume of PBST buffer was introduced to the ExoIII-HiBiT and GFP-ALFA-tag samples as a control. The resultant response data were normalized using Octet Data Analysis Studio 12.2.
Adeno-associated virus 2/8 (AAV 2/8) Preparation and Injections
C57BL/6JGpt-Rosa26tm1(CAG-LSL-Cas9-tdTomato)/Gpt mice were used for liver-specific gene knock-in and knock-out. AAV vectors carrying the U6-sgRNA (targeting the stop codon region of mYap1/Taz) and the donor contained the following elements: left and right homology arms, as well as linker and HiBiT, with TBG-Cre as the last element for liver-specific knock-in. AAV vector carrying TBG-Yap1-HiBiT was used for liver-specific expression of Yap1-HiBiT. AAV vector carrying the U6-sgRNA (targeting Mir122) and TBG-Cre for liver-specific knock-out. AAV vectors with Cre recombinase and no sgRNA were used as control viruses. The nucleotide sequences utilized are provided in Supplementary Data 1.
AAV vectors were packaged into serotype 2/8 capsids. For liver-specific knock-in, ~2 × 1012 vg per mouse of AAV 2/8 virus were injected via the tail vein into 6-week-old male C57BL/6JGpt-Rosa26tm1(CAG-LSL-Cas9-tdTomato)/Gpt mice. For tissue-specific expression of Yap1-HiBiT or tissue-specific gene knockout, 2–5×10¹¹ vg per mouse was administered. Control mice were injected with AAV 2/8 virus containing only TBG-Cre.
Immunoprecipitation
Cells (DLD1 TAZ-HiBiT, DLD1 TAZ-ALFA-tag, and DLD1 WT) were seeded in 10-cm dishes, trypsinized, and lysed in 1 mL of IP lysis buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 5% glycerol) containing protease inhibitor cocktail, pre-chilled on ice. After incubating on ice for 30 min with occasional mixing, the lysates were centrifuged at 12,000×g for 10 min at 4 °C, and the supernatants were collected. Protein concentrations were measured, and 100 µL of each sample was reserved as input. For immunoprecipitation, 800 µg of total protein was incubated with 1.5 µg of Flag-LgBiT-Tn5 or Flag-NbALFA-Tn5 protein at 4 °C for 3 h. Anti-FLAG Nanobody Magarose Beads (15 µL, Alpalifebio) were then added, and the samples were incubated overnight at 4 °C with rotation. The next day, the beads were washed, and the proteins were eluted in 45 µL of 1× loading buffer. Input samples were mixed with 5× loading buffer. Both input and IP samples were analyzed by western blotting.
Nano-Glo® HiBiT Lytic Detection System
To assess the activity of the NanoLuc large subunit (LgBiT) in fusion proteins, cells with stable knock-in of the HiBiT tag were enzymatically dissociated and resuspended. The cells were evenly divided into three groups, each further split into three replicates containing approximately 1×10⁵ cells per replicate. Experimental groups were treated with an equimolar amount of LgBiT or LgBiT-Tn5 Protein, while the control group received no additional protein. The Nano-Glo® HiBiT detection reagent (Promega, catalog number: N3040) was prepared by diluting LgBiT (1:100) or LgBiT-Tn5 Protein (1:100) and Nano-Glo® HiBiT Lytic Substrate (1:50) in an appropriate volume of room-temperature Nano-Glo® HiBiT Lytic Buffer, followed by gentle inversion to mix. Equal volumes of the cell suspension and detection reagent were then combined in each well. After incubation at room temperature for 10 min, luminescence was measured using a microplate reader with the following settings: LUM-EndPoint mode, single label detection, 700 nm blocker, top-read mode, measurement time of 1000 ms, and a default Z focus of 8.5 mm.
Western blotting
Trypsinized cell pellets were resuspended in lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% SDS, 0.125% sodium deoxycholate, and 1% Triton X-100 in Millipore water) containing protease inhibitor cocktail (Sigma). The liver tissue samples were homogenized and then lysed in a lysis buffer. Protein concentration was determined using a standard BCA assay (TIANGEN) and 30 μg per sample was separated by SDS-PAGE and transferred to a PVDF membrane (Millipore). Membranes were incubated with antibodies in TBS-Tween (0.2%) containing 5% milk at 4 °C overnight. Antibodies against YAP1 (Cell Signaling Technology, 14074), TAZ (Cell Signaling Technology, 72804), HiBiT (Promega, N7200) and actin (Abcam, ab20272) were used, and then with the corresponding horseradish peroxidase-labeled secondary antibody. Protein levels were measured using a standard ECL kit and images were acquired using a C-Series Imaging System (Azure Biosystems C500).
Stem-Loop RT-qPCR for miR-122
For detection of miR-122, total RNA was reverse-transcribed using stem-loop primer (RT miR-122: 5’- GTC GTA TCC AGT GCA GGG TCC GAG GTA TTC GCA CTG GAT ACG ACC AAA C-3’; RT U6: 5’-AAC GCT TCA CGA ATT TGC GT-3’). Specific oligonucleotide primers were used for miR-122 and U6 genes: miR-122 F: 5’-TGG AGT GTG ACA ATG GTG TT-3’ and R: 5’-CCA GTG CAG GGT CCG AGG T-3’; U6: F: 5’-CTC GCT TCG GCA GCA CA-3’, and R: 5’-AAC GCT TCA CGA ATT TGC GT-3’. RT reaction was carried out in Biometra T Gradient Thermocycler (16 °C for 30 min, 42 °C for 30 min, 85 °C for 5 min and taken out the sample at 4 °C). The amplification reactions were carried out and the expression levels of miR-122 were quantified as described above. miR-122 was normalized with U6 snRNA.
Histology and immunohistochemistry
Liver tissue samples were fixed in 4% paraformaldehyde and embedded in paraffin. For histopathological evaluation, sections were stained with hematoxylin and eosin (H&E) according to standard protocols. For detection of Ki-67 (1:200 dilution) and PCNA (1:200 dilution), immunostaining was performed via standard protocols. Antigen retrieval was performed for 20 min in citrate buffer by heating in a boiling water bath at 100 °C for 10 min. Endogenous peroxidase activity was blocked by incubating with 3% H2O2 for 10 min at room temperature. Then, tissue sections were blocked with 5% goat serum for 30 min and then incubated with primary antibodies overnight at 4 °C. After rinsing, the tissue sections were incubated with biotinylated goat anti-rabbit/mouse IgG for 10 min and streptavidin-peroxidase for 10 min. DAB solution was then added for colorization. After a brief rinse, the sections were stained with hematoxylin for nuclear staining. Finally, the sections were dehydrated with ethanol and xylene and sealed with a neutral resin. Images were acquired at 100×, 200×, and 400× magnifications using a Leica DM4B microscope. Samples in the negative control group were treated according to the same protocol, except that the primary antibody was replaced with the control IgG.
Oil red O staining
Liver tissue samples were fixed in 4% paraformaldehyde, dehydrated using a sucrose gradient, and embedded in O.C.T. compound. The embedded tissues were then sectioned into frozen sections. The sections were rinsed with phosphate-buffered saline (PBS) to remove any residual OCT. Subsequently, the sections were incubated in 60% isopropanol for 20 s and stained with freshly prepared Oil Red O working solution (Beyotime#C0157S) for 5 min at room temperature. After staining, the sections were briefly rinsed with 60% isopropanol to remove unbound stain, followed by thorough washing with distilled water. The nuclei were counterstained with hematoxylin for 10 s, and the sections were then rinsed with running tap water for 5 min to remove excess hematoxylin. Finally, the stained sections were mounted with a suitable aqueous mounting medium and examined under a Leica DM4B microscope.
Statistics & reproducibility
All statistical analyses were performed using GraphPad Prism 9.3 (San Diego, CA, USA). Bioinformatics analysis was performed with R (version 3.6.1) (http://www.R-project.org/). The number of biological replicates for each experiment is indicated in the corresponding figure legend. Unless otherwise specified, all values were calculated from at least three independent biological replicates. Unpaired t-test was used for all analyses. For all the statistical analyses between the control group and the experimental group, the results are shown in the figures as follows: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, and N.S. (not statistically significant).
No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Visualization
The genome browser tracks were created using pyGenomeTracks or IGV and further formatted in Adobe Illustrator. Volcano plot, heatmaps, boxplots, and GSEA plot were created using ggplot2 (version 3.5.1).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The accession number for the scAf-CUT&Tag, ATAC and Af-CUT&Tag sequencing and processed data reported in this paper is GEO: GSE283704, GSE283705, and GSE283706. The following publicly available datasets were used in this study: GSE180012 RNA-seq of mouse liver around the circadian clock after partial hepatectomy (PHx) or sham operation; GSE95135 RNA-seq of mouse liver regeneration after partial hepatectomy; GSE111805 RNA-seq of mouse livers during development and after miR-122 excision. The spatial transcriptomics data were obtained from the analytical platform available at https://db.cngb.org/stomics/lista. Source data are provided with this paper.
Code availability
The analysis pipeline used in this study is available on GitHub (https://github.com/xmuzhanglab/Af-CUT-Tag-analysis). The specific version of the code associated with this publication has been archived in Zenodo and is accessible via https://zenodo.org/records/1800690456.
References
Nakato, R. & Sakata, T. Methods for ChIP-seq analysis: a practical workflow and advanced applications. Methods 187, 44–53 (2021).
Park, P. J. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet 10, 669–680 (2009).
Casamassimi, A. & Ciccodicola, A. Transcriptional regulation: molecules, involved mechanisms, and misregulation. Int J. Mol. Sci. 20, 1281 (2019).
Wagh, K., Stavreva, D. A. & Hager, G. L. Transcription dynamics and genome organization in the mammalian nucleus: recent advances. Mol. Cell 85, 208–224 (2024).
Casamassimi, A., Federico, A., Rienzo, M., Esposito, S. & Ciccodicola, A. Transcriptome profiling in human diseases: new advances and perspectives. Int J. Mol. Sci. 18, 1652 (2017).
Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019).
Zentner, G. E., Kasinathan, S., Xin, B., Rohs, R. & Henikoff, S. ChEC-seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo. Nat. Commun. 6, 8733 (2015).
Skene, P. J. & Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, e21856 (2017).
Skene, P. J., Henikoff, J. G. & Henikoff, S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 13, 1006–1019 (2018).
Kaya-Okur, H. S., Janssens, D. H., Henikoff, J. G., Ahmad, K. & Henikoff, S. Efficient low-cost chromatin profiling with CUT&Tag. Nat. Protoc. 15, 3264–3283 (2020).
Bartosovic, M. & Castelo-Branco, G. Multimodal chromatin profiling using nanobody-based single-cell CUT&Tag. Nat. Biotechnol. 41, 794–805 (2023).
Stuart, T. et al. Nanobody-tethered transposition enables multifactorial chromatin profiling at single-cell resolution. Nat. Biotechnol. 41, 806–812 (2023).
Gopalan, S. & Fazzio, T. G. Multi-CUT&Tag to simultaneously profile multiple chromatin factors. STAR Protoc. 3, 101100 (2022).
Gopalan, S., Wang, Y., Harper, N. W., Garber, M. & Fazzio, T. G. Simultaneous profiling of multiple chromatin proteins in the same cells. Mol. Cell 81, 4736–4746.e4735 (2021).
Meers, M. P., Llagas, G., Janssens, D. H., Codomo, C. A. & Henikoff, S. Multifactorial profiling of epigenetic landscapes at single-cell resolution using MulTI-Tag. Nat. Biotechnol. 41, 708–716 (2023).
Khodabakhsh, F., Behdani, M., Rami, A. & Kazemi-Lomedasht, F. Single-domain antibodies or nanobodies: a class of next-generation antibodies. Int Rev. Immunol. 37, 316–322 (2018).
Edfors, F. et al. Enhanced validation of antibodies for research applications. Nat. Commun. 9, 4130 (2018).
Chaves, E. J. F. et al. Structure-based computational design of antibody mimetics: challenges and perspectives. FEBS Open Bio. 15, 223–235 (2024).
Mazhar, S. et al. Challenges and reinterpretation of antibody-based research on phosphorylation of Tyr(307) on PP2Ac. Cell Rep. 30, 3164–3170.e3163 (2020).
Kasahara, K. et al. Unveiling the structural mechanisms behind high affinity and selectivity in phosphorylated epitope-specific rabbit antibodies. J. Biol. Chem. 300, 107989 (2024).
Bock, I. et al. Detailed specificity analysis of antibodies binding to modified histone tails with peptide arrays. Epigenetics 6, 256–263 (2011).
Jones, I. K. A. & Streblow, D. N. Antibody-independent quantification of cytomegalovirus virion protein incorporation using HiBiT. Methods Mol. Biol. 2244, 213–232 (2021).
Boursier, M. E. et al. The luminescent HiBiT peptide enables selective quantitation of G protein-coupled receptor ligand engagement and internalization in living cells. J. Biol. Chem. 295, 5124–5135 (2020).
Arakawa, M. & Morita, E. Protein pull-down assay using HiBiT-tag-dependent luciferase activity measurement. Bio Protoc. 13, e4640 (2023).
Götzke, H. et al. The ALFA-tag is a highly versatile tool for nanobody-based bioscience applications. Nat. Commun. 10, 4403 (2019).
Akhuli, D., Dhar, A., Viji, A. S., Bhojappa, B. & Palani, S. ALIBY: ALFA Nanobody-Based Toolkit for Imaging and Biochemistry in Yeast. mSphere 7, e0033322 (2022).
Liu, M., Li, L., Jin, D. & Liu, Y. Nanobody-A versatile tool for cancer diagnosis and therapeutics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol 13, e1697 (2021).
Bai, Z. et al. Design of nanobody-based bispecific constructs by in silico affinity maturation and umbrella sampling simulations. Comput Struct. Biotechnol. J. 21, 601–613 (2023).
Larson, H. G. et al. A genome-edited ERα-HiBiT fusion reporter cell line for the identification of ERα modulators Via high-throughput screening and CETSA. Assay. Drug Dev. Technol. 19, 539–549 (2021).
Lu, Z. et al. Identification of G-quadruplex-interacting proteins in living cells using an artificial G4-targeting biotin ligase. Nucleic Acids Res 52, e37 (2024).
Galli, S. et al. DNA G-quadruplex recognition in vitro and in live cells by a structure-specific nanobody. J. Am. Chem. Soc. 144, 23096–23103 (2022).
Alwash, M. & Gariépy, J. Labeling cell surface receptors with ligand.BirA* Bispecifics. ACS Pharm. Transl. Sci. 5, 62–69 (2022).
Rosenberg, A. B. et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 (2018).
Cusanovich, D. A. et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015).
Carter, B. et al. Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq). Nat. Commun. 10, 3747 (2019).
Baddour, J. A., Sousounis, K. & Tsonis, P. A. Organ repair and regeneration: an overview. Birth Defects Res C. Embryo Today 96, 1–29 (2012).
Russell, J. O. & Camargo, F. D. Hippo signalling in the liver: role in development, regeneration and disease. Nat. Rev. Gastroenterol. Hepatol. 19, 297–312 (2022).
Moya, I. M. & Halder, G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 20, 211–226 (2019).
Hsia, O. et al. Targeted protein degradation via intramolecular bivalent glues. Nature 627, 204–211 (2024).
Schroer, A. B. et al. Platelet factors attenuate inflammation and rescue cognition in ageing. Nature 620, 1071–1079 (2023).
Wang, Q. et al. CoBATCH for high-throughput single-cell epigenomic profiling. Mol. Cell 76, 206–216.e207 (2019).
Zhu, C. et al. Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat. Methods 18, 283–292 (2021).
Bartosovic, M., Kabbe, M. & Castelo-Branco, G. Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat. Biotechnol. 39, 825–835 (2021).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Yu, G., Wang, L. G. & He, Q. Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015).
Chen, Y. et al. Acute liver steatosis translationally controls the epigenetic regulator MIER1 to promote liver regeneration in a study with male mice. Nat. Commun. 14, 1521 (2023).
Bou Khalil, M., Blais, A., Figeys, D. & Yao, Z. Lipin - The bridge between hepatic glycerolipid biosynthesis and lipoprotein metabolism. Biochim Biophys. Acta 1801, 1249–1259 (2010).
Jama, A. et al. Lipin1 plays complementary roles in myofibre stability and regeneration in dystrophic muscles. J. Physiol. 601, 961–978 (2023).
Koundouros, N. & Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 122, 4–22 (2020).
Briot, A. et al. Senescence alters PPARγ (Peroxisome Proliferator-Activated Receptor Gamma)-dependent fatty acid handling in human adipose tissue microvascular endothelial cells and favors inflammation. Arterioscler Thromb. Vasc. Biol. 38, 1134–1146 (2018).
Al-Gazally, M. E. et al. The role and mechanism of action of microRNA-122 in cancer: Focusing on the liver. Int Immunopharmacol. 123, 110713 (2023).
Rib, L. et al. Cycles of gene expression and genome response during mammalian tissue regeneration. Epigenetics Chromatin 11, 52 (2018).
Wang, Z. et al. ALFA nanobody-guided endogenous labeling. Nat. Chem. Biol. 21, 1992–2001 (2025).
Watson, J. L. et al. De novo design of protein structure and function with RFdiffusion. Nature 620, 1089–1100 (2023).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Xindong, W. et al. Af-CUT&Tag: a sensitive and antibody-free chromatin profiling method using genetically encoded tags and high-affinity binders fused to Tn5. Zenodo https://doi.org/10.5281/zenodo.18006903 (2025).
Xu, J. et al. A spatiotemporal atlas of mouse liver homeostasis and regeneration. Nat. Genet 56, 953–969 (2024).
Valdmanis, P. N. et al. miR-122 removal in the liver activates imprinted microRNAs and enables more effective microRNA-mediated gene repression. Nat. Commun. 9, 5321 (2018).
Acknowledgements
We thank all the members of Y.Z.’s laboratory, Dr. Sanford Markowitz at Case Western Reserve University, and Dr. Dongyin Guan at Baylor College of Medicine for their critical reading and valuable discussions. This work was supported by the National Key Research and Development Program of China (No. 2022YFA1105300 and No. 7100 to Y.Z.), the National Natural Science Foundation of China (No. 81772539 and No. 81972238 to Y.Z.), the Natural Science Foundation of Fujian Province (2024J01023 to Y.Z.), the Shenzhen Science and Technology Program (JCYJ20240813145615020 to Y.Z.), the Fundamental Research Funds for the Central Universities of China–Xiamen University (No. 20720180048 to Y.Z.), and the Innovation Research and Development Special Funds of the Municipality-Province-Ministry Co-constructed (GJZXHYSW-2024-02 to Z.L.). We also thank Dr. Xiaohong Ma, Xiufeng Sun, and Dr. Caiming Wu for their technical assistance.
Author information
Authors and Affiliations
Contributions
Conceptualization, Y.Z.; Methodology, X.W. and X.D.; Formal Analysis, X.W. and X.D., and H.S.; Investigation, X.W., X.D., L.Q., H.D., J.L., H.S., W.L., L.S., W.D., X.D, Y.H., B.L.; Writing – Original Draft, X.W., Y.Z.; Writing – Review & Editing, Y.Z., Z.L., and J.H.; Funding Acquisition, Y.Z.; Resources, Y.Z., Z. L. and J.H.; Supervision, Y.Z., and Z.L.
Corresponding authors
Ethics declarations
Competing interests
Y.Z., X.W., X.D., L.Q., and H.D. have submitted a patent application to Xiamen University pertaining to aspects of this work (application number 202410216797.2; published as CN 118184796 A). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hiroshi Nishina, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, X., Deng, X., Qiu, L. et al. Af-CUT&Tag: a sensitive and antibody-free chromatin profiling method using genetically encoded tags and high-affinity binders fused to Tn5. Nat Commun 17, 1746 (2026). https://doi.org/10.1038/s41467-026-68454-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68454-9
