
Abstract
Artemisinin-combination therapies (ACTs) are now recommended for the treatment of uncomplicated malaria caused by Plasmodium vivax, the parasite responsible for the majority of malaria infections outside of Africa. We sequence the genomes of 206 P. vivax parasites collected from Cambodian malaria patients and show that more than 80% of them carry a DNA deletion located immediately downstream of the multidrug resistance 1 gene (mdr1) protein-coding sequence. This 837 bp deletion overlaps with a different deletion present at low frequency in South American isolates, suggesting a functional role despite not altering the coding sequence of mdr1. Using RNA sequencing, we show that these deletions alter the transcripts expressed from mdr1 and result in mRNAs with different 3’ untranslated regions. In Cambodian isolates, the deletion was significantly associated with a higher level of mdr1 mRNA, a lower ex vivo susceptibility to mefloquine, and increased in frequency in Cambodia since the introduction of mefloquine as ACT partner drug. Overall, these findings indicate that a common deletion of a non-coding sequence affects the transcription, stability, or translation of mdr1 in P. vivax parasites and could mediate reduced susceptibility to antimalarial drug(s) currently used for the treatment of uncomplicated vivax malaria.
Similar content being viewed by others


Introduction
Malaria remains a major public health problem that threatens half of the world’s population1. The disease is caused by infections with unicellular parasites of the Plasmodium genus, including P. falciparum that causes most of the deaths associated with malaria, and P. vivax that is responsible for the majority of cases outside of Africa2. P. falciparum and P. vivax are often co-endemic and malaria infections are frequently treated indiscriminately. While global efforts have led to substantial reductions in the burden of malaria caused by these two species, the unique biological features of P. vivax continue to hamper its control relative to the progress made against P. falciparum. These features include the ability of P. vivax stages to persist in dormant liver stages, which can reactivate months after a treatment that clears the blood infection, causing relapse infections. P. vivax, because of its preference for reticulocytes, also often circulates at low levels in the bloodstream, making its detection and diagnosis more challenging than P. falciparum. Finally, the lack of robust in vitro culture system for P. vivax complicates many studies of this parasite, including the assessment of antimalarial drug resistance3 that often depends solely on patient studies and can be confounded by relapses and reinfections4,5.
Identification and validation of robust molecular markers for rapid and cost-effective surveillance of drug resistance is essential for improving malaria control and elimination efforts but has remained relatively unsuccessful for P. vivax, especially when compared to the progress in identifying P. falciparum drug resistance markers. Since the K76T mutation of the chloroquine resistance transporter (crt) gene is linked to chloroquine resistance in P. falciparum parasites, several studies have assessed its ortholog for a similar role in P. vivax. However, single nucleotide polymorphisms in crt have not associated with chloroquine resistance in field studies, nor in non-human primate models of P. vivax malaria6. Furthermore, engineered expression of P. vivax crt K76 or T76 polymorphisms showed no significant difference on their reductions of chloroquine accumulation in a Dictyostelium expression system7. On the other hand, some studies have suggested that increased expression of the crt gene, independent of specific point mutations, may have a role in mediating P. vivax chloroquine resistance. For example, a laboratory genetic cross in non-human primates showed linkage between increased crt transcription and an inherited chloroquine resistance phenotype8. Taken together, these findings suggest that gene regulation affecting the level of crt expression may be involved in the chloroquine responses of P. vivax malaria. Polymorphisms in the P. vivax multidrug resistance 1 (mdr1) gene, another important marker of antimalarial drug resistance in P. falciparum, have also been extensively examined for associations with antimalarial drug responses. Some studies have reported associations between specific mutations in mdr1, such as Y976F or G698S, and reduced sensitivity of P. vivax to chloroquine in ex vivo assays9,10. However, the exact role of mdr1 mutations and the extent of their contribution to chloroquine resistance remains unclear and sometimes contradictory3,9,11. Thus, while specific mutations of mdr1 may be involved in antimalarial drug responses, their effects might vary depending on the genetic background of P. vivax strains or other unknown factors. Finally, DNA duplications of the mdr1 gene have been suggested to influence antimalarial drug susceptibility12,13, similarly to findings in P. falciparum14,15,16.
Due to increasing reports of potential P. vivax resistance to chloroquine17,18,19,20, the first-line treatment for the blood stages of P. vivax since the 1950s, and the lack of robust molecular markers of resistance, the WHO now recommends using ACTs for treating uncomplicated vivax malaria worldwide, to both ensure efficacious clearance of blood stage P. vivax infections and to limit the emergence of drug resistance (although chloroquine is also recommended in areas without indication of chloroquine resistance)21. In Cambodia, for example, the standard of care for uncomplicated malaria (caused by any species) was switched from chloroquine to dihydroartemisinin-piperaquine in 2012 and then again to artesunate-mefloquine in 2016 (and was fully implemented in 2017).
We have recently reported that some Cambodian P. vivax parasites were cleared slowly after artesunate treatment22, which may enable them to outlast the short half-life of artesunate in the blood, especially in cases of incomplete patient adherence to the multi-day regimen. While we did not observe treatment failure in this study, this slow clearance upon artesunate treatment could theoretically facilitate the acquisition of resistance to the partner drugs and complicate malaria control and elimination efforts in Southeast Asia. Here, we analyze parasite DNA and RNA extracted from the blood of Cambodian vivax malaria patients and describe a common deletion immediately downstream of the mdr1 protein-coding sequence. We examine its consequences on mdr1 expression and on antimalarial drug susceptibility, as well as analyze 592 DNA samples collected between 2014 and 2024 to examine temporal changes in the frequency of this deletion in Cambodia.
Results
A region immediately downstream of the mdr1 protein-coding sequence is frequently deleted in Cambodian P. vivax isolates
We analyzed the genomes of 206 Cambodian P. vivax isolates collected between 2021 and 2023 and sequenced at high coverage (> 30X, Supplemental Data 1) and screened them for deletions and tandem duplications. In 30 out of the 206 isolates (14.5%), we identified the duplication of the dbp gene (PVP01_0623800) that has been previously reported from Cambodian parasites23. Many other sequence rearrangements occurred only in a few samples, but very few deletions and duplications were shared by many samples: in total, we only detected 26 deletions and 30 duplications present in more than 25% of the samples (Supplemental Data 2). These common sequence rearrangements typically involved genes belonging to multigene families, with 21 out of 26 deletions (81%) and 26 out of 30 duplications (87%) overlapping or neighboring genes annotated as PIR proteins, merozoite surface proteins (MSPs), serine-repeat antigens (SERAs), and tryptophan-rich proteins. The remaining sequence rearrangements involved Plasmodium exported proteins of unknown function, often PHIST proteins located in subtelomeric regions (Supplemental Data 2). The only sequence rearrangement frequently observed among the 206 Cambodian isolates that involved a single copy gene was a deletion located near the multidrug resistance 1 gene (mdr1, PVP01_1010900): 169 out of the 206 Cambodian P. vivax isolates (82%) carried a deletion of approximately 800 bp located immediately after the 3’ end of the mdr1 protein-coding sequence (Fig. 1).

Coverage plots of the mdr1 locus for four P. vivax samples: (top to bottom) a Cambodian isolate in which all parasites carried the non-deleted sequence (green), a Cambodian isolate containing clones with and without the mdr1 deletion (pink), a Cambodian sample in which all parasites contained the deletion (orange), and the NIH-1993-F3 clone derived from Salvador 1 which contains a different mdr1 deletion (blue). For each sample, the upper row displays the overall read coverage relative to the P01 reference sequence, while the lower row displays the coverage of reads flagged for incorrect insert size and indicative of a deletion. The red horizontal bar indicates the region used to determine the number of reads mapped within the deletion, while the horizontal green bar indicates the unaffected region used for comparison. Created in BioRender. Ko, K. (2025) https://BioRender.com/kcykrwm.
The analysis described above is only informative of the presence of a deletion: by only considering read pairs indicative of a deletion, this approach might overestimate the population frequency of the deletion if one sample contains multiple clones, with some carrying the deletion and some without. 93 of the 206 isolates sequenced (45%) were deemed polyclonal, consistent with previous reports in Cambodia (e.g., see refs. 24,25). We therefore evaluated whether some infections might be heterogeneous with regards to this deletion by comparing the sequence coverage in the deleted region with the coverage in a non-deleted section of the coding region of mdr1 (Fig. 1). Of the 169 samples carrying a mdr1 deletion, 146 (86%) were deemed homogenous for the deletion (i.e., all parasites carried the deletion, in orange in Fig. 1) and 23 (14%) heterogenous for the deletion (in pink in Fig. 1).
The mdr1 deletion is present on different haplotypes
We then reconstructed the protein-coding sequence of mdr1 using the genome sequence data generated from monoclonal isolates (n = 113) with or without the deletion. Surprisingly, we did not observe any clear clustering patterns separating samples with the deletion and those without using either the nucleotide sequence or the inferred amino acid sequence (Supplementary Fig. 1). In fact, we observed that the deletion was present on several distinct mdr1 haplotypes that were also present in P. vivax parasites without the deletion (i.e., different sequences carried both the deleted and non-deleted sequences). This lack of linkage disequilibrium between the protein-coding sequence of mdr1 and the downstream deletion suggests that either (i) the deletion occurred independently several times on different genetic backgrounds (with, apparently, the exact same boundaries), or (ii) that this deletion occurred some time ago and that recombination has re-shuffled it on different genetic backgrounds.
Genomic analyses reveal the presence of two independent and overlapping deletions downstream of the mdr1 protein-coding sequence
To evaluate if the mdr1 deletion observed in Cambodian P. vivax isolates was also present in other regions of the world, we re-analyzed 826 P. vivax samples that have been sequenced by the MalariaGEN project26 (Supplemental Data 3). This dataset contains whole genome sequence data for P. vivax parasites collected in 25 countries in Asia, South America, Africa, and Oceania. Overall, we detected seven putative duplications located within 2 kb of the annotated mdr1 gene (Supplementary Fig. 2, Supplementary Data 3), including a previously identified 35 kb duplication containing the entire mdr1 gene13. In addition, we detected read pairs consistent with a deletion downstream of mdr1 (and similar to the one we observed in our Cambodian isolates) in P. vivax isolates from Southeast Asia (Cambodia [122/169], Thailand [36/120], Vietnam [67/100], and Myanmar [3/8]), as well as in two of the 65 isolates from Colombia (Supplementary Data 3).
The identification of a similar deletion in two distant geographic locations raised the question of whether this deletion occurred once and spread from one region to the other or, alternatively, whether it derived from two independent events. To address this question, we examined the precise boundaries of the mdr1 deletion in Southeast Asian and South American samples using whole-genome sequence data from the Cambodian isolates and Colombian samples from MalariaGEN. We also included whole genome sequence data from the NIH-1993-F3 strain of P. vivax (a strain derived from the Salvador 1 strain initially isolated from a patient from El Salvador)27. The Colombian and NIH-1993-F3 samples appeared to carry the same deletion whose boundaries differed from all Cambodian samples and appeared to be smaller (Supplementary Fig. 3). To confirm this observation, we designed PCR primers surrounding the putative deletions (Supplementary Table 1) and amplified and sequenced DNA from Cambodian isolates carrying the deletion as well as from the NIH-1993-F3 clone. These analyses confirmed that the Cambodian P. vivax isolates carried the same 837 bp deletion compared to the P01 reference genome sequence, while NIH-1993-F3 carried a 487 bp deletion (Supplementary Fig. 4). Interestingly, both deletions shared one identical boundary, 25 bp after the 3’ end of the mdr1 coding sequence, while their differing ends occurred at a similar sequence “TGTACA” (Supplementary Fig. 4). The different boundaries of the deleted sequences indicated that these two deletions most likely occurred independently (once in South America and once in Southeast Asia) and the observation of two independent deletions at the same locus suggests that they might have been driven by natural selection to alter an important functional element.
The deletions downstream of mdr1 alter the mRNAs transcribed from the mdr1 gene and their expression level
We next analyzed RNA-seq data generated from 95 Cambodian samples (Supplementary Data 4) to determine if the deletion had any consequence on mdr1 mRNAs. While the deletion was located 25 bp after the end of the coding-sequence of mdr1, it led to a different mRNA being transcribed: in the parasites without the deletion, the 3’ UTR from mdr1 is ~800 bp long, while it is ~1300 bp long in parasites carrying the deletions and the nucleotide sequence is entirely different, except for the 25 bp immediately following the coding sequence (Fig. 2A). To examine if the South American deletion had a similar consequence on mdr1 mRNA, we reanalyzed published RNA-seq data generated from Salvador I (the parental strain of NIH-1993-F3)28 and observed a similar pattern: the DNA deletion led to the transcription of a different isoform of mdr1 with a different 3’ UTR (Fig. 2A).
To determine if the deletion was associated with a change in the level of mdr1 mRNA, we compared samples in which all P. vivax parasites carried the deletion (n = 61) to samples in which all parasites carried the non-deleted sequence (n = 19). After accounting for differences in mRNA length (see Materials and Method for details), we observed that P. vivax parasites carrying the deletion had a significantly greater expression of mdr1 than parasites without the deletion (unpaired t-test, p = 7.568 x 10−11) and displayed a nearly 2-fold increase in mRNA expression (Fig. 2B).

A Integrated Genome Viewer screenshot showing RNA-seq data mapping to the mdr1 locus from the P01 reference genome: the tracks show data from, top to bottom, a Cambodian isolate without the deletion, a Cambodian sample with the deletion, and the Salvador 1 strain. B Comparison of the normalized mdr1 expression between P. vivax isolates in which all clones carried the deletion (in blue, n = 61, min = 213.85, max = 1509.10, centre = 863.62, Q1 = 676.07, Q3 = 967.65, lower extreme=393.93, upper extreme = 1201.22) and isolates in which all clones have the non-deleted sequence (in yellow, n = 19, min = 129.29, max = 604.66, centre = 433.04, Q1 = 367.50, Q3 = 507.79, lower extreme = 300.01, upper extreme=604.66). We performed a two-sample unpaired t-test without adjustments for multiple comparisons (p = 7.568 × 10−11). The boxplot lines represent (from top to bottom) the first, second, and third quartiles, while the whiskers show the values within 1.5 times the interquartile range. Source data are provided as a Source Data file.
The mdr1 3’UTR deletion is possibly associated with differences in antimalarial drug susceptibility
Since MDR1 has been associated with antimalarial drug resistance in P. vivax12 and P. falciparum14,15,16,29, we then assessed whether the mdr1 deletion was associated with reduced antimalarial susceptibility. First, we tested if the deletion was statistically associated with differences in parasite clearance upon artesunate treatment using estimates of the slope half-life measured directly from the patients studied here22. Of note, 11 of the 167 infections (6.6%) analyzed showed a slope half-life greater than 5 hrs. We compared 138 infections where all parasites carried the deletion, to 29 infections where all parasites carried the non-deleted sequences but did not observe any differences in slope half-life (p = 0.2311, Fig. 3A). Next, we genotyped the deletion in 14 P. vivax infections for which mefloquine susceptibility was measured ex vivo. Interestingly, parasites carrying the deletion (n = 9) seemed to have a greater IC50 than parasites without the deletion (n = 5, p = 0.0496, Fig. 3B). Similarly, analyses conducted using IC90 estimates showed the same trend but did not reach statistical significance (n = 14, Supplementary Fig. 5).

A Distributions of slope half-life following in vivo artesunate treatment of patients infected with parasites with deletion (n = 138 infections) and without deletion (n = 29 infections). We performed two-sided unpaired t-tests without adjustments for multiple comparisons (p = 0.2311). Source data are provided as a Source Data file. B Distributions of IC50 of parasites with deletion (n = 9 infections) and without deletion (n = 5 infections) when exposed to mefloquine ex vivo. We performed two-sided unpaired t-tests without adjustments for multiple comparisons (p = 0.0496). Source data are provided as a Source Data file.
The mdr1 deletion increased in frequency among Cambodian P. vivax parasites since the introduction of mefloquine in ACTs
Mefloquine has been used in ACT as the standard-of-care in Cambodia for treating uncomplicated vivax malaria since 2016–201730. To determine whether the change in the frontline antimalarial drug was correlated with a change in the frequency of the mdr1 deletion in the Cambodian P. vivax population, we screened a total of 592 P. vivax isolates collected in Cambodia between 2014 and 2024. Overall, we observed a statistically significant increase in the proportion of isolates carrying the deletion over time (chi square test for trend, p = 9 × 10−13, Fig. 4), with ~30% of the parasites carrying the deletion before the introduction of mefloquine as standard-of-care compared to >60% carrying the deletion today (with most changes occurring between 2016 and 2018 and the proportion of isolates with the deletion remaining relatively stable afterwards). Note that this pattern remained significant even if only parasites from eastern Cambodia are analyzed (chi square test for trend, p = 0.0001, Supplementary Fig. 6).

The bars represent the proportion of P. vivax isolates collected in a given year that carry the deletion. The number of samples analyzed per year is indicated above each bar. The red arrow indicates the change in national treatment guidelines from DHA-PPQ to As-MQ. We observed a statistically significant increase in the proportion of P. vivax samples with the deletion over time (chi-square test for trend, p = 9×10−13). Source data are provided as a Source Data file. Created in BioRender. Ko, K. (2025) https://BioRender.com/wu4uobx.
Discussion
The multidrug resistance 1 gene in Plasmodium encodes an ATP-binding cassette transporter that is located on the membrane of the digestive vacuole where it regulates the flux of solutes through the membrane31. In P. falciparum, mdr1 has been shown to modulate susceptibility to various antimalarial drugs through coding polymorphisms altering the amino acid sequences and copy number variations14,15,16,29. Similarly in P. vivax, several studies have suggested that increased copy numbers of mdr1 reduces antimalarial drug susceptibility12,13. We showed here that mdr1 is unusually affected by deletions and duplications in P. vivax. While we observed many sequence rearrangements among Cambodian P. vivax isolates, almost all of them occurred in large multigene families and the only common sequence rearrangement affecting a single copy protein-coding gene in 206 Cambodian isolates occurred immediately downstream of mdr1. This uniqueness of mdr1 was further illustrated by a reanalysis of 592 P. vivax isolates from four continents that confirmed the presence of this deletion in Southeast Asian isolates but also revealed multiple, rarer, tandem duplications affecting this locus. Importantly, these analyses also revealed that a second deletion, overlapping but shorter, was present in some South American P. vivax isolates. The observation of two independent deletions affecting the same locus suggested that these deletions have functional consequences and may have been driven by positive selection.
In contrast to previous reports of mdr1 polymorphisms, this common deletion observed in Cambodian isolates (as well as the one present in South American parasites) did not affect the coding region of mdr1 but only modified its downstream DNA sequence. In addition, it was found on different genetic backgrounds and was linked to different mdr1 amino acid sequences. RNA sequencing analyses confirmed that the deletion altered the gene expression of mdr1 and produced transcripts with different 3’UTRs. While incompletely characterized in Plasmodium, 3’UTRs often play a critical role in mRNA stability in eukaryotes by binding RNAs and/or proteins that regulate mRNA stability and degradation32. Indeed, we showed that the presence of the deletion was associated with a two-fold increase in mdr1 mRNA levels, suggesting that the deleted region, or the newly appended 3’ UTR sequence, contains important elements for mRNA regulation. Additional laboratory studies will be necessary to fully understand the molecular mechanisms responsible for these changes in expression and evaluate the consequences of these variable 3’ UTR sequences on transcription or mRNA stability and decay, as well as their impact on mdr1 protein levels.
Similarly to the effect of increased mdr1 copy number in P. falciparum, and to a lesser extent in P. vivax, we showed that the deletion downstream of mdr1 was associated with a higher IC50 to mefloquine. While this association is compelling, it is important to note that it is based on a small number of isolates analyzed and relies on ex vivo assessment of drug susceptibility. It will therefore be important to confirm this association in a much larger cohort. In addition, it is worth noting that a lesser drug susceptibility (i.e., a higher IC50) is not to be directly equated with drug resistance and, as far as we know, there is no evidence of treatment failure in Cambodia. Nonetheless, it is puzzling that retrospective analyses of isolates collected in Cambodia since 2014 indicated that the frequency of the deletion increased after the introduction of mefloquine in ACT for uncomplicated vivax malaria in 2016–2018. Interestingly, we observed that the deletion was already present (although at lower frequency) in the oldest samples screened here, before the introduction of mefloquine in the standard-of-care for vivax malaria treatment. This observation, that is consistent with the lack of linkage disequilibrium between the deletion and the neighboring protein-coding sequence, could indicate that the deletion had been previously maintained in the P. vivax population, possibly due to “collateral” exposure of P. vivax parasites to mefloquine that has been extensively used against P. falciparum in Cambodia since 198333,34, or to advantages the mdr1 deletion could confer against other antimalarial drugs29,35,36,37. Additional studies, for example testing ex vivo the susceptibility of closely-related parasites transfected with various combinations of mdr1 coding sequences with or without the UTR deletion38 will be necessary to provide a better understanding of the relative contribution of (and possible interactions between) the mdr1 protein sequence, its gene expression and antimalarial drug susceptibility.
Our observations, combined with the recent report of delayed clearance upon artemisinin treatment22, could suggest that P. vivax parasites are becoming less susceptible to Artesunate-Mefloquine therapy in Cambodia. These results mirror the patterns that were observed for P. falciparum (also in Cambodia) in the last decades that led to the emergence and spread of parasites carrying multiple resistance alleles and threatened malaria control efforts in the Greater Mekong region and worldwide. While we did not observe treatment failure associated with these putative multidrug resistance phenotypes in our Cambodian patient cohort, these findings are worrying and beg for intensified monitoring and control efforts to ensure that P. vivax parasites remain susceptible to current antimalarials and to limit the spread of potentially resistant parasites that could threaten ongoing efforts to eliminate vivax malaria from the Greater Mekong region.
Methods
Sample collection
Parasite DNA and RNA samples were collected from participants in an open-label clinical trial designed to evaluate the efficacy of two regimens of primaquine39. Eligible individuals seeking treatment for malaria in the Kampong Speu province (Western Cambodia) and diagnosed with Plasmodium vivax by rapid diagnostic test were offered to participate in the study. All participants had uncomplicated vivax malaria (confirmed by PCR), were 15 years or older, and had hemoglobin concentration greater than 8 g/dL. Pregnant and breastfeeding women were excluded from this study. At enrollment (i.e., before drug treatment), we collected a venous blood sample from each participant and immediately stored 50 µL in trizol for RNA analyses and processed the rest of the blood on a cellulose column to remove white blood cells before storage at −80 °C for DNA analyses.
We also analyzed a biobank of DNA from 592 P. vivax infected individuals from the Ratanakiri, Mondolkiri and Kampong Speu provinces and collected between 2014 and 2024.
Whole genome sequencing and identification of sequence rearrangements in Cambodian isolates
We extracted parasite DNA from 230 leukocyte-depleted blood samples using the DNeasy blood and tissue kit (Qiagen) kit and prepared whole genome sequencing libraries using the NEBNext® Ultra™ II FS DNA Library Prep Kit for Illumina sequencing (NEB) according to the manufacturers’ instructions. We sequenced each library on an Illumina NovaSeq 6000 to generate 23–50 million paired-end reads of 100 bp per sample (Supplementary Data 1).
To identify sequence rearrangements throughout the P. vivax genome, we first used Hisat240 to map all reads to the P01 reference genome (version 67)41. We then used custom scripts to parse the resulting bam files and counted, for each sample, the number of read pairs with alignments indicative of deletions or tandem duplications per 100 bp non-overlapping window of the P01 genome13,42. Briefly, read pairs aligning in the correct orientation but distant from each other by at least 1000 bp were considered indicative of deletions (SAM flags 161/97 with insert size > = 1000 bp and SAM flags 81/145 with insert size < = −1000 bp), while read pairs aligning in incorrect orientation (tail to tail) and distant from each other by at least 1000 bp were considered indicative of tandem duplications (SAM flags 161/97 with insert size < = −1000 bp and SAM flags 81/145 with insert size > = 1000 bp). To determine if a given 100 bp window contained more reads indicative of a specific sequence rearrangement than expected by chance, we compared the number of such reads observed in one sample to the number expected under a Poisson distribution. All windows with a false discovery rate < 0.01 were considered significant. Note that this analytical pipeline enables detection of a sequence rearrangement even if only some of the clones present in one sample carry it.
We then evaluated, in samples where a mdr1 deletion was detected, whether all or only some of the clones within one sample carried the mdr1 deletion by comparing the number of reads mapped to a 400 bp portion within the mdr1 deletion (between positions 478,100 and 478,500 of the P01 genome version 67) to the number of reads mapped to a 400 bp portion of the mdr1 coding region that was not deleted or duplicated (between positions 480,000 and 480,400, Fig. 1). If the proportion of reads mapped to the deleted region represented less than 5% of the reads mapped to the control region, the sample was considered homogeneous for the deletion, while all other isolates were considered heterogeneous for the deletion (i.e., some clones carried the deletion and some clones carried the non-deleted sequence).
Determination of the clonality of the Cambodian isolates
To estimate whether each isolate was monoclonal, we first used GATK43 to call nucleotide variants from the whole genome sequence data. We masked regions of the genome containing multigene families and only considered nucleotide positions successfully sequenced at > 20X in at least 80% of samples and polymorphic in our dataset (only considering positions with two alleles). Overall, 195,699 nucleotides positions were deemed variable and used to determine the clonality of each infection with the R package moimix44. Samples with Fws > = 0.95 were considered monoclonal while samples with Fws < 0.95 were considered polyclonal.
Nucleotide and amino acid sequence analyses
For each Cambodian P. vivax isolate deemed to be monoclonal, we used bcftools mpileup and custom scripts to reconstruct the nucleotide sequence of the mdr1 protein-coding sequence using the gene coordinates from PlasmoDB. Any nucleotide positions sequenced at less than 20X was called as “N”, while all others positions were called using the most frequent allele (to account for possible sequencing errors and remaining, low-level, polyclonality). We then aligned these DNA sequences in MEGA1245 using MUSCLE45 and generated Neighbor-Joining trees based on the number of differences between sequences, from both the nucleotide and translated amino acid sequences.
Characterization of mdr1 sequence rearrangements in MalariaGEN samples
In order to characterize sequence rearrangements involving mdr1 in a worldwide dataset, we analyzed 826 MalariaGEN PV4 P. vivax isolates from 25 countries in Africa, Asia, Oceania and America26. We remapped all sequences generated from these samples onto a 60 kb region surrounding the mdr1 locus in the P01 reference genome (between positions 450,000 and 510,000 of chromosome 10) and screened these samples for deletions and tandem duplications by analyzing read pair orientation using the pipeline outlined above.
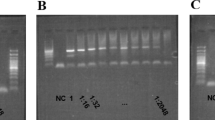
Validation of mdr1 deletions by PCR amplification and Sanger sequencing
To confirm the mdr1 deletions identified by whole genome analysis, we designed different sets of PCR primers to amplify the region surrounding the putative deletions downstream of mdr1 (Supplementary Table 1). We amplified DNA from three Cambodian samples identified as homogenous for the deletion, as well as from NIH-1993-F3, a monkey-adapted P. vivax strain derived from the Salvador 1 strain that, based on sequence data previously generated27, carries a deletion downstream of mdr1 similar to those observed in South-American isolates from MalariaGEN. PCR amplification was performed using the Q5 high-fidelity master mix with an annealing temperature of 67 °C and 35 cycles. The PCR products were loaded on a 1% agarose gel for 30 min and bands with the expected size were cut out, purified, and the DNA used for Sanger sequencing. The resulting DNA sequences were aligned to the P01 reference sequence to validate the deleted regions and their exact boundaries.
RNA sequencing and gene expression analysis
We extracted RNA from 138 whole blood samples collected from the same infections as used in the whole genome sequencing analysis. Briefly, we used phenol/chloroform to extract RNA from trizol samples and, after ribosomal RNA depletion and polyA selection (NEB), we prepared RNA-seq libraries using the NEBNext Ultra II Directional RNA Library Prep Kit (NEB). We sequenced each library on an Illumina NovaSeq 6000 and generated ~30–414 million paired-end reads of 75 bp per sample. We aligned all reads using Hisat240 to the P. vivax P01 genome with default parameters except for a maximal intron length set at 5000 bp. PCR duplicates were removed from all files using custom scripts. Out of the 138 samples, 128 had more than 500,000 unique reads mapped to P. vivax protein-coding genes and were analyzed further (Supplementary Data 4).
We tested whether isolates carrying a mdr1 deletion expressed mdr1 mRNA at a different level than isolates without deletion. To account for the transcript length difference caused by the deletion, we only counted reads mapped to the coding region of mdr1 (that is unaffected by the deletion) and normalized this count by the total number of reads mapped to the P. vivax genome for each sample. We then used a t-test to compare the normalized mdr1 expression of samples identified in our WGS analysis as being homogeneous for the deletion (n = 61) with those homogeneous for no deletion (n = 19).
Association between mdr1 deletion and antimalarial drug susceptibility
We first compared the susceptibility to artesunate of P. vivax isolates carrying the mdr1 deletion (n = 138) and isolates without the deletion (n = 29) based on genome sequence data and on PCR characterization of the presence/absence of the deletion. Artesunate susceptibility was expressed as the half-life of the parasite clearance slope determined in vivo from patients treated with artesunate22,39. We also characterized by PCR the absence/presence of the mdr1 deletion (Supplementary Table 1) in 14 P. vivax isolates for which the IC50 to mefloquine was determined ex vivo and tested whether these estimates were significantly associated with the deletion. Ex vivo mefloquine susceptibility assay was performed on freshly collected parasites using a ring-to-schizont maturation assay as previously described30. Briefly, P. vivax infected blood was collected in lithium heparin tubes and stored in a water bath at 37 °C until processing (within 2 h). The blood pellet was separated from plasma and passed through a NWF filter for leukocyte depletion. The RBC pellet was resuspended at 5% hematocrit and incubated in 96-well plates in supplemented IMDM based medium in presence of serial dilutions of mefloquine. Batches of mefloquine plates were validated using the P. falciparum 3D7 strain. Incubation was stopped when a majority of parasites reached schizont stage (> 4 nuclei visible) in the drug-free control (24–42 h depending on initial parasite stage). Each well was used to prepare a thick smear and was stained with Giemsa 5%. Schizonts were counted and normalized to the total number of parasites observed. Schizont counts were normalized to the drug-free controls and IC50s were calculated using a four-parameter logistic regression (Graphpad Prism).
Temporal changes in mdr1 deletion in Cambodian P. vivax
To measure changes in frequency of the mdr1 deletion in the P. vivax Cambodian population over time, we used the PCR assay described above to screen genomic DNA isolated from 592 P. vivax isolates that were collected by the Institut Pasteur of Cambodia in the provinces of Ratanakiri, Mondolkiri, and Kampong Speu between 2014 and 2024.
Ethics statement
Venous blood samples were collected from patients enrolled in an open-label clinical trial (NCT04706130). Ethical clearance was obtained by the National Ethics Committee for Health Research of Cambodia (158-NECHR, 06/29/2020) and the University of Maryland IRB (HP-00091095), and the study was overseen by the NIH Division of Microbiology and Infectious Diseases (Protocol 20-0010). All enrolled patients or their guardians provided written informed consent, and assent was obtained for all patients aged 15–18 years old.
Biobank DNA samples were collected from P. vivax infected individuals from the Ratanakiri, Mondolkiri and Kampong Speu provinces and approved by the National Ethics Committee for Health Research of the Ministry of Health of Cambodia (#364-NECHR, #038-NECHR, #197-NECHR, and #111-NECHR). All participants or their guardians provided informed consent.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All sequence data generated in this study have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive database under the accession codes PRJNA1310200 and PRJNA1305352. Source data are provided with this paper.
Code availability
Custom scripts are available at https://github.com/ko-katie/pvmdr1 and Zenodo (https://doi.org/10.5281/zenodo.17805214).
References
WHO. World Malaria Report 2024. (World Health Organization, Geneva, Switzerland, 2024).
Chu, C. S. & White, N. J. The prevention and treatment of Plasmodium vivax malaria. PLoS Med 18, e1003561 (2021).
Price, R. N., Auburn, S., Marfurt, J. & Cheng, Q. Phenotypic and genotypic characterisation of drug-resistant Plasmodium vivax. Trends Parasitol. 28, 522–529 (2012).
Popovici, J. et al. Frequent and complex relapses confound assessment of chloroquine resistance in Cambodian Plasmodium vivax. Int. J. Infect. Dis. 73, 75 (2018).
Buyon, L. E., Elsworth, B. & Duraisingh, M. T. The molecular basis of antimalarial drug resistance in Plasmodium vivax. Int J. Parasitol. Drugs Drug Resist 16, 23–37 (2021).
Nomura, T. et al. Evidence for different mechanisms of chloroquine resistance in 2 Plasmodium species that cause human malaria. J. Infect. Dis. 183, 1653–1661 (2001).
Sá, J. M. et al. Expression and function of pvcrt-o, a Plasmodium vivax ortholog of pfcrt, in Plasmodium falciparum and Dictyostelium discoideum. Mol. Biochemical Parasitol. 150, 219–228 (2006).
Sa, J. M. et al. Plasmodium vivax chloroquine resistance links to pvcrt transcription in a genetic cross. Nat. Commun. 10, 4300 (2019).
Suwanarusk, R. et al. Chloroquine resistant Plasmodium vivax: in vitro characterisation and association with molecular polymorphisms. PLoS One 2, e1089 (2007).
Zeng, W. et al. Molecular Surveillance and Ex Vivo Drug Susceptibilities of Plasmodium vivax Isolates From the China-Myanmar Border. Front Cell Infect. Microbiol 11, 738075 (2021).
Sá, J. M. et al. Plasmodium vivax: allele variants of the mdr1 gene do not associate with chloroquine resistance among isolates from Brazil, Papua, and monkey-adapted strains. Exp. Parasitol. 109, 256–259 (2005).
Suwanarusk, R. et al. Amplification of PVMDR1 associated with multidrug-resistant Plasmodium vivax. J. Infect. Dis. 198, 1558–1564 (2008).
Auburn, S. et al. Genomic Analysis Reveals a Common Breakpoint in Amplifications of the Plasmodium vivax Multidrug Resistance 1 Locus in Thailand. J. Infect. Dis. 214, 1235–1242 (2016).
Sidhu, A. B. et al. Decreasing pfmdr1 copy number in plasmodium falciparum malaria heightens susceptibility to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin. J. Infect. Dis. 194, 528–535 (2006).
Calçada, C. et al. Expansion of a Specific Plasmodium falciparum PfMDR1 Haplotype in Southeast Asia with Increased Substrate Transport. mBio 11, https://doi.org/10.1128/mBio.02093-20 (2020).
Price, R. N. et al. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet 364, 438–447 (2004).
Baird, J. K. et al. Resistance to chloroquine by Plasmodium vivax in Irian Jaya, Indonesia. Am. J. Tropical Med. Hyg. 44, 547–552 (1991).
Rieckmann, K. H., Davis, D. R. & Hutton, D. C. Plasmodium vivax resistance to chloroquine?. Lancet 2, 1183–1184 (1989).
Baird, J. K. Resistance to chloroquine unhinges vivax malaria therapeutics. Antimicrobial agents Chemother. 55, 1827–1830 (2011).
Price, R. N. et al. Global extent of chloroquine-resistant Plasmodium vivax: a systematic review and meta-analysis. Lancet Infect. Dis. 14, 982–991 (2014).
World Health Organization. WHO Guidelines for malaria. 462 (2024).
Tebben, K. et al. Cambodian Plasmodium vivax parasites with reduced hemoglobin digestion display delayed clearance upon artesunate treatment. medRxiv https://doi.org/10.1101/2025.03.09.25323469 (2025).
Hostetler, J. B. et al. Independent Origin and Global Distribution of Distinct Plasmodium vivax Duffy Binding Protein Gene Duplications. PLoS Negl. Trop. Dis. 10, e0005091 (2016).
Friedrich, L. R. et al. Complexity of Infection and Genetic Diversity in Cambodian Plasmodium vivax. PLoS Negl. Trop. Dis. 10, e0004526 (2016).
Popovici, J. et al. Genomic Analyses Reveal the Common Occurrence and Complexity of Plasmodium vivax Relapses in Cambodia. mBio 9, https://doi.org/10.1128/mBio.01888-17 (2018).
Adam, I. et al. An open dataset of Plasmodium vivax genome variation in 1,895 worldwide samples. Wellcome Open Res 7, 136 (2022).
Hazzard, B. et al. Single-cell analyses of polyclonal Plasmodium vivax infections and their consequences on parasite transmission. Nat. Commun. 15, 7625 (2024).
Carlton, J. M. et al. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature 455, 757–763 (2008).
Veiga, M. I. et al. Globally prevalent PfMDR1 mutations modulate Plasmodium falciparum susceptibility to artemisinin-based combination therapies. Nat. Commun. 7, 11553 (2016).
Roesch, C. et al. Impact of the first-line treatment shift from dihydroartemisinin/piperaquine to artesunate/mefloquine on Plasmodium vivax drug susceptibility in Cambodia. J. Antimicrob. Chemother. 75, 1766–1771 (2020).
Valderramos, S. G. & Fidock, D. A. Transporters involved in resistance to antimalarial drugs. Trends Pharm. Sci. 27, 594–601 (2006).
Mayr, C. What Are 3’ UTRs Doing? Cold Spring Harb. Perspect. Biol. 11, https://doi.org/10.1101/cshperspect.a034728 (2019).
Wongsrichanalai, C. & Meshnick, S. R. Declining artesunate-mefloquine efficacy against falciparum malaria on the Cambodia-Thailand border. Emerg. Infect. Dis. 14, 716–719 (2008).
Wongsrichanalai, C., Prajakwong, S., Meshnick, S. R., Shanks, G. D. & Thimasarn, K. Mefloquine–its 20 years in the Thai Malaria Control Program. Southeast Asian J. Trop. Med Public Health 35, 300–308 (2004).
Duraisingh, M. T. & Cowman, A. F. Contribution of the pfmdr1 gene to antimalarial drug-resistance. Acta Trop. 94, 181–190 (2005).
RTS,S/AS01 partially successful in preventing malaria in children. Hum. Vacc. Immunother. 11, 1298 (2015).
Khim, N. et al. Effects of mefloquine use on Plasmodium vivax multidrug resistance. Emerg. Infect. Dis. 20, 1637–1644 (2014).
Verzier, L. H., Coyle, R., Singh, S., Sanderson, T. & Rayner, J. C. Plasmodium knowlesi as a model system for characterising Plasmodium vivax drug resistance candidate genes. PLoS Negl. Trop. Dis. 13, e0007470 (2019).
Eng, V. et al. 14 days of high-dose versus low-dose primaquine treatment in patients with Plasmodium vivax infection in Cambodia: a randomised, single-centre, open-label efficacy study. Lancet Infect. Dis. https://doi.org/10.1016/S1473-3099(25)00033-7 (2025).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Auburn, S. et al. A new Plasmodium vivax reference sequence with improved assembly of the subtelomeres reveals an abundance of pir genes. Wellcome Open Res 1, 4 (2016).
Menard, D. et al. Whole genome sequencing of field isolates reveals a common duplication of the Duffy binding protein gene in Malagasy Plasmodium vivax strains. PLoS Negl. Trop. Dis. 7, e2489 (2013).
Auwera, G. V. D. & O’Connor, B. D. Genomics in the cloud: using Docker, GATK, and WDL in Terra. First edition. edn, (O’Reilly Media, 2020).
Lee, S. B., M. moimix: an R package for assessing clonality in high-througput sequencing data. (2016).
Kumar, S. et al. MEGA12: Molecular Evolutionary Genetic Analysis Version 12 for Adaptive and Green Computing. Mol. Biol. Evol. 41, https://doi.org/10.1093/molbev/msae263 (2024).
Acknowledgements
We thank all patients and health care workers involved in this study and the staff of the Malaria Research Unit at the Institut Pasteur of Cambodia and the staff of the National Center for Parasitology, Entomology and Malaria Control in Cambodia for their collaboration and sample collection. We also thank S. Ott, H. Bowen, L. Sadzewicz, and L. Tallon in Maryland Genomics at the University of Maryland School of Medicine for their support with Illumina sequencing. This study was supported by the NIH (R01AI146590 to DS), the NIH (R01AI173171, R01AI175134 and R61AI187100 to JP), the Pasteur International Unit PvESMEE (J.P.), the Division of Intramural Research of NIH (JMS and TEW), and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation – 549182383 to J.G.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization, J.P. and D.S.; Methodology, D.L., J.P. and D.S.; Investigation, K.K., K.T., T.A., A.O., J.G., V.E., R.E., N.K., L.B.F., J.S., and J.M.S.; Writing – Original Draft, K.K., J.P., and D.S.; Writing – Review & Editing, K.K., K.T., T.E.W., J.P., and D.S.; Funding Acquisition, T.E.W., J.P., and D.S.; Supervision, J.P. and D.S.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Sarah Auburn, who co-reviewed with Hidayat Trimarsanto; and Prashant Mallick for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ko, K., Tebben, K., Andrianinarivomanana, T. et al. A common DNA deletion altering the 3’UTR of mdr1 is associated with reduced mefloquine susceptibility in P. vivax parasites from Cambodian patients. Nat Commun 17, 1748 (2026). https://doi.org/10.1038/s41467-026-68456-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68456-7
