
Abstract
Neural crest cells (NCC) are a migratory progenitor cell population unique to vertebrates that contribute to nearly every organ system throughout the body. Disruptions in NCC development can result in congenital disorders (neurocristopathies). Yet, our understanding of the cellular mechanisms and signals that govern mammalian NCC formation remains poor. Here, we discovered nuclear receptor superfamily 6 group member 1 (NR6A1/GCNF/RTR) is a novel, critical regulator of mammalian NCC specification, formation and survival. Nr6a1 expression in mouse embryos spatiotemporally overlaps with early NCC development. NR6A1 loss-of-function perturbs anterior cranial NCC formation and survival, with complete agenesis of migratory NCC caudal to the first pharyngeal arch. Using targeted molecular and genomic approaches, we demonstrate that these phenotypes are associated with perturbation of NCC specification and epithelial-mesenchymal transition, and with persistent expression of pluripotency-associated factors. Supporting these observations, in vivo overexpression of Oct4 in gastrulating mouse embryos disrupts NCC specification and formation. Conditional temporal deletion revealed that Nr6a1 is required during mid-late gastrulation, demonstrating that the initiation of murine NCC specification likely occurs during gastrulation - earlier than previously thought, but in close alignment with the established timeline of NCC development in other vertebrate model organisms. These findings also reveal that the gold standard transgenic mouse line, Wnt1-Cre, is unsuitable for studying genetic function during NCC specification and formation. In summary, NR6A1 is essential for mammalian NCC development and may function during gastrulation as a bimodal switch modulating pluripotency-associated factors in the neuroepithelium, while concomitantly activating NCC specifiers and regulators of EMT.
Similar content being viewed by others


Introduction
Neural crest cells (NCC) are a heterogeneous migratory cell population. With varying degrees of potential, NCC contribute to nearly every tissue and organ system throughout the body and are therefore essential for proper development. During early embryogenesis, NCC are specified at the neural plate border, undergo an epithelial-mesenchymal transition (EMT), delaminate from the neuroepithelium and then migrate throughout the embryo. Depending on their axial level, NCC differentiate into cartilage, bone and connective tissues in the head and neck, neurons and glia of the peripheral nervous system, smooth muscle and cardiomyocytes in the heart, endocrine cells in many organs, and pigment cells in the skin, among many others1. Imbued with considerable inherent plasticity, NCC have served as a conduit for intraspecies and interspecies variation, and the evolution of anatomical novelties - particularly in the head and face2,3,4,5. However, perturbations in NCC development are responsible for a diverse class of congenital anomalies known as neurocristopathies6,7,8. Uncovering the mechanisms by which NCC regulate vertebrate development and evolution, and devising therapeutic approaches for preventing congenital neurocristopathies, requires a comprehensive understanding of the signals, switches and gene regulatory networks that govern each stage of NCC development.
Decades of research have elucidated many of the tissue interactions and signals that drive NCC specification and formation. NCC are induced at the neural plate border through contact-mediated interactions between the neural ectoderm, surface ectoderm and paraxial mesoderm9,10,11,12,13,14,15,16,17,18. Signals from these surrounding tissues include BMP, FGF and WNT which influence the expression of neural plate border genes such as, Tfap2a, Zic1, Msx1 and Pax 3/711,12,16,17,19,20,21,22,23,24,25,26,27,28,29,30. These inductive signals together with the neural plate border factors upregulate NCC-specifier genes Foxd3, Sox9, and Sox10 and EMT genes Snai1/2, Zeb2 and Twist1 to facilitate NCC specification, formation and delamination17,18,23,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46. However, many of these factors and their functions, which were uncovered in non-mammalian model organisms, appear dispensable in mouse47,48,49. For example, BMP4 and BMP7 function as critical inductive signals that regulate NCC specification in chicken, but when BMP4 and BMP7 are knocked out in mouse, either individually or in combination, NCC still form16,50,51,52,53,54,55. Similarly, BMP2 is also not required for NCC induction in the mouse, although it is important for NCC migration56. Neural plate border genes Pax3 and Pax7 are also expendable for NCC formation in mouse as is evident from single and double knockouts57,58,59. Finally, NCC still form, delaminate and migrate in mouse embryos even when EMT master regulators Snai1, Snai2, Zeb2 or Twist1 are knocked out60,61,62,63. These discrepancies between mammalian and non-mammalian species are surprising given the fundamental importance of NCC in vertebrate development, but may be attributable to developmental heterochronies, and differences in species-specific experimental approaches48. Thus, the gene regulatory network underpinning mammalian NCC formation has been difficult to define and remains poorly understood.
Previously, we uncovered nuclear receptor subfamily 6 group A member 1 (Nr6a1) as transcriptionally downregulated in a mouse model of the craniofacial neurocristopathy, Treacher Collins syndrome64, suggesting this nuclear receptor plays an unknown role in NCC development. While the greater nuclear receptor superfamily consists of 70 receptors organized into 7 subclasses, NR6A1 is in a subclass on its own. Nuclear receptors like NR6A1 contain an N-terminal domain, a DNA-binding domain and a ligand binding domain65,66,67. However, the ligand that interacts with NR6A1 is unknown and therefore NR6A1 is considered an orphan nuclear receptor. First isolated from testes and heart tissue, NR6A1 was originally named germ cell nuclear factor (GCNF) and retinoid receptor-related testis-specific receptor (RTR)68,69. Dynamically expressed during folliculogenesis and spermatogenesis, as well as in the embryo throughout pre- and post-implantation, Nr6a1 is critical for mouse embryo development and survival70,71,72,73. NR6A1 loss-of-function in mouse results in an open neural tube, axial truncation, cardiac defects, and failure of chorioallantoic fusion, which leads to embryonic lethality by embryonic day (E) 10.573.
In this work we investigate the role of Nr6a1 in NCC development—determining this orphan nuclear receptor is a novel pivotal regulator of murine NCC specification, formation and survival. We demonstrate that Nr6a1 is spatiotemporally expressed in the neuroepithelium and newly emigrating NCC indicating that Nr6a1 regulates NCC formation and survival. Indeed, through loss-of-function analyses in mice we show that Nr6a1 null embryos exhibit a deficiency in anterior cranial NCC, and complete agenesis of migrating NCC caudal to the first pharyngeal arch. These phenotypes are associated with downregulation of NCC specifiers (Foxd3, Sox9, Sox10) and EMT master regulators (Snail1, Zeb2), in concert with expansion of the neural stem cell marker Sox2, increased proliferation, and persistent expression of pluripotency-associated factors (Oct4 and Nanog) in the neuroepithelium. Using mouse embryonic stem cells (mESC), and human induced pluripotent stem cells (hiPSC) differentiated into NCC, as well as chromatin immunoprecipitation and multiomic approaches, we demonstrate that NR6A1 modulates the chromatin landscape and directly binds to putative DR0 motifs in the promoter regions of NCC, EMT and pluripotency-associated factors to regulate their expression. Furthermore, global temporal and conditional spatiotemporal deletion reveal that Nr6a1 is specifically required during mid-late gastrulation, suggesting that NCC specification in mouse embryos commences earlier than previously recognized. Consistent with this model, in vivo overexpression of Oct4 in gastrulating mouse embryos disrupts NCC specification and formation. Altogether, our work has uncovered NR6A1 as a novel regulator of mammalian NCC specification and formation. NR6A1 may therefore function as a bimodal switch that modulates the repression of pluripotency factors associated with neural stem cell maintenance and proliferation, while being required to activate a gene regulatory network of NCC specifiers and regulators of EMT.
Results
Spatiotemporal distribution of Nr6a1 mirrors NCC development
Treacher Collins syndrome is a neurocristopathy caused primarily by variants in the TCOF1 gene74,75,76. Characterized by downward slanting of the palpebral fissures, hypoplasia of the zygomatic complex, micrognathia and cleft palate74,75, previous studies of mouse and zebrafish models of Treacher Collins syndrome determined these characteristic craniofacial anomalies to be caused by a deficiency in NCC. Tcof1+/− haploinsufficiency results in diminished rRNA synthesis and ribosome biogenesis, as well as DNA damage, leading to increased p53-dependent apoptosis, which compromises the formation, proliferation and survival of NCC64,76,77,78,79,80. Transcriptomic analyses of E8.5 Tcof1+/− and control littermate embryos revealed numerous gene expression changes in association with the molecular and cellular pathogenesis of Treacher Collins syndrome64. We hypothesized that downregulated genes in this mouse neurocristopathy model might also be important for normal NCC development.
Interestingly, Nr6a1, which has been shown to regulate the transition of primitive neural stem cells to definitive neural stem cells81, was decreased two-fold in Tcof1+/− embryos compared to controls64. We therefore posited that Nr6a1 may also play a critical role in NCC development. Nr6a1 expression can be induced in embryonic stem cells at E4.5 and P19 cells when treated with retinoic acid and its expression pattern inversely correlates with Oct482,83. Furthermore, previous studies reported strong expression of Nr6a1 throughout the embryo at E7.5 before being restricted to the neural ectoderm and posterior of the embryo (E8.0-8.5)73.
To better characterize the spatiotemporal expression of Nr6a1 during NCC development, we performed a detailed temporal analysis by in situ hybridization on CD1 mouse embryos from E6.5 to 9.5 (Fig. 1). Nr6a1 expression is absent at E6.5, consistent with the known high levels of Oct4 present in the embryo at this stage (Fig. 1A). Nr6a1 expression commences between E6.5 and E7.5 and is observed broadly throughout the embryo (Fig. 1B). At E8.0, Nr6a1 is expressed in the neural plate and in the trunk progenitors required for axial elongation (Fig. 1C,D). Nr6a1 expression then becomes more pronounced along the entire neuraxis from E8.5-8.75 (Fig. 1E,F). Transverse histological sections of stained E8.5-E8.75 embryos showed Nr6a1 is dynamically expressed in the dorsal most region of the neuroepithelium, along the basal edge and in cells directly outside the neuroepithelium consistent with a pattern of newly delaminated NCC (Fig. 1E, arrows). At E9.5, Nr6a1 expression is maintained throughout the neuraxis, but with an absence in the posterior growth zone (Fig. 1F), consistent with the trunk to tail transition as we have previously reported84. Furthermore, we also previously showed that Nr6a1 expression becomes restricted to the somites at E10.5 before being downregulated by E12.584. Our expression analyses, therefore, indicated that Nr6a1 is expressed at the right time and in the right locations to potentially function in the specification, formation and early migration of NCC.

In situ hybridization of Nr6a1 (purple stain) in CD1 embryos from E6.5-9.5. A At E6.5, Nr6a1 is not expressed within the embryo or surrounding extraembryonic membranes. B At E7.5 Nr6a1 is expressed throughout the embryo proper. C At E8.0, Nr6a1 is predominantly expressed in the posterior of the embryo and throughout the forming neural folds. D Nr6a1 is more tissue restricted at E8.5 where it continues to be expressed in the posterior of the embryo and throughout the neuroepithelium. E A transverse section through the neural plate and first pharyngeal arch of an E8.75 embryo shows Nr6a1 is expressed along the basal edge and in the dorsal region of the neuroepithelium as well as in cells just outside of the neuroepithelium fanning out ventrally where NCC would be located. F At E9.5, Nr6a1 is expressed throughout the dorsal region of the embryo but is absent from the most distal tail bud region, matching previously published expression patterns84. Scale bar is 200 µm. A minimum of 5 embryos were assayed for each developmental stage.
NCC formation, delamination and survival require Nr6a1
Nr6a1 null mutant mice are embryonic lethal by E10.5 due to cardiac defects and a failure of chorioallantoic fusion73. Nr6a1−/− embryos also exhibit significant defects in axial elongation and tail bud formation73,84,85. To determine if Nr6a1 is required for NCC development, we evaluated the expression of NCC-specifier genes Foxd3, Sox9 and Sox10 in E8.75 Nr6a1−/− embryos compared to stage-matched control littermates, prior to embryonic lethality, using in situ hybridization chain reaction (HCR). Foxd3 is one of the earliest markers of NCC specification and it maintains the potency of NCC86 while also regulating cadherin turnover to facilitate delamination by EMT39. Compared to wildtype littermate controls, Foxd3 expression was significantly reduced in the Nr6a1 null embryos with only a minor cluster of cells expressing Foxd3 in the head (Fig. 2A,B, paired ratio t-test p = 0.0339).

A HCR of NCC and EMT marker genes (yellow) in Nr6a1 null and heterozygous stage-matched control embryos at E8.75. Foxd3, Sox9, Sox10, and Snai1 expression appear reduced in the Nr6a1 null embryo compared to controls. Twist1 is still expressed in expressed in migrating NCC. B Quantification and statistical analysis of the relative expression of each gene in Nr6a1 null and stage-matched controls based on the total fluorescence of the gene normalized to the total fluorescence of DAPI in each embryo. The expression of Foxd3, Sox9, Sox10 and Snai1 was down in Nr6a1 null embryos compared to controls consistent with the staining shown in the representative images in (A). Ratio paired t-test (two-tailed) determined the expression of Foxd3, Sox9, Sox10 and Snai1 were statistically significant while Twist1 was not. A total of 4 embryos across two litters was used in this quantification of each condition. C Traditional in situ hybridization of Zeb2 (purple stain) on an E8.75 Nr6a1 null embryo compared to a stage-matched control. Zeb2 is expressed throughout the cranial region and in the dorsal neural plate in the control embryos at E8.75. However, Zeb2 expression is restricted to the neural plate in the Nr6a1 null and is not found in the frontonasal region or arches suggesting its expression is also downregulated. D Beta-galactosidase staining (blue) of Nr6a1−/−;Wnt1-Cre;R26R-LacZ and littermate Wnt1-Cre;R26R-LacZ embryos at E9.5. Nr6a1 null embryos lack expression of LacZ caudal to the first pharyngeal arch indicating the absence of migrating NCC. E eYFP expression (green) in Nr6a1−/−;Wnt1-Cre;R26R-eYFP and littermate Wnt1-Cre;R26R-eYFP embryos at E9.0 illustrates the absence of NCC caudal to the first pharyngeal arch and fewer migrating NCC in the cranial region consistent with the LacZ stained embryos. Transverse histological sections through the first pharyngeal arch (PA1) in both embryos shows a reduction in migratory NCC in the null embryos. No NCC are present in the second arch (PA2) in the null embryo. A minimum of 4 embryos were assayed for each in situ hybridization marker and lineage tracing experiment.
Sox9 is also required for NCC formation and delamination and is expressed in pre-migratory and early migratory NCC, as well as later during their chondrogenic differentiation42,87,88. We could detect only a few cells expressing Sox9 in the craniofacial mesenchyme, with no labeled cells in the frontonasal prominences in Nr6a1 null embryos (Fig. 2A,B; paired ratio t-test p = 0.0190). Similarly, Sox10, a marker of migrating NCC40,43, was significantly reduced in Nr6a1−/− embryos with a complete absence of labeled cells in the first pharyngeal arch (Fig. 2A,B; paired ratio t-test p = 0.0300).
We next evaluated whether the expression of NCC EMT master regulators Snai1, Zeb2 and Twist1 was also disrupted in Nr6a1−/− embryos. Snai1 is typically expressed in neuroepithelial cells during NCC formation, and its expression persists in delaminating and migrating NCC31,32,36,89. However, Snai1 expression was reduced in Nr6a1−/− embryos, with an absence of Snai1 labeled cells in the first pharyngeal arch compared to the control stage-matched littermate (Fig. 2A,B; paired ratio t-test p = 0.0129). Zeb2 is also expressed in the neuroepithelium and migratory NCC37,63. Unfortunately, we were unable to detect Zeb2 by HCR or SABER-FISH, and therefore, instead performed traditional in situ hybridization to investigate the dynamics of Zeb2 expression. Although Zeb2 was expressed in the neural plate in Nr6a1−/− embryos, it was absent from the NCC derived facial and pharyngeal mesenchyme (Fig. 2C). Twist1, like Snai1 and Zeb2, is considered a master regulator of EMT. Interestingly, Nr6a1−/− embryos exhibit Twist1 expressing cells in the frontonasal prominences and pharyngeal arches (Fig. 2A,B), but previous studies have shown that Twist1 is not required for NCC formation and instead regulates NCC migration and differentiation during mouse embryo development60,90,91. Altogether, our gene expression analysis suggested Nr6a1 is required for the expression of NCC-specifier and EMT genes during development.
Gene expression is not necessarily an indicator of lineage, but the perturbations in NCC-specifier and EMT gene expression implied that Nr6a1 null embryos might be deficient in NCC formation and delamination. However, the expression of Twist1 suggested there were some mesenchymal cells present in the frontonasal prominences and pharyngeal arches. Therefore, to more conclusively assess the presence or absence of NCC in Nr6a1 null embryos, we performed lineage tracing by crossing the Wnt1-Cre transgene92 into the background of Nr6a1−/− embryos. Wnt1-Cre is expressed in the dorsal neuroepithelium encompassing the territory in which neural crest cells are formed. In agreement with this idea, E9.0-9.5 Nr6a1−/−;Wnt1-Cre;R26R-LacZ embryos completely lacked migrating NCC caudal to the first pharyngeal arch (Fig. 2D), but the presence of LacZ-positive cells within the frontonasal region and first pharyngeal arch in Nr6a1−/− embryos suggests some cells are still capable of delaminating and migrating from the midbrain and anterior hindbrain neuroepithelium (Fig. 2D). We observed identical cranial and trunk patterns using an eYFP reporter instead of LacZ (Fig. 2E). E9.0-9.5 Nr6a1−/−;Wnt1-Cre;R26R-eYFP embryos exhibited fewer migrating cranial NCC and complete absence of migrating NCC caudal to the first pharyngeal arch. Transverse histological sections of E9.0-9.5 Nr6a1−/−;Wnt1-Cre;R26R-eYFP embryos further illustrate the diminishment of eYFP-labeled putative NCC within the first pharyngeal arch and their complete absence from the second pharyngeal arch compared to controls (Fig. 2E). This is consistent with the expression of NCC-specifier and EMT genes as visualized by HCR (Fig. 2A), which had predicted Nr6a1−/− mutant embryos would have very few NCC in the head.
Given the expression of Twist1 in Nr6a1 null embryos, these eYFP-positive and LacZ-positive cells are likely to be mesenchymal (Fig. 2A). However, the downregulated expression of NCC-specifier and other EMT genes, and reduced number of labeled cells, raised questions about their long-term potential. TUNEL staining of E8.75 Nr6a1−/−;Wnt1-Cre;R26R-eYFP revealed visibly higher levels of cell death in Nr6a1 mutants compared to Wnt1-Cre;R26R-eYFP littermate controls (Supplemental Fig. 1). Given the Nr6a1 null embryos do not survive beyond E10.5, we wanted to confirm the cell death was specific to the NCC. We therefore counted the number of TUNEL-positive cranial NCC and non-NCC in E8.75 Nr6a1−/−;Wnt1-Cre;R26R-eYFP and control littermates well before embryonic lethality. Quantifying the percentage of cells in each population undergoing apoptosis confirmed a higher percentage of NCC undergoing apoptosis in the Nr6a1−/−;Wnt1-Cre;R26R-eYFP compared to controls (Supplemental Fig. 1B; one-way ANOVA, p = 0.0105; unpaired parametric t-test p = 0.0017). Furthermore, no statistical difference was observed in the level of apoptosis in non-NCC tissue in Nr6a1−/−;Wnt1-Cre;R26R-eYFP embryos compared to control littermates, thus revealing the cell death evident in Nr6a1−/− mutant embryos was NCC-specific. Altogether, these results indicate Nr6a1 is required for murine NCC formation and survival.
The neural stem cell to NCC transition requires Nr6a1
Nr6a1 is predominantly expressed in the neural ectoderm during early NCC development and is required for neural tube closure73. This raised the question of whether the deficiency in NCC formation and migration in Nr6a1 null embryos was a secondary consequence of abnormal specification and patterning of the neural ectoderm. Sox2 is a pan-neural marker of progenitor cells in the neural ectoderm and is sufficient to maintain their proliferation and stemness properties93,94. Through in situ hybridization, we observed that Sox2 is expressed in the open neural plate in Nr6a1 null embryos (Fig. 3A), indicating that pan-neural specification of the neural plate occurs in Nr6a1 mutants. Wnt1 and Pax3 are also expressed within the neural ectoderm, but specifically in dorsal domains that help establish dorsoventral patterning11,12. Their spatiotemporal patterns of expression remain unchanged in Nr6a1 null embryos compared to controls indicating that proper dorsal pattering is also established in the neural plate of Nr6a1 mutants.

A In situ hybridization of neuroepithelial markers (purple stain) in null and wildtype littermate control embryos at E9.0. Sox2, Wnt1 and Pax3 are all expressed in the Nr6a1 null embryos, indicating the neural ectoderm has formed and patterned. B Immunostaining of SOX2 (green), PAX3 (green) and pHH3 (red) on transverse histological sections at E9.0. SOX2 and PAX3 are expressed in the neural plate in both null and control embryos. SOX2 expression is expanded to dorsal regions of the neural plate. PAX3 expression is maintained in the Nr6a1 null, although no PAX3 positive migratory cells could be observed further demonstrating the NCC deficiency. More pHH3 staining can be seen in the neural plate of the Nr6a1 null embryos suggesting the neural ectoderm is maintained in a highly proliferative state. A minimum of 5 embryos were assayed for each in situ hybridization marker and cell proliferation assay.
Immunostaining of transverse sections confirmed SOX2 and PAX3 were expressed in the neural ectoderm of Nr6a1−/− embryos but also illustrated the increased thickness of the neural plate compared to controls (Fig. 3B). SOX2 is a key regulator of proliferating neural progenitor cells throughout the entire anterior-posterior axis of the central nervous system from neural plate stages of embryogenesis through to adulthood95. PAX3 also functions to maintain the neuroepithelium in a proliferative, undifferentiated state, allowing neurulation to proceed96. We therefore hypothesized that the thickened neuroepithelium in the Nr6a1 mutants was due to increased proliferation. Indeed, phospho-histone H3 (pHH3) immunostaining of transverse histological sections confirmed that Nr6a1−/− embryos exhibit considerably higher proliferation than littermate controls (Fig. 3B).
Additionally, we also observed that the expression of SOX2 persisted in the dorsolateral border region of Nr6a1−/− embryos compared to controls (Fig. 3B). Downregulation of Sox2 in the dorsolateral border of the neural ectoderm is essential for neural stem cells to differentiate into NCC and occurs as part of a SoxB (Sox1, Sox2) to SoxE (Sox9, Sox10) switch93,97. Furthermore, although the dorsal domain of PAX3 in the neural ectoderm appeared unchanged in Nr6a1 mutants, PAX3 expression was not observed outside of the neuroepithelium, consistent with the deficiency of migratory NCC in Nr6a1 mutants compared to littermate controls (Fig. 3B).
Thus, NR6A1 loss-of-function does not result in gross mis-patterning of the pan-neural or dorsal character of the neural ectoderm. Collectively, these results indicate the neural ectoderm forms and is appropriately patterned in Nr6a1 null embryos. However, the persistence of SOX2 expression in the dorsolateral border region together with elevated proliferation, suggests that the neural ectoderm in the Nr6a1 null mutants is maintained in a proliferative stem cell state which may restrict neural stem cells from differentiating into NCC.
To better understand how NR6A1 regulates NCC formation and survival, we initially performed microarray based transcriptomic analyses of E9.0 wildtype and Nr6a1 null mutant embryos. Consistent with our HCR results (Fig. 2A), the expression of NCC-specifier genes Foxd3, Sox9, and Sox10 was downregulated in Nr6a1 null embryos, together with EMT master regulators Snai1, Zeb2 and Twist1 (Table 1). Although the downregulation of these factors in the microarray did not reach statistical significance, this was likely due to the entire embryo being sampled, of which the NCC are but a relatively small portion at this stage of development. Therefore, we also performed bulk RNA-sequencing on only the anterior portion of the embryo, and again we observed considerable downregulation of NCC-specifier and EMT master regulator gene expression in Nr6a1−/− embryos compared to littermate controls (Table 1). In contrast, we found that pluripotency associated factors such as Oct4 and Nanog, were significantly upregulated in Nr6a1−/− embryos compared to littermate controls through each of the transcriptomic approaches (Table 1).
OCT4 and NANOG, together with SOX2, play essential coordinated roles in preserving the pluripotency and self-renewal capacity of ESCs and adult stem cells98,99,100. Interestingly, previous studies have shown that Nr6a1 directly represses Oct4 and Nanog during peri-implantation development82,83,101. To test this model in the context of NCC development, we performed a time course analysis of OCT4 expression using the Oct4-EGFP reporter mouse line102. Oct4-EGFP is strongly expressed and clearly observed in the epiblast at E7.5 but is then downregulated by E8.5 (Fig. 4A). We then crossed Oct4-EGFP into the background of Nr6a1 null embryos and observed persistent ectopic Oct4-EGFP expression throughout the neural ectoderm in E8.5 Nr6a1−/−;Oct4-EGFP embryos compared to control littermates (Fig. 4B-C). Although GFP is an indirect readout of OCT4 activity, persistent ectopic Oct4 expression has previously been reported in Nr6a1−/− embryos83. Collectively, these results indicate that OCT4 is typically downregulated in the neural ectoderm in association with NCC formation.

A Oct4-EGFP expression (green) at E7.5 (n = 5) and E8.5 (n = 5). Oct4 is highly expressed in the embryo proper at E7.5 according to high GFP expression but is downregulated by E8.5 as evidenced by the absence of GFP expression. B Oct4-EGFP and SOX10 expression in Nr6a1 null and control littermates. Oct4 is exogenously expressed in the null compared to control embryos which do not display Oct4 (GFP) expression. Notably, the pattern of SOX10 in the null suggests Oct4 is not expressed in the NCC that have managed to form. C Quantification and statistical analysis of Oct4-EGFP expression within NCC and neuroepithelial cells (NE) in Nr6a1 null (n = 3) and control littermates (n = 4) depicted by a boxplot. One-way ANOVA determined a statistical significance of <0.0001 across the cell populations. No statistical significance was found by unpaired parametric (two-tailed) t-test when comparing the control NCC and Nr6a1 null populations (difference between means is −5.692 + /- SEM 14.75) or the control NCC and control NE populations (difference between means is 15.62 + /- SEM 11.96). The Nr6a1 null embryos had a statistically significant increase in Oct4-EGFP expression in the NE cells compared to the NCC of the null that were SOX10+ (p = 0.0033, difference between means is 151.1 + /- SEM 24.14)) and compared to control NE cells (p = 0.0012, difference between means is 129.8 + /- SEM 19.77). This data demonstrates the maintenance of Oct4 expression in the Nr6a1 null and suggests that the SOX10+ cells in the null do not have Oct4 expression, consistent with the requirement for Oct4 downregulation in NCC formation. The bounds of each sample set are as follows: Control NCC (min=142.777, max=179.966, mean=165.8), Nr6a1 −/− NCC (min=138.352, max=184.154, mean=160.1), Control NE (min=168.913, max=206.145, mean=181.4), and Nr6a1 −/− NE (min=272.514, max=340.373, mean=311.2).
In further support of this idea, the expression of SOX10 did not align with the regions of persistent ectopic Oct4-EGFP activity, suggesting that Oct4 was downregulated in the few NCC that do form in Nr6a1−/− embryos (Fig. 4B-C). The region of persistent ectopic Oct4 expression notably aligns with the location where NCC formation is perturbed along the neuraxis. The persistent expression of pluripotency associated genes such as Sox2, Oct4 and Nanog, therefore suggest that the neural ectoderm in Nr6a1 null mutants is maintained in a proliferative stem cell state which restricts neural stem cells from differentiating into NCC in the head, and especially caudal to the first pharyngeal arch. Thus, Nr6a1 may be required for the transition of neural ectoderm or neural stem cells to NCC by repressing pluripotency and stem cell maintenance genes while concomitantly being required for the activation and expression of NCC specification and EMT genes.
NR6A1 modulates stem cell maintenance and NCC specification
NR6A1 is an orphan member of the nuclear receptor superfamily and shares the highest homology with COUP-TF, which functions as both a transcriptional repressor and activator68,101. NR6A1 binds to a double repeat of the consensus sequence AGGTCA with zero spacing (DR0) motif, to transcriptionally regulate target genes such as Oct4 and Nanog68,85,103. The downregulation of NCC specification and EMT genes in Nr6a1−/− embryos suggests NR6A1 could function by activating these genes during development in parallel with modulating the repression of pluripotency-associated genes. We therefore searched for this putative binding site or motif in NCC-specification and EMT genes to determine the potential for NR6A1 to directly regulate these genes. Sox9, Sox10, Snai1 and Zeb2 each possess DR0 binding sites in their promoter regions, which is consistent with downregulation of their expression in Nr6a1 null embryos.
To further understand how NR6A1 regulates NCC-specifier and EMT genes within the neural ectoderm during NCC development, we performed single cell multiomic analyses of E8.75 Nr6a1 null mutant and control littermate embryos. Combining single cell RNA-sequencing (scRNA-seq) with single nucleus transposase-accessible chromatin sequencing (snATAC-seq) allows for the assessment of differential gene expression and chromatin accessibility between cell populations in the Nr6a1 null and control littermate embryos. The scRNA-seq results from the controls was used to annotate the cell populations (Supplemental Fig. 2A-B). The neural plate border cells were identified based on high differential expression of Wnt1, Pax3/7 and Zic1104,105,106 (Supplemental Fig. 2A-B). NCC were annotated based on high differential expression of the NCC-specifier genes Foxd3, Sox9, Sox10, Crabp1, Snai1 and Zeb247,106,107,108 (Supplemental Fig. 2A-B). The neural ectoderm was identified by high differential expression of Sox2, Pax2, Otx2 and Sox3104,106,109 (Supplemental Fig. 2A-B). Finally, neural progenitors were annotated based on high expression of Olig2, Sox2 and Pax694,106,110,111 (Supplemental Fig. 2A-B). The same transcriptional signatures were used to identify the same specific cell populations within the Nr6a1−/− embryos.
Comparison of the Nr6a1−/− embryo scRNA-seq dataset to the control littermate scRNA-seq dataset revealed the NCC population was decreased by greater than 50% in the null, comprising 4% of the total cell population in the controls but only 1.4% in the Nr6a1−/− embryos (Supplemental Fig. 3A-C). The greater than 50% loss of NCC aligned with the results obtained from Nr6a1−/−;Wnt1-Cre;R26R-eYFP lineage tracing (Fig. 2E). Furthermore, the few cells that clustered as NCC in the mutant embryos exhibited a significant decrease in Sox10 expression, while in contrast the neural plate border (NPB) cells exhibited increased Oct4 and Nanog expression compared to control littermates, consistent with our previous microarray, bulk RNA-seq, HCR, immunostaining and transgenic reporter mouse analyses (Table 1).
Next, we evaluated changes in chromatin accessibility between Nr6a1−/− mutant embryos and control littermates (Supplemental Fig. 4). It is well established that NR6A1 directly binds to and represses Oct4. Therefore, we first evaluated the chromatin accessibility of Oct4 in the NCC and ectodermal populations in Nr6a1−/− mutant embryos compared to control littermates (Fig. 5A). NCC, neural ectoderm and neural plate border cells from Nr6a1−/− embryos exhibited increased chromatin accessibility compared to controls, consistent with NR6A1 loss-of-function preventing the repression of Oct4 (Fig. 5A). Furthermore, peaks illustrating the increased accessibility of open chromatin upstream of Oct4, aligned with the locations of putative DR0 bindings sites, demonstrating mechanistically how NR6A1 represses Oct4 expression (Fig. 5A).

For both (A and B), the snATAC-seq fragment density across each gene is shown alongside a violin plot displaying gene expression within each cell population. A map of the gene is shown below the fragment density to provide locational reference. Peak coverage is displayed below the gene and the ranges below the peak coverage label Nr6a1 DR0 binding sites along the genome. A Oct4 expression is increased in mutant cell populations compared to wild type as displayed by the violin plot. Consistent with the increased gene expression in the mutant cell populations, the fragment density if higher along Oct4 indicating the chromatin is more accessible along the gene. Finally, peak coverage overlaps with footprints of DR0 sites which aligns with the requirement of NR6A1 to downregulate Oct4 expression. B Sox10 expression is decreased in the mutant NCC population compared to wild type cells as displayed by the violin plot and matches our previous data showing Sox10 expression is downregulated in the null. Note that this is normalized to account for fewer NCC in the mutant. When comparing the density of fragments observed along Sox10 in the snATAC-seq data, there is a reduction in density in the null compared to control NCC. Further, peak coverage overlaps with DR0 bindings sites. Collectively, the data shows there is a reduction in chromatin accessibility along Sox10 in the null NCC compared to wild type which translates to decreased expression. It is possible this is due to a role of NR6A1 as a regulator of Sox10 expression according to the overlap in peak coverage and DR0 binding sites. Due to the loss of NR6A1 function, NR6A1 cannot bind to the DR0 sites along Sox10 in the null thus the chromatin remains inaccessible for transcription.
We next evaluated chromatin accessibility around Sox10, which is significantly decreased in Nr6a1−/− embryos (Fig. 5B). If NR6A1 regulates the transcription of Sox10, then we would expect Sox10 to be less accessible or no longer accessible in Nr6a1 null embryos. Indeed, Nr6a1−/− embryo NCC exhibited decreased chromatin accessibility at Sox10 compared to controls, consistent with NR6A1 being required for access and transcription of Sox10 (Fig. 5B). The DR0 sites overlapped with the Sox10 peaks as well, supporting the idea that NR6A1 binds to and regulates the transcription of Sox10 (Fig. 5B).
snATAC-seq demonstrated that NR6A1 can modulate the accessibility of pluripotency-associated and NCC-specifier genes like Oct4 and Sox10 respectively within NCC (Fig. 5). To validate NR6A1’s ability to directly bind to the DR0 binding sites we performed electrophoretic mobility shift assays (EMSA) and competitive inhibition assays. EMSA revealed the presence of shifted Zeb2 and Sox10 bands following the addition of NR6A1, indicating NR6A1 can directly bind to the DR0 sites in their promoter regions (Fig. 6A). For the competitive inhibition assay, wildtype and mutated DR0 site DNA were introduced in increasing amounts (Fig. 6B). We observed diminished binding in the presence of increasing wildtype DNA compared to mutated DR0 site DNA, supporting the ability of NR6A1 to selectively bind to the DR0 sites of Zeb2 and Sox10 (Fig. 6B).

A The ability for NR6A1 to bind to predicted sites of regulation on Zeb2 and Sox10 was analyzed by EMSA. When NR6A1 was present, a super-shifted band can be seen, representing positive interactions between NR6A1 and the predicted DR0 sequence. B Unlabeled wildtype sequences or sequences with mutated NR6A1 binding sites for Zeb2 and Sox10 were added in a dose-dependent manner resulting in reduced binding to labelled sequences in wildtype and minimal effect with mutant sequence. C A targeted ChIP assay (n = 3 biological replicates) was conducted in mESCs differentiated to NCC using antibodies against endogenous NR6A1 and binding was assessed by qPCR of predicted binding sites. NR6A1 binds to EMT factors, NCC-specifiers, and pluripotency factors in vivo with statistical significance as found by unpaired parametric (two-tailed) t-test for Zeb2 (p = 0.011; difference between means is 1.318 + /- SEM 0.2899), Snai1 (p = 0.01; difference between means is 1.327 + /- SEM 0.3017), Sox10 (p = 0.007; difference between means is 1.515 + /- SEM 0.2981) and Nanog (p = 0.016; difference between means is 1.346 + /- SEM 0.3363) when compared to control IgG. The bounds of each sample set are as follows: Control Zeb2-IgG (min=0.032664, max=0.24551, mean=0.1530), Zeb2-Nr6a1 (min=1.135916, max=2.033347, mean=1.471), Snai1-IgG (min=0.03831, max=0.24551, mean=0.1654), Snai1-Nr6a1 (min=1.074642, max=2.061731, mean=1.493), Snai2-IgG (min=0.257716389, max=0.312917921, mean=0.2798), Snai2-Nr6a1 (min=0.825795344, max=2.503343367, mean=1.521), Sox9-IgG (min=0.020247059, max=0.215215843, mean=0.1368), Sox9-Nr6a1 (min=0.885065535, max=6.163954403, mean=2.731), Sox10-IgG (min=0.012903052, max=0.012903052, mean=0.1319) Sox10-Nr6a1 (min=1.286860964, max=2.225078431, mean=1.647), Nanog-IgG (min=0.018631094, max=0.240457893, mean=0.1533), Nanog-Nr6a1 (min=1.030865555, max=1.030865555, mean=1.499).
To confirm NR6A1 binding occurs in vivo and to evaluate all of our NCC and EMT genes of interest, we performed a targeted ChIP assay using mESC that were differentiated into NCC. NR6A1 exhibited statistically significant binding of the DR0 sites in Zeb2, Snai1, and Sox10 (Fig. 6C). We also confirmed that NR6A1 can bind Nanog DR0 sites, which had previously been shown82, further supporting our results (Fig. 6C).
In conclusion, NR6A1 can modulate chromatin accessibility and bind to the DR0 sites found in the promoter regions of pluripotency-associated factors and NCC-specifier and EMT genes. NR6A1 transcriptionally represses pluripotency associated factors, but at the same time, downregulation of NCC-specifier and EMT genes in Nr6a1 null embryos indicates NR6A1 may function as a transcriptional regulator of NCC-specifier and EMT genes. Collectively these results indicate that NR6A1 governs the transition of neural stem cells to NCC by modulating the repression of stem cell maintenance and proliferation in the neural ectoderm, while concomitantly regulating the expression of NCC-specifier and EMT genes within the neural plate border and newly emigrating NCC.
NCC specification begins at mid-late gastrulation
Nr6a1 null embryos are embryonic lethal by E10.5 due to the failure of chorioallantoic fusion73. This early lethality prevents the assessment of a role for Nr6a1 in later NCC differentiation at all axial levels. However, given the important role of Nr6a1 in NCC formation, we posited that Nr6a1 should therefore be essential for craniofacial, cardiac, peripheral nervous system, and gastrointestinal development, as each of these tissues and organs depend heavily on NCC for their proper development. To bypass the early embryonic lethality of Nr6a1 null embryos, we conditionally deleted Nr6a1 from NCC, using Wnt1-Cre. Surprisingly, no phenotypic abnormalities were found in Nr6a1fl/fl;Wnt1-Cre embryos compared to controls (Supplemental Fig. 5). E18.5 Nr6a1fl/fl;Wnt1-Cre embryos appeared morphologically normal, and the NCC-derived craniofacial skeleton (evaluated by alizarin red and alcian blue staining), peripheral nervous system and enteric nervous system (evaluated by TUJ1 immunostaining) were indistinguishable from controls. Furthermore, the NCC-specific Nr6a1 knockouts were produced in Mendelian ratios and survived until adulthood with full reproductive capabilities. This indicated that NCC development was unaffected (Supplemental Fig. 5), which is in stark contrast to all of our other data clearly showing that Nr6a1 is required for NCC specification, formation and survival.
Recently, however, it was proposed that Wnt1-Cre excision occurs too late during neurulation to properly assess gene function during NCC specification and formation in mouse embryos48. Wnt1-Cre facilitates Cre excision of floxed alleles at approximately E8.5. This is important because it suggests that Nr6a1 may not be required for NCC development after E8.5. Furthermore, it also implies that Nr6a1 may instead be needed earlier in development to elicit its role in NCC specification and formation.
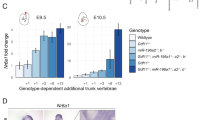
To test this idea, and establish when Nr6a1 is specifically required, we globally deleted Nr6a1 in a temporally specific manner using the tamoxifen inducible Cre line, ERT2-Cre112. Consistent with the Wnt1-Cre knockout, no morphological differences were observed when Nr6a1 was temporally excised with ERT2-Cre at E8.5. We then excised Nr6a1 via tamoxifen administration at progressively earlier half-day time points. Global excision at E7.75 or E7.25 resulted in no observable phenotypic differences in NCC gene expression in conditional Nr6a1 mutants compared to controls (Fig. 7). However, we discovered that global excision of Nr6a1 by E6.75 resulted in a phenotype matching the original Nr6a1 null embryos as evidenced by the downregulated expression of NCC-specifier and EMT genes (Fig. 7). This is consistent with our observations that Nr6a1 expression begins between E6.5 and E7.5 (Fig. 1). Thus, not only is Nr6a1 functionally required during mid-gastrulation but our data indicates that NCC specification commences during mid-gastrulation in mouse embryogenesis, which is considerably earlier than previously appreciated.

In situ hybridization of NCC markers (purple stain) on control (Nr6a1fl/fl) littermates and Nr6a1fl/fl;ERT2-Cre embryos treated with tamoxifen for excision of Nr6a1 at E7.25 and E6.75. Embryos with Nr6a1 excision by E7.25 and littermate controls show normal expression patterns of NCC and EMT markers. Once Nr6a1 is excised by E6.75, NCC and EMT marker expression is lost. Note that some images in this figure were reflected to match the overall orientation of all other images in the figure. A minimum of 3 control and 3 mutant embryos were assayed for each in situ hybridization marker.
NCC formation requires stem cell maintenance downregulation
Interestingly, it was recently proposed that NCC retain or re-activate pluripotency-associated genes to facilitate their characteristic capacity to generate a wide variety of cell and tissue derivatives113,114,115,116,117,118. However, our analyses of Nr6a1−/− mutant embryos indicate that the expression of pluripotency-associated factors including Oct4, Nanog and Sox2, persists in Nr6a1−/− mutant embryos, coinciding with deficient NCC generation. This suggests that retention of pluripotency-associated factors may actually restrict the generation of NCC from neural stem cells. Consistent with this idea, our analyses of wildtype Oct4-EGFP mouse embryos revealed that Oct4-EGFP expression in E7.5 mouse embryos is downregulated by E8.5, coinciding with the formation of NCC (Fig. 4A). In further support of these findings, our multiomic analyses revealed a similar absence of Oct4 and Nanog expression in E8.75 wildtype mouse embryos (Supplemental Fig. 2B).
To better substantiate whether Oct4 and other pluripotency and stem cell maintenance factors are downregulated in concert with NCC formation, we analyzed our scRNA-seq data of cranial tissues from pre-migratory and migratory NCC stage mouse embryos76,106,119. Sox2, Sox1 and Nr6a1 are highly expressed in the neural ectoderm, in contrast to Oct4 and Nanog (Fig. 8A, Supplemental Fig. 6). Furthermore, the expression of each of these genes is downregulated in the progenitor NCC population (Fig. 8A, Supplemental Fig. 6). In contrast, NCC-specification (Foxd3, Sox9, Sox10) and EMT (Snai1, Zeb2) genes are highly enriched in progenitor NCC as compared to the neural ectoderm, distinguishing these two populations (Fig. 8A, Supplemental Fig. 6). Other groups have reported a similar absence of pluripotency-associated factor expression during NCC development aligning with our data120. Therefore, to expand and further substantiate our assessment of the dynamics of Oct4, Nanog and Sox2 expression during development, we also analyzed a publicly available scRNA-seq dataset that covered gastrulation to neurulation stages in mouse embryos121. Complementary to our data, Oct4, Nanog and Sox2 were absent from the progenitor NCC cluster, and a definitive boundary was evident in the pseudotime trajectory separating the primitive neuroectoderm from progenitor NCC (Supplemental Fig. 7). Nr6a1 expression in this dataset aligned with our in situ hybridization data confirming that Nr6a1 is expressed in the epiblast during gastrulation, before becoming enriched in the neural ectoderm during neurulation (Supplemental Fig. 7).

A scRNA-seq of cranial tissue from Wnt1-Cre;R26R-eYFP and Mef2c-F10n-LacZ embryos shows a transcriptional signature isolating pluripotency-associated gene expression to neural ectodermal (NE) cells compared to NCC. Any expression of Oct4 and Nanog is restricted to a very small population of NE cells (see dot size for percent expressed). B In situ hybridization of NCC-specifier and EMT genes (purple stain) in Oct4/rtTA embryos treated with doxycycline (+Dox) as compared to untreated embryos (-Dox). Consistent with the Nr6a1 null embryos, NCC and EMT marker expression is no longer present through the cranial mesenchyme of the embryos, indicating a disruption in NCC formation. Note that some images in this figure were reflected to match the overall orientation of all other images in the figure. A minimum of 3 control and 3 Oct4 overexpressing embryos were assayed for each in situ hybridization marker.
To further evaluate pluripotency and stem cell maintenance factor expression during NCC formation, we measured by RT-qPCR, the dynamics of Oct4, Nanog, Sox2, Sox9 and Sox10 expression during mESC and hiPSC derivation of NCC (Supplemental Fig. 8A and C). Each approach revealed the absence of pluripotency-associated factor expression in the forming NCC populations, and this was strikingly exemplified by their immediate downregulation in hiPSC upon provision of NCC differentiation media (Supplemental Fig. 8A and C). Interestingly, Nr6a1 was also specifically elevated (days 1 and 2) in hiPSC during the switch between pluripotent stem cell maintenance, and NCC specification (Supplemental Fig. 8C). Immunostaining further substantiated the downregulation in expression of pluripotency-associated factors and upregulation of NCC-specifier gene expression (Supplemental Fig. 8B and D).
To conclusively show that downregulation of pluripotency and stem cell maintenance regulators is required for NCC formation, we overexpressed Oct4 in mouse embryos from E6.5 to E8.5 (during NCC specification and formation) using a doxycycline inducible system122 (Fig. 8B). The temporal initiation of Oct4 overexpression was designed to correlate with the temporal requirement for Nr6a1. Embryos overexpressing Oct4 presented with neural tube closure defects and craniofacial abnormalities, mimicking the Nr6a1 null phenotype. Moreover, the expression of NCC-specifier (Foxd3, Sox9, Sox10) and EMT (Zeb2, Snai1) genes in these Oct4 overexpressing embryos was downregulated, similar to that in Nr6a1 null mutant embryos (Fig. 8B). Downregulation of pluripotency and stem cell maintenance factor expression is therefore required for proper NCC formation.
Altogether, our data suggests that NR6A1 is a key regulator of the transition of neural stem cells to NCC by modulating the repression of pluripotency-associated genes while promoting the expression of NCC-specification and EMT genes during mid-late gastrulation.
Discussion
Despite decades of research, we have a poor understanding of the mechanisms, signals and gene regulatory networks that regulate mammalian NCC formation. In fact, to date, very few factors essential for mammalian NCC formation have been identified. Here, we demonstrate that NR6A1 is essential for NCC formation as a novel regulator of the transition of neural stem cells to NCC. Nr6a1 is dynamically expressed in the neural ectoderm and newly emigrating NCC, and NR6A1 loss-of-function results in a deficiency of anterior cranial NCC, and an absence of migrating NCC caudal to the first pharyngeal arch. This cellular phenotype occurs in association with the persistent expression of pluripotency and stem cell maintenance factors (Sox2, Oct4, Nanog), and the downregulated expression of NCC specifier (Foxd3, Sox9, Sox10) and EMT (Snai1, Zeb2) genes. Analysis of NR6A1 binding to DR0 sites in the promoter regions of pluripotency associated genes and NCC-specifier and EMT genes correlated with changes in chromatin accessibility, suggesting that NR6A1 directly regulates the activity of these factors. Thus, we propose that NR6A1 regulates murine NCC formation by modulating the repression of pluripotency and stem cell maintenance gene expression, while concomitantly inducing NCC specification and EMT genes, which collectively facilitates the transition of neural stem cells to NCC.
Our evaluation of Nr6a1 in NCC development through tissue-specific and temporally restricted excision allowed us to determine when Nr6a1 was precisely required to elicit its functions. Although Nr6a1 null mutant embryos exhibit clear defects in NCC development, conditional excision of Nr6a1, specifically in pre-migratory NCC using the gold standard Wnt1-Cre transgenic line, did not result in an observable phenotype. This was surprising, but it has been posited that Wnt1-Cre excision occurs too late to assess gene function during NCC specification and formation in mouse embryos48. Our work provides some of the first evidence in accord with this idea, since global temporal deletion revealed that Nr6a1 is required around E6.75-E7.0 to replicate the original Nr6a1 null mutant phenotype.
Interestingly, the onset of NCC specification was classically considered to occur during neurulation, after establishment of the three germ layers and subsequent subdivision of the ectoderm into neural ectoderm and non-neural ectoderm15. However, recent studies have suggested that NCC induction and specification begin earlier, during, or even preceding gastrulation9,113,123,124,125. Similar to these studies in avian and frog embryos9,125, our results imply that murine NCC specification also begins during mid-gastrulation. In support of this model and the temporal role of Nr6a1, it’s important to note that Nr6a1 has also been proposed to regulate the transition from primitive to definitive neural stem cells in the neural ectoderm during gastrulation81. Many of the genes identified in chicken, frog and fish NCC development have been suggested to be dispensable in mouse47,48,49. However, our findings that NCC specification in mouse may occur earlier than previously thought, and at a stage parallel to these other vertebrates, indicate that perhaps some of these genes need to be re-evaluated at earlier developmental stages for roles in early NCC development. Revisiting the role of these specification factors during early mouse development could demonstrate that the gene regulatory networks are more unified or conserved across vertebrates than currently thought. Evaluating the role of NR6A1 in NCC development in other model organisms would also provide further evidence for functional conservation across vertebrates. It has already been established that there is high Nr6a1 sequence conservation between zebrafish and mouse (98.5%), human (98.5%) and frog (97%) species126 and comparable Nr6a1 expression in oocytes and the neural tube127,128. Consistent with this data, conserved Nr6a1 function has recently been observed in mouse84, human and zebrafish with respect to axial elongation and the etiology and pathogenesis of oculo-vertebral-renal syndrome129.
The ability of NR6A1 to act as a transcriptional repressor of pluripotency-associated factors and activator of NCC-specifiers, combined with the deficiency of NCC in Nr6a1 mutants, therefore supports a model in which pluripotency and stem cell maintenance regulators must be downregulated for NCC specification and formation. However, some recently published studies have argued that NCC either retain or re-acquire the expression of pluripotency-associated factors such as Oct4 and, or Nanog underpins their formation, multipotency and other properties113,114,115,116,117,118,130. For instance, when Oct4 was knocked out in mouse embryos via ActinCreER at E7.5, this resulted in a smaller frontonasal mass at E9.5, which was interpreted to mean that Oct4 is required for ectomesenchyme differentiation of NCC118. However, a 10-fold increase in neural crest cell death and an 80% reduction in neural crest cell proliferation was observed in the frontonasal mesenchyme, and immunostaining against neural crest markers revealed that the proportions of the remaining cells expressing TFAP2α and ALX4 were normal. Furthermore, a similar trend was observed for the anterior cranial ganglia with TFAP2α and Sox10, although it did not reach statistical significance. Thus, it was concluded that Oct4 was dispensable for cranial neural crest cell induction and delamination. It’s also important to note that conditional knockout of Oct4 in the epiblast during early gastrulation affects EMT which could impact the generation of NCC131 and furthermore that EMT is associated with downregulation of pluripotency132.
Our analyses of Nr6a1 null embryos challenge the re-acquisition model and role for Oct4 in NCC specification and formation. We evaluated the expression of Oct4 during normal development using the Oct4-EGFP transgenic mouse line, which revealed that although Oct4 is strongly expressed in the epiblast at E7.5, it is downregulated by E8.5 coinciding with the formation of NCC. Furthermore, Oct4 expression persists ectopically in the neural ectoderm of Nr6a1−/−;Oct4-EGFP embryos in concert with deficient NCC formation. Subsequent evaluation of pluripotency-associated gene and stem cell maintenance factor expression during NCC development through analyses of our own and publicly available scRNA-seq datasets, each revealed a similar downregulation of Oct4 and Nanog expression in concert with NCC formation. Notably, by utilizing both Wnt1-Cre and Mef2c-F10N-LacZ embryos in our scRNA-seq, we were able to finely cluster the NCC populations based on the expression of one or both markers106. Wnt1-Cre demarcates dorsal neural ectoderm, pre-migratory and migratory NCC, whereas Mef2c-F10N-LacZ demarcates only migratory NCC92,133. Our scRNA-seq results matched those obtained in Xenopus where even when clustering with a bias towards re-activation of pluripotency, presumptive NCC do not express these factors120. Collectively, this data argues against the need for the retention of pluripotency-associated factor expression for proper NCC specification and formation.
To conclusively test the idea that downregulation of pluripotency-associated factor expression is required for NCC development, we overexpressed Oct4 in mouse embryos from E6.5-E8.5, coinciding with the specification and formation of NCC. This perturbed NCC formation, mimicking the Nr6a1 null mutant phenotype. Thus, persistent Oct4 expression inhibits NCC formation. However, although OCT4, NANOG and SOX2 comprise a core regulatory complex that maintains the pluripotency in various stem cells, it’s important to note that simply overexpressing Sox2 in the neuroepithelium of avian embryos represses NCC formation93,97. Furthermore, neurospheres selected for high levels of Sox2 expression fail to generate NCC upon transplantation into the neuroepithelium of chicken embryos97. Additionally, expressing Oct4 and Sox2 in late migratory NCC disrupts their differentiation114. Collectively, this corroborates the requirement to downregulate pluripotency-associated factors for proper NCC development, and our data indicate that NR6A1 is a key regulator of this process.
The duality of NR6A1 to both downregulate pluripotency-associated factors and activate NCC-specification and EMT genes suggests it functions as a bimodal switch. However, future studies will be necessary to determine how exactly NR6A1 activates NCC-specification gene expression and whether it is truly direct. NR6A1 lacks the canonical activation function-2 (AF-2) domain which bestows activation activity to nuclear receptors in the superfamily101,134. However, mutations in the H12 region of Nr6a1, where the AF2 domain is typically located in other nuclear receptors, demonstrated the ligand binding domain remains capable of undergoing conformational changes consistent with switching between activator and repressor functions101,135. Identification of NR6A1’s ligand with subsequent assays to assess direct transcriptional upregulation will help elucidate whether NR6A1 is indeed a bimodal switch capable of repression and activation like its most homologous nuclear receptor superfamily member, COUP-TF.
Regardless of whether NR6A1 directly regulates NCC-specifier and EMT genes, we have rigorously demonstrated that NR6A1 is required for proper NCC specification and formation. Interestingly, although the expression of NCC-specification and EMT genes is downregulated in Nr6a1−/− embryos, lineage tracing revealed that some Wnt1-Cre-positive cells delaminate and migrate into the facial prominences and first pharyngeal arch. These putative NCC cells ultimately undergo cell death as evidenced by TUNEL staining, demonstrating that NR6A1 is required for NCC survival. How these cells remain capable of delamination without the expression of EMT master regulators Snai1 and Zeb2 remains a mystery. However, it should be noted that individual knockouts of Snai1 and Zeb2 and even compound Snai1/2 double knockouts in mice do not prevent NCC specification and delamination61,62,63. In these scenarios, Snai1 and Zeb2 have been proposed to compensate for each other to justify the presence of delaminated NCC. However, in Nr6a1 null embryos, the expression of both of these factors is downregulated, and yet some putative NCC still delaminate from the neural ectoderm and migrate into the facial prominences and first pharyngeal arch. The expression of Twist1 in these cells indicates they are likely mesenchymal which could explain their migratory ability. However, Twist1 is not expressed by NCC in mouse embryos until after their delamination from the neural ectoderm, and consistent with this behavior, Twist1 loss-of-function does not affect NCC specification and delamination60,90,91. Thus, Twist1 expression in Nr6a1 null mutants does not explain the ability of some Wnt1-Cre;R26R-eYFP-positive cells to delaminate from the neural ectoderm. Interestingly, we recently discovered that a subpopulation of NCC can delaminate via a novel cell extrusion mechanism in parallel with canonical EMT119. This could explain the presence of delaminated migratory cells in Nr6a1 mutant embryos, however future experiments are needed to determine if this is indeed the case in addition to identifying specific markers of NCC extrusion.
The formation of some cranial NCC, and the striking absence of migrating NCC caudal to the first pharyngeal arch is intriguing and exemplifies known differences in NCC regulation along the body axis, such as is also the case with Pax3/759,136. Although future experiments will be needed to explore the differences in regulation between the cranial and trunk NCC populations, one explanation for the cranial-trunk disparity could be the presence and function of redundant orphan nuclear receptors in NCC development. Interestingly, Nr5a2, which maintains Oct4 expression, keeps NCC progenitors in an undifferentiated state, while priming them for later differentiation137. This contrasts with Nr6a1’s repression of Oct4 activity and promotion of NCC specification but illustrates the importance of investigating the roles of other orphan nuclear receptors in NCC development.
In summary, our work has demonstrated that pluripotency and stem cell maintenance factors need to be downregulated in association with the activation of NCC specification and EMT genes to facilitate the differentiation of neural stem cells to NCC. This transition in signaling is facilitated by NR6A1, a critical regulator of cell fate decisions73,81,103. NR6A1 is required during mid-late gastrulation for the specification, formation and survival of NCC. Altogether, our findings have expanded our knowledge of the regulatory signals and mechanisms driving NCC formation as well as the timing of NCC specification in mammals, deepening our overall understanding of NCC development.
Methods
Animal husbandry and embryo collection
All mice were housed in the Laboratory Animal Services Facility at the Stowers Institute for Medical Research under a 14-hour light/10-hour dark/light cycle, and experiments were performed according to IACUC animal welfare guidelines and approved protocol (#2022-014). Embryos were obtained by timed mating, with the morning of the vaginal plug defined as embryonic day (E)0.5 and genotyping of yolk sac tissue was performed as previously described73,138. For all experiments, both male and female embryos were included as there are no sex specific differences in NCC development. At least 5 embryos were used in each control and mutant group across two litters for each experiment, unless otherwise specified. The use of embryos from multiple litters ensures the findings were not litter specific.
For temporal excision of Nr6a1, pregnant dams were orally gavaged with 5 mg of Tamoxifen and 1 mg of progesterone dissolved in 100 µL of corn oil. Treatment was performed one day prior to the desired stage of allele excision as it takes at least 24 h for complete recombination to occur.
Oct4/rtTA mice were housed and maintained in the animal facility at the CBM (Madrid, Spain) in accordance with national and European legislation. Procedures were approved by the CBM Animal Welfare Committee and by the Area of Animal Protection of the Regional Government of Madrid (ref. PROEX 196/14).
To overexpress Oct4, CD1 females were mated with Oct4/rtTA (Col1a1-tetO-Oct4;R26R-M2rtTA) transgenic males (Jax stock #006911). Transgene expression was induced from E6.5 until the desired stage by replacing the drinking water of pregnant females with a 7.5% sucrose solution containing 0.5 mg/mL doxycycline. Embryos from non-treated mice of the same genotype were used as controls.
In situ hybridization
Following dissection, embryos were fixed in 4% paraformaldehyde for 2 hours at room temperature. Embryos were then dehydrated through an ascending series from 0.1% Tween in DEPC treated PBS (PBSTw) to 100% methanol, before being stored at −20 °C overnight and until further use. In situ hybridization was then performed with anti-sense digoxigenin-labeled mRNA riboprobes as previously described139. Briefly, embryos were gradually rehydrated from 100% methanol into PBSTw then bleached for 5 min in 6% hydrogen peroxide in PBSTw at room temperature. Two washes in PBSTw were then performed at room temperature followed by a 5 min digestion in 10 µg/mL proteinase K in PBSTw without rocking at room temperature. After three washes, each for 5 min in PBSTw without rocking at room temperature, the embryos were post-fixed in 4% PFA and 0.2% glutaraldehyde in PBSTw for 20 min at room temperature with rocking. Two additional washes in PBSTw were performed before gradually equilibrating the embryos in hybridization solution. The embryos were then left rocking at 68 °C in hybridization solution with 2 µg/µL of probe overnight. The next day, the embryos were repeatedly washed in PBSTw before being placed in a blocking solution of 10% lamb serum and 0.1% Roche blocking reagent in TBST for 90 min at room temperature. The embryos were then equilibrated in blocking solution with Roche Anti-Digoxigenin AP Fab fragments (1:2000) overnight at 4 °C. One day of TBST washes was performed followed by color development the next day. The embryos were washed in NTMT 3-times before developing in NTMT with 125 µg/mL BCIP and 250 µg/mL NBT at room temperature, protected from light, overnight. The embryos were then washed in PBSTw and re-fixed in 4% PFA to stop the reaction. Anti-sense and sense probes were run at the same time to ensure the signal was specific and not background. Embryos of the same stage were placed in the same tube to confirm lack of staining in E6.5 was real. Experiments were performed twice and a minimum of 5 embryos of each stage were evaluated each time.
Hybridized chain reaction (HCR)
HCR probes were commercially available and purchased from Molecular Instruments (V3.0) and performed according to the manufacturer’s protocol140 with the following modifications. E8.75 embryos were bleached in 6% hydrogen peroxide in PBSTw for 5 min at room temperature with rocking. The embryos were digested in 10 µg/mL proteinase K in PBSTw for 5 min at room temperature without rocking. 8 µL of probe was then added to 500 µL of Hybridization Buffer before adding 10 µL of each Hairpin. The embryos were mounted in 0.8% low melting point agarose for imaging and cleared with CE3D141.
Immunohistochemistry
For section immunohistochemistry, Nr6a1 litters were collected at E9.0 and fixed overnight in 1% PFA at 4 °C. Embryos were processed through a sucrose gradient, mounted in Tissue Tek OCT (VWR, West Chester, PA) and sectioned at 10 microns. Sections were rinsed 3 times with PBS 0.1% TritionX-100 (PBT) for 5 min and blocked in 10% goat serum (Invitrogen, Carlsbad, CA) in PBT for 1 hour at room temperature. Slides were incubated in primary antibody diluted in blocking solution at 4 °C overnight. The monoclonal antibodies SOX2 (R & D Systems, Minneapolis, MN) and pHH3 (Upstate/Millipore, Billerica, MA) were both used at 1:500. The antibody to PAX3 was obtained from the Developmental Studies Hybridoma Bank (Iowa City, IA 52242). TUNEL staining (Roche Life Sciences) was performed according to the manufacturer’s instructions. Slides were rinsed 3 times for 10 min in PBT at room temperature on a shaker and incubated in the appropriate Alexa secondary antibody (Molecular probes/Invitrogen, Carlsbad, CA) at 1:250 for 2 h at 4 °C. Sections were counterstained with a 1:1000 dilution of 2 mg/mL DAPI (Sigma, St. Louis, MO) in PBS for 5 min, followed by rinses in PBS before mounting slides with VectaShield (Vector Laboratories, Newark, CA). All images were collected using a Zeiss Axioplan microscope and processed using Illustrator (Adobe, San Jose, CA).
Immunostaining of whole embryos and cultured cells was performed as previously described142,143. For whole mount immunostaining a SOX10 antibody (1:500; Abcam, ab155279) was used together with GFP (1:1000; Abcam, ab13970). The WTC-11 human induced pluripotent stem cell line was obtained from the Coriell Institute (#GM25256). hiPSCs were plated in an iBidi 8-well plate (Ibidi #80826) and maintained as pluripotent stem cells or differentiated into NCC. Cells were fixed in 4% PFA (Alfa Aesar via VWR Cat. No. AA43368-9M) for 15 min at room temperature and then permeabilized in PBS + 0.3% Tx-100 (Millipore Sigma, #TX1568, PBST) for 20 min at room temperature. Cells were blocked using PBST + 0.5% BSA (Millipore Sigma, #2910) for 1–2 h at room temperature and incubated in primary antibody in blocking buffer overnight at 4 °C. The following primary antibodies were used on cells: Oct4 (R&D Systems, #MAB1759 1:50), Nanog (R&D Systems, #1997 1:40), Sox10 (R&D Systems, #2864 1:100), and Tfap2a (Invitrogen, #3B5 1:50). The cells were washed 3 times for 5 min with PBST and 0.5% BSA (Millipore Sigma, #2910) before incubating in secondary antibody and blocking solution. The following secondary antibodies were used at a 1:500 dilution: Donkey anti-Rabbit IgG (H + L) Alexa Fluor™ 546 (ThermoFisher #A10040), Donkey anti-Goat IgG (H + L) Alexa Fluor™ 488 (ThermoFisher, #A11055), and Donkey anti-Mouse IgG (H + L) Alexa Fluor™ 546 (ThermoFisher, #10036) at room temperature for 1-2 hours. The cells were then washed 3 times for 5 min with PBST and 0.5% BSA, mounted in Vectashield in DAPI (Vector Laboratories, #H-1200-10) and imaged. All images were individually acquired with the same settings, and brightness and contrast were adjusted the same throughout.
Microarray
E9.0 wildtype embryos and Nr6a1−/− embryos were isolated and total RNA was extracted from whole embryos using TRIzol reagent. Concentration and quality of RNA were determined by spectrophotometer and Agilent bioanalyzer analysis (Agilent Technologies, Inc., Palo Alto, CA). For array analysis, labeled mRNA targets were prepared with 150 ng total RNA using MessageAmp III RNA Amplification Kit (Applied Biosystems / Ambion, Austin, TX) according to the manufacturer’s specifications. Array analysis was performed using Affymetrix GeneChip Mouse Genome 430 2.0 Arrays processed with the GeneChip Fluidics Station 450 and scanned with a GeneChip Scanner 3000 7 G using standard protocols. Resulting CEL files were analyzed using RMA and limma in the R statistical environment84. The microarray data are publicly available in the Gene Expression Omnibus using accession number GSE166458.
Bulk RNA-sequencing
E8.75 Nr6a1−/− and wild type control littermates of between 9-11 somites were isolated and bisected into anterior (rostral to and including the first branchial arch) and posterior (caudal to and including the second branchial arch) samples. Total RNA was extracted from whole embryos using an RNA micro Kit. Concentration and quality of RNA were determined by bioanalyzer analysis. RNA-seq was run using single-end 50 bp sequencing reads with Illumina HiSeq 4000. FASTQ files were aligned with STAR, and EdgeR was used for differential expression analysis between mutant and wildtype84. The RNA-seq data are publicly available in the Gene Expression Omnibus using accession number GSE180427.
Single-cell RNA sequencing
Single-cell RNA sequencing of E8.5 Wnt1-Cre;R26R-eYFP and Mef2c-F10N-LacZ embryos, and subsequent bioinformatic analyses were performed as previously described76,106,119 and the data set is publicly available at the Gene Expression Omnibus using accession number GSE168351. Briefly, 10X Genomics Chromium Next GEM Single Cell 3’ Library +Gel Bead Kit (v3.1) was used on dissociated cells to generate libraries. Sequencing was performed with an Illumina NovaSeq6000 and 10X Genomic cell ranger was implemented to process the data. Normalization and downstream analysis all utilized Seurat (v3) in R. Clustering was performed using the shared nearest neighbor method first at a resolution of 0.05, then, within the NCC cluster at a resolution of 0.26. Finally, the pre-migratory NCC were then clustered at a resolution of 2.0 to produce the final 15 distinct NCC clusters. For single cell RNA sequencing gastrulation to neurulation time courses, the data was obtained and images generated from the interactive server: https://marionilab.cruk.cam.ac.uk/MouseGastrulation2018/ described in ref.121.
Multiomic single-cell RNA-sequencing and single-nucleotide ATAC-sequencing
Nr6a1−/− and wildtype control littermates were collected at E8.75. 3-4 embryos were used per genotype across two litters. Nuclei were isolated with the 10X Genomics Nuclei Isolation kit. Following isolation, the nuclei quality was assessed via bright field imaging and standard QC analysis. A total of 3200-3500 nuclei were captured per sample. Libraries were prepared and pooled such that the pooled gene expression and ATAC libraries could then be run on individual flow cells, resulting in 20,000 reads per nuclei for scRNA-seq and 25,000 reads per nuclei for snATAC-seq. Paired end sequencing was performed on an Illumina NextSeq 2000. Raw data was processed with 10X Cell Ranger and analysis was performed in R (v4.4.1) with Seurat (v5)144. Data was filtered using standard QC methods: ATAC reads greater than 50,000 and less than 500 were filtered out along with RNA reads greater than 25,000 and less than 1000. Cells with percent RNA mitochondria reads greater than 10% were filtered out. Finally, cells in which transcription start site enrichment was less than 1 or nucleosome signal greater than 2 were also filtered out. The final dataset analyzed following filtering and SCT normalization145 included 2987 mutant cells and 2728 wildtype cells. Wildtype and mutant cells were integrated together with Seurat’s CCA Integration146. Clustering was performed on the integrated dataset at a resolution of 0.5 using the find nearest neighbor method. Seurat FindMarkers was used to compare gene expression across clusters to aid in cell type annotation and to find differences between genotypes. Annotations from the scRNA-seq clustering were used to annotate the snATAC-seq. The data set is publicly available at the Gene Expression Omnibus using accession number GSE287235.
Electrophoretic mobility shift assay (EMSA) and competition binding assay
We produced NR6A1 protein in vitro using the TNT translation system (Promega). Oligo DNA 5’–hydroxyl end labeled with (γ-32P) ATP using T4 PNK system was used as a probe. EMSA experiments were performed at 30 °C for 30 min in 10 µL of EMSA buffer (10 mM HEPES, pH 7.5; 50 mM KCl; 1 mM EDTA, 5 mM MgCl2; 2 mM DTT; 10% Glycerol; Poly [dI-dC] 1 µg). For competition assays, unlabeled wildtype or mutated competitor DNA was added to the reaction at 5x, 10x or 50x concentrations relative to labelled probe. The total reactions were directly loaded onto a 5% acrylamide gel. Electrophoresis was carried out at 4 °C for 100 min at 300 V. The WT sequence for the competition assay was AGGTCA and the Mutant sequence was CTTGAC.
Mouse stem cell to primitive neural crest cell differentiation
KH2 cells were maintained on MEFs in KOSR media. Prior to initiating differentiation, the cells were adapted to feeder free conditions for 1 passage. For differentiation, 12,500 cells/cm2 were plated on Matrigel (Sigma-Adlrich) coated plates. Base differentiation media consisted of DMEM/F12 with Glutamax:Neurobasal 1:1 media containing 1X Embryomax, 2-mercaptoethanol, 0.5X N2, 0.5X, B27, and 1X NEAA. bFGF (10 ng/mL) (StemCell Technologies) and Heparin solution (0.0002%) (StemCell Technologies) were added to the media for the entirety of differentiation. After the first 4 days of differentiation, Bmp2 (10 ng/mL) was added to the culture until the cells were harvested.
Human induced pluripotent stem cell to neural crest cell differentiation
hiPSCs were obtained from the Allen Institute (GM25256). All experiments were performed according to the regulations of approved IBC registration HPCL-014) and protocol (IBC2003-24). Undifferentiated hiPSCs were maintained and passaged on plates coated with hESC-qualified Matrigel (Corning #354277) in mTeSR1 (Stem Cell Technologies #85850) media supplemented with 1% penicillin/streptomycin (ThermoFisher, #15070063). Cells were passaged approximately every 3–5 days (70–85% confluency) using Accutase (Gibco, #11105–01) to detach cells. Cells were then plated in mTeSR1 + 1% P/S and 10 μM Rock Inhibitor (Y-27632, StemCell Technologies, #72308). For NCC differentiation, hiPSCs were detached with Accutase (Gibco, #11105–01) and resuspended in STEMdiffTM Neural Crest Differentiation media (StemCell Technologies, #08610). Cells were plated at 7000 cells/cm2 in STEMdiffTM Neural Crest Differentiation media supplemented with 1% penicillin/streptomycin (ThermoFisher, #15070063) and 10 μM Rock Inhibitor (Y-27632, StemCell Technologies, #72308) for the first 24 h. Media was changed every day. On day 8 of differentiation, cells were harvested for RNA extraction or fixed for immunostaining.
Chromatin immunoprecipitation
ChIP experiments were performed as previously described147, using mouse KH2 stem cells that were differentiated into to NCC over 5 days. These experiments were repeated 3 times for rigor, reproducibility and statistical analysis. Antibodies used were: NR6A1 (Proteintech Ca. No. 12712-1-AP) and IgG control (Proteintech Ca. No. 30000-0-AP). Snai1 promoter sequence: CAGCGCCCAAAGGTCAGCAGCTCGGGGATG; Snai2 promoter sequence: AAGCCAAGTCGCCGTAGGTCACCTAGCGGAA; Sox9 Exon 3 sequence: GTTCCGGCCACCCACGGCCAGGTCACCTACAC; Sox9 3’ UTR sequence: GCTGTTCCCCGTGGAGGTCAGAGGGGAGAGGTA; Sox10 Exon 2 sequence: GTGAACTGGGCAAGGTCAAGAAGGAACAGCA; Zeb2 Intron 2 sequence: CTTGGCTCCAGCAGTGAGGTCAAGCCACAGCC; Zeb2 Intron 2 sequence 2: AGAGGTCATGTGAACCTCAGAGTCAGGCCCTCG.
Quantitative PCR
RNA was extracted from WTC-11 hiPSC or differentiated NCC using the Qiagen miRNeasy Micro Kit (Qiagen, #217084) with on-column DNase treatment (Qiagen, #79254). The Superscript III Kit (Invitrogen, 18080051) was used to synthesize cDNA for quantitative RT-PCR (qPCR) using random hexamer primers. qPCR was performed on ABI7000 (Thermo QuantStudio 7) using Perfecta Sybr Green (Quantbio # 95072-250). Primers are listed in supplemental Table 1. No template controls were run as negative controls. The ∆∆Ct method was used to calculate fold change. Student’s t-test and ANOVA were used for statistical analysis of n = 18 samples at each time point for each gene, and significance was determined based on p < 0.05.
Imaging and image analysis
Brightfield images of in situ hybridization embryos were captured as previously described106. In brief, images were taken on a Leica MZ16 microscope using a Nikon DS-Ri1 camera. Manual Z-stacks were acquired and then images were merged using Helicon Focus. Fluorescent images were taken on upright Zeiss LSM-700, inverted Zeiss LSM-800 equipped with a GaAsP detector and a Nikon CSU-W1 inverted spinning disk equipped with a sCMOS camera. Image analysis was performed using Fiji.
HCR imaging and quantification
All embryos were imaged using the same settings on a Nikon CSU-W1 inverted spinning disk with a 20x long working distance objective. Z-stacks of each embryo were max-projected in Fiji and the same contrast and brightness were used to generate the fluorescent patterns in Fig. 2A. To ensure equivalent tissue was analyzed during quantification, the same number of z-slices (2.5 µm per slice) were used to make a sum projection. A region of interest was drawn around the head of the embryo ending at the caudal edge of the first pharyngeal arch. The measure feature was used in Fiji to acquire the area, integrated density and mean of the signal for each channel/probe. An additional box region of interest was drawn next to the tissue in the empty space of the image to measure the mean intensity of the background for each channel/probe. The total corrected fluorescent signal was then calculated by taking the integrated density measurement and subtracting the area of the head that was measured, multiplied by the background. There can be differences in the number of cells in the head that are captured in the z-stack as well as distance from the objective that can affect signal. To account for these differences, the total corrected fluorescent signal of each probe was then divided by the total corrected fluorescent DAPI signal such that the final value graphed was the relative expression of each probe/gene in the cranial region of each embryo. Variation in development can occur across and within litters during early craniofacial development. Therefore, we chose to stage match our embryos to the closest developmental stage control littermate, and by evaluating the total corrected fluorescence for each probe over the total corrected DAPI fluorescence, and matching to the developmentally closest control littermate, we ensured the most precise quantitative comparisons. Finally, as the values are a ratio of total corrected fluorescence for each gene normalized to DAPI, a ratio paired t-test was performed to assess the statistical significance of the differences in relative fluorescent expression of each gene between control and Nr6a1 null embryos. Graphing and statistical analyses were performed in GraphPad Prism 10.
Quantification of cell death
Imaris was used to count the number of NCC and non-NCC labeled with TUNEL in a z-stack of each embryo. The spot count tool was used on a region of interest confined to the first pharyngeal arch and more rostral region of the head in the DAPI channel to count the total number of cells in the head. A surface was then generated of the same region using DAPI, followed by a mask of the surface using the TUNEL channel. The cells that were within the TUNEL masked region were then counted with the spot tool to identify all the cells in the head that were undergoing cell death. Next, a surface was generated using the GFP channel to capture all of the NCC. The GFP signal in the Wnt1-Cre;R26R-mTmG line is localized to the cell membrane, therefore, to count the cells that were GFP positive and thus NCC, we generated a mask of the GFP surface with DAPI. The DAPI-stained nuclei located in the GFP-positive region were then counted to determine the total number of NCC in the head. To determine the total number of NCC undergoing cell death, another mask was generated with the GFP surface using the TUNEL channel. Cells were once again counted using the spot tool. Please note that a cell is ~10 µm in diameter, therefore we chose a spot size of 8 µm for all spot counting steps assuming that the nuclei are slightly smaller than the overall size of a cell. To determine the number of non-NCC present in the measured cranial regions, we subtracted the total number of NCC from the total number of cells in the head. The total number of NCC with TUNEL staining was subtracted from the total number of cells in the head with TUNEL staining to find the total number of non-NCC undergoing cell death. We then calculated the percentage of TUNEL+ cells in each population and graphed the values with GraphPad Prism 10. We chose to evaluate the overall percentage of cell populations undergoing cell death as opposed to total cell numbers to account for any population size differences that might be found due to variation in developmental stages in Wnt1-Cre;R26R-mTmG embryo littermates. An un-paired parametric t-test was performed using GraphPad Prism to determine statistical significance.
Oct4-EGFP expression quantification
The average maximum intensity of GFP in the SOX10-positive NCC and neural ectoderm was measured using Fiji. A maximum projection was generated from the z-stack acquired of each embryo. A box was then drawn around 3 different NCC and 3 different neural ectoderm cells keeping the same size area each time. The measure feature was then used to obtain the mean intensity within each boxed cell. The three measurements for each cell population were then averaged to determine the average mean intensity for each cell population of each embryo. A total of 3 mutants and 4 controls were analyzed. Values were then graphed using GraphPad Prism 10. A one-way ANOVA was performed across the entire dataset and un-paired parametric t-tests were then performed to compare individual cell populations.
Statistical analysis and reproducibility
All experiments were performed at least three times to confirm results were reproducible, and sample sizes for each experiment are noted. Results were individually plotted with error bars representing standard error of the mean (SEM). Statistical analysis was performed by unpaired two-tailed Student’s t-test unless otherwise stated, and p-values < 0.05 were considered significant.
Skeletal staining of Wnt1-Cre conditional knockout
E18.5 embryos were anesthetized by immersion in ice cold PBS for at least 60 min until no reflex movements were observed following a pinch test. The embryos were then fixed in 95% ethanol for 1 hour and dehydrated in fresh ethanol overnight at 4 °C. The skin and viscera were manually removed, and the embryos were incubated in 70% ethanol/5% acetic acid for 30 min at room temperature, before staining overnight in alizarin red/alcian blue at room temperature as previously described148,149. The embryos were then washed in 70% ethanol/5% acetic acid for 30 min and quickly rinsed in water. Soft tissue digestion was performed in 2% KOH overnight for at least 6 h, followed by further digestion and clearing, initially in 0.25% KOH, and then through an increasing gradient of glycerol in 0.25% KOH before being stored in 50% glycerol.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All genomic data produced in this study are publicly available in the Gene Expression Omnibus (GEO). The microarray data accession number is GSE166458. The bulk RNA-seq data accession number is GSE180427. The scRNA-seq dataset accession number is GSE168351. Finally, the multiomic scRNA-seq and snATAC-seq data accession number is GSE287235. All remaining original data underlying this manuscript can be accessed from the Stowers Original Data Repository at http://www.stowers.org/research/publications/LIBPB-2530. Requests can also be made to the corresponding author. Source data are provided with this paper.
References
Le Douarin, N. M. & Dupin, E. The “beginnings” of the neural crest. Dev. Biol. 444, S3–S13 (2018).
Trainor, P. & Krumlauf, R. Plasticity in mouse neural crest cells reveals a new patterning role for cranial mesoderm. Nat. Cell Biol. 2, 96–102 (2000).
Trainor, P. & Krumlauf, R. Development. Riding the crest of the Wnt signaling wave. Science 297, 781–783 (2002).
Trainor, P. A. & Krumlauf, R. Patterning the cranial neural crest: hindbrain segmentation and Hox gene plasticity. Nat. Rev. Neurosci. 1, 116–124 (2000).
Trainor, P. A. & Krumlauf, R. Hox genes, neural crest cells and branchial arch patterning. Curr. Opin. Cell Biol. 13, 698–705 (2001).
Watt KENaT, P. A. Neurocristopathies: The Etiology and Pathogenesis of Disorders Arising from Defects in Neural Crest Cell Development. In: Neural Crest Cells. (Academic Press, 2014).
Vega-Lopez, G. A., Cerrizuela, S., Tribulo, C. & Aybar, M. J. Neurocristopathies: New insights 150 years after the neural crest discovery. Dev. Biol. 444, S110–S143 (2018).
Dash, S. & Trainor, P. A. The development, patterning and evolution of neural crest cell differentiation into cartilage and bone. Bone 137, 115409 (2020).
Basch, M. L. & Bronner-Fraser, M. Neural crest inducing signals. Adv. Exp. Med. Biol. 589, 24–31 (2006).
Bonstein, L., Elias, S. & Frank, D. Paraxial-fated mesoderm is required for neural crest induction in Xenopus embryos. Dev. Biol. 193, 156–168 (1998).
Dickinson, M. E., Selleck, M. A., McMahon, A. P. & Bronner-Fraser, M. Dorsalization of the neural tube by the non-neural ectoderm. Development 121, 2099–2106 (1995).
Goulding, M. D., Lumsden, A. & Gruss, P. Signals from the notochord and floor plate regulate the region-specific expression of two Pax genes in the developing spinal cord. Development 117, 1001–1016 (1993).
Mancilla, A. & Mayor, R. Neural crest formation in Xenopus laevis: mechanisms of Xslug induction. Dev. Biol. 177, 580–589 (1996).
Marchant, L., Linker, C., Ruiz, P., Guerrero, N. & Mayor, R. The inductive properties of mesoderm suggest that the neural crest cells are specified by a BMP gradient. Dev. Biol. 198, 319–329 (1998).
Selleck, M. A. & Bronner-Fraser, M. Origins of the avian neural crest: the role of neural plate-epidermal interactions. Development 121, 525–538 (1995).
Liem, K. F. Jr, Tremml, G., Roelink, H. & Jessell, T. M. Dorsal differentiation of neural plate cells induced by BMP-mediated signals from epidermal ectoderm. Cell 82, 969–979 (1995).
Alkobtawi, M., Pla, P. & Monsoro-Burq, A. H. BMP signaling is enhanced intracellularly by FHL3 controlling WNT-dependent spatiotemporal emergence of the neural crest. Cell Rep. 35, 109289 (2021).
Garcia-Castro, M. I., Marcelle, C. & Bronner-Fraser, M. Ectodermal Wnt function as a neural crest inducer. Science 297, 848–851 (2002).
Marchal, L., Luxardi, G., Thome, V. & Kodjabachian, L. BMP inhibition initiates neural induction via FGF signaling and Zic genes. Proc. Natl. Acad. Sci. USA 106, 17437–17442 (2009).
Suzuki, A., Ueno, N. & Hemmati-Brivanlou, A. Xenopus msx1 mediates epidermal induction and neural inhibition by BMP4. Development 124, 3037–3044 (1997).
Patthey, C. & Gunhaga, L. Signaling pathways regulating ectodermal cell fate choices. Exp. Cell Res 321, 11–16 (2014).
Wilson, S. I., Graziano, E., Harland, R., Jessell, T. M. & Edlund, T. An early requirement for FGF signalling in the acquisition of neural cell fate in the chick embryo. Curr. Biol. 10, 421–429 (2000).
Monsoro-Burq, A. H., Wang, E. & Harland, R. Msx1 and Pax3 cooperate to mediate FGF8 and WNT signals during Xenopus neural crest induction. Dev. Cell 8, 167–178 (2005).
de Croze, N., Maczkowiak, F. & Monsoro-Burq, A. H. Reiterative AP2a activity controls sequential steps in the neural crest gene regulatory network. Proc. Natl. Acad. Sci. USA 108, 155–160 (2011).
Luo, T., Lee, Y. H., Saint-Jeannet, J. P. & Sargent, T. D. Induction of neural crest in Xenopus by transcription factor AP2alpha. Proc. Natl. Acad. Sci. USA 100, 532–537 (2003).
Mitchell, P. J., Timmons, P. M., Hebert, J. M., Rigby, P. W. & Tjian, R. Transcription factor AP-2 is expressed in neural crest cell lineages during mouse embryogenesis. Genes Dev. 5, 105–119 (1991).
Rothstein, M. & Simoes-Costa, M. Heterodimerization of TFAP2 pioneer factors drives epigenomic remodeling during neural crest specification. Genome Res. 30, 35–48 (2020).
Schorle, H., Meier, P., Buchert, M., Jaenisch, R. & Mitchell, P. J. Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature 381, 235–238 (1996).
Zhang, J. et al. Neural tube, skeletal and body wall defects in mice lacking transcription factor AP-2. Nature 381, 238–241 (1996).
Basch, M. L., Bronner-Fraser, M. & Garcia-Castro, M. I. Specification of the neural crest occurs during gastrulation and requires Pax7. Nature 441, 218–222 (2006).
Aybar, M. J., Nieto, M. A. & Mayor, R. Snail precedes slug in the genetic cascade required for the specification and migration of the Xenopus neural crest. Development 130, 483–494 (2003).
Carl, T. F., Dufton, C., Hanken, J. & Klymkowsky, M. W. Inhibition of neural crest migration in Xenopus using antisense slug RNA. Dev. Biol. 213, 101–115 (1999).
LaBonne, C. & Bronner-Fraser, M. Neural crest induction in Xenopus: evidence for a two-signal model. Development 125, 2403–2414 (1998).
LaBonne, C. & Bronner-Fraser, M. Snail-related transcriptional repressors are required in Xenopus for both the induction of the neural crest and its subsequent migration. Dev. Biol. 221, 195–205 (2000).
Lander, R. et al. Interactions between Twist and other core epithelial-mesenchymal transition factors are controlled by GSK3-mediated phosphorylation. Nat. Commun. 4, 1542 (2013).
Nieto, M. A., Sargent, M. G., Wilkinson, D. G. & Cooke, J. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science 264, 835–839 (1994).
Nitta, K. R., Tanegashima, K., Takahashi, S. & Asashima, M. XSIP1 is essential for early neural gene expression and neural differentiation by suppression of BMP signaling. Dev. Biol. 275, 258–267 (2004).
Rogers, C. D., Saxena, A. & Bronner, M. E. Sip1 mediates an E-cadherin-to-N-cadherin switch during cranial neural crest EMT. J. Cell Biol. 203, 835–847 (2013).
Dottori, M., Gross, M. K., Labosky, P. & Goulding, M. The winged-helix transcription factor Foxd3 suppresses interneuron differentiation and promotes neural crest cell fate. Development 128, 4127–4138 (2001).
Aoki, Y. et al. Sox10 regulates the development of neural crest-derived melanocytes in Xenopus. Dev. Biol. 259, 19–33 (2003).
Spokony, R. F., Aoki, Y., Saint-Germain, N., Magner-Fink, E. & Saint-Jeannet, J. P. The transcription factor Sox9 is required for cranial neural crest development in Xenopus. Development 129, 421–432 (2002).
Cheung, M. & Briscoe, J. Neural crest development is regulated by the transcription factor Sox9. Development 130, 5681–5693 (2003).
Honore, S. M., Aybar, M. J. & Mayor, R. Sox10 is required for the early development of the prospective neural crest in Xenopus embryos. Dev. Biol. 260, 79–96 (2003).
Sato, T., Sasai, N. & Sasai, Y. Neural crest determination by co-activation of Pax3 and Zic1 genes in Xenopus ectoderm. Development 132, 2355–2363 (2005).
Hong, C. S. & Saint-Jeannet, J. P. The activity of Pax3 and Zic1 regulates three distinct cell fates at the neural plate border. Mol. Biol. Cell 18, 2192–2202 (2007).
Rogers, C. D., Jayasena, C. S., Nie, S. & Bronner, M. E. Neural crest specification: tissues, signals, and transcription factors. Wiley Interdiscip. Rev. Dev. Biol. 1, 52–68 (2012).
Trainor, P. A. Specification of neural crest cell formation and migration in mouse embryos. Semin Cell Dev. Biol. 16, 683–693 (2005).
Barriga, E. H., Trainor, P. A., Bronner, M. & Mayor, R. Animal models for studying neural crest development: is the mouse different? Development 142, 1555–1560 (2015).
Trainor, P. A. Specification and patterning of neural crest cells during craniofacial development. Brain Behav. Evol. 66, 266–280 (2005).
Dudley, A. T., Lyons, K. M. & Robertson, E. J. A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes Dev. 9, 2795–2807 (1995).
Luo, G. et al. BMP-7 is an inducer of nephrogenesis, and is also required for eye development and skeletal patterning. Genes Dev. 9, 2808–2820 (1995).
Selleck, M. A., Garcia-Castro, M. I., Artinger, K. B. & Bronner-Fraser, M. Effects of Shh and Noggin on neural crest formation demonstrate that BMP is required in the neural tube but not ectoderm. Development 125, 4919–4930 (1998).
Solloway, M. J. & Robertson, E. J. Early embryonic lethality in Bmp5;Bmp7 double mutant mice suggests functional redundancy within the 60A subgroup. Development 126, 1753–1768 (1999).
Trainor, P. A., Sobieszczuk, D., Wilkinson, D. & Krumlauf, R. Signalling between the hindbrain and paraxial tissues dictates neural crest migration pathways. Development 129, 433–442 (2002).
Winnier, G., Blessing, M., Labosky, P. A. & Hogan, B. L. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 9, 2105–2116 (1995).
Correia, A. C. et al. Bmp2 is required for migration but not for induction of neural crest cells in the mouse. Dev. Dyn. 236, 2493–2501 (2007).
Conway, S. J., Henderson, D. J. & Copp, A. J. Pax3 is required for cardiac neural crest migration in the mouse: evidence from the splotch (Sp2H) mutant. Development 124, 505–514 (1997).
Mansouri, A., Stoykova, A., Torres, M. & Gruss, P. Dysgenesis of cephalic neural crest derivatives in Pax7−/− mutant mice. Development 122, 831–838 (1996).
Zalc, A., Rattenbach, R., Aurade, F., Cadot, B. & Relaix, F. Pax3 and Pax7 play essential safeguard functions against environmental stress-induced birth defects. Dev. Cell 33, 56–66 (2015).
Bildsoe, H. et al. Requirement for Twist1 in frontonasal and skull vault development in the mouse embryo. Dev. Biol. 331, 176–188 (2009).
Murray, S. A. & Gridley, T. Snail family genes are required for left-right asymmetry determination, but not neural crest formation, in mice. Proc. Natl. Acad. Sci. USA 103, 10300–10304 (2006).
Van de Putte, T., Francis, A., Nelles, L., van Grunsven, L. A. & Huylebroeck, D. Neural crest-specific removal of Zfhx1b in mouse leads to a wide range of neurocristopathies reminiscent of Mowat-Wilson syndrome. Hum. Mol. Genet. 16, 1423–1436 (2007).
Van de Putte, T. et al. Mice lacking ZFHX1B, the gene that codes for Smad-interacting protein-1, reveal a role for multiple neural crest cell defects in the etiology of Hirschsprung disease-mental retardation syndrome. Am. J. Hum. Genet. 72, 465–470 (2003).
Jones, N. C. et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med 14, 125–133 (2008).
Porter B. A., Ortiz M. A., Bratslavsky G., Kotula L. Structure and function of the nuclear receptor superfamily and current targeted therapies of prostate cancer. Cancers. 11, (2019).
Duarte, J., Perriere, G., Laudet, V. & Robinson-Rechavi, M. NUREBASE: database of nuclear hormone receptors. Nucleic Acids Res. 30, 364–368 (2002).
Nuclear Receptors Nomenclature C. A unified nomenclature system for the nuclear receptor superfamily. Cell 97, 161–163 (1999).
Chen, F., Cooney, A. J., Wang, Y., Law, S. W. & O’Malley, B. W. Cloning of a novel orphan receptor (GCNF) expressed during germ cell development. Mol. Endocrinol. 8, 1434–1444 (1994).
Hirose, T., O’Brien, D. A. & Jetten, A. M. RTR: a new member of the nuclear receptor superfamily that is highly expressed in murine testis. Gene 152, 247–251 (1995).
Lan, Z. J., Xu, X., Chung, A. C. & Cooney, A. J. Extra-germ cell expression of mouse nuclear receptor subfamily 6, group A, member 1 (NR6A1). Biol. Reprod. 80, 905–912 (2009).
Katz, D., Niederberger, C., Slaughter, G. R. & Cooney, A. J. Characterization of germ cell-specific expression of the orphan nuclear receptor, germ cell nuclear factor. Endocrinology 138, 4364–4372 (1997).
Zhang, Y. L. et al. Expression of germ cell nuclear factor (GCNF/RTR) during spermatogenesis. Mol. Reprod. Dev. 50, 93–102 (1998).
Chung, A. C. et al. Loss of orphan receptor germ cell nuclear factor function results in ectopic development of the tail bud and a novel posterior truncation. Mol. Cell Biol. 21, 663–677 (2001).
Dixon, J., Trainor, P. & Dixon, M. J. Treacher Collins syndrome. Orthod. Craniofac Res. 10, 88–95 (2007).
Trainor, P. A., Dixon, J. & Dixon, M. J. Treacher Collins syndrome: etiology, pathogenesis and prevention. Eur. J. Hum. Genet. 17, 275–283 (2009).
Falcon, K. T. et al. Dynamic regulation and requirement for ribosomal RNA transcription during mammalian development. Proc. Natl. Acad. Sci. USA 119, e2116974119 (2022).
Noack Watt, K. E., Achilleos, A., Neben, C. L., Merrill, A. E. & Trainor, P. A. The Roles of RNA Polymerase I and III Subunits Polr1c and Polr1d in Craniofacial Development and in Zebrafish Models of Treacher Collins Syndrome. PLoS Genet. 12, e1006187 (2016).
Sakai, D., Dixon, J., Achilleos, A., Dixon, M. & Trainor, P. A. Prevention of Treacher Collins syndrome craniofacial anomalies in mouse models via maternal antioxidant supplementation. Nat. Commun. 7, 10328 (2016).
Watt, K. E. N., Neben, C. L., Hall, S., Merrill, A. E. & Trainor, P. A. tp53-dependent and independent signaling underlies the pathogenesis and possible prevention of Acrofacial Dysostosis-Cincinnati type. Hum. Mol. Genet. 27, 2628–2643 (2018).
Dixon, J. et al. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc. Natl. Acad. Sci. USA 103, 13403–13408 (2006).
Akamatsu, W., DeVeale, B., Okano, H., Cooney, A. J. & van der Kooy, D. Suppression of Oct4 by germ cell nuclear factor restricts pluripotency and promotes neural stem cell development in the early neural lineage. J. Neurosci. 29, 2113–2124 (2009).
Gu, P. et al. Orphan nuclear receptor GCNF is required for the repression of pluripotency genes during retinoic acid-induced embryonic stem cell differentiation. Mol. Cell Biol. 25, 8507–8519 (2005).
Fuhrmann, G. et al. Mouse germline restriction of Oct4 expression by germ cell nuclear factor. Dev. Cell 1, 377–387 (2001).
Chang, Y. C. et al. Nr6a1 controls Hox expression dynamics and is a master regulator of vertebrate trunk development. Nat. Commun. 13, 7766 (2022).
Chung, A. C., Xu, X., Niederreither, K. A. & Cooney, A. J. Loss of orphan nuclear receptor GCNF function disrupts forebrain development and the establishment of the isthmic organizer. Dev. Biol. 293, 13–24 (2006).
Mundell, N. A. & Labosky, P. A. Neural crest stem cell multipotency requires Foxd3 to maintain neural potential and repress mesenchymal fates. Development 138, 641–652 (2011).
Ng, L. J. et al. SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev. Biol. 183, 108–121 (1997).
Zhao, Q., Eberspaecher, H., Lefebvre, V. & De Crombrugghe, B. Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev. Dyn. 209, 377–386 (1997).
Sefton, M., Sanchez, S. & Nieto, M. A. Conserved and divergent roles for members of the Snail family of transcription factors in the chick and mouse embryo. Development 125, 3111–3121 (1998).
Tavares, A. T., Izpisuja-Belmonte, J. C. & Rodriguez-Leon, J. Developmental expression of chick twist and its regulation during limb patterning. Int J. Dev. Biol. 45, 707–713 (2001).
Gitelman, I. Twist protein in mouse embryogenesis. Dev. Biol. 189, 205–214 (1997).
Chai, Y. et al. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development 127, 1671–1679 (2000).
Wakamatsu, Y., Endo, Y., Osumi, N. & Weston, J. A. Multiple roles of Sox2, an HMG-box transcription factor in avian neural crest development. Dev. Dyn. 229, 74–86 (2004).
Graham, V., Khudyakov, J., Ellis, P. & Pevny, L. SOX2 functions to maintain neural progenitor identity. Neuron 39, 749–765 (2003).
Ellis, P. et al. SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev. Neurosci. 26, 148–165 (2004).
Sudiwala S. et al. Cellular mechanisms underlying Pax3-related neural tube defects and their prevention by folic acid. Dis. Model Mech. 12, (2019).
Remboutsika, E. et al. Flexibility of neural stem cells. Front Physiol. 2, 16 (2011).
Wang, J. et al. A protein interaction network for pluripotency of embryonic stem cells. Nature 444, 364–368 (2006).
Nichols, J., Smith, A. & Buehr, M. Rat and mouse epiblasts differ in their capacity to generate extraembryonic endoderm. Reprod. Fertil. Dev. 10, 517–525 (1998).
Avilion, A. A. et al. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 17, 126–140 (2003).
Hummelke, G. C. & Cooney, A. J. Germ cell nuclear factor is a transcriptional repressor essential for embryonic development. Front. Biosci. 6, D1186–D1191 (2001).
Lengner, C. J. et al. Oct4 expression is not required for mouse somatic stem cell self-renewal. Cell Stem Cell 1, 403–415 (2007).
Cooney, A. J., Hummelke, G. C., Herman, T., Chen, F. & Jackson, K. J. Germ cell nuclear factor is a response element-specific repressor of transcription. Biochem. Biophys. Res. Commun. 245, 94–100 (1998).
Simoes-Costa, M. & Bronner, M. E. Establishing neural crest identity: a gene regulatory recipe. Development 142, 242–257 (2015).
Sauka-Spengler, T. & Bronner-Fraser, M. A gene regulatory network orchestrates neural crest formation. Nat. Rev. Mol. Cell Biol. 9, 557–568 (2008).
Zhao, R. et al. Identification and characterization of intermediate states in mammalian neural crest cell epithelial to mesenchymal transition and delamination. Elife 13, (2024).
Stuhlmiller, T. J. & Garcia-Castro, M. I. Current perspectives of the signaling pathways directing neural crest induction. Cell Mol. Life Sci. 69, 3715–3737 (2012).
Prasad, M. S., Charney, R. M. & Garcia-Castro, M. I. Specification and formation of the neural crest: Perspectives on lineage segregation. Genesis 57, e23276 (2019).
Martik, M. L. & Bronner, M. E. Regulatory logic underlying diversification of the neural crest. Trends Genet. 33, 715–727 (2017).
Lee, S. K., Lee, B., Ruiz, E. C. & Pfaff, S. L. Olig2 and Ngn2 function in opposition to modulate gene expression in motor neuron progenitor cells. Genes Dev. 19, 282–294 (2005).
Ericson, J. et al. Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell 90, 169–180 (1997).
Indra, A. K. et al. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 27, 4324–4327 (1999).
Buitrago-Delgado, E., Nordin, K., Rao, A., Geary, L. & LaBonne, C. Neurodevelopment. Shared regulatory programs suggest retention of blastula-stage potential in neural crest cells. Science 348, 1332–1335 (2015).
Hovland, A. S. et al. Pluripotency factors are repurposed to shape the epigenomic landscape of neural crest cells. Dev. Cell 57, 2257–2272 e2255 (2022).
Lignell, A., Kerosuo, L., Streichan, S. J., Cai, L. & Bronner, M. E. Identification of a neural crest stem cell niche by Spatial Genomic Analysis. Nat. Commun. 8, 1830 (2017).
Pajanoja, C. et al. Maintenance of pluripotency-like signature in the entire ectoderm leads to neural crest stem cell potential. Nat. Commun. 14, 5941 (2023).
Patel, I. & Parchem, R. J. Regulation of Oct4 in stem cells and neural crest cells. Birth Defects Res. 114, 983–1002 (2022).
Zalc A. et al. Reactivation of the pluripotency program precedes formation of the cranial neural crest. Science 371, (2021).
Moore Zajic, E. L. et al. Cell extrusion drives neural crest cell delamination. Proc. Natl. Acad. Sci. USA 122, e2416566122 (2025).
Briggs J. A. et al. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science 360, (2018).
Pijuan-Sala, B. et al. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 566, 490–495 (2019).
Hochedlinger, K., Yamada, Y., Beard, C. & Jaenisch, R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell 121, 465–477 (2005).
Betters, E., Charney, R. M. & Garcia-Castro, M. I. Early specification and development of rabbit neural crest cells. Dev. Biol. 444, S181–S192 (2018).
Prasad, M. S., Charney, R. M., Patel, L. J. & Garcia-Castro, M. I. Distinct molecular profile and restricted stem cell potential defines the prospective human cranial neural crest from embryonic stem cell state. Stem Cell Res 49, 102086 (2020).
Steventon, B., Araya, C., Linker, C., Kuriyama, S. & Mayor, R. Differential requirements of BMP and Wnt signalling during gastrulation and neurulation define two steps in neural crest induction. Development 136, 771–779 (2009).
Braat, A. K. et al. Cloning and expression of the zebrafish germ cell nuclear factor. Mol. Reprod. Dev. 53, 369–375 (1999).
Adams, M. S., Gammill, L. S. & Bronner-Fraser, M. Discovery of transcription factors and other candidate regulators of neural crest development. Dev. Dyn. 237, 1021–1033 (2008).
Thisse, C. & Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59–69 (2008).
Neelathi, U. M. et al. Variants in NR6A1 cause a novel oculo vertebral renal syndrome. Nat. Commun. 16, 6111 (2025).
Scerbo, P. & Monsoro-Burq, A. H. The vertebrate-specific VENTX/NANOG gene empowers neural crest with ectomesenchyme potential. Sci. Adv. 6, eaaz1469 (2020).
Mulas, C. et al. Oct4 regulates the embryonic axis and coordinates exit from pluripotency and germ layer specification in the mouse embryo. Development 145, (2018).
Hamidi, S. et al. Mesenchymal-epithelial transition regulates initiation of pluripotency exit before gastrulation. Development 147, (2020).
Aoto, K. et al. Mef2c-F10N enhancer driven beta-galactosidase (LacZ) and Cre recombinase mice facilitate analyses of gene function and lineage fate in neural crest cells. Dev. Biol. 402, 3–16 (2015).
Greschik, H. & Schule, R. Germ cell nuclear factor: an orphan receptor with unexpected properties. J. Mol. Med (Berl.) 76, 800–810 (1998).
Greschik, H. et al. Characterization of the DNA-binding and dimerization properties of the nuclear orphan receptor germ cell nuclear factor. Mol. Cell Biol. 19, 690–703 (1999).
Van Ho, A. T. et al. Neural crest cell lineage restricts skeletal muscle progenitor cell differentiation through Neuregulin1-ErbB3 signaling. Dev. Cell 21, 273–287 (2011).
Chen, H. J. et al. Nuclear receptor Nr5a2 promotes diverse connective tissue fates in the jaw. Dev. Cell 58, 461–473 e467 (2023).
Lan, Z. J., Xu, X. & Cooney, A. J. Generation of a germ cell nuclear factor conditional allele in mice. Genesis 37, 172–179 (2003).
Nagy A. G. et al. Techniques for Visualizing Gene Products, Cells, Tissues and Organ Systems. In: Manipulating the Mouse Embryo. (Cold Spring Harbor Laboratory Press, 2003).
Choi, H. M. T. et al. Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145, (2018).
Li, W., Germain, R. N. & Gerner, M. Y. High-dimensional cell-level analysis of tissues with Ce3D multiplex volume imaging. Nat. Protoc. 14, 1708–1733 (2019).
Leung, A. W. et al. WNT/beta-catenin signaling mediates human neural crest induction via a pre-neural border intermediate. Development 143, 398–410 (2016).
Barlow, A. J., Dixon, J., Dixon, M. J. & Trainor, P. A. Balancing neural crest cell intrinsic processes with those of the microenvironment in Tcof1 haploinsufficient mice enables complete enteric nervous system formation. Hum. Mol. Genet. 21, 1782–1793 (2012).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Choudhary, S. & Satija, R. Comparison and evaluation of statistical error models for scRNA-seq. Genome Biol. 23, 27 (2022).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Smith, K. T., Martin-Brown, S. A., Florens, L., Washburn, M. P. & Workman, J. L. Deacetylase inhibitors dissociate the histone-targeting ING2 subunit from the Sin3 complex. Chem. Biol. 17, 65–74 (2010).
Nagy, A., Gertsenstein, M., Vintersten, K. & Behringer, R. Alizarin red staining of post-natal bone in mouse. Cold Spring Harb. Protoc. 2009, pdb prot5171 (2009).
Nagy, A., Gertsenstein, M., Vintersten, K. & Behringer, R. Alcian blue staining of the mouse fetal cartilaginous skeleton. Cold Spring Harb. Protoc. 2009, pdb prot5169 (2009).
Acknowledgements
The authors thank past and present members of the Trainor lab and Manzanares lab for their thoughtful insights, constructive feedback and discussions during the progress of this work. We are extremely grateful to Melissa Childers, Marina Thexton and the Laboratory Animal Services Core for their care and maintenance of our mouse colonies, and we appreciate Kaitlyn Petentler, Allison Scott and the Sequencing Core for their molecular biology support. We especially thank Dr. Austin Cooney for providing the Nr6a1+/− and Nr6a1fl/fl mice to establish our own lines. Nr6a1, Pax3, Snai1, Sox9, Sox10, Wnt1, and Zeb2 riboprobe plasmids were generously provided by Dr. Austin Cooney, Dr. Takayoshi Inoue, Dr. Angela Nieto, Dr. Trevor Williams, Dr. Martin Gassmann, Dr. Antony Gavalas, and Dr. Danny Huylebroeck, respectively. This work was supported by the Stowers Institute for Medical Research (PAT), a National Institute for Dental and Craniofacial Research F31 Ruth L. Kirschstein Predoctoral Individual National Research Service Award (DE032256; ELM), American Association for Anatomy Postdoctoral Scholar Awards (WAM and AA), and Asociacion Espanola Contra el Cancer Ideas Semilla grants PID2020-115755GB-I00 and PID2023-151742NB-I00 (MCIN/AEI/10.13039/501100011033; MM). The Centro de Biología Molecular is supported by an institutional grant from the Fundación Ramón Areces and is a Severo Ochoa Center of Excellence (grant CEX2021-001154-S, MICIN/AEI/10.13039/501100011033; MM).
Author information
Authors and Affiliations
Contributions
The paper was written by E.L.M.Z., W.A.M., J.F.D., S.B., A.A., M.L., M.M. and P.A.T.; experiments were designed by E.L.M.Z., W.A.M., J.F.D., S.B., D.S., A.A., R.Z., M.L., M.T. A.B., M.M. and P.A.T.; the research was performed by E.L.M.Z., W.A.M., J.F.D., S.B., D.S., A.A., R.Z., M.L., M.T., A.B., D.C., M.M. and P.A.T.; genomic analysis was performed by E.L.M.Z., W.A.M., A.A., D.S., R.Z., A.F.P. and C.S.; quantification and imaging analysis was performed by E.L.M.Z., W.A.M., D.S. and M.L.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Anne Monsoro-Burq and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Moore Zajic, E.L., Muñoz, W.A., Dennis, J.F. et al. NR6A1 is essential for neural crest cell specification, formation and survival. Nat Commun 17, 2140 (2026). https://doi.org/10.1038/s41467-026-68647-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68647-2
