
Abstract
Among the distinct adaptor protein (AP) complexes, AP-4 primarily functions as a non-clathrin-coated vesicle machinery essential for intracellular membrane trafficking. ARF1 is a master regulator of AP-4 membrane recruitment, but the underlying mechanism remains elusive. Here, we present the cryo-EM structures of soluble AP-4 and the AP-4/ARF1 complex. Unexpectedly, AP-4 adopts a dynamic equilibrium between closed and open conformations, caused by loose contacts between its medium subunit and central core. ARF1 binding induces only subtle changes in AP-4, which retains its conformational equilibrium. Mutations at the AP-4/ARF1 interface disrupt complex formation and impair ARF1-dependent membrane recruitment. Efficient membrane recruitment of AP-4 likely requires the synergistic engagement of ARF1 and cargoes. Disrupting the conformational flexibility of AP-4 interferes with this synergistic effect and compromises AP-4-mediated membrane trafficking. Our findings may redefine AP-4 as a conformationally dynamic complex modulated by cooperative interactions, providing insights into neurodevelopmental disorders associated with AP-4 dysfunction.
Similar content being viewed by others


Introduction
Intracellular membrane trafficking is a fundamental process essential for exocytotic and endocytic pathways, as well as the biogenesis of membrane organelles1,2,3,4,5. Transport cargoes, such as membrane proteins, are typically packaged into protein-coated vesicles that are generated at various organelle membranes to mediate membrane trafficking6,7,8,9. The adaptor protein (AP) complexes (AP-1 to AP-5 in mammals) and their structurally homologous COPI F-subcomplex constitute the core machinery of vesicular coat formation10,11,12,13. Together with accessory proteins, clathrin or alternative scaffolds, they play crucial roles in sorting membrane proteins throughout the intracellular endomembrane system10,11,14,15. All AP complexes are hetero-tetramers, comprising two large subunits, one medium-sized subunit, and one small subunit10,11,13. The COPI F-subcomplex shares this hetero-tetrameric architecture and is evolutionarily related with the AP family12. Based on the structural topology of each AP-related complex, the N-terminal trunk domains of the two large subunits associate with the medium and small subunits to form a characteristic core that recognizes the cytosolic tails of membrane proteins and incorporates them into transport vesicles6. In contrast, the C-terminal hinge and ear domains of the two large subunits extend from this central core and are accessible for the recruitment of scaffolding and accessory proteins, promoting the assembly of protein coats6. Most of the AP-related complexes, including the COPI F-subcomplex, are recruited to target membranes through interactions with membrane-anchored ADP-ribosylation factor (ARF) family small GTPases and/or membrane phosphoinositides, enabling them to function at various membrane compartments, including the trans-Golgi network (TGN), plasma membrane, tubular endosomes, and late endosomes10,11,12. Thus, the AP-related complexes orchestrate the assembly of protein coats and organize an intracellular network of coated vesicles to regulate membrane trafficking.
Among the AP-related complexes, AP-4 lacks the ability to interact with clathrin and is thought to assemble non-clathrin protein coats16,17,18, similar to AP-3 and AP-5 that can function without clathrin19,20. Although AP-4 is expressed at low levels in all cells and tissues, it appears to be particularly important in neurons and neuronal tissues. Loss of AP-4 function in humans results in a severe neurodevelopmental disorder characterized by progressive spasticity and intellectual disability, known as AP-4-deficiency syndrome18,21. Despite its lack of clathrin-binding capacity, AP-4 operates at the TGN and is recruited to membranes through association with the small GTPase ARF1, similar to AP-1, AP-3 and the COPI F-subcomplex, albeit via a distinct mechanism22. Once recruited to the TGN, AP-4 plays a primary role in sorting various membrane proteins, such as the autophagy-related lipid scramblase ATG9A23,24,25 and synaptic AMPA and δ2 receptors26,27. Moreover, AP-4 can directly interact with motor adaptors (such as RUSC1 and RUSC2) to link coated cargoes to microtubule-based motors for intracellular transport18,28.
ARF1 is a master regulator of AP-4 membrane recruitment to the TGN22. In addition to AP-4, the other AP-related complexes, AP-1, AP-3 and the COPI F-subcomplex, are recruited to target membranes also primarily through their association with ARF129,30. For AP-1, ARF1 binding not only mediates membrane recruitment but also induces a conformational switch from a closed, inactive state to an open, active state that enables cargo recognition31,32. Interestingly, this similar closed-to-open conformational transition also happens in AP-2, triggered by the association of membrane lipids and/or cargos33,34,35,36. In contrast, AP-3 adopts a flexible, open conformation prior to ARF1 association, with ARF1 binding likely initiating membrane recruitment37,38. Upon membrane recruitment, the engagement of AP-3 with membrane cargoes stabilizes its conformation and promotes binding to a second ARF1, which mediates AP-3 dimerization/multimerization for coat assembly38. Moreover, the COPI F-subcomplex contains two similar ARF1-binding sites that can regulate its membrane recruitment39,40. Intriguingly, AP-4 also exhibits an ability to bind two ARF1 molecules, i.e., one at the peripheral canonical binding site of its large subunit, and the other at the central core of its medium subunit, a configuration distinct from those of AP-1, AP-3 and the COPI F-subcomplex22. However, the mechanism underlying ARF1-mediated membrane recruitment of AP-4 remains unclear.
In this study, we determine the structures of soluble AP-4 and the AP-4/ARF1 complex. Unexpectedly, AP-4 exhibits a dynamic equilibrium between closed and open states prior to ARF1 association, distinct from AP-1 and AP-3. This conformational equilibrium is attributable to loose interactions between its medium subunit and central core. More intriguingly, ARF1 binding only induces subtle changes in AP-4, which remains in a conformational equilibrium. Mutations at the AP-4/ARF1 interface disrupt complex formation and impair ARF1-mediated membrane recruitment. Furthermore, we demonstrate that efficient membrane recruitment of AP-4 likely requires the synergistic engagement of ARF1 and cargoes, and disrupting the conformational flexibility of AP-4 interferes with this synergistic effect and compromises AP-4-mediated membrane trafficking.
Results
Biochemical characterization of the AP-4 core complex
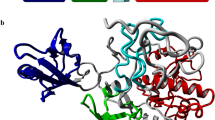
To investigate the mechanism underlying AP-4 membrane recruitment, we initiated this work with the biochemical characterization of AP-4, composed of the two large subunits ε and β4, the medium subunit μ4, and the small subunit σ4 (Fig. 1a). Based on the existing knowledge of the AP complexes, the hinge and ear domains of ε and β4 are flexible and unsuitable for structural studies, and the ARF1-binding sites of AP-4 were proposed to be located in the central core22. We then removed the hinge and ear domains of ε and β4, resulting in the AP-4 core complex, which includes the two trunk domains of ε and β4 and full-length μ4 and σ4 (referred to as the AP-4 core complex for clarity) (Fig. 1a).

a Domain organization of the AP-4 core complex. The AP-4 core complex contains the trunk domains of the two large subunits ε and β4, the medium subunit μ4, and the small subunit σ4. μ4 is composed of the N-terminal and C-terminal domains (μ4-NTD and μ4-CTD). The domain boundaries of each subunit and the fragments used for assembling the AP-4 core complex are marked. b Biochemical characterization of the hetero-tetrameric AP-4 core complex by the SEC-MALS assay, showing the stable complex formation of the four subunits. The inset shows the SDS-PAGE analysis of each component. The experiment is independently repeated three times (n = 3). Source data are provided as a Source Data file. c, d The cryo-EM density map and the atomic model of the μ4-CTD-docked AP-4 core complex. The N-terminal regions of ε (residues 1–113) and β4 (residues 1–26) are not visible in the density map, probably due to conformational flexibility. The inter-domain linker of μ4 (residues 140–183) and a number of internal loops are omitted from modeling. All domains and subunits are colored according to the same color scheme in panel (a). e, f The cryo-EM map and atomic model of the μ4-CTD-undocked AP-4 core complex. The N-terminal regions of ε (residues 1–53) and β4 (residues 1–10) are absent in the density map. Upon μ4-CTD undocking from the central core, the linker region and μ4-CTD (residues 137–453) become invisible, likely reflecting their intrinsic flexibility.
However, the individual expression of human AP-4 subunits (ε and β4) resulted in low solubility and instability in the baculovirus expression system. Therefore, we employed a co-expression strategy for all four subunits to mimic their native assembly context. However, the yield of the complex was insufficient for structural studies. To enhance the expression level of AP-4, we attempted to include its binding partner ARF1, which may facilitate the proper assembly and expression of AP-4 in the co-expression system. Fortunately, the inclusion of ARF1 (a truncated version lacking the N-terminal amphipathic helix) during co-expression significantly increased the expression level of AP-4, resulting in a sufficient yield for subsequent structural characterization. To obtain soluble AP-4, we then added a saturated amount of EDTA to remove magnesium, thereby dissociating ARF1 from the complex (Fig. 1b). With this co-expression and purification procedure, the hetero-tetrameric AP-4 core complex was successfully obtained and characterized by the size-exclusion chromatography coupled with multi-angle light-scattering (SEC-MALS) assay (Fig. 1b). As expected, the calculated molecular weight of AP-4 indicated a stoichiometry of 1:1:1:1 for the four subunits, confirming the formation of a hetero-tetrameric complex (Fig. 1b). Based on the high quality of the protein sample, we proceeded to perform cryo-electron microscopy (cryo-EM) analysis of the AP-4 core complex in its soluble form prior to ARF1 association.
Cryo-EM structure of the AP-4 core complex
We visualized the hetero-tetrameric AP-4 core complex using cryo-EM and attempted to determine its structure using the single-particle method. However, preliminary analysis of the cryo-EM data revealed the intrinsic flexibility of the core complex and produced a poor density map (see below for details), preventing the determination of its structure at high resolution. We then used the chemical cross-linking method to stabilize the complex and fix its conformation for cryo-EM studies. With the cross-linked protein sample, the cryo-EM data quality of AP-4 was significantly improved, enabling structural determination. Unexpectedly, during data processing, two distinct classes of particles were clearly observed (Supplementary Fig. 1a), indicating two different states of AP-4 in its soluble form. One class of particles showed a bowl-shaped conformation with an object docked in the center, while the other class exhibited a similar architecture but with a hollow center (Supplementary Fig. 1a). Based on previous structural studies of the AP complexes31,32,34,41, these two distinct conformations correspond to the closed and open states of the complex, with the C-terminal domain of μ4 (μ4-CTD) in the docked and undocked states, respectively.
The cryo-EM map of the AP-4 complex in the closed state, with the docking of μ4-CTD, was reconstructed at a resolution of 4.2 Å (Fig. 1c, d, Supplementary Figs. 1a, 2, 3a, b and Supplementary Table 1). The overall structure of the core complex consists of the two trunk domains of ε and β4, the medium subunit μ4, and the small subunit δ4. The two trunk domains of ε and β4 both adopt a super-helical conformation (with HEAT repeats), and their C-terminal ends merge to form a bowl-shaped structure (Fig. 1c, d). The N-terminal halves of ε and β4 capture σ4 and the N-terminal domain of μ4 (μ4-NTD), respectively, and wrap around them to form the central structural core of AP-4 (Fig. 1c, d). As expected, μ4-CTD is well docked into the central cavity formed between ε and β4 and tends to associate with the C-terminal halves of both subunits (Fig. 1c, d). On the other hand, the cryo-EM map of the core complex in the open state, with the undocking of μ4-CTD, was reconstructed at a resolution of 4.1 Å (Fig. 1e, f, Supplementary Figs. 1a, 2, 3a, c and Supplementary Table 1). The overall structure in the open conformation consists of the same four subunits and adopts a similar bowl-shaped conformation, while μ4-CTD becomes undetectable in the density map, possibly due to the intrinsic flexibility of this domain upon undocking from the central core (Fig. 1e, f). In contrast to the closed-state structure, the central cavity between ε and β4 in the bowl-shaped open structure is empty (Fig. 1c v.s. Figure 1e), indicating the undocking of μ4-CTD and aligning with the open state of AP-3, which exhibits a similar undocking conformation38 (see below structural comparisons).
Comparative analysis of the two reconstructed models of the core complex further revealed slight conformational differences. The distance measurements between β4-Y29 and σ4-Y94 in the two structures showed a more extended conformation in the closed state (Supplementary Fig. 4a), suggesting that the two opening ends of the bowled-shaped core can undergo certain conformational switching. However, given the chemical cross-linking method used for complex stabilization, the observed compactness of the central core in the open state might be caused by methodological artifacts. We reasoned that potential cross-linking between the two opening ends would restrict their flexibilities and induce a more compact conformation due to the empty central cavity (Supplementary Fig. 4a). In contrast, the central cavity in the closed state is occupied by μ4-CTD, which would preserve an expanded space between the two large subunits of the central core, thereby generating their enhanced N-terminal separation relative to the open state (Supplementary Fig. 4a).
To exclude potential cross-linking-induced artifacts, we reanalyzed the cryo-EM data of the non-cross-linked AP-4 core complex. As expected, similar to the data analysis for the cross-linked complex, two distinct classes of particles were observed (Supplementary Fig. 1b), indicating that the two conformations of the core complex are intrinsic properties rather than artifacts induced by the cross-linking method. Although the two cryo-EM maps of the complex were reconstructed at a much lower resolution due to poorer data quality (Supplementary Figs. 1b, 2, 3a and Supplementary Table 2), the two structures of the core complex (determined from the cross-linking data) could be well aligned into these maps (Supplementary Fig. 4b, c), demonstrating that the chemical cross-linking strategy induces negligible structural perturbations to the global architecture of the core complex. Notably, particle quantification revealed a marked reduction in the closed conformational population, i.e., while the cross-linked complexes maintained a 43.2% occupancy, the non-cross-linked specimens retained only 33.4% (Supplementary Fig. 1a, b). Therefore, our findings suggest that the observed conformational plasticity of μ4-CTD arises from its intrinsic dynamics, and the chemical cross-linking method can effectively stabilize the docked conformation of μ4-CTD.
Structural comparisons of the distinct conformations of the AP complexes
Based on the cryo-EM structures of the AP-4 core complex, AP-4 exhibits two distinct conformations in solution, in which the C-terminal domain of μ4 (μ4-CTD) adopts either a docked or an undocked state (Fig. 1c–f). Structural comparisons among the AP complexes (AP-1 to AP-4) suggest that these two structures of AP-4 correspond to the closed and open conformations of AP-1 and AP-2, respectively (Supplementary Fig. 5a–c). In the closed conformations of AP-1 and AP-2, μ1-CTD and μ2-CTD are docked into the central cavity of the bowl-shaped core, resembling the μ4-CTD-docked conformation of AP-4 (Supplementary Fig. 5a–c). In contrast, in their open conformations, μ1-CTD and μ2-CTD become undocked from the central cavity and tend to associate with the periphery of the structural core (Supplementary Fig. 5b, c), resulting in a compact architecture compared with the μ4-CTD-undocked state of AP-4. The conformational switch between the closed and open states of AP-1 and AP-2 is often triggered by membrane recruitment and/or cargo binding, which stabilizes their open conformations (Supplementary Fig. 5b, c). Moreover, the peripheral μ1-CTD and μ2-CTD in the open states are not fixed and can associate with different peripheral sites or dissociate from the structural core (Supplementary Fig. 5b, c), giving rise to multiple open conformations under certain conditions (such as binding of ARF1, membrane lipids and/or cargoes). Notably, AP-2 exhibits distinct open conformations and can adopt a μ2-CTD-undocked state during membrane recruitment (Supplementary Fig. 5c). In contrast, AP-4 can adopt both closed and open conformations in solution without accessory regulators (Supplementary Fig. 5a), highlighting its intrinsic conformational dynamics.
In contrast to AP-1 and AP-2, AP-3 is unable to adopt a closed conformation in solution and predominantly exists in an open conformation featured with an empty central cavity (Supplementary Fig. 5d). Interestingly, in the open conformation of AP-3, μ3-CTD is undocked from the central cavity and often absent from the core density, similar to the μ4-CTD-undocked conformation of AP-4 (Supplementary Fig. 5a, d). Moreover, the open conformation of AP-3 exhibits distinct configurations, including compact, intermediate and stretched states, and μ3-CTD can be transiently positioned at the periphery of the structural core under certain conditions (such as membrane recruitment and/or cargo binding) (Supplementary Fig. 5d). Collectively, the structural similarity between the open conformations of AP-3 and AP-4 suggests that they may share comparable intrinsic dynamic properties prior to membrane recruitment.
Inter-subunit interfaces for the closed conformation of the AP-4 core complex
The dynamic properties of μ4-CTD in the core complex prompted us to analyze the inter-subunit interfaces between μ4-CTD and the central core in detail. In the closed conformation of the core complex, μ4-CTD predominately anchors on the C-terminal half of β4 and loosely contacts μ4-NTD and σ4. Thus, the inter-subunit interfaces can be classified into two sites, the primary site (interface I) and the accessory site (interface II) (Supplementary Fig. 6a). Based on the extensive contacts between μ4-CTD and β4, interface I can be further divided into two sub-sites, interface Ia and Ib (Supplementary Fig. 6a). In interface Ia, R367 and Q369 from μ4-CTD establish a central hydrogen-bonding network with T468 and the backbone of S466 from β4; K324, E351 and R413 from μ4-CTD form an additional peripheral electrostatic network with D432, K465 and E395 from β4; and the bulky sidechain of F469 in the middle aligns well with the hydrogen-bonding network to stabilize this interface (Supplementary Fig. 6b). In interface Ib, V421, F423 and V440 from μ4-CTD make hydrophobic contacts with V359 from β4; H437 from μ4-CTD forms a hydrogen bond with T357 from β4; and the bulky sidechain of R425 from μ4-CTD leans against Y322 from β4 to lock this interface (Supplementary Fig. 6c). In interface II, E232 from μ4-CTD forms electrostatic interactions with R60 from μ4-NTD and R290 from β4, whereas R249 and D288 from μ4-CTD create potential electrostatic conflicts with K11 from μ4-NTD and E34 from σ4, respectively, which would disfavor the inter-subunit packing (Supplementary Fig. 6d).
Since AP-1 can adopt a similar closed conformation (Supplementary Fig. 5b), we next compared the inter-subunit interfaces for μ1/μ4-CTD between AP-1 and AP-4. In the closed conformation of the AP-1 core complex, the inter-subunit interfaces between μ1-CTD and the central core can be divided into similar sites, interface Ia and Ib and interface II (Supplementary Fig. 6e). In interface Ia, instead of anchoring on β1, μ1-CTD tightly associates with γ and forms extensive electrostatic and hydrophobic interactions with the C-terminal half of γ, which is distinct from interface Ia in AP-4 (Supplementary Figs. 6b, f, 7a). In interface Ib, μ1-CTD primarily forms hydrophobic interactions with β1, which is somewhat similar to interface Ib in AP-4 (Supplementary Figs. 6c, g, 7a), suggesting that this interface is highly conserved between AP-1 and AP-4. In interface II, μ1-CTD forms an electrostatic network with μ1-NTD and σ1, unlike the potential charge conflicts in interface II of AP-4 (Supplementary Figs. 6d, h, 7a). Taken together, the inter-subunit packings in AP-1 are more extensive, whereas those in AP-4 appear looser and contain potential charge conflicts, which may lead to the instability and dynamic conformations of μ4-CTD in the AP-4 core complex. However, given the limited resolution of the cryo-EM maps (Supplementary Fig. 1a), the putative electrostatic conflicts at the inter-subunit interface between μ4-CTD and the structural core are derived solely from structural modeling and should be considered as a testable hypothesis that awaits further investigations.
Biochemical assembly of the AP-4/ARF1 complex
Since the core complex mediates the association with ARF1 for membrane recruitment22, we assembled the AP-4/ARF1 complex using the AP-4 core complex for structural characterization. Based on the cryo-EM structures of the core complex, we then fused the medium subunit μ4 and the small subunit σ4 to the trunk domains of the two large subunits β4 and ε, respectively (forming the β4-μ4 and σ4-ε chains), to facilitate complex formation and simplify the purification procedure (Fig. 2a). Similar to the purification of the core complex, we co-expressed the β4-μ4 and σ4-ε chains along with the GTP-locked mutant of ARF1, ARF1(Q71L), using the baculovirus expression system and obtained the AP-4/ARF1 complex, with the co-elution of all three proteins in the analytical gel-filtration analysis (Supplementary Fig. 8a). Based on our experience with the core complex, we again used the chemical cross-linking method to stabilize the AP-4/ARF1 complex for structural studies. Moreover, the chemical cross-linking experiment showed that, in compassion to ARF1 alone, ARF1 could be clearly cross-linked with AP-4 (Supplementary Fig. 8b), confirming the formation of the AP-4/ARF1 complex for further cryo-EM analysis.

a Domain organization of the fused AP-4 core complex and ARF1. The domain boundaries of the fragments used for the complex assembly are marked. The point mutation Q71L in ARF1 is labeled. b, c The cryo-EM density map and the atomic model of the μ4-CTD-docked AP-4/ARF1 complex. The N-terminal region of ε (residues 1-48) is not visible in the density map, probably due to conformational flexibility. The inter-domain linker of μ4 (residues 140–183) and several internal loops in the core structure are omitted from modeling. All the domains and subunits are colored with the same color scheme in panel (a). d, e The cryo-EM map and atomic model of the μ4-CTD-undocked AP-4/ARF1 complex. The N-terminal region of ε (residues 1–50) is absent in the density map. Upon μ4-CTD undocking from the central core, the linker region and μ4-CTD (residues 136–453) become invisible, likely reflecting their intrinsic flexibility.
Cryo-EM structure of the AP-4/ARF1 complex
We next visualized the AP-4/ARF1 complex (with the cross-linked sample) using cryo-EM and attempted to determine its structure through the single-particle method. Interestingly, similar to the AP-4 core complex in its soluble form, two distinct categories of particles for the AP-4/ARF1 complex were also identified (Supplementary Fig. 1c), corresponding to the closed and open states of this complex (based on the structures of the core complex). During the cryo-EM data analysis, additional densities were clearly observed corresponding to the ARF1 molecule (Supplementary Fig. 1c). However, the cryo-EM maps of the AP-4/ARF1 complex in the closed and open states were reconstructed at lower resolutions of 6.8 Å and 6.6 Å, respectively (Fig. 2b–e, Supplementary Fig. 1c, 2, 3a and Supplementary Table 1). In the closed state, the core complex adopts a bowl-shaped conformation with the docking of μ4-CTD in the central cavity (Fig. 2b, c), while in the open state, the central cavity of the core complex is empty (Fig. 2d, e), in line with the structures of the core complex alone. Thus, the association of ARF1 is unlikely to have a significant impact on the μ4-CTD-mediated distinct conformations of the AP-4 core complex (see below for details).
Unexpectedly, in the two distinct states of the AP-4/ARF1 complex, one ARF1 molecule was identified associating with the N-terminal end of the large subunit ε (Fig. 2b–e), consistent with one of the two ARF1-binding sites in the AP-4 core complex identified in previous studies22. Moreover, this ARF1-binding site in the large subunit ε of AP-4 resembles the canonical ARF1-binding site in AP-1, AP-3 and the COPI F-subcomplex (in the large subunit γ, δ and γ, respectively) for their membrane recruitments (Supplementary Fig. 9). Unfortunately, due to the limited resolution of the cryo-EM maps, the nucleotide in this ARF1 molecule could not be resolved in the closed and open structures of the AP-4/ARF1 complex (Supplementary Fig. 1c). In contrast, the other ARF1-binding site in AP-1, AP-3 and the COPI F-subcomplex (in the other large subunit β1, β3 and β, respectively) is missing in the AP-4 core complex (Supplementary Fig. 9). Previous biochemical studies have demonstrated that AP-4 also possesses two ARF1-binding sites, i.e., one canonical site is located in the large subunit ε, while the other non-canonical site resides in μ4-CTD of the medium subunit μ4 (rather than in the large subunit β4)22. However, except for the canonical site in the large subunit ε, the second non-canonical ARF1-binding site in μ4-CTD was not determined since only one ARF1 molecule was found in the complex structure (Fig. 2b–e), possibly due to the low resolution of the cryo-EM map or the dynamic properties of μ4-CTD in the AP-4 core complex (see below ARF1-binding evaluation).
Multiple conformations of μ4-CTD in the AP-4 core and AP-4/ARF1 complexes
In the structures of the AP-4 core and AP-4/ARF1 complexes, a prominent feature is the dynamic conformations of μ4-CTD, which can dock onto or undock from the central core, likely inducing an equilibrium between the closed and open states of AP-4 (Fig. 3a). Moreover, the two distinct (μ4-CTD-docked/undocked) conformations of the central core are essentially similar between the AP-4 core and AP-4/ARF1 complexes (Fig. 3a), indicating that the association of ARF1 exerts minimal perturbation on the overall structure of the central core. To probe the dynamic conformations of μ4-CTD within the AP-4 core and AP-4/ARF1 complexes in solution, we employed the single-molecule fluorescence resonance energy transfer (smFRET) assay (Fig. 3b, c). For precise fluorescent probe labeling, a His₁₀-tag was engineered into specific solvent-exposed regions of μ4-CTD and the large subunit ε, allowing stable coordination of the Ni²⁺-NTA-functionalized Cy3/Cy5 dyes (Fig. 3d). The μ4-CTD and ε subunit were randomly labeled with Cy3 and Cy5, respectively, enabling the direct measurement of the interdomain distance between μ4-CTD and the central core. Moreover, to correlate the smFRET-derived distance measurements with the potential positional changes of μ4-CTD relative to the structural core, we constructed a schematic distance model between the two engineered His₁₀-tags based on SWISS-MODEL modelling using the distinct conformations of the AP complexes as structural templates42 (Fig. 3d and Supplementary Figs. 4d, 5).

a Superimposition of the atomic models of the AP-4 core and AP-4/ARF1 complexes in the μ4-CTD-docked and μ4-CTD-undocked states, respectively. In the μ4-CTD-docked state, the root-mean-square deviation (RMSD) is 0.76 Å across backbone atoms (left panel). In the μ4-CTD-undocked state, the RMSD is 1.38 Å across backbone atoms (right panel). b, c Single-molecule FRET (smFRET) distribution histograms of Cy3/Cy5-labeled AP-4 core-His10 in the soluble form (b) and ARF1-bound form (c). The left panels show the FRET efficiency distributions, revealing a high FRET population (peaked at E = 0.8), alongside a relatively broad distribution of lower FRET values (E < 0.3). The red curves represent probability distribution analysis (PDA) fits. The right panels depict the corresponding distance distributions between the labeled sites derived from the smFRET data. Source data are provided as a Source Data file. d A schematic diagram of the dynamic conformational states of the AP-4 core and AP-4/ARF1 complexes based on SWISS-MODEL (PDB templates: 9U9J; 9C5B; 6QH6) and smFRET results. The distance of the geometric centers between the two His10 tags, along with the fluorescence-labeling sites in μ4-CTD and ε are labeled. All subunits are colored with the same color scheme in Fig. 2a.
The smFRET results of the Cy3/Cy5-labeled AP-4-His10 core complex showed a high FRET population (peaked at E = 0.8), alongside a relatively broad distribution of lower FRET values (E < 0.3) (Fig. 3b), indicating that multiple conformations coexist between μ4-CTD and ε in the AP-4 core complex. Probability distribution analysis (PDA) was then applied to fit the smFRET histograms, converting them into distance distributions between the two labeled sites (Fig. 3b). The PDA-fitting data demonstrated that 24% of the population (with high FRET) exhibits a distance distribution centered at 49 Å, corresponding to the μ4-CTD-docked conformation (Fig. 3b). Notably, this distance does not fully align with that in the cryo-EM structure, likely due to the intrinsic flexibility of the loop regions together with the two engineered His10-tags for labeling (Fig. 3d and Supplementary Fig. 4d). On the other hand, the remaining populations (with low FRET) represent a wide range of unfixed states of μ4-CTD and are unexpectedly fitted with two distance distributions (rather than a simple open state), centered at 63 Å and 82 Å, respectively (Fig. 3b). Combined with structural modelling (Supplementary Fig. 4d), 51% of the population (distance distribution centered at 82 Å) corresponds to the μ4-CTD-undocked conformation, while 25% of the population (distance distribution centered at 63 Å) is likely in an intermediate state where μ4-CTD is partially undocked (Fig. 3d). Thus, these findings underscore the substantial conformational flexibility of μ4-CTD in the AP-4 core complex and support that μ4-CTD undergoes a dynamic equilibrium with multiple conformations (Fig. 3d).
The smFRET analysis of the AP-4/ARF1 complex (Cy3/Cy5-labeled μ4-CTD and ε subunit) revealed certain ARF1-induced conformational rearrangements and showed similar multiple states of μ4-CTD that align with its cryo-EM-resolved structural dynamics in the complex (Figs. 2, 3c). As expected, the PDA-fitting data also revealed three distance distributions centered at 47 Å, 63 Å, and 82 Å (Fig. 3c), corresponding to the closed (μ4-CTD-docked), partially closed, and extended (μ4-CTD-undocked) states of the complex, respectively (Fig. 3d). Interestingly, further comparative analysis revealed that ARF1 binding induces some population shifts between different conformations in the AP-4 core complex, i.e., the closed state occupancy decreased from 24% to 17%, while the extended conformation prevalence increased correspondingly from 51% to 57% (Fig. 3b, c). Thus, the association of ARF1 to the AP-4 core complex likely induces only a slight shift in the dynamic equilibrium without significantly affecting its overall structure (Fig. 3d).
To further characterize the conformational dynamics of μ4-CTD, we performed the 3D variability analysis (3DVA) in CryoSPARC using the refined particle dataset of the AP-4 core complex43. The analysis results were presented as a video, which clearly demonstrated that the observed flexibility of μ4-CTD arises from dynamic disorder. To facilitate visualization and illustrate the gradual conformational transitions captured by this analysis, several representative intermediate frames were extracted from the 3DVA output (Supplementary Fig. 4e). These representative frames highlighted the continuous range of motions sampled by μ4-CTD and provided the structural evidence supporting the smFRET biochemical data. Thus, we reasoned that the absence of resolvable density of μ4-CTD in the open states of the AP-4 core and AP-4/ARF1 complexes is possibly due to its dynamic conformations upon undocking from the central core (Fig. 3d).
Association of ARF1 to AP-4 in the membrane-anchored complex
Given the essential role of ARF1 in recruiting AP-4 to membranes, we aligned the ARF1 molecule in the AP-4/ARF1 complex with a membrane-docked ARF1 structure to create a model of the membrane-anchored complex (Fig. 4a). In the model of the μ4-CTD-docked complex, membrane docking positions the N-terminal regions of the ε and β4 subunits near the membrane interface, and notably, the identified target-binding site within μ4-CTD remains exposed and accessible for membrane cargoes in this configuration (Fig. 4a). In a similar membrane-anchored model of the μ4-CTD-undocked complex, μ4-CTD is released from the central core to associate with membrane cargoes (Fig. 4a). Based on these membrane-anchored complex models, both the closed and open conformational states of the AP-4/ARF1 complex are competent for membrane recruitment through ARF1 binding, with the dynamic docking and undocking of μ4-CTD likely playing a minimal role during the initial stages of AP-4 membrane recruitment.

a Structural alignments of the two AP-4/ARF1 complexes with a membrane-docked ARF1 to construct a membrane-anchored AP-4/ARF1 complex model. In two structural models, the structure of GTP-bound ARF1 (PDB: 2KSQ) is aligned with ARF1 in either the μ4-CTD-docked (left panel) or μ4-CTD-undocked (right panel) AP-4/ARF1 complex. The μ4-CTD/APP-peptide complex structure (PDB: 3l81) is shown in the middle panel, with the cargo-binding pocket highlighted. b Structural visualization of the AP-4-ε/ARF1 interface derived from the μ4-CTD-docked, μ4-CTD-undocked AP-4/ARF1 complex or AlphaFold3-predicted AP-4-ε/ARF1 model. The interface-related helix 3 (α3) and helix 5 (α5) in AP-4-ε are labeled. c, d A combined ribbon-and-stick model showing the interface between AP-4-ε and ARF1 in the μ4-CTD-docked (c) or μ4-CTD-undocked (d) AP-4/ARF1 complex. The Switch Ⅰ and Switch Ⅱ regions of ARF1 are labeled. e Strep-pull-down assay of ARF1 and the AP-4 core complex (wild-type and mutants). Protein samples were analyzed by SDS-PAGE and immunoblotting. The experiment is independently repeated three times (n = 3). Source data are provided as a Source Data file. f A schematic diagram of the liposome flotation assay used to detect ARF1-mediated membrane recruitment of the AP-4 core. After centrifuging, LUV (pink), streptavidin or streptavidin-ARF1 (green) and/or the AP-4 core complex (WT or L133E mutant, orange) were co-floated on top of the density gradients, and remaining unbounded proteins left at the bottom. g, k Flotation analyses using LUVs (69% DOPC, 5% DOPS, 10% DOPE, 14% Choline, 1% Rhodamine-PE and 1% Biotinyl Cap PE) incubated with (g) streptavidin-ARF1; (h) AP-4 core-WT; (i) streptavidin + AP-4 core-WT; (j) streptavidin-ARF1 + AP-4 core-WT; (k) streptavidin-ARF1 + AP-4 core-L133E. Each experiment is independently repeated three times (n = 3). Source data are provided as a Source Data file.
In both structures of the AP-4/ARF1 complex, ARF1 binds to a similar site at the N-terminal region of the large subunit ε, also consistent with the AlphaFold3-predicted AP-4-ε/ARF1 structural model (Fig. 4b). The interaction interface between ARF1 and ε is largely mediated by the canonical Switch I/II region of ARF1 and the two out-facing helices (α3 and α5) of ε (Fig. 4c, d). Although the limited resolutions of the two cryo-EM maps hindered precise residue-level mapping of the interface, key hydrophobic interactions could still be identified between the characteristic residues in the Switch I/II regions of ARF1 and α3/α5 of the ε subunit, with the helix α5 contributing more substantially to the packing interface (Fig. 4c, d). To evaluate the essential role of these contacts for assembling the AP-4/ARF1 complex, we introduced point mutations in both α3 and α5 of ε and examined the binding between ARF1 and the AP-4 core complex (Fig. 4e). Compared with wild-type AP-4, mutations in α5 (L132E and L133E) severely impaired ARF1 binding, while mutations in α3 (I99D/I102E) retained certain binding capacity (Fig. 4e), indicating that the helix α5 may serve as the primary site for ARF1 association. Collectively, our structural and biochemical data demonstrated direct interaction between the N-terminal region of ε and ARF1 in the AP-4/ARF1 complex.
To assess the role of ARF1 binding in AP-4 membrane recruitment, we performed the liposome flotation assay to evaluate the ability of membrane-bound ARF1 to recruit the AP-4 core complex (Fig. 4f). Under this experimental condition, the reconstituted liposomes (Large unilamellar vesicles, LUVs) provide a minimal membrane environment containing lipids that may be essential for AP-4 membrane recruitment. To generate ARF1-associated liposomes, biotinylated lipids were incorporated to capture streptavidin-tagged, GTP-locked ARF1(Q71L) on the membrane surface (Fig. 4f). Compared with liposomes lacking ARF1, the AP-4 core complex co-floated with ARF1-associated liposomes (Fig. 4g–k), indicating that membrane-bound ARF1 can efficiently recruit AP-4 to membranes. In contrast, the point mutation (L133E) in the ε subunit, which disrupts ARF1 binding, significantly reduced the co-flotation of AP-4 with ARF1-associated liposomes (Fig. 4g–k), supporting the critical role of the identified intermolecular interface between ARF1 and AP-4 in ARF1-mediated membrane recruitment. Taken together, these data demonstrated that the association of ARF1 with the AP-4 core complex is sufficient to drive its recruitment to membranes under a membrane-mimicking condition.
Previous studies demonstrated that, in addition to ε, the second ARF1-binding site is located in μ4-CTD22. To test whether ARF1 can bind to μ4-CTD, we further examined their interactions using the Strep-pull-down assay (Supplementary Fig. 10a). Consistent with previous studies, the binding between ARF1 and μ4-CTD could be detected and was less sensitive to mutations in the Switch I/II region of ARF1 (with the F51K mutation retaining binding capacity) (Supplementary Fig. 10a), confirming the existence of the second ARF1-binding site in μ4-CTD. Moreover, cross-linking mass spectrometry (XL-MS) analysis of the AP-4/ARF1 complex identified two specific cross-linked peptides between ARF1 and μ4-CTD (Supplementary Fig. 10b), further supporting their direct interactions. However, this ARF1-binding site was not observed in the structure of the AP-4/ARF1 complex, possibly due to its weak binding affinity or the dynamic properties of μ4-CTD, preventing stable association with ARF1 before membrane recruitment.
On the other hand, the other large subunit β1 in AP-1, β3 in AP-3 or β in the COPI F-subcomplex contains another ARF1-binding site32,38,40 (Supplementary Fig. 9). AlphaFold3-based structural predictions show a potential binding interface between ARF1 and β4 in AP-4 with relatively high pLDDT confidence scores and low predicted alignment error (PAE) values (Supplementary Fig. 11a–c). However, this predicted intermolecular interface contains pronounced charge repulsion between ARF1 and β4 (Supplementary Fig. 11d, e). To assess the predicted β4-ARF1 interface, we introduced point mutations in β4 (L82K/L85K) and checked the binding between ARF1 and the AP-4 core complex containing the β4 mutant (Supplementary Fig. 11d, f). As expected, mutations in β4 caused no detectable impairment on ARF1 binding (Supplementary Fig. 11f), indicating that β4 is dispensable for ARF1 association. Moreover, the mutations in β4 had the minimal impact on the structural folding and stability of the AP-4 core complex (Supplementary Fig. 11g, h). Taken together, based on structural analysis and biochemical data, the AP-4 core complex contains two ARF1-binding sites in the large subunit ε and the medium subunit μ4-CTD, instead of the large subunit β4, for assembling the membrane-anchored complex.
Synergistic effect of ARF1 and cargoes for efficient recruitment of AP-4
The structural modeling of the AP-4/ARF1 complex in distinct conformations for assembling the membrane-anchored complex suggests that μ4-CTD exists in a dynamic equilibrium between the docked and undocked states without association with membrane cargoes (Fig. 4a). The intrinsic flexibility of μ4-CTD tends to hinder its association with the second ARF1. Upon initial ARF1-mediated membrane recruitment, co-binding with membrane cargoes may restrain the dynamic states of μ4-CTD, fixing its conformation on the membrane in a manner favorable for efficient association with additional ARF1, which can in turn stabilize the overall membrane-anchored complex (Fig. 4a). Thus, the structural analysis of the two membrane-anchored complex models indicates the co-detection and synergistic engagement of ARF1 and membrane cargoes for efficient AP-4 membrane recruitment.
We tested this hypothesis by reconstituting AP-4 recruitment in vitro using recombinant proteins and a fluorescence microscopy-based bead assay44. In this assay, ARF1 was biotinylated and conjugated with Cy5 (Bio-ARF1-Cy5) to enable the streptavidin-mediated pull-down and fluorescence detection; the ATG9A peptide (one of the membrane cargoes of AP-4) was also biotinylated and conjugated with AF430 (AF430-ATG9A-Bio) for similar pull-down and detection; and the AP-4 core complex was only conjugated with Cy3 for fluorescence detection (AP-4-WT-Cy3) (Fig. 5a). The binding affinity between the ATG9A peptide and μ4-CTD was measured by the isothermal titration calorimetry (ITC) assay with the Kd of 1.1 ± 0.1 μM (Supplementary Fig. 12). To recapitulate the membrane recruitment process in vitro, streptavidin-coated beads were used as a biomimetic platform. These beads specifically bind to Bio-ARF1-Cy5 and AF430-ATG9A-Bio, enabling the co-localization and subsequent recruitment of the AP-4 core complex through the cargo (ATG9A) and regulator (ARF1) (Fig. 5a). The streptavidin-coated beads were pre-blocked with BSA to prevent the ATG9A peptide from diffusing into the porous matrix, thereby ensuring its efficient surface presentation.

a, b Representative confocal images showing the recruitment of AP-4-WT-Cy3 (a) and the AP-4-E395R-Cy3 mutant (b) to streptavidin beads coated with different ligands: no ligand (Group A), biotinylated ARF1-Cy5 (Group B), biotinylated AF430-ATG9A peptide (Group C), or a combination of biotinylated ARF1-Cy5 and AF430-ATG9A (Group D). Scale bar: 50 μm. c, d Schematic illustration of the experimental setup in which biotinylated ARF1-Cy5 (c) or AF430-ATG9A (d) was immobilized on beads and incubated with the AP-4 core complex (WT and the E395R mutant). Each experiment is independently repeated three times (n = 3), with the data collected from at least 10 beads (N ≥ 10) in each data set. Fluorescence intensities of bead-bound AP-4 (WT or E395R mutant) in Group B and Group C were normalized to Group A within each independent experiment. Statistical comparisons were performed on replicate means using a two-sided unpaired t test with Welch’s correction. Bars represent mean ± SD. **p-value < 0.01, ****p-value < 0.0001. Exact p-values are listed in the Source Data file. e Schematic illustration of the experimental setup in which biotinylated ARF1-Cy5 and AF430-ATG9A were co-immobilized on beads and incubated with the AP-4 core complex (WT and the E395R mutant). The expected fluorescence intensities of bead-bound AP-4 in Group D are calculated based on an addictive model, and then normalized and compared with the experimentally measured values in Group D. Each experiment is independently repeated three times (n = 3), with the data collected from at least 10 beads (N ≥ 10) in each data set. Fluorescence intensities of bead-bound AP-4 (WT or E395R mutant) in Group D were averaged within each independent experiment. Statistical comparisons were performed on replicate means between expected and measured intensities by using a two-sided paired t test. Bars represent mean ± SD. *p-value < 0.05, **p-value < 0.01. Exact p-values are listed in the Source Data file.
As the control, AP-4-WT-Cy3 was unable to associate with the beads (Fig. 5a). As expected, pre-incubation with Bio-ARF1-Cy5 or AF430-ATG9A-Bio individually promoted AP-4-WT-Cy3 recruitment to the beads (Figs. 5a, c, d), consistent with the above structural modeling of the membrane-anchored complex showing accessible ARF1-binding and cargo-binding sites (Fig. 4a). Under similar conditions, the beads were sequentially incubated with a sub-saturating amount of the ATG9A peptide followed by ARF1 to generate the ARF1/ATG9A-beads. Interestingly, the simultaneous presence of Bio-ARF1-Cy5 and AF430-ATG9A-Bio appeared to further enhance recruitment efficiency, as reflected by increased AP-4-WT-Cy3 fluorescence intensity on the beads (Fig. 5a). Given the limited bead surface area, the total occupancy of Bio-ARF1-Cy5 and AF430-ATG9A-Bio on the ARF1/ATG9A-beads (Group D in Fig. 5a) is approximately equal to the occupancy of each protein alone at saturation on their respective beads (Groups B and C in Fig. 5a). Under this assumption, the expected AP-4-WT-Cy3 fluorescence intensity on the beads could be calculated based on the additive contribution from Bio-ARF1-Cy5 and AF430-ATG9A-Bio occupancy (see detailed calculations in the Methods section). Comparison of the expected versus measured fluorescence intensity revealed a significant increase in AP-4 binding (Fig. 5e), indicating that the co-existence of ARF1 and the ATG9A peptide synergistically enhances the efficiency of AP-4 recruitment to the beads.
To assess whether the dynamic conformations of the AP-4 core complex (with flexible μ4-CTD) influence its recruitment by ARF1 and the ATG9A peptide, we introduced a point mutation (E395R) in the inter-subunit interface to disrupt the closed conformation (Supplementary Fig. 6b). As this mutation is located within the structural core without affecting on the binding of ARF1 or the ATG9A peptide, the E395R mutant (AP-4-E395R-Cy3) maintained high recruitment efficiency by either Bio-ARF1-Cy5 or AF430-ATG9A-Bio alone on the beads (Fig. 5b–d). In contrast, the E395R mutation markedly reduced the efficiency of AP-4-E395R-Cy3 recruitment to the ARF1/ATG9A-beads, as indicated by the measured fluorescence intensity being lower than the expected value (Figs. 5b, e). These results demonstrated that disruption of the dynamic conformations of the AP-4 core complex significantly impairs the synergistic recruitment by ARF1 and the ATG9A peptide.
Conformational flexibility of AP-4 is essential for membrane trafficking
Given the dynamic conformations of the AP-4 core complex are essential for the synergistic recruitment by ARF1 and the ATG9A peptide (Fig. 5), we further evaluated the potential cellular function of this conformational flexibility for AP-4-mediated membrane trafficking. Based on the structural analysis of the inter-subunit interface between μ4-CTD and the structural core, μ4-CTD primarily contacts the β4 subunit of the AP-4 core complex (Supplementary Fig. 6a). To disrupt this interface and impair the dynamic conformations of the AP-4 core complex, we introduced point mutations in the full-length β4 subunit (E395R, Y322R/V359S, and Y322R/V359S/E395R) (Supplementary Fig. 6b, c). For cellular studies, we generated β4 subunit knockout (AP-4B1 KO) HeLa cells using the CRISPR/Cas9 system and examined AP-4-mediated membrane trafficking of ATG9A by evaluating its co-localization with the TGN marker TGN46 (Fig. 6a, b and Supplementary Fig. 13). As expected, compared with control cells, AP-4B1 knockout markedly impaired ATG9A trafficking and resulted in its pronounced retention at the TGN, as indicated by increased co-localization with TGN46 (Fig. 6a, b). Re-expression of wild-type AP-4B1 largely rescued this trafficking defect, whereas expression of the mutant forms with disrupted inter-subunit interfaces failed to do so (Fig. 6c–g), indicating that the dynamic conformations of the AP-4 core complex are essential for AP-4-mediated membrane trafficking of ATG9A. Consistent with these cellular results, the bead-based assay showed that the E395R mutation in the inter-subunit interface severely impaired the efficiency of AP-4 recruitment to ARF1/ATG9A-coated beads (Fig. 5b, e), providing the potential mechanistic explanation for the observed trafficking defect resulting from disrupted conformational dynamics. Taken together, both biochemical and cellular data demonstrated the functional significance of the dynamic conformations of the AP-4 core complex.

a–f Representative images of HeLa WT cells (a) and AP-4B1-KO HeLa cells transiently transfected with plasmids encoding (b) mStayGold, (c) WT-mStayGold, (d) E395R-mStayGold, (e) Y322R/V359S-mStayGold, or (f) Y322R/V359S/E395R-mStayGold, followed by co-immunostaining for ATG9A and the trans-Golgi network (TGN) marker TGN46. Scale bars: overview, 10 μm; insets, 2 μm. For clarity, individual channels are pseudo-colored as cyan (TGN46), orange (ATG9A), green (mStayGold), and gray (nucleus), while the merged images show the blue (TGN46)/red (ATG9A)/green/gray combination, and the enlarged merged images display the blue/red combination. White arrows indicate the fluorescent signals corresponding to β4-mStayGold. White dashed lines in merged images indicate cell boundaries, and white solid box-marked regions are shown as enlarged views on the right. g Quantification of the colocalization between ATG9A and TGN46 was performed using Pearson’s correlation coefficient, based on 10-15 cells per condition. Three independent experiments were performed, and the average value from each experiment was used for statistical analysis (n = 3). Statistical significance was assessed using a one-way Welch’s ANOVA, followed by a two-sided Dunnett T3 multiple comparisons test comparing each experimental group to the control condition. Bars represent mean ± SD. **p-value < 0.01; ***p-value < 0.001. Exact p-values are listed in the Source Data file. h A working model for the ARF1-mediated membrane recruitment of the AP-4 core complex. Initially, the AP-4 core complex adopts multiple conformations with a dynamic equilibrium between the μ4-CTD-docked and μ4-CTD-undocked states. Either membrane-associated ARF1 or the membrane cargo ATG9A alone can bind to and recruit AP-4 to membranes, but neither is sufficient to assemble functional trafficking vesicles in the absence of the other. The coexistence of ARF1 and ATG9A synergistically enhances AP-4 membrane recruitment, and upon membrane recruitment, the simultaneous binding of ARF1 and ATG9A further stabilizes the membrane association of AP-4 through multiple interactions, enabling productive vesicle formation and efficient cargo transport.
Discussion
Among the distinct AP complexes, AP-4 assembles non-clathrin-coated vesicles and is recruited to target membranes through its association with ARF118,22, but the underlying mechanism remains unclear. In this study, we determine the structures of the AP-4 core and AP-4/ARF1 complexes, revealing several features for AP-4 and its membrane recruitment. Firstly, the AP-4 core exhibits a dynamic equilibrium between two distinct conformations before association with ARF1 (Fig. 1), which is somewhat distinct from other AP complexes (Supplementary Fig. 5). Secondly, the binding of ARF1 induces only slight conformational changes in the AP-4 core and the AP-4/ARF1 complex adopts two similar states (Fig. 2), which are both competent for the initiation of membrane recruitment (Fig. 4). Thirdly, an intermediate (partially closed) state of the AP-4 core and AP-4/ARF1 complexes identified through the smFRET assay supports the dynamic equilibrium of μ4-CTD with multiple conformations (Fig. 3). Fourthly, the efficient recruitment of AP-4 likely requires the coexistence of ARF1 and cargoes, and their synergistic engagement may ensure proper AP-4 membrane recruitment (Fig. 5). Finally, the conformational flexibility of the AP-4 core complex is essential for the synergistic recruitment and AP-4-mediated membrane trafficking (Figs. 5, 6). These findings provide mechanistic insights into the ARF1-mediated membrane recruitment of AP-4 to control membrane trafficking.
In the structures of the AP-4 core and AP-4/ARF1 complexes, μ4-CTD can dock onto or undock from the central core (Figs. 1, 2), representing a closed or an open state of the overall complex. Moreover, an intermediate state with partial undocking of μ4-CTD can be traced using the smFRET assay, supporting the dynamic conformational equilibrium of AP-4 (Fig. 3). This dynamic equilibrium induced by μ4-CTD in AP-4 is somewhat distinct from that in other AP complexes (Supplementary Fig. 5), e.g., in soluble AP-1 and AP-2, μ1-CTD and μ2-CTD tend to tightly associate with the central core and adopts a closed conformation31,41, while in soluble AP-3, μ3-CTD is unable to interact with the central core and exists in an open conformation37. Given that both AP-1 and AP-4 function at the TGN to mediate the trafficking of distinct membrane cargoes, AP-1 was selected for further structural comparison with AP-4 to delineate their detailed inter-subunit interfaces (Supplementary Fig. 6). In comparison to the closed conformation of AP-1, μ4-CTD of AP-4 only loosely contacts with the central core and exhibits potential charge conflicts (Supplementary Fig. 6), which may destabilize the association of μ4-CTD and lead to its dynamic conformations. Moreover, the linker between μ4-NTD and μ4-CTD of AP-4 is longer than the corresponding regions in AP-1, AP-2 and AP-3 (Supplementary Fig. 7b), which may also increase the intrinsic flexibility of μ4-CTD and contribute to its dynamic equilibrium. Nevertheless, the putative inter-subunit charge conflicts and the relatively longer internal linker within μ4 warrant further investigations.
In the AP-4/ARF1 complex, only one ARF1 molecule was found to associate with the central core (Fig. 2), although previous studies reported two ARF1-binding sites in AP-422. We further biochemically validated the direct interaction between ARF1 and μ4-CTD, which indeed contains the second ARF1-binding site (Supplementary Fig. 10). Unfortunately, this μ4-CTD-mediated association was unresolved in the structure of the AP-4/ARF1 complex. The possible explanation could be that, upon undocking from the central core, the structural dynamics and intrinsic flexibility of μ4-CTD would be unfavorable for stable association with ARF1 in solution, while in the docked state, μ4-CTD is somewhat sequestered in the central core and the potential ARF1-binding site might be inaccessible (Fig. 2). Based on the structural analysis of the AP-4 core and AP-4/ARF1 complexes and the smFRET data, the binding of ARF1 only triggers subtle conformational changes in the core complex, and μ4-CTD retains a dynamic equilibrium between the docked and undocked states (Fig. 3). This structural characteristic of μ4-CTD in AP-4 is distinct from that of μ1-CTD in AP-127, i.e., the binding of ARF1 to AP-1 leads to significant structural changes (Supplementary Figs. 5, 9). In contrast, since AP-3 adopts an open conformation with flexible μ3-CTD, the association of ARF1 with AP-3 has little impact on its overall structure (Supplementary Figs. 5, 9). Moreover, upon ARF1-mediated membrane recruitment, the recognition of membrane cargoes fixes μ3-CTD and rigidifies the overall conformation of AP-3, facilitating the binding of the second ARF1 for polymerization38. Under this condition with membrane cargoes, a similar cargo-ARF1 cooperation mechanism may also occur in AP-4, i.e., the association of membrane cargoes would fix μ4-CTD and then strengthen its binding to the second ARF1 for stable and efficient membrane recruitment (Fig. 6h). This cooperative hypothesis is in line with the synergistic effect of ARF1 and cargoes for higher efficient recruitment of AP-4 in the bead-based assay (Fig. 5). However, due to the lack of structural information regarding the second ARF1 in the AP-4/ARF1 complex, the detailed binding site and associating manner for the second ARF1 remain to be determined and the structure of the AP-4/ARF1 complex in a membrane context is worthy of further investigations.
The conformational flexibility of the AP-4 core complex is primarily induced by the dynamic conformations of μ4-CTD (Figs. 1, 2). To evaluate whether this conformational flexibility is essential for AP-4 membrane recruitment and cellular functions, we introduced point mutations in the inter-subunit interface between μ4-CTD and the structural core to disrupt the dynamic conformations of the AP-4 core complex (Supplementary Fig. 6). Interestingly, the point mutation in the inter-subunit interface severely decreased the synergistic effect of ARF1 and cargoes for AP-4 recruitment in the bead-based assay (Fig. 5). While the bead-based system serves only as a simplified proxy for membrane recruitment and recapitulates certain key aspects of AP-4 recruitment, additional membrane-associated factors may further modulate this process. Nevertheless, consistent with the bead-based AP-4 recruitment assay, similar interface mutations severely impaired AP-4-mediated trafficking of the membrane cargo ATG9A, leading to the cargo retention at the TGN (Fig. 6). Thus, all biochemical and cellular data support the functional importance of the conformational flexibility of the AP-4 core complex and underscore its essential role in membrane recruitment and trafficking.
Since AP-4 is essential for the membrane trafficking in neurons, genetic mutations of this complex lead to severe neuronal disorders with clinical phenotypes of progressive spastic paraplegia, intellectual disability, and microcephaly, collectively referred to as AP-4-deficiency syndrome18. We mapped the pathogenic and likely pathogenic point mutations onto the structure of the AP-4 core complex (Supplementary Fig. 14a). The disease-related mutations are not only enriched in the large subunit β4 and the small subunit σ4 but also prominently localized to the μ4-CTD sub-domain45 (Supplementary Fig. 14a). Interestingly, a disease-related mutation, R367Q, in μ4-CTD is located in the inter-subunit interface between μ4-CTD and β4, responsible for the docking of μ4-CTD (Supplementary Fig. 6). This mutation would disrupt the inter-subunit interface and impair the dynamic equilibrium of μ4-CTD between distinct states, suggesting that the multiple conformations of μ4-CTD could be physiologically relevant. Moreover, a recent study of this disease-related mutation identified a small molecule, rutin, which restores the cellular function of AP-4. Rutin can be docked into a small pocket near R367 to lock the inter-subunit interface and compensate for the negative impact of the R367Q mutation, based on the SWISS-MODEL homology model of AP-446 (Supplementary Fig. 14b). Consistently, the structure of the AP-4 core complex reveals a similar pocket around the inter-subunit interface between μ4-CTD and the central core that could potentially be targeted by rutin (Supplementary Fig. 14b). Thus, the dynamic conformations of μ4-CTD are likely essential for the physiological function of AP-4, and the structures of the AP-4 core and AP-4/ARF1 complexes may pave the way for further therapeutic interventions of AP-4-deficiency syndrome.
Finally, based on the structures of the AP-4 core and AP-4/ARF1 complexes, together with available biochemical data, a working model for ARF1-mediated membrane recruitment of the AP-4 core complex for membrane trafficking could be proposed (Fig. 6h). In this model, prior to ARF1 binding, the AP-4 core complex adopts multiple conformations and exists in a dynamic equilibrium between these distinct states, characterized by the docking and undocking of μ4-CTD (Fig. 6h). Either membrane-associated ARF1 or the membrane cargo ATG9A alone can bind to and recruit AP-4 to membranes, but neither is sufficient to assemble functional trafficking vesicles in the absence of the other (Fig. 6h). The coexistence of ARF1 and ATG9A synergistically enhances AP-4 membrane recruitment, thereby promoting the proper assembly of trafficking vesicles (Fig. 6h). During this process, the dynamic conformations of AP-4 facilitate the cooperative recognition of ARF1 and ATG9A, and disruption of the conformational dynamics of AP-4 severely impairs its membrane recruitment and trafficking activity (Fig. 6h). This model also suggests that the synergistic detection of ARF1 and ATG9A on membranes ensures the high specificity and efficiency of AP-4 recruitment, a process intrinsically supported by its conformational flexibility in solution. Upon membrane recruitment, the simultaneous binding of ARF1 and ATG9A further stabilizes the membrane association of AP-4 through multiple interactions, enabling productive vesicle formation and efficient cargo transport (Fig. 6h).
Methods
Plasmid construction
DNA sequences encoding the four subunits of the AP-4 complex: ε (UniProt ID: Q9UPM8), β4 (UniProt ID: Q9Y6B7), μ4 (UniProt ID: O00189), σ4 (UniProt ID: Q9Y587) and ADP-ribosylation factor 1 (ARF1) (UniProt ID: P84077) were amplified from a human cDNA library. For protein expression and purification, DNA sequences encoding the four subunits of the AP-4 core complex: ε-trunk (1–612), β4-trunk (1–568), μ4 and σ4 were individually cloned into a pFastBac™1 (Gibco, 10359016) vector, with ε and β4 fused with a C-terminal and N-terminal His8-Strep tag, respectively. Alternatively, a fusion strategy was introduced into the construct design. μ4 (1–453) was fused to the C-terminus of β4-trunk (1–576), and σ4 (1–143) was linked to the N-terminus of ε-trunk (1–612) that contains a C-terminal His8-Strep tag. The two fused fragments were then subcloned into a pFastBac™ Dual (Gibco, 10712024) polycistronic vector. DNA sequence encoding the GTP-locked Q71L mutant ARF1 (18–181), ARF1(Q71L), was cloned into the pFastBac™HTA (Gibco, 10584027) vector for the co-expression of the AP-4/ARF1 complex. Primers used in this study were listed in Supplementary Table 3.
Protein expression and purification
The AP-4 core complex and ARF1 were co-expressed in insect sf9 cells (Gibco,11496015) by using the Bac-to-Bac baculovirus expression system (Gibco,10359016). The plasmids were transformed into Escherichia coli DH10Bac cells (Gibco, 10359016) to acquire the bacmids. The P1 baculoviruses were obtained by transfecting the bacmids into sf9 cells using the Cellfectin II reagent (Gibco, 10362100), and the P2 baculoviruses were collected after 72 h of amplification from the P1 baculoviruses. For the large-scale production of the AP-4 core complex, the high density of sf9 cells (~ 2 × 106 cells/ml) were infected with the P2 viruses of four AP-4 subunits and ARF1 simultaneously at a total ratio of 0.75% (v/v). After ~ 72 h cultivation, the cells were harvested and suspended in the buffer containing 50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 5 mM MgCl2, 5 mM imidazole and 0.5 mM Guanosine triphosphate (GTP, Sigma-Aldrich, 36051-31-7). Recombinant proteins were purified by Ni2+-Sepharose™ 6 Fast Flow (Cytiva, 17531801) affinity column, followed by an extra affinity purification using the Strep-Tactin Sepharose resin (IBA Lifesciences, 2-1201-025). Upon adding 5 mM EDTA during this process, magnesium was removed, leading to the dissociation of ARF1 from the AP-4 core complex (verified by SDS-PAGE). Protein samples were concentrated and cross-linked by 0.5 mM disuccinimidyl glutarate (DSG, Thermo Fisher Scientific, 20593) at 25 °C for 30 min. The cross-linked AP-4 core complex was subsequently purified by the SuperoseTM6 Increase 10/300 GL column (Cytiva, 29091596) equilibrated with the buffer containing 50 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM EDTA. For the large-scale production of the AP-4/ARF1 complex, the expression and purification procedures were similar to that of the AP-4 core complex, except that the buffer condition for the AP-4/ARF1 complex was modified by adding 5 mM MgCl2, 0.5 mM GTP, without EDTA.
Size-exclusion chromatography coupled with multi-angle light scattering (SEC-MALS)
Protein samples (~ 1.5 mg/ml in 50 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM EDTA) were analyzed with static light scattering by injection of them into an Agilent FPLC system with a size-exclusion chromatography (SuperdexTM200 Increase 100/300) (Cytiva, 28990944) for MALS (WTC-030S5, Wyatt Technology). The chromatography system was coupled with an 18-angle light-scattering detector (DAWN HELEOS II, Wyatt Technology) and a differential refractive index detector (Optilab rEx, Wyatt Technology). Masses (molecular weights) were calculated with ASTRA (Wyatt Technology). Bovine serum albumin (Sigma) was used as the calibration standard.
Cryo-EM sample preparation and data collection
Aliquots of protein samples (the non-cross-linked or cross-linked AP-4 core complex, 0.5 mg/mL; the cross-linked AP-4/ARF1 complex, 0.35 mg/mL) were applied to the glow-discharged holey carbon grids (Electron Microscopy Sciences, Q250AR1.3, Quantifoil, Cu, R 1.2/1.3, 300 mesh) and was then plunge frozen in liquid ethane using a Vitrobot Mark IV robot (Thermo Fisher) set to 100% humidity, 16 ˚C, and blot time 9 s. Cryo-EM samples were imaged using a 300 kV Titan Krios G2 (Thermo Fisher Scientific) equipped with a K2 Summit direct electron detector (Gatan) and a GIF Quantum LS energy filter (Gatan). Images were collected at a nominal magnification of 130,000, yielding a final pixel size of 1.04 Å for super resolution mode. Dose fractionated movies were collected at a defocus range of − 1.2 μm to − 2.2 μm and an exposure rate of ~ 9.2 e- (pixel/s) with total exposure time 6.72 s frames and a total exposure of 60 electrons per Å2. SerialEM was used to automatically acquire the video stacks.
Cryo-EM data processing
All cryo-EM data processing steps were performed using cryoSPARC47. For the cross-linked AP-4 core complex, a total of 5264 video stacks were initially acquired. All particles were picked using Blob Picker, generating the data sets containing 1,345,730 particles. A number of rounds of two-dimensional (2D)-classification were performed, and the classes with structural features were chosen for Template Picker, yielding a total of 4,162,734 particles. The extensive 2D-classifications and ab initio reconstruction generated 5 classes of density maps, two of which exhibited typical structure features, such as α-helices, and were consequently used as the initial references. The significant difference between the two classes is that an appropriate β-sheet structure is docked at the center of the “core” in one class, while the density of the β-sheet structure is absent in the other class. The 3D-classification was further conducted and yielded two medium-resolution 3D-classes, which contain 208,640 (μ4-CTD-docked AP-4) and 275,020 (μ4-CTD-undocked AP-4) particles, respectively. The non-uniform refinement led to the final maps with the Gold Standard Fourier Shell Correlation (GSFSC) resolutions of 4.2 Å and 4.1 Å, respectively. For the non-cross-linked AP-4 core complex, a total of 689 video stacks were initially acquired. Particles were picked using Blob Picker, generating the data sets containing 636,338 particles. Several rounds of two-dimensional (2D)-classification were performed, resulting in a total of 39,525 particles. Further 3D-classification without alignment was conducted to distinguish the μ4-CTD-docked (13,194 particles) and μ4-CTD-undocked (26,331 particles) AP-4. The final reconstructed maps were resolved at a global resolution of 6.9 Å and 7.0 Å (GSFSC criterion), respectively.
For the cross-linked AP-4/ARF1 complex, a total of 4676 video stacks were initially acquired. Particles were picked using Template Picker, generating the data sets containing 3,708,344 particles. The extensive 2D-classifications and ab initio reconstruction generated 5 classes, similar to those of the cross-linked AP-4 core complex, two of which displayed favorable structural features, such as α-helices, and were consequently used as the initial references. Further 3D-classification without alignment was conducted, following local refinement, and gave rise to two low-resolution 3D-classes containing 13,372 (the μ4-CTD-docked AP-4/ARF1 complex) and 18,467 particles (the μ4-CTD-undocked AP-4/ARF1 complex), respectively. The CTF refinement and non-uniform refinement generated the final maps with the GSFSC resolutions at 6.8 Å and 6.6 Å, respectively.
The data-processing flowcharts for the cross-linked/non-cross-linked AP-4 core complex, as well as for the cross-linked AP-4/ARF1 complex, were summarized in Supplementary Fig. 1 to illustrate the complete stepwise pipelines. For 2D classification, representative class averages from multiple rounds of 2D classification were included, with the number or percentage of particles retained or discarded after each round indicated in the flowcharts (Supplementary Fig. 1a–c). For 3D classification and refinement, detailed steps from ab initio model generation to heterogeneous refinement were included, and several intermediate 3D maps were presented to visualize the evolution of particle classes and conformational features (Supplementary Fig. 1a–c). We performed the directional Fourier shell correlation (3D FSC) analysis to evaluate the anisotropy of the map resolution (Supplementary Fig. 2c). The 3D FSC results indicated that all cryo-EM maps display a certain degree of orientation preference, with more pronounced anisotropy observed in the cross-linked AP-4 core complex, as reflected by the relatively lower cFAR (conical FSC Area Ratio) value (Supplementary Fig. 2c).
AlphaFold predictions
All predictions were carried out by using the AlphaFold3 server (https://alphafoldserver.com). For predicting the AP-4-ε/ARF1 complex, the inputs were: AP-4-ε trunk (residues 1–612; UniProt ID: Q9UPM8), ARF1 (residues 18–181; UniProt ID: P84077), GTP and Mg²⁺. The AlphaFold3 model served only as a guide for cryo-EM model building and was not presented as a final structural model. pLDDT (predicted local distance difference test) score of the prediction was shown in Supplementary Fig. 15. For predicting the β4/ARF1 complex, the inputs include: β4 trunk (residues 1–568; UniProt ID: Q9UPM8), ARF1 (residues 18–181; UniProt ID: P84077), GTP and Mg²⁺. pLDDT score and PAE (predicted alignment errors) matrix of the prediction were shown in Supplementary Fig. 11. The input sequences for all predictions were provided in the Source Data file.
Cryo-EM model building and refinement
The μ4-CTD-docked/undocked AP-4 models were generated using the μ1-CTD-docked AP-1 core structure (PDB: 1W63) as a template. These initial models were rigid-body docked into their corresponding cryo-EM maps in UCSF Chimera48 to derive structural coordinates. The backbones and side chains of the AP-4 core complex models were manually built by mutating the template in Coot49, followed by iterative refinement with Coot and PHENIX49,50. For the μ4-CTD-docked/undocked AP-4/ARF1 complex, the initial models were constructed by combining the μ1-CTD-docked AP-1 core structure (PDB: 1W63) and the GTP-bound ARF1 structure extracted from the PDB entry 1J2J. These models were similarly docked into the cryo-EM maps using UCSF Chimera48. The AlphaFold3 model of the AP-4-ε/ARF1 complex was used to assist in assigning the intermolecular interface (Fig. 4b and Supplementary Fig. 15). The AlphaFold3 model predicted a binding interface between the Switch I/II regions of ARF1 and the α3/α5 helices of the ε subunit (Fig. 4b), which aligned well with the corresponding cryo-EM density map. Based on this structural alignment, rigid-body docking was performed to position the AP-4 core and ARF1 within the density map, yielding an initial complex model. Due to the low resolution of the AP-4/ARF1 complex maps, the densities corresponding to GTP and magnesium were absent, and these ligands were consequently omitted from the models. The backbones of the AP-4 core and ARF1 were manually adjusted in Coot49 and iteratively refined using Coot and PHENIX49,50 under secondary-structure and geometry restraints, with additional rigid-body domain restraints applied to prevent potential overfitting. All structural figures were prepared using UCSF ChimeraX51.
We generated the map-to-model FSC curves for all cryo-EM reconstructions by comparing the final refined atomic models against their corresponding maps (Supplementary Fig. 3a). Notably, the resolutions at the 0.143 cutoff in the corrected map-to-map FSC curves are essentially consistent with those at the 0.5 cutoff in the corresponding model-to-map FSC curves of the cross-linked AP-4 core complex (Supplementary Fig. 3a), supporting the reliability of our reconstructed atomic models.
Chemical cross-linking and in-gel digestion
The AP-4/ARF1 complex was cross-linked by 0.5 mM disuccinimidyl suberate (DSS, Thermo Fisher Scientific, 21655) at 25 °C for 30 min and purified by the SuperoseTM6 Increase 10/300 GL column (Cytiva, 29091596) equilibrated with the buffer containing 50 mM Tris-HCl, pH 7.5, 200 mM NaCl, 1 mM EGTA, 5 mM MgCl2, 0.5 mM GTP. The cross-linked complex samples were then analyzed by SDS-PAGE, and the gel bands were manually excised and dehydrated in 100% acetonitrile for 10 min and then dried in a speedvac for 15 min. After the reduction (with 10 mM DTT in 25 mM NH4HCO3 for 45 min at 56 °C) and alkylation (with 40 mM iodoacetamide in 25 mM NH4HCO3 for 45 min at room temperature in the dark) processes, the gel plugs were washed with 50% acetonitrile in 25 mM ammonium bicarbonate twice, dried using a speedvac, and digested with sequence-grade modified trypsin in 25 mM NH4HCO3 overnight at 37 °C, following chymotryptic digestion for 4 h. The enzymatic reaction was finally stopped by adding formic acid to a 1% final concentration, and the solution was then used for liquid chromatography mass spectrometry/mass spectrometry (LC-MS/MS) analysis.
Liquid chromatography mass spectrometry/mass spectrometry (LC-MS/MS) analysis
For the cross-linked AP-4/ARF1 complex samples, the peptide mixtures after digestion were analyzed by an Orbitrap Exploris 480 (Thermo Scientific) equipped with an Easy n-LC 1200 HPLC system (Thermo Scientific). The peptides were loaded onto a 100 μm (internal diameter, ID) × 2 cm fused silica trap column packed in-house with reversed phase silica (Reprosil-Pur C18 AQ, 5 μm, Dr. Maisch GmbH) and then separated on a 75 μm (internal diameter, ID) × 25 cm C18 column packed with reversed phase silica (Reprosil-Pur C18 AQ, 1.9 μm, Dr. Maisch GmbH). The peptides bounded on the column were eluted by a concave 73 min water-acetonitrile gradient from 4% to 95% solvent B (80% (v/v) acetonitrile, 0.1% (v/v) formic acid) at a flow rate of 300 nl/min. The MS analysis was performed with the FAIMS Pro interface (Thermo Scientific). With the data-dependent acquisition mode, the MS data were acquired at a high resolution 60,000 (m/z 200) across the mass range of 350–1500 m/z, and the target value was 3.00 E + 06 with a maximum injection time of 22 ms. The data dependent mode was selected as the cycle time mode, which was set as 2 s. The precursor ions were selected from each MS full scan with an isolation width of 1.6 m/z for fragmentation in the Ion Routing Multipole with normalized collision energy of 28%. Subsequently, the MS/MS spectra were acquired at resolution 15,000 at m/z 200, and the target value was 7.50 E + 04 with a maximum injection time of 22 ms. The raw data were analyzed with pLink2.3.73052 for the cross-linked peptide identification against target protein sequences. Some important searching parameters were set as followings: trypsin and chymotrypsin were used as enzymes and three missed cleavages were allowed for searching; the mass tolerance of precursor was set as 20 ppm, and the product ions tolerance was 20 ppm; the cysteine carbamidomethylation was selected as a fixed modification, but the methionine oxidation was as a variable modification. A 5% false discovery rate (FDR) cutoff was set at the spectral level for the peptide filter.
Pull-down assay
For the pull-down assay of the AP-4 core complex (with the mutations in ε-trunk and β4-trunk) and ARF1, the lysates of insect cells expressing the Strep-tagged AP-4 core complex (wild-type or its mutants) and the Escherichia coli lysates expressing Flag-tagged ARF1 (wild-type) were prepared using the RIPA lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with Protease Inhibitors Cocktail (Bimake, B14001). ARF1 was incubated with the AP-4 core complex (wild-type or its mutants) for 10 min in the pull-down buffer. 10 μl of Streptactin beads 4FF (Smart-Lifesciences, SA053005) were added, and the mixtures were rotated for 1 h at 4 °C. The beads were washed four times, and the bound protein samples were analyzed by SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. For immunoblotting, the Strep-tag antibody (HRP-conjugated, AF2927) and the Flag-tag antibody (HRP-conjugated, AF2855) were used. Chemiluminescent signals were detected using a Mini-Chemiluminescent Imager (MiniChemi 610 Plus, Beijing Sage Creation Science Co.).
For the pull-down assay of μ4-CTD (residues 184–453) and ARF1, the lysates of insect cells expressing Strep-tagged μ4-CTD and the Escherichia coli lysates expressing Flag-tagged ARF1 (wild-type or its mutants) were prepared using the RIPA lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with Protease Inhibitors Cocktail (Bimake, B14001). Subsequent experimental procedures followed those described above with two modifications, i.e., the beads incubated with μ4-CTD and ARF1 were washed five times (instead of four) with the pull-down buffer. The lysates of Strep-tagged GFP were used as a negative control.
Fluorescence labeling of proteins for smFRET
The His10-tags were introduced into the preconstructed plasmids, i.e., one was inserted between residues G387 and P388 of μ4, while the other (SGG-His10-GGS) was incorporated between residues K330 and S331 of ε. A PreScission protease (3 C protease) recognition sequence (LEVLFQ ↓ GP) was engineered between ε and its C-terminal His₈-Strep tag. The N-terminal His8-tag was deleted from the preconstructed β4 plasmid through molecular cloning. The C-terminal His8-Strep tag of the AP-4 core complex was cleaved with 3 C protease at 4 °C overnight and then removed by SuperdexTM200 100/300 with 1 × PBS. Subsequently, the cross-linked AP-4 core complex was incubated with HIS Lite™ Cy3 (AAT Bioquest, 12620) and HIS Lite™ Cy5 (AAT Bioquest, 12619) at a 1:1.5 molar ratio in the reaction buffer (50 mM Tris-HCl, pH 8.0, 200 mM NaCl, 5 mM MgCl₂). The excessive dyes were removed using a PD-10 desalting column packed with Sephadex G-25 resin (Cytiva, 17085101). The fresh dual-color labeled AP-4 core complex was immediately used for the smFRET experiment.
Confocal smFRET experiment
The confocal smFRET experiment was carried out on a home-built confocal microscope based on a Nikon Ti-U inverted microscope, similar to that used in our previous study53, except that the alternating-laser excitation (ALEX) setup was applied to identify the donor/acceptor dual-labeled species54.
The beams of a 532 nm laser and a 640 nm laser (Coherent) were combined using a dichroic mirror (ZT532, Chroma) and coupled into a single-mode optical fiber. An acousto-optical tunable filter (AOTF, Crystal Technology Inc) was used for wavelength selection and intensity modulation. The output beam from the fiber was collimated and directed to the back port of the Ti-U inverted microscope, with an excitation power of 50 μW for 532 nm and 30 μW for 640 nm. The laser was focused by a 100 × oil-immersion objective (N.A. = 1.4, Nikon) into the sample solution. The fluorescence emission was collected by the same objective, passed through a 50 μm pinhole and split into donor and acceptor channels by a dichroic mirror (T660LPXR, Chroma). The separated fluorescence emission signals were then focused into two avalanche photodiode detectors (SPCM-AQRH-14, Excelitas) through respective emission filters (Chroma, ET595/50 for Cy3 and ET690/50 for Cy5). The photon trace was recorded using a four-channel photon counting card (Coolight). The photons detected in both channels were assigned to one of four excitation-emission categories – \({I}_{{{\rm{Dex}}}}^{{{\rm{Dem}}}},{I}_{{{\rm{Dex}}}}^{{{\rm{Aem}}}},\,{I}_{{{\rm{Aex}}}}^{{{\rm{Dem}}}},\,{I}_{{{\rm{Aex}}}}^{{{\rm{Aem}}}}\) – based on their arrival time relative to the laser alternation sequence. The lasers were toggled between 532 nm and 640 nm every 100 μs, with photon collection performed in 10 μs bin intervals. The synchronization between laser alternation and data acquisition was achieved using an analog card (PCI-6733, National Instruments), which was connected to both the AOTF and the photon counting card. For analysis, the photons were binned every 500 μs for either donor excitation or acceptor excitation. To avoid potential excitation crosstalk, the first and last 10 μs of each alternation period were discarded. Bursts were identified using a threshold of \({I}_{{{\rm{Dex}}}}^{{{\rm{Dem}}}}+{I}_{{{\rm{Dex}}}}^{{{\rm{Aem}}}} > \)25 photons per 500 μs bin. The apparent FRET efficiency (Eapp) and the donor-acceptor stoichiometry (S) were calculated as follows:
where \({I}_{{{\rm{Dex}}}}^{{{\rm{Dem}}}},{I}_{{{\rm{Dex}}}}^{{{\rm{Aem}}}},\,{I}_{{{\rm{Aex}}}}^{{{\rm{Dem}}}},\,{I}_{{{\rm{Aex}}}}^{{{\rm{Aem}}}}\) are the fluorescence intensity of donor and acceptor emission upon donor or acceptor excitation respectively. Only events with a stoichiometry value S value between 0.30 and 0.65 were selected to generate the smFRET distribution histograms.
The dual-color labeled AP-4 core was diluted to approximately 100 pM in reaction buffer (50 mM Tris-HCl, pH 8.0, 200 mM NaCl, 5 mM MgCl₂) and loaded onto the coverslip for smFRET detection. To investigate the ARF1-induced conformational change of AP-4 core, 0.5 μM dual-color labeled AP-4 core was pre-incubated with 5 μM ARF1 and 5 mM GTP for 1 h at 4 °C. Subsequently, the AP-4/ARF1 complex (in buffer containing 50 mM Tris-HCl, pH 8.0, 200 mM NaCl, 5 mM MgCl₂, 0.5 mM GTP) were loaded onto the coverslip for smFRET detection. The smFRET data were collected for 10 min for each sample, with a total of thirteen replicates for the AP-4 core and eight replicates for the ARF1-bound AP-4. The resulting smFRET histograms were subjected to probability distribution analysis (PDA) to obtain the distance distribution between the dye labeling sites.
Probability distribution analysis (PDA) of smFRET
To determine the distance between the donor and acceptor dyes from smFRET data, we applied probability distribution analysis (PDA)55. This method calculates the theoretical probability of observing a given ratio of acceptor-to-donor photon counts, equivalent to the apparent smFRET efficiency. The background photon counts in both the donor and acceptor channels were assumed to be the Poisson distribution:
where the mean background 〈B〉 was 0.3 in our case. Meanwhile, the unknown conditional probability of detecting \({F}_{A}\) acceptor photons out of a total photon \(F\), was assumed to obey a binomial distribution:
where ε denotes the apparent FRET efficiency. This efficiency is related to the donor-acceptor distance \({R}_{{DA}}\) by:
with parameters α = 0.11, γ = 1.17 and Förster radius R0 = 60 Å. Together, these expressions form the core of the PDA framework, enabling the conversion of smFRET efficiency histograms into distance distributions between dye-labeled sites.
Liposome preparation
Individual lipids (Avanti Polar Lipids) were dissolved in chloroform at 10 mg/mL and stored at − 20 °C. Lipid mixtures with the following molar composition were used: DOPC (18:1 (Δ9-Cis) PC), DOPS (18:1 PS), DOPE 18:1 (Δ9-Cis) PE, Rhodamine-PE (18:1 Liss Rhod PE), 16:0 Biotinyl Cap PE. Lipids were mixed at the proper ratio (69 DOPC: 5 DOPS: 10 DOPE: 14 Chol: 1 Rhodamine-PE, 1 Biotinyl Cap PE) to a final concentration of 10 mM and dried under nitrogen, followed by vacuum for at least 3 h. Dried lipids were hydrated in buffer containing 50 mM Tris, pH 8.0, 150 mM NaCl, subjected to ten freeze–thaw cycles, and extruded through polycarbonate membranes (100 nm pore size; Whatman) to generate large unilamellar vesicles (LUVs).
Liposome flotation assay
The ARF1 (18–181) Q71L fragment was subcloned into the C-terminus of a pET-28a-streptavidin vector. For preparing proteoliposomes, 2 μM of purified streptavidin or streptavidin-ARF1 was incubated with liposome (2 mM of total lipids) in the reaction buffer containing 50 mM Tris, pH 8.0, 150 mM NaCl, 5 mM MgCl2, 0.5 mM GTP at 4 °C for 60 min. Subsequently, 0.5 μM of purified AP-4 core complex was added in a total reaction volume of 30 μl and incubated at 4 °C for 60 min. The sample was mixed with Sucrose in the reaction buffer to a final concentration of 40% Sucrose in a total volume of 130 µL and transferred to the bottom of an ultracentrifuge tube. The gradient was formed by overlaying 100 μL of 30% sucrose and 20 μL of reaction buffer on top. Samples were centrifuged using a SW 55 Ti rotor (Beckman Coulter; Boulevard Brea, CA) at 174,000 g for 60 min. Samples from the top and the bottom of the gradient (50 μl/fraction) were taken and analyzed by SDS-PAGE followed by silver staining.
Thermal denaturation assay
The thermal denaturation analysis of the AP-4 core wild type and the L82K/L85K mutant were performed on a Chirascan Plus (Applied Photo Physics) spectrometer using a 10 mm path-length quartz cell. The protein samples (~ 0.15 mg/ml) were in the PBS buffer. A buffer-only sample was used as the reference. The CD spectra were recorded from 10 to 90 °C at 222 nm by using a 1 °C/min temperature ramp with a 60 s delay. Data analysis was performed using Origin (Origin Lab).
Isothermal titration calorimetry (ITC) assay
Trx-His6-μ4-CTD was expressed in Escherichia coli BL21 (DE3) cells and purified with the buffer containing 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, and 5 mM 2-mercaptoethanol, and after removal of the Trx-His6 tag, the protein was further purified by a second step using a Superdex™75 26/60 column under the same buffer conditions. ITC experiments were performed using a MicroCalorimeter ITC200 (Microcal LLC) at 25 °C. μ4-CTD and the ATG9A peptide (QFDTEYQRLEAS, Sangon Biotech) were in the same buffer containing 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA and 5 mM 2-Mercaptoethanol. The titration processes were performed by injecting 20 × 2 µl aliquots of the ATG9A peptide loaded in the syringe (with the concentration of ~ 400 µM) into the stirred calorimeter cell containing μ4-CTD (with the concentration of ~ 40 µM) at time intervals of 120 s to ensure each titration peak fully returned to the baseline. Each experiment was repeated three times. The heat of dilution obtained by the titration of the ATG9A peptide into the buffer was subtracted. The titration data were fitted with the one-site binding model using ORIGIN 8.0.
Chemical modification and fluorescence labeling
The purified His-tagged ARF1(Q71L)-S147C mutant was treated with Biotin-X (succinimidyl ester, AAT Bioquest, 72040-63-2) following the established protocols for NHS-ester conjugation. Excessive Biotin-X were removed using the PD-10 desalting column packed with Sephadex G-25 resin (Cytiva, 17085101). The biotinylated ARF1(Q71L)-S147C mutant and the purified AP-4 core complex were incubated with Cy5 (Cytiva, PA25031) and Cy3 (Cytiva, PA23031) in the reaction buffer (50 mM Tris-HCl, pH 7.5, 200 mM NaCl, 1 mM EGTA, 5 mM MgCl2), respectively. The excessive dyes were removed using the NAP-5 desalting column packed with Sephadex G-25 resin (Cytiva, 17085301). Therefore, the surface-exposed cysteine residues in ARF1(Q71L) and the AP-4 core complex were labeled with Cy5 and Cy3, respectively. The ATG9A peptide (sequence: KQFDTEYQRLEASYSDK), biotinylated on the ε-amine group of the C-terminal lysine (Sangon Biotech, Shanghai), was reacted with AF430 NHS ester (Lumiprobe, 10820). The peptide-dye conjugate was purified by reversed-phase high-performance liquid chromatography (RP-HPLC) on a Dionex UltiMate 3000 system (Thermo Fisher Scientific, USA). Separation was carried out using a C18 column (4.6 × 150 mm, 5 µm) with solvent A (water) and solvent B (acetonitrile). A linear gradient of 0–50% solvent B was applied at 1 mL/min, and eluates were monitored at 214 nm. Fractions corresponding to peptide, peptide-dye conjugate, and free dye were collected separately and lyophilized to obtain purified products.
Fluorescence microscopy-based bead assay
Bead-based confocal assays were performed to evaluate the recruitment of AP-4 core complexes to streptavidin beads (Thermo Scientific™, 20361) coated with biotinylated ATG9A peptide and/or ARF1. Briefly, 20 µL of streptavidin beads were pre-blocked with BSA and washed sequentially in reaction buffer (50 mM Tris pH 8.0, 150 mM NaCl, 5 mM MgCl₂). Beads were divided equally into four groups: Group A: no ligand (control); Group B: 0.5 µg biotinylated ARF1 with 0.5 mM GTP (Sigma-Aldrich, 20-176); Group C: 0.5 µg biotinylated ATG9A peptide; Group D: sequential incubation with 0.2 µg ATG9A peptide, washing, and then 0.2 µg ARF1 with 0.5 mM GTP. After ligand coating, beads from each group were further divided into two subgroups and incubated with 2 μg fluorescently labeled AP-4 core (WT or β4-E395R mutant). Following incubation, beads were thoroughly washed to remove unbound protein.
Washed beads were transferred to wells of a 96-well glass-bottom plate (Greiner Bio-One, 655892) pre-filled with 100 µL of reaction buffer. Confocal imaging was performed using an Olympus FV3000 laser scanning confocal microscope equipped with a Plan Apochromat 20 × /0.8 objective (WD 0.6 mm) and 445 nm (AF430), 561 nm (Cy3), and 640 nm (Cy5) excitation lasers. Maximum fluorescence intensities (Imax) for AP-4, ARF1, and ATG9A were extracted from individual beads using the Radial Profile plugin in FIJI56. Mean background fluorescence was subtracted from each bead’s Imax to obtain the net fluorescence intensity Inet.
where Ibackground was defined as the mean intensity of a bead-free area in the same field of view. Each experiment was repeated three times independently, and all statistical analyses and data visualizations were performed in GraphPad Prism.
Modeling of individual and combined recruitment effects and statistical analysis
To assess whether ATG9A or ARF1 promotes AP-4 recruitment, fluorescence signals were normalized according to established procedures57. The fluorescence intensities of bead-bound AP-4 in Group B and Group C were normalized and compared to those in Group A. To evaluate whether ATG9A and ARF1 act synergistically in recruiting the AP-4 core, an additive model was established under the assumption of no interaction between ATG9A and ARF1. Given the consistent imaging parameters and absence of signal saturation, fluorescence intensity was considered linearly proportional to the amount of bound protein.
The increase in the mean AP-4 signal in Group B or Group C relative to the control group (Group A) was first calculated for each condition:
To account for differences in bait signal intensities between Group B and Group C, normalized recruitment efficiencies were defined as:
The expected additive AP-4 intensity for the dual-bait condition (Group D) was then calculated as:
The values for each sample were averaged between three independent replicates and plotted with the relative standard errors. For the quantification described above, statistical analysis was performed. Statistical significance of the difference between two samples was established by a two-sided paired t test. Statistical significance was indicated by * when p-value < 0.05 and ** when p-value < 0.01.
CRISPR/Cas9-mediated knockout of human AP-4B1 gene
HeLa cells (obtained from the American Type Culture Collection) with a knockout of the AP-4B1 gene were generated using the CRISPR/Cas9 system. A single guide RNA (sgRNA) targeting exon 5 of AP-4B1 (5’-ACGCTACCAACCCCGCAGTG-3’) was designed for high predicted efficiency and minimal off-target activity. The sgRNA was cloned into the PX458 vector, which co-expresses Cas9 and GFP. HeLa cells were transfected with 2.5 µg of the sgRNA-Cas9 construct using Lipofectamine 3000 (Thermo Fisher Scientific). After 48 h, GFP-positive cells were isolated by fluorescence-activated cell sorting (FACS; BD Influx), and single-cell clones were expanded. Genomic DNA was extracted, and the target locus was amplified by PCR. Frameshift mutations were identified by Sanger sequencing and analyzed using Synthego’s Inference of CRISPR Edits (ICE) tool. Successful AP-4B1 knockout clones were confirmed by immunoblotting with an anti-β4 antibody (CUSABIO, China).
Cell culture, immunofluorescence, imaging and data analysis
The DNA sequence encoding the β4 subunit was subcloned to the N-terminus of the mStayGold tag of the pcDNA3.1-mStayGold vector. Point mutations (E395R; Y322R/V359S and E395R/ Y322R/V359S) were introduced using overlap-extension PCR. The following antibodies were used in this assay: rabbit anti-ATG9A (Abcam, 1:100 for IF), sheep anti-human TGN46 (Bio-Rad, 1:1000 for IF), ABflo555-conjugated goat anti-rabbit (Abclonol, 1:1000 for IF), Alexa Fluor 647-conjugated Donkey anti-sheep (Abcam, 1:1000 for IF). HeLa cells (ATCC) were cultured in DMEM supplemented with 10 % (v/v) FBS and maintained at 37 °C. Transient transfection was performed using Lipofectamine 3000 (Thermo Fisher Scientific) following the manufacturer’s instructions. After 48 h, cells were fixed with pre-warmed 4% Paraformaldehyde (PFA) in PBS for 10 min at room temperature (RT). After permeabilization (0.1% saponin in PBS) and blocking (5% normal goat serum, 5% normal donkey serum, and 0.05% saponin in PBS), cells were incubated with the indicated primary (60 min at RT) and secondary antibodies (60 min at RT) diluted in 2.5% normal goat serum, 2.5% normal donkey serum, and 0.05% saponin in PBS. Nuclei were counterstained with 2 μg/mL Hoechst 33342 (Thermo Fisher Scientific) for 10 min at 37 °C.
Fluorescence images were acquired using an inverted laser scanning confocal microscope (Zeiss LSM 980 AxioObserver Z1/7) equipped with a Plan-Apochromat 63 × /1.40 NA oil immersion objective and controlled by Zeiss ZEN software (ZEN blue edition; Carl Zeiss Microscopy GmbH, Jena, Germany). Each experiment was repeated three times independently. Confocal acquisition settings were kept identical across all comparative experiments. Z-stack images (six optical sections, 1 µm step size) were acquired using LSM Plus (Zeiss) and processed as maximum intensity projections for visualization and quantification by using the Orthogonal Projection tool in ZEN, following established protocols58. Pearson’s correlation coefficients were calculated on the maximum-intensity projection images using the Coloc2 plugin in Fiji56, following established protocols. Thresholds of 20 and 40 were applied to the TGN46 channel and the ATG9A channel, respectively. Pixel values below these thresholds were considered background noise and excluded from analysis. Statistical significance was determined using a one-way Welch’s ANOVA, followed by a two-sided Dunnett T3 multiple comparisons test comparing each experimental group to the control condition. ns, not significant; **p-value < 0.01; ***p-value < 0.001.
Statistics and reproducibility
The SEC-MALS, immunoblotting assays and liposome flotation assays were independently repeated three times, yielding similar results. For bead-based experiments, the number of beads analyzed was indicated in the Source Data file, and sample sizes were determined based on technical feasibility. Cellular experiments were performed with standard sample sizes, and no data were excluded from analyses. Statistical analyses for bead-based and cellular experiments were provided in the figure legends of these experiments and the Source Data file.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The atomic coordinates of the AP-4 core complex have been deposited in the protein data bank with the accession codes: 9U9I (μ4-CTD-undocked) and 9U9J (μ4-CTD-docked). The atomic coordinates of the AP-4/ARF1 complex have been deposited in the protein data bank with the accession codes: 9U9R (μ4-CTD-undocked) and 9U9S (μ4-CTD-docked). The three-dimensional cryo-EM density maps of the AP-4 core complex have been deposited in the Electron Microscopy Data Bank with the accession codes: EMD-63965 (μ4-CTD-undocked) and EMD-63966 (μ4-CTD-docked. The three-dimensional cryo-EM density maps of the AP-4/ARF1 complex have been deposited in the Electron Microscopy Data Bank with the accession codes: EMD-63968 (μ4-CTD-undocked) and EMD-63969 (μ4-CTD-docked). The atomic coordinates used for the model building and docking of the AP-4 core and AP-4/ARF1 complexes were downloaded from the Protein Data Bank with the accession codes: 1J2J (ARF1), 1W63 (AP-1 core). The atomic coordinates used for structural comparisons and SWISS-MODEL building were downloaded from the Protein Data Bank with the accession codes: 4HMY, 6DFF, 2VGL, 7R4H, 7RW9, 2JKR, 2XA7, 6QH6, 7P3X, 7P3Y, 7P3Z, 9C5C, 9C5B, 9C58, 6QH5, 3TJZ, 5A1U. The mass spectrometry proteomics data of the AP-4/ARF1 complex have been deposited in the ProteomeXchange Consortium via the iProX partner repository with the dataset identifier PXD072343. The source data underlying Figs. 1b, 3b, c, 4e, g–k, 5c–e and 6g and Supplementary Figs. 8a, b, 10a, b, 11f and 13b are provided as a Source Data file. Source data are provided in this paper.
References
Bonifacino, J. S. & Glick, B. S. The mechanisms of vesicle budding and fusion. Cell 116, 153–166 (2004).
Rizo, J. & Sudhof, T. C. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices-guilty as charged? Annu. Rev. Cell Dev. Biol. 28, 279–308 (2012).
Hurley, J. H., Boura, E., Carlson, L. A. & Rozycki, B. Membrane budding. Cell 143, 875–887 (2010).
Brunger, A. T., Choi, U. B., Lai, Y., Leitz, J. & Zhou, Q. Molecular mechanisms of fast neurotransmitter release. Annu. Rev. Biophys. 47, 469–497 (2018).
Sudhof, T. C. & Rothman, J. E. Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474–477 (2009).
Paczkowski, J. E., Richardson, B. C. & Fromme, J. C. Cargo adaptors: structures illuminate mechanisms regulating vesicle biogenesis. Trends Cell Biol. 25, 408–416 (2015).
Dacks, J. B. & Robinson, M. S. Outerwear through the ages: evolutionary cell biology of vesicle coats. Curr. Opin. Cell Biol. 47, 108–116 (2017).
Traub, L. M. & Bonifacino, J. S. Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harb. Perspect. Biol. 5, a016790 (2013).
Robinson, M. S. Forty Years of Clathrin-coated Vesicles. Traffic 16, 1210–1238 (2015).
Dell’Angelica, E. C. & Bonifacino, J. S. Coatopathies: genetic disorders of protein coats. Annu. Rev. Cell Dev. Biol .35, 131–168 (2019).
Sanger, A., Hirst, J., Davies, A. K. & Robinson, M. S. Adaptor protein complexes and disease at a glance. J. Cell Sci. 132, https://doi.org/10.1242/jcs.222992 (2019).
Rout, M. P. & Field, M. C. The evolution of organellar coat complexes and organization of the eukaryotic cell. Annu. Rev. Biochem. 86, 637–657 (2017).
Owen, D. J., Collins, B. M. & Evans, P. R. Adaptors for clathrin coats: structure and function. Annu. Rev. Cell Dev. Biol. 20, 153–191 (2004).
Guardia, C. M., De Pace, R., Mattera, R. & Bonifacino, J. S. Neuronal functions of adaptor complexes involved in protein sorting. Curr. Opin. Neurobiol. 51, 103–110 (2018).
Kaksonen, M. & Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 19, 313–326 (2018).
Dell’Angelica, E. C., Mullins, C. & Bonifacino, J. S. AP-4, a novel protein complex related to clathrin adaptors. J. Biol. Chem. 274, 7278–7285 (1999).
Hirst, J., Bright, N. A., Rous, B. & Robinson, M. S. Characterization of a fourth adaptor-related protein complex. Mol. Biol. Cell 10, 2787–2802 (1999).
Mattera, R., De Pace, R. & Bonifacino, J. S. The role of AP-4 in cargo export from the trans-Golgi network and hereditary spastic paraplegia. Biochem. Soc. Trans. 48, 1877–1888 (2020).
Peden, A. A. et al. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J. Cell Biol. 164, 1065–1076 (2004).
Hirst, J. et al. The fifth adaptor protein complex. PLoS Biol. 9, e1001170 (2011).
Hirst, J., Irving, C. & Borner, G. H. Adaptor protein complexes AP-4 and AP-5: new players in endosomal trafficking and progressive spastic paraplegia. Traffic 14, 153–164 (2013).
Boehm, M., Aguilar, R. C. & Bonifacino, J. S. Functional and physical interactions of the adaptor protein complex AP-4 with ADP-ribosylation factors (ARFs. EMBO J. 20, 6265–6276 (2001).
Mattera, R., Park, S. Y., De Pace, R., Guardia, C. M. & Bonifacino, J. S. AP-4 mediates export of ATG9A from the trans-Golgi network to promote autophagosome formation. Proc. Natl. Acad. Sci. USA 114, E10697–E10706 (2017).
Davies, A. K. et al. AP-4 vesicles contribute to spatial control of autophagy via RUSC-dependent peripheral delivery of ATG9A. Nat. Commun. 9, 3958 (2018).
Ivankovic, D. et al. Axonal autophagosome maturation defect through failure of ATG9A sorting underpins pathology in AP-4 deficiency syndrome. Autophagy 16, 391–407 (2020).
Yap, C. C. et al. Adaptor protein complex-4 (AP-4) is expressed in the central nervous system neurons and interacts with glutamate receptor delta2. Mol. Cell Neurosci. 24, 283–295 (2003).
Matsuda, S. et al. Accumulation of AMPA receptors in autophagosomes in neuronal axons lacking adaptor protein AP-4. Neuron 57, 730–745 (2008).
Guardia, C. M. et al. RUSC2 and WDR47 oppositely regulate kinesin-1-dependent distribution of ATG9A to the cell periphery. Mol. Biol. Cell 32, ar25 (2021).
West, M. A., Bright, N. A. & Robinson, M. S. The role of ADP-ribosylation factor and phospholipase D in adaptor recruitment. J. Cell Biol. 138, 1239–1254 (1997).
Ooi, C. E., Dell’Angelica, E. C. & Bonifacino, J. S. ADP-Ribosylation factor 1 (ARF1) regulates recruitment of the AP-3 adaptor complex to membranes. J. Cell Biol. 142, 391–402 (1998).
Heldwein, E. E. et al. Crystal structure of the clathrin adaptor protein 1 core. Proc. Natl. Acad. Sci. USA 101, 14108–14113 (2004).
Ren, X., Farias, G. G., Canagarajah, B. J., Bonifacino, J. S. & Hurley, J. H. Structural basis for recruitment and activation of the AP-1 clathrin adaptor complex by Arf1. Cell 152, 755–767 (2013).
Kelly, B. T. et al. AP2 controls clathrin polymerization with a membrane-activated switch. Science 345, 459–463 (2014).
Jackson, L. P. et al. A large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex. Cell 141, 1220–1229 (2010).
Kovtun, O., Dickson, V. K., Kelly, B. T., Owen, D. J. & Briggs, J. A. G. Architecture of the AP2/clathrin coat on the membranes of clathrin-coated vesicles. Sci. Adv. 6, eaba8381 (2020).
Partlow, E. A., Cannon, K. S., Hollopeter, G. & Baker, R. W. Structural basis of an endocytic checkpoint that primes the AP2 clathrin adaptor for cargo internalization. Nat. Struct. Mol. Biol. 29, 339–347 (2022).
Schoppe, J. et al. Flexible open conformation of the AP-3 complex explains its role in cargo recruitment at the Golgi. J. Biol. Chem. 297, 101334 (2021).
Begley, M., Aragon, M. & Baker, R. W. A structure-based mechanism for initiation of AP-3 coated vesicle formation. Proc. Natl. Acad. Sci. USA 121, e2411974121 (2024).
Yu, X., Breitman, M. & Goldberg, J. A structure-based mechanism for Arf1-dependent recruitment of coatomer to membranes. Cell 148, 530–542 (2012).
Dodonova, S. O. et al. A structure of the COPI coat and the role of coat proteins in membrane vesicle assembly. Science 349, 195–198 (2015).
Collins, B. M., McCoy, A. J., Kent, H. M., Evans, P. R. & Owen, D. J. Molecular architecture and functional model of the endocytic AP2 complex. Cell 109, 523–535 (2002).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Punjani, A. & Fleet, D. J. 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol. 213, 107702 (2021).
Nguyen, T. N. et al. Unconventional initiation of PINK1/Parkin mitophagy by Optineurin. Mol. Cell 83, 1693–1709 (2023).
Ebrahimi-Fakhari, D. et al. Defining the clinical, molecular and imaging spectrum of adaptor protein complex 4-associated hereditary spastic paraplegia. Brain 143, 2929–2944 (2020).
Francisco, S. et al. Restoring adapter protein complex 4 function with small molecules: an in silico approach to spastic paraplegia 50. Protein Sci. 34, e70006 (2025).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Terwilliger, T. C. et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 (2008).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Chen, Z. L. et al. A high-speed search engine pLink 2 with systematic evaluation for proteome-scale identification of cross-linked peptides. Nat. Commun. 10, 3404 (2019).
Wu, S. et al. Kinetics of the conformational cycle of Hsp70 reveals the importance of the dynamic and heterogeneous nature of Hsp70 for its function. Proc. Natl. Acad. Sci. USA 117, 7814–7823 (2020).
Kapanidis, A. N. et al. Fluorescence-aided molecule sorting: analysis of structure and interactions by alternating-laser excitation of single molecules. Proc. Natl. Acad. Sci. USA 101, 8936–8941 (2004).
Kalinin, S., Felekyan, S., Antonik, M. & Seidel, C. A. Probability distribution analysis of single-molecule fluorescence anisotropy and resonance energy transfer. J. Phys. Chem. B 111, 10253–10262 (2007).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Abert, C. & Martens, S. Studies of Receptor-Atg8 Interactions During Selective Autophagy. Methods Mol. Biol. 1880, 189–196 (2019).
Kwon, O. S. et al. Exon junction complex dependent mRNA localization is linked to centrosome organization during ciliogenesis. Nat. Commun. 12, 1351 (2021).
Acknowledgements
We thank Xilin Miao, Dr. Jie Ren, Dr. Huaqing Cai and Dr. Junjie Hu for the help on the liposome flotation assay, Dr. Sarah Perrett for the help on the smFRET assay, Kewei Gu for the help on the cellular studies, and Jifeng Wang for the help on mass spectrometry. This work was supported by grants from the Brain Science and Brain-like Intelligence Technology – National Science and Technology Major Project (2022ZD0205800 to Y.Z.), the CAS Project for Young Scientists in Basic Research (YSBR-104 to W.F. and Y.Z.), and the National Natural Science Foundation of China (32571398 to W.F.). We acknowledge the support from the State Key Laboratory of Biomacromolecules and State Key Laboratory of Epigenetic Regulation and Intervention (to S.W.).
Author information
Authors and Affiliations
Contributions
Y.W., W.L., Y.Q., and S.W. conducted experiments and participated in the design and interpretation of the experiments. Specifically, Y.W. and W.L. conducted all biochemical and cellular experiments, Y.W. and Y.Q. performed structural studies, and Y.W. and S.W. performed the smFRET assay. L.H. provided the help on the smFRET data analysis. Y.Z. provided the guidance for the cryo-EM data collection and analysis. W.F. designed all the experiments, interpreted the data and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Wade Zeno and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Li, W., Qiu, Y. et al. Structural basis for the dynamic conformations of AP-4 and its association with ARF1. Nat Commun 17, 1897 (2026). https://doi.org/10.1038/s41467-026-68679-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68679-8
