
Abstract
The electrochemical C–N coupling of biomass-derived 5-hydroxymethylfurfural (HMF) with methylamine offers a promising sustainable route to value-added amines. However, achieving high selectivity remains challenging due to competing side reactions, including C = O hydrogenation and C–C dimerization. This paper describes tailoring the surface structure of Ag catalysts to modulate HMF adsorption and intermediate hydrogenation behavior and thus enhancing C–N coupling selectivity. Ag nanoparticles, predominantly exposing the (111) facet, exhibit higher selectivity compared with Ag nanocubes mainly enclosed by (100) planes. The Ag(111) facet favors the C–N coupling pathway by regulating both adsorption geometry and hydrogenation ability. In-situ Raman spectroscopy and density functional theory (DFT) calculations reveal that an η1(C)-aldehyde adsorption configuration, dominant on Ag(111), enhances carbonyl electrophilicity and facilitates C = O polarization, promoting nucleophilic attack by methylamine. In contrast, the η2(C,O)–aldehyde configuration, more common on Ag (100), stabilizes the C = O bond, limiting its reactivity. Moreover, Ag(111) shows enhanced imine hydrogenation activity, further improving amine selectivity. This study highlights the significance of facet-dependent adsorption modulation in steering reaction pathways and advancing sustainable electrochemical synthesis.
Similar content being viewed by others

Introduction
Amines are fine chemicals extensively applied in chemical, pharmaceutical, agrochemical, and material industries1,2,3. Among various amines, 5-[(methylamino)methyl]−2-furanmethanol (MAMF) serves as a key chemical for the synthesis of the pharmaceutical Ranitidine4. MAMF could be fabricated by the reductive amination of HMF with methylamine5,6. However, traditional production routes often rely on hazardous reducing agents and harsh reaction conditions, leading to high energy consumption and severe side reactions4,7. Electrochemical approaches enable controlled C–N bond formation through potential modulation and catalyst design, offering sustainable MAMF synthesis under ambient conditions with electrons as clean reductants8,9,10.
The electro-reductive amination of HMF can proceed either via a one-step direct reaction between HMF and methylamine or through a two-step route in which HMF first undergoes spontaneous condensation with methylamine to generate an intermediate imine, followed by its electrochemical reduction on the catalyst surface. However, during reductive conditions, the local pH near the cathode rises due to hydroxide accumulation11, making the imine intermediate susceptible to hydrolysis at the interface. This hydrolytic back-reaction regenerates HMF and methylamine, rendering the two-step pathway kinetically indistinguishable from the one-step process. Moreover, the two-step approach generally requires prolonged reaction times and additional separation steps, complicating the overall synthesis12,13. Unwanted side reactions may also occur: two imine molecules can couple to form diamine species, while the primary amine product may further condense with unreacted imine to yield a secondary imine that can be subsequently hydrogenated into a secondary amine. Moreover, imines may undergo polymerization or trimerization in solutions, leading to complex product mixtures, which have been reported previously (Supplementary Fig. 1)12. Considering these factors, the one-step electrolysis strategy was adopted and systematically investigated in this work.
In addition to reaction pathways, the hydrogenation ability of catalysts could also affect the product distribution of electrocatalytic C–N coupling13,14,15. Insufficient hydrogenation capability hinders the timely reduction of imine intermediate, facilitating its hydrolysis back to HMF and methylamine, thereby lowering overall selectivity toward MAMF. Comparative studies in thermal reductive amination systems have demonstrated that a moderate and directional hydrogenation ability is optimal for locking the rate-determining step at the selective hydrogenation of C=N, which maximizes primary amine formation and suppresses over-hydrogenation and other side reactions16.
Before the catalyst design, different metal electrocatalysts have been investigated for this reaction (Pt, Cu, Sn, Zn, and Ag)9,17. However, a lack of rational catalyst design has resulted in the easy occurrence of side reactions and low selectivity of C–N coupling18,19. To overcome these performance limitations, it is essential to understand the reaction from a mechanistic perspective. Previous studies have shown that enhancing the nucleophilic attack step is critical for effectively improving the overall selectivity of electrochemical C–N coupling reactions9,17. Taking the electrochemical C–N coupling of HMF and methylamine as an example, the reaction pathway involves multiple steps (Fig. 1)3,17. Typically, HMF could be activated by the catalyst, forming the absorbed HMF (HMF*)9,12. Then, the target product MAMF could be synthesized through two key steps13,14,15: (1) nucleophilic addition reaction of methylamine with the carbonyl group of HMF* to form the aldimine intermediate, and (2) subsequent reduction of the aldimine with protons from the electrolyte and electrons from the electrode to produce the MAMF (Fig. 1a). However, the hydrogenation and dimerization of HMF would compete with the desired C–N coupling reaction to form 2,5-bis(hydroxymethyl)furan (BHMF) and bis(hydroxymethyl)hydrofuroin (BHH), respectively, leading to low selectivity (Fig. 1b, c)16,20,21. Therefore, precise modulation of the aldehyde carbonyl reactivity and the catalyst’s hydrogenation ability to facilitate nucleophilic attack and subsequent imine conversion could be a useful strategy for enhancing the selectivity of electrochemical C–N coupling.

a MAMF, b BHMF, and c BHH.
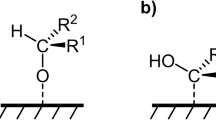
Tuning the adsorption behaviors of substrates on the catalyst surface has been proven essential for changing the electrochemical reaction pathways22,23. For example, the adsorption configuration of HMF is different on various metal electrodes, affecting the elongation of the aldehyde C=O bond, which impacts the reaction pathways24,25. This phenomenon provides a general idea of improving the selectivity for C–N coupling of HMF with methylamine by designing catalysts with appropriate substrate absorption configuration26,27,28. Adjusting the hydrogenation ability of the intermediate could further affect the product distribution. Among different catalyst design strategies29,30,31, facet engineering could precisely control substrate adsorption configurations and hydrogenation owing to the specific atomic arrangements and electronic structures of a certain facet32,33. For example, Koper et al. revealed that facet-dependent adsorption configurations of organic ketones govern electrochemical reduction selectivity on Pt surfaces34. Acetophenone was horizontally adsorbed on Pt(111) with the compacted adsorption of the phenyl ring, enhancing carbonyl reduction. In contrast, the vertical adsorption configuration of 4-acetylpyridine on Pt(100) increases the distance between the carbonyl functional group and the catalyst surface, thereby suppressing reduction activity compared to acetophenone. Therefore, these facet-dependent adsorption effects highlight a general structure-selectivity relationship, underscoring that a mechanistic understanding of how adsorption configurations govern intermediate activation and hydrogenation is essential for rationally designing catalysts with improved selectivity for the electrochemical C–N coupling of HMF with methylamine.
In this work, Ag catalysts with well-defined structures and preferentially exposed facets are fabricated to investigate the impact of the adsorption behaviors of substrates and hydrogenation ability of intermediates on the selectivity of the C–N coupling reaction9,35. Ag nanoparticles (Ag NP), mainly enclosed by Ag(111) facet, exhibited the highest Faradaic efficiency (FE) of 89.2% and a selectivity of 90.2% at –0.45 V vs. reversible hydrogen electrode (RHE), outperforming most previous reported catalysts19,36,37,38,39. Combined with in situ Raman spectroscopy results and density functional theory (DFT) calculations, we propose that Ag NPs favor the η1(C)-aldehyde adsorption configuration and enhanced imine hydrogenation ability, which facilitates nucleophilic attack by methylamine and conversion of imine intermediates, while Ag nanocubes (Ag NC), dominated by (100) facets, favor the η2(C, O)-aldehyde configuration that stabilizes the C=O bond and promotes competing hydrogenation or dimerization. These findings suggest that selective C–N coupling is closely related to the facet-dependent adsorption behavior and possibly the cooperative effects of multiple surface sites.
Results and discussion
To rationalize the choice of the one-step C–N coupling pathway, comparative electrolysis experiments were conducted using an Ag foil to evaluate the selectivity between direct coupling and the two-step route via the pre-formed imine intermediate. The direct electrochemical coupling between HMF and methylamine exhibited an FE of 62.3%, significantly higher than the 39.8% obtained for the imine-mediated process. In contrast, the two-step reaction yielded a more complex product distribution (Supplementary Fig. 2), likely due to secondary condensation leading to diamine or triamine species. To further elucidate the role of adsorption properties in governing C–N coupling activity, a series of metal foils (Fe, Ni, Cu, Pt, Zn, Sn, Ag, Cd, and In) with distinct adsorption affinities toward HMF were systematically examined17. Among those metals, Ag demonstrates relatively good electrocatalytic performance in the C–N coupling reaction, achieving the highest partial current density of 13 mA cm–2 and FE of 62.3% for MAMF production due to the moderate HMF adsorption strength among tested metal foils (Supplementary Fig. 3)16,40. To further investigate the impact of adsorption behavior of HMF on the electrochemical C–N coupling, Ag NP and Ag nanocubes (Ag NC) with varied exposed facets and surface electronic structures were successfully fabricated. Scanning electron microscopy (SEM) images revealed that Ag NP exhibits a nearly spherical morphology with an average diameter of approximately 30 nm, whereas Ag NC exhibits cubic shapes (Fig. 2a, b). Transmission electron microscopy (TEM) characterization (Fig. 2c–f and Supplementary Fig. 4) further confirms that Ag NP and Ag NC exhibit distinct morphologies. High-resolution TEM (HRTEM) images and the corresponding fast Fourier transform (FFT) patterns (Fig. 2d, f) provide evidence of the surface structures (Supplementary Fig. 5). Ag NP exhibits a lattice spacing of 0.24 nm, corresponding to the (111) crystallographic plane of Ag. On the other hand, Ag NC shows a lattice spacing of 0.21 nm, which corresponds to the (200) plane of Ag41. Additionally, the primary diffraction peaks in the X-ray diffraction (XRD) patterns confirm that Ag NP and Ag NC mainly expose the (111) and (100) facets, respectively (Fig. 2g). X-ray photoelectron spectroscopy (XPS) characterization (Fig. 1h) was conducted to confirm that Ag in these catalysts exists in the metallic state32,42. These results demonstrate the successful synthesis of Ag catalysts with well-controlled crystal structures.

a, b SEM images and corresponding size distributions of as-prepared Ag NP (The inserted figure represents the size distribution) (a) and Ag NC (The inserted figure represents the size distribution) (b). c–f TEM (c, e) and HRTEM (d, f) images of Ag NP (c, d) and Ag NC (e, f) (the inserted figures in (d) and (f) represent magnified images of the corresponding measured areas). g XRD patterns of Ag NP and Ag NC (JCPDF#04–0783). h XPS spectra of as-prepared Ag NP and Ag NC. Source data are provided as a Source Data file.
Electrochemical activity tests for the C–N coupling reaction of HMF with methylamine were conducted in a three-electrode H-cell (Supplementary Fig. 6), using a 0.5 M phosphate buffer solution as the electrolyte (see the Methods for more details)9,12. The liquid and gaseous products were quantified by liquid chromatography (LC) and gas chromatography (GC), respectively (Supplementary Figs. 7–14). The methylamine concentration was optimized, and 1000 mM was selected according to the performance tests (Supplementary Fig. 15). Linear sweep voltammetry (LSV) curves (Fig. 3a and Supplementary Fig. 16) indicate that both Ag NP and Ag NC exhibit enhanced current densities upon the addition of HMF, suggesting that the electrochemical C–N coupling of HMF with methylamine requires a lower overpotential compared to the hydrogen evolution reaction (HER). Ag NP achieves a current density of ~20 mA cm–2 at –0.45 V vs. RHE, which is higher than that of Ag NC (~14 mA cm–2). Notably, the Ag NC exhibits a higher current density of HMF reduction as the potential becomes more negative (lower than –0.45 V), suggesting that direct HMF reduction might be more favorable on Ag NC.

a LSV curves of Ag NP and Ag NC in the electrolyte (phosphate buffer solution with methylamine) with or without 20 mM HMF. b, c Product FEs and MAMF selectivity as a function of potential over Ag NP (b) and Ag NC (c). (d) Potential-dependent production rate of MAMF over Ag NP and Ag NC. e, f Stability tests of Ag NP (e) and Ag NC (f). The error bars represent the standard deviation of three parallel repeated results. All tests were performed in an electrolyte of pH 11.0 ± 0.2 using an electrode with a surface area of 1.5 cm2, without iR correction. Source data are provided as a Source Data file.
Ag NP and Ag NC with different surface structures exhibit distinct selectivity and product distributions in the electrochemical C–N coupling reaction of HMF and methylamine (Fig. 3b, c). Tests at a wide range of potentials (–0.35 V to –0.75 V vs. RHE) demonstrate that Ag NP shows better performance in both selectivity and FE of electrochemical C–N coupling compared to Ag NC. The highest FE of 89.2% could be achieved by Ag NP with a selectivity of 90.2% at –0.45 V vs. RHE (Fig. 2b), which outperforms most previously reported C–N coupling systems (Supplementary Table S1)9,17,18,19,36. Regarding the distribution of products, Ag NC shows a higher FE of BHH than Ag NP at various voltages (Fig. 3c), with more BHH produced at low potentials (–0.35 V and –0.45 V vs. RHE). Furthermore, Ag NC exhibits a rising trend in BHMF production even in the presence of methylamine at higher potentials (–0.55 V to –0.75 V vs. RHE). These results indicate that, on Ag NCs, the aldehyde group of HMF preferentially undergoes hydrogenation to BHMF or dimerization to BHH, which is likely attributable to the strong interaction between the catalyst surface and the C=O moiety16,33,40. Additionally, introducing BHMF into the electrolyte with methylamine at reductive potentials eliminates the possibility of MAMF production from BHMF (Supplementary Fig. 17), indicating that these two reactions are competitive on Ag catalysts. Furthermore, Ag NP possesses a slightly smaller electrochemically active surface area (ECSA) than Ag NC (Supplementary Fig. 18), suggesting that Ag NP has a better intrinsic performance. Therefore, the higher production rate of MAMF on Ag NP suggests that the adsorption behavior on Ag NP favors the C–N coupling process (Fig. 3d). During stability tests, Ag NP shows the steady production of MAMF with a consistent FE (>85%) over 24 h (Fig. 3e), higher than that of the Ag NC (Fig. 3e, f). The morphology and crystal structure of the Ag catalysts remain unchanged (Supplementary Figs. 19 and 20), suggesting the importance of the exposed facet for the promoted activity. The results above indicate that the adsorption behaviors of HMF seem to be essential for selectivity. To validate the generality of our findings, we broadened the substrate scope to several furfural derivatives and varied the nitrogen source. With 2-furaldehyde and 5-methylfurfural as substrates and methylamine as the nucleophile, Ag NP delivered FEs of 63.0% and 81.0% toward the corresponding amines, whereas Ag NC afforded lower FEs of 50.4% and 69.1%, respectively (Supplementary Fig. 21a, b). Replacing methylamine with ammonia as the nitrogen source in the coupling reaction with HMF preserved a high MAMF FE of 65.3% on Ag NP, higher than that of 29.1% on Ag NC (Supplementary Fig. 21c). Taken together, these results validate a facet-governed adsorption-reactivity paradigm. The Ag NP surface favors the η1(C)-aldehyde configuration, translates into favorable product selectivity across distinct furfural substrates and nitrogen sources, indicating the versatility of facet engineering for electrochemical amination (Supplementary Fig. 21d). To further support this hypothesis, it is necessary to elucidate the intrinsic reaction mechanism of C–N coupling and how the HMF adsorption behavior affects the performance.
To further investigate the reaction mechanism of electrochemical C–N coupling, kinetics studies were carried out on Ag catalysts. The Tafel slopes (Fig. 4a, b) for Ag NP (157 mV dec–1) and Ag NC (168 mV dec–1) are lower compared to blank counterparts of Ag NP and Ag NC (179 mV dec–1 and 182 mV dec–1). This reduction in Tafel slopes demonstrates that the electrochemical C–N coupling of HMF with methylamine is kinetically more favorable than HER. Notably, the smaller Tafel slope of Ag NP than Ag NC implies more favorable kinetics of Ag NP for C–N coupling12,28,34. This result is further supported by the electrochemical impedance spectroscopy (EIS) study (Fig. 4c and Supplementary Fig. 22). The Ag NPs exhibit a reduced charge-transfer resistance (R = 12.4 Ω) compared to Ag NCs (R = 18.7 Ω), indicating enhanced interfacial electron transfer efficiency during the C–N coupling process43,44. Furthermore, electrochemical experiments with H/D kinetic isotope effect (KIE) studies were conducted on the Ag NP catalyst. The cathodic shift of 145 mV (Fig. 4d) suggests that the proton-coupled electron transfer step is significant for the C–N coupling reaction, indicating the important role of proton dynamics in the reduction of the HMF intermediate and C–N bond formation17,36,45,46. Besides, time-resolved 1H NMR spectroscopic characterization of the reaction mixture was performed under operando electrochemical conditions (Supplementary Fig. 23). The results show that the imine intermediate remains at a consistently low concentration throughout the electrochemical reaction, exhibiting a gradual decline as electrolysis proceeds. This behavior can be attributed to the continuous conversion and hydrolysis of the imine at the electrode–electrolyte interface, where the elevated local pH promotes hydrolytic decomposition. In contrast, the imine concentration remains nearly constant in the absence of applied potential, confirming that electrolysis induces its consumption (Supplementary Fig. 24). According to Fick’s first law, we have calculated the local alkalinity during the reaction47. It was found that the local pH elevated to nearly 12.6 at –0.45 V vs. RHE (Supplementary Fig. 25a, see the Methods for more detials). To further verify this observation, the imine was synthesized independently and introduced into electrolyte solutions with pH 11.0 and 12.6. The imine remained relatively stable at pH 11.0 but underwent hydrolysis under pH 12.6 conditions (Supplementary Fig. 25). Considering the enhanced local alkalinity near the cathode during electrolysis, these results substantiate our rationale for adopting a one-step C–N coupling strategy and highlight the critical importance of controlling the interfacial adsorption configuration and hydrogenation ability to achieve high selectivity36,48,49.

a, b Tafel plots of HER and C–N coupling over Ag NP (a) and Ag NC (b). c Comparison of Nyquist plots for HER and C–N coupling over Ag NP and Ag NC at –0.45 V vs. RHE. d Comparison of LSV curves on Ag NP for C–N coupling reaction in the electrolyte with H2O and D2O. e Partial current density and FE of MAMF as a function of the charges passed. f The relationship between HMF concentration and partial current density of MAMF, and the corresponding FE of MAMF. The error bars represent the standard deviation of three parallel repeated results. All tests were performed in an electrolyte of pH 11.0 ± 0.2 using an electrode with a surface area of 1.5 cm2, without iR correction. Source data are provided as a Source Data file.
Kinetic analysis of the reaction process shows that a decline in FE for C–N coupling correlates with the concentration decrease of HMF as consumed during electrolysis (Fig. 4e), accompanied by a rise in HER activity. This trend further supports the conclusion that the C–N coupling reaction follows the inner-sphere route17,36. Further validation is provided by the dependence of the MAMF partial current density on the HMF concentration. In the low-concentration regime (Fig. 4f), a logarithmic slope of 0.62 is observed, which is close to the theoretical value of 0.5 predicted by Langmuir–Hinshelwood kinetics for bimolecular surface reactions. This agreement suggests that the surface adsorption of HMF becomes a key rate-limiting factor under these conditions.5,49. At higher HMF concentrations, side reactions like polymerization and reduction increase, inhibiting the C–N coupling reaction23. These findings collectively demonstrate that HMF adsorption modulated by catalyst surface properties directly affects the competition between C–N coupling and side reactions.
To clarify the adsorption behavior of HMF on Ag NP and Ag NC, spectroscopic studies were carried out. Before the in situ experiments for the C–N coupling reaction, the Raman spectra of HMF, methylamine, and possible products (MAMF and BHMF) were first collected to confirm key peaks for further experiments (Supplementary Fig. 26). Raman spectroscopy enables precise detection of adsorption-induced spectral perturbations, including frequency shifts and intensity variation, by leveraging its high sensitivity to molecular vibrational modes50. Notably, Ag NP and NC exhibit distinct spectral responses to HMF adsorption (Fig. 5a). For Ag NP, peaks at 976 cm–1 and 1526 cm–1 are attributed to the C–C stretching of the furan ring. The peak at 1022 cm–1 is attributed to the C–H stretching of the furan ring. These results show that HMF is adsorbed on Ag NP with the horizontally tilted furan ring51,52. This adsorption configuration shows that the C–C end of the furan ring is closer to the catalyst surface on Ag NP, with the absence of the peak at 718 cm–1 attributed to C–O–C stretching. For Ag NC, the appearance of the peak at 718 cm–1 and the disappearance of peaks at 976 cm–1 and 1526 cm–1 confirm that the C–O–C end of the furan ring interacts more closely with the catalyst surface53. The peak at 1002 cm–1 is attributed to the hydroxymethyl group54,55. The absence of a peak at 1526 cm–1 on Ag NC could suggest that Ag NC preferentially adsorbs the oxygen-containing bonds (C–O–C of the furan ring, C–OH of the alcohol, and C=O of the aldehyde)56. These results show that the adsorption configuration of HMF is strongly dependent on the Ag catalysts, possibly owing to the different surface structures.

a Comparison of the adsorption configuration of HMF on Ag NP (red curve) and Ag NC (purple curve). The Raman spectrum of pure HMF (blue curve) serves as the reference. b The adsorption energy of HMF with corresponding adsorption configurations on Ag NP and Ag NC. c, d In situ Raman studies for C–N coupling at various potentials on Ag NP (c) and Ag NC (d). e The schematic diagrams for electrochemical C–N coupling of HMF and methylamine on the Ag NP and Ag NC. Source data are provided as a Source Data file. The model information for DFT calculations is provided in Supplementary Data 1.
The varied adsorption configurations of HMF could be further supported by the results of density functional theory (DFT) calculations (Fig. 5b). DFT results reveal that HMF preferentially adopts the η¹(C)-aldehyde adsorption mode on Ag NP, where the furan ring tilts horizontally and the aldehyde group of HMF coordinates monodentate to the catalyst surface through the carbonyl carbon atom. Additionally, Ag NC stabilizes the η2(C,O)-aldehyde configuration, in which the C atom and the O atom of the carbonyl group are together bonded to the surface. These distinct adsorption modes could influence the interaction strength and orientation of the C=O group with catalytic sites57,58. Furthermore, the calculated adsorption energy difference (Fig. 5b) indicates that the thermodynamic stabilization of the η1(C)-aldehyde configuration, which concurrently improves both the activation efficiency and adsorption stability of the C=O group during prolonged electrolysis35,59. Therefore, it is proposed that the Ag catalysts affect the selectivity of the C–N coupling reaction by controlling the adsorption mode of the aldehyde group of HMF.
To further understand the mechanism of structure-influenced adsorption configurations in steering C–N coupling reaction, in situ Raman spectroscopy investigations were carried out at the potentials from –0.15 V to –0.75 V vs. RHE with an interval of 0.1 V (Fig. 5c, d). Adsorbed species on the Ag surface are observed under reduction conditions. The C–C, C–H vibrations of the furan ring observed at open circuit potential (Fig. 5c) could all be found at a similar Raman shift as those found in Fig. 4a, suggesting that HMF is adsorbed on Ag NP via η1(C)-aldehyde configuration in the electrochemical C–N coupling process. The peaks for aldehyde (1391 cm–1 and ~1670 cm–1) and furan ring (1523 cm–1) are observed from –0.15 V to –0.75 V vs. RHE, indicating a stabilized adsorption on Ag NP at a wide potential range (Fig. 5c)60. For Ag NC, peaks of HMF appear at 718 cm–1, 1002 cm–1, and 1506 cm–1, consistent with η2(C, O)-aldehyde adsorption mode (Fig. 5d). Control experiments without HMF are investigated under the same applied potential. The peak at ca. 1300 cm–1 (Supplementary Fig. 27), attributed to the wagging of –NH260,61 and stable adsorption of methylamine, eliminates the concerns about the identification of adsorption configurations and the influence of methylamine adsorption.
Upon applying the negative potential, peaks at 1452 cm–1 attributed to symmetric stretching of the C–N bond are detected at –0.15 V to –0.75 V vs. RHE, demonstrating the generation of MAMF (Supplementary Fig. 26c). The surface-enhanced Raman scattering (SERS) effect amplifies the furan ring vibration at 1562 cm–1 in generated MAMF46, demonstrating that the reaction occurs preferentially near the Ag NP surface rather than in bulk solution. Additionally, the relatively lower intensity of the peak of the C–N bond in MAMF on Ag NC (peak at 1452 cm–1) corresponds to the lower coverage of MAMF under operando conditions25,47, implying that the reaction did not favor the C–N coupling (Fig. 5e). In particular, the vibration peak at 1589 cm–1, assigned to the hydrogenation product BHMF (Supplementary Fig. 26d), is absent in the Raman spectra of Ag NP (Fig. 5d), indicating that the η1(C)-aldehyde adsorption configuration effectively suppresses the competing hydrogenation pathway. However, the peak of BHMF was observed on Ag NC from –0.25 V to –0.65 V vs. RHE, indicating that the C–N coupling reaction was inhibited by the hydrogenation of HMF.
The Raman peak at 1678 cm–1 attributed to the stretching mode of C=O in HMF (blue curve in Fig. 5a) shows a 3 cm–1 redshift on Ag NP (Fig. 5c)50,62. The redshift is assigned to the adsorption of HMF through the carbonyl π* antibonding orbital and indicates the weakening of C=O bond on Ag NP25,63. Compared to Ag NP, the 9 cm–1 redshift of C=O on Ag NC indicates a stronger interaction (Fig. 5d)50,53. Furthermore, the relatively larger stark tuning (Supplementary Fig. 28) of Ag NC demonstrates the excessive interaction of aldehyde53,64, leading to the cleavage of C=O to form BHH or BHMF16,28. This divergence in binding geometry directly modulates the polarization degree of the C=O group. The η1(C)-aldehyde adsorption on Ag NP induces moderate electron withdrawal from the carbonyl moiety compared to the η2(C, O)-aldehyde adsorption on Ag NC, thereby enhancing its susceptibility to nucleophilic attack by absorbed methylamine (Fig. 1a) rather than the hydrogenation (Fig. 1b) and dimerization (Fig. 1c)16,26.
To further exclude how the adsorption configuration affects the selectivity, DFT calculations were carried out. The results reveal that the nucleophilic attack of methylamine on adsorbed HMF over Ag NP exhibits a Gibbs free energy barrier of –0.28 eV that is lower than –0.14 eV on Ag NC. Considering potential contribution from the trace facet of (110), the C–N coupling energy barrier on Ag (110) was also considered. It showed that the C–N coupling energy barrier is –0.17 eV, higher than that on Ag(111). The hydrogenation barrier of the imine is also calculated. Ag(111) surface exhibits the lowest hydrogenation energy barrier (0.07 eV), markedly lower than those on Ag(100) (0.24 eV) and Ag(110) (0.13 eV). Such a low barrier facilitates the rapid hydrogenation of the imine intermediate, thereby accelerating the overall conversion and enhancing the C–N coupling selectivity (Supplementary Fig. 29)23,65. As a result, Ag NP stabilizes η¹(C)-aldehyde adsorption configuration with optimal interaction of C=O for C–N coupling, whereas Ag NC favors η2(C, O)-aldehyde configurations that steer reactions toward hydrogenation.
These spectroscopic and computational results highlight that the adsorption configuration of HMF could play a crucial role in affecting the selectivity of electrochemical C–N coupling. As shown in Fig. 5e, HMF tends to absorb on the Ag NP via η1(C)-aldehyde configuration. Due to the moderate adsorption strength of C=O, nucleophilic attack of methylamine would occur preferentially. Thereafter, hydrogen atom transfer (HAT) would transfer H* on the electrode to the absorbed organic species rapidly to converting imine to respective amines48,56. The mechanism of Ag NP gives clear evidence that η1(C)-aldehyde adsorption configuration favors generating the C–N bond and suppressing the side reactions. Furthermore, the stronger polarization of aldehyde C=O on Ag NC contributes to straight HAT to form BHH and BHMF rather than coupling with methylamine. Given the reaction pathways for C–N coupling, MAMF would not be generated once the straight HAT to aldehyde happens (Fig. 1). The specific structures of catalyst surfaces impact the C–N coupling selectivity by controlling HMF adsorption configurations and imine hydrogenation that differentially activate the C=O group for the nucleophilic attack28,62. Therefore, manipulating the adsorption configuration of HMF and the hydrogenation of imine on Ag catalysts could enhance the FE of electrochemical C–N coupling of HMF with methylamine.
In this work, we rationalized the choice of a one-step C–N coupling pathway by directly comparing it with the conventional two-step process via the imine intermediate. Building on this understanding, we achieved highly efficient electrochemical C–N coupling of HMF with methylamine by tailoring the adsorption configuration of HMF on Ag catalysts predominantly exposing the (111) facet. A high FE of 89.2% for MAMF with a selectivity of 90.2% could be achieved by Ag NP with exposed Ag(111) facet. Kinetics studies reveal that the C–N coupling follows an inner-sphere mechanism, where the adsorption behavior of HMF and hydrogenation of imine could play a crucial role in affecting the product distribution. In situ Raman spectroscopy and DFT calculations reveal that the η1(C)-aldehyde adsorption configuration of HMF on Ag NP leads to an optimal interaction strength of the functional aldehyde group with the catalyst surface. Besides, the better imine hydrogenation on Ag NP further helps to convert the imine to the respective amines. Thus, the polarization of the C=O could be optimized to facilitate the C–N bond formation while suppressing side reactions such as hydrogenation and dimerization. This work offers new insights into how substrate adsorption configuration regulates the selectivity of electrochemical C–N coupling between HMF and methylamine. These findings could potentially guide the design of efficient catalysts for electrochemical organic synthesis. Future efforts should focus on enabling green substrates, creating pathways to more valuable products, and designing scalable systems like the membrane electrode assembly (MEA), which will collectively pave the way for the industrial deployment of electrochemical C–N coupling technology.
Methods
Materials
AgNO3 (>99.8%), methylamine (40 wt.% in H2O), and acetone (HPLC) were purchased from Tianjin Jiangtian Chemical Technology Co., Ltd. (China). 5-Hydroxymethylfurfural (99%), CF3COOAg (98%) and polyvinylpyrrolidone (PVP, Mv = 55,000–58,000, 98%) were purchased from Meryer (Shanghai) Biochemical Technology Co., Ltd. K2HPO4 (98%), 5-[(methylamino)methyl]-2-furanmethanol (95%), 2,5-bis(hydroxymethyl)furan (98%) were purchased from Bide Pharmatech Ltd. NaOH (99%), ethylene glycol (HPLC), acetonitrile (HPLC) and NaCl (99%) were purchased from Aladdin Industrial Co. Ltd. NaHS (68–72%), ammonium formate (>99%) were purchased from Energy Chemical (Anhui, China). Sodium citrate (>99%) was purchased from J&K Scientific Ltd. L (+)-Ascorbic acid was purchased from Tianjin Guangfu Technology Development Co. Ltd. (China). KOH (96%) was purchased from Macklin Inc. (Shanghai, China). All gases used in this paper were from Air Liquide Co. Ltd., and all reagents except 5-[(methylamino)methyl]-2-furanmethanol (MAMF) were used directly without further purification.
Synthesis of Ag NP and Ag NC
Ag NP was synthesized using L (+) -ascorbic acid as the reductant and sodium citrate anhydrous as the stabilizer. Typically, 0.23 g ascorbic acid and 0.77 g sodium citrate anhydrous were dissolved in 200 mL of deionized water, and the pH of the solution was adjusted to 11 by adding 0.1 mol L–1 NaOH solution66. Subsequently, 4 mL of 0.5 mol L–1 aqueous solution of AgNO3 was added dropwise under continuous stirring (900 rpm) in a 30 °C water bath. The solution immediately changed from colorless to black, indicating the formation of Ag nanoparticles. After 150 min of reaction, the product was collected by centrifugation at 9000 rpm for 5 min, washed repeatedly with ethanol and deionized water, and finally redispersed after a second centrifugation at the same speed to obtain purified Ag NP.
In a standard synthesis for Ag NC, 25 mL of ethylene glycol (EG) was added into a flask and heated for 40 min under magnetic stirring in an oil bath set to 142 °C. A stable temperature of approximately 138 °C was measured for the reaction solution. Other reagents were separately dissolved in EG and sequentially introduced into the flask using a pipette. Specifically, 0.3 mL of NaHS solution (3.5 mM) was added first. After 4 min, 2.5 mL of NaCl solution (3 mM) was introduced, followed by the addition of 6.25 mL of PVP solution (20 mg mL–1) 2 min later. After another 2 min, 2 mL of CF3COOAg solution (282 mM) were introduced. Finally, 2.5 mL of NaBr solution (3 mM) was added after 5 min of delay. Note that the NaHS solution was prepared right before its injection. During the entire process, the flask was capped with a glass stopper except during the addition of reagents. After growth for 15 min, the synthesis was quenched by immersing the flask in an ice-water bath. The solution was split into four portions, and 125 mg of PVP was dissolved in each fraction. This is necessary in order to prevent aggregation during the washing steps. The products were precipitated with acetone and collected by centrifugation at 8000 rpm for 6 min, followed by washing with DI water and centrifuging at 12 000 rpm for 10 min twice to remove the remaining precursor, EG, and excessive PVP67. Since the use of NaBr during the synthesis, the product solution was centrifuged twice at 9000 rpm for 10 min to separate the insoluble AgBr precipitate from the Ag NC solution.
Fabrication of electrodes
For the preparation of electrodes, 30 mg of catalyst (Ag NP or Ag NC) with 160 μL of Nafion (5 wt.%) were dispersed in 3 mL of ethanol. After 40 min of sonication in an ice water bath, the catalyst ink was sprayed onto carbon paper at a set temperature of 78 °C. The loadings of the catalysts were controlled to be 1 mg cm–2.
Activity evaluation
The electrochemical performances of all the samples were tested using the Auto LAB electrochemical analyzer and carried out in an H-type cell. The anolyte (20 mL of buffer solution) and the catholyte (20 mL of buffer solution containing 20 mM HMF and methylamine) were separated by a bipolar membrane (BPM, Fumasep FBM-PK, Fumatech). Since the pKa of CH3NH3+ is about 10.6, adjusting the solution pH to 11 ± 0.2 ensures a significant portion of the amine to present as a base form to react with HMF to form aldimine. The electrolyte was freshly prepared the same day before the experiment. The 0.5 M phosphate buffer solution was prepared by using K2HPO4 and adjusting the pH to 11.0 with KOH. And pH of the solution was monitored before (11.0 ± 0.2) and after (11.1 ± 0.3) adding the methylamine to make sure no obvious change was observed.
The catholyte was stirred for 10 min with Ar protection before electrolysis. Carbon rod and Ag/AgCl (saturated KCl) electrodes were used as the anode and the reference electrode, respectively. The carbon paper of the working electrode was cut into a size of 2 cm2, and the geometric surface area for activity tests is 1.5 cm2. The back side of the carbon paper is covered with Kapton tape. All the tests for electrochemical C–N coupling of HMF with methylamine were conducted under magnetic stirring at 1200 rpm. The electrochemical impedance spectroscopy (EIS) measurements were performed with a 5 mV amplitude and a frequency range from 10,000 to 0.1 Hz. All the potentials in this paper were provided without iR corrected and converted to the reversible hydrogen electrode (RHE) through the following equation:
Product identification and quantification
During the reaction, the catholyte was continually stirred at 1200 rpm with Ar as the protective gas and carrier gas (flow rate: 5 sccm). The products were quantified after the amount of electrons flowing through the cathode reached 20 C as controlled by a potentiostat (Autolab PGSTAT204, Metrohm). Gaseous products were analyzed with online gas chromatography (GC7890B, Agilent Technologies, Inc.). For quantification of liquid products, the collected catholyte was first subjected to rotary evaporation at 40 °C to remove residual methylamine. Subsequently, the obtained liquid products were diluted fivefold and analyzed by high-performance liquid chromatography (HPLC, Agilent Technologies 1200 Infinity) equipped with a variable wavelength detector (VWD). The products were analyzed with a C18 column. The mobile phase is acetonitrile/DI water/5% ammonium formate (V/V/V = 6:1:3) solution with a flow rate of 0.5 mL min–1. The formation of products was further ensured by mass spectrometry (MS) and 1H nuclear magnetic resonance (1H NMR) spectroscopy.
The FE, selectivity and production rate are calculated according to Eqs. (2) – (6):
where the \({{n}}_{{\rm{MAMF}}}\), and \({{n}}_{{\rm{BHMF}}}\) represent the mole amounts of MAMF and BHMF after the electrolysis. The \({n}_{{\rm{HMF}}}^{0}\) represents the mole amount of HMF before reaction. The \({y}_{{\rm{HMF}}}\) represents the yield of HMF. Q refers to the total charge. F is Faraday’s constant. Scatalyst is the geometric area of the catalyst loaded on the electrode, and t is the reaction time.
Characterizations
Scanning electron microscopy (SEM, Regulus 8100) was employed to characterize the morphology of the catalysts. Transmission electron microscopy (TEM, JEOL JEM-2100F) was used to characterize the structure of the catalysts. X-ray powder diffraction (XRD; Bruker D8 Focus, Cu Kα radiation (λ = 1.54056 Å) at 40 kV and 40 mA) and X-ray photoelectron spectroscopy with an Al Kα X-ray source (XPS, EscaLab MK) were applied to characterize the structure and valence states of the as-prepared catalysts. XRD spectra were collected over a 2θ range of 10–80° at a scanning speed of 10°/min. The binding energy was calibrated using the C 1 s photoelectron peak at 284.6 eV as the reference.
In situ Raman spectroscopy
In situ Raman experiments were carried out in a custom-designed H-cell electrolyzer, using 0.5 M phosphate buffer solution as the electrolyte. The in situ Raman samples of Ag NP and Ag NC were prepared on Ag foil. The electrode was encased in a PEEK fitting, with an exposed geometric surface area of 1 cm2. A platinum wire and an Ag/AgCl electrode (saturated KCl, Gaossunion) were used as the counter and the reference electrode, respectively. Electrochemical measurements were carried out with a potentiostat CompactStat.e20250, IVIUM. The Raman tests were performed on a Confocal Raman Microscopy (RENISHAW, inVia reflex) equipped with a 532 nm He-Ne laser. The signal acquisition time is 51 s for each Raman spectrum.
Calculation of local pH
Potassium phosphite (K2HPO3) is derived from phosphorous acid (H3PO3), a diprotic acid possessing only two acidic protons (pKa1 ≈ 1.3, pKa2 ≈ 6.7). Under strongly alkaline conditions (pH = 11.0), the phosphite species exists almost entirely in its fully deprotonated form (HPO32–), far well outside its effective buffering range. As a result, the phosphite electrolyte provides negligible buffering capacity. Consequently, the local pH increase near the cathode is governed primarily by the generation and diffusion of OH– produced during electrochemical reactions. For the OH– generation flux, \({{J}_{{\rm{OH}}}}^{-}\), each electron transferred at the cathode approximately produces one OH– ion. Therefore,
where i is the current density and F is the Faradic constant (96495 C mol–1).
One-dimensional steady-state diffusion balance model was adopted in this manuscript. Fast ion coupling (electroneutral diffusion) was assumed. Therefore,
where the parameters could be used as19: bulk pH = 11.0, \({C}_{{\rm{bulk}}}\) = 10–3M; diffusion layer thickness δ = 100 µm; Deff = 5.27 × 10–9 m2 s–1; pHsurf = 14 + log10\((\frac{{C}_{{\rm{surface}}}}{M})\).
Computational model
Ag(111) and Ag(100) surface was modeled by a (3√3×3√3)R30° supercell and a 6×5 supercell, respectively. All surface models were composed of 4 layers of atoms with 2 uppermost layers relaxed. The metal slabs were separated by a ~ 20 Å vacuum space. The Brillouin zone was sampled at a 3 × 3 × 1 k-point mesh with Γ-centered for hexagonal supercells and Monkhorst-Pack for orthorhombic supercells. The Gibbs free energy under ambient conditions was corrected using a computational hydrogen electrode (CHE) model68.
DFT calculations
Density functional theory (DFT) calculations were performed utilizing the Vienna Ab Initio Simulation Package (VASP) in conjunction with the projector augmented wave (PAW) method69,70. The revised Perdew–Burke–Ernzerhof (RPBE)71 exchange-correlation functional was applied, and the dispersion effects were accounted for by employing the semi-empirical D3 method72,73. For this method, the consideration of a Hubbard U parameter is typically not necessary. The cutoff energy was set to 450 eV. The convergence threshold of electronic and force was set to 10−6 eV atom–1 and 0.02 eV Å–1, respectively. The Methfessel-Paxton smearing was applied with a smearing width of 0.15 eV. Related information is attached in Supplementary Data 1.
Data availability
The data that support the findings of this study are available within the paper and supplementary information. Additional data are available from the corresponding author on request. Source data are provided with this paper.
References
Froidevaux, V. et al. Biobased amines: from synthesis to polymers; present and future. Chem. Rev. 116, 14181–14224 (2016).
Kim, J. E. et al. Electrochemical C–N bond formation for sustainable amine synthesis. Trends Chem. 2, 1004–1019 (2020).
Li, J., Zhang, Y., Kuruvinashetti, K. & Kornienko, N. Construction of C-N bonds from small-molecule precursors through heterogeneous electrocatalysis. Nat. Rev. Chem. 6, 303–319 (2022).
Chandrashekhar, V. G. et al. Reductive amination, hydrogenation and hydrodeoxygenation of 5-hydroxymethylfurfural using silica-supported Cobalt-nanoparticles. ChemCatChem 14, e202101234 (2022).
Bender, M. T., Yuan, X., Goetz, M. K. & Choi, K.-S. Electrochemical hydrogenation, hydrogenolysis, and dehydrogenation for reductive and oxidative biomass upgrading using 5-hydroxymethylfurfural as a model system. ACS Catal. 12, 12349–12368 (2022).
Rosatella, A. A., Simeonov, S. P., Frade, R. F. M. & Afonso, C. A. M. 5-Hydroxymethylfurfural (HMF) as a building block platform: biological properties, synthesis and synthetic applications. Green. Chem. 13, 754–793 (2011).
Chen, W. et al. One pot synthesis of pharmaceutical intermediate 5-dimethylaminomethyl-2-furanmethanol from bio-derived carbohydrates. J. Biobased. Mater. Bio. 10, 378–384 (2016).
Meyer, T. H., Choi, I., Tian, C. & Ackermann, L. Powering the future: how can electrochemistry make a difference in organic synthesis? Chem 6, 2484–2496 (2020).
Roylance, J. J. & Choi, K.-S. Electrochemical reductive amination of furfural-based biomass intermediates. Green. Chem. 18, 5412–5417 (2016).
Gong, J. et al. Power-to-X: lighting the path to a net-zero-emission future. ACS Sustain. Chem. Eng. 9, 7179–7181 (2021).
Welch, A. J. et al. Operando local pH measurement within gas diffusion electrodes performing electrochemical carbon dioxide reduction. J. Phys. Chem. C. 125, 20896–20904 (2021).
Ji, K. et al. Electrocatalytic hydrogenation of 5-hydroxymethylfurfural promoted by a Ru1Cu single-atom alloy catalyst. Angew. Chem. Int. Ed. 61, e202209849 (2022).
Luo, D. et al. Intrinsic mechanism of active metal dependent primary amine selectivity in the reductive amination of carbonyl compounds. J. Catal. 395, 293–301 (2021).
Irrgang, T. & Kempe, R. Transition-metal-catalyzed reductive amination employing hydrogen. Chem. Rev. 120, 9583–9674 (2020).
Komanoya, T. et al. Electronic effect of ruthenium nanoparticles on efficient reductive amination of carbonyl compounds. J. Am. Chem. Soc. 139, 11493–11499 (2017).
Lee, D. K. et al. The impact of 5-hydroxymethylfurfural (HMF)-metal interactions on the electrochemical reduction pathways of HMF on various metal electrodes. ChemSusChem 14, 4563–4572 (2021).
Schiffer, Z. J., Chung, M., Steinberg, K. & Manthiram, K. Selective electrochemical reductive amination of benzaldehyde at heterogeneous metal surfaces. Chem. Catal. 3, 100500 (2023).
Zhang, M. et al. Designed TiS2 nanosheets for efficient electrocatalytic reductive amination of biomass-derived furfurals. Green. Chem. 24, 9570–9578 (2022).
Fukushima, T. & Yamauchi, M. Electrosynthesis of amino acids from biomass-derivable acids on titanium dioxide. Chem. Commun. 55, 14721–14724 (2019).
Anibal, J. & Xu, B. Electroreductive C–C coupling of furfural and benzaldehyde on Cu and Pb surfaces. ACS Catal. 10, 11643–11653 (2020).
Nilges, P. & Schröder, U. Electrochemistry for biofuel generation: production of furans by electrocatalytic hydrogenation of furfurals. Energy Environ. Sci. 6, 2925–2931 (2013).
Zhou, P. et al. Electrochemical hydrogenation of furfural in aqueous acetic acid media with enhanced 2-methylfuran selectivity using CuPd bimetallic catalysts. Angew. Chem. Int. Ed. 61, e202117809 (2022).
Zhang, W. et al. Facet dependence of electrocatalytic furfural hydrogenation on palladium nanocrystals. Chin. J. Catal. 43, 3116–3125 (2022).
Wu, Y. et al. Selective electroreduction of 5-hydroxymethylfurfural to dimethylfuran in neutral electrolytes via hydrogen spillover and adsorption configuration adjustment. Adv. Mater. 36, 2307799 (2024).
Anibal, J., Malkani, A. & Xu, B. Stability of the ketyl radical as a descriptor in the electrochemical coupling of benzaldehyde. Catal. Sci. Technol. 10, 3181–3194 (2020).
Chen, S. et al. How catalysts and experimental conditions determine the selective hydroconversion of furfural and 5-hydroxymethylfurfural. Chem. Rev. 118, 11023–11117 (2018).
Sitthisa, S. & Resasco, D. E. Hydrodeoxygenation of furfural over supported metal catalysts: a comparative study of Cu, Pd and Ni. Catal. Lett. 141, 784–791 (2011).
Yu, J. et al. Electroreductive coupling of benzaldehyde by balancing the formation and dimerization of the ketyl intermediate. Nat. Commun. 13, 7909 (2022).
Gong, J. & Luque, R. Catalysis for production of renewable energy. Chem. Soc. Rev. 43, 7466–7468 (2014).
Wu, X. et al. Multi-site catalysis of high-entropy hydroxides for sustainable electrooxidation of glucose to glucaric acid. Energy Environ. Sci. 17, 3042–3051 (2024).
Hu, C. et al. Synergism of geometric construction and electronic regulation: 3D Se-(NiCo)S/(OH) nanosheets for highly efficient overall water splitting. Adv. Mater. 30, 1705538 (2018).
Dong, H. et al. Electrochemical epoxidation enhanced by C2H4 activation and hydroxyl generation at the Ag/SnO2 interface. Nat. Commun. 16, 1901 (2025).
Zhu, W. et al. Enhanced CO2 electroreduction on neighboring Zn/Co monomers by electronic effect. Angew. Chem. Int. Ed. 59, 12664–12668 (2020).
Li, L., Wang, P., Shao, Q. & Huang, X. Metallic nanostructures with low dimensionality for electrochemical water splitting. Chem. Soc. Rev. 49, 3072–3106 (2020).
Xiao, Y. et al. Comprehensive study addressing the challenge of efficient electrocatalytic biomass upgrading of 5-(hydroxymethyl)furfural (HMF) with a CH3NH2 ionic liquid on metal-embedded Mo2B2 MBene nanosheets. Small 19, 2302271 (2023).
Kim, T. et al. Mechanistic investigation of electrocatalytic reductive amination at copper electrode. Chem. Commun. 59, 4818–4821 (2023).
Lan, J. et al. Efficient electrosynthesis of formamide from carbon monoxide and nitrite on a Ru-dispersed Cu nanocluster catalyst. Nat. Commun. 14, 2870 (2023).
Jia, S. et al. Synthesis of hydroxylamine via ketone-mediated nitrate electroreduction. J. Am. Chem. Soc. 146, 10934–10942 (2024).
Chen, W. et al. Catalyst selection over an electrochemical reductive coupling reaction toward direct electrosynthesis of oxime from NOx and aldehyde. J. Am. Chem. Soc. 146, 6294–6306 (2024).
Luo, X. et al. Optimization of electrochemical reduction of biomass derived 5-hydroxymethylfurfural (HMF): a volcano plot and bimetallic catalysts. ChemSusChem 17, e202400723 (2024).
Liu, S., Sun, C., Xiao, J. & Luo, J.-L. Unraveling structure sensitivity in CO2 electroreduction to near-unity CO on silver nanocubes. ACS Catal. 10, 3158–3163 (2020).
Sanghez de Luna, G. et al. AgCu bimetallic electrocatalysts for the reduction of biomass-derived compounds. ACS Appl. Mater. Interfaces 13, 23675–23688 (2021).
Magdić Košiček, K., Kvastek, K. & Horvat-Radošević, V. Hydrogen evolution on Pt and polyaniline modified Pt electrodes—a comparative electrochemical impedance spectroscopy study. J. Solid State Electr. 20, 3003–3013 (2016).
Deng, W. et al. Unraveling the rate-determining step of C2+ products during electrochemical CO reduction. Nat. Commun. 15, 892 (2024).
Pan, X.-Q. et al. Promoting electrocatalytic hydrogenation of 5-hydroxymethylfurfural using buffer electrolytes as proton-donating motifs: Theoretical predictions and experimental validations. Appl. Catal. B Environ. 323, 122191 (2023).
Bell, S. E. J. et al. Towards reliable and quantitative surface-enhanced Raman Scattering (SERS): from key parameters to good analytical practice. Angew. Chem. Int. Ed. 59, 5454–5462 (2020).
Zhang, J. et al. Determination of 5-hydroxymethylfurfural (5-HMF) in milk products by surface-enhanced Raman spectroscopy and its simulation analysis. Spectrochim. Acta A Mol. Biomol. Spectrosc. 279, 121393 (2022).
Chadderdon, X. H. et al. Mechanisms of furfural reduction on metal electrodes: distinguishing pathways for selective hydrogenation of bioderived oxygenates. J. Am. Chem. Soc. 139, 14120–14128 (2017).
Cheng, D. et al. Why do CuAl catalysts outperform in CO2 electro-reduction to C2H4? Sci. China Chem. 68, 763–771 (2025).
Bondue, C. J. & Koper, M. T. M. Electrochemical reduction of the carbonyl functional group: the importance of adsorption geometry, molecular structure, and electrode surface structure. J. Am. Chem. Soc. 141, 12071–12078 (2019).
Yu, W. et al. Theoretical and experimental studies of the adsorption geometry and reaction pathways of furfural over FeNi bimetallic model surfaces and supported catalysts. J. Catal. 317, 253–262 (2014).
Grass, M. E., Rioux, R. M. & Somorjai, G. A. Dependence of gas-phase crotonaldehyde hydrogenation selectivity and activity on the size of Pt nanoparticles (1.7–7.1 nm) supported on SBA-15. Catal. Lett. 128, 1–8 (2009).
Bondue, C. J., Calle-Vallejo, F., Figueiredo, M. C. & Koper, M. T. M. Structural principles to steer the selectivity of the electrocatalytic reduction of aliphatic ketones on platinum. Nat. Catal. 2, 243–250 (2019).
Xiong, K., Wan, W. & Chen, J. G. Reaction pathways of furfural, furfuryl alcohol and 2-methylfuran on Cu(111) and NiCu bimetallic surfaces. Surf. Sci. 652, 91–97 (2016).
Solomon, J. L., Madix, R. J. & Stöhr, J. Orientation and absolute coverage of furan and 2,5-dihydrofuran on Ag(110) determined by near edge x-ray absorption fine structure and X-ray photoelectron spectroscopy. J. Phys. D Appl. Phys. 94, 4012–4023 (1991).
Qin, Z. et al. Tuning the microenvironment of immobilized molecular catalysts for selective electrochemical CO2 reduction. Chem. Sci. 16, 5872–5879 (2025).
Greenler, R. G., Snider, D. R., Witt, D. & Sorbello, R. S. The metal-surface selection rule for infrared spectra of molecules adsorbed on small metal particles. Surf. Sci. 118, 415–428 (1982).
Pang, S. H., Román, A. M. & Medlin, J. W. Adsorption orientation-induced selectivity control of reactions of benzyl alcohol on Pd(111). J. Phys. Chem. C. 116, 13654–13660 (2012).
Lin, X. et al. Data-driven design of single-atom electrocatalysts with intrinsic descriptors for carbon dioxide reduction reaction. Trans. Tianjin Univ. 30, 459–469 (2024).
Bhardwaj, S., Rai, S., Sau, T. K. & Singh, H. Theoretical studies of Raman scattering properties of methylphosphine and methylamine adsorbed on gold clusters. Vib. Spectrosc. 76, 38–47 (2015).
Dai, W. et al. Synthesis of a two-dimensional covalent organic monolayer through dynamic imine chemistry at the air/water interface. Angew. Chem. Int. Ed. 55, 213–217 (2016).
Heidary, N. & Kornienko, N. Operando Raman probing of electrocatalytic biomass oxidation on gold nanoparticle surfaces. Chem. Commun. 55, 11996–11999 (2019).
Zhang, G. et al. Efficient CO2 electroreduction on facet-selective copper films with high conversion rate. Nat. Commun. 12, 5745 (2021).
Schlucker, S. Surface-enhanced Raman spectroscopy: concepts and chemical applications. Angew. Chem. Int. Ed. 53, 4756–4795 (2014).
Lin, X. et al. Machine learning-assisted dual-atom sites design with interpretable descriptors unifying electrocatalytic reactions. Nat. Commun. 15, 8169 (2024).
Qin, Y. et al. Size control over spherical silver nanoparticles by ascorbic acid reduction. Colloids Surf. A Physicochem. Eng. Asp. 372, 172–176 (2010).
Ruditskiy, A. & Xia, Y. Toward the synthesis of Sub-15 nm Ag nanocubes with sharp corners and edges: the roles of heterogeneous nucleation and surface capping. J. Am. Chem. Soc. 138, 3161–3167 (2016).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab-initio total energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Hammer, B., Hansen, L. B. & Nørskov, J. K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 59, 7413–7421 (1999).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Phys. D Appl. Phys. 132, 154104 (2010).
Acknowledgements
The authors acknowledge the National Key R&D Program of China (2021YFA1501503), the National Natural Science Foundation of China (22121004, 22250008, 22361142838, 22038009), the Energy Revolution S&T Program of Yulin Innovation Institute of Clean Energy (YIICE E412040705), the Haihe Laboratory of Sustainable Chemical Transformations (24HHWCSS00013), the Program of Introducing Talents of Discipline to Universities, and the Xplorer Prize for financial support.
Author information
Authors and Affiliations
Contributions
J.L.G. and P.Z. conceived and supervised the project. D.B.L., J.Y., X.L.Y., P.Z., and T.W. carried out the experiments, analyzed the data, and wrote the manuscript. Z.A.M. and Z.J.Z. carried out the DFT calculations. C.X.W. helped with the in situ Raman tests. J.Q.Z. helped with the electrochemical test. All the authors participated in the writing of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lai, D., Yu, J., Ma, ZA. et al. Electrochemical C–N coupling via adsorption modulation: selective synthesis of amines from biomass-derived 5-hydroxymethylfurfural. Nat Commun 17, 1892 (2026). https://doi.org/10.1038/s41467-026-68734-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68734-4
