
Abstract
Hierarchical immobilization of multicomponent biomacromolecules has been demonstrated to be a favorable protocol for constructing advanced biopharmaceuticals, but still with formidable challenges. Herein, we innovate a multicomponent synergistic immobilization approach combining an in-situ method with a structural-transformation-promoted dynamic exchange method in a flexible zeolitic pyrimidine framework (ZPF) platform to construct multicomponent biopharmaceuticals. In-depth mechanism investigation reveals that special functional groups (e.g., carboxyl, sulfhydryl, imidazolyl) on protein surface can significantly promote the dynamic structural transformation of ZPF and allow for the efficient immobilization of proteins on the surface of ZPF crystals via dynamic exchange approach. By combining with the in-situ method, which can immobilize biomacromolecules in the inner part of ZPF crystals, multicomponent biomacromolecules can be regionally localized within ZPF crystals. This controlled spatial preparation provides substantial advantages over conventional core-shell architectures for cascading enzyme immobilization and enables the development of advanced anticancer agents demonstrating enhanced therapeutic efficacy for female Balb/c nu mice. Moreover, this platform can also generate highly efficient hierarchical antibacterial formulations to significantly enhance the wound-healing process of male Sprague-Dawley (SD) rats. This study provides a revolution to immobilize multicomponent biomacromolecules and represents an innovative and effective platform of biopharmaceuticals with immense potential for anticancer and antibacterial formulations.
Similar content being viewed by others


Introduction
Biological macromolecules1,2,3,4,5, including proteins, nucleic acids, and polysaccharides, are essential and fundamental large molecules that play critical roles in sustaining life6,7, regulating organismal functions8,9, and facilitating the transmission of genetic information10,11. Investigating the synergistic use of biomacromolecules is of great significance for accumulating scientific theories and technological advances in a wide range of fundamental sciences12,13,14. However, the utilization of such biomacromolecules faces several challenges. For instance, packaging multiple biomacromolecules into one system with a rational design to achieve a synergistic effect remains challenging due to the intrinsically varied characteristics of each molecule15,16,17. Furthermore, after encapsulation, maintaining stability and biocompatibility of the whole system under various conditions, such as varied temperature, pH, and ionic strength, also requires considerable effort, particularly for in vivo applications18,19,20. The applications of these multicomponent biomacromolecules are diverse, including enzyme immobilization and the development of biopharmaceuticals, such as therapeutic agents targeting cancer or bacterial infections21,22. To ensure effective immobilization and delivery, carriers that provide protection, stabilization, and controlled release of the biomacromolecules are often employed23,24,25. However, conventional immobilization strategies face challenges, including biomacromolecule inactivation, poor carrier stability, and limited versatility26,27,28,29. Particularly, the immobilized enzyme carrier often causes the enzyme activity to decrease, and the ordered enzyme distribution inside the carrier is often challenging to achieve30. Furthermore, it is difficult to adjust the position of different components in their carriers31. To address these issues, it is urgently essential to develop carriers and immobilization methods that can facilitate the positioning, proportion, and quantification of multicomponent biomacromolecules to achieve a synergistic effect.
Over the past two decades, metal-organic frameworks (MOFs) have emerged as one of the most attractive functional supramolecular materials due to their adaptable and defined structures, high porosity, tunable pore size, and customizable functionality32,33,34. MOFs have demonstrated great potential in diverse research fields, including gas adsorption and separation35, chemical sensing36, heterogeneous catalysis37, and especially the immobilization of biomacromolecules for biopharmaceuticals38. The immobilization methods of MOFs for biomacromolecules mainly include pore adsorption, covalent grafting, and in-situ encapsulation39,40,41,42,43. Among them, the in-situ immobilization method, which simulates the phase separation of natural biological macromolecules, offers the significant advantage of being capable of immobilizing biological molecules regardless of their size. However, previously reported MOFs capable of enzyme immobilization under ambient aqueous conditions remain exceedingly rare (<10)32. Furthermore, conventional methodologies (e.g., core-shell methods) suffer mass transfer barriers and lack precise control over biomolecular spatial distribution within these materials, which presents significant challenges, particularly for multicomponent enzyme systems. Recently, our group has developed a dynamic exchange strategy (also named dynamic defect generation strategy) for enzyme immobilization, utilizing the dissociation equilibrium of MOFs mediated by enzymes44. This approach enables the uniform loading of enzymes onto the material surface, circumventing the size limitations of biomacromolecules. Thus, combining the in-situ and dynamic exchange strategies in one system can achieve the regional distribution of multicomponent biomacromolecules and facilitate their immobilization and function, but this has not yet been realized.
In this study, aiming to address the challenges associated with the spatial immobilization of multicomponent biomacromolecules, we have developed a multicomponent synergistic approach integrating in-situ and dynamic exchange methods to construct multicomponent biopharmaceutical formulations within a flexible MOF structure synthesized under ambient aqueous conditions—ZPF-8 (ZPF = zeolitic pyrimidine framework). The resulting bio-composites could realize a hierarchical localization of multicomponent proteins, hence serving as highly efficient anticancer or wound-healing biopharmaceuticals (Fig. 1). The discovery and utilization of ZPF-8 advance the design and synthesis of versatile platforms for combining multicomponent biomacromolecules and establish the guiding principles for developing biopharmaceutical platforms.

Schematic illustration figures were created in BioRender. Yi Tan, X. (2025) https://BioRender.com/ehu871i.
Results and discussion
Immobilization of biomacromolecules by the in-situ method
Pyrimidine plays a crucial role in biological systems, not only as an essential base in nucleic acids but also as a participant in metabolic processes and genetic information transmission45. Furthermore, it exhibits significant potential for applications in the pharmaceutical domain46. Our group has reported a flexible zinc-based zeolitic pyrimidine framework (ZPF-2), which can be synthesized at room temperature in an aqueous solution, hence benefiting the immobilization of enzymes via an in-situ method (Fig. 2a)47. That is, enzymes can be immobilized during the formation process of ZPF-2 crystals. Single-crystal X-ray diffraction (SCXRD) characterization suggested that the actual crystal structure of ZPF-2 fitted with the theoretical simulation (Figs. 2a, S1, Table S1). Notably, this is the inaugural instance where the structure of ZPF-2-Zn can be precisely determined by using single-crystal data. The results of Powder X-ray diffraction (PXRD), Thermal gravimetric analysis (TGA), Scanning electron microscope (SEM) images, N2 and CO2 adsorption-desorption of the synthesized ZPF-2 were also found to be consistent with the literature (Figs. 2a, S2–S4)47. To explore the ability of ZPF-2 to immobilize enzymes, we then used ZPF-2 to immobilize enzymes (e.g., glucose oxidase (GOx)) via an in-situ assembly approach (GOx@ZPF-2, @ = in-situ immobilization)48. GOx@ZPF-2 retained the initial PXRD pattern and SEM morphology of ZPF-2 (Fig. S5, S6), while Fourier transform infrared spectroscopy (FT-IR) spectra showed the characteristic peaks of GOx in GOx@ZPF-2 (Fig. S7). To investigate the distribution of GOx, ZPF-2 was synthesized in FITC-tagged GOx solution, and a Super-Resolution Imaging System (SIM) was conducted. The SIM images demonstrated that FITC-labeled GOx was uniformly distributed inside the GOx@ZPF-2 (Fig. S8).

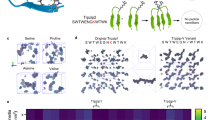
a Synthesis, structure, and calculated and experimental Powder X-ray diffraction (PXRD) patterns of ZPF-2. b Optical images of the single crystal and structural transformation from ZPF-2 to ZPF-8. c Structure, and calculated and experimental PXRD patterns of ZPF-8. Atom colors: zinc, light blue; carbon, gray; nitrogen, blue; oxygen, red; fluorine, green.
Immobilization of biomacromolecules by the dynamic exchange method
Subsequently, we further explored whether ZPF-2 could undergo the traditional dynamic exchange approach to achieve surface immobilization of enzymes, which could further construct a multi-enzyme regional distribution system. However, when ZPF-2 was immersed directly in the solution of GOx, we found that it barely immobilized the enzyme, indicating that ZPF-2 was challenging to be triggered by the enzyme molecules to dissociate and create defects for enzyme immobilization. Interestingly, we discovered that after treating ZPF-2 with acetone, THF, DMSO, 1,4-dioxane, or ethyl acetate, PXRD results revealed that ZPF-2 could transform into a phase (named ZPF-8) exhibiting a distinct PXRD pattern (Fig. 2c, S9). SCXRD characterization suggested ZPF-8 has nearly the same coordinate environment as ZPF-2 but crystallizes in higher symmetry, i.e., the cubic I-43m space group (a = b = c = 15.8963 Å, α = β = γ = 90°, V = 4016.9 Å3) (Fig. 2b, Table S1). The topological structure and coordination environment of ZPF-8 are almost identical to those of the well-known ZIF-8 (Figs. S10, S11). A face-to-face comparison of the structures of ZPF-2 and ZPF-8 found differences between the coordinates of the coordinate bonds and dihedral angles of ligands with 6-membered rings (Fig. S12). These results indicate that upon stimulation with specific solvents, the F-pymo ligand can rotate, thus leading to structural transformation (Fig. 2b). It should be noted that it belongs to a single-crystal-to-single-crystal transformation process, not the typical dissolve-regenerate pathway (Fig. 2b).
The transition process from ZPF-2 to ZPF-8 was then investigated in an in situ fashion. PXRD patterns of the transition states showed the gradual disappearance of the ZPF-2 peaks and the appearance of ZPF-8 (Fig. S13). Quantitative analysis of each transition state showed the deceleration of these transformations until all ZPF-2 microsized crystals transformed to ZPF-8 (Fig. S14). Interestingly, by simply immersing ZPF-8 in the water for a certain time (e.g., 24 h), ZPF-8 could transform back to ZPF-2. Both processes could be performed repeatedly upon altering the solvents for five cycles (Fig. S15). These interesting structure transformation processes were then investigated in depth via FT-IR spectra (Fig. S16). After immersing ZPF-8 in water, the characteristic peaks of Zn–H2O (γ(Zn–O) at 495 cm−1 and ρw(H2O) at 557 cm−1) appeared initially and then disappeared gradually (Fig. S17)49. These phenomena proved that the Zn–N bonds in ZPF-8 could be partially dissociated upon the attack of water molecules. Then, they reformed after extending the immersion time, forming a more stable structure in the water environment, i.e., ZPF-2. TGA traces and SEM images for ZPF-8 showed similar results with ZPF-2 (Figs. S18, S19), while its CO2 adsorption-desorption isotherm showed a typical type I isotherm and adsorbed ~40 cm3/g of CO2 at ~100 kPa, indicative of a more rigid structure (Fig. S20). Besides, the transformed ZPF-2 from ZPF-8 showed a feature very similar to that of pristine ZPF-2, suggesting complete reversibility of the structural transformation (Figs. S16–S20).
The dynamic nature and mild structural transformation conditions of ZPF-8 encouraged us to try the immobilization of biomacromolecules. ZPF-8 was immersed in an aqueous solution of GOx as a demonstration. A time-dependent loading curve was displayed (Fig. 3a), and the loading amount increased gradually with the extension of the loading time and approached a saturation of 0.12 g/g after 20 min. The PXRD pattern of the formed product (named as GOx⊂ZPF-2, ⊂ = dynamic exchange immobilization) confirmed the successful transition from ZPF-8 to ZPF-2 (Fig. 3b). FT-IR also showed the characteristic peaks of GOx in GOx⊂ZPF-2 (Fig. S21). To further investigate the distribution of GOx, ZPF-8 was soaked in FITC-tagged GOx solution for different times. The SIM images demonstrated that FITC-labeled GOx was mainly embedded in the outer layer of ZPF-2 particles (Fig. 3a), and the thickness of GOx entry increased with increasing loading time (Fig. S22).

a Loading capability and relative activity of ZPF-2 with different loading times toward glucose oxidase (GOx), with Super-Resolution Imaging System (SIM) image and illustration of FITC-GOx⊂ZPF-2. b PXRD patterns of GOx⊂ZPF-2. c SIM image and illustration of GOx⊂ZPF-2, and cycle stability of GOx⊂ZPF-2 and GOx@ZPF-2 for glucose oxidation. d Illustration of the structural-transformation-promoted dynamic exchange method to immobilize enzymes, which cannot be achieved via the conventional dynamic exchange strategy. @ = in-situ method; ⊂ = dynamic exchange method. All error bars mean ± s.d. received from three independent experiments (n = 3 independent experiments).
Accordingly, one can reason that the disintegration and reorganization process of the framework would be the driving force facilitating the encapsulation of GOx during the structural transformation from ZPF-8 to ZPF-2. To further explore the mechanism of enzyme encapsulation during immobilization, we also monitored the changes in soaking solutions via 1H Nuclear Magnetic Resonance (1H NMR) (Fig. S23). After the same soaking time in GOx solution, the ligand signal of ZPF-8 rose significantly (nearly two times compared with pure D2O), while pristine ZPF-2 released almost no ligands into D2O. This phenomenon indicated the generation of more defects on MOFs and the coordinate bonds mediated by GOx, which should come from the interaction between functional groups on proteins with metal ions. To prove this, 19 natural amino acids (except Tyr, which had a low solubility) were screened to identify which functional groups (-R) could promote this process via different immobilization efficiencies of ZPF-8. The results showed that the significantly high loading capacities of Cys (0.14 g/g), Glu (0.035 g/g), Asp (0.083 g/g), and His (0.10 g/g) suggested that besides predicted carboxyl, sulfhydryl and imidazolyl in proteins could also possibly promote the enzyme immobilization (Table S2, Fig. S24). To further prove this conclusion, seven glycine-based dipeptides and four polypeptides with different sizes were also tested, suggesting that the immobilization efficiencies had a positive correlation with the ratio of \(\frac{{{\rm{Cys}}}+{{\rm{Glu}}}+{{\rm{Asp}}}+{{\rm{His}}}}{{{\rm{total}}}\; {{\rm{amino}}}\; {{\rm{acids}}}}\) in these peptides (Tables S3, S4, Figs. S25, S26). These comparisons proved that after “activating” ZPF-2 into ZPF-8 and then immersing it in enzyme solution, proteins could generate more competitive coordination against original ligands during the dynamic structural transformation and enzyme immobilization process. Finally, along with original ligand release and defect generation, the coordination and immobilization of enzymes within the MOF structures were achieved.
To study the effect of soaking time on the catalytic performance of the encapsulated enzyme, we investigated the relationship between enzyme activity and loading time (Fig. S27). The activity increased gradually with the extension of the loading time and approached its highest at ~20 min. Notably, GOx⊂ZPF-2 could fully retain the free enzyme activity (Fig. 3a). Theoretically, immobilized enzymes on the surface of particles would greatly promote catalytic performance because they could expose more active enzymes from the material surface (Fig. 3d)50. To highlight the advantages of this dynamic structural transformation approach, we also used GOx@ZPF-2 as a comparison (Fig. 3c). It was found that the encapsulated enzymes retained 90% of their activity (loading amount: 0.15 g/g). All these findings further demonstrate that the dynamic transformation induces the formation of more defects in the material, resulting in a higher enzyme loading compared to the conventional defect generation strategy (0.12 vs 0.04 g/g), which exhibits meaningless enzymatic activity due to insufficient loading capacity (Fig. 3d). Compared with free GOx, GOx⊂ZPF-2 retained over half of its initial activity under various harsh conditions (e.g., GOx⊂ZPF-2 exposure to 70 °C, remaining activity = ~100%, trypsin: 56%; while free GOx only retained <15% under the same treating conditions) (Fig. S28, S29). Furthermore, GOx⊂ZPF-2 could maintain approximately 80% of its catalytic activity after 10 recycles and retain its crystallinity and morphology (the initial activity (defined as 100%) corresponds to the conversion rate measured during the inaugural catalytic cycle, Figs. 3c, S30). However, only 25% of the remaining activity of GOx@ZPF-2 was observed. It is possible because enzymes encapsulated inside the crystal catalyze the substrate, and then the produced H2O2 accumulates and partially damages the carrier from the inside (Fig. S31). These results indicated that enzyme⊂ZPF-2 is an excellent enzyme immobilization platform with superior biocatalytic performance and superior cyclic catalytic capacity.
In practical applications, multi-enzyme systems are often required to achieve advanced catalytic functions, presenting significant challenges for rational construction and optimization51. For example, GOx can catalyze the oxidation of glucose to gluconic acid while simultaneously generating hydrogen peroxide (H₂O₂), which subsequently serves as a substrate for catalase (CAT)52. Co-immobilization of these two enzymes could effectively verify the feasibility of enzyme cascade reactions. To systematically explore the influence of spatial protein distribution on cascade reaction efficiency, we developed a synergistic immobilization strategy. Initially, GOx was incorporated into the ZPF-2 via an in-situ synthesis method (i.e., GOx@ZPF-2), followed by selective immobilization of CAT onto these composite structures through a dynamic exchange method (during the transformation from GOx@ZPF-2 to GOx@ZPF-8, the enzymatic activity of GOx was fully retained), thereby establishing a GOx@CAT⊂ZPF-2 cascade system (Fig. S32). This synergistic strategy exploits the complementary characteristics of both immobilization methods: the in-situ embedded GOx remains stably anchored within the crystal core through multiple coordination interactions established during framework assembly, while the subsequent dynamic exchange process—triggered by the ZPF-8 → ZPF-2 transformation—generates surface defects and coordination vacancies that selectively capture CAT at the particle surface. This spatially programmed architecture creates an optimal substrate channeling pathway where glucose diffuses inward to react with core-embedded GOx, and the generated H₂O₂ is immediately consumed by the surface-localized CAT, minimizing intermediate accumulation and preventing oxidative damage to both enzymes and the framework structure. The hierarchical localization was confirmed by SIM images (Fig. S33). The catalytic performance of this hierarchical design was rigorously compared with two approaches: (I) a traditional core-shell configuration (GOx@ZPF-2@CAT@ZPF-2)53,54 and (II) simultaneous co-immobilization of both enzymes via dynamic exchange (GOx&CAT⊂ZPF-2) (Fig. S34). Under standardized conditions, maintaining equivalent enzyme loading capacities and ratios (GOx:CAT = 1:1) across all configurations, the GOx@CAT⊂ZPF-2 system exhibited superior catalytic efficiency, achieving >200% relative activity compared to the alternative materials (Fig. S35). These results convincingly demonstrate the critical importance of optimizing both the regional cascade distribution of enzymes and simultaneously maximizing enzyme-substrate interactions for enhanced catalytic performance. The recyclability of the optimized GOx@CAT⊂ZPF-2 system was evaluated through ten consecutive catalytic cycles. The system maintained approximately 80% of its initial activity, demonstrating substantial operational stability attributed to the rapid consumption of the generated H₂O₂ intermediate via CAT (Fig. S36).
The hierarchical immobilization strategy demonstrated above extends beyond enzymatic cascade catalysis to encompass diverse multicomponent biomedical systems requiring precise spatiotemporal coordination. In advanced therapeutic applications, distinct biomolecular components must function in orchestrated sequences—targeting moieties and therapeutic proteins in tumor therapy must operate sequentially to achieve site-specific accumulation followed by localized drug action55, while in wound healing applications, antimicrobial agents and tissue repair factors necessitate programmed release profiles to combat infection before promoting regeneration56. These complex biomedical scenarios uniformly demand spatially compartmentalized delivery systems that can regulate the sequential presentation of multiple bioactive components, thereby maximizing therapeutic efficacy while minimizing off-target effects.
Construction of multicomponent anti-tumor biopharmaceuticals
The starvation therapy for cancer, as an approach to treating malignant tumors, could consume the glucose reserves of tumor cells to eliminate cancer cells57. We then tried to combine in-situ and dynamic exchange methods to construct multicomponent synergistic formulations. For example, transferrin (Tf), known for its strong selective binding to the abundant transferrin receptors on the surface of tumor cells, can be utilized for targeted drug delivery to tumor cells. By employing Tf in a carrier, GOx can be directly transported into cancer cells, inducing apoptosis through intracellular glucose consumption58. So we incorporated GOx into ZPF-2, then immobilized Tf onto these composite structures, forming GOx@Tf⊂ZPF-2 multicomponent synergistic system (Figs. 4a, b, S37, S38). The hierarchical localization was confirmed by utilizing FITC-labeled GOx and RhB-labeled Tf, which was conducted via SIM images (Fig. 4c). The pH-responsive property of GOx@Tf⊂ZPF-2 revealed remarkable stability at physiological pH (7.4)—exhibiting merely 15% GOx release after 5 h (Fig. S39). Conversely, under acidic conditions (cancer cell’s lysosomal environment), material rapidly released GOx (a notable 90% at pH 4.6, 5 h)—indicating the carrier’s response capability to environmental stimuli and promoting the bioactive interaction between GOx and glucose within the tumor microenvironment.

a Illustration of the synthesis of GOx@Tf⊂ZPF-2 (Tf = transferrin). b PXRD patterns of GOx@Tf⊂ZPF-2, GOx@ZPF-8, GOx@ZPF-2, calculated ZPF-2, and calculated ZPF-8. c SIM images of GOx@Tf⊂ZPF-2. Each experiment was independently repeated three times, and the results were all similar. d Cytotoxicity of protein@ZPF-2 toward HeLa cells. 1: Tf⊂ZPF-2; 2: GOx@ZPF-2; 3: GOx&Tf@ZPF-2; 4: GOx@Tf⊂ZPF-2; 5: Tf@GOx⊂ZPF-2. IC50: 90.00, 66.79, 57.72, 39.77, and 66.64 μg/mL. e Cytotoxicity of protein@ZPF-2 toward HeLa cells. I: Tf⊂ZPF-2; II: GOx@Tf⊂ZPF-2; III: GOx&5-FU@Tf⊂ZPF-2 (5-FU = 5-fluorouracil). All error bars mean ± s.d. received from three independent experiments (n = 3 independent experiments). The significance was calculated by one-way ANOVA using Tukey’s multiple comparisons test. ****p < 0.0001.
The cytotoxicity of GOx@Tf⊂ZPF-2 was evaluated in vitro via MTT assay. This study involved two representative cancer cell lines (HeLa and MCF-7) and a normal cell line (3T3) to assess the cytotoxicity. Compared to the control ZPF-2, which exhibited half-maximum inhibitory concentration (IC50) values of 58.05 μg/mL, the viability of HeLa cells notably decreased when treated with GOx@Tf⊂ZPF-2 (IC50 = 39.77 μg/mL). This suggests that the encapsulation of GOx and Tf within the nanoparticles enhanced the toxicity against the cancer cells (Fig. S40). Conversely, negligible cytotoxicity was observed in 3T3 cells within the nanoparticle concentration range of 0–40 μg/mL (Fig. S41). The study also explored other counterpart multicomponent synergistic systems created through a similar method. Remarkably, adjusting the protein configuration within the nanoparticles could increase the values of IC50 (Fig. 4d). These results confirm that the enhanced cytotoxic effect is primarily due to the specific hierarchical distribution of the anti-tumor multicomponent synergistic system. To further support these findings, FITC-labeled materials were analyzed to assess cellular uptake capabilities through a confocal laser scanning microscope (CLSM) (Fig. S42). The images revealed a pronounced increase in green fluorescence in cells, indicating that surface-incorporated Tf significantly enhances cellular uptake by effectively targeting transferrin receptors, thereby leading to a marked accumulation of the material in HeLa cells.
The rational design of nanoparticle components and their ratios is crucial for optimizing cancer therapy. By immobilizing both proteins within the ZPF-2 structure, we could also adjust the GOx/Tf ratio to optimize anti-tumor efficiency (Fig. S43). The optimized GOx@Tf⊂ZPF-2 nanoparticles (GOx: Tf = 1.1: 1) yielded the lowest IC50 values of 39.77 μg/mL and 68.00 μg/mL for HeLa and MCF-7 cells, respectively. Additionally, we tested the universality of this approach by integrating two different anticancer agents—5-fluorouracil (5-FU) and apoptin caspase 3—into similar systems (Fig. S44). The assembled composites demonstrated significant anti-tumor activity, surpassing their counterparts without Tf. Furthermore, we combined cancer-starving therapy with small-molecule drugs to enhance the killing effect of the combined formulation on tumor cells, via incorporating 5-FU into GOx@Tf⊂ZPF-2 to assess its anti-tumor properties (Figs. 4e, S45). Synthesized GOx&5-FU@Tf⊂ZPF-2 nanoparticles exhibited remarkable cytotoxicity at lower concentrations against both HeLa and MCF-7 cells (IC50 = 25.38 or 54.01 μg/mL), confirming a significant enhancement in the combined anti-tumor effects via the design and regulation of this hierarchical multicomponent synergistic system.
After assessing the anti-tumor effect in vitro, the multicomponent synergistic tumor inhibition effect of GOx&5-FU@Tf⊂ZPF-2 was evaluated using a subcutaneous HeLa tumor-bearing female Balb/c nu mouse model (Fig. 5a). Throughout the therapeutic period (11 days), negligible fluctuations in mice’s body weights were observed following various treatments (Fig. S46). Digital photographs and spider plots visually confirmed that the tumors in the GOx&5-FU@Tf⊂ZPF-2 group were nearly eliminated, and the corresponding tumor-bearing ratio had decreased significantly, verifying the prominent multicomponent synergistic therapeutic outcome (Figs. 5b, c, S47). The relative tumor volume curves for different groups are presented in Fig. 5d, and the corresponding tumor growth inhibition (TGI) rates were calculated, while the GOx&5-FU@Tf⊂ZPF-2 group was found to be the highest (73.26%) (Fig. S48). In contrast, other control groups showed negligible tumor suppression or continued growth, comprehensively demonstrating the synergistic efficacy of our strategy. To investigate the therapeutic mechanism, an H&E staining assay was performed to investigate the histological damage and apoptosis levels in the tumors. As shown in Fig. S49, severe morphological changes and apoptosis were observed in the tumors of GOx&5-FU@Tf⊂ZPF-2 group. In contrast, no significant damage were detected in the tumors treated with the counterpart groups, further validating the synergistic effects mediated by the multicomponent system. These results clearly demonstrate the excellent therapeutic outcome exerted by GOx&5-FU@Tf⊂ZPF-2, which can be attributed to several factors. Primarily, Tf is utilized for targeted drug delivery to tumor cells. Subsequently, GOx mediates the competitive consumption of tumorous glucose while, simultaneously, 5-FU directly kills cancer cells. In summary, GOx&5-FU@Tf⊂ZPF-2, as a multicomponent synergistic anti-tumor system, potently inhibits tumor growth through hierarchical localization.

a The treatment strategy toward HeLa tumor-bearing mouse by employing GOx&5-FU@Tf⊂ZPF-2. b Photos of the tumor-bearing mice with different treatments on Day 0 and Day 11. c Photos of tumor dissection. d Relative tumor volume after different treatments. I: control; II: 5-FU; III: GOx@Tf⊂ZPF-2; IV: GOx&5-FU@ZPF-2; V: GOx&5-FU@Tf⊂ZPF-2. All error bars mean±s.d. received from three independent experiments (n = 4 rats). Schematic illustration figures were created in BioRender. Yi Tan, X. (2025) https://BioRender.com/ehu871i.
Construction of multicomponent anti-bacteria biopharmaceuticals for wound healing
Skin, the largest sensory organ, serves as a protective barrier for the human body against external damage. Wounds on the skin frequently develop in various situations, and the blood or blood clot in the early phases of hemostasis provides a nourishing culture media for the growth of invading bacteria59. The above promising multicomponent synergistic anti-tumor results have also inspired the further construction of multicomponent synergistic anti-bacteria formulations to form composite agents for application in wound healing. By adjusting the distribution of the components within the system, it is possible to target the various phases of wound healing. Extraordinary, nisin, known for its strong inhibitory effect on many gram-positive bacteria, can be utilized in the initial inflammation phase to eliminate invading bacteria60. Zn2+ in MOFs can promote the proliferation of fibroblasts and collagen synthesis, which is conducive to the growth of granulation tissue61. Additionally, trypsin (Tps) can provide powerful support for the maturation phase by eliminating inflammatory secretions, breaking down denatured proteins, and promoting tissue regeneration62. Considering the above positive factors, Tps@Nisin⊂ZPF-2 multicomponent synergistic system could be formed for wound healing (Figs. 6a, S50, S51). The hierarchical localization was confirmed by SIM images (Fig. S52). The in vitro antibacterial activity of Tps@Nisin⊂ZPF-2 was evaluated using minimum inhibitory concentration (MIC) standard tests against Staphylococcus aureus (ATCC 29213). Under the same nisin concentration (MIC of free nisin = 50 mg/L), the antibacterial effects of Tps@Nisin⊂ZPF-2 were found to be very close to free nisin, while an insignificant antibacterial effect was observed for ZPF-2 (Fig. S53). The results demonstrated that our reversible deformation strategy did not affect the antimicrobial properties of the encapsulated nisin. These preliminary results showed the feasibility of applying this synergistic immobilization strategy to build antibacterial formulations through hierarchical localization for wound healing.

a Illustration of the synthesis of Tps@Nisin⊂ZPF-2 (Tps = trypsin). b Schematic diagram of the infected wound model with treatment project. c Microphotographs of the infected wound on different days (0, 3, 7, 10, and 14). d Schematic diagram of the wound-healing process of I ~ V groups. e Wound healing rate compared to the initial wounds. I: control; II: Nisin; III: ZPF-2; IV: Nisin⊂ZPF-2; V: Tps@Nisin⊂ZPF-2. All error bars mean ± s.d. received from three independent experiments (n = 4 rats). The significance was calculated by one-way ANOVA using Tukey’s multiple comparisons test. **p < 0.01. ***p < 0.001.****p < 0.0001. Schematic illustration figures were created in BioRender. Yi Tan, X. (2025) https://BioRender.com/ehu871i.
To further evaluate the wound healing capacity of Tps@Nisin⊂ZPF-2 in vivo, experiments were conducted on the dorsum of male Sprague-Dawley (SD) rats (Fig. 6b). Results showed the wound contraction for Tps@Nisin⊂ZPF-2 was superior to other groups, exhibiting the best wound healing capacity even on the 3rd day (Fig. 6c, e). The percentages of wound area closure at different healing times were calculated, revealing that after 7th days, wounds treated with Tps@Nisin⊂ZPF-2 achieved significant closure of ~95%, compared to the control, which showed ~75% wound closure. Remarkably, after 14th day of treatment with Tps@Nisin⊂ZPF-2, the wounds healed completely, with 100% wound contraction, demonstrating a highly improved wound healing ability. These results indicate that multicomponent synergistic anti-bacteria systems can be beneficial and effective for accelerating wound healing, due to the multicomponent synergies in Tps@Nisin⊂ZPF-2.
Histopathological studies were carried out to further compare the wound healing efficiencies via hematoxylin-eosin (H&E) and Masson’s staining (Figs. S54, S55). After 7th day, wounded tissues in the control group still exhibited numerous immune cell infiltrations. Surprisingly, for Tps@Nisin⊂ZPF-2, inflammatory cell infiltration was suppressed, and a significantly higher number of fibroblasts was observed around the impaired region. After 14th day, compared to the irregular structure in control, the wounds treated with Tps@Nisin⊂ZPF-2 showed higher density and volume fraction of collagen, greater regularity of both epithelium and connective tissue with more fibroblasts, a complete epithelium structure, and regenerated hair follicles and blood vessels. Tissue of Tps@Nisin⊂ZPF-2 group exhibited excellent wound healing characteristics, facilitating a faster and more effective re-epithelization process, connective tissue remodeling and arrangement, and the formation of a more regular structure with increased hair follicle formation. This can be attributed to the excellent multicomponent synergistic properties of Tps@Nisin⊂ZPF-2, promoting overall wound healing.
In conclusion, we have demonstrated the construction of a dynamic zeolitic pyrimidine framework to enable precise and efficient regulation of multicomponent combinations of biomacromolecules. This flexible MOF possesses the ability to either immobilize biomacromolecules via an in situ method or via a structural-transformation-promoted dynamic exchange method. The structural transformation process and enzyme immobilization mechanism were unveiled in-depth via various characterizations, including SCXRD, in situ PXRD, FT-IR, etc. We found that the Zn-N bonds in the ZPF structure could be partially dissociated, then reversibly reformed upon the attack of water molecules, accompanied by ligand movement, rotation, and recombination. This structural transformation allows for the immobilization of enzymes on the outer part of the framework, which is challenging to achieve by the traditional immobilization strategies. Our findings suggest that proteins can produce more competitive coordination from their sulfhydryl, carboxyl, and imidazolyl functional groups, ultimately promoting enzyme immobilization. The resulting biocatalysts exhibited superior biocatalytic performance, protective capabilities, and superior cyclic catalytic capacity. Notably, by combining the in-situ method with the dynamic exchange method in one system, various multicomponent biomacromolecule formulations with hierarchical localization can be constructed, such as cascading enzyme immobilization, anti-tumor and anti-bacteria multicomponent formulations. For instance, cascading immobilized enzyme preparations demonstrated a two-fold enhancement in catalytic activity compared to conventional core-shell structured formulations. Furthermore, a multicomponent biopharmaceutical with co-immobilization of transferrin, GOx and 5-FU led to enhanced therapeutic performance, confirmed by in vitro and in vivo anti-tumor experiments. Further, by incorporating the antibacterial nisin and the wound healing-promoting trypsin, advanced antibacterial formulations for wound healing were successfully constructed through hierarchical localization, while simultaneously utilizing the components of the ZPF to significantly enhance the wound healing process through in vitro and in vivo experiments. This study enriches the toolbox for biomacromolecule immobilization and provides a synergistic approach for fabricating multicomponent biopharmaceuticals.
Methods
General
Except for the special description, chemicals like Zn(NO3)2·6H2O (Innochem, AR), 5-fluoro-2-hydroxypyrimidine (Innochem, 97%), 5-FU (Innochem, 99%), Nisin (Innochem, 98%), organic solvents (Innochem, AR), and biological reagents like GOx (Solarbio, 10KU), CAT (Solarbio, BR), transferrin (Solarbio, 98%), trypsin (Solarbio, BR) throughout the experiments, were purchased from commercial sources and used as received without further purification. All cell lines certification is provided by the ATCC company from which they originated. Animal experiments on anti-tumor were approved by the Animal Research Committee of Nankai University, China (Approval Number: 2023-SYDWLL-000641), and were carried out strictly according to the Guide for the Care and Use of Laboratory Animals (National Research 119 Council, 1996). Animals used in wound healing experiments complied with the National Research Council’s Guide for the Care and Use of Laboratory Animals, and these procedures were reviewed and approved by the Committee for the Care and Use of Animals at the Tianjin University of Science & Technology in China (Approval Number: SYXK2023-0003).
Characterization
SCXRD data were collected at 120 K via an Oxford Cryo stream system on a SuperNova (Mo) X-ray Source with a micro-focus sealed X-ray tube. The structure was solved and refined using Olex2 with ‘XS’ and ‘XL’ plug-in. Hitachi UH-5300 UV-vis spectrophotometer was used to record the catalytic process. The absorbance value was measured with the SpectraMax M2e microplate reader. Powder X-ray diffraction (PXRD) patterns of all materials were collected at ambient temperature with a Rigaku dmax 2500 diffractometer operated at 40 kV and 40 mA using Cu Kα (λ = 1.5418 Å) radiation, with a scan speed of 1 sec/step, a step size of 0.02° in 2θ, and a 2θ range of 5° to 40°. Crude data were subjected to baseline correction and analyzed by Jade 6. The low-pressure CO2 adsorption and desorption isotherms were measured at 195 K using a Micromeritics ASAP 2460 instrument. Before gas adsorption measurements, the samples were outgassed at 95 °C for 24 h before the measurements. Fourier transform infrared spectrophotometer (FT-IR) spectra of all materials were obtained using a Thermo Fisher spectrometer with ATR modes. The confocal images experiments were performed on a Zeiss Elyra 7 Super-Resolution Imaging System (SIM) with a 63× objective oil lens. The Confocal laser scanning microscopy (CLSM) images were recorded on Leica TCS SP8.
Synthesis of ZPF-8
Zn(NO3)2·6H2O (1 mmol) was dissolved in deionized water (5 mL), and 5-fluoro-2-hydroxypyrimidine (2 mmol dissolved in 5 mL of water) was added under stirring. The clear solutions were kept at room temperature for about 5 min, and then the pH was raised to 4.5 by adding 1 M base (NaOH, KOH, or NH3·H2O). The precipitate was harvested by centrifugation at 4000 x g for 1 min and then washed twice with deionized water to obtain ZPF-2. As-synthesized ZPF-2 powder was immersed in THF for 24 h to obtain ZPF-8.
Synthesis of GOx⊂ZPFs
GOx solution (3 mg/mL) was prepared by dissolving GOx in 50 mM MES buffer (pH = 6.0). 15 mg as-synthesized ZPF-8 was ultrasonically dispersed in 1 mL GOx solution, and incubated in a 37 °C shaker for 1, 2, 4, 7, 10, 15, 20, 30, and 60 min, respectively. The solid was collected by centrifuge and washed with DI water twice. The supernatants were collected to determine the amount of immobilized GOx in MOFs. The uptake amount was determined by the Bradford method. The loading efficiency (LE%) and loading capacity (LC, g/g) were calculated as follows:
Catalytic performance of GOx, and GOx⊂ZPFs composites
The activity of GOx was determined through the 3,5-dinitrosalicylic acid (DNS) method, which is used to detect the concentration of glucose. 100 μg GOx (or GOx⊂ZPFs composites containing 100 μg GOx) were added into 0.91 mL aqueous solution. After that, 90 μL of glucose solution (50 mM) was added. After 7 min, 30 μL of the supernatant was taken and mixed with 60 μL DNS solution. The mixture was heated at 100 °C for 5 min. Hitachi UH-5300 UV-vis spectrophotometer was used to record the catalytic process. After cooling to room temperature, the final glucose concentrations were monitored by measuring the absorbance at 540 nm and compared with the standard curve to calculate the final glucose concentration.
Synthesis of GOx@Tf⊂ZPF-2
3 mg GOx was added to the aqueous solutions of 5-fluoro-2-hydroxypyrimidine (0.2 M, 667 μL). Zinc nitrate hexahydrate was dissolved in deionized water (0.2 M, 333 μL). These two aqueous solutions were mixed and placed at room temperature for 20 min. The solid was collected by centrifugation and washed with DI water and DMSO twice, respectively. As-synthesized GOx@ZPF-2 powder was immersed in DMSO and incubated at room temperature on a shaker for 20 min to obtain GOx@ZPF-8, then collected by centrifugation. Transferrin solution (3 mg/mL) was prepared by dissolving transferrin in 50 mM HEPES buffer (pH = 7.0). As-synthesized GOx@ZPF-8 was ultrasonically dispersed in 1 mL transferrin solution, and incubated at room temperature on a shaker for 20 min to obtain GOx@Tf⊂ZPF-2. The solid was collected by centrifugation and washed with DI water twice. The supernatants were collected to determine the amount of immobilized GOx or transferrin in MOFs. The uptake amount was determined by the Bradford method.
In vivo anticancer therapeutic evaluation
Tumor (HeLa) bearing Balb/c nu mice (female, 6 weeks of age) were randomly divided into 4 groups (n = 5). When the tumor volume reached about 100 mm3, the mice were intratumorally injected with Normal saline (100 µL), 5-FU (156 μg/mL, 100 µL), GOx@Tf⊂ZPF-2, GOx&5-FU@ZPF-2, and GOx&5-FU@Tf⊂ZPF-2 (the corresponding 5-FU was 156 μg/mL, 100 μL), respectively. Prior to the initiation of the experiment, the rats were anesthetized with isoflurane (3 % to 4 % for induction and 2 % to 2.5 % throughout the surgical procedure) to ensure the maintenance of a stable state. The tumor volume and body weight of mice were recorded every other day. The tumor volume was calculated as follows: V = W2×L/2, where W and L represented the tumor width and length, respectively. The mice were sacrificed 11 days post-treatment, and the tumors were collected and photographed. The tumor growth inhibition (TGI) rate was calculated according to the equation.
Where VC represents the tumor volume of the control group, and VT represents the tumor volume after the treatments.
Simultaneously, the tumors of the mice were collected and used for histological and H&E staining assay, and the tumor size of all experimental animals did not exceed 20 mm as stated in the ethical review documents.
Wound healing assay
A full-layer skin defect infection model was used to evaluate the wound healing of Sprague-Dawley (SD) rats (male, 180–200 g, 6–8 weeks of age), and 25 rats were randomly divided into 5 groups. Prior to the initiation of the experiment, the rats were anesthetized with isoflurane (3% to 4% for induction and 2% to 2.5% throughout the surgical procedure) to ensure the maintenance of a stable state. After anesthesia, shave the back and disinfect it with medical alcohol. A circular full-layer excision wound (8 mm diameter) was produced with a biopsy perforator (Japan, BP-80F). Each wound was inoculated with 50 μL Staphylococcus aureus suspension (OD600 = 0.3). Leave the bacterial suspension on the wound for 30 min. They were treated with PBS (Control), nisin, ZPF-2, Nisin⊂ZPF-2, and Tps@Nisin⊂ZPF-2, respectively. After that, the wound area was immobilized with sterile gauze. Wound photos were taken on days 0, 3, 7, 10 and 14, and the wound size was quantified by Image J software. The healing process takes about 14 days. Freshly regenerated skin tissue was collected on days 7 and 14. These tissues were immobilized in 4% PFA solution for 48 h. Wound healing was evaluated by H&E and Masson staining. Sex was considered in the study design, and detailed information was described in the manuscript.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Center, under deposition numbers CCDC 2172065 (1), 2172121 (2). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other relevant data generated and analysed during this study, which include experimental, spectroscopic, crystallographic and computational data, are included in this article and its supplementary information, or available from the corresponding authors on request. Source data are provided with this paper.
References
Aguirre-Ramirez, M., Silva-Jimenez, H., Banat, I. M. & De Rienzo, M. A. D. Surfactants: physicochemical interactions with biological macromolecules. Biotechnol. Lett. 43, 523–535 (2021).
Moraitakis, G., Purkiss, A. G. & Goodfellow, J. M. Simulated dynamics and biological macromolecules. Rep. Prog. Phys. 66, 383–406 (2003).
Russo Krauss, I., Merlino, A., Vergara, A. & Sica, F. An overview of biological macromolecule crystallization. Int. J. Mol. Sci. 14, 11643–11691 (2013).
Mirkin, N. & Moreno, A. Advances in crystal growth techniques of biological macromolecules. J. Mex. Chem. Soc. 49, 39–52 (2005).
Amini, N. et al. Microorganism-derived biological macromolecules for tissue engineering. Front. Med-PRC 16, 358–377 (2022).
Hasnain, S. S. Synchrotron techniques for metalloproteins and human disease in the post-genome era. J. Synchrotron Radiat. 11, 7–11 (2004).
Huang, H. Y. et al. Terahertz spectral properties of glucose and two disaccharides in solid and liquid states. iScience 25, 104102 (2022).
Honjoh, S. & Nishida, E. Two sides of lifespan regulating genes: pro-longevity or anti-longevity? J. Biochem. 149, 381–388 (2011).
Soultanas, P. & Janniere, L. The metabolic control of DNA replication: mechanism and function. Open Biol 13, 230220 (2023).
Eckenroth, B. E. et al. Remote mutations induce functional changes in active site residues of human DNA polymerase β. Biochemistry 56, 2363–2371 (2017).
Franchi, M. & Gallori, E. A surface-mediated origin of the RNA world: biogenic activities of clay-adsorbed RNA molecules. GENE 346, 205–214 (2005).
Chaves, R. C., Dahmane, S., Odorico, M., Nicolaes, G. A. F. & Pellequer, J. L. Factor Va alternative conformation reconstruction using atomic force microscopy. Thromb. Haemostasis 112, 1167–1173 (2014).
Ehlers, W. & Wagner, A. Multicomponent modelling of human brain tissue: a contribution to the constitutive and computational description of deformation, flow and diffusion processes with application to the invasive drug-delivery problem. Comput. Methods Biomech. Biomed. Eng. 18, 861–879 (2015).
Banerjee, A. & Howarth, M. Nanoteamwork: covalent protein assembly beyond duets towards protein ensembles and orchestras. Curr. Opin. Biotechnol. 51, 16–23 (2018).
Goor, O., Hendrikse, S. I. S., Dankers, P. Y. W. & Meijer, E. W. From supramolecular polymers to multicomponent biomaterials. Chem. Soc. Rev. 46, 6621–6637 (2017).
Landman, N. & Kim, T. W. Got RIP? Presenilin-dependent intramembrane proteolysis in growth factor receptor signaling. Cytokine Growth F. R. 15, 337–351 (2004).
Pourjavadi, A., Doulabi, M., Soleyman, R., Sharif, S. & Eghtesadi, S. A. Synthesis and characterization of a novel (salep phosphate)-based hydrogel as a carrier matrix for fertilizer release. React. Funct. Polym. 72, 667–672 (2012).
Lu, Y. Identification and Roles of Photosystem II Assembly, Stability, and Repair Factors in Arabidopsis. Front. Plant Sci. 7, 168 (2016).
Farag, M. R. & Alagawany, M. Erythrocytes as a biological model for screening of xenobiotic toxicity. Chem. Biol. Interact. 279, 73–83 (2018).
Li, Y. et al. Controlling the functional performance of emulsion-based delivery systems using multicomponent biopolymer coatings. Eur. J. Pharm. Biopharm. 76, 38–47 (2010).
Liao, J. Y. et al. Co-immobilization of two-component hydroxylase monooxygenase by functionalized magnetic nanoparticles for preserving high catalytic activity and enhancing enzyme stability. Int. J. Biol. Macromol. 164, 3163–3170 (2020).
Mohsin, N. U. et al. Cyclooxygenase-2 (COX-2) as a target of anticancer agents: a review of novel synthesized scaffolds having anticancer and COX-2 inhibitory potentialities. Pharmaceuticals 15, 1471 (2022).
Samanta, D. & Sarkar, A. Immobilization of bio-macromolecules on self-assembled monolayers: methods and sensor applications. Chem. Soc. Rev. 40, 2567–2592 (2011).
Avnir, D., Coradin, T., Lev, O. & Livage, J. Recent bio-applications of sol-gel materials. J. Mater. Chem. 16, 1013–1030 (2006).
Kuchler, A., Yoshimoto, M., Luginbuhl, S., Mavelli, F. & Walde, P. Enzymatic reactions in confined environments. Nat. Nanotechnol. 11, 409–420 (2016).
Piana, S. & Bilic, A. The nature of the adsorption of nucleobases on the gold [111] surface. J. Phys. Chem. B 110, 23467–23471 (2006).
Ladaviere, C., Delair, T., Domard, A., Pichot, C. & Mandrand, B. Covalent immobilization of biological molecules to maleic anhydride and methyl vinyl ether copolymers - A physico-chemical approach. J. Appl. Polym. Sci. 71, 927–936 (1999).
Bentley, M. D., Roberts, M. J. & Harris, J. M. Reductive amination using poly(ethylene glycol) acetaldehyde hydrate generated in situ: applications to chitosan and lysozyme. J. Pharm. Sci. 87, 1446–1449 (1998).
Maghraby, Y. R., El-Shabasy, R. M., Ibrahim, A. H. & Azzazy, H. M. E. Enzyme immobilization technologies and industrial applications. ACS Omega 8, 5184–5196 (2023).
Katsimpouras, C. & Stephanopoulos, G. Enzymes in biotechnology: critical platform technologies for bioprocess development. Curr. Opin. Biotechnol. 69, 91–102 (2021).
Magro, M. et al. Nanotechnology-based strategies to develop new anticancer therapies. Biomolecules 10, 735 (2020).
Zhang, X. et al. A historical overview of the activation and porosity of metal–organic frameworks. Chem. Soc. Rev. 49, 7406–7427 (2020).
Liang, J. & Liang, K. Nano-bio-interface engineering of metal-organic frameworks. Nano Today 40, 101256 (2021).
Feng, L., Wang, K.-Y., Lv, X.-L., Yan, T.-H. & Zhou, H.-C. Hierarchically porous metal–organic frameworks: synthetic strategies and applications. Natl. Sci. Rev. 7, 1743–1758 (2020).
Li, J.-R., Kuppler, R. J. & Zhou, H.-C. Selective gas adsorption and separation in metal–organic frameworks. Chem. Soc. Rev. 38, 1477–1504 (2009).
Kreno, L. E. et al. Metal–organic framework materials as chemical sensors. Chem. Rev. 112, 1105–1125 (2012).
Huang, W., Huang, S., Chen, G. & Ouyang, G. Biocatalytic metal-organic frameworks: promising materials for biosensing. ChemBioChem 23, e202100567 (2022).
Ge, X., Wong, R., Anisa, A. & Ma, S. Recent development of metal-organic framework nanocomposites for biomedical applications. Biomaterials 281, 121322 (2022).
Wang, C. E. & Liao, K. M. Recent advances in emerging metal- and covalent-organic frameworks for enzyme encapsulation. ACS Appl. Mater. Interfaces 13, 56752–56776 (2021).
Sha, F. et al. Unveiling the critical role of spatial organization on enzymatic cascade reactions in a crystalline framework with hierarchical porosity. ACS Mater. Lett. 7, 409–416 (2025).
Hsu, P.-H. et al. Rapid fabrication of biocomposites by encapsulating enzymes into Zn-MOF-74 via a mild water-based approach. ACS Appl. Mater. Interfaces 13, 52014–52022 (2021).
Liao, F.-S. et al. Shielding against unfolding by embedding enzymes in metal–organic frameworks via a de novo approach. J. Am. Chem. Soc. 139, 6530–6533 (2017).
Shieh, F.-K. et al. Imparting functionality to biocatalysts via embedding enzymes into nanoporous materials by a de novo approach: size-selective sheltering of catalase in metal–organic framework microcrystals. J. Am. Chem. Soc. 137, 4276–4279 (2015).
Feng, Y. et al. A dynamic defect generation strategy for efficient enzyme immobilization in robust metal-organic frameworks for catalytic hydrolysis and chiral resolution. Angew. Chem. Int. Ed. 62, e202302436 (2023).
Chandel, N. S. Nucleotide metabolism. Cold Spring Harbor Perspect. Biol. 13, a040592 (2021).
Prachayasittikul, S. et al. Roles of pyridine and pyrimidine derivatives as privileged scaffolds in anticancer agents. Mini-Rev. Med. Chem. 17, 869–901 (2017).
Galli, S. et al. Polymorphic coordination networks responsive to CO2, moisture, and thermal stimuli: porous cobalt(II) and zinc(II) fluoropyrimidinolates. Chem. - Eur. J. 14, 9890–9901 (2008).
Li, M. et al. Fabricating covalent organic framework capsules with commodious microenvironment for enzymes. J. Am. Chem. Soc. 142, 6675–6681 (2020).
Zhu, K., Gao, Y. & Li, G. Synthesis, crystal structure, and fluorescence properties of a zinc(II) coordination polymer. Synth. React. Inorg. M. 41, 785–790 (2011).
An, H. et al. Incorporation of biomolecules in metal-organic frameworks for advanced applications. Coord. Chem. Rev. 384, 90–106 (2019).
Shi, J. et al. Bioinspired construction of multi-enzyme catalytic systems. Chem. Soc. Rev. 47, 4295–4313 (2018).
Ma, Y. et al. Nanoclustered cascaded enzymes for targeted tumor starvation and deoxygenation-activated chemotherapy without systemic toxicity. ACS Nano 13, 8890–8902 (2019).
Man, T. et al. Hierarchically encapsulating enzymes with multi-shelled metal-organic frameworks for tandem biocatalytic reactions. Nat. Commun. 13, 305 (2022).
Fan, W., Yu, Z., Appadoo, D., Liang, K. & Liang, J. Enhancing multi-enzyme cascade activity in metal-organic frameworks via controlled enzyme encapsulation. Small 21, 2503059 (2025).
Zhang, Y. et al. Hierarchical targeting nanoplatform for nir-ii photoacoustic imaging and photo-activated synergistic immunotherapy of glioblastoma. Adv. Funct. Mater. https://doi.org/10.1002/adfm.202419395 (2025).
Cheng, Y. et al. Hierarchical acceleration of wound healing through intelligent nanosystem to promote multiple stages. ACS Appl. Mater. Interfaces 11, 33725–33733 (2019).
Fan, W. et al. Glucose-responsive sequential generation of hydrogen peroxide and nitric oxide for synergistic cancer starving-like/gas therapy. Angew. Chem. Int. Ed. 56, 1229–1233 (2017).
Shen, Y. et al. Transferrin receptor 1 in cancer: a new sight for cancer therapy. Am. J. Cancer Res. 8, 916–931 (2018).
Caldwell, M. D. Bacteria and antibiotics in wound healing. Surg. Clin. N. Am. 100, 757–776 (2020).
Mouritzen, M. V., Andrea, A., Qvist, K., Poulsen, S. S. & Jenssen, H. Immunomodulatory potential of Nisin A with application in wound healing. Wound Repair Regen. 27, 650–660 (2019).
Chasapis, C. T., Ntoupa, P. S. A., Spiliopoulou, C. A. & Stefanidou, M. E. Recent aspects of the effects of zinc on human health. Arch. Toxicol. 94, 1443–1460 (2020).
Villalba-Rodríguez, A. M. et al. Nanoclay/polymer-based hydrogels and enzyme-loaded nanostructures for wound healing applications. Gels 7, 59 (2021).
Acknowledgments
This work is supported by financial support from the National Natural Science Foundation of China (22022808, Y. C., 21502139, X. M.), the National Key Research and Development Program of China (2021YFC2102100, Y. C.), Haihe Laboratory of Synthetic Biology (22HHSWSS00008, Y. C.), China Postdoctoral Science Foundation (General Program, 2022M721700, M. Y., 2023M731440, X. M.), 2023 Talent Development Special Funds Support Project of Jinan City (202333020, X. M.), Joint Fund for Innovation and Development of Natural Science Foundation of Shandong Province (ZR2023LSW017, X. M.) and The Fundamental Research Funds for the Central Universities (Nankai university, Y. C.). The authors extend their gratitude to Ms. Wang Fanghui from Scientific Compass (www.shiyanjia.com) for providing invaluable assistance with the ICP-OES/MS analysis.
Author information
Authors and Affiliations
Contributions
Y.C., Z.Z. and M.Y. conceived and designed the project. R.S., J.G., J.Y., and Z.D. performed the catalytic experiments. L.X., A.Z. and Y.S. performed the anti-tumor experiments. X.M. and L.W. performed the wound healing experiments. M.Y. and X.M. performed the other experiments, analyzed the results, and wrote the manuscript with contributions from all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, M., Meng, X., Shi, R. et al. Multicomponent synergistic immobilization via in-situ and dynamic exchange strategies for constructing hierarchical biopharmaceuticals. Nat Commun 17, 2208 (2026). https://doi.org/10.1038/s41467-026-69009-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69009-8
