
Abstract
Electrochemical C − N coupling is an appealing approach for sustainable urea synthesis, while it is technically challenging due to the complex reaction mechanism and the spatiotemporal mismatch between C- and N- intermediates. Here, inspired by click chemistry, we design a hierarchical click-site catalyst (Se−InOx) that enables an efficient sequential-chain coupling pathway for urea electrosynthesis, achieving a urea yield rate of 254.94 mmol h−1 g−1, Faradaic efficiency of 78.61%, >85% Nurea-selectivity and 100% Curea-selectivity. Mechanistic studies reveal that Se−InOx as the first click-site can selectively adsorb NO3− and hydrogenate it to stable *NO2, while inhibiting CO2 adsorption at this stage. The surface-anchored *NO2 then acts as the second click-site to click couple with CO2, forming the key *CO2NO2 intermediate. This sequential-chain coupling strategy effectively resolves the spatiotemporal mismatch between N- and C- intermediates, thereby maximizing the suppression of side-reactions and enhancing C − N coupling selectivity. Techno-economic analysis and scalable synthesis validate the feasibility of this approach, providing a blueprint for high-selectivity multicomponent electrosynthesis.
Similar content being viewed by others

Introduction
Urea (CO(NH2)2), with annnual production exceeding 180 million tons, is an indispensable nitrogen-based fertilizer and a key intermediate in pharmaceuticals and fine chemicals1,2,3,4. Its industrial production via the Haber−Bosch (350−550 °C, 150−350 bar) and Bosch−Meiser (150−200 °C, 150−250 bar) processes is highly energy-intensive5,6,7,8, consuming 1–2% of global energy and emitting over 200 million tons of CO2 annually (equivalent to 1.2 tons of CO2 emissions per ton of urea, Fig. 1a)9,10,11. Electrochemical urea synthesis (urea electrosynthesis) from waste-derived nitrate (NO3−) and anthropogenic CO2 under ambient conditions offers a sustainable alternative to fossil-dependent thermochemical processes, with the potential for net-negative carbon emissions (up to −0.6 tons of CO2 per ton of urea, Fig. 1b)3,8,9,12,13,14,15,16,17,18,19,20,21. However, this route is technically limited by low activity and selectivity.

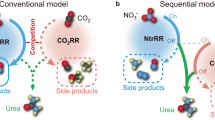
a Comparison of traditional urea production via the industrial Bosch−Meiser process versus sustainable electrosynthesis driven by renewable electricity. The conventional Bosch−Meiser process operates under harsh conditions, consuming substantial energy and resulting in large CO2 emissions. In contrast, the electrochemical approach powered by renewable energy provides a sustainable and carbon-negative alternative, directly converting waste-derived nitrate and anthropogenic CO2 into urea under ambient conditions. b Net CO2 emissions associated with the Bosch−Meiser process compared to sustainable electrochemical synthesis. c Mechanistic differences between conventional electrocatalytic processes and the hierarchical click catalysis strategy introduced herein. Traditional methods suffer significantly from competitive adsorption of reactants, spatial-temporal mismatches, and undesired side-reactions. Our proposed hierarchical click catalysis approach overcomes these limitations by introducing sequential and non-competitive hierarchical active sites, enabling precise adsorption and sequential coupling of nitrate and CO2-derived intermediates. This effectively suppresses side-reactions and substantially enhances catalytic activity and selectivity toward C−N coupling to urea. Colour codes: C, grey; N, blue; O, red; H, white.
Recent advances in urea electrosynthesis have demonstrated promising improvements in activity and selectivity through the design of synergistic catalytic strategies9,14,15,16,22,23,24,25,26,27 such as bonded Fe−Ni pairs15, alternating CuWO4 dual-metal sites16 and heterostructured Cu−Bi bimetallics22. While these studies underscore the immense promise of sustainable urea production, major challenges remain as follows: (i) a complex 16-electron transfer involving two C−N coupling steps results in broad product distributions16,17,23; (ii) spatial separation of active sites leads to spatiotemporal mismatches between C- and N- intermediates (Fig. 1c)19,22; and (iii) insufficient regulation over the cross-adsorption of NO3−, CO2 and their corresponding N- and C- intermediates causes undesired cross-contamination14,28. These factors collectively promote undesired CO2 reduction (CO2RR) and nitrate reduction (NO3RR) side-reactions that suppress C − N coupling and limit urea selectivity9,12,15,18. These challenges underscore the urgent need for new catalytic strategies capable of orchestrating C−N coupling with high precision and selectivity.
Click chemistry, awarded the 2022 Nobel Prize in Chemistry, offers a highly modular and selective framework for assembling complex molecules under mild conditions with high efficiency and minimal by-products29,30,31. Inspired by its ability to precisely stitch together small molecular units, we propose a hierarchical click-site strategy for urea electrosynthesis, in which two reactants, NO3− and CO2, undergo sequential-chain coupling. The first click-site selectively anchors and activates either NO3− or CO2 while excluding the other, followed by a second click-site that promotes targeted coupling with the remaining reactant via a nucleophilic attack on the surface-bound intermediate. This hierarchical click-site strategy, inspired by click chemistry, enables precise sequential-chain adsorption of NO3− and CO2 on a single active site, addressing key challenges in C−N coupling selectivity by mitigating (i) competitive intermediate adsorption, (ii) spatiotemporal mismatches, and (iii) interfacial cross-contamination. Despite its conceptual promise, this approach remains unexplored in urea electrosynthesis due to two major challenges: (i) the difficulty of achieving selective adsorption of only one feedstock at the first click-site, given the comparable affinity of most catalysts for both NO3− and CO2; and (ii) the instability of the second click-site, which is often over-hydrogenated or prematurely desorbed.
To overcome these challenges, we systematically screened representative catalysts from CO2RR, NO3RR and C−N coupling, identifying InOx as a promising base due to its weak CO2 adsorption via O-terminal (*OCO) configurations, disfavoring *CO formation while preferentially adsorbing and stabilizing nitrate-derived *NO2 (Supplementary Fig. 1)32,33,34,35,36,37. Guided by molecular orbital theory (Supplementary Fig. 2)38, substituting lattice oxygen with a larger and less electronegative element is expected to increase electron density at the active centers and induce moderate lattice distortion, thereby making CO2 adsorption thermodynamically unfavorable. Following this rationale, we replaced lattice O with Se, yielding a Se-modulated InOx catalyst (Se−InOx) with enhanced *NO2 stabilization and fully suppressed CO2 binding. In this system, Se−InOx (Fig. 1c) serves as the first click site, selectively adsorbing NO3− and converting it to stable *NO2 without interacting with CO2, while suppressing undesired NO3RR and CO2RR. The surface-bound *NO2 then functions as second click site, facilitating the selective nucleophilic attack of CO2 at the N site of *NO2 to form the key *CO2NO2 intermediate. This sequential-chain coupling aligns C- and N- intermediates both spatially and temporally, enabling precise C−N bond formation while minimizing cross-contamination and by-product generation. Overall, the hierarchical click-site concept enables selective and sustainable urea electrosynthesis, establishing a generalizable framework for high-selectivity multicomponent electrosynthesis.
Results
Design of hierarchical click-site catalysts
Effective implementation of hierarchical click catalysis requires sequential and selective adsorption of CO2 and NO3−, wherein CO2 adsorption is selectively suppressed while NO3− is preferentially adsorbed and subsequently transformed into a stable *NO2 during the first click step. To identify suitable host for this function, we initially screened several metals commonly employed in CO2RR. Metals such as Cu, Fe, Zn, and Ag exhibited strong CO2 adsorption via carbon-terminal (*COO) modes (Fig. 2a), leading to complex intermediates and products, such as methane, methanol, ethylene, and ethanol, etc.32,33. Conversely, metals such as Bi, Sn, and particularly In, exhibit weaker CO2 adsorption via the oxygen-terminal (*OCO) mode, predominantly yielding formate (HCOOH)34. Among these, In-based catalysts, represented by In2O3, exhibit the weakest CO2 affinity35,36,37. Meanwhile, the intrinsically high activation barrier from *NO2 to *NOOH in NO3RR favors the stabilization of *NO2 (Supplementary Fig. 1), positioning In2O3 as a promising candidate for hierarchical click-site design. However, pristine In2O3 still retains measurable CO2RR activity, necessitating further electronic modification to completely suppress undesired CO2 adsorption36,37. Based on molecular orbital theory (Supplementary Fig. 2)38, enhancing the local electron density around In centers can weaken interactions with the electrophilic CO2 molecule. To this end, we substituted lattice oxygen with larger and less electronegative anions (S, Se) to modulate the local electronic environment of the In2O3. Electron localization function (ELF) analysis confirmed the enhanced electron density around In atoms upon doping with S and Se (Fig. 2b). This result is quantitatively supported by charge redistribution analysis (Fig. 2c and Supplementary Figs. 3 and 4), manifesting as an increase in electron density around In atoms from 1.24 (InOx) to 1.83 (Se−InOx). Structural distortion indices (Di) further revealed progressive lattice distortion from InOx (Di=0.014) to S−InOx (Di=0.039) and Se−InOx (Di=0.048, Fig. 2d). These distortions shift electronic states toward the Fermi level (Fig. 2e), weakening intrinsic In−O covalent bonds, thus enabling selective adsorption and minimizing undesired side-reactions.

a Comparison of CO2 adsorption strength across representative metal-based catalysts. b Electron location function maps for InOx, S−InOx, and Se−InOx. c Charge variations at In and doping sites (X=O, S, Se) in InOx, S−InOx, and Se−InOx. d Corresponding crystal structures illustrating lattice distortions, with Di denoting distortion indices. e Effect of atomically dispersed Se on the electronic environment of In sites within Se−InOx. f, g Comparative CO2 adsorption energies (f) and ICOHP analysis of In−OCO2 bond (g) for InOx, S−InOx, and Se−InOx. h, i Comparative NO3− adsorption energies (h) and corresponding ICOHP analysis of In−ONO3− bond strengths (i) for InOx, S−InOx, and Se−InOx. j, k Schematic illustrations of CO2 and NO3− adsorption behaviors on pristine InOx and Se−InOx surfaces. Colour codes: C grey, O red, N blue, H white, In orchid, S yellow, Se light green, active site white bar. Source data are provided as a Source data file.
Density functional theory (DFT) calculations were conducted to validate the modulation of surface adsorption behavior. Substitution of lattice oxygen with S or Se rendered CO2 adsorption thermodynamically unfavorable, with the adsorption energy shifting from −0.26 eV (InOx) to +0.17 eV (S−InOx) and +0.27 eV (Se−InOx), respectively (Fig. 2f). Integrated crystal orbital Hamilton population (ICOHP) analyses revealed decreased In−OCO2 bonding strength on Se−InOx surfaces, confirming the suppression of CO2 adsorption (Fig. 2g and Supplementary Fig. 5-7). In contrast, NO3− adsorption progressively strengthened from InOx to S−InOx and Se−InOx, as indicated by increased covalency in In−ONO3− interactions observed via ICOHP analysis, projected density of states (PDOS), and differential charge density mapping (Fig. 2h, i and Supplementary Figs. 8–10). As expected, the Se−InOx displayed the strongest *NO2 adsorption and the highest hydrogenation barrier toward *NOOH formation (Supplementary Fig. 11), effectively preventing the over-reduction to NH3 by-product. Collectively, these results indicate that Se−InOx acts as an efficient hierarchical click-site, selectively adsorbing NO3− and stabilizing it as *NO2, while completely suppressing CO2 adsorption and thereby minimizing undesired side-reactions (Fig. 2j, k).
Characterization of hierarchical click-site catalysts
The Se−InOx catalyst was synthesized via a two-step pyrolysis process. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images revealed that Se−InOx consisted of porous nanoparticles with an average diameter of ~30 nm, comparable to InOx and S−InOx (Fig. 3a and Supplementary Figs. 12–17). Powder X-ray diffraction (XRD) analysis (Fig. 3a) confirmed retention of the cubic In2O3 phase (PDF#71-2195) after S or Se incorporation. High-resolution aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) demonstrated distinct lattice fringes of 0.25 nm indexed to the (040) and (004) planes along the [100] zone axis (Fig. 3b, c), in agreement with simulated HR-TEM and electron diffraction patterns (Supplementary Figs. 18–21). Atomic-resolution STEM-energy dispersive X-ray spectroscopy (STEM-EDS) mapping distinctly revealed atomically dispersed Se uniformly substituting for O atoms around In sites (Fig. 3d), corroborated by X-ray photoelectron spectroscopy (XPS, Supplementary Figs. 22 and 23). Elemental analysis determined the Se content as ~2.92 at%, comparable to metal single-atom concentrations corresponding to a near-stoichiometric atomic dispersion39,40. Similar substitution levels were observed for S–InOx (Supplementary Table 1). Complementary integrated differential phase contrast (iDPC) imaging revealed local lattice distortions arising from the substitution of larger-radius Se atoms in place of smaller O atoms within the InOx lattice (Fig. 3e and Supplementary Figs. 24–26), consistent with previous distortion analysis and further confirmed by geometric phase analysis (GPA) strain (Supplementary Fig. 27)41. Based on these findings, an atomic model was constructed (Fig. 3f), highlighting how Se substitution at O lattice sites induced lattice rearrangements, modifying the local electronic environment of the In active sites and thereby influencing catalytic performance. We performed X-ray Absorption Near Edge Structure (XANES) and Extended X-ray Absorption Fine Structure (EXAFS) measurements at the In K-edge to examine local coordination and oxidation state changes on the InOx, S−InOx, and Se−InOx catalysts (Supplementary Fig. 28−31 and Supplementary Table 3). The XANES spectra (Supplementary Fig. 30a) show that the absorption edges of three samples lie between those of metallic In and In2O3, and are close to that of In2O3, suggesting the oxidation state is slightly below +3 in all samples. Notably, with the incorporation of S and Se, a gradual redshift of the edge was observed, indicating a gradual reduction in the In oxidation state. This trend is attributed to the larger atomic radii and lower electronegativities of S and Se, which promote electron localization around In centers and reduce the symmetry of In−O coordination, thereby enhancing electronic back-donation to In atoms. The non-phase-corrected FT-EXAFS spectra (Supplementary Fig. 30b) display a sharp first-shell peak for pristine InOx, whereas broader and more asymmetric peaks appear in S−InOx and Se−InOx, indicating increased local disorder and the presence of additional In−S or In−Se bonds. This interpretation is further supported by wavelet transform (WT) analysis (Supplementary Fig. 31), where the maximum intensity for Se−InOx (~4.60 Å−1) exceeds that of S−InOx (~4.20 Å−1) and InOx (~4.00 Å−1), reflecting contributions from heavier scatterers. EXAFS fitting (Supplementary Table 3) reveals that all three samples maintain a similar In coordination number (N ≈ 6), with partial substitution of In−O by In−S or In−Se (N ≈ 0.6) in the doped catalysts. These comparable coordination environments establish a solid basis for subsequent structure-property analysis. Collectively, these findings confirm that S and Se incorporation induces subtle local distortions and partially modulates the In oxidation state, without drastically altering the overall coordination geometry.

a XRD patterns of InOx, S−InOx and Se−InOx; inset of (a) shows a TEM image of Se−InOx. b, c Aberration-corrected HAADF-STEM images along the [100] direction; inset of (c) shows an intensity line scan for the atoms within the yellow boxed area. d Atomic-level STEM-EDS element mapping images of In + O + Se, In + Se, In + O and HAADF + O + Se. e iDPC−STEM image along the [100] direction; wherein yellow and red circles indicate positions of In and O/Se, respectively, and arrows indicate the distortion. f Graphical representation of InOx and Se−InOx, wherein blue, pink, and purple dots represent In, O, and Se atoms, respectively. g In-situ ATR−SEIRAS results on InOx and Se−InOx surfaces saturated with CO2, NO3− and CO2 + NO3− under operando potentials. h Experimental (Exp.) and simulated (Sim.) EPR spectra. i FEHCOOH and FENO2- on InOx, S−InOx and Se–InOx surfaces at 50 mA cm−2. j Proposed catalytic mechanism of hierarchical click catalysis on the Se−InOx surface. Colour codes: C grey, O red, N blue, H white, In orchid, Se light green. Source data are provided as a Source data file.
Subsequently, in-situ attenuated total reflection infrared (ATR-SEIRAS) spectroscopy under CO2-saturated conditions showed pronounced CO2 adsorption on InOx but negligible signals on Se−InOx (Fig. 3g and Supplementary Fig. 32), consistent with theoretical predictions of suppressed CO2 affinity (Fig. 2f, g). In contrast, under conditions with only NO3− present, Se–InOx exhibited markedly enhanced adsorption and stabilization of *NO2 species. Notably, simultaneous introduction of CO2 and NO3− resulted in the detection of a red-shifted CO2 adsorption peak and a blue-shifted *NO2 signal on the Se−InOx surface (Fig. 3g and Supplementary Fig. 33), suggesting direct interaction and electron transfer between adsorbed CO2 and *NO2, thus forming the crucial *CO2NO2 intermediate. Electron paramagnetic resonance (EPR) spectroscopy confirmed signals for *CO2NO2 and *CONHx intermediates (Fig. 3h), further supporting direct C−N coupling on Se−InOx. Complementary electrochemical characterization reinforced the spectroscopic findings (Fig. 3i). Based on these results, we propose a hierarchical click-site catalytic mechanism proceeding via a sequential-chain coupling pathway (Fig. 3j): (i) Se doping activates adjacent In sites as first click-sites, which selectively adsorb NO3− and rapidly convert it into stable *NO2 without competing CO2 adsorption. (ii) These *NO2 species then function as second click-sites, selectively coupling with CO2 at the N-center to generate the pivotal *CO2NO2 intermediate. This sequential-chain design effectively suppresses side-reactions associated with CO2RR and NO3RR, thereby overcoming the intrinsic challenges of competitive adsorption and spatiotemporal mismatch (Supplementary Fig. 34).
Enhanced urea selectivity on hierarchical click-site catalysts
The urea electrosynthesis performance of hierarchical click-site catalysts was evaluated using gas-diffusion electrodes loaded with catalysts in a flow electrolysis cell with CO2-saturated 0.1 M KHCO3 + 0.1 M KNO3 electrolyte (Supplementary Fig. 35). Se−InOx with 2.92 at% Se displayed the highest urea FE, yield rate and Jurea among samples with varying Se contents (1.16, 2.92 and 6.77 at%) and was thus selected for subsequent evaluation (Supplementary Figs. 36–44 and Table 2). As shown in Fig. 4a, b, urea yield rate and FE increased progressively with S and Se incorporation, particularly Se−InOx achieved a competitive urea yield rate and FE at −0.4 V (vs. RHE) with 69.29 ± 7.48 mmol h−1 g−1 and 62.44 ± 2.85%, surpassing S−InOx (49.45 ± 6.51 mmol h−1 g−1 and 44.15 ± 3.50%) and significantly outperforming InOx (14.85 ± 2.11 mmol h−1 g−1 and 10.02 ± 1.12%, Supplementary Tables 4–6). In addition, the S/Se co-doped sample (S1.16Se1.64 − InOx) exhibited intermediate urea electrosynthesis activity between that of S−InOx and Se−InOx, further corroborating the electronic modulation effect of Se doping on the In active sites (Supplementary Figs. 45–49 and Table 1). Full product analysis showed similarly low NH3 and NO2− yield rates among the three catalysts, while HCOOH production predominated on InOx but was strongly suppressed on Se−InOx with enhanced urea performance (Supplementary Figs. 50, 51 and Tables 7–9). These observations, supported by partial current densities (Jproduct) and Tafel slopes (Supplementary Figs. 52–58), are consistent with theoretical and in-situ spectroscopic evidence showing that Se introduction promotes C−N coupling while suppressing CO2RR and NO3RR side-reactions. Control experiments confirmed that urea formation required the co-presence of CO2 and NO3− (Fig. 4c), affirming that urea arose from their co-electroreduction. Moreover, Se−InOx exhibited stable electrochemical performance over 10 consecutive cycles (Fig. 4d), with negligible variation in urea yield rate, Jurea, and FE. Post-reaction characterizations (XRD, HAADF-STEM, STEM mapping, diffraction, and XPS, Supplementary Figs. 59–62 and Table 1) confirmed that the atomic-scale structural integrity, homogeneous elemental distribution, and original electronic configuration after prolonged electrochemical operation of Se−InOx, suggesting good electrochemical durability and structural stability.

a, b Urea yield rates and FEs of Se−InOx, S−InOx, and InOx in CO2-saturated 0.1 M KHCO3 + 0.1 M KNO3 electrolyte. c Comparison of urea yield rates for Se−InOx under different gas/electrolyte conditions. d Cycling stability test of Se−InOx over 10 consecutive electrolysis cycles at −0.4 V (vs. RHE). e Effect of NO3− concentration on the FE distribution of all products. f FEs of all products for Se−InOx, S−InOx, and InOx at −0.4 V (vs. RHE) in CO2-saturated 0.1 M KHCO3 + 0.15 M KNO3 electrolyte. g, h N−selectivity (g) and C−selectivity (h) for Se−InOx, S−InOx, and InOx in CO2-saturated 0.1 M KHCO3 + 0.15 M KNO3 electrolyte. i Comparison of optimal urea yield rates, FEs, Jurea, Nurea−selectivity, and Curea−selectivity for Se−InOx, S−InOx, and InOx. j Comparison of urea yield rates and FEs of Se−InOx with other catalysts reported in the literatures. Electrolyte pH: 6.8 ± 0.5. All potentials are without iR compensation. Error bars represent the standard deviation from three independent measurements. Source data are provided as a Source data file.
The first click-site requires sufficient NO3− to generate adequate *NO2 on the catalyst surface, while the second click-site necessitates a comparable amount of CO2 to effectively couple with *NO2. Further, optimized urea selectivity was achieved by balancing NO3− and CO2 availability at the catalytic interface through controlled adjustment of NO3– concentrations (c(NO3−)) and CO2 flow rate. At c(NO3−) below 0.15 M (0.05 and 0.10 M), HCOOH and H2 were detected as the main by-product alongside urea, with minor amounts of NO2− and NH3 (Fig. 4e). Increasing the c(NO3−) to 0.15 M resulted in the highest urea FE of 78.61%, with HCOOH undetected and minimal production NO2–, NH3 and H2 production (Fig. 4e), indicating an optimal dynamic balance of NO3− and CO2 at the catalyst surface. Further increasing c(NO3−) resulted in increased NO2− and NH3 formation, suggesting over-hydrogenation to NH3 by-product, consistent with a concurrent decrease in H2 evolution. We further evaluated the effect of CO2 flow rate at fixed c(NO3−) = 0.15 M (Supplementary Fig. 63). At low flow rates (≤40 mL min−1), urea FE decreased markedly, while NO3RR by-products became dominant, likely due to limited CO2 accessibility at the catalyst-electrolyte interface. This imbalance led to *NO2 accumulation, followed by desorption or over-hydrogenation to NH3. Increasing the flow rate to 80 mL min−1 significantly enhanced urea FE and suppressed NO2− and NH3 formation, thereby mitigating side reactions (Supplementary Fig. 63). Notably, HCOOH remained undetectable throughout. These findings define 0.15 M NO3− and 80 mL min−1 CO2 as the optimal conditions for achieving high urea selectivity on hierarchical click-site catalysts. Even under optimal interfacial conditions, InOx favored CO2RR (FEHCOOH = 41.28%, CHCOOH–selectivity = 94.42%, Fig. 4f–h and Supplementary Figs. 64 and 65) over NO3RR (FENH3+NO2- = 20.85%, NNH3+NO2-–selectivity = 73.38%) and C−N coupling (FEurea = 19.52%, Nurea−selectivity = 26.62%, Curea−selectivity = 5.58%). Conversely, S−InOx and Se−InOx notably suppressed CO2RR and NO3RR side-reactions. Notably, Se−InOx achieved a urea FE of 78.61%, Nurea−selectivity of 85.32%, and Curea−selectivity of 100% (Figs. 4f–h and Supplementary Figs. 64 and 65), alongside significantly reduced NH3 formation (FENH3 = 0.53%, NNH3−selectivity = 0.58%) and completely inhibits HCOOH formation (FEHCOOH = 0.00%, NHCOOH−selectivity = 0.00%). Thus, hierarchical click-site engineering endowed Se−InOx with competitive urea yield rate, FE, Jurea, and Nurea/Curea−selectivities (Fig. 4i), demonstrating promising performance among reported electrocatalysts (Fig. 4j and Supplementary Table 10).
Mechanistic verification
To elucidate the underlying mechanism of urea electrosynthesis, DFT calculations (Figs. 5a–f, Supplementary Fig. 66, Data 1 and Table 11) and in-situ spectroscopic analyses were conducted (Fig. 5g–j). As shown in Fig. 5b, two major uphill steps dominate the urea formation pathway, i.e., the initial *NO2 and CO2 coupling to form *CO2NO2 (*NO2 + CO2 → *CO2NO2), identified as the likely rate-determining step (RDS), and the subsequent protonation of *CO2NH2 to *COOHNH2. Notably, the second coupling step occurred spontaneously. Se incorporation significantly lowered the energy barrier for *CO2NO2 formation from 0.98 eV (InOx) to 0.55 eV (Se−InOx), and for *CO2NH2 protonation from 0.72 to 0.45 eV, suggesting a more favorable energy landscape. All catalysts also showed a thermodynamic preference for *NO2 + CO2 → *CO2NO2 over *OCHO + H → *OCHOH and *NO2 + H → *NOOH (Fig. 5d–f and Supplementary Figs. 67–69), with Se−InOx showing the largest energy gap (0.87 and 2.78 eV), compared to S−InOx (0.20 and 2.59 eV) and InOx (0.04 and 1.37 eV), which may help reduce HCOOH and NH3 by-products. CI-NEB calculations suggest that CO2 diffusion and subsequent coupling with anchored *NO2 proceeds with reduced barriers of 0.87, 0.66, and 0.48 eV for InOx, S−InOx, and Se−InOx, respectively (Supplementary Fig. 70). The resulting *CO2NO2 intermediates are predicted to exhibit progressively shorter C−N bond lengths (1.81 to 1.61 Å) and higher ICOHP values (2.08 to 3.50; Supplementary Figs. 71a and 72), implying stronger C−N covalent bonding upon Se doping. COHP analysis further reveals an upward shift of the antibonding orbital, which may enhance bond stability (Supplementary Fig. 71b). The negligible role of *NH2- and *NH2OH-based coupling with CO2 highlights the efficiency and specificity of the *NO2 + CO2 pathway (Supplementary Fig. 73). Additionally, Se−InOx showed the lowest H2O dissociation barrier, which could facilitate proton supply for subsequent hydrogenation of *CO2NO2 to urea (Supplementary Figs. 74–80).

Urea electrosynthesis reaction pathway diagrams on S−InOx (a) and Se−InOx (c). b Calculated free energy diagram for urea electrosynthesis. Comparison of energy barriers from *NO2 to *NOOH and *CO2NO2 on InOx (d), S−InOx (e), and Se−InOx (f). Electrochemical in-situ ATR-SEIRAS spectra on InOx (g), S−InOx (h), and Se−InOx (i) in CO2-saturated 0.1 M KHCO3 + 0.1 M KNO3 electrolyte. j Electrochemical in-situ EPR spectra on Se−InOx in CO2-saturated 0.1 M KHCO3 + 0.1 M KNO3 electrolyte (Sim. = simulated). k Schematic representation of hierarchical click-site mechanism: Se-induced tuning of In electronic structure enables selective NO3− adsorption and stabilization of *NO2 (1st click-site), followed by spatially guided CO2 adsorption to form *CO2NO2 (2nd click-site), ultimately driving selective C − N coupling toward urea. Colour codes: C grey, O red, N blue, H white, In orchid, S yellow, Se light green. Source data are provided as a Source data file.
In-situ ATR-SEIRAS further confirmed these insights (Fig. 5g–i). On InOx surface (Fig. 5g), strong *HCOO signals at ~1650 cm−1 was increased with negative potential, consistent with dominant CO2RR42. Concurrently, a *NO2 signal at ~1200 cm−1 diminished with increasing potential, suggesting poor *NO2 stability on InOx43. The absence of *COOHNH2 and *C−N signals implies that *NO2 conversion led primarily to NH3, as evidenced by the increasing *NH2 signal18. Therefore, CO2RR and NO3RR are independent and dominant on the InOx surface. For S−InOx, *HCOO signals are suppressed, while *NHx/*NH2 and NH2OH bands are enhanced, indicating improved NO3− adsorption and conversion to NH3 compared to InOx (Fig. 5h)17. Notably, the *NO2 signal decreased at −0.3 V (vs. RHE) and gradually transformed into *CO2NO2, accompanied by minor *COOHNH2 and *C−N signals, suggesting improved urea electrosynthesis. Se−InOx showed no *HCOO signal, and negligible *NHx, *NH2, and NH2OH signals, indicating complete suppression of CO2RR and significant inhibition of NO3RR. Instead, a clear transformation of *NO2 to *CO2NO2 at −0.2 V (vs. RHE) was observed (Fig. 5i), with rapid growth of the *CO2NO2 signal and accompanying *C−N and *COOHNH2 signals, indicating a preference for C−N coupling reactions on Se−InOx. 11,44 These findings were further supported by a series of control experiments, demonstrating sequential-chain coupling pathways at hierarchical click-sites (Supplementary Fig. 81).
In-situ EPR spectra further provided additional support for the sequential-chain coupling pathway on Se−InOx (Fig. 5j). The signal intensity of *CO2NO2 increased with applied potential, in agreement with the ATR-SEIRAS findings, confirming the role of *NO2 and CO2 in the C−N coupling mechanism. Based on these results, we propose a hierarchical click-site catalysis mechanism via a sequential-chain coupling pathway (Fig. 5k), where Se-induced electronic modulation of In sites selectively activates of NO3− to form *NO2 without CO2 adsorption, constituting the first click step. The adsorbed *NO2, along with the catalyst, facilitates selective coupling with CO2 as the second click step. The resulting *CO2NO2 undergoes hydrogenation to urea with minimal side-reactions, aided by efficient H2O dissociation, providing protons. To probe the catalyst behavior under operating conditions, we performed in-situ electrochemical XAS at the In K-edge on the InOx, S−InOx, and Se−InOx catalysts. As shown in Supplementary Figs. 82–87, shifting the applied potential from open-circuit potential (OCP) to −0.6 V (vs. RHE) resulted in only minor edge shifts across all samples, indicating that the In centers retain relatively stable electronic structures during operation. These subtle changes reflect moderate electron transfer between In sites and the adsorbed intermediates involved in C−N coupling, CO2RR, and NO3RR, without inducing significant redox transitions. Additionally, FT-EXAFS spectra show that the In−S and In−Se coordination environments in S−InOx and Se−InOx remain essentially unchanged under applied potentials (Supplementary Tables 12–14), demonstrating the structural robustness of these heteroatom-doped environments during electrocatalytic operation.
Techno-economic evaluation and scale-up of urea electrosynthesis
To enhance the economic viability of urea electrosynthesis, we increased the electrolyte concentration to CO2-saturated 1 M KHCO3 + 0.1 M KNO3, leveraging the known benefits of high K+ concentration in accelerating C−N coupling via improved proton/electron transfer11,45. This high K⁺ concentration significantly enhanced the urea yield rate and Jurea, achieving peak values of 254.94 mmol h−1 g−1 and 21.87 mA cm−2 (Fig. 6a and Supplementary Fig. 88), which were 2.7 times those of low K+ concentration, respectively. A techno-economic analysis (TEA, Fig. 6b and Supplementary Table 15) showed that the minimum cost of urea production using Se−InOx is US$1.01 kg−1 at an electricity price of US$0.03 kWh−1, close to market levels but highlighting limited competitiveness at higher electricity costs. However, the production cost could be reduced to as low as US$0.16 kg−1, if electricity cost is reduced to US$0.01 kWh−1, enabled by sustainable energy sources and by-products are valorized46. Subsequently, a financial net present value (FNPV) analysis was conducted, amortizing capital and operational costs over a 20-year lifespan. Profitability can only be achieved at electricity prices of US$0.01 kWh−1 and current densities ≥50 mA cm−2 (Fig. 6c and Supplementary Fig. 89), with payback periods decreasing substantially from 19-years at 50 mA cm−2 (FNPV = US$82,923) to just 6-years at 100 mA cm−2 (FNPV = US$7,141,881). Despite the gradual decline in FE at higher current densities, the economic value of by-products ensured cost recovery and profitability.

a Urea yield rate and FE of Se−InOx under different current densities in CO2-saturated 1 M KHCO3 + 0.1 M KNO3 electrolyte. b The production cost per kg of urea at electricity prices of US$0.03 and US$0.01 kWh−1. c FNPV of Se-InOx as a function of current density at an electricity price of US$0.01 kWh−1. d Sensitivity analysis of single-variable impact on urea production cost; Bars show the cost change when each parameter is varied by ±25% around the baseline value (in parentheses). Vertical dashed line: baseline cost ($1.01 kg−1urea). e Cost-reduction roadmap based on continuous tuning of key economic parameters. f Long-term electrolysis performance over 20 h in a 5*5 cm2 flow-type electrolyzer using CO2-saturated 1 M KHCO3 + 0.1 M KNO3 electrolyte; the inset of (f) shows the isolated urea product and the corresponding 1H NMR and 13C NMR. Electrolyte pH: 7.8 ± 0.4. Source data are provided as a Source data file.
To provide guidelines for future development, univariate sensitivity analysis (Fig. 6d) identified key cost drivers as cell voltage, electricity price, FE, equipment, and labor cost, while water and maintenance costs had negligible impact. Specifically, reducing electrolyzer voltage to 1.5 V (e.g., via alternative anodic small molecule oxidation reactions) could lower costs by US$0.30 kg−1. Decreasing electricity costs (expansion of renewable energy) to US$0.01 kWh−1 and increasing FE to 85% (catalyst optimization based on the hierarchical click-site strategy) could further lower the cost by US$0.28 and US$0.16 kg−1 (Supplementary Figs. 90–93), respectively. Collectively, these improvements could decrease urea production costs from US$1.01 kg−1 to US$0.24 kg−1 even without considering by-product revenues. Finally, the scalability of Se−InOx was validated in 20 h continuous operation within a 5 × 5 cm2 flow electrolyzer under CO2-saturated 1 M KHCO3 + 0.1 M KNO3 electrolyte, maintaining stable current density (Fig. 6f and Supplementary Fig. 94). The solid product, isolated by concentration, benzene extraction, and subsequent removal of benzene, yielded 1.102 g of pure urea solid, which was confirmed by 1H and 13C NMR characterization (inset of Fig. 6f)21. Based on the mass of the isolated urea, the average urea yield rate and FE over 20 h were calculated as 183.70 mmol h−1 g−1 and 35.28%, respectively. Compared to the small-scale setup, the slightly reduced performance is attributed to increased ohmic resistance, mass transport limitations, and unavoidable losses during purification, challenges typical in scaled-up systems. Nonetheless, the Se–InOx demonstrated robust activity and selectivity under scaled conditions, affirming its promise for practical applications. The life cycle assessment (LCA) shows that when powered by the average U.S. grid (0.12 kgCO2 MJ−1), the urea electrosynthesis route generates higher greenhouse gas (GHG) emissions (15.76 kgCO2 kg−1urea) than the conventional Bosch−Meiser process (1.90 kgCO2 kg−1urea), mainly due to the high electricity demand of the electrochemical cell (Supplementary Fig. 95 and Tables 16 and 17). Under low-carbon renewable electricity (0.003 kgCO2 MJ−1), the GHG intensity drops significantly to 0.63 kgCO2 kg−1urea urea, which is lower than that of the conventional route under comparable renewable scenarios (1.81 kgCO2 kg−1urea), highlighting the potential for emission reductions within a gate-to-gate boundary. In line with the conservative LCA framework adopted in this study, the results reflect process-level emissions at the plant gate and do not imply permanent CO2 sequestration or net carbon removal. Nevertheless, the use of renewable electricity markedly enhances the environmental performance of the electrochemical pathway, supporting its promise for sustainable chemical manufacturing.
Discussion
In summary, we have developed a hierarchical click-site catalysis strategy via a sequential-chain coupling pathway to overcome the long-standing challenge of low selectivity in urea electrosynthesis from NO3− and CO2. Mechanism studies demonstrate that Se−InOx, constructed by uniformly dispersed Se on CO2-inert InOx surface, serves as the first click-site to selectively adsorb NO3− and stabilize it as *NO2. This adsorbed *NO2 together with the catalyst, then acts as the second click-site to sequential-chain coupling CO2, forming the key *CO2NO2 intermediate, which undergoes rapid hydrogenation to urea. This hierarchical click-site design effectively resolves the spatiotemporal mismatch between N- and C- intermediates, achieving precise C − N coupling while effectively preventing cross-contamination and undesirable by-products. As a result, Se−InOx achieves a urea yield rate of 254.95 mmol h−1 g−1, a FE of 78.61%, >85% Nurea−selectivity, and 100% Curea−selectivity, outperforming most previously reported materials. TEA and urea powder production, together with gate-to-gate LCA results under renewable electricity scenarios, suggest the feasibility of scalable, cost-effective, and lower-emission deployment. This work provides a generalizable framework for achieving high selectivity in complex electrochemical transformations and presents a promising route toward sustainable nitrogen and carbon valorization.
Methods
Chemicals
Indium(III) chloride tetrahydrate (InCl3·4H2O, 97%), cadmium acetate dihydrate (Cd(Ac)2·2H2O, ≥98.0%), 2,5-thiophenedicarboxylic acid (C6H4O4S, 2, 5-TDC, 99%), selenium powder (Se, ~100 mesh, ≥99.5%), thioacetamide (C2H5NS, ≥99.0%), potassium nitrate (KNO3, ≥99.0%), isopropanol (C3H8O, 99.5%), diacetylmonoxime (C4H7NO2, ≥99.0%), phosphoric acid (H3PO4, >85% in H2O), sulfuric acid (H2SO4, 95%–98%), iron(III) chloride (FeCl3, ≥99.99%), thiosemicarbazide (TSC, 99%), ethylenediaminetetraacetic acid disodium salt (EDTA, 99%), urease (~1 U/mg), potassium bicarbonate (KHCO3, ≥99.95% trace metals basis, 99.7–100.5% dry basis), ammonium chloride (NH4Cl, ≥99.5%), hydrochloric acid (HCl, 37%), para-(dimethylamino)benzaldehyde (C9H11NO, 99%), salicylic acid (C7H6O3, ≥99.0%), sodum citrate dihydrate (C6H5Na3O7, ≥99.0%), sodium hypochlorite (NaClO, available chlorine 4.00–4.99%), sodium hydroxide (NaOH, 97%, powder), sodium nitroferricyanide(III) dihydrate (Na2Fe(CN)5NO·2H2O, 99%), ethanol (CH3CH2OH) and Nafion 115 were purchased from Sigma-Aldrich (Singapore) and used without further purification unless otherwise noted. High-purity CO2 (≥99.999%) and Argon (Ar, ≥99.999%) gases were purchased from Air Liquide (Singapore). All aqueous solutions were prepared with high-purity de-ionized water (DI-water, resistance 18 MΩ cm-1).
Synthesis of InOx
To synthesize InOx, an In-based metal-organic framework (In-MOF) was constructed using 2, 5-TDC as the organic linker. In a representative procedure, 0.5 g of InCl3·4H2O and 0.5 g of Cd(Ac)2·2H2O were dissolved in 200 mL of ethanol and combined with 100 mL of an ethanolic solution containing 1 g of 2,5-TDC. The mixed solution was maintained at 60 °C for 12 h to allow for MOF crystallization. The solid product was collected by centrifugation, sequentially washed with ethanol and deionized water to remove residual reactants, and then dried under vacuum. The resulting In-MOF precursor was thermally treated at 800 °C for 4 h in a muffle furnace. Post-calcination, the material was washed with deionized water and lyophilized to obtain InOx.
Synthesis of Se-InOx
Selenium incorporation was achieved by positioning 0.3 mmol of Se powder upstream and 100 mg of InOx downstream within an alumina crucible. The system was sealed in a tubular furnace under argon and heated to 400 °C at a heating rate of 20 °C min−1 and kept warm for 2 h. This step was repeated three times to promote homogeneous Se dispersion across the surface. The obtained solid was purified via triple deionized water washing and drying. The final sample was labeled Se−InOx−2.92 (abbreviated as Se−InOx). Analogous procedures with adjusted selenium amounts (0.1 mmol and 1 mmol) yielded Se−InOx−1.16 and Se−InOx−6.77, respectively.
Synthesis of S-InOx
Similarly, S−InOx was synthesized by replacing Se powder with C2H5NS under the same experimental conditions.
Physical characterization
The morphology of the as-prepared samples was characterized using a FESEM (JEOL 7800 F), TEM. High-resolution imaging was conducted using a Titan G2 60-300 HRTEM equipped with spherical and chromatic aberration correctors, and a FEI Titan G3 20-80 for HAADF-STEM analysis. Elemental composition was determined by ICP-MS (Thermo XSERIES 2), EDS, and elemental mapping (150 ~ 230 Mx at 200 kV on STEM). Crystal structures were identified by XRD (Bruker D8 Advance, 9 kW, 40 kV, 40 mA) using Cu Kα radiation (λ = 1.5418 Å). XPS (Thermo ESCALAB 250XI, energy range: 0−1400 eV) was employed to evaluate surface chemical states. EPR spectral signals were collected using Bruker EMX plus-9./12 spectrometer operating at X-band (9.85 GHz). FTIR spectra were recorded using a Thermo Fisher Nicolet iS 50 spectrometers to detect organic functional groups. UV−vis absorption spectra were obtained with an Agilent Cary 60 spectrophotometer. Additionally, the 1H NMR spectra were obtained using a superconducting-magnet NMR spectrometer (Bruker AVANCE III HD, 600 MHz).
In-situ ATR-SEIRAS characterization
In-situ ATR-SEIRAS measurements were carried out on a Nicolet™ iS50 FT-IR spectrometer equipped with a specially designed reaction cell and a mercury cadmium telluride (MCT) detector cooled with liquid nitrogen. A 20 mm Au-coated Si hemispherical prism (MTI Corporation) was used both as the catalyst support and the internal reflection element. A Pt foil, an Ag/AgCl electrode (4 M KCl), and a CO2-saturated 0.1 M KNO3 + 0.1 M KHCO3 solution served as the counter electrode, reference electrode, and electrolyte, respectively. Catalyst ink was prepared following the electrochemical protocol and drop-cast on the prism at a loading of 1 mg cm−2. In-situ ATR-FTIR spectra were recorded while scanning the working electrode potential from OCP to −0.6 V (vs. RHE) at a scan rate of 5 mV s−1. During measurements, pure CO2 gas (99.999% purity) was continuously flowed into the reaction chamber, and spectral changes were monitored in the range of 4000 − 1000 cm−1.
In-situ EPR characterization47,48
In-situ EPR measurements were conducted at room temperature using a continuous-wave (CW) Bruker EMX plus-9/12 spectrometer operating at X-band (9.85 GHz). A custom-designed quartz flat cell (Supplementary Fig. 96), comprising four ports, was employed to establish a three-electrode electrochemical configuration. The cell featured a 1 mm-thick flat window, optimized to reduce signal attenuation due to excessive solution thickness. The working electrode consisted of a flexible indium tin oxide (ITO) substrate (0.125 mm, ~10 Ω) loaded with catalyst (1 mg cm−2), precisely cut to align with the central window region. A platinum wire served as the counter electrode, while an Ag/AgCl electrode was used as the reference. The electrolyte—comprising 0.1 M KNO3 and 0.1 M KHCO3 supplemented with 10 mM DMPO—was purged with CO2 for 20 min before use and continuously supplied during operation. EPR spectra were recorded using a 100 kHz modulation frequency and 10 dB microwave power, across a 200 G sweep range. Each scan lasted 60 s, and final spectra were averaged from two consecutive measurements for improved resolution. Spin-trapping experiments using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) were additionally performed on a Bruker EMX Micro EPR spectrometer under identical conditions. Spectral simulations were carried out using the EasySpin software (v6.0.0) integrated in MATLAB R2020a49.
The X-ray absorption fine structure (XAFS) and in-situ XAFS characterization
The X-ray absorption fine structure (XAFS) spectra of the samples were collected at BL11B beamline of Shanghai Synchrotron Radiation Facility (SSRF). The SSRF is a third-generation synchrotron radiation light source composed of a 3.5 GeV electron storage ring, a full energy booster, and a 150 MeV linear accelerator. The electron-beam current during testing is 200 mA. The BL11B beamline used a bending magnet as the photon source. The experiments were performed using the double-crystal monochromator (DCM) with a Si (311) crystal. The corresponding reference samples and the samples for ex-situ experiments were collected in transmission mode. The samples for in-situ experiments were collected in fluorescence mode using a Lytle detector.
In-situ XAFS measurement was conducted with a homemade H-type cell with Nafion 115 to separate the anode and the cathode. This cell contains three electrodes with a carbon foil coated by catalyst as the working electrode, Pt mesh as the counter electrode, and Ag/AgCl as the reference electrode, respectively. The catalyst was drop-cast on the carbon foil and allowed to dry. A small window was cut out on the cathode side and sealed with Kapton film to allow fluorescence signals to pass from the electrode to the detector. The electrolyte was obtained by bubbling CO2 into the 0.1 M KNO3 and 0.1 M KHCO3 aqueous solution for 30 min before testing. The XAFS spectra were collected under open-circuit potential and applied potentials. The acquired XAFS data were processed according to the standard procedures using the ATHENA module of Demeter software packages. To obtain the quantitative structural parameters around central atoms, least-squares curve parameter fitting was performed using the ARTEMIS module of Demeter software packages.
The following EXAFS equation was used:
Electrochemical characterizations
The catalyst ink was prepared by dispersing 5 mg of the as-synthesized material and 30 µL of 5 wt% Nafion solution into 1 mL of a mixed solvent (isopropanol: deionized water = 4:1, v/v), followed by ultrasonication for 30 min to ensure homogeneity. A 40 µL of the resulting ink was transferred onto a 1.0 × 1.0 cm2 GDL, achieving a mass loading of 0.2 mg cm−2. Urea electrosynthesis from NO3− and CO2 was conducted within a three-electrode flow-type electrolytic cell connected to a CHI 760E electrochemical workstation under ambient laboratory conditions (25 ± 1 °C). The synthesized catalyst on GDL served as the working electrode, a GDL as the counter electrode, and a saturated Ag/AgCl/KCl electrode as the reference. A Nafion 115 membrane (thickness ~127 μm, 2 × 2 cm2) was employed as the separator. Prior to use, the Nafion 115 membrane was sequentially treated in 5 wt% H2O2 at 80 °C for 3 h, rinsed with deionized water at 80 °C for 1 h, soaked in 0.5 M H₂SO₄ at 80 °C for 2 h and again at 100 °C for 8 h, followed by additional rinsing in deionized water at 80 °C and thorough washing to ensure cleanliness. The electrolyte, consisting of KNO3 and 0.1 M KHCO3, was freshly prepared on the day of each experiment and used on the same day. The gas (CO2 or Ar) and electrolyte (30 mL) were fed at rates of 80 mL and 40 min−1, respectively. Before the electrochemical test, the electrolyte was thoroughly purged with Ar or CO2 flow for a period of 30 min. During electrolysis, the catholyte and anolyte were directed into their respective chambers, separated by the Nafion membrane, and subsequently routed into individual reservoirs. CO2 was introduced into the cathode chamber through PTFE tubing, facilitating its interaction with the catalyst surface. The flow of the catholyte and anolyte was maintained using a peristaltic pump. Gas-phase products were analyzed using gas chromatography after flow stabilization. Chronoamperometric measurements were performed at various potentials for 1200 s, while maintaining a constant Ar or CO2 flow within the electrochemical system. All potentials were converted to the reversible hydrogen electrode (RHE) scale using the formula: ERHE = EAg/AgCl + 0.0592 × pH + 0.197. The Ag/AgCl (saturated KCl) reference electrode was calibrated in H2-saturated 0.5 M H2SO4 using a clean Pt wire as both working and counter electrodes. The open-circuit potential was recorded and taken as 0 V (vs. RHE) after stabilization. The resistance was measured by EIS at open circuit potential (100 kHz–0.1 Hz, 5 mV), giving an average value of 2.7 ± 0.8 Ω. Unless otherwise stated, all reported voltages are without iR compensation. The Tafel slopes were determined using the Tafel equation: η = b log Jurea + a, where η is potential (V), Jurea is partial current density of urea production (mA cm−2), and b is the corresponding Tafel slope (mV dec−1). Catalyst durability was assessed over ten successive chronoamperometric cycles at −0.4 V (vs. RHE).
Large-scale electrolysis and urea isolation
Under identical conditions, electrolysis was carried out in 1 M KHCO3 + 0.1 M KNO3 electrolyte using a constant current method across a range of 2−100 mA cm−2. To obtain high-purity urea, a scaled-up electrolysis was performed in a 5 × 5 cm2 flow cell continuously for 20 h. The resulting electrolyte was concentrated and subjected to benzene extraction to isolate urea3. After solvent removal, the solid urea product was obtained and confirmed by both H and C NMR spectroscopy.
Determination of urea
Method 1-Diacetylmonoxime (DAMO) colorimetry17: Briefly, 1 mL of electrolyte was mixed with 2 mL of acid−ferric solution (comprising 100 mL concentrated phosphoric acid, 300 mL concentrated sulfuric acid, 600 mL deionized water, and 100 mg ferric chloride). Then, 1 mL of a DAMO-thiosemicarbazide (TSC) solution (5 g DAMO and 100 mg TSC in 1 L deionized water) was added. The mixture was heated to 100 °C for 20 min and cooled to room temperature. Absorbance was measured at 525 nm using a UV−vis spectrophotometer, and urea concentrations were calculated based on a standard calibration curve. Method 2-Urease enzymatic decomposition14: 0.5 mL of a urease solution (5 mg mL−1, contained 0.1 g ethylenediaminetetraacetic acid disodium salt (EDTA) and 0.49 g K2HPO4 per 100 mL) was added to 4.5 mL of electrolyte, followed by incubation at 40 °C for 40 min. NH3 concentrations were determined before and after decomposition using the indophenol blue method, and urea concentration was obtained by subtracting the pre-decomposition NH3 content from the post-reaction result.
Determination of NH3
The concentration of NH3 produced in the electrolyte was determined using the indophenol blue method39. After electrolysis, 2 mL of the electrolyte was sequentially mixed with: 2 mL of 1 M NaOH containing 5 wt% sodium citrate and 5 wt% salicylic acid as solution A, 1 mL of 0.05 M NaClO as solution B, and 0.2 mL of 1 wt% sodium nitroprusside (C5FeN6Na2O) as solution C. The solution was kept in the dark at ambient temperature for 2 h, followed by UV−vis absorption measurement in the range from 550 to 750 nm.
Determination of NO2 −
NO2− in the electrolyte was quantified using the Griess reagent method14. A 200 μL aliquot of Griess reagent was added to 5 mL of electrolyte, heated to 100 °C for 1 min, and cooled to room temperature. The absorbance at 540 nm was recorded, and nitrite concentration was obtained by referencing a standard calibration curve.
Determination of other liquid-phase products
Liquid-phase products were analyzed by 1H NMR spectroscopy using dimethyl sulfoxide (DMSO) as the internal standard. Dimethyl sulfoxide (DMSO) was used as an internal standard, allowing for accurate determination of the liquid product yield rate.
Determination of gas-phase products
Gas-phase products were analyzed using a gas chromatograph (GC) equipped with both a thermal conductivity detector (TCD) and a flame ionization detector (FID), with Ar used as the carrier gas.
Calculaions for yield rate and FE
The detailed yield rates have been calculated by using the following equation:
The detailed FEs have been calculated as follows:
where F is the Faradic constant (96485.3 C mol−1), c is concentration, V is the volume of electrolyte, t is the reaction time and A is the catalyst area and Q is the total charge passed through the working electrode.
Calculaions for selectivity
The C-selectivity for CO2-to-urea and CO2-to-HCOOH conversions was calculated as follows:
where n(C-product)C is the number of moles of C in C-product, and n(total)C is the total number of moles of C atoms in the products from CO2RR.
Similarly, the N−selectivity for NO3−-to-urea, NO3−-to-NO2- and NO3−-to-NH3 conversion was calculated using the following equation:
where n(N-product)N is the number of moles of nitrogen in as-produced urea, and n(total)N is the total number of moles of N atoms in the products from NO3RR
DFT calculation details
First-principles calculations based on spin-polarized density functional theory (DFT) were performed using the Vienna Ab initio Simulation Package (VASP)50, with the exchange-correlation interactions were described by the Perdew-Burke-Ernzerhof (PBE) functional within the generalized gradient approximation (GGA)51. To model surface reactions, periodic slab models were constructed with a 15 Å vacuum layer along the non-periodic direction to eliminate spurious interactions between periodic layers. The plane-wave basis set was truncated at a cutoff energy of 450 eV, and Brillouin zone integration was sampled using a Monkhorst-Pack k-point grid of 2 × 2 × 1 for structural optimizations. Convergence thresholds were set to10−5 eV for total energy and 0.01 eV Å−1 for atomic forces, ensuring high precision in geometry relaxations. The adsorption energy was calculated according to the Gibbs free energy difference between the product and reactant, and Gibbs free energy correction, including entropy and the zero-point energy correction, was taken into consideration at 298.15 K. For the intermediate species of H+, its Gibbs free energy was calculated as half of H2. The averaged length of In-X (X=O, S, Se) and distortion index of polyhedron analyses were implemented to present the change of local structure. The distortion index (D) was calculated as the following equation:
where \({l}_{i}\) and \({l}_{{ave}}\) represent the distance from the central atom In to the ith coordinating atom (O, or S, or Se) and the average bond length of the polyhedron, respectively. Electron localization function52 and Bader charge methods were employed to obtain the distribution and transfer of electron. The crystal orbital Hamilton population (COHP)53 calculations were performed to reveal the chemical bonding and antibonding mechanism. For the dissociation of H2O, the climbing-image nudged elastic band (CI-NEB)54 method was used to obtain the dissociation barrier of H2O.
Techno-economic evaluation
A comprehensive TEA was performed to assess the feasibility of urea electrosynthesis via the co-reduction of CO2 and NO3− under industrially relevant conditions. The assessment included energy consumption estimation based on Faraday’s law, process-scale current and voltage inputs, and material balances for all relevant species (urea, NH3, HCOOH, CO2, H2O, and O2). Capital costs were calculated based on DOE H2A guidelines, including stack and balance-of-plant components, with depreciation modeled over a 20-year plant lifetime. Operating costs accounted for electricity, water usage, membrane separation, and labor. Sensitivity analyses were conducted to evaluate the influence of current density, FEurea, cell voltage, and electricity price on financial net present value (FNPV) and urea production cost. Detailed parameters, assumptions, and numerical results are provided in the Supplementary Information.
Data availability
All data supporting the findings of this study are available within the article and its Supplementary Information. Additional datasets are available from the corresponding authors upon request. Source data are provided with this paper.
References
Zhang, X. et al. Managing nitrogen for sustainable development. Nature 528, 51–59 (2015).
Wang, J. et al. Synchronous recognition of amines in oxidative carbonylation toward unsymmetrical ureas. Science 386, 776–782 (2024).
Huang, D.-S. et al. Electrosynthesis of urea by using Fe2O3 nanoparticles encapsulated in a conductive metal–organic framework. Nat. Synth. 3, 1404–1413 (2024).
Zhu, X., Zhou, X., Jing, Y. & Li, Y. Electrochemical synthesis of urea on MBenes. Nat. Commun. 12, 4080 (2021).
McKie, D. Wöhler’s ‘synthetic’ urea and the rejection of vitalism: a chemical legend. Nature 153, 608–610 (1944).
Li, J., Zhang, Y., Kuruvinashetti, K. & Kornienko, N. Construction of C-N bonds from small-molecule precursors through heterogeneous electrocatalysis. Nat. Rev. Chem. 6, 303–319 (2022).
Mao, C., Byun, J., MacLeod, H. W., Maravelias, C. T. & Ozin, G. A. Green urea production for sustainable agriculture. Joule 8, 1224–1238 (2024).
Chen, C. et al. Coupling N2 and CO2 in H2O to synthesize urea under ambient conditions. Nat. Chem. 12, 717–724 (2020).
Luo, Y. et al. Selective electrochemical synthesis of urea from nitrate and CO2 via relay catalysis on hybrid catalysts. Nat. Catal. 6, 939–948 (2023).
Wu, Q., Dai, C., Meng, F., Jiao, Y. & Xu, Z. J. Potential and electric double-layer effect in electrocatalytic urea synthesis. Nat. Commun. 15, 1095 (2024).
Wang, Y. et al. Electron deficiency is more important than conductivity in C-N coupling for electrocatalytic urea synthesis. Angew. Chem. Int. Ed. 63, e202410938 (2024).
Wei, X. et al. Oxygen vacancy-mediated selective C-N coupling toward electrocatalytic urea synthesis. J. Am. Chem. Soc. 144, 11530–11535 (2022).
Li, Y. et al. Sequential co-reduction of nitrate and carbon dioxide enables selective urea electrosynthesis. Nat. Commun. 15, 176 (2024).
Xu, M. et al. Kinetically matched C-N coupling toward efficient urea electrosynthesis enabled on copper single-atom alloy. Nat. Commun. 14, 6994 (2023).
Zhang, X. et al. Identifying and tailoring C-N coupling site for efficient urea synthesis over diatomic Fe-Ni catalyst. Nat. Commun. 13, 5337 (2022).
Zhao, Y. et al. Efficient urea electrosynthesis from carbon dioxide and nitrate via alternating Cu-W bimetallic C-N coupling sites. Nat. Commun. 14, 4491 (2023).
Hu, Q. et al. Pulsed co-electrolysis of carbon dioxide and nitrate for sustainable urea synthesis. Nat. Sustain. 7, 442–451 (2024).
Lv, C. et al. Selective electrocatalytic synthesis of urea with nitrate and carbon dioxide. Nat. Sustain. 4, 868–876 (2021).
Gong, C. et al. Selective electrocatalytic synthesis of urea using entangled iron porphyrins in covalent organic frameworks. Nat. Synth. 4, 720–729 (2025).
Li, H. et al. Ligand engineering towards electrocatalytic urea synthesis on a molecular catalyst. Nat. Commun. 15, 8858 (2024).
Xiong, H. et al. Urea synthesis via electrocatalytic oxidative coupling of CO with NH3 on Pt. Nat. Catal. 7, 785–795 (2024).
Song, X. et al. Urea synthesis via coelectrolysis of CO2 and nitrate over heterostructured Cu-Bi catalysts. J. Am. Chem. Soc. 146, 25813–25823 (2024).
Zhao, C. et al. Tailoring activation intermediates of CO2 initiates C-N coupling for highly selective urea electrosynthesis. J. Am. Chem. Soc. 147, 8871–8880 (2025).
Chen, X. et al. Amorphous bismuth-tin oxide nanosheets with optimized C-N coupling for efficient urea synthesis. J. Am. Chem. Soc. 146, 13527–13535 (2024).
Zhan, P. et al. Efficient electrosynthesis of urea over single-atom alloy with electronic metal support interaction. Angew. Chem. Int. Ed. 63, e202409019 (2024).
Ye, W. et al. A strongly coupled metal/hydroxide heterostructure cascades carbon dioxide and nitrate reduction reactions toward efficient urea electrosynthesis. Angew. Chem. Int. Ed. 63, e202410105 (2024).
Xia, B. Y. et al. Remote carbon monoxide spillover improves tandem urea electrosynthesis. Angew. Chem. Int. Ed. 64, e202421266 (2025).
Wei, X. et al. Lattice oxygen-driven co-adsorption of carbon dioxide and nitrate on copper: a pathway to efficient urea electrosynthesis. J. Am. Chem. Soc. 147, 6049–6057 (2025).
Meng, G. et al. Modular click chemistry libraries for functional screens using a diazotizing reagent. Nature 574, 86–89 (2019).
Agrahari, A. K. et al. Cu(I)-catalyzed click chemistry in glycoscience and their diverse applications. Chem. Rev. 121, 7638–7956 (2021).
Bauer, D., Sarrett, S. M., Lewis, J. S. & Zeglis, B. M. Click chemistry: a transformative technology in nuclear medicine. Nat. Protoc. 18, 1659–1668 (2023).
Tan, X. et al. Recent advances in innovative strategies for the CO2 electroreduction reaction. Energy Environ. Sci. 14, 765–780 (2021).
Chang, B. et al. Electrochemical reduction of carbon dioxide to multicarbon (C2+) products: challenges and perspectives. Energy Environ. Sci. 16, 4714–4758 (2023).
Yang, S. et al. Halide-guided active site exposure in bismuth electrocatalysts for selective CO2 conversion into formic acid. Nat. Catal. 6, 796–806 (2023).
Albani, D. et al. Semihydrogenation of acetylene on indium oxide: proposed single-ensemble catalysis. Angew. Chem. Int. Ed. 56, 10755–10760 (2017).
Shang, H. et al. Design of a single-atom indium+-N4 interface for efficient electroreduction of CO2 to formate. Angew. Chem. Int. Ed. 59, 22465–22469 (2020).
Qiu, C. et al. MOF-transformed In2O3-x@C nanocorn electrocatalyst for efficient CO2 reduction to HCOOH. Nano-Micro Lett 14, 167 (2022).
Zhang, X., Guo, S.-X., Gandionco, K. A., Bond, A. M. & Zhang, J. Electrocatalytic carbon dioxide reduction: from fundamental principles to catalyst design. Mater. Today Adv. 7, 100074 (2020).
Sun, Y. et al. Biomimetic design of a dynamic M-O-V pyramid electron bridge for enhanced nitrogen electroreduction. J. Am. Chem. Soc. 146, 7752–7762 (2024).
Sun, Y. et al. Biomimetic elastic single-atom protrusions enhance ammonia electrosynthesis. Angew. Chem. Int. Ed. 64, e202418095 (2024).
Sun, Y. et al. Main-group metal-nonmetal dynamic proton bridges enhance ammonia electrosynthesis. Angew. Chem. Int. Ed. 63, e202412426 (2024).
Sun, Y. et al. Direct parallel electrosynthesis of high-value chemicals from atmospheric components on symmetry-breaking indium sites. Proc. Natl. Acad. Sci. USA 121, e2409620121 (2024).
Wang, Y. et al. Dynamic reconstruction of two-dimensional defective Bi nanosheets for efficient electrocatalytic urea synthesis. Angew. Chem. Int. Ed. 63, e202318589 (2024).
Dai, Z. et al. Surface engineering on bulk Cu2O for efficient electrosynthesis of urea. Nat. Commun. 16, 3271 (2025).
Tu, X. et al. A universal approach for sustainable urea synthesis via intermediate assembly at the electrode/electrolyte interface. Angew. Chem. Int. Ed. 63, e202317087 (2024).
Shin, H., Hansen, K. U. & Jiao, F. Techno-economic assessment of low-temperature carbon dioxide electrolysis. Nat. Sustain. 4, 911–919 (2021).
Dong, K. et al. H2O2-mediated electrosynthesis of nitrate from air. Nat. Synth. 3, 763–773 (2024).
Seif-Eddine, M. et al. Operando film-electrochemical EPR spectroscopy tracks radical intermediates in surface-immobilized catalysts. Nat. Chem. 16, 1015–1023 (2024).
Stoll, S. & Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55 (2006).
Kresse, G. & Furthmiiller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Silvi, B. & Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 371, 683–686 (1994).
Dronskowski, R. & Blöchl, P. E. Crystal orbital hamilton populations (COHP). energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 97, 9617–8624 (1993).
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Arias, T. A. & Hennig, R. G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 140, 084106 (2014).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant 52125202, U24A2065 to J.Z). Q.Wu and Z.Xu thank the financial support from the National Research Foundation, Singapore, and A*STAR (Agency for Science, Technology and Research) under its LCER Phase 2 Programme Hydrogen & Emerging Technologies FI, Directed Hydrogen Programme (Award No. U2305D4003 to Z.J.X.). We thank the Shanghai Synchrotron Radiation Facility of BL11B (31124.02.SSRF.BL11B) for the assistance on XAFS measurements.
Author information
Authors and Affiliations
Contributions
Z.J.X, J.Z., and Y.S. conceived and designed the research; Y.S. and Y.H. synthesized the samples and performed the characterizations; Y.S., M.Z., and W.Z. carried out the electrochemical measurements; Y.S. and X.Z. performed and analyzed the in-situ results; Y.S. carried out the XAFS measurements and data analyais; M.T. and Y.S. performed and analyzed the DFT calculations. Y.S. wrote the manuscript. Z.J.X and Q.W. revised the manuscript. Y.S., J.Z., and Z.J.X discussed, and all authors co-wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mark Mba Wright and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, Y., Tian, M., Wu, Q. et al. Sequential-chain coupling over hierarchical click-sites enables highly selective urea electrosynthesis. Nat Commun 17, 2388 (2026). https://doi.org/10.1038/s41467-026-69207-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69207-4
