
Abstract
Most colorectal cancers (CRCs) are insensitive to cancer immunotherapy due to the tumor-promoting activities and immunosuppressive effects of cancer-associated fibroblasts (CAFs), which profoundly drive cancer progression. Here, we provide a treatment strategy targeting CAFs to remodel the structural and immune tumor microenvironment of CRC based on magnetic nanoparticles. These nanoparticles not only induce the enhanced ferroptosis of CAFs through the synergistic effect of iron and copper ions but also ameliorate the immunosuppressive tumor microenvironment, simultaneously breaking down physical obstructions and activating the immune system. Mechanistically, these nanoparticles regulate the intrinsic signaling pathways of CAFs and alter chemokine secretion to improve the maturation of dendritic cells and the activation and function of CD8+ T cells, eventually resulting in boosting the immune response to inhibit CRC growth. Moreover, we verify the effectiveness of this strategy in the patient-derived xenograft and organoid models, further illustrating its universality and promising clinical translational potential. Collectively, we show that our magnetic nanoparticles targeting CAFs can reprogram the tumor microenvironment of CRC by breaking down physical barriers and alleviating immunosuppression, offering promising avenues for the therapeutic strategy in clinical CRC management.
Similar content being viewed by others

Introduction
During the process of colorectal cancer (CRC) initiation, progression, and metastasis, cancer-associated fibroblasts (CAFs), as the most abundant stromal cells in the tumor microenvironment (TME), play a crucial role in providing a scaffold and nutrients for cancer cell growth1,2,3. Clinically, 85% of CRC identified as microsatellite stable CRC is resistant to cancer immunotherapy largely because of CAFs-mediated structural obstacles4 and immunosuppression5. On the one hand, CAFs typically induce immune-related multidrug resistance of CRC due to their endocytosis and permeation obstruction towards drugs4. Meanwhile, CAFs can assist tumor cells in evading immune surveillance by remodeling the extracellular matrix (ECM), secreting cytokines such as transforming growth factor (TGF)-β via an autocrine or paracrine manner6. Moreover, the ECM remodeling capacity of CAFs mediates T cell trapping through increased matrix rigidity and enhanced resistance to T cell movement7,8. On the other hand, CAFs induce immunosuppression through their biological functions5, including suppressing dendritic cell (DC) differentiation and inducing T cell exhaustion6,8,9,10. Hence, targeting CAFs is a very reasonable and promising therapeutic strategy in the treatment of CRC.
At present, the conventional strategies targeting CAFs in CRC mainly include drugs or antibodies directly targeting CAFs11, inhibitors targeting CAFs-secreted growth factors, cytokines, or exosomes12, and reprogramming CAFs to normal fibroblasts (NFs)13. However, the ineluctable bottlenecks of the aforementioned strategies still exist in the translational applications, thanks to inefficiency caused by CAFs-induced heterogeneity and cumulative side effects14,15,16,17. Hence, an advanced treatment needs to be urgently developed to break through these obstacles.
Nanotherapy, an emerging treatment, has been extensively applied in cancer theranostics. However, most nanomaterials directly targeting cancer cells often have poor therapeutic efficacy, especially for advanced cancers, largely due to CAFs. By secreting high levels of ECM and increasing solid pressure and interstitial fluid pressure, CAFs limit the extravasation and diffusion of nanomaterials in tumor stroma18,19,20. Meanwhile, due to the preferential localization near blood vessels21, the phagocytosis of nanomaterials by CAFs is much greater than that by cancer cells, resulting in only a small amount of nanomaterials accumulated in cancer cells22,23. Therefore, nanomaterials designed to target CAFs can effectively overcome the above challenges. In the diverse range of nanomaterials, magnetic nanoparticles (NPs) represented by Fe3O4 NPs with excellent biocompatibility can act as photothermal therapy (PTT) and chemodynamic therapy (CDT) agents due to their good photothermal conversion properties and peroxidase-like activity24,25,26,27,28, which can be further optimized by the introduction of copper ions29.
In this work, we prepare Cu@Fe3O4 magnetic NPs consisting of human trace elements with the core-shell nanostructure to target CAFs of CRC for remodeling the structural and immune TME. Based on the intrinsic physicochemical properties of NPs, we provide a nanotherapy strategy targeting CAFs to significantly inhibit CRC growth by not only inducing ferroptosis of CAFs, thereby reducing pro-tumorigenic paracrine signaling, but also remodeling the TME, and alleviating CAF-mediated immunosuppression. Specifically, we find that NPs induce enhanced ferroptosis of CAFs through reactive oxygen species (ROS) generation caused by iron ions and glutathione peroxidase 4 (GPX4) downregulation caused by copper ions. Meanwhile, NPs modulate the secretion of chemokines, such as C-C motif chemokine ligand (CCL) 3 and C-X-C motif chemokine ligand (CXCL) 12, promoting DCs maturation and T cell activation, thereby enhancing T cell function and ultimately generating an immune-activated TME (Fig. 1a). Moreover, pronounced antitumor effects are observed in two kinds of subcutaneous tumor mouse models, a genetically engineered mouse model (GEMM), a patient-derived xenograft (PDX) model, and a patient-derived organoid (PDO) model, which more closely simulate the clinical tumor environment, further demonstrating the broad applicability and translational potential of our strategy as a next-generation CRC therapy.

a Schematic illustration of the antitumor process of NPs through inducing the ferroptosis of CAFs and remodeling the CAFs-related immune TME. b Individuals in the GSE17538 cohort are stratified into low and high CAF groups based on MCP-counter derived CAF abundance by the optimal cutoff value. Box plots depict the median (centre line), interquartile range (25th-75th percentiles; box), and 1.5 × interquartile range (whiskers). For the low group: min = 7.682, Q1 = 8.675, median = 8.873, Q3 = 9.165, max = 9.414 (n = 134 patients); for the high group: min = 9.424, Q1 = 9.547, median = 9.673, Q3 = 9.826, max = 10.854 (n = 98 patients). Statistical significance is assessed using a two-sided Mann-Whitney U test. c Kaplan-Meier curves of 232 colon cancer patients from the GSE17538 cohort stratified by CAF abundance assessed using the MCP-counter method. Tick marks indicate censored events. P values are calculated using a two-sided log-rank test, and hazard ratios (HRs) with 95% confidence intervals are estimated using univariate Cox proportional hazards regression. Source data are provided as a Source Data file.
Results
NPs induced synergistically-enhanced ferroptosis of CAFs
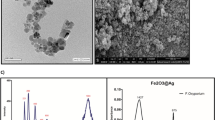
In our work, to evaluate the impact of CAFs on CRC patient survival, tumor samples from GSE17538 and GSE87211 were grouped by CAF scores using microenvironment cell populations-counter (MCPcounter), estimating the proportion of immune and cancer cells (EPIC), and estimation of stromal and immune cells in malignant tumor tissues using expression data (ESTIMATE)30,31,32,33,34. Higher CAF levels were consistently linked to poorer survival outcomes across all methods (Fig. 1b, c, and Supplementary Fig. 1). In addition, the impact of sex on CAF scores was evaluated in the GSE17538, GSE87211, and a single-cell RNA-sequencing (scRNA-seq) dataset E-MTAB-8017. The results consistently indicated that sex had no significant association with CAF abundance in the TME (Supplementary Fig. 2a–e). Based on these findings, sex was not included in the design of subsequent experiments. Firstly, in terms of NPs preparation, Cu@Fe3O4 NPs (simplified as NPs) were synthesized by thermal decomposition in the oil phase, consisting of a core of Cu NPs (~10 nm diameter) with a shell of Fe3O4 NPs (~15 nm thick) (Supplementary Fig. 3). Then, in terms of CRC patients-derived fibroblast lines preparation, NFs from three normal colonic tissues and CAFs from three CRC specimens were successfully isolated (Fig. 2a) and CAFs were identified by the higher expression of their characteristic markers35 (Fig. 2b). In vitro cytotoxicity assay revealed that CAFs were more sensitive to NPs compared to NFs (Fig. 2c) partly due to the higher NPs internalization by CAFs than that by NFs (Fig. 2d, and Supplementary Fig. 4a, b). Calcein-AM/PI double staining assay indicated that NPs combining 808 nm laser irradiation led to nearly complete cell death of CAFs, demonstrating the remarkable PTT effect of NPs (Fig. 2e), which was completely opposite in NFs (Supplementary Fig. 4c, d). Moreover, the most generation of ROS in CAFs was also observed in the NPs+Laser group (Supplementary Fig. 4e–h), and the better CDT effect of NPs combined with laser was also observed towards CAFs than NFs (Supplementary Fig. 4i–l). Similar results were also proved in the CRC cell line HCT116 and noncancerous colon cell line NCM460 (Supplementary Fig. 5).

a Images displaying the isolation of NFs from adjacent normal tissues and CAFs from CRC tumor tissues. b Western blot confirming successful isolation of NFs from three normal colonic tissues and CAFs from three CRC specimens, validated by antibodies against fibroblast activation protein-α (FAP), Vimentin, and α-smooth muscle actin (SMA); β-actin as a loading control. c Cytotoxicity assessment of NPs in NFs and CAFs for 24 h at varying concentrations. d Bio-TEM images of NFs and CAFs incubated with NPs for 24 h; Red dashed boxes marking NPs location. e Bright-field and fluorescent images of CAFs stained with Calcein-AM (live cells, green fluorescence) and PI (dead cells, red fluorescence) after treatment with phosphate buffer saline (PBS), Laser, NPs, and NPs+Laser. PBS as control (NC) (left); Statistical analysis of cell viability (right). f, g Fluorescent images of (f) intracellular Fe2+ levels and (g) LPO in CAFs treated without or with NPs for 24 h; Nuclei stained with Hoechst 33342 (blue). h Western blot of GPX4 expression in CAFs treated with increasing concentrations of NPs for 24 h; β-actin as a loading control. i Relative cell viability of CAFs treated with NPs alone, NPs with DFO, NPs with TM, and NPs with both DFO and TM for 24 h by CCK8 assay. j, k Fluorescent images of (j) intracellular Fe2+ levels and (k) LPO in CAFs treated as in (i); Nuclei stained with Hoechst 33342 (blue). l Western blot of GPX4 expression in CAFs treated as in (i); β-actin as a loading control. n = 3 independent experiments per group in (c–g) and (i–k). Western blot experiments in (b, h, l), FerroOrange and Liperfluo staining experiments in (f, g, j, k) are repeated independently three times with similar results. Data are presented as means ± standard deviation (SD). Statistical analyses are performed using one-way analysis of variance (ANOVA) with multiple comparisons (e). Source data are provided as a Source Data file.
Given the significantly higher cytotoxicity of Cu@Fe3O4 NPs compared to Fe3O4 NPs alone (Supplementary Fig. 6a), we focused on the roles of Fe²⁺ and Cu²⁺ in exploring the mechanisms underlying NPs-induced cell death. NPs treatment led to a marked increase in Fe²⁺ levels in CAFs (Fig. 2f, and Supplementary Fig. 6b, c), accompanied by significant lipid peroxidation (LPO) (Fig. 2g, Supplementary Fig. 6d), suggesting that NPs elevated intracellular Fe²⁺ within the labile iron pool, enhancing iron metabolism to induce ferroptosis in CAFs36. Additionally, NPs treatment resulted in a dose-dependent reduction of GPX4 expression (Fig. 2h), consistent with previous reports that Cu²⁺ suppressed GPX4 expression, thereby promoting ferroptosis37. The cytotoxic effects of NPs were significantly attenuated by the Fe²⁺ chelator deferoxamine (DFO) and the Cu²⁺ chelator tetraethylenepentamine (TM), with combined treatment yielding an additive effect (Fig. 2i). DFO notably reduced NPs-induced Fe²⁺ elevation, while TM did not affect Fe²⁺ levels (Fig. 2j, Supplementary Fig. 6e, f). Both DFO and TM inhibited NPs-induced LPO, and their combination further reduced LPO levels in CAFs (Fig. 2k, and Supplementary Fig. 6g). Fe²⁺ modulation had minimal impact on GPX4 expression, whereas Cu²⁺ chelation significantly restored GPX4 levels (Fig. 2l). These findings indicated that NPs, following lysosomal uptake, elevated intracellular Fe²⁺ and Cu²⁺ levels, simultaneously regulating iron metabolism and GPX4 pathway to induce synergistically-enhanced ferroptosis.
Moreover, similar experiments were designed in CRC cells. Cu@Fe₃O₄ NPs exhibited a stronger cytotoxic effect on HCT116 cells compared to Fe₃O₄ NPs (Supplementary Fig. 7a). An increase in intracellular Fe²⁺ levels, a significant accumulation of lipid peroxides, and a progressive decrease in GPX4 expression were observed as NPs concentration increased, suggesting that CRC cell death was induced via iron metabolism and the GPX4 pathway (Supplementary Fig. 7b–g). Moreover, co-chelation of Fe²⁺ and Cu²⁺ restored cell viability more effectively than the chelation of Fe²⁺ or Cu²⁺ alone (Supplementary Fig. 7h). Further analysis of Fe²⁺ content, LPO, and GPX4 expression upon individual or combined chelation of Fe²⁺ and Cu²⁺ revealed that Fe primarily mediated ferroptosis through iron metabolism, whereas Cu exerted its effect predominantly via the GPX4 pathway, collectively inducing ferroptosis in tumor cells (Supplementary Fig. 7i-n).
NPs regulated multiple signaling pathways of CAFs in vitro
To evaluate the impact of NPs targeting CAFs on CRC cells, we collected conditioned media (CM) from NFs and CAFs without/with NPs treatment and assessed the effects of these CM on the proliferative and migratory potential of HCT116 cells (Fig. 3a). Compared to NFs-derived CM, CAFs-derived CM significantly enhanced the proliferation, migration, and expression of epithelial-mesenchymal transition (EMT) related markers of HCT116 cells. However, this pro-tumorigenic effect was markedly suppressed after NPs treatment in CAFs (Fig. 3b–d).

a Schematic illustration of cell viability and transwell migration assays using HCT116 cells treated with supernatants from NFs, CAFs, or NPs-treated CAFs. b Transwell migration images presenting the number of migrated HCT116 cells treated with CM from NFs, CAFs, or NPs-treated CAFs for 24 h. c Cell viability of HCT116 cells treated as in (b) by the CCK8 assay. d Western blot of E-cadherin, N-cadherin, and Vimentin expression in HCT116 cells treated with CM from NFs, CAFs, or NPs-treated CAFs for 24 h; β-actin as a loading control. e, f Bubble plots of (e) GO and (f) KEGG pathway enrichment analyses for differentially expressed genes (DEGs) in NPs-treated CAFs compared to controls. g, h MSigDB Hallmark GSEA analysis showing enrichment of (g) chemokine activity and (h) ferroptosis pathways in NPs-treated CAFs compared to controls. ES: enrichment score; NES: normalized enrichment score. i Heatmap and histogram displaying gene expression levels of DEGs (log₂|FC | ≥ 1) from three chemokine-related GO pathways (GO:0016493, GO:0048020, GO:0008009) in NPs-treated CAFs compared to controls, ranked by differential expression. j QRT-PCR quantification of CCL3, CXCL12, and CCR7 expression in NPs-treated CAFs compared to controls. k, l Anti-human ELISA quantification of (k) CCL3 and (l) CXCL12 concentrations in supernatants from CAFs treated without or with NPs for 24 h. m Western blot of MAPK, p-MAPK (Thr180/Tyr182), NF-κB, p-NF-κB (Ser536), AKT, and p-AKT (Ser473) expression in CAFs treated without or with NPs for 24 h; β-actin as a loading control. n = 3 independent experiments in (b, c) and (j–l). Western blot experiments in (d, m) are repeated independently three times with similar results. Data are presented as means ± SD. Statistical analyses are performed using one-way ANOVA with multiple comparisons for (b, c); and two-tailed unpaired Student’s t-test for (j–l). Source data are provided as a Source Data file.
As the intrinsic molecular signaling pathways of CAFs can not only regulate their physiological proliferation and programmed cell death but also influence the interaction of CAFs with surrounding tumor cells, dysregulated pathways of CAFs treated with NPs were explored by RNA-sequencing (RNA-seq), with identification of 2380 significantly altered genes (1382 upregulated, 998 downregulated) (Supplementary Fig. 8a). Gene Ontology (GO) analysis of differentially expressed genes in NPs-treated CAFs revealed significant upregulation of chemokine receptor binding and chemokine activity, alongside reductions in cell growth, migration, and adhesion (Fig. 3e, Supplementary Fig. 8b). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis highlighted that ferroptosis and key signaling pathways such as tumor necrosis factor (TNF), nuclear factor ĸB (NF-ĸB), janus tyrosine kinase (JAK)-signal transducer and activator of transcription (STAT), and mitogen-activated protein kinase (MAPK) were upregulated, while TGF-β, wingless-related integration site (Wnt), and phosphatidylinositol 3-kinase (PI3K)-protein kinase B (Akt) pathways were downregulated (Fig. 3f, and Supplementary Fig. 8c). Pathways enriched by Gene Set Enrichment Analysis (GSEA) further showed high concordance with GO and KEGG results, emphasizing the critical roles of these pathways in reducing the pro-tumorigenic effects of CAFs upon NPs treatment (Fig. 3g, h, and Supplementary Fig. 8d–f). Given the well-established roles of TGF-β and Wnt pathways in CAFs17,38, we focused on three chemokine-related GO pathways. Pro-inflammatory chemokines (e.g., CCL2, CCL3, CCL7, CXCL8, CXCL10, and CXCL11) and C-C motif chemokine receptor (CCR) 7 were significantly upregulated, contributing to immune cell recruitment, antigen presentation, and antitumor responses38,39,40,41,42,43. Conversely, CXCL12, which supported CRC proliferation and immunosuppression6,42, was downregulated (Fig. 3i). According to RNA-seq findings, transcriptional alignment of CCL3, CXCL12, and CCR7 (Fig. 3j) and secreted protein levels of CCL3 and CXCL12 (Fig. 3k, l) were also further validated. Additionally, Western blot results substantiated the activation of MAPK and NF-κB pathways, along with attenuated AKT pathway, corroborating the transcriptional data (Fig. 3m).
The immunoregulatory effect of NPs towards CAFs in vivo
We established two kinds of CAFs-related cancer mouse models (CT26 and BALB/3T3 cells, MC38 and NIH3T3 cells) to explore the immunoregulatory effect of NPs towards CAFs (Fig. 4a). The growth rate of CRC cells co-inoculated with CAFs (as CT26 + BALB/3T3 group) was much faster than that of only CRC cells (as CT26 group), and NPs treatment presented satisfactory suppression effects on both the CT26 group and the CT26 + BALB/3T3 group. Considering the larger pre-treated tumor sizes of the CT26 + BALB/3T3 group compared to the CT26 group, the tumor inhibition efficiency of NPs in the CT26 + BALB/3T3 group was reasonably considered higher than that in the CT26 group (Fig. 4b–d). The consistent results were also observed in MC38-corresponding four groups (Supplementary Fig. 9a–c). Meanwhile, the good biosafety and biocompatibility of NPs in vivo were proved by metabolic performance assay29, coagulation and hemolysis assay (Supplementary Fig. 9d–e), and body weight curves (Fig. 4e, and Supplementary Fig. 9f). Furthermore, compared to the CT26 group, co-implantation with BALB/3T3 cells decreased CCL3 and increased CXCL12 expression in vivo and NPs treatment reversed these chemokine alterations through significantly elevating CCL3 and suppressing CXCL12, which resulted in transforming the immunosuppressive TME into a more active state (Fig. 4f). Hence, we subsequently made a comprehensive and systematic evaluation on the immunomodulatory effect of NPs. In the CT26 + BALB/3T3 group, the costimulatory molecules CD80 and CD86 in DCs around tumor tissues or in the spleen were slightly down-regulated compared to the CT26 group, indicating CAFs’ inhibitory effect on DC activation. However, NPs treatment up-regulated CD80 and CD86 in both the CT26 group and the CT26 + BALB/3T3 group, accelerating DC activation and leading to functional outcomes like DC maturation and enhanced antigen presentation (Fig. 4g, Supplementary Fig. 10). Although there was no significant change in the population of CD8+ T cells around tumor tissues or in the spleen in these four groups (Supplementary Fig. 11a–d), the activation and function of CD8+ T cells presented a surprising improvement after NPs administration, which were indicated by the up-regulated expression of T-bet/Eomesodermin (Eomes) (Fig. 4h, and Supplementary Fig. 11e–j) and interferon (IFN)-γ/TNF-α/CD107a (Fig. 4i, and Supplementary Fig. 12), respectively. All these results demonstrated the impairment of CD8+ T cell activation and function caused by CAFs, which were reversed visibly through the NPs treatment.

a Schematic illustration of the treatment strategy and related immune assessment in vivo based on NPs for the CT26 and the CT26 + BALB/3T3 group. b Individual tumor growth curves and (c) average tumor growth curves of the CT26 and the CT26 + BALB/3T3 group without or with the treatment of NPs. d Representative images of mice bearing CT26 cells and CT26 + BALB/3T3 cells without or with the treatment of NPs. e Body weight curves of CT26 cells- and CT26 + BALB/3T3 cells-bearing mice without or with the treatment of NPs. f Quantified expression of CCL3 and CXCL12 in homogenized tumor tissues from the CT26 and the CT26 + BALB/3T3 groups without or with the treatment of NPs. g Flow cytometry and corresponding statistical analysis of CD11c+MHC II+CD80+ and CD11c+MHC II+CD86+ cells (in total CD45+ cells) around tumor tissues of mice in four groups. h Statistical analysis of flow cytometry of CD3+CD8+Eomes+ and CD3+CD8+T-bet+ cells around tumor tissues of mice in four groups. i Statistical analysis of flow cytometry of CD3+CD8+CD107a+, CD3+CD8+IFN-γ+, and CD3+CD8+TNF-α+ cells around tumor tissues of mice in four groups. n = 5 mice per group in (b–e) and n = 3 independent experiments per group in (f–i). Data are presented as means ± SD. Statistical analyses are performed using two-way ANOVA with multiple comparisons for (c); and one-way ANOVA with multiple comparisons for (f–i). Source data are provided as a Source Data file.
The targeting specificity of AS1411 in different cells
To improve the efficiency, specificity, and biosafety of NPs targeting CAFs in vivo, we integrated scRNA-seq and subcellular protein localization datasets to identify optimal cell surface targets. Approximately twenty reported nano-drug surface markers were examined, and nine of them were significantly upregulated in CAFs according to scRNA-seq data (Supplementary Fig. 13a). Among fibroblast subclusters, nucleolin (NCL) showed the broadest distribution and highest expression, followed by CD44. Conversely, CD44 was predominantly expressed in phagocytic myeloid cells (Fig. 5a-c), suggesting potential enhancement of clearance and reduced tumor accumulation of NPs. Nucleolin, although mainly localized to the nucleolus and nucleus, is known to translocate to the cell surface and is markedly upregulated in CRC44,45, representing a more specific and suitable target. In the further subdivided NF and CAF subpopulations, NCL mRNA expression was significantly higher in CAFs than in NFs (Supplementary Fig. 13b–d), and western blot analysis showed that nucleolin protein levels were markedly elevated in the three CAF lines compared with the three NF lines (Supplementary Fig. 13e). To determine nucleolin localization, immunofluorescence (IF) assays of living cells (for membrane nucleolin) and fixed cells (for nuclear nucleolin) were performed. Nucleolin membrane expression was strongest in CRC CAFs and tumor cells but minimal in macrophages and nearly undetectable in NFs, epithelial cells, and endothelial cells (Fig. 5d). To further validate the specific targeting of AS1411 binding to nucleolin and off-target effects evaluation of NPs, we isolated different types of cells from the in situ TME of the subcutaneous tumor model and detected their uptake of NPs. As shown in Fig. 5e–k and Supplementary Fig. 14a–i, B cells, T cells, and endothelial cells hardly engulfed NPs and NPs-AS1411, while DCs and macrophages had minimal phagocytosis of NPs and NPs-AS1411. As expected, CAFs had the highest uptake of NPs, followed by tumor cells. Moreover, due to the targeting specificity of AS1411, both CAFs and tumor cells had a significant increase in phagocytosis of NPs-AS1411. Moreover, the results of Supplementary Fig. 14j–k confirmed that both NPs and NPs-AS1411 were mainly distributed in the liver and kidney, and NPs-AS1411 were more enriched in the tumor site than NPs, further demonstrating the good metabolizability of NPs and the effectiveness of AS1411 targeting. All the results suggested the enhanced targeting specificity and biosafety of NPs, especially NPs-AS1411.

a Uniform manifold approximation and projection (UMAP) plot showing the main cell types identified in the scRNA-seq dataset E-MTAB-8107 with specimens from 7 CRC patients. b Bubble heatmap showing marker genes across 8 clusters from (a). Dot size indicates fraction of expressing cells, colored according to z-score normalized expression levels. c UMAP plot showing the expression of 9 selected surface binding targets in different cell clusters. d Representative fluorescent images showing nucleolin expression on the plasma membrane (living cells, left panel) and within the nucleus (fixed cells, right panel) of different cells. β-catenin was stained as the marker of cell membrane, and cell nuclei were counterstained with Hoechst 33342 or DAPI. e–k Statistical analysis of flow cytometry of the uptake of NPs and NPs-AS1411 in different cells including (e) B cells, (f) T cells, (g) DCs, (h) macrophages, (i) tumor cells, (j) fibroblasts, and (k) endothelial cells of the in situ tumors in the subcutaneous tumor model. n = 3 independent experiments in (e–k). Immunofluorescent experiments (d) are repeated independently three times with similar results. Data are presented as means ± SD. Statistical analyses are performed using one-way ANOVA with multiple comparisons for (e–k). Source data are provided as a Source Data file.
The synergistic treatment effect of NPs in vivo
Taking the excellent physicochemical properties, including photothermal conversion properties, peroxidase-like activity, and immunoregulatory effect of NPs into account, a synergistic therapy strategy (i.e., PTT/CDT/immunotherapy) for CRC treatment was implemented based on the schematic illustration in Fig. 6a. Referring to previous results29, laser irradiation was applied to the mice with NPs and NPs-AS1411 treatment 24 h after injection, with the maximum enriched amounts of NPs at the tumor sites. As expected, compared to the other five groups, NPs-AS1411 treatment combined with the 808 nm laser irradiation presented the most satisfactory suppression effects on CT26 cells co-inoculated with BALB/3T3 cells and MC38 cells co-inoculated with NIH3T3 cells (Fig. 6b, c, and Supplementary Fig. 15a, b). Meanwhile, the body weight curves (Supplementary Fig. 15c, d), blood routine test (Supplementary Fig. 15e), liver and kidney function assay (Supplementary Fig. 15f), and hematoxylin and eosin (H&E) staining results (Supplementary Fig. 15g) demonstrated no differences in these six groups after different treatments, intensely revealing the good in vivo biosafety of this strategy again. Moreover, the NPs+Laser group showed the most CCL3 and the least CXCL12 expression in tumors, suggesting the strongest immune response (Fig. 6d, e). Among the six groups, the NPs-AS1411+Laser group exhibited the highest expression of CD80 and CD86 of DCs (Fig. 6f, g, and Supplementary Fig. 16), in accordance with the highest expression of T-bet and Eomes (Fig. 6h, and Supplementary Fig. 17) as well as IFN-γ, TNF-α, and CD107a in CD8+ T cells around tumor tissues or in the spleen (Fig. 6i, and Supplementary Fig. 18). All these results suggested NPs-mediated initiation of the strongest immune response and amelioration of CRC immunosuppression. To better simulate the process of CRC occurrence and development, as well as to more accurately substantiate the therapeutic efficacy of NPs and NPs-AS1411 against CRC, a kind of colorectal cancer genetically engineered mouse model (GEMM), namely the Apc-KO mouse model, was employed for further evaluation. Mice were injected with NPs or NPs-AS1411 intraperitoneally three times at 13, 14, and 15 weeks of age, with PBS-treated Apc-KO and wild-type (WT) C57BL/6J mice as controls. At 18 weeks, tumor progression in the colon and small intestine was evaluated (Fig. 6j). Both NPs and NPs-AS1411 markedly reduced intestinal tumor number and burden, increased colon length, and partially restored body weight, with stronger effects in the NPs-AS1411 group (Fig. 6k–l, Supplementary Fig. 19a–e). H&E staining confirmed significant alleviation of adenocarcinoma lesions (Fig. 6m, Supplementary Fig. 19d). ELISA assays revealed elevated CCL3 and reduced CXCL12 levels in peritoneal lavage fluid, particularly after NPs-AS1411 treatment (Fig. 6o). Immunohistochemistry (IHC) showed enhanced CD8⁺ T cells and CD86⁺ DCs infiltration, indicating robust immune activation mediated by NPs, especially NPs-AS1411 (Fig. 6n, and Supplementary Fig. 19e).

a Schematic illustration of the treatment strategy based on NPs for CT26 + BALB/3T3 cells-bearing mice. b Individual tumor growth curves and (c) average tumor growth curves of CT26 + BALB/3T3 cells-bearing mice in six groups. d, e Quantified expression of (d) CCL3 and (e) CXCL12 in homogenized tumor tissues of CT26 + BALB/3T3 cells-bearing mice in six groups. f, g (f) Flow cytometry and (g) corresponding statistical analysis of CD11c+MHC II+CD80+ and CD11c+MHC II+CD86+ cells around tumor tissues of CT26 + BALB/3T3 cells-bearing mice in six groups. h Statistical analysis of flow cytometry of CD3+CD8+Eomes+ and CD3+CD8+T-bet+ cells around tumor tissues of CT26 + BALB/3T3 cells-bearing mice in six groups. i Statistical analysis of flow cytometry of CD3+CD8+CD107a+, CD3+CD8+IFN-γ+, and CD3+CD8+TNF-α+ cells around tumor tissues of CT26 + BALB/3T3 cells-bearing mice in six groups. j Schematic depicting the administration of NPs in Apc-KO transgenic mice. Apc-KO mice were treated with NPs or NPs-AS1411 at the indicated time points (weeks 13-15). WT mice and PBS-injected Apc-KO as control groups. k Representative images of the opened colon from WT mice and Apc-KO mice treated with PBS, NPs and NPs-AS1411. Tumors were indicated with red arrows. Scale bar, 1 cm. l Quantification of tumor numbers in four groups. m H&E staining on colonic sections in four groups. Scale bar, 100 μm. n IHC staining of CD8a and CD86 on colonic sections in four groups. Scale bar, 100 μm. o Quantified expression of CCL3 and CXCL12 in peritoneal lavage fluid of mice in four groups. n = 6 mice per group in (a–c, j–l) and n = 3 independent experiments per group in (d–i, m–o). Data are presented as means ± SD. Statistical analyses are performed using two-way ANOVA with multiple comparisons for (c); and one-way ANOVA with multiple comparisons for (d, e, g–i, l, o). Source data are provided as a Source Data file.
The tumor inhibitory effect of NPs on patient-derived CRC models
Adopting the same treatment strategy in the PDX model, the NPs-AS1411 group showed the most significant temperature rise at tumor sites after the laser irradiation, which proved the excellent photothermal conversion performance of NPs and the effective active targeting of AS1411 again (Fig. 7a). Meanwhile, the NPs-AS1411+Laser group also expectedly exhibited the strongest tumor-suppressive effect in the PDX model with tumors that almost disappeared (Fig. 7b, c). In addition, this strategy had no effect on the body weight of the PDX model (Supplementary Fig. 20a). The histochemical analysis of tumors indicated the more extensive necrosis appeared with severe cell shrinkage and loss of contact in the NPs-AS1411+Laser group compared to the other five groups (Supplementary Fig. 20c). The expression of carcinoembryonic antigen (CEA) and Ki67 as epithelial cells markers and α-SMA and Vimentin as CAFs markers were gradually decreased with the continuous enrichment of strategies, indicating the inhibition effect on both CRC cells and CAFs (Supplementary Fig. 20d). To evaluate the simultaneous effects of NPs on CAFs and cancer cells, co-cultured cells from the PDX model were treated with NPs and laser irradiation after confirming their cellular characteristics (Supplementary Fig. 20b). Primary CRC cells surrounded by CAFs were identified by CEA and Ki67 and CAFs were identified by α-SMA and Vimentin (Fig. 7d). In co-culture system, the dose of NPs that effectively eliminated CAFs had limited direct cytotoxicity against CRC cells. However, when active targeting capability and NPs-triggered PTT were introduced, the cytotoxic effects of NPs on both CAFs and tumor cells were markedly enhanced (Fig. 7e).

a Real-time thermal infrared images of the PDX model after intravenous injection of PBS, NPs, and NPs-AS1411 for 24 h under 808 nm laser irradiation. b Representative images of tumors of the PDX model in six groups. c Average tumor growth curves of the PDX model in six groups. d Bright-field and fluorescent images of CEA, Ki67, α-SMA, and Vimentin expression in co-cultured tumor cells and CAFs from PDX model tumor tissues. e Bright-field and fluorescent images of co-cultured tumor cells and CAFs from PDX model tumor tissues stained with Calcein-AM (live cells, green fluorescence) and PI (dead cells, red fluorescence) after treatment with PBS, Laser, NPs, NPs-AS1411, NPs + Laser, and NPs-AS1411 + Laser. f Bright-field and H&E staining images of organoids from normal colonic epithelia and CRC. g H&E staining and immunohistochemical images of expression of Ki67, CDX2, CK20, CK7, and CEA of normal colonic organoids and CRC organoids. h, i Confocal bright-field and fluorescent images of Calcein-AM (live cells, green fluorescence) and PI (dead cells, red fluorescence) staining in (h) normal colonic organoids and (i) CRC organoids after treatment with PBS, Laser, NPs, NPs-AS1411, NPs + Laser, and NPs-AS1411 + Laser. n = 5 mice per group in (a–c). Immunofluorescent experiments (d, e), H&E and immunohistochemical experiments (f, g), and Calcein-AM/PI staining experiments (h, i) are repeated three times with similar results. Data are presented as means ± SD. Statistical analyses are performed using two-way ANOVA with multiple comparisons for (c). Source data are provided as a Source Data file.
To better conform to the real pathological environment of clinical patients, organoids were derived from cancerous and adjacent normal colonic tissues. Normal colonic organoids exhibited thinner lumens, uniformed cell organization, and better differentiation compared to CRC organoids (Fig. 7f), which were confirmed by consistent marker expressions in these organoids with their respective source tissues (Fig. 7g, and Supplementary Fig. 20e). Additionally, the cytotoxic effects of NPs at CAF-eliminating doses on CRC organoids were further enhanced by the active targeting of aptamer AS1411 and laser irradiation, while these treatments caused nearly no damage to normal colonic organoids. It was worth noting that the targeting ability of AS1411 could increase the enrichment of NPs in tumor tissues, thus exhibiting stronger tumor inhibitory effects in CRC organoids, regardless of whether laser irradiation is present or not (Fig. 7h, i).
Discussion
To address the issue of CRC insensitivity to immunotherapy caused by CAFs46,47,48, we developed a strategy targeting CAFs to ameliorate the immunosuppressive TME through both attenuating CAFs-induced tumor-promoting function and activating the immune system. Existing CAFs-targeting nanomaterials like liposomes and lipids49,50,51, focused only on drug delivery function, neglecting their properties. Hence, we made NPs with photothermal and catalytic abilities to kill CAFs. Interestingly, compared to the traditional Fe3O4 NPs, the incorporation of Cu element further enhanced the physical and chemical properties of NPs, thereby enabling them to have a stronger tumor-killing function (Supplementary Fig. 21a-b). Moreover, both Fe3O4 and Cu@Fe3O4 NPs exhibited excellent biocompatibility in vivo (Supplementary Fig. 21c). Active targeting of the discovered marker AS1411 and the natural phagocytic selectivity of cells, with CAFs showing higher phagocytic efficiency for NPs than NFs and cancer cells, ensured NPs’ good in vivo biosafety. It is worth mentioning that the design of our NPs can effectively avoid off-target effects commonly seen in nanotherapy. Macrophages are generally recognized as the principal cellular subgroup responsible for NP uptake and the resulting off-target effects52,53,54,55. Firstly, we optimized the physicochemical properties of the NPs, including the size and surface chemistry, to reduce macrophage phagocytosis. NPs larger than 100 nm are more prone to phagocytosis by macrophages, whereas our Cu@Fe3O4 NPs, with an average diameter of approximately 20 nm, preferentially accumulate in tumor tissues via the enhanced permeability and retention (EPR) effect. Moreover, surface modification of Cu@Fe3O4 with DSPE-PEG-NH₂ forms a hydrated layer that reduces protein adsorption and consequently decreases macrophage uptake significantly52,53,54,56,57,58,59. Then, identifying an efficient molecule that targets CAFs and tumor cells is also a powerful means to avoid off-target effects. Based on this strategy, the scRNA-seq and subcellular protein localization results indicated nucleolin, a multi-compartment-shuttling protein, as a more specific and suitable targeting molecule, which was highly expressed in both CAFs and tumor cells. Hence, AS1411 binding to nucleolin was selected for attachment to the surface of the NPs to enhance the specific targeting. Moreover, in vivo cellular uptake experiments in mice demonstrated distinct phagocytic efficiencies among various cell types, further confirming our conclusions. Both CAFs and tumor cells had a more significant increase in phagocytosis of NPs-AS1411. Collectively, these findings indicated that the specific targeting property of AS1411 could significantly alleviate off-target effects, thereby achieving improved biosafety in vivo.
Regarding mechanisms, though previous studies noted magnetic NPs’ ferroptosis effect on cancer cells, there’s no relevant report on CAFs yet. We found that NPs induced ferroptosis in CAFs via Fe2+-mediated ROS generation and Cu2+-mediated GPX4 downregulation. Our bioinformatics results suggested some signaling pathways (e.g., Wnt, TGF-β, MAPK, NF-κB, PI3K-Akt) potentially involved in regulating ferroptosis in CAFs, as they’re linked to ferroptosis in cancer cells36,60,61,62,63,64. This result interests us and will be further explored.
However, in some preclinical models and clinical trials, simply depleting CAFs through genetic manipulation, drug targeting, or specific antibodies may paradoxically convert the tumor microenvironment into a pro-tumorigenic state and even increase the risk of tumor dissemination65,66,67. Hence, the advantages of our strategy were further highlighted. Our NPs have multiple biological effects, inducing the death of CAFs, activating antitumor immunity, suppressing CAF-mediated tumor-promoting function, and directly killing CRC cells, along with established mechanisms associated with tumor cell dissemination and metastasis68. Specifically, we found that the treatment of NPs not only effectively reduced the viability of CAFs, but that the conditioned medium from NP-treated CAFs significantly suppressed CRC cell proliferation, migration, and EMT. This phenomenon was associated with the marked inhibition of Wnt and TGF-β paracrine signaling by NPs in CAFs, and these pathways are known to promote tumor proliferation, metastasis, and the formation of an immunosuppressive TME8,69,70,71. Another critical factor promoting tumor cell escape and metastasis is the presence of a local immunosuppressive microenvironment68. Interestingly, our results showed that NPs-treated CAFs released abundant chemokines such as CCL3 to remodel the immune TME through maturing DCs, activating CD8+ T cells, and enhancing CD8+ T cell function, which could significantly activate antitumor immunity. Together with the direct cytotoxicity of NPs against CRC cells, these findings suggested that targeting CAFs with NPs could help prevent tumor cell escape.
It is worth mentioning that we developed the Apc-KO mouse model, PDX model, and PDO model to verify the effectiveness of our strategy in addition to conventional subcutaneous tumor mouse model. The Apc-KO mouse model could exhibit spontaneous gastrointestinal neoplasia, which was closer to the real process of disease development in clinical CRC patients. At the tumor site of the primary CRC, the investigation of the anti-tumor effect and immunoregulatory role of NPs was even more convincing. PDX models, with tumor cells sourced from patients, offered a superior means to assess the efficacy of NPs in treating CRC. Further, in vitro co-culturing of CRC cancer cells and CAFs from PDX tissue allowed direct observation of the sensitivity of stromal and epithelial cells to NPs-induced killing, facilitating treatment strategy optimization. Moreover, the 3D culture system derived from normal colonic organoids and CRC organoids, bearing a closer resemblance to in vivo organ structures, more effectively gauged NPs’ tumor-killing potency and cytotoxicity to normal tissues. These phenomena further underlined the universality of and the potential of our strategy as the next-generation antitumor therapy clinically. In conclusion, we designed a promising strategy targeting CAFs to remodel immune TME, which provided the theoretical basis for the clinical treatment of CRC.
Methods
Ethics statement
All patient samples used in the present study were obtained from individuals undergoing surgical resection at the Department of Gastrointestinal Surgery IV, Peking University Cancer Hospital & Institute. None of the patients had received chemotherapy, radiotherapy, or immunotherapy prior to surgery. The study was conducted in accordance with the principles of the Declaration of Helsinki and was approved by the Ethics Committee of Peking University Cancer Hospital & Institute (approval no. 2020KT127). Written informed consent was obtained from all participants. All animal experiments were performed in strict accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of Peking University, Beijing, China (MSE-HouYL-1). As approved by IACUC (MSE-HouYL-1), when measuring tumor size, the maximum diameter of no more than 20 mm was strictly as the upper limit for tumor measurement in all animal experiments and the final tumor volumes did not exceed 2000 mm³ or 10% of the animal’s body weight. The tumor volume calculation formula adopted the most widely accepted and used standard formula V = 1/2 * L * W2 (V is the volume, L is the longest diameter, W is the width).
Synthesis of monodisperse Cu@Fe3O4 NPs
Cu@Fe3O4 NPs (simplified as NPs) were synthesized as follows: copper acetylacetonate (Cu(acac)2) (1 mmol, J & K), 1octadecene (ODE) (46.875 mmol, Alfa Aesar), oleic acid (2 mmol, Alfa Aesar), and oleamine (OAm) (10 mmol, J & K) were mixed in a fournecked flask. The solution was heated to 120 °C and kept for 1 h to remove the organic impurities under the mild N2 flow. When the temperature reached 180 °C, Fe(CO)5 (5 mmol) was injected into the reaction system and kept for 10 min. Then, the solution was heated to 280 °C and kept for 2 h. At room temperature, the solution was washed with acetone and hexane three times, and the products were kept in trichloromethane.
Synthesis of Cu@Fe3O4-AS1411 NPs
As performed previously72, NPs were modified with DSPE-PEG-NH2. 1-Ethyl-3-(3-(dimethylamino) propyl) urea hydrochloride (EDC) (1.9 mg, Aldrich, 99%) and N-hydroxysuccinimide (NHS) (2.2 mg, Aldrich, 98%) were mixed with the aforementioned NPs (20 mg) in an aqueous solution for 4-8 h. AS1411 (60 μg) was added to the reaction system and mixed for 24 h. The products were washed three times and kept in an aqueous solution.
Characterization
Transmission electron microscopy (TEM) images were obtained from the FEI Tecnai T20 microscope. High-resolution TEM (HRTEM) images were obtained from the FEI Tecnai F30 microscope. X-ray photoelectron spectroscopy (XPS) measurements were performed on an imaging X-ray photoelectron spectrometer using Al Kα radiation (Axis Ultra DLD, Kratos Analytical Ltd.). All the collected spectra were calibrated with the contaminated C 1 s peak at 284.8 eV and were analyzed using CasaXPS software (2.3.12 Dev7). X-ray diffraction (XRD) patterns were carried out using a Rigaku DMAX-2400 X-ray diffractometer equipped with Cu Kα (λ = 0.15405 nm) radiation. The concentrations of Fe were quantified using an inductively coupled plasma-atomic emission spectrometer (ICP-AES, Prodigy 7, Leeman, USA).
Cell culture
Human colonic epithelial cell line NCM460, human CRC cell line HCT116, murine CRC cell lines CT26 and MC38, the human monocytic leukemia cell line THP-1, and murine fibroblast cell lines (BALB/3T3 and NIH3T3) were originally obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and China Infrastructure of Cell Line Resources, Institute of Basic Medical Sciences, and Chinese Academy of Medical Sciences. Immortalized human colonic microvascular endothelial cells (HCMEC, BL12-H) and human pulmonary microvascular endothelial cells (HPMEC, HTX2255) were obtained from Otwo Biotech (Shenzhen, China). All cells were cultured in either Dulbecco’s modified eagle medium (DMEM) or RPMI-1640 supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin in a humidified environment at 37 °C with 5% CO2. All the reagents for cell culture were purchased from Invitrogen.
Human subjects
Fresh CRC tissues and matched adjacent normal tissues used for the isolation and culture of fibroblasts and organoids were obtained from patients undergoing surgical resection at the Department of Gastrointestinal Surgery IV, Peking University Cancer Hospital & Institute. NF lines were established from the adjacent normal tissues of three patients (NF1, 58-year-old male; NF2, 63-year-old male; NF3, 54-year-old female), and a normal colonic organoid was derived from the adjacent normal tissue of a 66-year-old male patient. CAF lines were generated from primary CRC tissues of three patients (CAF1, 61-year-old male; CAF2, 55-year-old female; CAF3, 71-year-old male), and a CRC PDO as well as a PDX model were established from tumor tissues of two patients (PDO, 52-year-old female; PDX, 72-year-old male). Written informed consent was obtained from all participants. All human tissue samples were used exclusively for the isolation of fibroblast lines, organoids, and PDX models in this study, and no residual tissues were retained.
Isolation and culture of NFs and CAFs
CRC tissues and adjacent normal tissues (located at least 10 cm from the tumor margin) were surgically obtained and transferred to the laboratory under sterile conditions. After PBS washing and removal of necrotic areas, the tissues were minced into ~1 mm fragments. These fragments were explanted in 6 cm culture dishes containing DMEM/Nutrient Mixture F-12 (DMEM/F12) supplemented with 10% FBS and 1% penicillin-streptomycin. Migrating fibroblasts were allowed to expand until reaching 70–80% density, then harvested and enzymatically digested for further expansion. Three NFs from normal colonic tissues and three CAFs from CRC samples were successfully isolated, with NF1 and CAF1 validated and selected for further experiments.
Preparation of conditioned medium
Primary fibroblasts (NFs or CAFs) were seeded and cultured in complete growth medium until reaching approximately 80-90% confluence. Prior to collecting the CM, cells were gently washed twice with sterile PBS, and the culture medium was replaced with fresh medium. Fibroblasts were then incubated for 24-48 h at 37 °C in a humidified atmosphere containing 5% CO₂ to allow accumulation of secreted factors. The supernatant was collected and centrifuged at 300 g for 10 min to remove floating cells and debris, followed by filtration through a 0.22 µm sterile filter. For CAFs treated with NPs, the supernatant was additionally centrifuged at 12,000 g for 10 min to remove any residual nanoparticles. The resulting CM was aliquoted and stored at −80 °C until use. Before application, CM was thawed on ice and mixed 1:1 with fresh complete medium for the subsequent treatment of CRC cells.
In vitro cytotoxicity assay
The cytotoxicity of NPs was evaluated using the Cell Counting Kit-8 (CCK-8; DOJINDO, Tokyo, Japan). Briefly, NFs, CAFs, NCM460, and HCT116 cells were seeded in a 96-well plate to reach 70–80% density. The cells were treated with NPs at varying Fe concentrations and incubated for 24 h. After washing, CCK-8 reagent was added to each well, and absorbance at 450 nm was measured using a microplate reader (Bio-Rad 680) after 2 h incubation. Each experiment was repeated in triplicate. Cytotoxicity was calculated as a percentage relative to untreated controls, and IC50 values were determined by fitting dose-response curves using nonlinear regression analysis in GraphPad Prism. All subsequent cell experiments were conducted at the following IC50 concentrations: 12.26 μM (95% CI: 11.35-13.90 μM) for NFs, 2.63 μM (95% CI: 2.36-2.89 μM) for CAFs, 7.08 μM (95% CI: 6.87–7.30 μM) for NCM460 cells, and 4.62 μM (95% CI: 4.25–5.00 μM) for HCT116 cells.
Bio-TEM characterization
Bio-TEM was utilized to determine the subcellular distribution of the NPs in NFs, CAFs, NCM460, and HCT116 cells. NFs, CAFs, NCM460, and HCT116 cells were seeded into a 6-well culture plate (105 cells per well). When the cell density reached 80% - 90%, the cells were then incubated with NPs for 24 h. The treated cells were washed three times with PBS, fixed using paraformaldehyde and osmium tetroxide, and then dehydrated with ethanol. Eventually, the treated cells were embedded in Spurr resin and sectioned to 70 nm in thickness.
Double staining of living/dead cells assay
The viability of cultured cells and organoids was assessed using the Calcein-AM/PI Double Stain Kit (40747ES76, Yeasen, Shanghai, China). NFs, CAFs, NCM460, and HCT116 cells were seeded into a 24-well culture plate (2 × 104 cells per well), achieving approximately 70% density. Normal colonic and CRC organoids were seeded in 3D low-attachment cell culture plates (Thermo Scientific). Indicated cells or organoids were then incubated with NPs for 24 h. After washing out the free NPs with PBS, the fresh culture medium was added. Laser (808 nm, 0.25 W/cm2) was then used to irradiate the cells or organoids for 5 min. The staining method was directed by the instructions. Each experiment was repeated in triplicate. Stained cells and organoids were visualized using an inverted microscope (Olympus IX71) or a confocal microscope (Zeiss LSM 880, Germany).
Intracellular ROS assay
Intracellular ROS production was determined by the ROS Assay Kit (S0033, Beyotime, China). NF, CAF, NCM460, and HCT116 cells were seeded into a 24-well culture plate (2 × 104 cells per well). When the cell density reached 70%, the cells were then incubated with NPs for 24 h. After washing out the free NPs with PBS, the fresh culture medium was added. Laser (808 nm, 0.25 W/cm2) was then used to irradiate the cells for 5 min. The staining method was directed by the instructions. Each experiment was repeated in triplicate. The cells were finally visualized using an inverted microscope (Olympus IX71) or detected through flow cytometry using a BD Fortessa flow cytometer (BD Biosciences).
Measurement of intracellular ferrous ions
The intracellular content of ferrous ions was assessed both qualitatively and quantitatively using the FerroOrange Kit (Dojindo, Japan) and the Ferrous Ion Content Assay Kit (BC5415, Solarbio, China), following the manufacturer protocols. Each experiment was repeated in triplicate.
For qualitative analysis, indicated cells were washed twice with PBS and incubated with 1 μM of the FerroOrange fluorescent probe at 37 °C for 30 min. The cells were then visualized using a confocal laser scanning microscope (Zeiss LSM 880, Germany), and fluorescence intensity was quantified using ImageJ software.
For quantitative analysis of Fe²⁺ content, treated cells were ultrasonically lysed in an ice bath, and the lysates were centrifuged at 4 °C for 10 min. The resulting supernatants were analyzed using the Ferrous Ion Content Assay Kit, with the absorbance measured at 593 nm using a microplate reader. A standard curve was generated based on standard samples, and the relative intracellular Fe2+ levels were calculated accordingly.
Liperfluo staining for LPO
Cells were assessed using Liperfluo staining (Dojindo, Japan). Cells were incubated with 10 μM Liperfluo in the dark at 37 °C for 30 min. After incubation, cells were washed twice with PBS and visualized using a confocal laser scanning microscope (Zeiss LSM 880, Germany), and fluorescence intensity was quantified using ImageJ software. Each experiment was repeated in triplicate.
Immunofluorescence
To examine protein expression on the plasma membrane, living cell IF staining was performed. Cells were washed twice with PBS and incubated with a primary antibody at 4 °C for 60 min without fixation, followed by an appropriate fluorophore-conjugated secondary antibody. After washing, nuclei were counterstained with Hoechest33342 and imaged immediately under a confocal microscope. For conventional IF, cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.5% Triton-X 100 for 10 min at 4 °C, and blocked with 1% bovine serum albumin for 1 h at room temperature. Cells were then incubated overnight at 4 °C with primary antibodies and with the corresponding secondary antibody for 1 h at room temperature the next day. Primary antibodies used were as follows Ki67 (Abcam, ab16667, 1:200), CEA (Huabio, ET1705-52, 1:500), α-SMA (STARTER, S0B2103, 1:200), Vimentin (Huabio, ET1610-39, 1:500), Nucleolin (Abcam, ab129200, 1:200), and CTNNB1 (Proteintech, 51067-2-AP,1:200). Following primary antibody incubation, cells were washed three times with PBS for 5 min. Secondary antibody incubation was performed for 1 h with Goat Anti-Rabbit IgG (Alexa Fluor 488) (Abcam, ab150077, 1:200) and Goat Anti-Rabbit IgG (Alexa Fluor 594) (Abcam, ab150080, 1:200). Images were captured using an inverted microscope (Olympus IX71) or a laser scanning confocal microscope (Zeiss LSM 880, Germany).
ELISA
Supernatants from treated CAFs homogenized mouse tumor tissues, as well as the peritoneal lavage fluid of mice, were collected. CCL3 and CXCL12 levels in CAF supernatants and homogenized mouse tumor tissues were measured using the anti-human ELISA kits (CCL3 for SEKH-0246, and CXCL12 for SEKH-0310, Solaribio) and anti-mouse ELISA kits (CCL3 for SEKM-0112, and CXCL12 for SEKM-0050, Solaribio), respectively, following manufacturer protocols. All experiments were conducted in triplicate.
Animals and subcutaneous cancer mouse models
Mice were housed under standard specific pathogen-free (SPF) conditions with a 12-h light/12-h dark cycle, an ambient temperature of 22 ± 1 °C, and a relative humidity of 50 ± 10%. BALB/c mice and C57BL/6 J mice (female, 6-8 weeks) were purchased from Vital River Laboratory Animal Technology Co. Ltd (Beijing, China). For the subcutaneous cancer mouse model, CT26 cells (2 × 106 cells in 0.1 ml of saline) or CT26 cells (1.95 × 106 cells in 0.1 ml of saline) mixed with BALB/3T3 cells (0.05 × 106 cells) were injected subcutaneously into BALB/c mice at the root of the right hind legs. Similarly, MC38 cells (2 × 106 cells in 0.1 ml of saline) or MC38 cells (1.95 × 106 cells in 0.1 ml of saline) mixed with NIH3T3 cells (0.05 × 106 cells) were injected subcutaneously into C57BL/6 J mice at the root of the right hind legs. All animal experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of Peking University, Beijing, China.
Apc-KO mouse model
A genetically engineered colorectal cancer mouse model, the Apc-KO strain (C57BL/6JCya-Apcem2/Cya; C001511, Cyagen Biosciences, Saiye Biotechnology Co., Ltd., Guangzhou, China), was employed to evaluate the in vivo therapeutic efficacy of NPs. This model is established by deleting the mutation cluster region (MCR) of the Apc gene using CRISPR/Cas9-mediated gene editing. Homozygous Apc-KO mice are embryonically lethal, whereas heterozygous mice spontaneously develop multiple intestinal adenomas that progress to adenocarcinomas in both the small intestines and colon. For treatment, heterozygous Apc-KO male mice received intraperitoneal injections of either NPs or NPs-AS1411 at 13, 14, and 15 weeks of age, with WT mice and PBS-treated Apc-KO mice serving as controls. All mice were euthanized at 18 weeks of age, and tumor progression in the colon and small intestine was systematically evaluated.
Patient-derived xenograft (PDX) models
Surgically resected CRC tissues were implanted subcutaneously into the dorsal flanks of 6-8-week-old NOD/SCID mice (Hua-Fu-Kang Corporation, Beijing, China). Tumor xenografts were allowed to grow for 1-3 months, depending on the tumor growth rate. Successfully engrafted tumors were expanded and retransplanted into additional NOD/SCID mice for evaluation of the therapeutic effects.
Isolation, establishment, and maintenance of patient-derived organoids (PDOs)
PDOs were established as previously described73. Briefly, CRC tissues were minced and digested with collagenase and dispase. While adjacent normal tissues were treated with ethylenediaminetetraacetic acid (EDTA) to release crypts. Following filtration and washing, tumor cells or crypts were resuspended in ECM droplets for culture. Organoid passage and maintenance utilized the Human Colonic Organoid Kit (Biogenous, K2003-HC) for normal colonic organoids and the Colorectal Cancer Organoid Kit (Biogenous, K2103-CR) for CRC organoids.
Organoid viability assay
Normal colonic organoids and CRC organoids were treated with NPs or NPs-AS411 first. After 6–8 h, the supernatant was removed and replaced with fresh culture medium, followed by laser irradiation 4–6 h. After 24 h, CellTiter-Glo 3D reagent (G9681; Promega, Madison, WI) was added to each well at a volume equal to that of the culture medium. The organoids were lysed by vigorous mixing, and the plate was incubated at room temperature for 30 min to stabilize the luminescent signal. Luminescence was measured using a microplate reader (Bio-Rad 680), and organoid viability was calculated relative to untreated controls. All experiments were conducted in triplicate.
In vivo treatment experiments
For CT26 cells- and CT26 + BALB/3T3 cells-bearing mouse models in four groups, as the schematic diagram showed (Fig. 4a), NPs (20 mg/kg, 200 μl) were intravenously injected into mice after 14, 17, and 20 days of cell inoculation, respectively. Similarly, for MC38 cells- and MC38 + NIH3T3 cells-bearing mouse models in four groups, the same treatment was administrated, with the second and third injections repeated after 20 and 26 days of cell inoculation, respectively.
For CT26 + BALB/3T3 cells-bearing mouse model in six groups, as the schematic diagram showed (Fig. 5c), NPs (20 mg/kg, 200 μl) and NPs-AS1411 (20 mg/kg, 200 μl) were intravenously injected into mice after 14 days of cell inoculation. After 24 h postinjection, the tumors of mice were exposed to 808 nm laser irradiation (0.25 W/cm2) for 5 min. An infrared thermal imaging instrument (FLIR A325SC camera) was used to record the temperature detection and thermal image. The 808 nm high-power multimode pump laser (Shanghai Connect iber Optics Co.) was used for the NIR laser. The above injection and laser irradiation were repeated after 17 and 20 days of cell inoculation, respectively. Similarly, for MC38 cells- and MC38 + NIH3T3 cells-bearing mouse models in six groups, the same treatment was administrated, with the second and third injections and laser irradiation repeated after 20 and 26 days of cell inoculation, respectively.
For the PDX mouse model in six groups, NPs (20 mg/kg, 200 μl) and NPs-AS1411 (20 mg/kg, 200 μl) were intravenously injected into mice after 22 days of tissue inoculation. After 24 h postinjection, the tumors of mice were exposed to 808 nm laser irradiation (0.25 W/cm2) for 5 min. The above injection and laser irradiation were repeated after 34 and 46 days of cell inoculation, respectively.
Histological evaluation
PDX mice with different treatments were sacrificed at the end of treatment, and the tissues (heart, liver, spleen, lung, kidney, and tumor) were selected, followed by fixing with 4% paraformaldehyde. The tissues were embedded in paraffin and sectioned at 5 mm. H&E staining was performed for histological evaluation. The slides were observed under an optical microscope.
Immunohistochemistry
The paraffin sections of tumor tissues in mice were prepared at the end of treatment. Immunohistochemistry was performed as described previously74. Paraffin-embedded tissue sections were deparaffinized, rehydrated, and antigen-recovered. Sections were then blocked with 5 % serum/PBS for 30 min, incubated with primary antibodies at 4 °C overnight, and followed by a reaction using an immunohistochemistry staining kit (Yeasen, 36311ES50). Primary antibodies were used including Ki67 (Abcam, ab16667, 1:500), CEA (Huabio, ET1705-52, 1:200), CDX2 (Huabio, ET1605-4, 1:200), CK20 (Huabio, EM1901-97, 1:200), CK7 (Huabio, EM0702, 1:200), α-SMA (STARTER, S0B2103, 1:200), Vimentin (Huabio, ET1610-39, 1:200), CD8a (Servicebio, GB15068-100, 1:500), and CD86 (Servicebio, GB150054-100,1:500).
Flow cytometry
Flow cytometry was performed using a BD Fortessa flow cytometer (BD Biosciences) and data were analyzed using FlowJo 10 software (TreeStar). All experiments were conducted in triplicate. Monoclonal antibodies against mouse were purchased from Biolegend as followed: CD45 (103111, 1:200), CD3 (100335, 1:200), CD4 (100405, 1:200), CD8a (100733, 1:200), T-bet (644809, 1:200), Eomes (157703, 1:200), CD107a (121613, 1:100), IFN-γ (113603, 1:200), TNF-ɑ (506323, 1:200), CD80 (104707, 1:200), CD86 (105027, 1:200), MHC II (107605, 1:200), MHC II (107607, 1:200), CD11c (117317, 1:200), CD11b (101215, 1:200), F4/80 (111603, 1:200), CD19 (152407, 1:200), CD31 (102409, 1:200), CD45 (103131, 1:200), Ep-CAM (118205, 1:200). Monoclonal antibodies against mouse were purchased from Absin as followed: α-SMA (abs172181, 1:200).
Uptake assay of NPs by different cells
According to the different color matching of antibodies marking the different cells, NPs and NPs-AS1411 were linked with the appropriate fluorescence molecules (FITC fluorescence molecules) for subsequent flow cytometry analysis. Splenocytes or cells around tumors were cocultured with NPs or NPs-AS1411. The specific experimental steps were performed as described previously75. To assess uptake into primary cells, single cell suspensions from the total spleen or tumors were used. Splenocytes or cells around tumors were cultured, and NPs or NPs-AS1411 were added and co-incubated with splenocytes or cells around tumors. Lastly, the uptake of NPs or NPs-AS1411 by different cells was analyzed by flow cytometry. All experiments were conducted in triplicate.
CD8+ T cell function assay
Splenocytes (2 × 106) or lymphocytes around tumors were collected. BD GolgiStop™ reagent (BD Biosciences) was used to inhibit intracellular protein transport, and meanwhile, APC-conjugated anti-CD107a antibody or respective control isotypes were added at the beginning of incubation. After co-culturing for 4 h, cells were harvested and stained with the indicated antibodies, fixed, permeabilized with Cytofix/Cytoperm Buffer (BD Biosciences), and then stained with CD107a, IFN-γ, and TNF-ɑ antibody. All experiments were conducted in triplicate.
RNA extraction and quantitative real-time PCR
Total RNA was extracted utilizing TRIzol reagent (Thermo Fisher Scientific, Carlsbad, USA) and subsequently reverse transcribed into cDNA using the GoScript™ Reverse Transcriptase (Promega, Madison, USA). The obtained cDNAs were then subjected to quantitative real-time PCR (qRT-PCR) analysis using SYBR Green PCR Master Mix (Toyobo Life Science, Osaka, Japan) on an ABI 7500 Real-time PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.), with each reaction conducted in triplicate. GAPDH was used as the internal control gene for normalization, and the relative gene expression was analyzed using the 2−ΔΔCt method. The primers of CCL3, CXCL12, CCR7, and GAPDH were as follows:
CCL3 forward: AGTTCTCTGCATCACTTGCTG,
CCL3 Reverse: CGGCTTCGCTTGGTTAGGAA;
CXCL12 forward: ATTCTCAACACTCCAAACTGTGC,
CXCL12 Reverse: ACTTTAGCTTCGGGTCAATGC;
CCR7 forward: TGAGGTCACGGACGATTACAT,
CCR7 Reverse: GTAGGCCCACGAAACAAATGAT;
GAPDH forward: TGCACCACCACCTGCTTAGC,
GAPDH Reverse: GGCATGGACTGTGGTCATGAG.
Western blot
Cells (105 cells per well) were seeded in a 6-well plate and incubated with NPs for 24 h once reaching 80%-90% density, and then lysed in RIPA buffer (Cell Signaling Technology). Western blot analysis was performed using standard protocols76. Primary antibodies from Cell Signaling Technology were as followed: Akt (4691 T, 1:1000), Pho-Akt (Ser473) (4060 T, 1:1000), NF-κB (p65) (8242 T, 1:1000), Pho-NF-κB (Ser536) (3033 T, 1:1000), MAPK (p38) (8690 T, 1:1000), Pho-MAPK (Thr180/Tyr182) (4511 T, 1:1000), E-Cadherin (3195 s, 1:1000), N-Cadherin (13116 s, 1:500), and Vimentin (5741 s, 1:500). Other primary antibodies used were Nucleolin (Abcam, ab129200, 1:1000), and β-actin (Sigma-Aldrich, A1978, 1:2000). All experiments were conducted in triplicate.
RNA-seq analysis
CAFs were seeded to 5 × 106 cells reaching 80%-90% density and then incubated with or without NPs for 24 h. The treated cells were washed three times with PBS and then harvested for total RNA extraction with a total RNA purification kit (GeneMarkbio). RNA purity and quantification were assessed using a NanoDrop 2000 spectrophotometer (Thermo Scientific, USA). RNA integrity was confirmed with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Subsequently, libraries were constructed using the VAHTS Universal V6 RNA-seq Library Prep Kit, following the manufacturer’s instructions. Transcriptome sequencing and subsequent bioinformatic analysis were conducted by OE Biotech Co., Ltd. (Shanghai, China).
Bioinformatic analysis
Whole human genome microarray data were obtained from GEO datasets GSE1753834 and GSE87211233 and analyzed by using the “GEOquery” R package77. The infiltration levels of CAFs were evaluated using the MCP-counter, EPIC, and ESTIMATE algorithms30,31,32, via the “IOBR” package78. The optimal cut-off values for CAF abundance were ascertained by the “surv_cutpoint” function in the “survminer” followed by Kaplan–Meier survival analysis with “survival” and “survminer” with log-rank test for p-values.
To further analyze the effects of NPs on CAFs, RNA-sequencing data of CAFs with different treatments were further explored. DEGs with log2 |fold change (FC)| ≥ 1 and adjusted P-value ≤ 0.05 were identified by the “limma” package79. and functional enrichment was performed through GO, KEGG, and Hallmark GSEA using clusterProfiler and enrichplot. Data visualization was conducted with ggplot2 and pheatmap80,81,82.
For single-cell analysis, the CRC dataset E-MTAB-8107 was downloaded and processed by using the “Seurat” R package83 (R software, version 4.3.3). Cells with >200 and <6000 detected genes and <15% mitochondrial reads were retained, followed by normalization, PCA, clustering (resolution 0.1–1), and UMAP visualization., Major cell types were annotated based on canonical markers including epithelial cells (EPCAM, KRT884), myeloid cells (CD14, CD6884), endothelial cells (PECAM1, VWF85), fibroblasts (COL1A1, COL1A286), enteric glial cells (S100B87, PLP184), B cells (CD79A, CD79B88), T cells (CD3D, CD3E89), and mast cells (KIT, CPA388). Fibroblast subsets were further reclustered into CAFs and NFs using markers DCN, IGFBP6, and MFAP590.
Statistical analysis and reproducibility
All experiments were performed at least three times independently. Quantitative data were presented as means ± SD. Statistical significance between two groups was assessed using a two-tailed unpaired Student’s t-test. Comparisons across more than two groups were performed using one-way ANOVA followed by appropriate post hoc tests to determine pairwise differences. For experiments involving two independent factors, such as treatment type and time, two-way ANOVA was applied to evaluate the main effects of each factor as well as their interaction. A two-tailed p < 0.05 was considered statistically significant. Western blot, immunofluorescence, FerroOrange and Liperfluo staining, as well as H&E staining and IHC experiments were performed at least three times as independent experiments with similar results, and representative images are shown. Statistical analyses were performed using GraphPad Prism (version 10.0). All schematic diagrams in the article were generated by PowerPoint or Adobe Illustrator.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq data generated in this study have been deposited in the Gene Expression Omnibus database under accession number GSE301220. The microarray gene expression profiling data sets (GSE17538 and GSE87211) are downloaded from Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) database. The scRNA sequencing data were retrieved from the EMBL-EBI ArrayExpress database under accession number E-MTAB-8107 (https://www.ebi.ac.uk/biostudies/arrayexpress). All data supporting this work are available within the Article, Supplementary Information, or Source Data file. Source data are provided with this paper.
Code availability
The code used for the analysis of public GEO datasets, bulk RNA-seq data, and single-cell RNA-seq data in this study is available on GitHub (https://github.com/hoshino-tech/Nano-Shuren-Zaozao-2025) and has been archived in Zenodo at https://zenodo.org/records/1796043291.
References
Chhabra, Y. & Weeraratna, A. T. Fibroblasts in cancer: unity in heterogeneity. Cell 186, 1580–1609 (2023).
Sahai, E. et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 20, 174–186 (2020).
Chen, Y., McAndrews, K. M. & Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 18, 792–804 (2021).
Linares, J. et al. Long-term platinum-based drug accumulation in cancer-associated fibroblasts promotes colorectal cancer progression and resistance to therapy. Nat. Commun. 14, 746 (2023).
Hegde, P. S. & Chen, D. S. Top 10 challenges in cancer immunotherapy. Immunity 52, 17–35 (2020).
Kobayashi, H. et al. Cancer-associated fibroblasts in gastrointestinal cancer. Nat. Rev. Gastro Hepat. 16, 282–295 (2019).
Joyce, J. A. & Fearon, D. T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 348, 74–80 (2015).
Huang, T. X. et al. Targeting cancer-associated fibroblast-secreted WNT2 restores dendritic cell-mediated antitumour immunity. Gut 71, 333–344 (2022).
Pei, L. P. et al. Roles of cancer-associated fibroblasts (CAFs) in anti- PD-1/PD-L1 immunotherapy for solid cancers. Mol. Cancer 22, 29 (2023).
Khaliq, A. M. et al. Refining colorectal cancer classification and clinical stratification through a single-cell atlas. Genome Biol. 23, 113 (2022).
Saw, P. E., Chen, J. N. & Song, E. W. Targeting CAFs to overcome anticancer therapeutic resistance. Trends Cancer 8, 527–555 (2022).
Holmgaard, R. B. et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 6, 47 (2018).
Ferrer-Mayorga, G. et al. Vitamin D receptor expression and associated gene signature in tumour stromal fibroblasts predict clinical outcome in colorectal cancer. Gut 66, 1449–1462 (2017).
Öhlund, D. et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med 214, 579–596 (2017).
Alkasalias, T., Moyano-Galceran, L., Arsenian-Henriksson, M. & Lehti, K. Fibroblasts in the tumor microenvironment: shield or spear? Int. J. Mol. Sci. 19, 1532 (2018).
Maia, A., Schöllhorn, A., Schuhmacher, J. & Gouttefangeas, C. CAF-immune cell crosstalk and its impact in immunotherapy. Semin. Immunopathol. 45, 203–214 (2023).
Wu, F. L. et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Tar. 6, 218 (2021).
Khawar, I. A., Kim, J. H. & Kuh, H. J. Improving drug delivery to solid tumors: Priming the tumor microenvironment. J. Controlled Release 201, 78–89 (2015).
Minchinton, A. I. & Tannock, I. F. Drug penetration in solid tumours. Nat. Rev. Cancer 6, 583–592 (2006).
Barbazan, J. et al. Cancer-associated fibroblasts actively compress cancer cells and modulate mechanotransduction. Nat. Commun. 14, 6966 (2023).
Yuan, S. J. et al. Transforming cancer-associated fibroblast barrier into drug depots to boost chemo-immunotherapy in “shooting fish in a barrel” pattern. Acs Nano 17, 13611–13626 (2023).
Miao, L. et al. The Binding site barrier elicited by tumor associated fibroblasts interferes disposition of nanoparticles in stroma-vessel type tumors. Acs Nano 10, 9243–9258 (2016).
Guo, J., Zeng, H. T. & Chen, Y. Emerging nano drug delivery systems targeting cancer-associated fibroblasts for improved antitumor effect and tumor drug penetration. Mol. Pharm. 17, 1028–1048 (2020).
Zhou, Z. G. et al. Iron/iron oxide core/shell nanoparticles for magnetic targeting MRI and near-infrared photothermal therapy. Biomaterials 35, 7470–7478 (2014).
Gao, L. Z. et al. Intrinsic peroxidase-like activity of ferromagnetic nanoparticles. Nat. Nanotechnol. 2, 577–583 (2007).
Wang, S. R. et al. Magnetic nanostructures: rational design and fabrication strategies toward diverse applications. Chem. Rev. 122, 5411–5475 (2022).
Wang, S. R. et al. A catalytic immune activator based on magnetic nanoparticles to reprogram the immunoecology of breast cancer from “cold” to “hot” state. Adv. Healthc. Mater. 11, e2201240 (2022).
Wang, S. R. & Hou, Y. L. New types of magnetic nanoparticles for stimuli-responsive theranostic nanoplatforms. Adv. Sci. 11, 2305459 (2024).
Wang, S. R. et al. Amelioration of systemic antitumor immune responses in cocktail therapy by immunomodulatory nanozymes. Sci. Adv. 8, eabn3883 (2022).
Becht, E. et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 17, 218 (2016).
Racle, J., de Jonge, K., Baumgaertner, P., Speiser, D. E. & Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 6, e26476 (2017).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013).
Hu, Y. et al. Colorectal cancer susceptibility loci as predictive markers of rectal cancer prognosis after surgery. Gene Chromosome Canc 57, 140–149 (2018).
Smith, J. J. et al. Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology 138, 958–968 (2010).
Zhao, Z. H., Li, T. M., Yuan, Y. & Zhu, Y. M. What is new in cancer-associated fibroblast biomarkers? Cell Commun. Signal 21, 96 (2023).
Yao, F. et al. A targetable LIFR-NF-κB-LCN2 axis controls liver tumorigenesis and vulnerability to ferroptosis. Nat. Commun. 12, 7333 (2021).
Xue, Q. et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy 19, 1982–1996 (2023).
Ozga, A. J., Chow, M. T. & Luster, A. D. Chemokines and the immune response to cancer. Immunity 54, 859–874 (2021).
Wang, C. X., Wang, J. Y., Zhu, Z. C., Hu, J. L. & Lin, Y. Spotlight on pro-inflammatory chemokines: regulators of cellular communication in cognitive impairment. Front. Immunol. 15, 1421076 (2024).
Sokol, C. L. & Luster, A. D. The Chemokine System in Innate Immunity. Cold Spring Harb. Perspect. Biol. 7, a016303 (2015).
Zhou, C. J., Gao, Y., Ding, P. L., Wu, T. & Ji, G. The role of CXCL family members in different diseases. Cell Death Discov. 9, 212 (2023).
Cambier, S., Gouwy, M. & Proost, P. The chemokines CXCL8 and CXCL12: molecular and functional properties, role in disease and efforts towards pharmacological intervention. Cell. Mol. Immunol. 20, 217–251 (2023).
Salem, A., Alotaibi, M., Mroueh, R., Basheer, H. A. & Afarinkia, K. CCR7 as a therapeutic target in Cancer. Bba-Rev. Cancer 1875, 188499 (2021).
Wu, D. M. et al. Phosphorylation and changes in the distribution of nucleolin promote tumor metastasis via the PI3K/Akt pathway in colorectal carcinoma. FEBS Lett. 588, 1921–1929 (2014).
Hovanessian, A. G. et al. Surface expressed nucleolin is constantly induced in tumor cells to mediate calcium-dependent ligand internalization. PLoS One 5, e15787 (2010).
Le, D. T. et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372, 2509–2520 (2015).
Feig, C. et al. Targeting CXCL12 from FAP-expressing carcinomaassociated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. Usa. 110, 20212–20217 (2013).
Costa, A. et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 33, 463–479 (2018).
Zhao, L. et al. Fibroblast activation protein-based theranostics in cancer research: A state-of-the-art review. Theranostics 12, 1557–1569 (2022).
Sonju, J. J., Dahal, A., Singh, S. S. & Jois, S. D. Peptide-functionalized liposomes as therapeutic and diagnostic tools for cancer treatment. J. Controlled Release 329, 624–644 (2021).
Miao, L. et al. Targeting tumor-associated fibroblasts for therapeutic delivery in desmoplastic tumors. Cancer Res. 77, 719–731 (2017).
Blanco, E., Shen, H. & Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 33, 941–951 (2015).
Gustafson, H. H., Holt-Casper, D., Grainger, D. W. & Ghandehari, H. Nanoparticle uptake: the phagocyte problem. Nano Today 10, 487–510 (2015).
Jia, J. et al. Interactions between nanoparticles and dendritic cells: from the perspective of cancer immunotherapy. Front Oncol. 8, 404 (2018).
Hinze, C. & Boucrot, E. Endocytosis in proliferating, quiescent and terminally differentiated cells. J. Cell Sci. 131, jcs216804 (2018).
Perrault, S. D., Walkey, C., Jennings, T., Fischer, H. C. & Chan, W. C. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 9, 1909–1915 (2009).
Hoshyar, N., Gray, S., Han, H. & Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomed. (Lond.) 11, 673–692 (2016).
Alexis, F., Pridgen, E., Molnar, L. K. & Farokhzad, O. C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 5, 505–515 (2008).
Qie, Y. et al. Surface modification of nanoparticles enables selective evasion of phagocytic clearance by distinct macrophage phenotypes. Sci. Rep. 6, 26269 (2016).
Wang, X. Y., Tan, X., Zhang, J. P., Wu, J. P. & Shi, H. J. The emerging roles of MAPK-AMPK in ferroptosis regulatory network. Cell Commun. Signal 21, 200 (2023).
Yi, J. M., Zhu, J. J., Wu, J., Thompson, C. B. & Jiang, X. J. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl. Acad. Sci. Usa. 117, 31189–31197 (2020).
Wang, Y. et al. Wnt/beta-catenin signaling confers ferroptosis resistance by targeting GPX4 in gastric cancer. Cell Death Differ. 29, 2190–2202 (2022).
Chen, H. D. et al. SMAD4 endows TGF-β1-induced highly invasive tumor cells with ferroptosis vulnerability in pancreatic cancer. Acta Pharmacol. Sin. 45, 844–856 (2024).
Kim, D., Kim, W. D., Kim, S. K., Moon, D. H. & Lee, S. J. TGF-β1-mediated repression of SLC7A11 drives vulnerability to GPX4 inhibition in hepatocellular carcinoma cells. Cell Death Dis. 11, 406 (2020).
Özdemir, B. C. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734 (2014).
Rhim, A. D. et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 25, 735–747 (2014).
Hofheinz, R. D. et al. Stromal antigen targeting by a humanised monoclonal antibody:: An early phase II trial of sibrotuzamab in patients with metastatic colorectal cancer. Onkologie 26, 44–48 (2003).
Lambert, A. W., Pattabiraman, D. R. & Weinberg, R. A. Emerging Biological Principles of Metastasis. Cell 168, 670–691 (2017).
Hao, Y., Baker, D. & Ten Dijke, P. TGF-beta-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 20, 2767 (2019).
Liang, J., Pan, Y., Yang, J., Zeng, D. & Li, J. WNT signaling in cancer: molecular mechanisms and potential therapies. Mol. Biomed. 6, 83 (2025).
Holmgaard, R. B. et al. Targeting the TGFbeta pathway with galunisertib, a TGFbetaRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 6, 47 (2018).
Ju, Y. M. et al. Monodisperse Au-Fe2C janus nanoparticles: an attractive multifunctional material for triple-modal imaging-guided tumor photothermal therapy. Acs Nano 11, 9239–9248 (2017).
van de Wetering, M. et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945 (2015).
Wang, S. R. et al. LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer. Ther. Adv. Med. Oncol. 11, 1758835919843736 (2019).
Wagner, J. et al. Mesoporous silica nanoparticles as pH-responsive carrier for the immune-activating drug resiquimod enhance the local immune response in mice. ACS Nano 15, 4450–4466 (2021).
Wang, S. R. et al. TAZ promotes cell growth and inhibits Celastrol-induced cell apoptosis. Biosci. Rep. 36, e00386 (2016).
Sean, D. & Meltzer, P. S. GEOquery: a bridge between the gene expression omnibus (GEO) and BioConductor. Bioinformatics 23, 1846–1847 (2007).
Zeng, D. Q. et al. IOBR: multi-omics immuno-oncology biological research to decode tumor microenvironment and signatures. Front. Immunol. 12, 687975 (2021).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Bindea, G. et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 (2009).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. Usa. 102, 15545–15550 (2005).
Yu, G. C., Wang, L. G., Han, Y. Y. & He, Q. Y. clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. Omics-a J. Integr. Biol. 16, 284–287 (2012).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Li, R. et al. Single-cell transcriptomic analysis deciphers heterogenous cancer stem-like cells in colorectal cancer and their organ-specific metastasis. Gut 73, 470–484 (2024).
Zheng, X. et al. Single-cell transcriptomic profiling unravels the adenoma-initiation role of protein tyrosine kinases during colorectal tumorigenesis. Signal Transduct. Target Ther. 7, 60 (2022).
Wang, F. et al. Single-cell and spatial transcriptome analysis reveals the cellular heterogeneity of liver metastatic colorectal cancer. Sci. Adv. 9, eadf5464 (2023).
Qi, J. et al. Single-cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat. Commun. 13, 1742 (2022).
Zhou, Y. et al. Single-cell multiomics sequencing reveals prevalent genomic alterations in tumor stromal cells of human colorectal cancer. Cancer cell 38, 818–828 (2020).
Chu, X. et al. Integrative single-cell analysis of human colorectal cancer reveals patient stratification with distinct immune evasion mechanisms. Nat. Cancer 5, 1409–1426 (2024).
Chen, B. N. et al. The molecular classification of cancer-associated fibroblasts on a pan-cancer single-cell transcriptional atlas. Clin. Transl. Med. 13, e1516 (2023).
Shuren, W., Zaozao, W. & Chenxin Wu. hoshino-tech/Nano-Shuren-Zaozao-2025: Amelioration of Colorectal Cancer-Associated Fibroblasts-Induced Immunosuppressive Microenvironment by Ferroptosis-Based Nanotherapy. Zenodo: https://zenodo.org/records/17960432 (2025).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22575006 (S. W.), 52201198 (S. W.), 52027801 (Y. H.), 92263203 (Y. H.), 82573616 (Z. W.), 82073357 (Z. W.), 82573356 (X. S.), 82171720 (X. S.)), the Beijing Natural Science Foundation-Haidian Primitive Innovation Joint Fund (L232015 (S. W.)), the National Key R&D Program of China (2022YFA1203902 (Y. H.)), the China-Germany Collaboration Project (M-0199 (Y. H.)), Clinical Medicine Plus X-Young Scholars Project of Peking University, the Fundamental Research Funds for the Central Universities (PKU2024LCXQ014 (S. W.), PKU2025PKULCXQ040 (S. W.)), Science Foundation of Peking University Cancer Hospital (JC202405 (Z. W.)), Open Project funded by Key Laboratory of Carcinogenesis and Translational Research, Ministry of Education (2025 Open Project-5 (X. S.)), and Science Foundation of Peking University Cancer Hospital & Institute (XKFZ2606 (X. S.)).
Author information
Authors and Affiliations
Contributions
S.W., Z.W., X.S., and Y.H. conceived the study. S.W. and Z.W. designed and performed the experiments. S.W., Z.W., C.W., R.M., and Y.P. analyzed the data. C.Z. provided the clinical samples. S.W. and Z.W. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Nick Peake and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, S., Wang, Z., Wu, C. et al. Amelioration of colorectal cancer-associated fibroblasts in immunosuppressive microenvironment by ferroptosis-based nanotherapy. Nat Commun 17, 2778 (2026). https://doi.org/10.1038/s41467-026-69462-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69462-5
