
Abstract
Atherosclerosis (AS) is a leading cause of cardiovascular and cerebrovascular disease and is driven by lipid accumulation and chronic inflammation within arterial plaques. Foam cells play a central role in plaque growth and destabilization, yet their precise diagnosis and targeted regulation remain challenging. Here we present a hierarchical nanoagent that integrates multimodal imaging with selective foam cell intervention for improved AS management. The molecular probe enables near-infrared II fluorescence, photoacoustic, and magnetic resonance imaging, providing complementary information on plaque location, depth, and morphology. To achieve targeted therapy, the probe is co-encapsulated with atorvastatin using responsive nanocarriers camouflaged with macrophage membranes and equipped with a foam cell-targeting peptide. This hierarchical strategy enables precise targeting to inflamed plaques and foam cells, where controlled drug release promotes lipid efflux, reduces inflammatory signaling, and enhances plaque stabilization in female AS mouse models. This integrated imaging-guided therapeutic platform holds great promise for precise diagnosis and treatment of cardiovascular disease.
Similar content being viewed by others
Introduction
Cardiovascular diseases (CVDs) remain the leading global cause of morbidity and mortality, accounting for nearly half of all deaths worldwide annually1. Among these, atherosclerosis (AS) stands out as the most prevalent and clinically significant, representing a chronic inflammatory disorder characterized by the progressive, often asymptomatic accumulation of lipid-laden plaques within the arterial walls2,3,4. Over time, these plaques can rupture, precipitating catastrophic cardiovascular events such as myocardial infarction, stroke, and sudden cardiac death5,6. Early detection and precise monitoring of AS are paramount for facilitating timely therapeutic interventions, mitigating symptoms, and halting disease progression7,8,9. However, current diagnostic modalities are limited in their capacity to detect early-stage lesions with the requisite sensitivity and specificity, thereby hindering effective clinical management.
Traditional plaque imaging techniques, such as intravascular ultrasound and optical coherence tomography suffer from low sensitivity, weak functional signals, restricted imaging depth (typically 1–3 mm), and invasiveness, which significantly restrict their broader clinical application10. In contrast, magnetic resonance imaging (MRI), with its non-invasive nature and absence of ionizing radiation, offers a promising alternative for detecting AS plaques11,12. However, conventional small-molecule MRI contrast agents, including gadolinium-based compounds, are characterized by low specificity, rapid systemic clearance, and suboptimal image quality, which undermine diagnostic precision13,14,15. These limitations, coupled with the extended acquisition time required for high-resolution imaging further limit their utility for real-time monitoring and detection of smaller plaques16,17. To address these challenges, integrating MRI with other complementary imaging modalities emerges as a promising approach to enhance diagnostic accuracy and sensitivity by providing more comprehensive, high-resolution pathological insights18,19.
Optical imaging, renowned for its high sensitivity, real-time capabilities, cost-effectiveness, and accessibility, offers a valuable complementary tool to MRI20,21. In particular, the second near-infrared (NIR-II) window (1000-1700 nm) has been explored for optical imaging recently, which can minimize tissue autofluorescence and light scattering, thereby enabling superior tissue penetration and enhanced signal-to-noise ratio22,23,24. Furthermore, photoacoustic (PA) imaging, a rapidly advancing non-invasive modality, has garnered increasing attention in the biomedical field25,26. PA imaging offers deeper tissue penetration (up to several centimeters) and high spatial resolution, surpassing traditional fluorescence imaging27,28. Despite these promising attributes, the sensitivity of PA imaging remains a notable challenge29. Given the complex and heterogeneous pathological microenvironment of AS, there is a pressing need for multimodal imaging strategies that leverage the strengths of each imaging modality to provide more comprehensive insights into disease diagnosis, treatment, and therapeutic outcome evaluation30,31. In this regard, single-molecule probes capable of integrating multiple imaging functions hold substantial promise, offering distinct advantages over traditional mixed-mode systems. However, optimizing the performance of each imaging modality within a single molecule poses a significant challenge, as different imaging components often interfere with one another32,33. Currently, reports of single-molecule probes that simultaneously enable MRI, NIR-II fluorescence, and PA imaging remain rare, highlighting the need for further efforts to overcome these obstacles and fully exploit the potential of such multimodal imaging platforms in AS diagnosis.
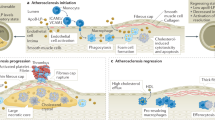
Foam cells, along with the inflammatory responses they provoke, play pivotal roles in the pathogenesis of AS11. These lipid-laden macrophages accumulate within atherosclerotic plaques, where they contribute significantly to plaque instability and progression34. Foam cells are central to necrotic core formation and act as potent producers of pro-inflammatory cytokines, which further recruit immune cells to the plaque site to amplify the inflammatory cascade35,36,37,38. Moreover, foam cells express surface molecules such as CD47, a “don’t eat me” signal, which allows them to evade phagocytosis and clearance by the immune system39,40,41. Given their crucial involvement in the disease process, the precise imaging and regulation of foam cells are vital for both accurate detection and effective treatment of AS. However, the complex microenvironment of atherosclerotic plaques makes targeted imaging and effective modulation of foam cells challenging42,43. Inefficient plaque accumulation, limited penetration into the plaque core, and nonspecific targeting of foam cells associated with conventional imaging agents and drugs usually compromise imaging outcomes and lead to undesirable side effects44,45. These limitations undermine the effectiveness of both diagnostic and therapeutic strategies in managing AS. Consequently, there is an urgent need to develop advanced, multi-tiered targeting strategies that facilitate more precise imaging of pathological foam cells and enable efficient therapeutic interventions.
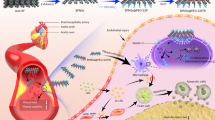
In this work, we present a hierarchical nanoplatform for precise multimodal imaging and targeted intervention of AS (Fig. 1). We engineered a high-performance multimodal molecular probe that enabled simultaneous MRI, NIR-II fluorescence, and PA imaging. Through rational molecular design, the signals for each imaging modality were optimized, offering a robust imaging system. In particular, NIR-II fluorescence imaging, with its high sensitivity and real-time visualization capabilities, facilitated rapid localization of the disease and dynamic, in situ tracking of plaque progression. MRI, known for its deep tissue penetration, enabled precise detection of plaque location and volume, aiding in the assessment of plaque stability. PA imaging, with its superior spatial resolution, supplied detailed insights into plaque morphology. By integrating these modalities, this molecular probe offers a versatile platform for comprehensive AS evaluation, which also holds great potential for guiding subsequent therapeutic interventions and offering in situ assessment of therapeutic outcomes. This probe, along with the therapeutic agent atorvastatin (AT), was encapsulated in responsive nanocarriers, which were subsequently coated with biomimetic macrophage membranes and functionalized with a phosphatidylserine-targeting peptide, thereby creating a hierarchical targeting platform. This engineered nanosystem exploits the adhesive bioligands on macrophage membranes for selective targeting of inflamed endothelium, while the targeting peptide further directs the system to foam cells within plaques. The AT released in foam cells promoted lipid efflux and reduced the expression of CD47 demonstrated that the targeted manipulation of foam cells phagocytosis significantly inhibited plaque progression, alleviated the inflammatory burden, and reinforced plaque stability in AS modeled mice. By combining hierarchical targeting, multidimensional visualization, and on-demand manipulation of pathological cells, this platform provides a novel paradigm for advanced management of AS.

A multimodal molecular probe was designed for simultaneous MRI, NIR-II fluorescence, and PA imaging, with optimized signal enhancement for each modality. The probe, along with AT, was encapsulated in responsive nanocarriers, coated with biomimetic macrophage membranes, and functionalized with a phosphatidylserine-targeting peptide. This engineered nanosystem exploits the adhesive bioligands on macrophage for selective targeting of inflamed endothelium, while the targeting peptide directs the system to foam cells within plaques. The ROS-responsive AT release within foam cells promotes lipid efflux and reduces CD47 expression, enhancing macrophage-mediated phagocytosis. Targeted manipulation of foam cell phagocytosis significantly inhibited plaque progression and stabilized plaques in AS mouse models.
Results
Design and property of molecular probes
As shown in Fig. 2a, a low-bandgap molecule was conjugated with Gd-DOTA to create the targeted molecular probe with multimodal imaging capabilities. The probe was designed by synthesizing a donor-acceptor (D-A) type chromophore, in which fluorene and benzobisthiadiazole (BBT) served as the donor and acceptor units, respectively. The electron-rich, highly fluorescent fluorene groups enhance the probe’s brightness, while the methoxy-substituted thiophene ring plays a dual role: facilitating electron transfer and extending the conjugation length. Furthermore, the steric hindrance introduced by the methoxy group induces a distorted molecular geometry, which prevents aggregation-caused quenching and further improves the fluorescence brightness46. The incorporation of highly conjugated and electron-withdrawing BBT unit results in a long-wavelength-response NIR emitter (FTB), which is ideal for both fluorescence and PA imaging. To endow FTB with MRI properties, Gd-DOTA was conjugated to the carboxyl group of FTB via an amidation reaction. The large Gd-DOTA moiety is expected to avoid strong intermolecular interactions, such as π-π stacking, leading to brighter fluorescence emission. Moreover, the Gd-DOTA substitution could also provide some room for molecular motion in aggregate state, facilitating PA transformation47. Additionally, this conjugation effectively restricts the flipping rate of the gadolinium ion compared to small gadolinium molecules, which in turn enhances the relaxation time and longitudinal relaxivity, resulting in an amplified MRI signal48. Detailed syntheses and characterizations of FTBGd are provided in the Supporting Information (Supplementary Figs. 1–16).

a Synthesis and chemical structure of FTB-Gd. b Absorption and PL spectra of FTBGd in THF. c Plots of PL peak intensity versus water fraction in THF/water mixture, where I0 and I represent the PL intensity in pure THF and THF/water mixtures with various water fractions. d PL spectra of FTB and FTBGd in THF/water mixture with 95% water fraction. e The r1 relaxivities and T1-weighted MR images of FTBGd. f Particle size distribution and morphology (insert TEM) of FA-P@MC. Scale bar: 100 nm. g The average sizes and zeta potentials of FA-P, FA-P@M and FA-P@MC (n = 3 independent experiments). h Western blots of FA-P, macrophage membrane and FA-P@MC. i Excitation-emission mapping of FA-P@MC. j In vitro PA spectra of FA-P@MC. k Corresponding PA intensity of various concentrations of FA-P@MC at 790 nm. Inset shows the PA intensity of FA-P@MC under PA laser excitation (n = 3 independent experiments). l Representative T1-weighted MR images of (i) FA-P@MC (FTB), (ii) Gd-DOTA and (iii) FA-P@MC (FTBGd) and quantitative analysis of MR intensity in the right picture (n = 3 independent experiments). m Size change of FA-P@MC exposed to H2O2 (1.0 mM) for 12 h. Scale bar: 250 nm. All data are presented as mean ± SD. For b–f, h–j, m experiment was repeated three times independently with similar results. For l statistical significance was determined using one-way ANOVA for multiple comparisons. Source data are provided as a Source Data file.
The absorption and photoluminescence (PL) spectra of FTBGd in THF revealed maximal absorption and emission at 776 nm and 984 nm, respectively (Fig. 2b), indicating great potential for NIR-II fluorescence and PA imaging. To investigate the emission characteristics in aggregate states, FL spectra of FTB and FTBGd were recorded in THF/water mixtures with varying water fractions (fw). As shown in Fig. 2c and Supplementary Fig. 17, FTB exhibited a 5.2-fold enhancement in PL intensity at high fw (95%), while FTBGd demonstrated an even greater 8.6-fold amplification under the same conditions, highlighting the favorable aggregation-induced emission (AIE) feature of FTBGd. Unlike conventional fluorophores that exhibit aggregation-caused quenching effect, AIE-active molecules are non-emissive or weakly emissive in solution but become highly fluorescent upon aggregation. Furthermore, a direct comparison of their PL intensities in the aggregate state (fw = 95%) revealed that FTBGd emitted fluorescence over five times stronger than FTB (Fig. 2d). This remarkable enhancement can be attributed to the anti-quenching effect of Gd-DOTA block in FTBGd, which effectively mitigates fluorescence quenching. Additionally, the PA spectrum of FTBGd was further examined, which exhibited a strong correlation with its absorption profile and exhibited a maximum PA signal around 780 nm (Supplementary Fig. 18). To assess the MRI property, we measured the MR signal intensity of both FTBGd, FTB, and Gd-DOTA at an equal concentration (0.05 mM). As shown in Supplementary Fig. 19, FTBGd exhibited a bright MR signal, whereas FTB, lacking Gd-DOTA substitution, produced negligible MR signals, confirming that the MR contrast originates from Gd-DOTA moiety. Furthermore, at the same Gd content, FTBGd displayed significantly enhanced MR signal intensity compared to Gd-DOTA alone. This improvement can be attributed to the larger molecular size of Gd-DOTA conjugate, which not only restricts gadolinium’s molecular tumbling rate but also enhances its ability to amplify MR contrast through increased longitudinal relaxivity49. Additionally, the MR signal intensity of FTBGd was found to increase accordingly with probe concentration (Fig. 2e). The stability of FTBGd as a multimodal imaging probe was further evaluated by exposing its solution to common cellular and in vivo interferents, including hydrogen peroxide (H2O2), superoxide (O2•−), cysteine (Cys), glutathione (GSH), and various metal ions. Notably, FTBGd maintained stable FL and MR signals in the presence of these interferents (Supplementary Fig. 20), demonstrating its resistance to external influences.
Fabrication and characterization of nanoagents
To develop a biocompatible and multifunctional platform, FTBGd and therapeutic drug AT were co-encapsulated within a ROS-responsive polymer matrix, yielding the water-soluble nanoprobe FA-P. AT, a widely used HMG-CoA reductase inhibitor, has been employed for CVD management. Recent studies indicate that AT can also exert local effects within vascular walls and immune cells, including modulation of cholesterol efflux and anti-inflammatory activity50,51. Based on these properties, AT was incorporated in this work to locally modulate lipid metabolism and foam cell formation. Foam cells typically exhibit elevated ROS levels due to their uptake of oxLDL and impaired mitochondrial function. Thus, we utilized poly (1,4-phenyleneacetone dimethylene thioketal) (PPADT), a ROS-responsive polymer in which thioketal (TK) bonds undergo cleavage and oxidation to sulfone in the presence of ROS, to serve as both a nanocarrier for ROS neutralization and a controlled drug-release system within ROS-enriched foam cells52. The synthesis and characterization of PPADT are detailed in Supplementary Fig. 21. FA-P, co-loaded with FTBGd and AT, were formulated via the nanoprecipitation method. As confirmed by dynamic light scattering (DLS) and transmission electron microscopy (TEM) analyses, FA-P exhibited a spherical morphology with an average hydrodynamic diameter of 138 nm (Supplementary Fig. 22a). To enhance the targeting capability, the nanoprobes were further cloaked with macrophage cell membranes and modified with phosphatidylserine (PS)-targeting peptides (CLIKKPF), resulting in the formation of FA-P@M and FA-P@MC, respectively53,54. The biomimetic macrophage membranes, equipped with natural adhesion molecules such as α4β1 integrins, facilitate interactions with activated endothelial cells and inflamed atherosclerotic plaques55,56. Meanwhile, the PS-targeting peptide promotes the subsequent targeting to the exposed PS on foam cells within the plaque. TEM and DLS measurements revealed that FA-P@M exhibited a spherical morphology with an average diameter of approximately 156 nm (Supplementary Fig. 22b), with a thin membrane layer approximately 10 nm on the surface. Similarly, FA-P@MC was coated with a membrane layer, and the further incorporation of PS-targeting peptides resulted in a slight increase in particle size to 163 nm (Fig. 2f). Furthermore, the zeta potential of FA-P@M decreased to −13.1 mV and then increased to −5.8 mV for FA-P@MC (Fig. 2g), which could be attributed to the introduction of negatively charged cell membranes and positively charged PS-targeting peptides57. The peptide coating density on FA-P@MC was estimated, and the NPs synthesized from different batches exhibited good reproducibility (Supplementary Fig. 23). Additionally, Western blotting analysis confirmed the presence of membrane proteins inherited from the macrophage in both the macrophage cell membrane and FA-P@MC (Fig. 2h). The colloidal stability of FA-P@MC was further assessed, and the size remained largely unchanged over 3 days in a physiologically relevant medium containing 10% fetal bovine serum (FBS) (Supplementary Fig. 24).
We next investigated the photophysical properties of FA-P@MC. The maximum absorption of FA-P@MC was observed at 790 nm, with emission spanning from 850 to 1250 nm within the NIR-II region (Supplementary Fig. 25), aligning well with the spectra of FTBGd probe. Excitation-emission mapping also revealed that FA-P@MC could be efficiently excited by NIR light, with strong fluorescence emission in the NIR-II spectral region above 1000 nm (Fig. 2i). Their potential for PA imaging was further examined, and the nanoprobe exhibited maximal PA signal at approximately 790 nm (Fig. 2j), corresponding to its absorption spectrum. Notably, the PA intensity displayed a linear relationship with the concentration of FA-P@MC, highlighting their potential for quantitative analysis (Fig. 2k). The PA signal demonstrated excellent stability, with minimal change in the intensity under continuous 790 nm laser excitation (inset of Fig. 2k). We also compared the photostability of FA-P@MC with the FDA-approved contrast agent, indocyanine green (ICG). The results showed that FA-P@MC exhibited remarkable stability and resistance to photobleaching, while the absorption intensity of ICG significantly decreased with light exposure (Supplementary Fig. 26). Subsequently, MRI analysis demonstrated that FA-P@MC generated strong MR signals, with the intensity nearly 3 times greater than the clinically used Gd-DOTA (Fig. 2l). These findings further validated that the rational design and integration of Gd-DOTA with the probe not only preserved the individual imaging signals but also enhanced their performance, leading to the development of a robust multimodal contrast agent.
Next, we investigated the ROS-triggered release process. As shown in Fig. 2m, in the presence of H2O2, DLS and TEM measurements revealed that FA-P@MC underwent substantial breakdown, resulting in both small and large aggregates. Furthermore, upon the H2O2-induced degradation, the MR signal of FA-P@MC increased by approximately 1.5-fold (Supplementary Fig. 27), whereas the non-responsive control nanoplatform, FA@MC (prepared without the ROS-responsive polymer), exhibited minimal changes in signal intensity in the presence of ROS. The observed increase in MR signal intensity following FA-P@MC decomposition was likely attributed to enhanced interaction between water and the paramagnetic Gd-DOTA center, thereby shortening the water molecule retention time and improving T1 relaxation. Additionally, the release of AT from FA-P@MC was quantified using high-performance liquid chromatography (HPLC). Based on previous reports58,59, FA-P@MC were incubated with different concentrations of H2O2 to mimic the oxidative microenvironment of atherosclerotic lesions. First, in the presence of H2O2, AT release increased over time, showing a rapid release compared to the untreated control (Supplementary Fig. 28). Under identical H2O2 conditions, FA-P@MC displayed slightly lower drug release than FA-P lacking membrane coating (Supplementary Fig. 29), indicating that the membrane coating only modestly influenced the H2O2-responsive release. We also included a physiological H2O2 concentration (2.5 µM) as a control condition. At this level, cumulative release from both FA-P and FA-P@MC remained below 10% over 36 h (Supplementary Fig. 30b). This result indicated the drug release was limited under basal physiological conditions but markedly enhanced in high H2O2 environments. Furthermore, pre-treatment with catalase to decompose H2O2 (1.0 mM) effectively inhibited drug release from both FA-P and FA-P@MC, providing evidence that the release process is H2O2-dependent (Supplementary Fig. 30c). Beyond H2O2, we further examined the responsiveness of FA-P@MC to other common ROS in atherosclerotic plaques, including hydroxyl radicals (•OH) and superoxide anions (O2•−). As shown in Supplementary Fig. 31, both ROS effectively triggered the AT release from FA-P@MC. These findings demonstrate that FA-P@MC are responsive to multiple ROS species, confirming their ROS-sensitive degradation and release properties.
In vitro cell investigation
The safety profile of nanoprobe is paramount for their application in bioimaging. In this context, we first assessed the biocompatibility of FA-P@MC prior to their use in in vivo imaging studies. Cytotoxicity assays were initially conducted using HUVECs and NIH 3T3 cells (Supplementary Fig. 32a). No cytotoxicity was observed in either cell line, even at a concentration of 100 µM, thereby confirming their low toxicity. Furthermore, to investigate the potential effects of FA-P@MC on cellular membrane integrity, we performed a lactate dehydrogenase (LDH) assay, a widely accepted indicator of plasma membrane damage. The absence of substantial LDH release across all tested concentrations (ranging from 10 to 100 µM) in any of the cell lines further confirmed the cytocompatibility of FA-P@MC (Supplementary Fig. 32b).
We then focused on assessing the targeting efficacy of FA-P@MC towards foam cells, which is a key component of atherosclerotic plaques. Foam cells form when macrophages uptake lipid material, and during this process, PS externalization becomes a distinctive feature of their pathophysiology, rendering PS an ideal target for selective binding. We then evaluated the targeting capability of the macrophage membrane-coated, PS-binding peptide-modified nanoprobe toward foam cells. Following incubation with various formulations, foam cells were visualized using confocal laser scanning microscopy (CLSM). The results clearly demonstrated that foam cells internalized substantially more FA-P@M compared to FA-P that lacked macrophage membrane coating after 4 h of incubation at 37 °C (Fig. 3a). Furthermore, the incorporation of PS-targeting peptide in FA-P@MC resulted in a 4.1-fold increase in cellular uptake compared to FA-P and a 1.6-fold increase compared to FA-P@M (Fig. 3b). Additionally, the FA-P@MC prepared from different batches showed comparable cellular internalization, indicating consistent foam cell–targeting capability across batches (Supplementary Fig. 33). These results suggested the potential of engineered macrophage membrane-coated delivery systems to enhance targeting.

a CLSM images and b quantitative analyses of foam cells upon incubation with FA-P, FA-P@M or FA-P@MC for 4 h (n = 5 independent experiments). The cell nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI; blue fluorescence). Scale bar: 20 μm. c Schematic illustration of in vitro transwell migration assays to evaluate NP transmigration across inflamed HUVECs and subsequent uptake by foam cells. Created in BioRender. Li, W. (2026) https://BioRender.com/xdjn9re. d Representative CLSM images and e quantitative analyses of NP fluorescence signals in foam cells (n = 3 independent experiments). Scale bar: 20 μm. f Representative flow cytometry analysis of NP fluorescence in foam cells in different groups. g CLSM images and h quantified results of DCF fluorescence intensity in foam cells after the treatment of different formulations (n = 5 independent experiments). Scale bar: 100 μm. i Flow cytometry analysis of DCF fluorescence intensity in different groups. In g–i the ROS levels were evaluated using ROS probe, DCFH-DA. j Optical microscopy images of foam cells in different groups, stained with Oil Red O (ORO). Scale bar: 50 μm. k Quantitative analysis of the intracellular ORO by measuring the optical density at 492 nm across various experimental groups (n = 5 independent experiments). All data are expressed as mean ± SD. For f, i experiment was repeated three times independently with similar results. For b, e, h, k statistical significance was determined using one-way ANOVA for multiple comparisons. Source data are provided as a Source Data file.
We subsequently evaluated the transmigration of the nanoparticles (NPs) across inflamed HUVECs and their uptake by foam cells through in vitro transwell migration assays. The transwell model comprised a monolayer of HUVECs in the upper chamber and foam cells in the lower chamber (Fig. 3c). HUVECs in the upper chamber were subjected to the following treatments: (1) LPS + FA-P, (2) LPS + FA-P@M, (3) LPS + FA-P@MC, and (4) FA-P@MC. The internalization of NPs by foam cells in the lower chamber was subsequently assessed using CLSM and flow cytometry. As shown in Fig. 3d, the NP signal in the foam cells of the FA-P-treated group was faint, suggesting limited passive transendothelial migration of the unmodified NPs. In contrast, FA-P@M exhibited a marked increase in NP signal in the basolateral chamber, indicative of enhanced endothelial permeability and foam cell uptake following macrophage membrane coating. The enhancement in NP transmigration is likely attributed to the presence of cell surface proteins, such as integrins, on the macrophage membrane, which could facilitate the efficient passage of encapsulated NPs across the inflamed endothelial barrier. Remarkably, FA-P@MC exhibited markedly superior endothelial crossing and foam cell targeting capabilities, as evidenced by the strongest NP fluorescence observed in foam cells. As depicted in Fig. 3e, quantitative analysis revealed that the quantities of NPs migrating across the endothelial layer into the basolateral foam cells were 5.24- and 1.88- times higher compared to the FA-P and FA-P@M groups, respectively. Moreover, in the absence of LPS pre-treatment to induce inflammatory conditions, there was a significant reduction in the permeability of FA-P@MC, highlighting the enhanced endothelial traversal and foam cell uptake under inflamed conditions. Flow cytometry analysis further corroborated the CLSM findings (Fig. 3f and Supplementary Fig. 34), confirming that the macrophage membrane coating and PS-binding peptide modification significantly facilitated the transport of NPs across the inflamed endothelial barrier and their subsequent internalization by foam cells.
In vitro anti-inflammatory and immunomodulatory effects
AS is a chronic inflammatory disease, with foam cell formation playing a pivotal role in its progression11. Foam cells not only contribute to lipid deposition but also act as key contributors to inflammation in atherosclerotic lesions. FA-P@MC were formulated by co-assembling a ROS-responsive polymer with AT, which have the potential to scavenge ROS, reduce lipid accumulation, and mitigate inflammation. To evaluate the therapeutic efficacy of FA-P@MC, an in vitro model of foam cell was established using oxLDL-treated macrophages. After 24 h of incubation with oxLDL (50 µg/mL), a pronounced oxidative stress response was observed in foam cells, as detected by the ROS fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA) (Fig. 3g, h). In contrast, FA-P@M and FA-P@MC treatment led to a reduction in DCF fluorescence, with the most pronounced effect observed in the FA-P@MC-treated cells. Flow cytometry analysis further demonstrated that FA-P@MC were more effective than FA-P@M in alleviating oxidative stress (Fig. 3i and Supplementary Fig. 35), potentially due to their enhanced internalization by foam cells.
Subsequently, we evaluated the ability of FA-P@MC to reduce the oxLDL uptake and inhibit foam cell formation. RAW264.7 macrophages exposed to 50 μg/mL oxLDL for 24 h exhibited significant intracellular lipid droplet accumulation and the formation of foam cells, as verified by Oil Red O (ORO) staining (Fig. 3j, k). Importantly, FA-P@MC treatment markedly suppressed foam cell formation in comparison to other groups. Using the DiI-labeled oxLDL (DiI-oxLDL), fluorescence microscopic analysis further confirmed that FA-P@MC markedly inhibited oxLDL uptake by RAW264.7 cells (Supplementary Fig. 36a, b). Mechanistic studies revealed that FA-P@MC treatment downregulated the expression of scavenger receptors CD36 and LOX-1, which were responsible for oxLDL internalization (Supplementary Fig. 36c–f). To assess the cholesterol efflux, we measured cholesterol levels in the culture supernatant of treated foam cells. We found that the cholesterol content in the supernatant of FA-P@MC group was significantly increased (Supplementary Fig. 37a). Immunofluorescence staining further showed the upregulation of cholesterol transporters SR-BI and ABCA1 after FA-P@MC treatment (Supplementary Fig. 37b–e). We also performed functional cholesterol efflux assays in foam cells using BODIPY-labeled cholesterol with ApoA-I (ABCA1-dependent) or HDL (SR-BI/ABCG1-associated) as acceptors. FA-P@MC enhanced cholesterol efflux to both acceptors compared with other nanoformulations, indicating engagement of these transporters in promoting efflux. Pharmacological inhibition of ABCA1 markedly reduced FA-P@MC-enhanced efflux to ApoA-I, whereas SR-BI inhibition reduced efflux to HDL (Supplementary Fig. 38), consistent with their contribution under our experimental conditions. Parallel studies in foam cells induced from bone marrow–derived macrophages (BMDMs) showed similar FA-P@MC–enhanced efflux, which was attenuated by ABCA1 or SR-BI inhibition (Supplementary Fig. 39), supporting involvement of these pathways without suggesting exclusivity.
Collectively, these results indicated that FA-P@MC not only limited lipid uptake but also enhanced cholesterol efflux via upregulation of efflux transporters, thereby suppressing foam cell formation. We then proceeded to evaluate whether FA-P@MC could also mitigate cellular inflammation in foam cells. The PBS-treated foam cells displayed elevated nitric oxide (NO) levels, indicative of an inflammatory response (Supplementary Fig. 40). Compared to free AT alone, FA-P showed improved efficacy in reducing cellular NO production, likely due to the enhanced intracellular delivery of AT via the nanocarriers. Furthermore, treatment with FA-P@M or FA-P@MC significantly reduced NO production in foam cells, with FA-P@MC exhibiting the most potent effect in suppressing NO generation. These results highlighted that FA-P@MC not only inhibited foam cell formation but also alleviated the inflammatory response within foam cells.
Efferocytosis is the process by which macrophages engulf and eliminate dead or dying cells, including foam cells, which is essential for preventing plaque necrosis and instability60,61. However, in atherosclerotic lesions, the efficiency of efferocytosis diminishes over time due to the chronic inflammatory environment and cellular dysfunction. Notably, the upregulation of CD47, a “don’t eat me” signal, on the surface of foam cells inhibits their clearance by macrophages. As a result, foam cells can persist in the plaque, contributing to the formation of a necrotic core and heightening the plaque’s vulnerability to rupture. The oxidative and inflammatory milieu in atherosclerotic plaques promote the upregulation of CD47 on foam cells, thus we next sought to investigate whether the ROS-scavenging, AT-loaded, and foam cell-targeting nanoformulations could enhance efferocytosis by modulating CD47 expression. First, CD47 expression in foam cells across different treatment groups was evaluated by Western blotting and confocal microscopy. As shown in Fig. 4a–c, foam cells exhibited a high level of CD47 expression, which was consistent with previous reports39,40,41. However, following treatment with nanoformulations, particularly FA-P@MC, the expression of CD47 on foam cells was significantly reduced. To study the ROS-dependent regulation of CD47, we employed specific ROS scavengers, including catalase and mannitol, to selectively eliminate H2O2 and •OH, respectively. The effective removal of each ROS was first verified using corresponding fluorescent probes (Supplementary Fig. 41a). We found that the presence of these scavengers partially attenuated FA-P@MC–induced CD47 downregulation, likely due to the reduced AT release. In line with this, the ROS-nonresponsive control NPs (FA@MC) exhibited weak effect on CD47 expression (Supplementary Fig. 41b, c). Collectively, these findings suggested the ROS-responsive CD47 downregulation effect of FA-P@MC. Then we explored whether the reduction in CD47 expression on foam cells could potentially enhance their phagocytosis by macrophages. As illustrated in Fig. 4d, bone marrow-derived macrophages (BMDMs) were harvested and applied as phagocytic cells, while oxLDL-induced RAW264.7 cells served as foam cells. CLSM images revealed that significantly more foam cells treated with FA-P@MC were engulfed by BMDMs compared to those treated with PBS (Fig. 4e). Flow cytometry analysis further confirmed that the efferocytosis rate of foam cells in the FA-P@MC group (~68%) was substantially higher than in the PBS, Free AT, A-P, FA-P, and FA-P@M groups by 8.92, 2.97, 2.02, 2.16, and 1.26 times, respectively (Fig. 4f, g and Supplementary Fig. 42). Additionally, for the non-ROS-responsive control FA@MC, weak CD47 downregulation and limited phagocytosis were observed (Supplementary Fig. 43). These results suggest that ROS-enhanced drug release contribute to the subsequent CD47 downregulation and phagocytosis increase. These findings indicate that FA-P@MC promoted the phagocytosis of foam cells by macrophages, suggesting their potential to promote plaque regression and stabilization.

a Immunofluorescence images of CD47 in foam cells from various treatment groups. Scale bar: 50 μm. b Quantitative analysis of the CD47 expression under different treatment conditions (n = 3 independent experiments). c Western blot analysis of CD47 protein expression in foam cells treated with various formulations. d Schematic illustration of in vitro assessment of the ability of various nanoformulations in promoting foam cell efferocytosis. Created in BioRender. Li, W. (2026) https://BioRender.com/8ci0v9l. e Representative confocal microscopy images showing the colocalization of BMDMs and foam cells treated with different formulations. Scale bar: 50 μm. f The phagocytosis of foam cells by BMDMs determined by flow cytometry. g Quantification of foam cell phagocytosis by BMDMs according to flow cytometry analysis (n = 3 independent experiments). All data are expressed as mean ± SD. For c, e experiment was repeated three times independently with similar results. For b, g statistical significance was determined using one-way ANOVA for multiple comparisons. Source data are provided as a Source Data file.
In vivo biosafety and pharmacokinetics of FA-P@MC
Prior to conducting in vivo bioimaging studies, the biosafety profile and pharmacokinetics of FA-P@MC were evaluated. Healthy C57BL/6 mice were administered PBS or FA-P@MC (10 mg kg−1) by intravenous injection twice weekly for 2 weeks. Histological analysis of major organs, including the heart, liver, spleen, lung, and kidneys (Supplementary Fig. 44), revealed no pathological abnormalities or inflammatory lesions in the FA-P@MC-treated mice. Furthermore, comprehensive blood panel tests indicated that the hematological parameters of the mice treated with FA-P@MC remained within normal physiological ranges (Supplementary Fig. 45). Additionally, liver biomarkers, such as alanine transaminase (ALT) and aspartate aminotransferase (AST), as well as kidney function markers, including blood urea nitrogen (BUN) and creatinine, showed no significant differences between the PBS group and FA-P@MC-treated group (Supplementary Fig. 46). Moreover, after 7 weeks of continuous administration, no obvious abnormalities were detected in the major organs, routine blood parameters, or hepatorenal function indicators (Supplementary Fig. 47), indicating their favorable long-term biocompatibility. Additionally, no significant hemolysis was observed, even at a concentration of 100 μM (Supplementary Fig. 48). Taken together, these results strongly highlighted the favorable biocompatibility of FA-P@MC, positioning it as a promising candidate for in vivo AS imaging and therapeutic applications.
Subsequently, the in vivo circulation profiles of FA-P@MC were investigated. The NPs were intravenously injected into mice, and blood samples were collected at designated time points post-injection to measure the fluorescence signal of the NPs in circulation. As shown in Supplementary Fig. 49, free AT was rapidly cleared from the bloodstream, exhibiting a half-life of ~15 min. In contrast, FA-P@MC demonstrated a significantly extended circulation time, comparable to that of FA-P@M. These findings indicated that the incorporation of foam cell-targeting peptides on the macrophage membrane surface did not substantially alter the circulation time of the NPs. FA-P@MC exhibited prolonged circulation profiles compared to free drug, highlighting their enhanced propensity to accumulate at atherosclerotic sites and sustain therapeutic efficacy. The biodistribution of FA-P@MC was systematically assessed in major organs (liver, spleen, kidneys, heart, and lung) at multiple time points after administration. The results showed that the nanoparticles were cleared from these organs by day 7 (Supplementary Figs. 50, 51). Analysis of fecal and urinary signals also suggested that FA-P@MC was primarily cleared via the biliary pathway within 1 week (Supplementary Fig. 52).
Correlation between multimodal imaging signals and plaque burden severity
Prior to employing the multimodal nanoprobe for in vivo AS diagnosis, we first assessed the ability of FA-P@MC to detect plaques and differentiate plaque severities (Fig. 5a). This preliminary evaluation aimed to determine whether the multimodal nanoprobe could serve as a reliable indicator for the assessment of plaque progression. The atherosclerotic model used here was induced by feeding apolipoprotein E-deficient (ApoE−/−) mice a high-fat diet for 12 weeks, followed by intravenous injection of FA-P@MC nanoprobe. At 12 h post-injection, the aorta was harvested and first subjected to NIR-II fluorescence imaging using a custom-built NIR-II imaging system. As shown in Fig. 5b, distinct signals were observed across different arterial regions, and the aorta was segmented into regions A through D based on NIR-II fluorescence intensity. Following this, PA and MRI imaging were performed on each segment using dedicated PA and MRI systems. Consistent with the NIR-II fluorescence findings, both PA and MRI signal intensities showed a progressive increase from segment A to segment D (Fig. 5c, d). These results confirmed the consistency of signal intensity across the three imaging modalities.

a Schematic illustration of ex vivo imaging and histological analysis of the aorta following intravenous administration of the probe in AS mice, validating its capability for plaque detection and progression monitoring. Created in BioRender. Li, W. (2026) https://BioRender.com/fjkdy86. b NIR-II FL image of the aorta, with an excitation wavelength of 808 nm and emission collected using a 1000 nm long-pass filter. The aortic sample was sectioned into four segments (A to D) based on the NIR-II fluorescence signal intensity. c Corresponding PA images at 790 nm excitation and T1-weighted MR images for different aortic segments. d Quantification of T1-weighted MR signal intensity (red line) and PA intensity (blue bars) for the corresponding aortic segments. e Histological analysis of different aortic segments using H&E and ORO staining, with plaques outlined by solid black curves. Scale bars: 200 μm. f Quantification of the mean plaque area and necrotic core area based on H&E staining (n = 3 independent experiments). g Measurement of mean fibrous cap thickness from H&E images (n = 3 independent experiments). h Confocal fluorescent images of different segments stained with anti-F4/80 antibody and DCFH-DA, indicating macrophage infiltration (orange fluorescence), and ROS levels (green fluorescence), respectively. Plaques are marked by yellow dotted curves. Scale bars: 100 μm. i Comparative analysis of NIR-II fluorescence signal intensity, PA signal, MR signal, necrotic area, ROS levels, and macrophage infiltration levels across different aortic segments (n = 3 independent experiments). All data are expressed as mean ± SD. For b–e, h, experiment was repeated three times independently with similar results. Source data are provided as a Source Data file.
To validate the correlation between imaging signal intensities and plaque severity, ORO and hematoxylin and eosin (H&E) staining were performed on these aortic segments. ORO staining, widely used to assess lipid deposition in plaques, revealed that segment A showed no signs of plaque formation, while the extent of plaque areas gradually increased in segments B, C, and D (Fig. 5e), reflecting a progression in plaque severity. Key pathological parameters of the plaques in segments A through D, including the necrotic area, the proportion of the necrotic core relative to the total plaque area, and fibrous cap thickness, were quantified according to the H&E-stained images. As anticipated, the necrotic area increased progressively from segment B to segment D (Fig. 5f), while fibrous cap thickness diminished (Fig. 5g), indicating an escalation in plaque instability from segments B to D. Macrophage infiltration and ROS levels, both key indicators of plaque vulnerability, were evaluated via immunofluorescence staining with an anti-F4/80 antibody and DCFH-DA, respectively. Fluorescence signals for both anti-F4/80 and ROS exhibited a progressive increase from segment B to segment D, indicating enhanced macrophage infiltration and ROS accumulation as plaque severity advanced (Fig. 5h). Notably, a strong correlation was observed between the NIR-II fluorescence, PA, and MRI signal intensities and the severity of the plaques (Fig. 5i). Stronger signal intensities were detected in the regions with more advanced and vulnerable plaques, highlighting the potential of the multimodal nanoprobe to accurately delineate plaque lesions and act as a non-invasive method for assessing plaque severity. The ability of the multimodal nanoprobe to detect plaque progression is likely attributed to its targeting affinity for foam cells, a hallmark of plaque lesions, coupled with its high sensitivity and robust signal strength under each imaging modalities.
In vivo, in situ multimodal imaging of AS
We proceeded to investigate the in vivo real-time diagnostic capabilities of FA-P@MC. Atherosclerotic models were induced in ApoE−/− mice by administering a high-fat diet, after which the mice were intravenously administrated with equal doses of FA-P@C, FA-P@M, or FA-P@MC to compare the imaging performance across different nanoformulations. NIR-II fluorescence imaging was performed at various time points following the administration of the nanoprobes. As shown in Fig. 6a, b and Supplementary Fig. 53, in mice treated with FA-P@C (FA-P modified with PS-targeted peptides), weak fluorescence was initially detected in the carotid artery at 3 h post-injection, which peaked at 6 h and gradually decreased as the probes were metabolized. The macrophage membrane-camouflaged biomimetic FA-P@M demonstrated enhanced plaque accumulation, with peak enrichment occurring at 12 h. During the progression of AS, macrophages actively migrate toward atheromatous plaques via inflammatory chemotaxis. The macrophage membrane-coated NPs, inheriting membrane proteins from macrophages, thereby exhibited a natural binding affinity for inflamed plaques. FA-P@MC, which further incorporated the CLIKKPF peptide to bind PS expressed on activated and senescent foam cells, resulted in significantly enhanced plaque accumulation—nearly double that observed with the FA-P@M group. We also conducted a competitive blocking experiment in which free CLIKKPF peptide was injected prior to FA-P@MC administration. Pre-injection of the peptide reduced plaque accumulation of FA-P@MC to the levels comparable to non-targeted FA-P@M (Supplementary Fig. 54). This result suggests that PS-binding peptide interactions promote plaque targeting. Moreover, the fluorescence images acquired at 12 h in FA-P@MC group displayed the highest signal-to-noise ratio (SNR) (more than 100) in comparison with FA-P@C (~23.3) and FA-P@M (~56.8) group (Fig. 6c). Ex vivo fluorescence imaging further demonstrated that FA-P@MC accumulated substantially more in the aortic root and thoracic aorta compared to FA-P@C and FA-P@M (Fig. 6d, e), validating the in vivo results. Major organs, including the heart, liver, lungs, spleen, and kidneys, from mice injected with either FA-P@M or FA-P@MC were harvested to assess the biodistribution of the nanoprobes. No significant differences in organ distribution were observed between the two groups (Fig. 6f, g).

a Representative NIR-II fluorescence images of carotid plaques at various time points following i.v. injection of PBS, FA-P@C, FA-P@M or FA-P@MC. Scale bars: 1 cm. b Quantitative analyses of NIR-II fluorescence intensity in carotid plaques across different treatment groups (n = 3 mice). c The corresponding SNR of fluorescence signals in different treatment groups at 6 h and 12 h post i.v. injection (n = 3 mice). d Fluorescence images of isolated carotid samples and e) corresponding quantification of NIR-II fluorescence intensity in isolated blood vessels at 12 h after treatment with PBS, FA-P@C, FA-P@M or FA-P@MC (n = 3 mice). Scale bar: 5 mm. f Ex vivo fluorescence images of major organs (heart, liver, spleen, lung, and kidneys) collected from atherosclerotic mice after injection of FA-P@M or FA-P@MC. Scale bar: 5 mm. g Quantitative analysis of FL intensity in these organs (n = 3 mice). In a, d, and f, NIR-II fluorescent images were acquired with an excitation wavelength of 808 nm and emission collected using a 1000 nm long-pass filter. All data are expressed as mean ± SD. For c, e statistical significance was determined using one-way ANOVA for multiple comparisons. Source data are provided as a Source Data file.
To visualize the biodistribution of AT, we labeled AT with Cy5, and prepared FA-P@M and FA-P@MC loaded with Cy5-AT. Twelve hours after intravenous injection, the aorta was collected for ex vivo fluorescence imaging. In the AT alone group, only weak fluorescence was detected in the aorta, consistent with the rapid clearance of small-molecule AT. Importantly, FA-P@MC exhibited substantially higher accumulation in aortic plaques compared with the non-targeted FA-P@M and free AT groups (Supplementary Fig. 55). To further support these observations, AT content in different tissues was quantified via homogenization and extraction. Consistently, plaque tissues from the FA-P@MC group contained approximately 12.7-fold higher drug levels than free AT and about 2-fold higher than FA-P@M (Supplementary Fig. 56). No significant differences in AT signals were detected in other major organs, including the liver, spleen, or kidneys, between the FA-P@MC and FA-P@M groups. Additionally, confocal microscopy of atherosclerotic aortic root sections revealed enhanced foam cell targeting by FA-P@MC in vivo. As shown in Supplementary Figs. 57, 58, the proportions of Cy5⁺F4/80⁺BODIPY⁺ foam cells in plaques were increased in mice treated with FA-P@MC compared with other groups. Additionally, the Cy5-labeled FA-P@MC signals exhibited obvious co-localization with PS, CD68, and PLIN2, supporting their preferential association with PS-exposing foam cells (Supplementary Fig. 59). These observations highlighted the enhanced targeting efficiency of FA-P@MC for atherosclerotic plaques and foam cells, which is crucial for advancing both the diagnostic accuracy and therapeutic efficacy of the nanoprobe.
Given the remarkable in vitro PA imaging capabilities and plaque-targeting properties of FA-P@MC, the in vivo non-invasive PA diagnostic performance was further assessed. At 12 h after administering equivalent doses of FA-P@C, FA-P@M, or FA-P@MC, anatomical localization was assessed through two-dimensional grayscale ultrasound imaging of the carotid artery, captured in both transverse and sagittal sections views. Compared to the PBS group, the carotid artery in FA-P@C group exhibited a faint PA signal upon excitation at 790 nm, suggesting that foam cell-targeting alone was insufficient for effective plaque lesion targeting. The FA-P@M-treated mice exhibited stronger PA signals in the carotid plaques, attributed to the macrophage membrane biomimetics that enhanced plaque accumulation. Interestingly, plaques in the carotid arteries of mice treated with FA-P@MC were distinctly detected with intense PA signals (Fig. 7a, b), suggesting that the combination of macrophage membrane coating and foam cell-targeting peptide modification significantly boosted NPs enrichment in the plaques. Quantification of PA intensity revealed that plaques from FA-P@MC-injected mice exhibited PA signals approximately 4.9 times and 1.8 times stronger in the transverse section than those seen in the FA-P@C and FA-P@M groups, respectively, and 6.1 times and 2.1 times stronger in the sagittal section compared to the FA-P@C and FA-P@M groups (Fig. 7c, d). FA-P@MC enabled enhanced PA imaging of carotid plaques with strong contrast relative to normal tissue, showing the highest SNR compared to other groups (Fig. 7e).

Representative PA/US images of the carotid arteries in atherosclerotic mice, shown in a transverse and b sagittal section views, 12 h post-treatment with different formulations. The regions of interest (ROIs, indicated by the white square) highlight the right carotid arteries. Scale bars: 3 mm. Quantitative analysis of PA signal intensity in carotid plaques following various treatments in c the transverse section and d sagittal section (n = 3 mice). e The corresponding SNR of PA signals quantified according to the images in a and b (n = 3 mice). f MR images of the carotid arteries in atherosclerotic mice acquired before and 12 h after injection of Gd-DOTA, FA-P@M or FA-P@MC. g Quantitative analysis of MR signal intensity in carotid plaques after different treatments (n = 3 mice). All data are expressed as mean ± SD. For c, d, g statistical significance was determined using one-way ANOVA for multiple comparisons. Source data are provided as a Source Data file.
In addition to PA imaging, the nanoprobe with concurrent incorporation of Gd complex can be further employed for in vivo MRI, enabling comprehensive detection of AS. As a proof of concept, in vivo MRI of AS was performed before and at 12 h after the injection of Gd-DOTA, FA-P@M or FA-P@MC into atherosclerotic ApoE−/− mice, with a focus on the carotid region. As shown in Fig. 7f, a pronounced heterogeneous T1-MRI signal enhancement was observed in both the left and right common carotid artery walls at 12 h post-injection of FA-P@MC, compared to pre-injection images, indicating the presence of plaque formation in these regions. Furthermore, mice injected with FA-P@MC exhibited a significant increase in MRI signals, which was 2.1- and 3.2-fold higher than those treated with FA-P@M and Gd-DOTA (Fig. 7g). These results highlighted the potential of FA-P@MC for both PA and MRI-based plaque detection. The nanoprobe’s favorable multi-modal imaging performance was attributed to its superior AS-targeting capability, as well as the amplified signal intensity in each imaging modality. Considering the complementary strengths of each technique—such as high sensitivity, spatial resolution, and anatomical detail—the combination of NIR-II fluorescence, PA, and MRI imaging facilitates precise, non-invasive assessment of plaque progression, providing a comprehensive overview of plaque characteristics.
In vivo therapeutic efficacy of FA-P@MC in AS
Building on the encouraging results outlined above, we further conducted a thorough investigation into the in vivo therapeutic efficacy of FA-P@MC in treating AS. As detailed in the treatment protocol (Fig. 8a), ApoE−/− mice were initially fed a high-fat diet for 8 weeks to induce the development of atherosclerotic plaques at arterial bifurcations. Following this, the atherosclerotic mice were randomly assigned to six treatment groups, including PBS, free AT, A-P, FA-P, FA-P@M, and FA-P@MC. Each group received intravenous injections of the designated treatment twice a week while maintaining the high-fat diet for an additional 8 weeks. At the end of the treatment period, the entire aortas were excised, and the effectiveness of each treatment in reducing atherosclerotic plaque size was assessed using ORO staining of the aortic arch, which highlighted lipid accumulation (Fig. 8b). As shown in Fig. 8c, the PBS group exhibited the largest ORO-positive area (~38.5%), indicating significant plaque formation. Treatment with free AT resulted in a moderate reduction in plaque size, with the ORO-positive area decreasing to approximately 26.0%. Both A-P and FA-P groups showed slight therapeutic improvements over free AT, likely due to enhanced delivery of AT via the nanocarriers. Mice treated with FA-P@M, which utilized macrophage membrane biomimicry for targeted delivery to inflammatory plaques, displayed a significantly smaller plaque area (~12.4%) compared to those treated with A-P and FA-P. Notably, the FA-P@MC treatment led to a dramatic reduction in the ORO-positive plaque area to approximately 7.5%, exhibiting the smallest ORO-stained lesions in the aortas. The average plaque-to-total aortic area ratio in the FA-P@MC group was 5.1-, 3.5, 2.5-, 2.4-, and 1.6- times lower than those in the PBS, AT, A-P, FA-P, and FA-P@M groups, respectively. These results highlighted the potent anti-atherosclerotic efficacy of FA-P@MC. A consistent trend was observed in the ORO-stained frozen sections of aortic root, with FA-P@MC demonstrating the most pronounced anti-atherosclerotic effect (Supplementary Fig. 60). Additionally, FA-P@MC could effectively promote cholesterol efflux, lowering cholesterol levels in the plaques (Supplementary Fig. 61). Subsequently, we examined the composition of atherosclerotic plaques in different experimental groups through histochemical and immunohistochemical analyses. As shown in Fig. 8d, H&E staining of the aortic root plaques revealed that plaques in both the PBS and AT-treated groups predominantly consisted of acellular, lipid-rich necrotic cores. While treatment with A-P, FA-P, or FA-P@M reduced the size of the necrotic core, the overall inhibition was limited. In contrast, the FA-P@MC-treated group exhibited a significant reduction in both plaque size and necrotic core area, outperforming all other groups.

a Schematic of the experimental procedure for AS modeling, treatment administration, and outcome evaluation in ApoE−/− mice. Created in BioRender. Li, W. (2026) https://BioRender.com/w6tdj6p. b Representative images of ORO-stained aortas and c corresponding quantitative analyses following different treatments (n = 6 mice). Scale bars: 5 mm. d Representative H&E-stained images and e corresponding quantitative analyses of plaque area across different treatment groups (n = 6 mice). Scale bar: 500 μm. f Representative DHE-stained images and g corresponding quantitative analyses of plaque DHE fluorescence across different treatment groups (n = 6 mice). Scale bar: 500 μm. h Western blot analysis of CD47 protein expression in aortas from mice treated with various formulations. i Immunofluorescence staining images showing the CD47 expression and CD68-positive macrophages in aortic sinus sections across different groups. Scale bar: 25 μm. j Quantitative analyses showing the MFI of CD47 under different treatment conditions (n = 6 mice). All data are expressed as mean ± SD. For h experiment was repeated three times independently with similar results. For c, e, g, j statistical significance was determined using one-way ANOVA for multiple comparisons. Source data are provided as a Source Data file.
FA-P@MC inhibit CD47 expression and inflammatory burden, and modulate immune cell infiltration in plaques
We further explored the underlying mechanism by which FA-P@MC exerted their anti-atherosclerotic effects. The excessive production of ROS within plaques contributes to local tissue damage and perpetuates inflammation. To assess the ROS levels within the plaques, aortic root sections were stained with the ROS-sensitive fluorescent probe dihydroethidium (DHE) following treatment with various formulations (Fig. 8f). Compared to the intense DHE-positive staining observed in the aortic sections of PBS-treated mice, treatment with different nanoformulations resulted in notable reductions in ROS-positive areas. These findings aligned with the ROS-scavenging properties of the polymeric carriers, as demonstrated in our in vitro studies. Remarkably, the DHE staining in the aortic root sections of the FA-P@MC-treated group was significantly lower, showing a 4.6- to 1.9-fold reduction compared to other groups (Fig. 8g), emphasizing their potent ROS scavenging ability within plaques.
Next, we assessed the anti-inflammatory and immunomodulatory efficacy of FA-P@MC in atherosclerotic mice. Foam cells are lipid-laden macrophages that play a pivotal role in the formation and progression of atherosclerotic plaques. Foam cells contribute to plaque inflammation by secreting pro-inflammatory cytokines (such as TNF-α, IL-6 and IL-1β) and chemokines that recruit additional immune cells, thereby perpetuating a cycle of inflammation. As a statin drug, AT facilitates the removal of excess cholesterol from foam cells, thereby alleviating the lipid burden within these cells. Furthermore, our in vitro studies indicated that the combination of polymer carrier’s anti-ROS properties with AT’s anti-inflammatory effects could downregulate CD47 expression on foam cells. This, in turn, enhances the immune clearance of foam cells, reshaping the inflammatory and immune landscape within the plaque microenvironment. The expression of CD47 in plaque tissue across different treatment groups was then assessed. Western blot analysis confirmed that FA-P@MC notably decreased CD47 levels in plaque tissue (Fig. 8h). Additionally, the immunofluorescence images indicated that the plaques from the PBS-treated control group exhibited relatively high levels of the “don’t eat me” molecule, CD47, which allowed apoptotic foam cells to evade macrophage-mediated clearance (Fig. 8i, j). In contrast, FA-P@MC treatment resulted in a significant reduction in CD47 expression within the plaques. The enhanced foam cell modulation exhibited by FA-P@MC, in comparison to other nanoformulations, aligns with its improved affinity for atherosclerotic plaques and foam cell-targeting capacity, which together facilitate the targeted delivery of the ROS-scavenging polymer and AT into plaque foam cells for synergistic treatment.
Subsequently, the potential of FA-P@MC intervention to mitigate immune cell infiltration and inflammatory responses was investigated. Since the recruitment of monocytes and macrophages to the plaque is closely linked to plaque progression and disease severity, inhibiting their recruitment can help attenuate plaque development.62,63 Immunostaining staining for anti-CD68 antibodies demonstrated that FA-P@MC significantly reduced macrophage accumulation within plaques in the aortic arch, showing superior efficacy over other formulations (Supplementary Fig. 62). Similarly, immunohistochemical staining for monocytes using an anti-CD14 antibody revealed a notable decrease in monocyte presence within the plaques following FA-P@MC treatment. In particular, the CD14-positive staining in aortic root sections from the FA-P@MC group was approximately 1.5%, significantly lower than those observed in the PBS (11.7%), free TA (9.1%), A-P (6.4%), FA-P (6.1%), and FA-P@M (3.7%) groups (Supplementary Fig. 63). T lymphocytes, recruited by antigen-presenting cells, have been reported to play an important role in driving lesion inflammation64. During plaque progression, CD4+ and CD8+ T cells typically increase and release pro-inflammatory cytokines, whereas regulatory T cells (Tregs) remain relatively low, providing insufficient anti-inflammatory regulation. In the FA-P@MC–treated group, flow cytometry analysis revealed that the proportions of infiltrating CD8+ and CD4+ T cells within plaques were reduced by approximately 4.68- and 2.53-fold, respectively, compared with the model group (Supplementary Figs. 64, 65). Conversely, the proportion of CD25+Foxp3+ Treg cells was increased in the FA-P@MC group relative to other groups. Key pro-inflammatory cytokines, such as TNF-α, IL-6 and IL-1β, are known to be overexpressed by activated immune cells. To assess the impact of various treatments on these cytokines, their serum levels were quantified using ELISA. As shown in Supplementary Fig. 66, inflammatory marker levels were significantly high in the PBS group. The treatment with A-P, FA-P, or FA-P@M resulted in a modest reduction of these cytokine concentrations. Remarkably, FA-P@MC once again exhibited the most pronounced suppression of inflammatory cytokine expression compared to other groups. Collectively, these results demonstrated that FA-P@MC could effectively attenuate AS progression through a combination of targeted drug delivery, oxidative stress reduction, and immune-inflammatory modulation.
Therapeutic impact on enhancing plaque stability
AS is a leading cause of cardiovascular disease, with plaque stability serving as a critical determinant of patient prognosis65. The therapeutic efficacy of various formulations was further assessed by evaluating plaque vulnerability. The expression of metalloproteinase-9 (MMP-9) is closely associated with plaque instability65. Unlike the pronounced positive MMP-9 staining observed in the aortic root sections from the PBS group, administration with FA-P@MC resulted in a significant reduction66 in MMP-9 expression (Fig. 9a). Specifically, MMP-9 staining in the FA-P@MC group decreased by 2.0- to 5.9-fold compared to other treatment groups (Fig. 9b). Furthermore, Masson’s trichrome staining revealed a considerable increase in collagen content around the plaques and thicker fibrous caps in the FA-P@MC-treated group, thereby strengthening the structural integrity of plaques (Fig. 9c, d). Previous studies have demonstrated the role of vascular smooth muscle cell (VSMC) proliferation in enhancing plaque stability and reducing the risk of rupture. To assess VSMC accumulation, α-smooth muscle actin (α-SMA) staining was performed. As indicated in Fig. 9e, f, an increase in VSMC presence was observed in the plaques treated with FA-P@MC, suggesting the formation of a more robust fibrous cap and improved plaque stability. Additionally, angiogenesis is often a sign of plaque vulnerability, linked to heightened inflammation, plaque rupture, and an increased risk of thrombosis. CD31 immunohistochemistry staining revealed that FA-P@MC treatment significantly reduced the number of perivascular CD31+ neovessels (Supplementary Fig. 67). The observed increases in collagen and α-SMA, alongside reductions in MMP-9 expression and angiogenesis, collectively suggested the therapeutic efficacy of FA-P@MC in enhancing plaque stability. Taken together, these results indicated that FA-P@MC not only halted plaque progression but also fostered a more resilient plaque environment, thereby lowering the potential risk of plaque rupture.

a Representative immunohistochemical images of MMP-9-stained aortic root sections and b quantitative analyses of MMP-9-positive areas in different groups (n = 6 mice). c Representative images of Masson’s trichrome-stained aortic root sections and d quantitative analyses of Masson’s trichrome-positive areas in different groups (n = 6 mice). e Representative immunohistochemical images of α-SMA-stained aortic root sections and f quantitative analyses of α-SMA-positive areas in different groups (n = 6 mice). Scale bars for all immunohistochemical images are 500 μm. g Representative NIR-II fluorescence images and h corresponding NIR-II intensity of the carotid arteries in atherosclerotic mice before and after either PBS or FA-P@MC treatment (n = 3 mice). Images were acquired with an 808 nm excitation wavelength and a 1000 nm long-pass emission filter. Scale bars: 1 cm. i Representative PA/US images and j corresponding PA intensity of the carotid arteries in atherosclerotic mice, presented in both sagittal and transverse views, before and after either PBS or FA-P@MC treatment (n = 3 mice). Scale bar: 3 mm. k Representative H&E-stained images and l corresponding quantitative analyses of plaque area after either PBS or FA-P@MC treatment (n = 3 mice). Scale bar: 500 μm. m Representative immunohistochemical images of α-SMA-stained aortic root sections and n quantitative analyses of α-SMA-positive areas after either PBS or FA-P@MC treatment (n = 3 mice). Scale bar: 500 μm. All data are expressed as mean ± SD. For b, d, f statistical significance was determined using one-way ANOVA for multiple comparisons; for h, j, l, n statistical significance was determined using two-tailed Student’s t-test. Source data are provided as a Source Data file.
Non-invasive monitoring of therapeutic outcomes using the theranostic nanoprobe
The feasibility of the multimodal nanoprobe to monitor plaque progression following treatment was further evaluated. At the end of the treatment regimen, mice were intravenously injected with FA-P@MC nanoprobe, and NIR-II fluorescence imaging was first conducted at 12 h post-injection. As illustrated in the representative NIR-II fluorescence images (Fig. 9g), the carotid artery of AS mice in the PBS group displayed a strong NIR-II fluorescence signal, indicative of severe plaque development. In contrast, mice treated with FA-P@MC exhibited only a faint NIR-II fluorescence signal in the carotid artery, suggesting a reduction in plaque formation following treatment (Fig. 9h). Further PA imaging was performed to gain deeper insights into the progression of plaque development. PA imaging was carried out at 12 h post-injection of the nanoprobe under 790 nm laser excitation. Compared to the PBS group, a marked reduction in PA signal was observed from the aortas of AS mice following FA-P@MC treatment (Fig. 9i, j). The PA imaging results mirrored those obtained from NIR-II fluorescence imaging, with both showing trends consistent with H&E analysis and plaque stability assessment of isolated aortic roots section (Fig. 9k–n), which also validated that plaque progression was substantially attenuated after FA-P@MC treatment. Taken together, these results demonstrated that FA-P@MC not only effectively mitigated AS and increase plaque stability but also served as a powerful multimodal imaging agent for real-time evaluation of treatment response and plaque progression, providing valuable insights for precision therapy.
Discussion
In this study, we present a versatile theranostic nanoplatform that integrates multimodal imaging-based precision diagnosis with synergistic therapeutic activity for AS. Through rational molecular design, we developed a multimodal molecular probe capable of concurrently generating robust long-wavelength NIR-II fluorescence, PA, and MRI signals. The large Gd-DOTA moiety could avoid strong intermolecular quenching interactions, leading to brighter fluorescence emission. Moreover, it also facilitated molecular motion in aggregate state, being favorable for PA transformation. The conjugation of Gd-DOTA and conjugated chromophore effectively restricted the flipping rate of the gadolinium ion, and enhanced the relaxation time and longitudinal relaxivity, resulting in an amplified MRI signal. By exploiting the complementary strengths of each imaging modality, this integrated probe facilitated precise, non-invasive assessment of plaque progression, providing a comprehensive assessment of plaque characteristics. Notably, unlike conventional multifunctional systems that typically amalgamate several disparate components, our meticulously designed organic theranostic probe integrates strong NIR-II fluorescence, PA, and MRI imaging within a single, unified molecular entity. This streamlined architecture ensures defined structural integrity and improved reproducibility. The multimodal molecular probe, coupled with a AT-based therapeutic, was further encapsulated within a ROS-responsive polymer carrier to enable ROS scavenging and on-demand release. By subsequently camouflaging the nanoprobe with macrophage membranes and functionalizing it with a PS-targeting peptide, this design harnesses the biomimetic affinity of macrophages for inflamed plaques and the targeting of foam cells via the PS-binding peptide. This hierarchical targeting approach enhanced nanoprobe accumulation at atherosclerotic lesions, demonstrating superior targeting efficiency compared to control formulations with isolated targeting mechanisms. The enhanced targeting ability of FA-P@MC is crucial for both the precision of diagnostic imaging and the effectiveness of therapeutic interventions.
In a high-cholesterol diet-fed ApoE−/− mouse model of AS, the nanoprobe enabled real-time, sensitive detection of atherosclerotic lesions in vivo by simultaneously generating multimodal imaging signals, while also differentiating plaques at various stages of severity. For feasibility assessment, carotid AS was mainly employed to evaluate the performance of our probe, given its common use in proof-of-concept studies and relevance to systemic AS and coronary artery disease. The preliminary experiments indicated that the platform-based NIR-II fluorescence imaging could visualize coronary lesion in ApoE⁻/⁻ mice (Supplementary Fig. 68). Histological analyses confirmed the presence of coronary plaques and lipid accumulation, with NIR-II signal correlating with plaque burden. These findings suggested the potential of extending this multimodal imaging strategy to coronary applications. While our findings present a promising proof-of-concept in animal models, translating this platform to the clinic will require extensive future studies to comprehensively evaluate its efficacy, safety, and overall feasibility in humans. Beyond its precise diagnostic capabilities, the nanoplatform demonstrated potent therapeutic effects against AS. Notably, it significantly mitigated the ROS-enriched microenvironment and reduced CD47 expression on foam cells, facilitating their efferocytosis and subsequent clearance via macrophage endocytosis. FA-P@MC treatment effectively halted plaque progression, suppressed intraplaque inflammation, curtailed immune cell recruitment, and fostered a more stable plaque environment, thereby reducing the risk of plaque rupture. Regarding the mechanistic studies, although our experiment results suggested that ROS scavenging and AT release facilitated CD47 downregulation, lipid efflux, and macrophage-mediated phagocytosis of foam cells, together contributing to the therapeutic effects of our nanoplatform on plaque regression, we acknowledge that additional concurrent mechanisms may also be involved in the overall therapeutic outcome. The treatment outcomes were further monitored using real-time NIR-II fluorescence and PA imaging, which were in strong agreement with ex vivo ORO staining and histopathological analysis. Furthermore, the nanoagent demonstrated excellent in vivo biocompatibility and minimal toxicity to the circulatory and major organ systems, confirming its safety for AS therapy. In summary, the tailor-engineered nanoplatform facilely integrates multimodal molecular imaging with synergistic therapeutic strategies, offering significant potential to advance the precise diagnosis and management of cardiovascular diseases.
Methods
Materials
All chemicals and reagents were provided by commercial sources. Dulbecco’s Modified Eagle’s Medium (DMEM) culture medium, fetal bovine serum (FBS), and penicillin streptomycin were purchased from Gibco-BRL (Grand Island, NY, USA). Cell culture dishes and confocal dishes were purchased from Corning Inocorporated Co., Ltd (Beijing, China). 3-(4,5-Dimethylthiazil-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was obtained from Saiguo biotech Co., Ltd (BioFROXX, Germany). Griess reagent, reactive oxygen species assay kit (DCFH-DA), Oil Red O Staining Kit (ORO), nitric oxide assay kit (DAF-FM DA), and Masson’s Trichrome Staining Kit were purchased from Beyotime Biotechnology (Shanghai, China). DiI, DiD, Carboxyfluorescein diacetate, and succinimidyl ester (CFSE) were obtained from Beyotime Biotechnology (Shanghai, China). Cy5-labelled AT was obtained from Xi’an Qiyue Biotechnology Co., Ltd. Hematoxylin-eosin staining solution (alcohol soluble) and LDH Cytotoxicity Assay Kit were purchased from Leagene Biotechnology Co., Ltd (Beijing, China). Centrifugal filters were obtained from Merck Millipore Co., Ltd (Merck, America). Immunofluorescence analysis: Anti-CD47 (ABclonal, A1838, 1:200), recombinant anti-CD68 antibody (Abcam, ab283654, 1:200), anti-CD36 antibody (Huabio, ET1701-24, 1:100), anti-LOX-1 antibody (Huabio, ET1706-31, 1:100), anti-SR-BI antibody (Abcam, #ab217318, 1:100), anti-ABCA1 antibody (Huabio, R1510-42, 1:100), Goat anti-Rabbit IgG H&L (Alexa Fluor® 488) (1:1000, Abcam, #ab150077), and Goat anti-Rabbit IgG H&L (Alexa Fluor® 594) (Cell Signaling Technology, #8889, 1:100,). Immunohistochemical analysis: Recombinant anti-MMP9 antibody (Abcam, ab283575, 1:200), α-SMA (Abcam, ab5694, 1:200), Anti-CD31 antibody (Abcam, ab222783, 1:200), Anti-CD14 (Abcam, ab182032, 1:200). TNF-α ELISA kit, IL-6 ELISA kit and IL-1β ELISA kit were purchased from Dakewe Co., Ltd (Beijing, China). OxLDL (YB-002) and HDL (YB-003) were purchased from Yiyuan biotechnology Co., Ltd. (Guangzhou, China). BODIPY-cholesterol (HY-125746), Probucol (HY-B0388), ApoA-I (HY-P72833), and BLT-1 (HY-116767) were obtained from MedChemExpress (MCE).
Cell lines and animals
The RAW264.7 murine macrophage cell line and the mouse embryonic fibroblasts line (NIH 3T3) were purchased from the Chinese Academy of Sciences Cells Bank (Shanghai, China). The human umbilical vein endothelial cell line (HUVEC) was purchased from Cyagen Biosciences (Guangzhou, China) Inc. All cells were cultivated in a humidified atmosphere at 37 °C with 5% of CO2.
C57BL/6 mice (6 weeks, female) were purchased from Beijing Vital River Laboratory Animal Technology. ApoE−/− mice (6 weeks, female) were purchased from SPF (Beijing) Biotechnology Co., Ltd. High-fat diet (HFD) feeding of AopE−/− mice began at 6 weeks of age. All mice were housed in the same specific pathogen-free (SPF) facility and maintained under identical environmental parameters, including cage type, temperature (25 °C), humidity, handling procedures, and a 12-h light/dark cycle. All model groups received the same HFD throughout the study, and food and water were provided ad libitum, with diet replaced twice weekly. The high-fat diet used in this study was sourced from SPF (Beijing) Biotechnology Co., Ltd., catalog number SFD004. The diet has an energy density of 4.7 kcal/g, with 41% of total kcal from fat, 42% from carbohydrate, and 17% from protein. By weight, the diet contained 21.2% lard, 49.1% carbohydrates, 19.8% protein, and 0.2% cholesterol. Carbohydrates in the formulation are primarily composed of corn starch, sucrose, and maltodextrin, and the fiber component contains 31 g/kg crude fiber based on 90% dry matter. The primary sources of fat are lard (42% saturated fatty acids, 48% monounsaturated fatty acids, 10% polyunsaturated fatty acids) and soybean oil (15% saturated fatty acids, 24% monounsaturated fatty acids, 61% polyunsaturated fatty acids). The diet contains 0.2% cholesterol by weight (equivalent to 2 g/kg), and sodium cholate is not included in the formulation. All procedures involving animals were conducted by the guidelines set by the Tianjin Committee of Use and Care of Laboratory Animals, and approved by the Animal Ethics Committee of Nankai University (2024-SYDWLL-000693).
Characterizations
1H and 13C nuclear magnetic resonance (NMR) spectra were acquired using a Bruker-DPX 400 spectrometer. High-resolution mass spectra (HRMS) were performed on a Bruker AutoflexIII LRF200-CID mass spectrometer in matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mode. The absorption spectra were detected using a Shimadzu UV-1800 spectrometer. Edinburgh FS5 was used to measure the photoluminescence (PL) spectra. Transmission electron microscope (TEM) images were captured using a Talos L120C G2 instrument by FEI, Czech. Dynamic light scattering (DLS) measurements were performed on a Malvern Zeta sizer Nano ZS-90. PA imaging was conducted on a commercial small-animal optoacoustic tomography system (Vevo LAZR, FujiFilm VisualSonics, America). A short-wavelength infrared (SWIR) imaging system (Wuhan Grand-imaging Technology Co., LTD) was utilized for NIR-II fluorescence imaging of animals and organs. A bruker 9.4T BioSpec 94/30 MRI installations for small animal magnetic resonance imaging test.
Fabrication of nanoformulations
First, RAW264.7 cells were dispersed in the membrane protein extraction reagent (Beyotime, China) containing 1 mM PMSF and cooled down at 4 °C for 30 min, and subsequently broken by ultrasound (80 W) for 10 min. The mixture was repeatedly frozen and thawed at −80 °C and 37 °C for 3 times, and then centrifuged at 3000 × g for 10 min. The resulting supernatant was centrifugated at 12,000 × g × 10 min, followed by a final centrifugation at 100,000 × g for 1 h to obtain membrane in precipitates. The membrane was stored at −80 °C for further use.
The targeting molecule (DSPE-PEG-CLIKKPF) was synthesized according to previous reports28,67. Briefly, CLIKKPF was reacted with DSPE-PEG-maleimide via thiol-Michael addition reaction in water/methanol solution (90:10, v/v) for 8 h at room temperature. Then the residual CLIKKPF peptide was removed by centrifugal filters (Merck Millipore Co., Ltd, Shanghai China) and the final product DSPE-PEG-CLIKKPF was obtained by lyophilizing.
The molecular probe and AT-loaded nanoformulations were fabricated using a modified nanoprecipitation technique. The synthesis was carried out at 20 °C, where FTBGd (1 mg), AT (1 mg), DSPE-PEG (2 mg), and PPADT (4 mg) were dissolved in 1 mL of DMSO under constant stirring. After achieving complete dissolution, the resulting stock solution was gradually introduced into 15 mL of deionized water under vigorous stirring. During nanoprecipitation, DSPE-PEG was incorporated as a stabilizing surfactant. Owing to its amphiphilic structure, DSPE-PEG anchors its hydrophobic lipid tail within the nanoparticle core while presenting a hydrophilic PEG corona on the surface, thereby enhancing colloidal stability. To eliminate the organic solvent, the mixture underwent dialysis against water for 12 h using a dialysis membrane with an MWCO of 3500 Da, ultimately producing the final solution, termed FA-P. The encapsulation efficiencies of FTBGd and AT were found to be 63.7% and 59.5%, respectively. To synthesize FA-P@M, a solution of FA-P (100 μL, 1 mg mL‒1 based on FTBGd) was combined with 0.5 mg of membrane and subjected to ultrasound treatment (100 W, 50 Hz) for 5 min. The mixture was then repeatedly extruded through a micro-extruder with a 200 nm pore size to form FA-P@M. Uncoated membranes were eliminated by centrifugation at 3000 × g, followed by washing until no residual protein was detected in the supernatant. The obtained FA-P@M was stored at 4 °C for future use. After obtaining FA-P@M, FA-P@MC was prepared by incorporating DSPE-PEG-CLIKKPF into its lipid membrane.
Drug release from FA-P@MC
The AT release profile from FA-P@MC or FA-P was investigated under varying H2O2 levels (0, 2.5 μM, 0.1 mM, 1.0 mM, or 1.0 mM + catalase). Prior to the release studies, both nanoformulations were confirmed to exhibit similar particle sizes and identical drug loading, ensuring a valid comparison. 2 mg of FA-P@MC or FA-P was suspended in 5 mL of deionized water and sealed inside a dialysis bag (MWCO = 10,000). This bag was subsequently submerged in 15 mL of buffer solution and placed in a shaker set to 37 °C with a rotation speed of 30 × g. At predetermined time points, 2 mL of the incubation medium was withdrawn for HPLC analysis, and an equivalent volume of fresh medium was added back to the buffer solution. The percentage of cumulative AT release was determined using the equation: Cumulative drug release (%) = (Mt/M0) × 100%, where Mt represents the drug amount released at time t, and M0 corresponds to the initial drug content in FA-P@MC. All release experiments were performed in triplicate.
The H2O2 levels in plaque tissues were measured using a commercially available Hydrogen Peroxide Content Assay Kit (Solarbio, BC3595), following the manufacturer’s protocol. Briefly, freshly isolated plaque tissues were homogenized in the supplied buffer on ice and centrifuged, and the resulting supernatants were used for analysis. Absorbance was measured at 415 nm using a UV–Vis spectrophotometer (SHIMADZU), and H2O2 concentrations were calculated based on a standard curve generated with known H2O2 standards. The results are reported as mean ± SD (n = 6 individual plaque samples), yielding an H2O2 level of 0.34 ± 0.05 mM, corresponding to approximately 0.34 µmol per gram of wet tissue.
Cellular uptake behavior
Cellular uptake of different formulations by foam cells was evaluated. To generate foam cells, RAW264.7 cells were exposed to a medium supplemented with 50 μg/mL oxLDL. Following this, the medium was substituted with fresh medium containing DiI-labeled nanoparticles—FA-P, FA-P@M, or FA-P@MC—at an FTGd concentration of 50 µM. After incubation, the cells underwent three PBS washes, were fixed using 4% tissue fixative for 15 min, and then washed three additional times with PBS. The nuclei were stained with DAPI for 10 min, followed by another PBS wash to eliminate residual dye. Finally, confocal laser scanning microscopy (CLSM) (LSM 800 with Airyscan, ZEISS, Germany) was utilized for imaging, and analysis was conducted using ZEN 2012 software.
The transmigration of the NPs across inflamed HUVECs and their subsequent uptake by foam cells were evaluated using in vitro transwell migration assays. HUVECs were cultured in the upper chamber of a Transwell insert (0.3 μm) and exposed to 1 μg/mL LPS for 24 h to induce an inflammatory response. At the same time, RAW264.7 cells (1 × 105 per well) were seeded in the lower chamber of a 24-well plate and treated with 50 μg/mL oxLDL for 24 h to form foam cells. Subsequently, FA-P, FA-P@M, or FA-P@MC (50 µM based on FTGd) were added to the upper chamber and incubated for 4 h. After incubation, the foam cells were rinsed three times with PBS and stained with DAPI for 15 min. Imaging was carried out using CLSM, and flow cytometry (BD LSR Fortessa, USA) was used for quantitative analysis.
In vitro safety evaluation
The cytotoxicity of FA-P@MC was evaluated in vitro using MTT assays with NIH 3T3 and HUVEC cell lines. In brief, the cells were plated in a 96-well plate at a concentration of 1 × 105 cells/mL. After 24 h of incubation, the medium was replaced with fresh medium containing nanoparticles at varying concentrations (0, 20, 40, 60, 80, and 100 µM). Following another 24-h incubation period, 100 µL of MTT solution was added to each well. After 4 h, the MTT solution was removed, and 150 µL of DMSO was added to each well, followed by shaking at room temperature. The optical density at 570 nm was then recorded using a microplate reader. For the LDH cytotoxicity assay, cells were exposed to various concentrations of the nanoparticle formulation. LDH release was then quantified using an LDH Cytotoxicity Assay Kit (Leagene Biotechnology Co., Ltd.) following the manufacturer’s instructions.
In vitro anti-inflammatory effect
Macrophages were simultaneously treated with 50 μg/mL ox-LDL and different drug formulations (free AT, A-P, FA-P, FA-P@M, and FA-P@MC) at a concentration of 50 µM for 24 h. After the treatment, the cells were washed three times with PBS. To assess intracellular ROS, the cells were incubated with 10 µM of DCFH-DA in the dark. After removing the excess probe, the cells were further incubated at 37 °C for 30 min. Confocal microscopy was employed with 488 nm excitation and 530 ± 20 nm emission to observe the dichlorofluorescein (DCF) fluorescence resulting from the reaction of DCFH-DA with ROS. The release of nitric oxide (NO) from cells was assessed using the fluorescent NO probe, DAF-FM DA. The cells were treated with 50 μg/mL ox-LDL and various drug formulations (free AT, A-P, FA-P, FA-P@M, and FA-P@MC) at a concentration of 50 µM for 24 h, followed by three PBS washes. The NO probe, DAF-FM DA (final concentration: 5 μM), was added, and the cells were incubated at 37 °C for 30 min, after which they were washed three times with PBS. Confocal microscopy was employed to visualize the cells. Statistical analysis and graphing were conducted using ZEN 2012 (Version 1.1.2.0).
Inhibition of lipid uptake and foam cell formation
RAW264.7 cells were seeded in confocal dishes at 1 × 105 cells per dish and allowed to adhere overnight. Cells were stimulated with 1 µg mL−1 of LPS for 6 h, followed by treatment with different formulations for an additional 12 h. Subsequently, cells were incubated with 50 μg mL−1 of DiI-oxLDL for 4 h. The cells were then washed with PBS, and fixed with 4% paraformaldehyde. Fluorescence imaging was performed using CLSM to evaluate the cellular uptake of oxLDL. The expressions of CD36 and LOX-1 were evaluated by immunofluorescence using primary antibodies against CD36 and LOX-1, followed by incubation with fluorescently labeled secondary antibodies.
For foam cell formation and lipid accumulation assays, RAW264.7 cells were seeded in 12-well plates at 1 × 105 cells per well. Cells were then treated with 50 μg mL−1 of oxLDL in combination with various formulations for 12 h. For Oil Red O (ORO) staining, the foam cells were treated with the freshly prepared ORO working solution for 15 min and then rinsed with 60% isopropanol. Images of the cells were captured using confocal microscopy. After imaging, ORO was extracted using isopropanol, and its absorbance was measured at 492 nm using a microplate reader. Cholesterol efflux was quantified by collecting culture supernatants post-treatment. The expressions of SR-BI and ABCA1 were evaluated by immunofluorescence using primary antibodies against SR-BI and ABCA1, followed by incubation with fluorescently labeled secondary antibodies.
For functional cholesterol efflux assay, foam cells were loaded with 1 µg/mL BODIPY-labeled cholesterol (HY-125746, MedChemExpress) for 6 h to allow intracellular labeling. After washing, cells were treated with FA-P@MC or relevant control formulations in the presence of ApoA-I (10 µg mL−1, ABCA1-dependent) or HDL (40 µg mL−1, SR-BI/ABCG1-associated, YB-003, Yiyuan biotechnology) as cholesterol acceptors in serum-free medium for 24 h. As a control, cells were pre-incubated with pharmacological inhibitors of ABCA1 (probucol, 20 µM) or SR-BI (BLT-1, 1 µM) for 2 h prior to efflux to assess the contribution of these transporters. Fluorescence intensity was determined in both supernatant and cell lysate using a microplate reader (excitation/emission 485/510 nm). The percent of cholesterol efflux was calculated as (fluorescence in the supernatant/the sum of the fluorescence in the medium and the fluorescence remaining in the cells) × 100%. Parallel cholesterol efflux studies were also conducted in foam cells induced from bone marrow–derived macrophages (BMDMs). Bone marrow cells were flushed from femurs and tibias of 8-week-old C57BL/6 mice, filtered through a 70 µm strainer, and plated in RPMI 1640 supplemented with 10% FBS, 1% penicillin-streptomycin, and 20ng/mL M-CSF. Cells were differentiated for 7 days at 37 °C with 5% CO2 and used for cholesterol efflux assays. Macrophage differentiation was confirmed by F4/80 immunostaining.
CD47 expression analysis and in vitro efferocytosis evaluation
RAW264.7 cells were incubated with 50 μg/mL oxLDL for 24 h, followed by treatment with different NPs for an additional 24 h. Cells were first harvested and lysed in RIPA buffer supplemented with protease and phosphatase inhibitors. Protein concentration was determined using a BCA assay, and equal amounts of protein were mixed with 4 × loading buffer and denatured by heating at 95 °C for 10 min in a metal bath. Samples were then separated by 10% SDS-PAGE and electrotransferred onto a 0.4 μm PVDF membrane. The membrane was blocked with BSA in TBST (Tris-buffered saline with 0.1% Tween-20) for 1 h at room temperature to minimize nonspecific binding. The membrane was then incubated overnight at 4 °C with a primary antibody against CD47, while GAPDH was used as a loading control. Following three washes with TBST, membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. Protein bands were visualized using an enhanced chemiluminescence detection system, and band intensities were quantified using ImageJ software to determine relative CD47 expression levels.
For phagocytosis study, bone marrow–derived macrophages (BMDMs) were labeled with CFSE to serve as phagocytic cells, while oxLDL-treated RAW264.7 cells were used as foam cells. BMDMs and foam cells were co-cultured for 6 h. Following incubation, the medium was discarded, and the cells were washed with PBS, then fixed with 4% formaldehyde for 15 min. Fluorescence images were acquired using CLSM. For flow cytometry, treated cells were washed with PBS, detached, and subsequently analyzed on a BD LSR Fortessa (USA). Flow cytometric data were analyzed using FlowJo version 10.8.1.
In vivo biosafety and pharmacokinetics analysis
To assess the potential toxicity of FA-P@MC, 6-week-old healthy C57BL/6 mice were intravenously injected with PBS or FA-P@MC (10 mg kg-1) by intravenous injection twice weekly for 2 weeks (Day 0–14). One week after treatment (Day 22), the mice were euthanized, and blood samples were collected for analysis. For long-term biosafety evaluation, mice received weekly intravenous injections of either PBS or FA-P@MC (10 mg/kg based on FTBGd) for 7 weeks (Day 0–49). On Day 50, hematological parameters, including white blood cell count, red blood cell count, hemoglobin concentration, hematocrit, mean red blood cell volume, platelet count, mean corpuscular hemoglobin content, and mean corpuscular hemoglobin concentration, were measured using an automated hematology analyzer. Serum samples were obtained to evaluate various biochemical markers in the blood, such as alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and creatinine concentrations. Additionally, key organs, including the heart, liver, spleen, lungs, and kidneys, were collected for histopathological examination. In brief, tissues were fixed in 4% paraformaldehyde (PFA), embedded in paraffin, and sliced into 5 μm sections for H&E staining. The stained tissue sections were subsequently analyzed using a digital microscope (Leica QWin).
For pharmacokinetic evaluation, 6-week-old C57BL/6 mice were given FA-P@MC intravenously at a dose of 10 mg/kg. Blood samples were taken at different time intervals (0, 4, 8, 12, 24, 48, and 72 h) from the inner eye socket following intravenous injection of different nanoparticles. The fluorescence intensity of the blood samples was quantified using a SWIR imaging system (Wuhan Grand-imaging Technology Co., LTD) to assess the plasma signals of the nanoparticles. The clearance of free AT in the bloodstream was determined by HPLC analysis. To study the in vivo metabolic fate and clearance routes, FA-P@MC was labeled with the near-infrared dye DiR. Fecal and urinary samples were collected at days 1, 3, and 7 after intravenous administration, and their fluorescence signals were monitored using IVIS.
Multimodal imaging and analysis of the aorta ex vivo
To perform ex vivo fluorescence and PA imaging of the aorta, ApoE−/− mice were fed a high-fat diet (HFD; SPF Biotechnology Co., Ltd., Beijing) for 12 weeks (Day 0–84) to induce atherosclerosis. On Day 85, atherosclerotic mice were intravenously administered FA-P@MC. Twelve hours after injection, the mice were euthanized, and the aortas were excised for ex vivo fluorescence and PA imaging to evaluate the probe’s ability to detect plaques and differentiate plaque severity. NIR-II fluorescence imaging was performed using a short-wave infrared (SWIR) system with 808 nm excitation and emission collected through a 1000 nm long-pass filter. PA imaging was acquired at 790 nm excitation using a VisualSonics Vevo LAZR multimodal imaging system (FUJIFILM VisualSonics, Canada). For fluorescence imaging, the laser power was 50 mW cm−2; for PA measurements, pulse energy = 20 mJ cm−2, frequency = 30 MHz, PA gain = 35 dB, pulse repetition rate = 20 Hz, and duration of pulsed laser = 7 ns. 790 nm was selected for PA imaging as it coincides with the probe’s absorption peak and yields strong PA responses. Endogenous absorbers such as hemoglobin exhibit reduced absorption in this spectral region compared to their strong visible-band absorption, and water shows low absorption at this wavelength before its absorption increases at longer wavelengths. For fluorescence imaging, 808 nm light was employed as the excitation source because the probe has strong absorption at this wavelength and can be efficiently excited using the widely available commercial 808 nm laser. This wavelength selection ensures sufficient excitation efficiency while maintaining compatibility with commonly available NIR laser. MRI was carried out on a 9.4 T small-animal MRI scanner (BioSpec 94/30, Bruker).
To validate the correlation between imaging signal intensities with plaque severity, the aorta was subsequently embedded in optimal cutting temperature (OCT) compound, frozen at −80 °C for 30 min, and sliced into 10 μm sections. For ORO staining, a working solution was prepared by diluting 6 mL of the stock solution with 4 mL of deionized water. Cryosections of the aorta were incubated with 10 µL of this solution for 2–5 min, then washed with 60% isopropanol and deionized water. Hematoxylin was used as a counterstain for 5 min. For immunofluorescence, sections were rinsed, blocked, and incubated with anti-F4/80 primary antibody (1:500, Bio-Rad, MCA497GA), followed by washing and incubation with fluorescently labeled secondary antibody for 60 min at room temperature. Nuclei were counterstained with DAPI for 10 min. Images were acquired using a Zeiss fluorescence microscope (FITC: excitation at 488 nm; DAPI: excitation at 405 nm). For ROS staining, aorta sections were treated with DCFH-DA (10 µM) for 60 min at 37 °C, followed by a 10-min DAPI incubation at 37 °C. Fluorescent images of DCFH-DA were captured using a laser confocal microscope (DCFH-DA: excitation at 488 nm; DAPI: excitation at 405 nm).
In vivo NIR-II fluorescence, PA, and MR imaging
AopE-/- mice were fed a HDF for 8 weeks (Day 0–56), and then received intravenous injections of FA-P@C, FA-P@M, or FA-P@MC (150 µL, 4 mg/kg based on FTBGd) via the tail vein on day 57. For the competitive blocking control, mice were pretreated with free CLIKKPF peptide (2 mg/kg, i.v.) 4 h prior to FA-P@MC administration on Day 57. At predefined time points post nanoformulation injection (0, 3, 6, 12, 24, and 36 h post-injection), mice were anesthetized with 2% isoflurane and then imaged using a SWIR system equipped with an InGaAs scientific-grade camera and a 1000 nm long-pass filter. To validate plaque visualization, a surgical incision was made to expose the carotid arteries. To evaluate drug distribution and plaque-targeting efficiency, atherosclerotic mice were intravenously administered Cy5-labeled AT, Cy5-AT-loaded FA-P@M, or Cy5-AT-loaded FA-P@MC on Day 57. Twelve hours post-injection, major organs and the aorta were harvested for ex vivo fluorescence imaging using an IVIS system. For quantitative analysis, tissues were weighed, homogenized, and centrifuged to collect supernatants. Cy5 fluorescence intensity in the supernatants was measured using a spectrofluorometer, and background fluorescence from untreated control tissues was subtracted. The AT content in each tissue was calculated based on a standard curve generated from known concentrations of Cy5-AT under identical conditions. To study lesion distribution, plaque tissues were cryosectioned, fixed with 4% PFA, permeabilized with 0.1% Triton X-100, and blocked with 5% BSA. Foam cells were identified by immunostaining with anti-F4/80 and counterstaining with BODIPY to visualize intracellular lipids. Additional staining was performed for PS (Sigma-Aldrich, 05-719, 1:200), CD68 (Abcam, ab283654, 1:200), and PLIN2 (Huabio, ET1704-17, 1:200) to evaluate co-localization with FA-P@MC. Images were acquired using a confocal laser scanning microscope under identical settings for all groups, and co-localization was analyzed using ImageJ.
PA imaging was conducted on living mice on day 57 (12 h post-injection of different nanoprobe) using the VisualSonics Vevo LAZR multimodal system (Canada) under the following parameters: pulse energy = 20 cm−2, wavelength range = 790 nm, frequency = 30 MHz, PA gain = 35 dB, pulse repetition rate = 20 Hz, and duration of pulsed laser = 7 ns. Ultrasound and PA signals were acquired simultaneously, and merged PA/US images were generated by overlaying pseudo-colored PA signals onto grayscale ultrasound images. MRI was performed on a 9.4T Bruker BioSpec 94/30 system before and 12 h post-injection of Gd-DOTA, FA-P@M, or FA-P@MC (150 µL, 4 mg/kg based on FTBGd) (day 57). Mice were anesthetized with 2% isoflurane and positioned in the MRI coil for baseline and post-injection scans. T1-weighted axial images were acquired for analysis.
In vivo therapeutic efficacy study
ApoE−/− mice were initially fed a high-fat diet for 8 weeks to induce the development of atherosclerotic plaques at arterial bifurcations (experimental days 0–56). On day 57, the atherosclerotic mice were randomly assigned to six treatment groups (n = 6 mice per group), including PBS, free AT, A-P, FA-P, FA-P@M, and FA-P@MC (at a dose of 3 mg/kg AT). Each group received intravenous injections of the designated treatment twice a week (days 57, 61, 64, 68, 71, 75, 78, 82, 85, 89, 92, 96, 99, 103, 106, and 110) while maintaining the high-fat diet for an additional 8 weeks (Day 57–112). A standard chow-fed control group was included for comparison. For the evaluation of therapeutic effects via in situ imaging at the end of treatment, mice were intravenously administered the FA-P@MC nanoprobe on Day 113. NIR-II fluorescence and PA imaging were performed 12 h post-injection following the procedure described above. After imaging, mice were euthanized, and the entire aortas were carefully excised for subsequent analyses (Day 113). Atherosclerotic plaque burden was evaluated by ORO staining of the aortic arch, following established protocols as described above, to quantify lipid-rich plaque areas. Histological assessment of aortic root sections was performed after paraffin embedding. Sections were stained with H&E to examine general tissue morphology, while dihydroethidium (DHE) staining was applied to assess ROS levels within plaques. Immunofluorescence staining was carried out to evaluate the expression of CD47 and CD68, markers of phagocytosis and macrophage content, respectively. Plaque stability was further assessed by immunohistochemical staining of aortic root sections with Masson’s trichrome for collagen content, MMP-9 for matrix degradation, CD31 for angiogenesis, and α-SMA for smooth muscle cell content. Quantitative analysis of stained sections was performed using Image-Pro Plus 6.0. The inflammation was evaluated by collecting fresh blood post-treatment, centrifuging to obtain serum, and measuring TNF-α and IL-6 levels using ELISA kits according to the manufacturer’s instructions. Immune cell profiling within plaques was performed by isolating plaque tissue, enzymatically digesting to obtain single-cell suspensions, and analyzing CD4+ (CD3+CD4+), CD8+ (CD3+CD8+), and regulatory T (Treg) cell (CD25+Foxp3+) populations using flow cytometry.
Statistical analysis
All data were presented as mean value ± the standard deviation of independent experiments. One-way ANOVA and two-way was utilized for statistical analysis. Values of *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001 were applied to annotate statistical significance.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The remaining data are available within the Article, Supplementary Information or Source Data file. Source data are provided with this paper.
References
Roth, G., Mensah, G. & Fuster, V. The global burden of cardiovascular diseases and risks: a compass for global action. J. Am. Coll. Cardiol. 76, 2980–2981 (2020).
Libby, P. The changing landscape of atherosclerosis. Nature 592, 524–533 (2021).
Grover, S. P. & Mackman, N. Tissue factor in atherosclerosis and atherothrombosis. Atherosclerosis 307, 80–86 (2020).
Wang, Y., Li, G., Yang, L., Luo, R. & Guo, G. Development of innovative biomaterials and devices for the treatment of cardiovascular diseases. Adv. Mater. 34, 2201971 (2022).
Hafiane, A. Vulnerable plaque, characteristics, detection, and potential therapies. J. Cardiovasc. Dev. Dis. 6, 26 (2019).
Moreno, P. R. Vulnerable plaque: definition, diagnosis, and treatment. Cardiol. Clin. 28, 1–30 (2010).
Chen, W. et al. Macrophage-targeted nanomedicine for the diagnosis and treatment of atherosclerosis. Nat. Rev. Cardiol. 19, 228–249 (2022).
Grodecki, K. et al. Phenotyping atherosclerotic plaque and perivascular adipose tissue: signalling pathways and clinical biomarkers in atherosclerosis. Nat. Rev. Cardiol. 22, 443–455 (2025).
Wu, Z. et al. An enzymatic cleavage-triggered minimally invasive nanosensor for urine-based detection of early atherosclerosis. Sci. Adv. 11, eadu7614 (2025).
Zhang, S. et al. Targeting the microenvironment of vulnerable atherosclerotic plaques: an emerging diagnosis and therapy strategy for atherosclerosis. Adv. Mater. 34, e2110660 (2022).
Mulder, W. J. M., Jaffer, F. A., Fayad, Z. A. & Nahrendorf, M. Imaging and nanomedicine in inflammatory atherosclerosis. Sci. Transl. Med. 6, 239sr1 (2014).
Gemma, A. F. et al. Noninvasive plaque imaging to accelerate coronary artery disease drug development. Circulation 146, 1712–1727 (2022).
Lin, Y. H. et al. Vascular and hepatic enhancements at MR imaging: comparison of Gd-EOB-DTPA and Gd-DTPA in the same subjects. Clin. Imag. 38, 287–291 (2014).
Kim, B. H. et al. Large-scale synthesis of uniform and extremely small-sized iron oxide nanoparticles for high-resolution T1 magnetic resonance imaging contrast agents. J. Am. Chem. Soc. 133, 12624–12631 (2021).
Peldschus, K. et al. Comparison of the high relaxivity Gd chelates P1152 and Gd-BOPTA for contrast-enhanced MR angiography in rabbits at 1.5 Tesla and 3.0 Tesla. J. Magn. Reson. Imaging 32, 459–465 (2010).
Lu, K. et al. PEGylated ultrasmall iron oxide nanoparticles as MRI contrast agents for vascular imaging and real-time monitoring. ACS Nano 19, 3519–3530 (2025).
Mézquita, A. J. V. et al. Clinical quantitative coronary artery stenosis and coronary atherosclerosis imaging: a Consensus Statement from the Quantitative Cardiovascular Imaging Study Group. Nat. Rev. Cardiol. 20, 696–714 (2023).
Boehm, K. M., Khosravi, P., Vanguri, R., Gao, J. & Shah, S. P. Harnessing multimodal data integration to advance precision oncology. Nat. Rev. Cancer 22, 114–126 (2022).
Chen, N. et al. Simultaneous head-mounted imaging of neural and hemodynamic activities at high spatiotemporal resolution in freely behaving mice. Sci. Adv. 11, eadu1153 (2025).
Jiang, Y. & Pu, K. Molecular probes for autofluorescence-free optical imaging. Chem. Rev. 121, 13086–13131 (2021).
Yang, Y., Jiang, Q. & Zhang, F. Nanocrystals for deep-tissue in vivo luminescence imaging in the near-infrared region. Chem. Rev. 124, 554–628 (2024).
Antaris, A. L. et al. A small-molecule dye for NIR-II imaging. Nat. Mater. 15, 235–242 (2016).
Yang, Y. et al. Fluorescence-amplified nanocrystals in the second near-infrared window for in vivo real-time dynamic multiplexed imaging. Nat. Nanotechnol. 18, 1195–1204 (2023).
Zhang, Y. et al. Microenvironment-activatable probe for precise NIR-II monitoring and synergistic immunotherapy in rheumatoid arthritis. Adv. Mater. 36, 2409661 (2024).
Wang, L. V. & Hu, S. Photoacoustic tomography: in vivo imaging from organelles to organs. Science 335, 1458–1462 (2012).
Weber, J., Beard, P. C. & Bohndiek, S. E. Contrast agents for molecular photoacoustic imaging. Nat. Methods 13, 639–650 (2016).
Liu, J., Cheng, P., Xu, C. & Pu, K. Molecular probes for in vivo optical imaging of immune cells. Nat. Biomed. Eng. 9, 618–637 (2025).
Song, J. et al. Near-infrared-II photoacoustic imaging and photo-triggered synergistic treatment of thrombosis via fibrin-specific homopolymer nanoparticles. Nat. Commun. 14, 6881 (2023).
Mallidi, S., Luke, G. P. & Emelianov, S. Photoacoustic imaging in cancer detection, diagnosis, and treatment guidance. Trends Biotechnol. 29, 213–221 (2011).
Khera, R. et al. Transforming cardiovascular care with artificial intelligence: from discovery to practice: JACC state-of-the-art review. J. Am. Coll. Cardiol. 84, 97–114 (2024).
Wang, S., He, H., Mao, Y., Zhang, Y. & Gu, N. Advances in atherosclerosis theranostics harnessing iron oxide-based nanoparticles. Adv. Sci. 11, 2308298 (2024).
Ni, J.-S. et al. A photoinduced nonadiabatic decay-guided molecular motor triggers effective photothermal conversion for cancer therapy. Angew. Chem. Int. Ed. 59, 11298–11302 (2020).
Song, J., Wang, H., Meng, X., Li, W. & Qi, J. A hypoxia-activated and microenvironment-remodeling nanoplatform for multifunctional imaging and potentiated immunotherapy of cancer. Nat. Commun. 15, 10395 (2024).
Canfrán-Duque, A. et al. Macrophage-derived 25-hydroxycholesterol promotes vascular inflammation, atherogenesis, and lesion remodeling. Circulation 147, 388–408 (2023).
Engelen, S. E., Robinson, A. J. B., Zurke, Y.-X. & Monaco, C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: how to proceed? Nat. Rev. Cardiol. 19, 522–542 (2022).
Moore, K. J., Sheedy, F. J. & Fisher, E. A. Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 13, 709–721 (2013).
Fan, J. et al. CKIP-1 limits foam cell formation and inhibits atherosclerosis by promoting degradation of Oct-1 by REGgamma. Nat. Commun. 10, 425 (2019).
Tabas, I. & Bornfeldt, K. E. Macrophage phenotype and function in different stages of atherosclerosis. Circ. Res. 118, 653–667 (2016).
Kojima, Y. et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 53, 86 (2016).
Cao, Y. et al. Sonodynamic therapy promotes efferocytosis via CD47 down-regulation in advanced atherosclerotic plaque. Int. Heart J. 63, 131 (2022).
Logtenberg, M. E. W., Scheeren, F. A. & Schumacher, T. N. The CD47-SIRPα immune checkpoint. Immunity 52, 742–752 (2020).
Allahverdian, S., Chaabane, C., Boukais, K., Francis, G. A. & Bochaton-Piallat, M. L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 114, 540–550 (2018).
Allahverdian, S., Chehroudi, A. C., McManus, B. M., Abraham, T. & Francis, G. A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 129, 1551–1559 (2014).
Lobatto, M. E., Fuster, V., Fayad, Z. A. & Mulder, W. J. Perspectives and opportunities for nanomedicine in the management of atherosclerosis. Nat. Rev. Drug Discov. 10, 835 (2011).
Dvir, T., Timko, B. P., Kohane, D. S. & Langer, R. Nanotechnological strategies for engineering complex tissues. Nat. Nanotechnol. 6, 13–22 (2011).
Li, B. et al. Photothermal therapy of tuberculosis using targeting pre-activated macrophage membrane-coated nanoparticles. Nat. Nanotechnol. 19, 834–845 (2024).
Xu, C. et al. Molecular motion and nonradiative decay: towards efficient photothermal and photoacoustic systems. Angew. Chem. Int. Ed. 61, e202204604 (2022).
Zhang, C. et al. Magnetic-susceptibility-dependent ratiometric probes for enhancing quantitative MRI. Nat. Biomed. Eng. 9, 671–685 (2025).
Meng, Q., Wu, M., Shang, Z., Zhang, Z. & Zhang, R. Responsive gadolinium(III) complex-based small molecule magnetic resonance imaging probes: design, mechanism and application. Coord. Chem. Rev. 457, 214398 (2022).
Greenwood, J., Steinman, L. & Zamvil, S. S. Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat. Rev. Immunol. 6, 358 (2006).
Koushki, K. et al. Anti-inflammatory action of statins in cardiovascular disease: the role of inflammasome and toll-like receptor pathways. Clin. Rev. Allergy Immunol. 60, 175–199 (2020).
Wilson, D. S. et al. Orally delivered thioketal nanoparticles loaded with TNF-α−siRNA target inflammation and inhibit gene expression in the intestines. Nat. Mater. 9, 923–928 (2010).
Gao, C. et al. Treatment of atherosclerosis by macrophage-biomimetic nanoparticles via targeted pharmacotherapy and sequestration of proinflammatory cytokines. Nat. Commun. 11, 2622 (2020).
Ji, M. et al. In vivo fluorescent labeling of foam cell-derived extracellular vesicles as circulating biomarkers for in vitro detection of atherosclerosis. J. Am. Chem. Soc. 146, 10093–10102 (2024).
Guo, L. et al. Biomimetic macrophage membrane and lipidated peptide hybrid nanovesicles for atherosclerosis therapy. Adv. Funct. Mater. 32, 2204822 (2022).
Anghelache, M. et al. Biomimetic nanocarriers of pro-resolving lipid mediators for resolution of inflammation in atherosclerosis. Adv. Healthc. Mater. 13, 2302238 (2024).
Zhou, S., Sun, X. & Liang, G. Activatable peptide–AIEgen conjugates for cancer imaging. Chem. Sci. 16, 5369–5382 (2025).
Cheng, J. et al. A targeting nanotherapy for abdominal aortic aneurysms. J. Am. Coll. Cardiol. 72, 2591–2605 (2018).
Wang, Y. et al. Targeted therapy of atherosclerosis by a broad-spectrum reactive oxygen species scavenging nanoparticle with intrinsic anti-inflammatory activity. ACS Nano 12, 8943–8960 (2018).
Adkar, S. S. & Leeper, N. J. Efferocytosis in atherosclerosis. Nat. Rev. Cardiol. 21, 762–779 (2024).
De Meyer, G. R. Y., Zurek, M., Puylaert, P. & Martinet, W. Programmed death of macrophages in atherosclerosis: mechanisms and therapeutic targets. Nat. Rev. Cardiol. 21, 312–325 (2024).
Döring, Y., van der Vorst, E. P. C. & Weber, C. Targeting immune cell recruitment in atherosclerosis. Nat. Rev. Cardiol. 21, 824–840 (2024).
Zhang, S. et al. Targeting the microenvironment of vulnerable atherosclerotic plaques: an emerging diagnosis and therapy strategy for atherosclerosis. Adv. Mater. 34, 2110660 (2022).
Fernandez, D. M. et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 25, 1576–1588 (2019).
Stone, P. H., Libby, P. & Boden, W. E. Fundamental pathobiology of coronary atherosclerosis and clinical implications for chronic ischemic heart disease management—the plaque hypothesis: a narrative review. JAMA Cardiol. 8, 192–201 (2023).
Sharifi, M. A. et al. ADAMTS-7 modulates atherosclerotic plaque formation by degradation of TIMP-1. Circ. Res. 133, 674–686 (2023).
Li, W. et al. BBB pathophysiology independent delivery of siRNA in traumatic brain injury. Sci. Adv. 7, eabd6889 (2021).
Acknowledgements
This work was financially supported by the NSFC (824B2057 (J.S.), T2422030 (W.L.), 32471462 (J.Q.), 82572413 (W.L.), and 82172081 (J.Q.)), the CAMS Initiative for Innovative Medicine (2024-I2M-TS-027 (W.L.), and 2025-I2M-XHJC-046 (W.L.)), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2021-RC350-006 (W.L.) and 2025-JKCS-02 (W.L.)), Tianjin Key Science and Technology Project (24ZXZSSS00270 (J.Q.)), and the Fundamental Research Funds for the Central Universities (2024-RC320-07 (W.L.) and 63253263 (J.Q.)).
Author information
Authors and Affiliations
Contributions
W.L. and J.Q. designed the research; J.S., X.K., S.Y., X.M., and Z.S. performed the research; J.S., X.K., W.L., and J.Q. analyzed the data; J.S., W.L., and J.Q. wrote the paper with input from all authors. W.L. and J.Q. obtained funding for data collection and analysis, supervised the study and provided strategic guidance.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, J., Kang, X., Yang, S. et al. A hierarchical theranostic nanoagent for multimodal imaging and targeted foam cell intervention in atherosclerosis. Nat Commun 17, 3794 (2026). https://doi.org/10.1038/s41467-026-70463-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70463-7
