
Abstract
Celiac disease (CeD) is an immune-mediated condition driven by dietary gluten resulting in small intestinal mucosal inflammation and injury, along with myriads of symptoms. The only treatment is a lifelong gluten-free diet (GFD) and although most patients improve, the restriction can lead to nutrient deficiencies including fiber. Duodenal microbiota is known to be altered in CeD, but whether and how the microbial fiber metabolism may be affected is unknown. Here we show that CeD patients had impaired microbial fiber metabolism in the small intestine which associated with depletion of the fiber degrading taxa, Prevotella spp, independent of treatment with the GFD. Colonization of germ-free mice with Prevotella spp increased small intestinal short chain fatty acids (SCFA). In gluten-sensitized mice expressing the celiac risk gene, HLA-DQ8, an inulin-supplemented diet facilitated microbial saccharolytic function and SCFA production to accelerate mucosal healing in the small intestine during the GFD. The results support clinical investigations of dietary fiber supplementation and microbial fiber degradation to enhance responses to the GFD in CeD.
Similar content being viewed by others


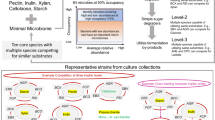
Introduction
Celiac disease (CeD) is an immune-mediated disorder driven by related prolamin proteins termed “gluten” in wheat, rye, and barley1. The disease affects the proximal small intestine, leading to intraepithelial lymphocyte infiltration, villus blunting, and crypt hyperplasia. CeD has autoimmune traits, including HLA-DQ2 or -DQ8 association, anti-tissue transglutaminase 2(TG2) autoantibodies, and immune-mediated intestinal epithelial cell destruction1,2. While about 30% of the global population is genetically predisposed, only 2–3% of those at risk develop the condition3,4. This, tied to the rapid growth in incidence in the past decades, suggests that there are environmental determinants in addition to gluten5,6.
Longitudinal studies in genetically at-risk populations suggest that gastrointestinal infections at early life, especially when combined with high gluten intake, are associated with a higher risk of developing CeD6,7,8. Cross-sectional studies have demonstrated differences in microbiota composition and function in the small intestine of active CeD9,10,11. Indeed, diet is a major driver of microbiota establishment and function, and recent studies have suggested that higher consumption of fiber-rich foods such as vegetables is associated with lower odds of developing celiac autoimmunity5,12. A recent reanalysis of the large longitudinal cohort study, The Environmental Determinants of Diabetes in the Young (TEDDY), suggests an association between increased fiber intake in early life (<3 years) and reduced risk of CeD13. In the colon, short-chain fatty acids (SCFAs; such as acetate, propionate, and butyrate) are produced mainly by microbial fiber metabolism and have been linked to gastrointestinal health through improved motility, anti-inflammatory activity, and microbiome modulation. These effects are primarily mediated through G-protein coupled receptors, GPR41 and GPR43, which are widely expressed on immune and epithelial cells14,15. However, the role of microbial fiber metabolism and SCFA production in the small intestine in the context of CeD remains understudied.
The only current treatment for CeD is the lifelong elimination of gluten. While a gluten-free diet (GFD) is effective in most patients, mucosal healing is slow and often impeded by rapid reactivation of memory cells, likely related to accidental gluten exposure. Although the GFD is usually low in fiber16, especially when composed of processed gluten-free products, little is known about the fiber metabolic capacity of CeD microbiota along the gastrointestinal tract. Indeed, increased fiber intake is often recommended to CeD patients on GFD to manage constipation, without mechanistic insight into microbial saccharolytic capacity or its impact on inflammation.
Our study reveals that patients with active and treated CeD had impaired microbial fiber metabolism in the small intestine independently of differences in fiber intake. Fiber-degrading taxa depleted in the CeD duodenum include Prevotellaceae family members17. Colonization of germ-free NOD/DQ8 mice with Prevotella spp increased SCFA in the small intestine. In gluten-sensitized NOD/DQ8 mice, the soluble fiber inulin accelerated mucosal healing by supporting saccharolytic activity of the small intestinal microbiota. Our results support intervention with dietary fibers, such as inulin, in CeD patients alongside the GFD, to improve mucosal healing, so long as patients harbor the necessary microbial function.
Results
Patient demographics
A total of 16 CeD patients diagnosed within the previous 3 months (newly diagnosed CeD), 11 GFD-treated-CeD patients (T-CeD; on at least >2 years of GFD) and 26 healthy controls were recruited for the study. Details on participant demographics, clinical characteristics and samples collected are summarized in Table 1.
Microbial saccharolytic function in the small intestine was lower in CeD than in controls, independent of GFD
Microbiota composition of duodenal aspirates was assessed using 16S rRNA gene sequencing. Duodenal α-diversity was lower in CeD than in controls or T-CeD (Fig. 1a and Supplementary Fig. 1a), and β-diversity indicated profiles clustered distinctly between the three groups (Fig. 1b). In the newly diagnosed CeD patients, Rothia sp, Granulicatella sp, Leptotrichia wadei, F0058, and Ruminococcus torques group sp relative abundance was higher, while Oribacterium asaccharolyticum, Bergeyella, Alloprevotella, Treponema, Granulicatella elegans, Porphyromonas endodontalis, Prevotella sp, Prevotella pallens, and Actinobacillus sp were lower than in healthy controls. The T-CeD group had higher Rothia sp, Ruminococcus torques group sp, Faecalibacterium prausnitzii, unclassified Veillonellaceae, Rothia mucilagenosa, Ruminococcus bromii, Gemella parahemolysans, Agathobacter sp, unclassified Eubacterium group, unclassified P5D1.392, Blautia massiliensis, Subdoligranulum sp, Eubacterium hallii group sp, Dialister, Fusobacterium nucleatum, Dorea longicatena, Dorea sp, Blautia sp, Haemophilus sp, Anaerostipes hadrus, Lachnoclostridium sp, Ruminococcus sp, Romboustia sp, and Fusicatenibacter saccharivorans and lower relative abundance of Eubacterium nodatum group sp, Gemella sanguinis, Porphyromonas endodontalis, Prevotella pallens, Mogibacterium sp, Johnsonella sp, and Staphylococcus sp compared with healthy controls (Supplementary Fig. 1b).

a Faith’s Phylogenetic Diversity α-diversity of duodenal aspirate microbiota (Ctrl vs CeD, p = 0.0003). b Aitchinson distance β-diversity plot of microbial profiles from duodenal aspirates from controls. Samples were distinctly separated by group using PERMANOVA (p < 0.001; Ctrl vs CeD p = 0.013, Ctrl vs T-CeD p = 0.013, CeD vs T-CeD p = 0.003. c Cumulative abundance of microbial glycoside hydrolase genes in the total PICRUSt2 predicted metagenome from duodenal aspirates. d Relative abundance of α-amylase (K07405) and fructan β-fructosidase (K03332) gene copies from duodenal aspirates (Ctrl vs CeD p = 0.0002). e PICRUSt2 predicted metagenomic contribution of top bacterial genera to genes of interest. Data for (a, c, d) were compared using one-way ANOVA followed by Tukey’s test. For all data, healthy controls (Ctrl; blue, n = 17), celiac disease (CeD; red, n = 7), and treated celiac disease (T-CeD; green, n = 7), and sex was represented by females (squares) and males (circles).
After sequencing, functional predictions using PICRUSt218 revealed no differences in total number of glycosidase hydrolase genes (EC 3.2.1) between CeD, T-CeD, and controls (Fig. 1c). However, specific microbial genes encoding starch-degrading α-amylase (EC 3.2.1.1; K07405) and fructan β-fructosidase (EC 3.2.1.80; K03332), an inulin-like fiber degrading enzyme19, were lower in CeD and T-CeD versus controls (Fig. 1d). The predicted contributions to these genes from the top genera are visualized in Fig. 1e, indicating that the top 4 predicted contributors belonged to Prevotellaceae family, which were less abundant in CeD and T-CeD versus controls (Supplementary Fig. 2a, b). To confirm the PICRUSt2-predicted results, we performed qRT-PCRs for the highest-abundance bacterium identified at the species level (Prevotella 7 melaninogenica) for genes K07405 and K03332. We found that K07405 had a similar RNA expression profile as the predicted gene content, with reduced expression in CeD compared with controls. No statistical differences were observed for K03332 (Supplementary Fig. 2c). Overall, microbial alterations associated with CeD were characterized by lower predicted microbial saccharolytic function, independently of treatment with the GFD.
Diet does not fully explain lower microbial saccharolytic function or gastrointestinal symptoms in CeD
Diet was investigated to determine whether altered microbial predicted function was associated with fiber consumption. In most participants, fiber intake was below the minimum recommendation set by Health Canada (25 g/day)20 based on a food frequency questionnaire (FFQ21), with 86% of T-CeD being fiber-deficient (Fig. 2a). Within the total fiber consumed, there were no differences in the proportion of soluble or insoluble fibers between groups (Fig. 2b). To compare dietary patterns at time of sample collection, plant DNA present in fecal samples was analyzed using FoodSeq22, which indicated lower observed plant richness (total plant species) in T-CeD group compared with controls (Fig. 2c), although Shannon diversity was similar (Fig. 2d). The post-hoc analysis of overall dietary plant composition revealed a significant difference between T-CeD and controls (Fig. 2e), characterized by patterns such as opposing loadings of Oryza sativa (rice) and Poaceae (rye, wheat family), consistent with the elimination of gluten-containing foods. Overall, food sequencing patterns demonstrated GFDs were highly individualized (Supplementary Fig. 3). Patient-reported gastrointestinal symptom scores (GSRS; total score and individual sub-scores) did not correlate with fiber intake, suggesting no obvious relationship with symptoms. Thus, while T-CeD participants had a shift in plant DNA signatures, overall fiber intake was not significantly different from CeD or controls.

a Estimated total daily fiber consumption in controls (blue, n = 5), active CeD (red, n = 6), and T-CeD (green, n = 7). The dashed line represents the minimum daily recommended intake from Health Canada, 25 g fiber/day. b Calculated soluble and insoluble fractions of estimated total fiber in healthy controls (blue, n = 5), active CeD (red, n = 6), and T-CeD (green, n = 7). c Number of distinct plant taxa in healthy controls (blue, n = 23), CeD (red, n = 15), and T-CeD (green, n = 11); (Control vs T-CeD: β = 5.98 ± 2.78, p < 0.05). d Shannon α-diversity of plant taxa in healthy controls (blue, n = 23), CeD (red, n = 15), and T-CeD (green, n = 11). e Principal component analysis of dietary plant composition in healthy controls (blue, n = 23), CeD (red, n = 15), and T-CeD (green, n = 11). Vectors identify top taxa driving variation with red emphasis on Oryza sativa and Poaceae. Overall dietary plant composition was influenced by patient group (PERMANOVA; df = 2, F = 2.04, R² = 0.080, p = 0.001), and specifically between control vs T-CeD: PERMANOVA; df = 1, F = 3.94, R² = 0.110, adjusted p < 0.05. f Fecal SCFA in healthy controls (blue, n = 18), CeD (red, n = 11), and T-CeD (green, n = 8). For SCFA determinations, each symbol represents the average of two technical replicates. Total fecal SCFA Ctrl vs CeD p = 0.00019. Individual fecal SCFA Ctrl vs CeD p = 0.00014). For all data, multiple groups were compared using one-way ANOVA followed by Tukey’s test, and sex was represented by females (squares) and males (circles).
To assess microbial metabolism of fiber in our cohorts, fecal SCFA were measured as a marker of fiber fermentation using gas chromatography mass spectrometry (GC/MS). CeD patients had lower total SCFA compared with controls (Fig. 2f). Specifically, acetic acid levels were lower in CeD compared to controls and T-CeD, while butyric acid levels were lower in both CeD and T-CeD groups as compared to controls. Overall, T-CeD had the lowest fiber intake of the 3 groups, yet these patients had modestly higher SCFA levels than CeD. The results suggest T-CeD partially recovers microbial saccharolytic capacity, despite the sub-optimal amount of fiber ingested by these patients, perhaps due to improvements in digestion and motility.
Inulin accelerates recovery of gluten-immunopathology in NOD/DQ8 mice on GFD
Mice with transgenic expression of the HLA-DQ8 gene on a non-obese diabetic background (NOD/DQ8) were sensitized with digested gluten by oral gavage23 to determine the timing of mucosal recovery from gluten-immunopathology after 2, 6, 10, and 24 weeks of GFD (Supplementary Fig. 4a). CD3+ IELs, villus-to-crypt (V/C) ratios and composite VCIEL scores24 normalized by week 10 of GFD in the gluten-sensitized mice (Supplementary Fig. 4b, c, d). However, small intestinal antibodies against gliadin or TG2 remained elevated in some mice (Supplementary Fig. 5a, b).
Once it was determined that sensitized NOD/DQ8 mice normalized histological parameters at week 10 of GFD, the effect of fiber supplementation was investigated. Another cohort of sensitized NOD/DQ8 mice (Fig. 3a) was given GFD with no added fiber (NAF), or supplemented with prebiotic substrates, either inulin (52 g/kg) or HylonVII (136 g/kg) (described in Table 2), for 6 weeks and clinically monitored. Compared with non-sensitized controls (naïve), sensitized mice had higher CD3+ IELs (Fig. 3b), and lower V/C ratios (Fig. 3c) and lower composite VCIEL scores (Fig. 3d). Supplementation with inulin during GFD improved all histological parameters versus the no-added fiber group. HylonVII did not normalize IELs, but mice had higher V/C ratios versus the no-added fiber group. Neither of the fiber diets normalized antibodies in intestinal content at 6 weeks of follow-up. However, all mice became seronegative after 12 weeks of inulin or HylonVII (Supplementary Fig. 5c, d).

a Schematic protocol for gluten sensitization, wheat diet, and different fibers supplemented during recovery on GFD. Naïve (non-sensitized controls, no added fiber GFD, white n = 9), no added fiber (gray n = 6), inulin (blue n = 6) and HylonVII (yellow n = 6). b CD3+ intraepithelial lymphocytes numbers after six weeks of recovery by diet (n = 9 naïve controls and 6/experimental group). c Villus to-crypt ratio after six weeks of recovery by diet (n = 9 naïve controls and 6/experimental group), Naïve vs NAF p = 0.000009. d Composite VCIEL scores after six weeks of recovery by diet (n = 9 naïve controls and 6/experimental group), where the dashed line represents the baseline determined from naïve controls. Naïve vs NAF p = 0.000002; Naïve vs HylonVII p = 0.0001. e Representative histological images of jejunal tissue after six weeks of recovery by diet. For all data, multiple groups were compared using one-way ANOVA followed by Tukey’s test.
Inulin increases small intestinal SCFA without major changes in microbial profiles
Inulin supplementation during GFD, but not HylonVII, was associated with higher SCFA, primarily acetic acid, in the small intestine of SPF NOD/DQ8 mice, compared with no added fiber (Fig. 4a). There was no difference in microbial α-diversity between no added fiber, inulin, and HylonVII fed mice after 6 weeks (Supplementary Fig. 6a). However, HylonVII fed mice clustered separately from the no added fiber and inulin groups as evaluated by microbial β-diversity (Fig. 4b). Specifically, HylonVII had higher Helicobacter, Alistepes, Aneroplamasma, Intestinimonas, Candidatus saccharimonas, Streptococcus, Gemella and Escherichia/Shigella and lower abundance of Roseburia, Turicibacter, Parasutterella, Desulfovibrio, Dorea, Lachnoclostridium, Muribaculum, Enterorhabdus, A2, Parabacteroides, Aerotruncus, and Marvinbyantia than no added fiber controls. Inulin, which did not affect β-diversity compared with the no-added fiber diet, had higher Faecalibaculum, Gemella, Staphylococcus, Coriobacteriaceae UCG.002, and lower abundance of Intestinimonas, Alistepes, Oscillibacter, Parabacteroides, Aerotruncus, and Marvinbryantia versus the no-added fiber control GFD (Supplementary Fig. 6b). When all groups were considered, higher Escherichia/Shigella (Fig. 4c) and lower Turicibacter and Muribaculum relative abundance were associated with reduced small intestinal SCFAs (Fig. 4d).

a Small intestinal SCFA after 6 weeks of recovery with no added fiber, GFD, inulin (IN), or HylonVII (RS). Each dot represents the average of two technical replicates. b Aitchinson distance β-diversity plot of microbial profiles from mice after six weeks of recovery on no added fiber (NAF) GFD (gray), inulin (blue) and HylonVII (yellow). Samples from HylonVII were distinct from GFD and inulin groups using PERMANOVA (p < 0.01). c Relative abundances of Turicibacter, Muribaculum, and Escherichia/Shigella in the duodenum after 6 weeks of recovery, where each symbol represents the relative abundance of duodenal bacteria in one mouse. d Small intestinal SCFA levels positively correlated with abundances of Turicibacter and Muribaculum, and negatively correlated with Escherichia/Shigella by single-tailed Pearson’s correlation. For (a, c), multiple groups were compared using one-way ANOVA followed by Tukey’s test. For all data n = 6/group.
Inulin and Prevotella spp boost SCFA production in the small intestine
To further investigate small intestinal microbial fiber metabolism in vivo, we first compared SCFA production in germ-free (GF) versus SPF NOD/DQ8 mice in response to inulin (Fig. 5a). Inulin increased total small intestinal SCFA in SPF versus GF mice (Fig. 5b), confirming that fiber metabolism required microbiota presence. This was driven by increases in acetic acid, with no differences in propionic or butyric acids. As expected, the magnitude of SCFA increase was 10 times higher in feces than in the small intestine, also evidencing differences in the main SCFA produced between the two sites (Supplementary Fig. 7). Next, to investigate host responses to local SCFA availability, expression of SCFA receptors, GPR41 and GPR43, was assessed along the intestinal tract. Overall, in SPF mice, inulin increased the mean Gpr41 and Gpr43 expression at most small intestinal sites, including the duodenum (Table 3). No differences in SCFA receptors were observed in GF mice after an inulin-supplemented diet.

a Schematic protocol for diet intervention in non-sensitized germ-free (GF, squares) and specific pathogen-free (SPF, circles) NOD/DQ8 mice were fed either GFD with no added fiber (gray) or supplemented with inulin (blue) for 3 weeks (n = 5/group). b Small intestinal SCFA after 3 weeks of no added fiber GFD (white) or inulin diet (blue) in GF (squares) and SPF (circles) mice. Multiple groups were compared using one-way ANOVA followed by Tukey’s test. Individual SI AA GF + NAF vs SPF+Inulin p = 0.00009; GF+Inulin vs SPF+Inulin p = 0.00021). c Schematic protocol for Prevotella colonization of GF NOD/DQ8 mice (n = 4/group). Mice were given inulin diet and colonized by oral gavage with either Prevotella cocktail (triangles) or saline (squares; vehicle control) on days 1 and 3. d Small intestinal SCFA of GF (square) and Prevotella spp. (triangles) colonized mice after three weeks of diet. Data for total SI SCFA was compared using a 2-tailed Students t Test and data for individual SI SCFA were compared using two-way ANOVA followed by Šidák’s multiple comparison test. Individual SI AA GF+Inulin vs P+Inulin p = 0.0002. AA Acetic acid, PA Propionic acid, BA Butyric acid.
Finally, based on our observation that fiber-degrading bacteria from the Prevotellaceae family were depleted in the duodenum of CeD patients, GF NOD/DQ8 mice were colonized with a cocktail of 10 Prevotellaceae members (Fig. 5c and Table 4). Prevotella-colonized mice, but not GF mice, supplemented with inulin had higher small intestinal SCFA (Fig. 5d). The results demonstrate that small intestinal bacteria, such as Prevotella spp, can produce SCFA from dietary fiber in the small intestine when the appropriate substrate is present.
Discussion
Management of chronic gastrointestinal conditions through dietary approaches is common and is related to the ability of some food substrates to modulate gut microbiota composition or function in a beneficial15 or detrimental manner25. Fiber and its metabolites produced in the lower gastrointestinal tract as a result of microbial metabolism are frequently associated with improved gastrointestinal health14,15, although current dietary guidelines do not always provide evidence-based recommendations for specific diseases. In CeD, inflammation occurs in low-biomass sites, primarily in the duodenum26. Thus, the impact of fiber metabolism on gluten-driven inflammation has not been investigated, despite a high fiber intake often being recommended to newly diagnosed patients alongside the only current therapy, a strict GFD. Here, we report that key fiber-degrading bacteria are depleted in the small intestine of patients with CeD both before and after treatment with a GFD, impairing microbial saccharolytic activity. Furthermore, in genetically predisposed mice sensitized with gluten, we identify a fiber-specific effect that increases small intestinal SCFA production and accelerates recovery from gluten immunopathology after GFD.
Given that a GFD can often be low in fiber, especially when relying on processed gluten-free products, CeD patients are advised to increase fiber intake through diet or supplements, to avoid symptomatic complications such as constipation16,27. There was no relationship between estimated fiber consumption and symptoms in our cohort, although many participants were already consuming lower than recommended fiber. Our study was not designed to investigate fiber tolerance in humans with CeD, as has been done previously in inflammatory bowel disease28,29, but to investigate the impact of diet on microbial saccharolytic function in the small intestine. Results from different large-scale population-based prospective studies have identified a potential role for fiber in CeD management beyond constipation. The data from the Norwegian Mother, Father, and Child Cohort Study (MoBa) indicated that higher maternal fiber consumption was associated with a lower risk of CeD in offspring30, which parallels the findings from the TEDDY cohort, where increased fiber during early life reduced risk as well13. While these studies identified interesting epidemiological associations, they did not investigate underlying mechanisms. Using a cohort of healthy controls, newly diagnosed CeD patients and T-CeD >2 years on a GFD, we show here that many individuals had lower than recommended fiber intake, which tends to further decrease when they adopt a GFD. This fiber deficit is consistent with the Canadian Community Health Survey, which reported inadequate median fiber intake in over 35,000 healthy Canadians31. To further investigate differences in food intake between cohorts objectively, the plant DNA from fecal samples was sequenced. Specific taxa such as Poaceae (wheat, rye family) and Oryza sativa (rice) were identified as opposing pattern drivers between controls and T-CeD. Whole-grain flours derived from wheat, barley, rye, or oats contain considerably more fiber than rice32. Notably, the FoodSeq analysis was used for the first time as a complementary metric to estimate differences in dietary plant intake between unrestricted diets and gluten-free diets through fecal DNA profiling, not to assess ecological dynamics, which is common to microbial DNA profiling. Despite similar abundance and diversity of plant DNA in feces, CeD patients had lower total SCFA than healthy controls. T-CeD had the highest estimated fiber deficiency, and yet demonstrated a partial increase in SCFA compared with the untreated CeD group, which aligns with previous studies showing lower SCFA in untreated CeD children versus patients with partial restoration after the GFD33,34. The reason for this is unclear but could relate to some recovery of microbial function after improvement of small intestinal function and reduced inflammation in T-CeD, as evidenced by increased relative abundance of Faecalibacterium prausnitzii, Ruminococcus bromii, and Blautia sp, which are taxa associated with gut health and immunity through SCFA production35,36,37. Thus, our data support the hypothesis that the lower saccharolytic function in CeD is not due solely to a lack of dietary substrate but rather a primary microbial deficiency. These results thus support future microbial therapy interventions, which could be supplemented by dietary recommendations in this population.
Microbial changes in CeD have been described as factors that can contribute to disease initiation, perpetuation, and resolution5,38. Our results are consistent with previous descriptions that duodenal microbiota profiles differ between active CeD, T-CeD and healthy subjects. Although diagnostic and universal signatures have not been identified due to individual and methodological variability, some taxa are more commonly observed across different studies9. The use of higher resolution sequencing techniques in low biomass samples, such as duodenal aspirates, is not always possible due to the risk of contamination by host DNA. Our analysis using 16S rRNA gene sequencing, a technique previously validated for the small intestine9 found that members of the Prevotellaceae family were less abundant in CeD and T-CeD versus controls, resulting in lower predicted microbial saccharolytic function, independently of treatment with a GFD. Prevotellaceae, a fiber-degrading family39, has been previously reported to be lower in duodenal biopsies of both children and adults with CeD compared with controls11. Prevotellaceae is a functionally diverse family, and some promising species have been involved in immunomodulation within the gastrointestinal tract and shown to ameliorate systemic autoimmunity in experimental autoimmune encephalomyelitis, models of collagen-induced arthritis, and diabetes, with positive effects in phase one trials for autoimmune skin disease40,41.
To further investigate this mechanistically, we used NOD/DQ8 mice, which express moderate immunopathology after sensitization with gluten and a bacterial adjuvant23,42,43. In this previously validated model, severity can be modulated depending on the microbial community mice are colonized with44,45. Here, we demonstrate that an inulin-supplemented diet increased small intestinal SCFA in SPF NOD/DQ8 mice, specifically acetate, without significantly altering microbiota composition compared with HylonVII, and that this was associated with faster histological resolution, compared with the no-added-fiber GFD. It is possible that inulin is more vulnerable to rapid partial digestion in the small intestine compared with HylonVII, a type-2 resistant starch made of linear α−1,4 and branching α−1,6 linkages. Inulin supplementation was associated with increased relative abundance of bacterial taxa associated with fiber fermentation and SCFA production, such as Turicibacter and Muribaculum, which were positively associated with small intestinal SCFA46, while a higher relative abundance of the potentially proinflammatory taxa, such as Escherichia/Shigella47,48 was observed in HylonVII-fed mice. The latter could explain the lower degree of histopathology improvement versus inulin. The relative abundance of Prevotella spp was low in the mouse small intestine and was not significantly affected by either fiber diet. Thus, we designed a mechanistic experiment by colonizing germ-free mice with a collection of Prevotella spp. from our biobank of human-derived bacteria49,50. These species were all confirmed to carry the genes of interest for α-amylase (K07405) and fructan-β-fructosidase (K03332), and in colonized mice, they increased small intestinal SCFA upon inulin supplementation. The Prevotellaceae family contains diverse strain-level variation, which contributes to different microbial metabolic profiles in humans51. Certain species, such as Prevotella copri, are highly influenced by diet and can alter their genome expression based on the host diet when fiber is deprived52. In this case, Prevotellaceae represents a known fiber- degrading family that can be present in the oral and duodenal microbiota at high abundance. Whether there are other microbial probiotic candidates for future therapeutics that can improve saccharolytic activity in the small intestine remains to be investigated. A previous seminal study postulated that long-term fiber inadequacy could cause loss of fiber-degrading taxa in the gut, which might not be restored by increased fiber without supplemental microbiota53. Thus, while some gluten-free products are now formulated with a higher fiber content54, overall, our results indicate the need to optimize both substrate and microbial metabolizers during the GFD, likely in a personalized manner. Our dietary intervention was designed to investigate the effect of fiber on the small intestinal microbiome and how that related to intestinal healing in genetically susceptible sensitized mice, not to determine the relevant dose of fiber sufficient to increase microbial activity, promote healing, or impact symptoms in human CeD patients. Our study should encourage future clinical investigation with prospective trials to further elucidate the relationship between fiber and gastrointestinal symptoms in CeD.
Our study has limitations. Our cohort was small and exploratory. The FFQ was not specifically designed to assess the GFD, and not all participants completed the questionnaire. However, we supplemented the FFQ with an objective dietary sequencing method, FoodSeq, which investigated plant DNA in fecal samples at the time of sample collection. There was limited availability of relevant small intestinal samples for microbial metabolic analysis. We could not measure small intestinal SCFA directly in human duodenal aspirates, which is technically challenging due to the patient preparation (e.g., overnight fasting) and low concentrations of fiber metabolites in the sample. Instead, we predicted the capacity for microbial small intestinal fiber metabolism in the human aspirates using PICRUSt2 and performed mechanistic validation studies in mice with humanized microbiota. For microbiome analysis, next-generation sequencing such as shotgun metagenomics may provide more robust functional analysis than 16S rRNA gene sequencing, especially if they include jejunum or ileum, but could not be completed in our low-biomass duodenal aspirates due to high levels of host contamination55,56. We used a previously validated 16S rRNA gene sequencing protocol9, and performed qRT-PCR for targeted measurement of gene expression. Finally, the NOD/DQ8 mouse employed here requires sensitization and the use of a microbial adjuvant and is well suited for the study of environmental triggers or modulators of disease severity and fulfills the validity criteria of animal models57, such as the ability to produce mechanistic insight for clinical trial validation. This translational study provides unique insight into the relevance of fiber supplementation in improving gluten-specific immunity through microbial metabolism, a finding that warrants further evaluation in clinical trials using human-equivalent doses.
In summary, the duodenum constitutes a unique microbial niche that should be considered distinct from the rest of the intestine when investigating the role of fiber metabolism in the prevention and treatment of diseases that affect the small bowel primarily. Our study provides clinical evidence that the small intestinal microbiota in CeD contains less fiber-degrading bacteria, such as Prevotellaceae, independently of diet and provides preclinical evidence that certain fibers could be used as adjuvant therapy for the GFD to promote mucosal healing, when metabolizing taxa are present.
Methods
Human participants
Patients attending the McMaster University Celiac Clinic were recruited after signing informed consent. New cases of CeD (consuming gluten) were diagnosed based on positive serological markers, the presence of either HLA-DQ2 or HLA-DQ8, and evaluation of duodenal biopsies (n = 16). Treated CeD (T-CeD) included patients with a verified diagnosis, who were on GFD > 2 years and attending the clinic as part of their follow-up (n = 11). Healthy controls included patients attending the McMaster University Digestive Diseases Clinic with a history of gastroesophageal reflux disease without other symptoms, and/or having confirmed normal endoscopy and biopsy, negative celiac serology, and negative Helicobacter pylori. They also did not fulfill the criteria for disorders of gut-brain interaction (DGBI) based on Rome IV criteria58. All participants were asked to complete a validated food frequency questionnaire (Victoria Cancer Council Dietary Questionnaire for Epidemiological Studies v2) to assess their habitual diet, and a validated gastrointestinal symptoms ratings score (GSRS)59,60 to assess symptoms. No participant had prescribed proton pump inhibitors, antibiotics, probiotics, or fiber supplementation at the time of sample collection. Sex, but not gender information, was collected and included in the analysis. The study was registered at clinicaltrials.gov (ID: NCT06532110) and approved by the Hamilton Integrated Research Ethics Board (HiREB # 15311). For details, see Supplementary Data 1.
Human sample collection
Fecal samples were collected and stored in anaerobic packages. After transport to the hospital, the samples were aliquoted in an anaerobic chamber (90% CO2, 5% H2, 5% N2)61. Duodenal aspirates were collected during endoscopy, mixed with 10% glycerol and aliquoted in aerobic conditions62. All samples were frozen at −80 °C until analysis.
Mouse model
Breeding pairs of non-obese diabetic AB° DQ8 (NOD/DQ8) mice63 were originally received from Dr. Joseph Murray (Mayo Clinic, USA)43. Female and male specific-pathogen-free (SPF) mice were bred and housed in the McMaster Central Animal Facility (CAF) and were 8–10 weeks old and sex-matched for all experimental groups. SPF mice were housed in clean rooms, with ad libitum water and an irradiated grain-based diet formulated without gluten (GFD, TD.05620, Envigo; Table 2). The rooms had a 12-h dark/light cycle, and at room temperature and ambient humidity was maintained between 40–60%. Germ-free (GF) NOD/DQ8 mice were generated by two-stage embryo transfer and housed within the McMaster Axenic Gnotobiotic Unit (AGU) in flexible film isolators with free access to autoclaved food and water44. Diets were autoclaved to ensure sterility. Mice were sacrificed under anaesthesia (Isofluorane) using cervical dislocation before necropsy and tissue collection. All experiments were conducted with approval from the McMaster University Animal Care Committee and McMaster Animal Research Ethics Board (AREB) under the Animal Utilization Protocol (AUP# 210930).
Gluten sensitization and dietary interventions
To minimize litter and cage effects, littermates were distributed across treatment groups using random assignment to sex-balanced cages before sensitization with gluten using an established protocol where mice were gavaged with 25 μg cholera toxin and 1 mg of pepsin/trypsin-digested gliadin once a week for 3 weeks23. Mice were then switched from a gluten-free diet (TD.05620) to a calorically balanced gluten-containing wheat-based diet (TD.200056) for 3 weeks, and finally returned to the GFD for recovery over 2, 6, 10, or 24 weeks. Age-matched naïve controls (non-sensitized on GFD with no gluten exposure) were used to determine normal and baseline values and compare against experimental groups. Mice receiving fiber-supplemented diets were transitioned over the first week with a 50:50 mixture of no added fiber GFD and either 52.8 g/kg inulin (IN, TD.2100884) diet or 136 g/kg HylonVII (RS, TD.210885) to allow the mice to acclimatize. The total fiber content in the supplemented diets was increased to approximately 15% from a grain-based chow formula with ~7.5% dietary fiber. Diet composition is listed in Table 2 and was calculated based on allometric species nutritional requirements and not direct mass-to-mass conversions64. Mice were monitored for signs of distress, changes in food consumption, weight gain, and fecal output through the duration of the diet intervention study.
Evaluation of gluten-immunopathology
Cross sections from the proximal small intestine were fixed in 10% formalin and embedded in paraffin for blinded evaluation using brightfield microscopy (Olympus) using open-source QuPath software65. The CD3+ intraepithelial lymphocytes (IELs) were quantified by immunohistochemical staining23,66. The primary antibody, polyclonal rabbit antihuman CD3 (A045229-2; Agilent), was diluted 1:1000 (1 μL in 1 mL of 1% BSA in PBS) and incubated overnight. CD3+ IELs were visualized with 3,3’-diamonobenzideine (DAB+ substrate chromogen system, K346811-2; Agilent) and counterstained with Mayer’s hematoxylin (S330930-2; Agilent) for 60 s. CD3+ IELs were quantified in 5 randomly chosen villi tips and reported as the number of IELs per 100 enterocytes. The same paraffin-embedded sections were stained with hematoxylin and eosin for villus-height and crypt-depth measurements for the villus-to-crypt ratio (V/C). Ten villi and adjacent crypt pairs were randomly selected and measured. An equally weighted composite VCIEL scoring system was used to combine histological measures and create a standardized baseline around the naïve control group24. At necropsy, the small intestine was washed out and contents collected in 0.05 M EDTA in PBS with soy trypsin inhibitor (0.1 mg/mL; T9128-1G; Sigma) and phenylmethanesulfonyl fluoride (0.8 mM; P7626; Sigma-Aldrich) to prevent protein degradation. Direct ELISA was performed to quantify anti-TG2 or anti-gliadin IgA antibodies in mouse luminal contents23,66. 96-well plates were coated with either transglutaminase in PBS (0.1 μg per well; T5398-2UN; Sigma-Aldrich) or gliadin dissolved in 70% ethanol and diluted in PBS (5 μg per well; G3375; Sigma-Aldrich). Non-specific binding was blocked with 2% BSA in PBS-Tween before a 2-h incubation of 50 μL small intestinal wash solution in triplicate. Detection with 1 h incubation with HRP-conjugated secondary anti-mouse IgA antibody (diluted 1:5000; 1040-05; Southern Biotech) followed by TMB substrate (Agilent) reaction for 20 min before measuring absorbance at 450 nm using a Spectramax M3 plate reader (Molecular Devices). OD values were normalized to the total amount of protein in the sample using a protein quantification assay (Bio-Rad) according to the manufacturer’s instructions.
Prevotellaceae cocktail colonization
Ten isolated strains from the Prevotellaceae family were obtained from the collection of Dr. Michael Surette’s Lab (McMaster University49,50), based on their reduced abundance in CeD (Table 4). Isolates’ identities were confirmed with MALDI-TOF and cultured anaerobically in BHI-3 for 48 h with agitation. NOD/DQ8 mice were gavaged with PBS (negative control) or approximately 1 million cells of each strain combined, given on days 1 and 3 of the inulin diet.
Metabolite determinations with gas chromatography mass-spectrometry
A targeted selected ion monitoring (SIM) gas chromatography mass-spectrometry (GC-MS) protocol was used to quantify SCFA in the small intestinal washes and feces collected at end point. Samples were extracted with 10% isobutanol before the addition of 125 µL of 20 mM NaOH and 400 µL of chloroform. After mixing, 400 µL of the upper aqueous phase was transferred to a new tube before the addition of 80 µL isobutanol, 100 µL pyridine, and 70 µL H2O. Finally, 50 µL of isobutyl-chloroformate and 150 µL of hexane were added, mixed, and the upper aqueous phase was transferred to pre-slit autosampler vials. GC-MS measurements were carried out on an Agilent 5975 C inert mass selective detector (MSD) equipped with an Agilent 7890 A GC and an Agilent 7693 autosampler (Agilent, Santa Clara, CA, USA), in the Centre for Microbial Chemical Biology (CMCB) at McMaster University. Separation of the analytes was achieved on an Agilent (Santa Clara, CA, USA) DB-1MS UI (30 m x 0.25 mm × 0.25 µm) column. The injector, ion source, quadrupole and the GC-MS interface temperatures were 260, 230, 150, and 280 °C, respectively. The flow rate of helium carrier gas was kept at 1 mL/min. 1 µL of derivatized sample was injected in duplicate with a 4.5 min solvent delay time and split ratio of 10:1. The initial column temperature was 40 °C and held for 5 min, ramped to 165 °C at the rate of 10 °C/min, and then increased to 310 °C at the rate of 100 °C/min and kept at this temperature for 3 min. The total run time was 22 min. The ionization was carried out in the electron impact (EI) mode at 70 eV. The MS data were acquired in the SIM mode using GC-MS MassHunter and MS quantitative analysis. The identification of compounds was confirmed by the injection of pure standards and comparison of the retention time and target ions.
RNA extraction and quantitative reverse-transcription PCR (RT-qPCR)
Mouse tissue samples from the duodenum, jejunum, ileum, and proximal colon were collected in RNALater (Invitrogen) before RNA extraction using the RNEasy Mini kit (Qiagen) according to the manufacturer’s instructions. Extracted RNA was used for cDNA synthesis using iScript Reverse Transcriptase (Bio-Rad) with SsoFast EvaGreen Supermix (Bio-Rad) according to the manufacturer’s instructions. The qPCR cycling consisted of initial denaturation at 95 °C for 3 min, followed by 40 cycles of denaturation at 95 °C for 10 s and annealing/extension at 65 °C for 30 s. GAPDH was used as an endogenous control for normalization of gene expression, and data were reported as the fold change in expression in response to inulin compared to GFD alone. Validated primer sequences for mouse GPR41 (142345193c1) and GPR43 (22122727a1) were obtained from Primer Bank50. Bacterial RNA was extracted from human duodenal aspirate samples using a Stool Total RNA purification kit (Norgen Biotek) and cDNA was synthesized using SuperScriptTM IV Reverse Transcriptase (Invitrogen) with random hexamer primers (Thermo Scientific) according to manufacturer instructions. RNA quality was measured using the differential ratio amplicons (RAMP) method of the bacterial glutamine synthetase A transcript (glnA)67 as previously reported for feces61,68. Samples with negative RAMP were removed from analysis. Primers for α-amylase (K07405) and fructan β-fructosidase (K03332) were designed using Primer3 using the following sequences for K07405 (5’−3’: AGGGAATGTTGACAGAGGGC and AAGCGGAAGTTGTCACTTGC) and K03332 (5’−3’ CAGCGTATCTCTGGGCAGAA and AGATGATGGCCCCTTCTCCA). The qRT-PCR cycling consisted of initial denaturation at 95 °C for 3 min, followed by 45 cycles of denaturation at 95 °C for 10 s, annealing at 62 °C for 30 s, and extension at 72 °C for 30 s, and normalized to the expression of glnA. Relative quantitation was performed using standard curves from serial dilutions of the PCR product of P. melaninogenica K07405 and K0333269.
Microbiota sequencing and predicted microbial function
After genomic DNA extraction, an established protocol consisting of a 2-stage nested polymerase chain reaction (PCR) to amplify the variable regions 3–4 of the 16S rRNA gene from low biomass samples was used to sequence the mouse duodenum and human aspirates using the MiSeq Illumina platform9. Sequences were then processed in R, version 4.4.2, using the package Divisive Amplicon Denoising Algorithm 2 (DADA270) and the SILVA reference database, version 138.171. FastTree 272 was used to calculate a phylogenetic tree of sequences, and data were explored using the phyloseq package73. Functional predictions based on 16S rRNA sequences were made using PICRUSt218, annotated using the Kyoto Encyclopedia of Genes and Genomes database74. Beta-Diversity was calculated using the Aitchison distance75, and differences between groups were calculated using PERMANOVA. Taxonomic and functional microbial differences were evaluated using a generalized linear mixed model with a negative binomial distribution76 with adjustment for age and sex as confounding variables for patient data. Statistical differences between groups were identified using estimated marginal means using the emmeans package77. 16S rRNA gene sequences were deposited in Sequence Read Archive (PRJNA1256891).
FoodSeq (Sequencing, bioinformatic pipeline, and diversity analysis)
We performed DNA extraction on randomized fecal samples using the DNeasy PowerSoil Pro kit (Qiagen) following the manufacturer’s instructions. We prepared libraries using a two-step PCR process78 using trnL g and h primers79,80,81. Primary PCR amplification was conducted as previously described81. Secondary PCR amplification contained 5 μL each of 2.5 μM forward and reverse indexing primers, 25 μL of 2X KAPA HiFi HotStart Ready Mix, 0.5 μL of 100X SYBR Green I, 9.5 μL nuclease-free water, and 5 μL of primary PCR product diluted 1:100 in nuclease-free water. Amplicon libraries were then cleaned and pooled as previously described81. Paired-end sequencing was carried out on an Illumina MiniSeq using a 300-cycle Mid kit (Illumina). We demultiplexed, trimmed, filtered, and denoised sequence reads using a QIIME282 pipeline v. 2023.5.1 with DADA270 plugin. Sequence reads were aligned to a custom trnL reference database81 (available at https://github.com/LAD-LAB/mbpipeline/blob/main/reference/trnLGH.fasta), and taxonomic assignments were made using assignSpecies() in the DADA2 R package v. 1.30.0. We calculated observed richness and Shannon diversity using estimate_richness() in the phyloseq73 R package v. 1.46.0 for each sample after removing taxa unassigned to any dietary species in our reference database. Principal component analysis was performed using prcomp(), centered and not scaled, on CLR-transformed relative abundance data. A distance matrix was then generated using Euclidean distance calculations. Prior to PERMANOVA, homogeneity of dispersion was assessed using betadisper(). PERMANOVA was performed using adonis2() in the vegan R package v. 2.6.6.183 with 999 permutations, adjusting for age, sex, ethnicity/race, with posthoc pairwise comparisons using pairwiseAdonis() v. 0.4.184, adjusting p-values using the Bonferroni method. Age was significant in the PERMANOVA model (df = 1, F = 1.41, R2 = 0.028, p = 0.039) but showed significant dispersion (p < 0.05), indicating heterogeneous variance in dietary composition across ages, and was therefore interpreted with caution due to the assumption violation. The trnL gene sequences were deposited in the Sequence Read Archive (PRJNA1256896)
Statistical analysis
Data are presented for each individual sample (human or mouse). Graphpad Prism Version 10.4.2 was used for most statistical analyses and graph creation. R version 4.4.2 was used for the analysis of gene sequences. In Prism, all data were first tested for statistical outliers for exclusion (ROUT, Q = 1%). For analysis in R, outliers were excluded if they exceeded 3 times the interquartile range. Data normality was then checked using the Kolmogorov-Smirnov test, such that non-parametric tests were used for non-normally distributed data. For comparisons between two groups, the Student’s t test or the nonparametric Mann–Whitney was used. For three or more groups, one-way analysis of variance (ANOVA) was used with Tukey’s post hoc test, or the nonparametric Kruskal-Wallis followed by Dunn’s test. A minimum threshold of p < 0.05 was used to determine statistically significant effects.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data produced in this study are available in the NCBI database. The data generated to create the main figures is uploaded to Figshare [https://figshare.com/s/3a4043d02b3d58d8cade]. The gene sequences have been uploaded to the Sequence Read Archive: PRJNA1256891 [https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1256891], 16S rRNA gene sequences for both human and mouse microbiota. PRNJA1256896 [https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1256896] trnl gene sequences (FoodSeq).
References
Catassi, C., Verdu, E. F., Bai, J. C. & Lionetti, E. Coeliac disease. Lancet 399, 2413–2426 (2022).
Oberhuber, G., Granditsch, G. & Vogelsang, H. The histopathology of coeliac disease: time for a standardized report scheme for pathologists. Eur. J. Gastroenterol. Hepatol. 11, 1185–1194 (1999).
King, J. A. et al. Incidence of celiac disease is increasing over time: a systematic review and meta-analysis. Am. J. Gastroenterol. 115, 507–525 (2020).
Gujral, N., Freeman, H. J. & Thomson, A. B. R. Celiac disease: prevalence, diagnosis, pathogenesis and treatment. World J. Gastroenterol. 18, 6036–6059 (2012).
Galipeau, H. J., Hinterleitner, R., Leonard, M. M. & Caminero, A. Non-host factors influencing onset and severity of celiac disease. Gastroenterology 167, 34–50 (2024).
Jiang, H., Zhang, X., Zhou, Y., Jiang, C. & Shi, Y. Infection, antibiotic exposure, and risk of celiac disease: a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 35, 557–566 (2020).
Kemppainen, K. M. et al. Factors that increase the risk of celiac disease autoimmunity after a gastrointestinal infection in early life. Clin. Gastroenterol. Hepatol. 15, 694–702.e5 (2017).
Mårild, K., Kahrs, C. R., Tapia, G., Stene, L. C. & Størdal, K. Infections and risk of celiac disease in childhood: a prospective nationwide cohort study. Am. J. Gastroenterol. 110, 1475–1484 (2015).
Constante, M. et al. Biogeographic variation and functional pathways of the gut microbiota in celiac disease. Gastroenterology 163, 1351–1363.e15 (2022).
Caminero, A. et al. Duodenal bacteria from patients with celiac disease and healthy subjects distinctly affect gluten breakdown and immunogenicity. Gastroenterology 151, 670–683 (2016).
Nistal, E. et al. Differences of small intestinal bacteria populations in adults and children with/without celiac disease: effect of age, gluten diet, and disease. Inflamm. Bowel Dis. 18, 649–656 (2012).
Barroso, M. et al. Dietary patterns after the weaning and lactation period are associated with celiac disease autoimmunity in children. Gastroenterology 154, 2087–2096.e7 (2018).
Hård Af Segerstad, E. M. et al. Early dietary fiber intake reduces celiac disease risk in genetically prone children: insights from the TEDDY study. Gastroenterology https://doi.org/10.1053/j.gastro.2025.01.241 (2025).
Gill, P. A., van Zelm, M. C., Muir, J. G. & Gibson, P. R. Review article: short chain fatty acids as potential therapeutic agents in human gastrointestinal and inflammatory disorders. Aliment Pharmacol. Ther. 48, 15–34 (2018).
Gill, S. K., Rossi, M., Bajka, B. & Whelan, K. Dietary fibre in gastrointestinal health and disease. Nat. Rev. Gastroenterol. Hepatol. 18, 101–116 (2021).
Pinto-Sanchez, M. I., Blom, J.-J., Gibson, P. R. & Armstrong, D. Nutrition assessment and management in celiac disease. Gastroenterology 167, 116–131.e1 (2024).
Prasoodanan, P. K. et al. Western and non-Western gut microbiomes reveal new roles of Prevotella in carbohydrate metabolism and the mouth–gut axis. NPJ Biofilms Microbiomes 7, 77 (2021).
Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688 (2020).
Tanno, H. et al. Characterization of fructooligosaccharide metabolism and fructooligosaccharide-degrading enzymes in human commensal butyrate producers. Gut Microbes 13, 1869503 (2021).
Trumbo, P., Schlicker, S., Yates, A. A. & Poos, M. Dietary reference intakes for energy, carbohydrate, fiber, fat, fatty acids, cholesterol, protein and amino acids. (Commentary). J. Am. Diet. Assoc. 102, 1621 (2002).
Hebden, L., Kostan, E., O’Leary, F., Hodge, A. & Allman-Farinelli, M. Validity and reproducibility of a food frequency questionnaire as a measure of recent dietary intake in young adults. PLoS ONE 8, e75156 (2013).
Petrone, B. L. et al. A pilot study of metaproteomics and DNA metabarcoding as tools to assess dietary intake in humans. Food Funct. 16, 282–296 (2025).
Galipeau, H. J. et al. Sensitization to gliadin induces moderate enteropathy and insulitis in nonobese diabetic-DQ8 mice. J. Immunol. 187, 4338–4346 (2011).
Syage, J. A. et al. A composite morphometric duodenal biopsy mucosal scale for celiac disease encompassing both morphology and inflammation. Clin. Gastroenterol. Hepatol. 22, 1238–1244.e3 (2024).
Gibson, P. R., Halmos, E. P. & Muir, J. G. Review article: FODMAPS, prebiotics and gut health-the FODMAP hypothesis revisited. Aliment Pharmacol. Ther. 52, 233–246 (2020).
Murray, J. A. et al. Mucosal atrophy in celiac disease: extent of involvement, correlation with clinical presentation, and response to treatment. Clin. Gastroenterol. Hepatol. 6, 186–193 (2008).
Chang, L. et al. American gastroenterological association-american college of gastroenterology clinical practice guideline: pharmacological management of chronic idiopathic constipation. Gastroenterology 164, 1086–1106 (2023).
Armstrong, H. K. et al. Unfermented β-fructan fibers fuel inflammation in select inflammatory bowel disease patients. Gastroenterology 164, 228–240 (2023).
Kuang, R. & Binion, D. G. Should high-fiber diets be recommended for patients with inflammatory bowel disease? Curr. Opin. Gastroenterol. 38, 168–172(2022).
Lund-Blix, N. A. et al. Maternal fibre and gluten intake during pregnancy and risk of childhood celiac disease: the MoBa study. Sci. Rep. 10, 16439 (2020).
Bédard, M. & Ledrou, I. Canadian Community Health Survey Cycle 2.2, Nutrition (2004). https://publications.gc.ca/pub?id=9.650206&sl=0 (Health Canada, 2006).
Joye, I. J. Dietary fibre from whole grains and their benefits on metabolic health. Nutrients 12, 3045 (2020).
Raffaella, D. C. et al. Different Fecal microbiotas and volatile organic compounds in treated and untreated children with celiac disease. Appl. Environ. Microbiol. 75, 3963–3971 (2009).
Di Cagno, R. et al. Duodenal and faecal microbiota of celiac children: molecular, phenotype and metabolome characterization. BMC Microbiol. 11, 219 (2011).
Martín, R. et al. Faecalibacterium: a bacterial genus with promising human health applications. FEMS Microbiol. Rev. 47, fuad039 (2023).
Kim, Y.-J., Jung, D.-H. & Park, C.-S. Important roles of Ruminococcaceae in the human intestine for resistant starch utilization. Food Sci. Biotechnol. 33, 2009–2019 (2024).
Holmberg, S. M. et al. The gut commensal Blautia maintains colonic mucus function under low-fiber consumption through the secretion of short-chain fatty acids. Nat. Commun. 15, 3502 (2024).
Caminero, A. et al. Potential role of microbes in antigen-based food sensitivities. Semin Immunol. 79, 101982 (2025).
Tett, A., Pasolli, E., Masetti, G., Ercolini, D. & Segata, N. Prevotella diversity, niches and interactions with the human host. Nat. Rev. Microbiol. 19, 585–599 (2021).
Bodkhe, R. et al. Human gut-derived commensal suppresses the generation of T-cell response to gliadin in humanized mice by modulating gut microbiota. Anaerobe 68, 102237 (2021).
Balakrishnan, B. et al. Small intestinal-derived Prevotella histicola simulates biologic as a therapeutic agent. Sci. Rep. 14, 29217 (2024).
Karell, K. et al. HLA types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: results from the European Genetics Cluster on Celiac Disease. Hum. Immunol. 64, 469–477 (2003).
Marietta, E. et al. A new model for dermatitis herpetiformis that uses HLA-DQ8 transgenic NOD mice. J. Clin. Investig. 114, 1090–1097 (2004).
Galipeau, H. J. et al. Intestinal microbiota modulates gluten-induced immunopathology in humanized mice. Am. J. Pathol. 185, 2969–2982 (2015).
Caminero, A. et al. Duodenal bacterial proteolytic activity determines sensitivity to dietary antigen through protease-activated receptor-2. Nat. Commun. 10, 1198 (2019).
Lynch, J. B. et al. Gut microbiota Turicibacter strains differentially modify bile acids and host lipids. Nat. Commun. 14, 3669 (2023).
Baltazar-Díaz, T. A. et al. Escherichia/Shigella, SCFAs, and metabolic pathways-the triad that orchestrates intestinal dysbiosis in patients with decompensated alcoholic cirrhosis from Western Mexico. Microorganisms 10, 1231 (2022).
Hu, J. et al. Correlation between altered gut microbiota and elevated inflammation markers in patients with Crohn’s disease. Front. Immunol. 13, 947313 (2022).
Lau, J. T. et al. Capturing the diversity of the human gut microbiota through culture-enriched molecular profiling. Genome Med. 8, 72 (2016).
Whelan, F. J. et al. Culture-enriched metagenomic sequencing enables in-depth profiling of the cystic fibrosis lung microbiota. Nat. Microbiol. 5, 379–390 (2020).
Abdelsalam, N. A., Hegazy, S. M. & Aziz, R. K. The curious case of Prevotella copri. Gut Microbes 15, 2249152 (2023).
De Filippis, F. et al. Distinct genetic and functional traits of human intestinal <em>Prevotella copri</em> strains are associated with different habitual diets. Cell Host Microbe 25, 444–453.e3 (2019).
Sonnenburg, E. D. et al. Diet-induced extinctions in the gut microbiota compound over generations. Nature 529, 212–215 (2016).
González, M. P. et al. Determination and comparison of fat and fibre contents in gluten-free and gluten-containing flours and breads: nutritional implications. Foods https://doi.org/10.3390/foods14050894 (2025).
Jovel, J. et al. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front. Microbiol. 7, 2016 (2016).
Caminero, A. et al. Credible inferences in microbiome research: ensuring rigour, reproducibility and relevance in the era of AI. Nat. Rev. Gastroenterol. Hepatol. https://doi.org/10.1038/s41575-025-01100-9 (2025).
Britton, R. A. et al. Taking microbiome science to the next level: recommendations to advance the emerging field of microbiome-based therapeutics and diagnostics. Gastroenterology 167, 1059–1064 (2024).
Sperber, A. D. et al. Worldwide prevalence and burden of functional gastrointestinal disorders, results of the Rome Foundation global study. Gastroenterology 160, 99–114.e3 (2021).
Svedlund, J., Sjödin, I. & Dotevall, G. GSRS—A clinical rating scale for gastrointestinal symptoms in patients with irritable bowel syndrome and peptic ulcer disease. Dig. Dis. Sci. 33, 129–134 (1988).
Kulich, K. R. et al. Reliability and validity of the gastrointestinal symptom rating scale (GSRS) and quality of life in reflux and dyspepsia (QOLRAD) questionnaire in dyspepsia: a six-country study. Health Qual. Life Outcomes 6, 12 (2008).
Santiago, A. et al. Crohn’s disease proteolytic microbiota enhances inflammation through the PAR2 pathway in gnotobiotic mice. Gut Microbes 15, 2205425 (2023).
Rueda, G. H. et al. Oral tryptophan activates duodenal aryl hydrocarbon receptor in healthy subjects: a crossover randomized controlled trial. Am. J. Physiol. Gastrointest. Liver Physiol. 326, G687–G696 (2024).
Kudva, Y. C. et al. Modulation of insulitis and type 1 diabetes by transgenic HLA-DR3 and DQ8 in NOD mice lacking endogenous MHC class II. Hum. Immunol. 63, 987–999 (2002).
Rucker, R. Allometric scaling: comparison of interspecies nutritional relationships and requirements. J. Nutr. 152, 2626–2627 (2022).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017).
Bruno, L. et al. Evaluating the effects of chronic oral exposure to the food additive silicon dioxide on oral tolerance induction and food sensitivities in mice. Environ. Health Perspect. 132, 27007 (2024).
Cholet, F., Ijaz, U. Z. & Smith, C. J. Differential ratio amplicons (Ramp) for the evaluation of RNA integrity extracted from complex environmental samples. Environ. Microbiol. 21, 827–844 (2019).
Czerucka, D. & Rampal, P. Diversity of Saccharomyces boulardii CNCM I-745 mechanisms of action against intestinal infections. World J. Gastroenterol. 25, 2188–2203 (2019).
Makui, H., Soares, R. J., Jiang, W., Constante, M. & Santos, M. M. Contribution of Hfe expression in macrophages to the regulation of hepatic hepcidin levels and iron loading. Blood 106, 2189–2195 (2005).
Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 (2013).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
Aitchison, J., Barceló-Vidal, C., Martín-Fernández, J. A. & Pawlowsky-Glahn, V. Logratio analysis and compositional distance. Math. Geol. 32, 271–275 (2000).
Zhang, X. et al. Negative binomial mixed models for analyzing microbiome count data. BMC Bioinforma. 18, 4 (2017).
Lenth, R. emmeans: Estimated marginal means, aka least-squares means. R package version 1.7.2. (2022).
Gohl, D. M. et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol. 34, 942–949 (2016).
Taberlet, P. et al. Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res. 35, e14 (2007).
Reese, A. T. et al. Using DNA metabarcoding to evaluate the plant component of human diets: a proof of concept. mSystems 4, e00458–19 (2019).
Petrone, B. L. et al. Diversity of plant DNA in stool is linked to dietary quality, age, and household income. Proc. Natl. Acad. Sci. USA. 120, e2304441120 (2023).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Oksanen, J. et al. Vegan community ecology package, version 2.0–8: R package version 3.4.0. http://vegan.r-forge.r-project.org/ (2024).
Martinez Arbizu, P. PairwiseAdonis: Pairwise multilevel comparison using adonis. R package version 0.4.1 (2020).
Acknowledgements
The authors thank McMaster’s AGU staff for their support with germ-free mouse breeding and gnotobiotic experiments. The authors also thank the CeD volunteers who participated in this study. The work was funded by a CIHR grant MOP#168840 to E.F.V., who holds a Tier 1 Canada Research Chair. M.W. received the JA Campbell Young Investigator Award from Celiac Canada. This work was funded in part by the Chan Zuckerberg Initiative, Schmidt Sciences, Gerber, Nature-Springer, Burroughs Wellcome Fund Pathogenesis of Infectious Disease Award, Duke Microbiome Center, Duke Clinical and Translational Science Award, and National Institute of Diabetes and Digestive and Kidney Diseases (R01-DK116187, R01-DK128611, and T32DK007568). PB holds the GlaxoSmithKline chair in Gastroenterology. AC and DA hold the Douglas Family Chairs in Intestinal Research and Nutrition Research, respectively. We would like to thank Nikki Henriquez for training and supporting M.W. in the CMCB.
Author information
Authors and Affiliations
Contributions
M.W. and E.F.V. conceptualized the study. M.W. performed most of the experiments and analyzed the data. M.C. completed bioinformatics analysis for microbiota data and assisted with molecular analysis. H.J.G. completed germ-free experiments within the AGU. J.B. processed and analysed FFQ data. G.R. facilitated access to clinical samples and demographic information from patients attending the McMaster adult Celiac Clinic with M.I.P.S. and gastroscopy with P.B. and D.A. Fecal FoodSeq analysis was conducted by D.K.S., S.J., and L.A.D., who also supported writing those sections. J.A.M. sent NOD/DQ8 mice. N.E. and M.G.S. provided Prevotella strains and valuable expertise on microbiota experiments and analysis. M.W., H.J.G., A.C., and E.F.V. wrote the manuscript. All of the authors discussed the results and editing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare the following competing interests: M.W. is a co-chair for the International Society for the Study of Celiac Disease Young Investigator committee. H.J.G. is a member of the Research Committee for the Society for the Study of Celiac Disease. L.A.D. consults for City of Hope on dietary assessment technology and is a member of the Global Grants for Gut Health Colloquium (Nature Portfolio/Yakult). D.A. is the Co-founder and Chief Innovation Officer of A.I. VALI Inc.; a member of the advisory board for Cinclus Pharma, Crohn’s and Colitis Canada, Phathom Pharmaceuticals, and Sanofi; a board member of the Canadian Digestive Health Foundation, International Working Group for the Classification of Oesophagitis; has received speaker’s fees from Ferring and Takeda; and research funding from Nestle Health Sciences, Weston Family Foundation, INOVAIT, and Digital Health Innovation Fund. J.A.M. reports grants from: National Institutes of Health, Department of Defence, Takeda Pharmaceutical, Sanofi, Oberkotter Foundation, and 9 Meters Biopharma; Teva; and Consultancy Fees from Immunic Therapeutics, Chugai Pharma, Alimentiv, Vaccitech PLC, Dr Falk, Sanofi Aventis, FCPM III, Forte Biosciences, Zag Bio, LoopLab Bio, AnaptysBio Inc., Kerna Ventures, Catalys Pacific LLC, GI Reviewers LLC; and has received Royalties from Evelo Biosciences. J.A.M. also shares in Entero Healthcare and Minnesota Medical Technologies. M.I.P.S. received funding from Takeda, ProventionBio, and Celiac Canada, and is a member of Celiac Canada’s professional advisory board. E.F.V. is the secretary of the International Society for the Study of Celiac Disease and a member of the Advisory Board of the Biocodex Foundation. M.C., J.B., G.H.R., N.E., D.K.S., S.J., M.G.S., P.B., and A.C. declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Daniel Agardh, Angelica Ahrens and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wulczynski, M., Constante, M., Galipeau, H.J. et al. Small intestinal microbial fiber metabolism dysfunction in celiac disease. Nat Commun 17, 2698 (2026). https://doi.org/10.1038/s41467-026-70644-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70644-4
