
Abstract
Point defects critically govern the properties of two-dimensional semiconductors, yet their atomic-scale characteristics in the emerging MoSi2N4 family remain unexplored. Here we combine atomic-resolution scanning transmission electron microscopy and first-principles calculations to systematically investigate point defects in monolayer semiconducting WSi2N4 and MoSi2N4. We identify ten distinct defect types, with Si-for-top-N antisites (SiN(t)), double-middle-N divacancies (VN(m)2), and double-top-N divacancies (VN(t)2) being the most abundant. These defects induce mobility modulation and bandgap reduction — in some cases leading to complete band closure and insulator-to-metal transitions — and, in certain configurations, give rise to spin-polarized bands with localized magnetic moments. Additionally, we observe low-dimensional assemblies formed via defect self-organization, including 2D SiN(t) networks and 1D Si2Mo chains. Our findings establish fundamental defect–property relationships and provide insights for defect-driven engineering of electronic and magnetic states in 2D WSi2N4 and MoSi2N4 semiconductors.
Similar content being viewed by others

Introduction
Point defects, which are ubiquitous in crystalline materials, have a significant influence on the electronic, optoelectronic, thermal, magnetic, and catalytic properties, particularly for 2D materials due to the reduced dimensionality1,2,3,4,5,6,7,8,9,10,11,12,13,14. Usually, point defects act as scattering centers of electrons and phonons, leading to a great degradation of electron mobility, thermal conductivity, and photoelectric properties of 2D materials1,3,4,9,12,15,16,17,18,19. On the other hand, some unique or improved properties could be achieved by introducing point defects. For example, S vacancies in MoS2 can induce localized states, enabling MoS2 a carrier hopping transport phenomenon10; Pt vacancies in PtSe2 introduce localized magnetic moments, giving rise to the Kondo effect2; the Frenkel point defects and S vacancies greatly promote the hydrogen evolution reaction activity of monolayer MoS26,11. Thus, understanding the point defects in 2D materials is essential not only for the controlled growth of perfect crystals but also for tailoring their properties to meet various applications.
The MoSi2N4 family is an emerging class of van der Waals (vdW) layered 2D materials with a sandwich-structured septuple-atomic-layer configuration20,21. Such a unique structure enables MoSi2N4-like materials many interesting properties21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38, such as remarkable electrical, optical, valley-contrasting properties, piezoelectricity, ferroelectricity, magnetism, and catalytic properties. Meanwhile, the complex compositions and structures of the MoSi2N4 family enable the formation of a wider variety of point defects compared to graphene, h-BN, and transition metal dichalcogenides (TMDCs), providing greater degrees of freedom for tailoring the properties. Monolayer MoSi2N4 and WSi2N4 are the first two materials that have been synthesized by chemical vapor deposition (CVD) in the MoSi2N4 family20,21. Both of them are p-type 2D semiconductors with intrinsically higher carrier mobility, higher mechanical strength, higher thermal conductivity, and better stability than most 2D semiconductors20,21,22,39. Moreover, monolayer WSi2N4 has been predicted to possess higher carrier mobilities and thermal conductivity than monolayer MoSi2N421,28. Although point defects have been predicted to have a significant influence on the properties of MoSi2N4-like materials38,40,41,42,43, the atomic structures of point defects remain experimentally unknown, leaving a critical gap in our understanding of their role in shaping material properties.
Here, we studied the point defects in monolayer MoSi2N4 and WSi2N4 using aberration-corrected scanning transmission electron microscopy (STEM) combined with first-principles calculations, with a focus on monolayer WSi2N4 because it has higher tolerance to electron beam irradiation than MoSi2N4 (Supplementary Fig. 1). Four kinds of vacancies and six kinds of antisite substitutions were identified, among which the Si-for-top-N antisites (SiN(t)), double-middle-N divacancies (VN(m)2), and double-top-N divacancies (VN(t)2) exhibited higher concentrations. First-principles calculations show that these point defects reduce the bandgap of monolayer WSi2N4: some completely close the bandgap, resulting in an insulator-to-metal transition; some introduce spin-polarized bands, leading to local magnetic moments. Intriguingly, two low-dimensional defect configurations of 2D SiN(t) networks and 1D double-Si-for-Mo antisite (Si2Mo) chains were identified in monolayer MoSi2N4, both exhibiting unique electronic characteristics.
Results and discussion
Intrinsic structure of monolayer WSi2N4 and MoSi2N4
Both WSi2N4 and MoSi2N4 monolayers were synthesized by CVD20. Atomic force microscopy (AFM) characterization of the MoSi2N4 and WSi2N4 samples transferred onto SiO2 substrates confirms their monolayer nature. The measured thicknesses are approximately 1.21 nm for MoSi2N4 and 1.19 nm for WSi2N4 (Supplementary Fig. 2), which align with the reported values for monolayers20. X-ray photoelectron spectroscopy (XPS) characterization confirms the existence of W/Mo, Si, and N elements in WSi2N4/MoSi2N4 (Supplementary Figs. 3 and 4). It is worth noting that island-like multilayers can form under an increased NH3 flow. Such intentionally grown thick samples were used to reveal the cross-sectional atomic structure of WSi2N4.
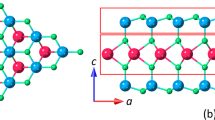
Figure 1a, b are optical and bright-field TEM images of CVD-grown WSi2N4 crystals, respectively, showing their triangular morphology. Figure 1c exhibits an annular dark field (ADF)—STEM image of monolayer WSi2N4, in which W and Si atoms are arranged into a projective hexagonal ring. Since Z-contrast ADF intensity is related to Z1.7 (Z, atomic number)44, W atoms are much brighter than Si atoms. To visualize the locations of N atoms, the integrated differential phase contrast (iDPC)—STEM technique was adopted. As the intensity of the iDPC-STEM image is approximately proportional to Z, the heavy and light elements can be captured simultaneously45. The iDPC-STEM image clearly demonstrates that the top N atoms are precisely positioned at the center of W-Si2 projective hexagonal rings (Fig. 1d). In contrast, the bilayer and trilayer WSi2N4 display six-membered ring structures with distinct central features depending on the number of stacking layers (Supplementary Fig. 5).

a, b Optical (a) and bright-field transmission electron microscopy (TEM) (b) images of CVD-grown monolayer WSi2N4 crystal. c, d In-plane ADF- (c) and iDPC- (d) STEM images by Gaussian filter. e Cross-sectional iDPC-STEM image of a multilayer WSi2N4 sample. f Schematic diagram of the atomic structure of monolayer WSi2N4. Steel blue, orange, and blue spheres denote W, Si, and N atoms, respectively. Scale bars: 2 μm (a, b); 5 Å (c–e).
We have tried to acquire cross-sectional STEM images of monolayer WSi2N4. However, the inherent flexibility of monolayers led to severe bending and distortion during cross-sectional sample preparation, which prevented the resolution of atomic arrangements. Thus, we prepared multilayer WSi2N4 samples to analyze the cross-sectional atomic structure of WSi2N4. As shown in Fig. 1e, the W, Si, and N atoms form a septuple-atomic-layer configuration. The above STEM results confirm that WSi2N4 has the same crystalline structure as the reported MoSi2N4 monolayer20 (Supplementary Fig. 6). Figure 1f presents the atomic schematic diagrams of monolayer WSi2N4, determined from the above characterizations.
Structure and concentration of point defects
Unlike single-atomic-layer graphene, WSi2N4 exhibits a stereo structure with overlapped atoms of Si and N, increasing the difficulty of discerning point defect configurations. The structural determination of point defects requires the imaging of light N atoms, heavy W atoms, and Si atoms. However, iDPC-STEM is highly sensitive to variations in sample contaminants and image defocus46,47,48,49, which can introduce artifacts that may compromise the accuracy of structural analysis. Additionally, the lighter atoms result in a low signal-to-noise ratio in ADF-STEM imaging, especially when the materials contain heavier atoms50. Meanwhile, the local fluctuation of the sample also diminishes the accuracy of structural analysis for both ADF-STEM and iDPC-STEM50,51,52,53,54,55,56. Thus, we simultaneously employed iDPC- and ADF-STEM imaging, complemented by iDPC- and ADF-STEM simulations and first-principles calculations, to identify the point defects in monolayer WSi2N4.
Figures 2 and 3 show ten main point defect configurations that were observed in monolayer WSi2N4, including four vacancies and six antisite substitutions. The vacancies include VN(m)2, VN(t)2, double-Si-and-single-middle-N trivacancies (VSi2N(m)) and W vacancies (VW) (Fig. 2); the antisite substitutions include SiN(t), Si-for-middle-N antisites (SiN(m)), double-Si-for-W antisites (Si2W), Si-for-W antisites (SiW), W-for-Si antisites (WSi), and W-for-top-N antisites (WN(t)) (Fig. 3). Notably, the ADF- and iDPC-STEM simulations of corresponding point defects are in good line with the experimental observations (Figs. 2a–d and 3a–d). Furthermore, the ADF line scan profiles across defective areas show good agreement between experimental and simulated images (Figs. 2e and 3e). These consistent results validate the reliability of our defect assignments. The obtained atomic models of point defects are presented in Figs. 2f and 3f. Similarly, identical point defects were observed in monolayer MoSi2N4, as shown in Supplementary Figs. 7 and 8.

a–d ADF- (a), simulated ADF- (b), iDPC- (c), and simulated iDPC-STEM (d) images of four distinct vacancy point defects: VN(m)2, VN(t)2, VSi2N(m), and VW. Defect sites are encircled by dashed lines. Arrows in (a, b) indicate the line scan regions in experimental and simulated ADF-STEM images. e ADF-STEM line scan profiles across defective areas marked by arrows in (a, b). The defect sites are indicated by arrows. f Corresponding atomic models of the vacancy point defects. Gray, blue, and silver spheres denote W, Si, and N atoms, respectively. Defect sites are marked by dashed circles. A Gaussian filter was applied to all ADF-STEM images. Scale bars: 5 Å.

a–d ADF- (a), simulated ADF- (b), iDPC- (c), and simulated iDPC- (d) STEM images of six distinct antisite point defects: SiN(m), SiN(t), Si2W, SiW, WSi, and WN(t). Defect sites are encircled by dashed lines. Arrows in (a, b) indicate the line scan regions in experimental and simulated ADF-STEM images. e ADF-STEM line scan profiles across defective areas marked by arrows in (a, b). The defect sites are indicated by arrows. f Corresponding atomic models of the antisite point defects. Gray, blue, and silver spheres denote W, Si, and N atoms, respectively. Defect sites are marked by dashed circles. A Gaussian filter was applied to all ADF-STEM images. Scale bars: 5 Å.
Notably, the absence of a single N atom in WSi2N4 does not produce a detectable contrast change in the ADF- and iDPC-STEM images, which prevented unambiguous identification of VN(t) and VN(m) (Supplementary Fig. 9) and distinguishing the defect sets of VSi/VSiN(m)/VN(m)2 (Supplementary Fig. 10) and VSi2/VSi2N(m) (Supplementary Fig. 11). Moreover, due to the larger Z difference between W and Si/N, WSi exhibits similar contrast with WN(m) (Supplementary Fig. 12). To identify these defects, we calculated the formation energies of various point defects by first-principles calculations under the experimentally determined Si-rich conditions (Fig. 4, Supplementary Fig. 13, Supplementary Table 1, and see Supplementary Note 1 for calculation details). The obtained formation energies follow the trend VN(m)2 < VSiN(m) < VSi, VSi2N(m) < VSi2, and WSi < WN(m) (Supplementary Fig. 14). Thus, we assign these defects as VN(m)2, VSi2N(m), and WSi, rather than VSi (VSiN(m)), VSi2, and WN(m), respectively. It is worth noting that adatoms are highly mobile under electron beam irradiation and thus are relatively straightforward to distinguish compared to lattice defects.

a, b Large field-of-view ADF- (a) and iDPC- (b) STEM images of monolayer WSi2N4. Different types of point defects are marked by color-coded circles, with the corresponding defect types listed below the images. c The experimentally determined concentrations (histogram) and corresponding calculated formation energies (triangles) of point defects in monolayer WSi2N4. The error bars represent the standard deviation of the mean values. In the histogram, the statistical counts for VN(m)2 may include both VSiN(m) and VSi configurations due to their nearly identical contrast. Although VN(t) and VN(m) are key point defects expected to have high concentration, they remain unresolved in our STEM imaging. For reference, we have included their theoretical formation energies in the histogram. Scale bars: 1 nm.
Figure 4a, b shows large field-of-view STEM images of monolayer WSi2N4, clearly demonstrating the diversity of point defect structures present within a single sample. We then counted the concentration of the ten kinds of point defects by statistically analyzing 140 atomic-resolution STEM images, encompassing 6600 defects. As shown in Fig. 4c, the most prevalent defects are SiN(t), VN(m)2, and VN(t)2. This is in sharp contrast to CVD-grown MoS2 monolayers, where only sulfur vacancy (VS) was frequently detected9,57. The frequent occurrence of multiple types of point defects in WSi2N4, including vacancies and antisite substitutions, reveals its inherently more complex structural configuration. The SiN(t) antisite and VN(m)2 vacancy exhibit the highest defect concentrations ‒ (2.12 ± 0.002) × 1013 cm−2 and (1.14 ± 0.005) × 1013 cm−2, respectively ‒ which are comparable to that of VS in CVD-grown MoS2 (1.2 × 1013 cm−2)9. In contrast, the concentrations of other defects, such as VSi2N(m) and VW, are much lower. It should be noted that the statistical analysis of VN(m)2 may inadvertently include VSi and VSiN(m) due to their nearly identical image contrast, even though the VN(m)2 configuration is thermodynamically most stable and dominant. This leads to a potential overestimation of the true VN(m)2 concentration. Similarly, the statistical analysis of WSi possibly includes the case of WN(m).
Furthermore, it should be noted that W-containing vacancy complexes do exist. For instance, Supplementary Fig. 15 shows the absence of two Si atoms and two N(t) atoms around the missing W atom. Owing to the multiple atomic configurations of W-containing vacancy complexes and their inherently low concentration, all instances were included in the statistical analysis of VW. For simplicity, the term tungsten vacancies (VW) was used representatively to present the statistical results.
To understand the formation of the observed defects, we first conducted in-situ STEM experiments at 300 kV to rule out the influence of electron beam irradiation. During the observation for normal photography with only one brief irradiation event, no new point defects were formed. After ~33 s STEM image preview, only a limited number of new nitrogen vacancies emerged (Supplementary Fig. 16), along with the migration of a few tungsten adatoms. Continued exposure of 102 s led to the occasional ejection of W atoms from the lattice at a few sites (Supplementary Fig. 17). Furthermore, electron energy loss spectroscopy (EELS) confirmed the absence of O in the WSi2N4 (Supplementary Fig. 18). These results collectively demonstrate the robust stability of monolayer WSi2N4 under STEM imaging. Moreover, in our experiments, we frequently moved the samples to different locations. Each position was subjected to no more than 5 s preview and only one image acquisition. Therefore, the observed defects intrinsically originate from the CVD growth process rather than being caused by electron beam irradiation.
Furthermore, we systematically evaluated the stability of point defects in monolayer WSi2N4 under various experimental conditions. First, defect concentration statistics remained consistent at different STEM beam currents (~5–20 pA), with minor variations attributable to sample heterogeneity (Supplementary Fig. 19 and Supplementary Table 2). Second, defects showed no measurable change after two months of ambient exposure (Supplementary Fig. 20). Third, electrical biasing at 1 V induced no detectable alteration in defect types or concentrations (Supplementary Fig. 21). Finally, thermal cycling up to 500 °C likewise preserved the defect configurations (Supplementary Fig. 22). Collectively, these results confirm that the identified point defects are structurally and chemically stable under the employed imaging conditions, upon ambient exposure, as well as under moderate electrical and thermal stress.
As shown in Fig. 1f, monolayer WSi2N4 consists of seven elementally distinct atomic layers. The formation of point defects during its growth is highly sensitive to the CVD atmosphere. In our experiments, a quartz tube served as Si source, creating a Si-rich environment. Meanwhile, the supply of N was limited because a higher NH3 flow rate would cause undesirable etching of the Cu growth substrate. Under such Si-rich and N-deficient environments, the formation of Si-substitute defects and N vacancies is thermodynamically favored. This is because excess silicon can substitute atoms in various layers while maintaining coordination symmetry, and N deficiency promotes vacancy formation across all four N layers. Experimentally, the dominant point defects observed include antisites (e.g., SiN(t), SiN(m), Si2W, and SiW) and vacancy complexes (e.g., VN(m)2 and VN(t)2), consistent with these expectations. The increasing concentration of SiW and Si2W defects with higher Si supply (Supplementary Fig. 23) further supports the preliminary understanding of the formation mechanism of defects.
Moreover, the observed point defect concentrations correlate with their calculated formation energies (Fig. 4c). In WSi2N4, the formation energies of vacancies are generally higher than those of antisite substitutions, similar to the case of MoSi2N443. Thus, antisite substitutions are more frequently observed in comparison with vacancies. This is in sharp contrast to the case of MoS2, where vacancies are generally more stable than antisite substitutions9,57. Notably, the concentration of VN(m)2 and VN(t)2 are relatively high despite their higher formation energy, even surpassing that of antisite substitutions with lower formation energy. Two primary factors may account for this observation. First, the formation energies of single VN(t) and VN(m) vacancies (3.89 eV and 3.21 eV, respectively) are approximately half those of their double-vacancy counterparts, VN(t)2 and VN(m)2 (8.31 eV and 6.18 eV). This near-additivity behavior suggests weak coupling between the two single vacancies in VN(t)2 and VN(m)2. Consequently, a high concentration of single vacancies could facilitate the formation of double vacancies through statistical pairing. Second, the N(t) atoms at the outermost surface are more susceptible to damage during TEM sample preparation, including transfer and heat treatment. It is worth noting that once a VN(t) forms near an N(m) atom, the computed energy barrier for N(m) moving towards the adjacent VN(t) is relatively low compared to that for W or Si escaping into the vacuum (Supplementary Fig. 24). This suggests that N(m) atoms are susceptible to environmental disturbances in the presence of N(t) vacancies, which may further contribute to an increased concentration of VN(m)2 defects.
In certain cases, a discrepancy arises between the experimentally derived defect concentration and the corresponding calculated formation energy, such as the case of WSi. There are several reasons for such a discrepancy. First, the formation energies of point defects are strongly influenced by chemical potential, which may differ between experimental and calculation conditions58. Second, our computational modeling was confined to point defects within a 4 × 4 × 1 supercell (already large for the DFT calculations), which corresponds to an unrealistically high defect concentration (~0.74 nm−2) compared to the much lower experimental concentration (~0.01 nm−2). Third, our calculations assume charge-neutral defects, but the possibility of charged defects cannot be excluded58. Fourth, while we assume defect-induced charge distribution is delocalized across the lattice, the potential impact of charge localization on formation energy remains difficult to rule out58.
Influence of point defects on properties
We then conducted first-principles spin-polarized density functional theory (DFT) calculations to explore the influence of the observed point defects on the electronic properties of monolayer WSi2N4 (Fig. 5, Supplementary Table 1). Pristine monolayer WSi2N4 is a 2D semiconductor with an indirect bandgap of 2.09 eV. The presence of point defects reduces the band gaps to 0.64–1.26 eV depending on the types of point defects. The presence of VW, Si2W, WSi, and WN(t) defects induces a change in the bandgap type from indirect to direct. Notably, the introduction of VN(t)2, VSi2N(m), SiN(m), SiN(t), and SiW defects completely closes the bandgap, resulting in insulator-to-metal transitions. Additionally, VN(t), VN(m)2, VN(t)2, VSi2N(m), WSi, and WN(t) defects introduce spin-polarized bands, leading to local magnetic moments. For vacancies, the magnetic moment in VN(m)2 originates from the three surrounding W atoms; similarly, the magnetic moment in VSi2N(m) arises from the three N(t) atoms on the upper and lower surfaces; VN(t) has a magnetic moment of 0.51 μB, which is attributed to the three surrounding Si atoms; VN(t)2 has a magnetic moment of 1.02 μB, which stems from the three Si atoms on the upper and lower surfaces. Notably, the magnetic moment of VN(t)2 is twice that of VN(t), indicating the weak coupling effect between the two aligned N(t) atoms. For antisite substitutions, WN(t) and WSi exhibit magnetism derived from the antisite W atoms. Particularly, WN(t) possesses the largest magnetic moment of 2.11 μB among all the point defects.

The Fermi energy is aligned to 0 eV. The blue and orange lines represent the spin-up and spin-down channels, respectively.
We further calculated the orbital-resolved density of states (DOSs) to identify the orbital characters of in-gap states (Supplementary Fig. 25). The contribution from vacancies was obtained by introducing pseudo atoms into the positions of vacancies. It is found that for VN(m)2, VSi2N(m), VW, SiN(t), Si2W, and SiW, the in-gap states are predominantly composed of W orbitals. While for VN(t) and VN(t)2, the in-gap states are primarily composed of Si orbitals. For WSi and WN(t), the in-gap states are mainly attributed to the defect orbitals. These in-gap states induced by point defects strongly modulate the electronic and magnetic structure of pristine WSi2N4 (Fig. 5 and Supplementary Table 1) and therefore change the associated optical properties. For instance, the presence of VN(t) decreases the bandgap by inducing the in-gap states, which introduce additional optical absorption within the bandgap (Supplementary Fig. 26). In particular, these in-gap states are rather localized, which is manifested by the appearance of flat bands within the bandgap (Fig. 5). These flat bands significantly increase the hole effective mass at Γ from 1.5 me (pristine) to 15.3 me and thus decrease the hole mobility from 411 cm2 s−1 V−1 (pristine) to 2 cm2 s−1 V−1 according to the deformation potential theory9,59. In contrast, the introduction of SiN(t) reduces the Fermi energy, resulting in an almost rigid upward shift of the band structure and triggering an insulator-to-metal transition (Fig. 5). The crossing of the maximum valence band with the Fermi energy facilitates optical transitions within the infrared regime (Supplementary Fig. 26). Furthermore, the presence of SiN(t) significantly increases hole mobility, reaching a value of 740 cm2 s−1 V−1.
To correlate the theoretical calculation results, we used atomic-scale scanning tunneling microscopy (STM) to probe the impact of point defects on the local electronic structure of monolayer WSi2N4. The surface tunneling properties of STM allowed us to identify VN(t) and SiN(t) defects from topographic images (Fig. 6). Scanning tunneling spectroscopy (STS) measurements on a defect-free region (Fig. 6a, b) revealed a bandgap of ~2.05 eV, consistent with our DFT prediction (2.09 eV). In contrast, STS on defect sites showed significantly reduced band gaps (Fig. 6c, d), with values approaching the calculated gap (0.84 eV) for VN(t) defects. The discrepancy between the easy identification of VN(t) by STM and its limited representation in STEM images is due to the distinct imaging mechanisms and sensitivities of the two techniques. An additional defect site exhibited a metallic signature, which we identify as the SiN(t) defect—consistent with our theoretical prediction of its metallic character (Fig. 6e, f). These results confirm the decisive influence of point defects on the electronic properties of WSi2N4.

a, b A STM topographic image (7 nm × 7 nm) (a) and the dI/dV spectrum acquired from the defect-free site (b), which is highlighted by a red circle in (a). Bias voltage U = +2.3 V, tunneling current I = 500 pA. E denotes the band gap. c, d A STM topographic image (8 nm × 8 nm) (c) and the dI/dV spectra acquired from three defect sites (d), which are highlighted by different colored arrows in (c). U = +2.3 V, I = 500 pA. e, f A STM topographic image (8 nm × 8 nm) (e) and the dI/dV spectra acquired from another defect site (f), which is highlighted by a pink arrow in (e). U = +1.5 V, I = 1000 pA. The spectra were offset vertically for better visualization. a, c Represent partially overlapping regions, with contrast adjusted to clearly distinguish defect sites from the pristine lattice.
We further measured the ultraviolet and visible (UV-Vis) absorption spectra of three distinct WSi2N4 samples and obtained their corresponding Tauc plots (Supplementary Fig. 27): the original sample, the sample annealed in Ar at 300 °C for 12 h, and the sample annealed in air at 300 °C for 12 h. The absorption data showed similar band gaps for the original sample (~2.1 eV) and the air-annealed sample (~2 eV), indicating that the point defects exhibit oxidation resistance in air at the evaluated temperature. In contrast, the sample annealed in Ar exhibited a noticeably reduced bandgap of ~1.8 eV, indicating an increase in defect concentration. In our work, the annealed samples were used to obtain high-quality STEM images for defect concentration analysis. Taking the VN(t)2 defect as an example, argon annealing led to a significantly higher defect concentration compared to air annealing (Supplementary Fig. 28), consistent with the more pronounced bandgap reduction.
The comprehensive atomic-scale catalog of point defects in monolayer WSi2N4, together with their tunable properties, offers significant opportunities for device engineering. For example, the substantially increased hole mobility by the SiN(t) defect suggests its utility for channel optimization. Defects that induce an insulator-to-metal transition, such as VN(t)2, SiN(t), SiN(m), and SiW, when strategically incorporated at contact interfaces, may lower the Schottky barrier height, reduce contact resistance, and enhance on-state current density. Similarly, the SiW antisite defects introduce a shallow acceptor state near the valence band maximum, enabling effective p-type doping for threshold voltage tuning. Conversely, in-gap states induced by VN(t) defects promote trap-assisted tunneling, potentially increasing the off-state leakage current and reducing the switching ratio. Consequently, optimizing field effect transistor performance—balancing high mobility, low leakage, and low contact resistance—will require precise spatial defect engineering: suppressing channel-degrading defects, leveraging mobility-enhancing defects in the active region, and potentially incorporating beneficial defects at contact interfaces.
Furthermore, although pristine WSi2N4 exhibits high intrinsic thermal conductivity, a trait well-suited for transistor applications, high concentrations of defects such as VN(m)2 and SiN(t) are expected to substantially reduce the lattice thermal conductivity through mass-contrast and strain-field scattering, as observed in other 2D systems60,61,62. Such reduction is highly advantageous for thermoelectric applications, where a lowered lattice thermal conductivity directly enhances the thermoelectric figure of merit ZT, particularly when coupled with defect-tuned electronic properties, for example, the metallic states introduced by SiN(t) defects.
Configurations and properties of defect aggregations
Beyond isolated point defects in monolayer WSi2N4 and MoSi2N4, we observe a propensity for defect aggregation in these materials. Figure 7a–c and Supplementary Fig. 29 exhibit typical ADF images of double, triple, and multiple SiN(t) aggregations in MoSi2N4 and WSi2N4 monolayers, respectively. It is worth noting that SiN(t) could even form a 2D network in certain areas of MoSi2N4 monolayer (Fig. 7c). Simulated ADF images of SiN(t) networks are in accordance with the experimental observations (Fig. 7d). Geometric phase analysis (GPA) indicates the absence of lattice strain across the SiN(t) 2D networks and MoSi2N4 matrixes (Supplementary Fig. 30). Structurally, the SiN(t) 2D network is a periodic, planar crystalline defect characterized by the aggregation of collective SiN(t) antisite defects on one side of the MoSi2N4 monolayer, analogous to a stacking fault.

a, b ADF-STEM images of two (a) and three (b) SiN(t) aggregations. Each individual SiN(t) is highlighted by a yellow circle. c, d ADF-STEM image of 2D SiN(t) network (c) and the corresponding ADF-STEM simulation (d). The orange dashed arrow in (c), and the blue dashed arrow in (d) mark the regions used for line intensity profiling, with the resulting profile overlaid in (d). Representative SiN(t) sites in these regions are indicated by yellow arrows. e, f Electronic band structure (e) and orbital-resolved DOSs (f) of 2D SiN(t) network. g Low-magnification ADF-STEM image of a 1D Si2Mo chain denoted by yellow arrows. h, i High-magnification ADF-STEM image (h) and corresponding ADF-STEM simulation (i) of the 1D Si2Mo chain in the region marked by the yellow box in (g). The orange dashed arrow in (h), and the blue dashed arrow in (i) mark the regions used for line intensity profiling, with the resulting profile overlaid in (i). Representative Si2Mo sites in these regions are indicated by yellow arrows. j, k Electronic band structure (j) and the orbital-resolved DOSs (k) of 1D Si2Mo chain. Scale bars: 5 Å (a–d, h, i); 5 nm (g). A Gaussian filter was applied to the ADF-STEM images in (g, h).
Taking MoSi2N4 as an example, we constructed defect aggregation models containing one, two, and three SiN(t) defects within a 4 × 4 × 1 configuration of the MoSi2N4 unit cell (Supplementary Figs. 31, 32) and calculated their corresponding binding energies. The configurations with SiN(t) antisite defects aggregated on one side of the monolayer MoSi2N4 exhibit higher stability (Supplementary Fig. 33). The increased binding energies with the quantity of SiN(t) located on one side suggest that SiN(t) defect aggregation is energetically favorable (Supplementary Figs. 33 and 34). Moreover, the formation of 2D SiN(t) network triggers electronic structure reconstruction in the MoSi2N4 system (Fig. 7e). Similar to isolated SiN(t) point defects43, the introduction of 2D SiN(t) network induces bandgap closure. Systematic analysis of DOSs demonstrates that the electronic states near the Fermi level in 2D SiN(t) network are predominantly contributed by the 3p orbitals of Si atoms (Fig. 7f).
Additionally, we observed a 1D point defect chain embedded in monolayer MoSi2N4 (Fig. 7g), where no lattice strain was detected as well (Supplementary Fig. 35). Careful examination of defect configurations in an enlarged ADF-STEM image revealed a 1D Si2Mo chain (Fig. 7h), in good agreement with corresponding ADF image simulation (Fig. 7i). This Si2Mo chain is a linear crystalline defect that separates the single-crystal MoSi2N4 monolayer into two parts without a misorientation angle, analogous to a 0° grain boundary in 2D materials. We constructed the 1D chain-like models containing one, two, three, and four Si2Mo within a 4 × 4 × 1 configuration of the MoSi2N4 unit cell (Supplementary Fig. 36) and calculated their binding energies. Similar to the 2D SiN(t) aggregations, the binding energies of these 1D defect aggregations increase as the quantity of Si2Mo increases (Supplementary Fig. 37), indicating the increasing energetic stability for linear assembly of Si2Mo. Upon introducing this 1D chain into MoSi2N4, the bandgap of the system significantly decreases from 1.72 eV to 0.40 eV (Fig. 7j). Further DOSs analysis reveals that the 1D chain-induced in-gap states near the Fermi level are dominated by the d-orbitals of Mo atoms (Fig. 7k).
The thermodynamic stability of the observed 2D SiN(t) networks and 1D Si2Mo chains, as confirmed by our binding‑energy calculations, is consistent with an in‑situ assembly mechanism during CVD growth rather than post‑growth migration. The high‑temperature growth environment provides sufficient atomic mobility and a concurrent supply of all constituent elements, allowing direct incorporation of multi‑site defects into the extending lattice. The seamless embedding and crystallographic alignment of these extended structures further indicate a growth‑governed process. First‑principles migration‑barrier calculations additionally show that post‑growth rearrangement of isolated SiN(t) or Si2Mo defects—even in the presence of nearby vacancies—requires overcoming high energy barriers (> 4 eV) (Supplementary Fig. 38), rendering large‑scale diffusion kinetically unfeasible under ambient conditions. Collectively, the structural coherence, energetic trends, and calculated kinetic barriers support a kinetically enabled, growth‑mediated pathway for the formation of these low‑dimensional defect aggregates.
In summary, by combining atomic-resolution STEM imaging and first-principles calculations, we revealed point defect configurations in monolayer semiconductors WSi2N4 and MoSi2N4 and their influence on material properties. Ten distinct types of lattice point defects, comprising four vacancies and six antisite substitutions, were identified. Taking monolayer WSi2N4 as a representative system, we demonstrated that these defects substantially modulate electronic structure, induce magnetism, and alter optical responses. Furthermore, we identified two defect aggregation configurations in monolayer MoSi2N4: 2D SiN(t) networks and 1D Si2Mo chains, which exhibit significant influence on the electronic band structure. This work provides crucial insights into the defect-property correlation of monolayer semiconducting WSi2N4 and MoSi2N4, which is essential for controlled synthesis and property engineering of such materials and paves the way for their applications in nanoelectronics, spintronics, and catalytic systems.
Methods
Sample preparation
The monolayer WSi2N4 and MoSi2N4 samples were first grown on Cu/W and Cu/Mo substrates, respectively, by the CVD method20. Then, they were transferred onto the TEM grids coated with silicon nitride membranes by the chemical etching method with the help of a polymethyl methacrylate (PMMA) layer. After that, the TEM grids were immersed in hot acetone to dissolve the PMMA layer and subsequently annealed at 300 °C for 12 h in air to remove hydrocarbon residues from the sample surfaces before inserting them into the electron microscope.
Structural characterization and simulation
The optical microscopy image was obtained on Nikon LV100D. The thicknesses of samples were measured using an atomic force microscope (AFM, MultiMode 8, Bruker, Inc.). Optical absorption spectra were recorded using a UV-Vis spectrophotometer (JASCO V-770).
The BF-TEM image was obtained on a FEI Tecnai F20 at 200 kV. The ADF- and iDPC-STEM imaging of monolayer WSi2N4 and MoSi2N4 was performed on a FEI Titan Cube Themis G2 300 instrument equipped with a high-brightness field-emission gun (X-FEG), double spherical aberration corrector, and a monochromator. The accelerating voltage was 300 kV. For imaging the lattice point defects, the camera length was 230 mm; the collection semi-angle was 25–152 mrad for ADF imaging, while 6−24 mrad for iDPC imaging; the major beam current was set to ~ 9 pA. The STEM image acquisition and preview were typically conducted under the conditions of 5 μs dwell time with 2048 × 2048 pixels and 1.32 × 107 e- nm−2 electron dose and 1 μs dwell time with 1024 × 1024 pixels and 6.6 × 105 e- nm−2 electron dose, respectively. For imaging the SiN(t) 2D network and Si2Mo 1D chain in MoSi2N4, the camera length was 145 mm; the collection semi-angle was 37–200 mrad for ADF imaging; the beam current was set to ~22 pA.
To ensure image quality, annealing treatment was carried out for all samples to remove surface contaminants as thoroughly as possible before STEM imaging. To improve the image contrast and signal-to-noise ratio of Si substitutions in the 2D networks and 1D chains, we selected strictly flat zones in monolayer MoSi2N4 samples and extended the image exposure time to 10 μs dwell time with 2048 × 2048 pixels. The stability comparison for monolayer WSi2N4 and MoSi2N4 was conducted on JEOL ARM 200 F at 200 kV.
The ADF- and iDPC-STEM images were simulated using ToTEM63 or Dr. Probe64 software based on the multi-slice method. The simulation parameters were in accordance with the experimental values, disregarding any aberrations. The atomic models were optimized by DFT. To compensate for structural distortions (elongation or compression) in experimental images caused by sample drift, equivalent deformations were intentionally introduced into the simulated images to achieve precise atom-to-atom alignment.
For the statistical analysis of defect concentration, we acquired a large number of STEM images – each with a typical area of 9.45 × 9.45 nm2—from random locations across multiple WSi2N4 samples. The types of point defects were identified by combining STEM image contrast (ADF and iDPC) with image simulations. For VN(m)2 and SiN(t) point defects with inherently relatively high concentration, we used CalAtom software to perform the high-throughput statistical analysis65. The defect positions were identified by analyzing the intensity variations in ADF images. Other types of point defects were directly counted manually due to the limited quantity. The defect concentration was determined by normalizing the number of defects to the corresponding imaging area. Due to the low concentration, the datasets for VW and VSi2N(m) defects exhibited a discrete distribution. Therefore, a weighted averaging method was used to estimate their concentrations. For other defects, to minimize experimental statistical errors, Gaussian fitting was applied to the datasets to obtain the mean (μ) and variance (σ), yielding the final concentration values (Supplementary Fig. 39).
For the biasing- and heating-STEM experiments, an in-situ thermoelectric holder (Protochips Inc.) was used. Biasing was performed in constant voltage mode using a four-probe electrical chip, with the voltage increased to 1 V in steps of 0.1 V/s. Heating was conducted with a two-probe thermal chip, where the temperature was raised from 25 °C to 500 °C at a ramp rate of 0.5 °C/s. The thermal cycling procedure comprised three stages: (1) heating to 500 °C for ∼1 h, (2) cooling to room temperature for ∼0.5 h, and (3) reheating to 500 °C for ∼1 h.
STM measurements
The monolayer WSi2N4 sample was initially introduced into a combined scanning tunneling microscope–molecular beam epitaxy (STM-MBE) system (Unisoku Co., USM1500) and annealed at ~300 °C for 12 h under ultrahigh vacuum to achieve an atomically clean surface. The base pressure during annealing was maintained at 1 × 10⁻10 Torr. After annealing, the sample was transferred to a low-temperature STM stage without breaking the vacuum. All the STM and STS experiments were conducted at liquid nitrogen temperature (~78 K). The STM topographic images were acquired under the constant-current mode using a PtIr tip, while the STS spectra were collected via a lock-in amplification technique with an AC modulation of 60 mV at a frequency of 879 Hz.
Theoretical calculations
First-principles calculations were carried out using the Vienna Ab initio Simulation Package66,67. The generalized gradient approximation of Perdew–Burke–Ernzerhof 68 was used for the exchange-correlation functional. The interaction between nuclei and valence electrons was described by the pseudopotential within the framework of the projector augmented wave method69. The plane wave cutoff was set to 450 eV. To model the point defects, a supercell slab of 4 × 4 × 1 unit cell with a vacuum width of 20 Å was employed. The electronic optimization was performed until the total energy difference between two iterations was less than 10−6 eV. The structural relaxation was terminated when the force on each atom was smaller than 0.01 eV/Å. More computational details on the calculations of defect formation energy and binding energy are provided in Supplementary Note 1.
Data availability
The Source Data underlying the figures of this study are available at https://doi.org/10.6084/m9.figshare.31314547. All raw data generated during the current study are available from the corresponding authors upon request.
References
Liang, Q., Zhang, Q., Zhao, X., Liu, M. & Wee, A. T. S. Defect engineering of two-dimensional transition-metal dichalcogenides: applications, challenges, and opportunities. ACS Nano 15, 2165–2181 (2021).
Ge, J. et al. Magnetic moments induced by atomic vacancies in transition metal dichalcogenide flakes. Adv. Mater. 33, 2005465 (2021).
Hong, J., Jin, C., Yuan, J. & Zhang, Z. Atomic defects in two-dimensional materials: from single-atom spectroscopy to functionalities in opto-/electronics, nanomagnetism, and catalysis. Adv. Mater. 29, 1606434 (2017).
Hu, Z. et al. Two-dimensional transition metal dichalcogenides: interface and defect engineering. Chem. Soc. Rev. 47, 3100–3128 (2018).
Wang, S., Robertson, A. & Warner, J. H. Atomic structure of defects and dopants in 2D layered transition metal dichalcogenides. Chem. Soc. Rev. 47, 6764–6794 (2018).
Wang, X. et al. Single-atom vacancy defect to trigger high-efficiency hydrogen evolution of MoS2. J. Am. Chem. Soc. 142, 4298–4308 (2020).
Avsar, A. et al. Probing magnetism in atomically thin semiconducting PtSe2. Nat. Commun. 11, 4806 (2020).
Li, H. et al. Activating and optimizing MoS2 basal planes for hydrogen evolution through the formation of strained sulphur vacancies. Nat. Mater. 15, 48–53 (2016).
Hong, J. et al. Exploring atomic defects in molybdenum disulphide monolayers. Nat. Commun. 6, 6293 (2015).
Qiu, H. et al. Hopping transport through defect-induced localized states in molybdenum disulphide. Nat. Commun. 4, 2642 (2013).
Xu, J. et al. Frenkel-defected monolayer MoS2 catalysts for efficient hydrogen evolution. Nat. Commun. 13, 2193 (2022).
Yu, Z. et al. Towards intrinsic charge transport in monolayer molybdenum disulfide by defect and interface engineering. Nat. Commun. 5, 5290 (2014).
Jiang, J. et al. Defect engineering for modulating the trap states in 2D photoconductors. Adv. Mater. 30, 1804332 (2018).
Ren, W. et al. The 2D materials roadmap. 2D Materials. 13, 021501 (2026).
Min, J., Kim, J. H. & Kang, J. Chalcogen vacancy engineering of two-dimensional transition metal dichalcogenides for electronic applications. ACS Appl. Nano Mater. 7, 26377–26396 (2024).
Amani, M. et al. Near-unity photoluminescence quantum yield in MoS2. Science 350, 1065–1068 (2015).
Kwon, Y. A. et al. Wafer-scale transistor arrays fabricated using slot-die printing of molybdenum disulfide and sodium-embedded alumina. Nat. Electron. 6, 443–450 (2023).
Hao, F., Fang, D. & Xu, Z. Mechanical and thermal transport properties of graphene with defects. Appl. Phys. Lett. 99, 041901 (2011).
Yu, X., Guijarro, N., Johnson, M. & Sivula, K. Defect mitigation of solution-processed 2D WSe2 nanoflakes for solar-to-hydrogen conversion. Nano Lett. 18, 215–222 (2018).
Hong, Y.-L. et al. Chemical vapor deposition of layered two-dimensional MoSi2N4 materials. Science 369, 670–674 (2020).
Zhou, T., Xu, C. & Ren, W. The van der Waals MoSi2N4 materials family. Nat. Rev. Mater. 10, 907–928 (2025).
Latychevskaia, T., Bandurin, D. A. & Novoselov, K. S. A new family of septuple-layer 2D materials of MoSi2N4-like crystals. Nat. Rev. Phys. 6, 426–438 (2024).
Guo, S.-D., Zhu, Y.-T., Mu, W.-Q., Wang, L. & Chen, X.-Q. Structure effect on intrinsic piezoelectricity in septuple-atomic-layer MSi2N4 (M = Mo and W). Comput. Mater. Sci. 188, 110223 (2021).
Guo, S.-D., Zhu, Y.-T., Mu, W.-Q. & Ren, W.-C. Intrinsic piezoelectricity in monolayer MSi2N4 (M = Mo, W, Cr, Ti, Zr and Hf). Epl-Europhys. Lett. 132, 57002 (2020).
Zhong, T., Ren, Y., Zhang, Z., Gao, J. & Wu, M. Sliding ferroelectricity in two-dimensional MoA2N4 (A = Si or Ge) bilayers: high polarizations and moire potentials. J. Mater. Chem. A 9, 19659–19663 (2021).
Bafekry, A. et al. MoSi2N4 single-layer: a novel two-dimensional material with outstanding mechanical, thermal, electronic and optical properties. J. Phys. D Appl. Phys. 54, 155303 (2021).
Bafekry, A. et al. Band-gap engineering, magnetic behavior and dirac-semimetal character in the MoSi2N4 nanoribbon with armchair and zigzag edges. J. Phys. D: Appl. Phys. 55, 035301 (2022).
Mortazavi, B. et al. Exceptional piezoelectricity, high thermal conductivity and stiffness and promising photocatalysis in two-dimensional MoSi2N4 family confirmed by first-principles. Nano Energy 82, 105716 (2021).
Yu, J., Zhou, J., Wan, X. & Li, Q. High intrinsic lattice thermal conductivity in monolayer MoSi2N4. New J. Phys. 23, 033005 (2021).
Wang, Q. et al. Efficient ohmic contacts and built-in atomic sublayer protection in MoSi2N4 and WSi2N4 monolayers. Npj 2D Mater. Appl. 5, 71 (2021).
Li, S. et al. Valley-dependent properties of monolayer MoSi2N4, WSi2N4, and MoSi2As4. Phys. Rev. B 102, 235435 (2020).
Yang, C., Song, Z., Sun, X. & Lu, J. Valley pseudospin in monolayer MoSi2N4 and MoSi2As4. Phys. Rev. B 103, 035308 (2021).
Cao, L., Zhou, G., Wang, Q., Ang, L. K. & Ang, Y. S. Two-dimensional van der Waals electrical contact to monolayer MoSi2N4. Appl. Phys. Lett. 118, 013106 (2021).
Nandan, K., Ghosh, B., Agarwal, A., Bhowmick, S. & Chauhan, Y. S. Two-dimensional MoSi2N4: an excellent 2-D semiconductor for field-effect transistors. IEEE. Trans. Electr. Dev. 69, 406–413 (2022).
Qian, W., Chen, Z., Zhang, J. & Yin, L. Monolayer MoSi2N4-x as promising electrocatalyst for hydrogen evolution reaction: a DFT prediction. J. Mater. Sci. Technol. 99, 215–222 (2022).
Zhao, J. et al. Stacking engineering: a boosting strategy for 2D photocatalysts. J. Phys. Chem. Lett. 12, 10190–10196 (2021).
Huang, J., Li, P., Ren, X. & Guo, Z.-X. Promising properties of a sub-5-nm monolayer MoSi2N4 transistor. Phys. Rev. Appl. 16, 044022 (2021).
Zang, Y. et al. Activating electrocatalytic hydrogen evolution performance of two-dimensional MSi2N4 (M = Mo, W): a theoretical prediction. Phys. Rev. Mater. 5, 045801 (2021).
He, C. et al. Unusually high thermal conductivity in suspended monolayer MoSi2N4. Nat. Commun. 15, 4832 (2024).
Bafekry, A. et al. Tunable electronic and magnetic properties of MoSi2N4 monolayer via vacancy defects, atomic adsorption and atomic doping. Appl. Surf. Sci. 559, 149862 (2021).
Xiao, C. et al. Adsorption behavior of environmental gas molecules on pristine and defective MoSi2N4: possible application as highly sensitive and reusable gas sensors. ACS Omega 7, 8706–8716 (2022).
Chi, Y. et al. Effect of point defect-induced surface active sites of MoSi2N4 on performance of photocatalytic CO2 reduction. Appl. Catal. A-Gen. 650, 118975 (2023).
Ma, H. et al. Chemical environment dependent stabilities, electronic properties and diffusions behaviors of intrinsic point defects in novel two-dimensional MoSi2N4 monolayer. Appl. Surf. Sci. 592, 153214 (2022).
Krivanek, O. L. et al. Atom-by-atom structural and chemical analysis by annular dark-field electron microscopy. Nature 464, 571–574 (2010).
Yucelen, E., Lazic, I. & Bosch, E. G. T. Phase contrast scanning transmission electron microscopy imaging of light and heavy atoms at the limit of contrast and resolution. Sci. Rep. 8, 2676 (2018).
Bosch, E. G. T. & Lazić, I. Analysis of depth-sectioning STEM for thick samples and 3D imaging. Ultramicroscopy 207, 112831 (2019).
Liu, Z. et al. Two-dimensional superconducting MoSi2N4(MoN)4n homologous compounds. Natl. Sci. Rev. 10, nwac273 (2023).
Lazic, I., Bosch, E. G. T. & Lazar, S. Phase contrast STEM for thin samples: integrated differential phase contrast. Ultramicroscopy 160, 265–280 (2016).
Wang, H. et al. The development of iDPC-STEM and its application in electron beam sensitive materials. Molecules 27, 3829 (2022).
Pennycook, S. J. Z-contrast STEM for materials science. Utramicroscopy 30, 58–69 (1989).
Sha, H., Cui, J. & Yu, R. Deep sub-angstrom resolution imaging by electron ptychography with misorientation correction. Sci. Adv. 8, eabn2275 (2022).
Cui, J., Yao, Y., Wang, Y. G., Shen, X. & Yu, R. C. Origin of atomic displacement in HAADF image of the tilted specimen. Ultramicroscopy 182, 156–162 (2017).
Held, J. T., Duncan, S. & Mkhoyan, K. A. Effects of small-angle mistilts on dopant visibility in ADF-STEM imaging of nanocrystals. Ultramicroscopy 177, 53–57 (2017).
Maccagnano-Zacher, S. E., Mkhoyan, K. A., Kirkland, E. J. & Silcox, J. Effects of tilt on high-resolution ADF-STEM imaging. Ultramicroscopy 108, 718–726 (2008).
Hong, J., Koshino, M., Xu, H., Feng, Q. & Suenaga, K. STEM imaging artifacts with three-fold astigmatism in monolayer transition metal dichalcogenides. Appl. Phys. Lett. 116, 243102 (2020).
Burger, J., Riedl, T. & Lindner, J. K. N. Influence of lens aberrations, specimen thickness and tilt on differential phase contrast STEM images. Ultramicroscopy 219, 113118 (2020).
Zhou, W. et al. Intrinsic structural defects in monolayer molybdenum disulfide. Nano. Lett. 13, 2615–2622 (2013).
Freysoldt, C. et al. First-principles calculations for point defects in solids. Rev. Mod. Phys. 86, 253–305 (2014).
Bardeen, J. & Shockley, W. Deformation potentials and mobilities in non-polar crystals. Phys. Rev. 80, 72–80 (1950).
Fu, C. et al. Realizing high figure of merit in heavy-band p-type half-Heusler thermoelectric materials. Nat. Commun. 6, 8144 (2015).
Chen, Z. et al. Lattice dislocations enhancing thermoelectric PbTe in addition to band convergence. Adv. Mater. 29, 1606768 (2017).
Yang, J., Meisner, G. P. & Chen, L. Strain field fluctuation effects on lattice thermal conductivity of ZrNiSn-based thermoelectric compounds. Appl. Phys. Lett. 85, 1140–1142 (2004).
Yuan, P. J. et al. ToTEM: a software for fast TEM image simulation. J. Microsc. 287, 93–104 (2022).
Barthel, J. Dr. Probe: a software for high-resolution STEM image simulation. Ultramicroscopy 193, 1–11 (2018).
Zhang, Q., Zhang, L. Y., Jin, C. H., Wang, Y. M. & Lin, F. CalAtom: a software for quantitatively analysing atomic columns in a transmission electron microscope image. Ultramicroscopy 202, 114–120 (2019).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 52188101 (W.R.), 52272050 (Z.L.), 52422112 (P.L.), 52472052 (C.X.), 12374183 (S.-C.L.), and 92165205 (S.-C.L.)), the LiaoNing Revitalization Talents Program (No. XLYC2201003 (W.R.), XLYC2403170 (C.X.)), Youth Innovation Promotion Association CAS (No. 2021000185) (Z.L.), Young Talents Project of Shenyang National Laboratory for Materials Science (No. 2019000191) (Z.L.), the National Key Research and Development Program of China (Grant No. 2021YFA1400403) (S.-C.L.), the Natural Science Foundation of Jiangsu Province (No. BK20233001) (S.-C.L.), and the Innovation Program for Quantum Science and Technology (Grant No. 2021ZD0302800) (S.-C.L.). We thank Prof. Lili Zhang for her essential hardware support in heating- and biasing-STEM experiments, and Dr. Yao Guo for the valuable discussions on the influence of defects on device performance.
Author information
Authors and Affiliations
Contributions
W.R. conceived and supervised the project, J.T. and Z.L. performed TEM measurements and analyses, Y.C., P.L., and X.-Q.C. performed theoretical simulations, D.Y., C.H., and C.X. grew samples. Y.-K.W. and S.-C.L. performed STM and STS measurements and analyses. Z.L., J.T., Y.C., P.L., and W.R. analyzed data and wrote the manuscript. All the authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tong, J., Cao, Y., Wang, YK. et al. Point defects in monolayer WSi2N4 and MoSi2N4. Nat Commun 17, 4319 (2026). https://doi.org/10.1038/s41467-026-70946-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70946-7
