
Abstract
While chemotherapy-induced tumor cell death is known to modulate the local immune landscape, its systemic impact on distant bone marrow—a site essential for immune cell maturation—remains underexplored. Here, we show that gemcitabine chemotherapy induces inflammatory caspase-1-dependent pyroptosis in epithelial cancer cells (epiCaspase-1). Despite its inflammatory nature, epiCaspase-1-mediated cell death is non-immunogenic. Clinically, elevated expression of an epiCaspase-1 gene signature correlates with worse patient outcomes. Mechanistically, epiCaspase-1 triggers the noncanonical release of IL-1α through NINJ1 lytic pores, remotely skewing bone marrow hematopoiesis towards granulocyte-monocyte progenitors and mature neutrophil output. This systemic reprogramming elevates the neutrophil-to-lymphocyte ratio (NLR) in both peripheral blood and the local tumor microenvironment. Pharmacological inhibition of caspase-1 and IL-1α disrupts this cascade, normalizes hematopoiesis, and recalibrates NLR by promoting intratumoral CD8+ T cell infiltration and activation, ultimately enhancing chemotherapeutic efficacy. These findings challenge the assumption that inflammatory pyroptosis is inherently immunogenic; instead, it can reshape systemic immune landscape towards a neutrophil-dominant inflammation in the chemotherapy context.
Similar content being viewed by others


Introduction
Prior studies investigating the effects of systemic chemotherapy on solid tumors have primarily emphasized its direct cytotoxic effects on epithelial cancer cells (i.e., the primary therapeutic target) and downstream local influence on the tumor microenvironment. One such mechanism connecting cancer cell death to immunomodulation is immunogenic cell death1,2,3, which enhances adaptive immune cell recruitment and activation within the local tumor immune microenvironment (TIME)4. Immunogenic cell death or stress is characterized by the extracellular release and cell surface expression of damage-associated molecular patterns (DAMPs)4. While immunostimulatory DAMPs activate antigen-presenting dendritic cells to prime a CD8+ T cell response, our recent work also reveal the existence of inhibitory DAMPs (iDAMPs), which dampen anti-tumoral immune response at the local TIME5,6. The net outcome of immunogenic cell death reflects a dynamic balance between immunostimulatory DAMPs and iDAMPs. However, the role of other innate immune cells—particularly neutrophils, the first responders to tissue injury and abundant constituents of the TIME—in modulating immunogenic cell death remains largely unexplored.
Clinically, immunogenic cell death enhances the efficacy of certain chemotherapies and contributes to improved treatment outcomes7. While lytic cell death has been widely regarded as immunogenic, particularly with the advent of small-molecule agonists that elicit anti-tumoral immune activities, emerging evidence challenges this notion, highlighting its context-dependent nature. Notably, certain lytic cell death mechanisms, such as ferroptosis and necroptosis in cancer cells, can paradoxically suppress immune responses8 and foster an immunosuppressive local TIME9. These observations raise critical questions about the molecular pathways that dictate whether cancer cell death promotes or dampens anti-tumoral immunity, a research area that remains poorly understood and warrants further investigation. A particularly underexplored axis is the role of inflammatory caspase-1-mediated pyroptosis in modulating immunogenic cell death. Inflammasome-mediated activation of caspase-1 and pyroptosis were originally identified as a fundamental self-defense mechanism for eliminating infected host cells during pathogenic infection8. This lytic cell death process, characterized by plasma membrane rupture, ionic gradient imbalance, increase in osmotic pressure, cell swelling, and eventual cellular self-destruction, has been extensively studied in myeloid cells such as monocytes and macrophages. Indeed, chemotherapy-induced inflammasome activation in these myeloid cells has shown divergent effects, capable of both promoting10 and dampening11 anti-cancer immunity. However, the role of caspase-1-mediated pyroptosis in epithelial cancer cells, particularly in orchestrating immunogenic cell death and influencing chemotherapy response, remains largely undefined. To our knowledge, this represents a critical gap in understanding of how pyroptosis determines immunogenic versus tolerogenic cell death.
Current investigations into the immunological effects of chemotherapy on solid tumors have predominantly centered around alterations within the local TIME. In contrast, the bone marrow—the central site of hematopoiesis where hematopoietic stem and progenitor cells give rise to the immune cell repertoire, including both myeloid (e.g., neutrophils) and lymphoid (e.g., T lymphocytes) lineages12—remains an underexplored yet critical player in shaping systemic anti-tumoral immunity. Hematopoietic reprogramming occurring within the bone marrow can profoundly shape the intratumoral immune landscape and alter immune cell ratios, which in turn dictate treatment responses13. Clinically, an elevated neutrophil-to-lymphocyte ratio (NLR) in peripheral blood has been consistently associated with poor prognosis across multiple solid malignancies13, whereas a lower NLR often predicts favorable survival outcomes in patients receiving neoadjuvant chemotherapy. Despite these correlations, the mechanistic underpinnings by which chemotherapy-induced tumor cell death modulates the dynamic interplay between neutrophils and T lymphocytes within the local TIME remains insufficiently understood. Even less is known about its systemic effects on the bone marrow, a distal yet pivotal organ where immune cells are continuously produced and undergo maturation. Therefore, deciphering such mechanistic connections is expected to uncover a previously overlooked therapeutic axis by linking bone marrow hematopoiesis to the local TIME landscape, with potential to improve chemotherapy and immunotherapy responses.
Our current study addresses this gap by delineating a mechanistic axis linking intratumoral epiCaspase-1-dependent pyroptosis to distal bone marrow reprogramming, which is responsible for shaping both systemic and intratumoral NLR, and ultimately, influencing chemotherapy response. Specifically, chemotherapy-induced activation of epiCaspase-1 in primary tumors trigger the noncanonical release of IL-1α via NINJ1-mediated pores, which remotely skews hematopoiesis towards granulocyte-monocyte progenitor production to elevate intratumoral NLR. Notably, although inflammatory caspase-1 is conventionally recognized for its host protective role in myeloid cells during infection14, our findings uncover a role for epiCaspase-1 under chemotherapeutic stress. Importantly, inflammatory pyroptosis is not inherently immunogenic; rather, it orchestrates a systemic hematopoiesis skew in the bone marrow, shaping the immune landscape towards pro-tumorigenic, neutrophil-dominant inflammation. Collectively, these findings expand the focus beyond the local TIME to encompass a distal microenvironment (i.e., the bone marrow) as a key immunoregulatory site governing systemic responses to cytotoxic chemotherapy. Furthermore, we establish IL-1α as a second iDAMPs molecule within this emerging family that, in addition to its localized effects within the TIME9, exerts distal immunomodulatory activity at the bone marrow to dampen anti-tumoral immune responses.
Results
Chemotherapy-induced epiCaspase-1 correlates with neutrophil-dominant inflammation and poor clinical outcome
Gemcitabine, a first-line standard-of-care chemotherapy for multiple solid malignancies, including muscle-invasive bladder cancer, is well established to exert its cytotoxic effects through caspase-3-mediated apoptosis in cancer cells. However, whether gemcitabine can also trigger alternative inflammatory cell death pathways—particularly, inflammatorycaspase-1-mediated pyroptosis—remains understudied. To interrogate this possibility, we performed transcriptomic analyses on twenty patients with chemotherapy-resistant bladder cancer, leveraging matched pre- and post-treatment tissues15. Gene set enrichment analysis (GSEA) revealed a significant enrichment of not only the apoptotic pathway (Supplementary Fig. 1a), but also the NOD-like receptor pathway, a major upstream sensor of the inflammasome and caspase-1 activation (Fig. 1a). Conversely, other cell death pathways, including ferroptosis and necroptosis, were not enriched in these chemo-resistant cancer patients (Supplementary Fig. 1b). To validate these clinical findings using experimental models, we performed RNA sequencing on gemcitabine-treated or vehicle-treated human bladder cancer cells (Fig. 1b). Mirroring the clinical transcriptomic findings, GSEA confirmed an enrichment of the NOD-Like receptor pathway, with substantial overlap in key pathway genes (Fig. 1c), corroborating the clinical relevance of the inflammasome-caspase-1 axis in the chemotherapy context.

Gene set enrichment analysis (GSEA) demonstrating the enrichment of NOD-Like Receptor pathway in patients with chemo-resistant bladder cancer, comparing matched pre- and post-chemotherapy cancer tissues (n = 20) (a) and gemcitabine treated T24 (3 biological replicates) (b). c Heatmap of common differentially expressed genes from (a) and (b). Scale bar represents normalized expression. Column represents genes and row represents matched patients pre- and post- treatment/ independent T24 samples. d Representative of three western blot replicates evaluating the caspase-1 pathway components [full length (GSDMD-FL) and cleaved N-terminus gasdermin D (GSDMD-N)] in T24, G69, and 4T1 after gemcitabine chemotherapy treatment at 0.5 μM, 120 μM and 0.1 μM, respectively. e Representative fluorescent microscopy evaluating caspase-1 dimerization using caspase-1-specific bimolecular fluorescence complementation (BiFC, green) reporter, and dsRed2-mito7 (red) in T24. f Representative real-time imaging demonstrating the temporal increase of caspase-1 dimerization in T24 and G69 cancer cells during chemotherapy (1 of 3 biological replicates shown here). g Representative scanning electron microscopy displaying cellular morphology of cancer cells undergoing apoptotic (top panel) and lytic cell death (bottom panel), respectively, in response to gemcitabine chemotherapy. Lactate dehydrogenase (LDH) release assay upon gemcitabine and co-treatment with the caspase-1 inhibitor VRT-043198 (40 μM) in T24 (h), G69 (i), and 4T1 (j) cancer cells (3 biological replicates per model). k qPCR showing relative IL1B mRNA expression post LPS priming (4 biological replicates). l Representative of three western blot replicates of molecular markers characterizing caspase-1 dependent lytic cell death after LPS priming and nigericin treatment. m Methodology for the derivation of the CASCADE signature (Chemotherapy-Activated Signaling reflecting Caspase-1 Associated Death Effect) Kaplan-Meir survival curve of patients with high and low CASCADE expression score in TCGA Bladder Cancer (BLCA) (n = 404) (n) and GSE25066 breast cancer cohort (n = 374) (o). p 10X Xenium spatial profiling in responder (n = 1) and non-responder (n = 1) patient tissues. Images represent full panel (left panel, ROI: yellow box), magnified ROI (middle panel; yellow box in full panel), and single channels (right panels). q UMAP analysis of epithelial cell clusters from patient tissues in (p), showing eleven distinct sub-clusters (left panel) and the relative expression of CASCADE signature in each sub-cluster (right panel). Bubble plot illustrating the relative expression CASCADE signature in each sub-clusters; clusters 4, 5 and 10 are designated as CASCADEhigh while clusters 1, 6, 9 are designated as CASCADElow (bottom). A total of 44,935 cells were analyzed. r Bar plot showing the proportions of CASCADEhigh (red) and CASCADElow (blue) epithelial clusters in responder (n = 1) and non-responder (n = 1) patients. s CIBERSORTx neutrophil signature score of CASCADEhigh and CASCADElow patients in TCGA BLCA (n = 404). Statistics: Ordinary one-way ANOVA followed by Dunnett’s multiple comparisons test (h–k), log-rank test (n, o) and two-tailed Wilcoxon rank sum test (s) were performed. Error bars represent standard error of mean (s.e.m.) unless otherwise stated. Source data are provided as a Source Data file.
Further molecular characterization confirmed chemotherapy-induced epiCaspase-1 activation in human bladder, murine bladder5, and breast epithelial cancer cells. Western blot analysis revealed that gemcitabine induces the protein expression of inflammatory caspase-1 and proteolytic cleavage of its key substrate, gasdermin D (GSDMD) (Fig. 1d), alongside the extracellular release of other caspase-1 substrates and hallmark cytokines of inflammatory cell death, i.e., interleukin-1β and interleukin-1816 (Supplementary Fig. 1c). Real-time imaging using an established caspase-1-specific bimolecular fluorescence complementation (BiFC, GFP) reporter for visualizing caspase-1 dimerization17 revealed progressive epiCaspase-1 activation in cancer cells upon gemcitabine treatment. This activation was observed through BiFC reporter dimerization and the accumulation of GFP signal (Fig. 1e, f, Supplementary Fig. 1d, white arrows) over the dsRed2-Mito7 reporter, which was used to monitor transfection efficiency. Interestingly, GFP-positive cancer cells displayed a swelling morphology characteristic of lytic cell death, in stark contrast to classical apoptosis observed in GFP-negative cancer cells (Fig. 1e, white arrows). Scanning electron microscopy corroborated these findings, revealing two morphologically distinct cell death phenotypes: (1) classic apoptotic “beads-on-a-string”18 (Fig. 1g; top panel), and (2) a swelling morphology indicative of lytic cell death (Fig. 1g; bottom panel). We next assessed whether epiCaspase-1 activation in this context depended on the inflammasome by knocking down PYCARD in T24 cells (T24 shASC). Knockdown efficiency was confirmed by both qPCR (Supplementary Fig. 1e) and western blotting (Supplementary Fig. 1f). To this end, we observed that gemcitabine-induced IL-1β (Supplementary Fig. 1g) and Lactate Dehydrogenase (LDH) (Supplementary Fig. 1h) release were markedly reduced in T24 shASC cells, indicating that inflammasome signaling is involved in epiCaspase-1 activation in response to chemotherapy.
To rigorously validate that chemotherapy-induced lytic cell death is epiCaspase-1 dependent, we employed an array of functional assays that capture distinct facets of the inflammatory cell death process. First, flow cytometry co-staining of cancer cells with Annexin-V (AV) and 7AAD revealed a significant expansion of the lytic cell fraction (AV-7AAD+) following gemcitabine treatment, highlighting the presence of lytic cell death (Supplementary Fig. 1i, j). Further corroborating this phenomenon, the LDH release assay—widely recognized as a gold-standard indicator of membrane rupture—demonstrated a marked increase in LDH release across multiple cancer cell models following gemcitabine treatment (Fig. 1h–j; black vs red bars). Co-treatment with the caspase-1 inhibitor VRT-043198 (40 μM) significantly attenuated LDH release (Fig. 1h–j; red vs light blue bars), confirming that epiCaspase-1 is functionally important for gemcitabine-induced lytic cell death. We also treated G69 with a panel of chemotherapies belonging to different classes, namely: cisplatin (platinum—a backbone chemotherapy for many solid cancers), doxorubicin (anthracycline), mitoxantrone (topoisomerase II inhibitor), and ML162 (ferroptosis inducer) to assess the generalization of this phenomenon. Cisplatin seems to induce a higher degree of lytic cell death and is epiCaspase-1 dependent, as demonstrated by a decrease in LDH when co-treated with the caspase-1 inhibitor, while other drugs elicit a varying degree of lytic cell death (Supplementary Fig. 1k). These results provided further rationale to study the role of epiCaspase-1-dependent lytic cell death in the context of chemotherapy treatment. Collectively, these findings advance our understanding of gemcitabine-induced cytotoxic effects and provides insight into the conventional view that apoptosis is the dominant mode of cell death, as these result reveal that gemcitabine elicits a heterogeneous spectrum of cell death, including an epiCaspase-1-mediated lytic cell death.
Given that chemotherapy orchestrates multiple cell death pathways, we sought to isolate the specific contribution of epiCaspase-1-dependent pyroptosis from other cell death modalities. To achieve this, we sought to develop a unique gene signature that reflects the downstream effects and biology of epiCaspase-1-driven pyroptosis in cancer cells—designated as CASCADE (Chemotherapy-Activated Signaling reflecting Caspase-1-Associated Death Effect). We employed a two-step induction model previously validated from other cell types19 (Supplementary Fig. 1l). Human bladder cancer cells were first stimulated with lipopolysaccharide (LPS), a canonical TLR4 agonist, to initiate inflammasome priming, which yielded peak IL1B mRNA expression at 3 h (Fig. 1k). This was followed by nigericin treatment to trigger epiCaspase-1 activation. Using this approach, we successfully recapitulated robust epiCaspase-1 mediated pyroptosis, as confirmed by real-time imaging validation of Sytox Green uptake and the characteristic swelling morphology of lytic cell death (Supplementary Fig. 1m). We next performed integrated analyses using (i) flow cytometry to quantify the lytic cell fraction (AV-PI+) (Supplementary Fig. 1n), and (ii) immunoblotting for hallmark pyroptosis mediators, including cleaved caspase-1 and GSDMD N-terminus (Fig. 1l). Both assays confirmed robust activation of epiCaspase-1-dependent pyroptosis. With this model established, we performed bulk RNA sequencing on three biological replicates of LPS/nigericin- and vehicle-treated cancer cells, identifying 2,212 differentially expressed genes between the groups (Fig. 1m). Using a clinical cohort (GSE87304)20,21 as the training dataset, we refined this gene set by Cox proportional hazard regression analysis for their association with overall survival (p < 0.05, HR > 1), yielding 61 significant genes that defined the CASCADE signature (Fig. 1m, Supplementary Table 1).
Notably, CASCADEhigh tumors demonstrated significant prognostic value correlating with poor overall survival not only in the training cohort (Supplementary Fig. 2a), but also in the TCGA BLCA cohort (Fig. 1n), and multiple other independent bladder cancer (GSE32894, GSE48276) (Supplementary Fig. 2b) and breast cancer (GSE25066) (Fig. 1o) patient datasets. Extending the analysis to pan-cancer TCGA datasets, CASCADEhigh tumors correlated with poorer overall survival across other malignancies, including Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma (CESC), Brain Lower Grade Glioma, Lung Squamous Cell Carcinoma, Mesothelioma (MESO), Glioblastoma Multiforme (GBM), Stomach Adenocarcinoma (STAD), Thyroid Carcinoma (THCA), and Uveal Melanoma (UVM) (Supplementary Fig. 2c). These findings establish that CASCADEhigh patients correlate with poor clinical outcome as a generalized phenomenon across multiple malignancies.
To further establish CASCADEhigh tumor cells as key determinants of chemotherapy response and modulators of the immune microenvironment, we performed spatial transcriptomic profiling on tissues obtained from two patients with bladder cancer: a responder and a non-responder, using the 10X Genomics Xenium Human Immuno-Oncology Panel, coupled with key CASCADE signature genes (Fig. 1p, Supplementary Fig. 2d). Both patients were initially diagnosed with T2 muscle-invasive bladder cancer and underwent four cycles of gemcitabine-cisplatin chemotherapy. Following treatment, the responder’s tumor had down-staged to T0, while the non-responder progressed to T3b.
Spatial transcriptomic profiling reaffirmed our transcriptomic findings from Fig. 1n–o (Supplementary Fig. 2a–c). UMAP clustering of epithelial cancer cells revealed eleven distinct tumor cell subclusters, with populations 4, 5, and 10 designated as CASCADEhigh tumor populations, whereas populations 1, 8, and 9 were designated as CASCADElow tumor populations (Fig. 1q). Intriguingly, the chemotherapy-non-responder exclusively harbored CASCADEhigh tumor cells, while the responder patient primarily harbored CASCADElow tumor cells (Fig. 1p, r). This remarkable divergence underscores a potential association between CASCADEhigh tumor cells and chemoresistance. Unbiased analysis of the immune cell landscape further revealed a significantly elevated neutrophil presence in the non-responder compared with the responder (Fig. 1p). Given that a high NLR–an indicator of neutrophil dominant inflammation and impaired adaptive immunity–had reported association with poor prognosis and reduced therapy response, these findings implicate an immunomodulatory role for CASCADEhigh tumor cells. Supporting this hypothesis, deconvolution of the TCGA BLCA dataset using CIBERSORTx22 confirmed significantly enriched neutrophil signatures in CASCADEhigh patients relative to CASCADElow patients (Fig. 1s). Despite the clinical value of NLR, its molecular underpinnings remain unknown. Our findings highlight epiCaspase-1-driven CASCADEhigh tumor cells as potential drivers of neutrophil-dominant inflammation by shifting NLR dynamics toward an unfavorable immune landscape, warranting further mechanistic investigation.
epiCaspase-1 suppresses immunogenic cell death in a classical vaccination assay in vivo
Building on these findings, we next investigated the functional consequences of chemotherapy-induced epiCaspase-1 activation in shaping downstream immunologic responses. While caspase-1 activation in innate immune cells is well-documented in the context of infection23, its role within epithelial tumor cells in the context of chemotherapy treatment remains poorly understood. Specifically, we sought to determine whether epiCaspase-1-dependent pyroptosis has a role in immunomodulation, recognizing that the mode of tumor cell death can be a key determinant of therapeutic outcomes. To evaluate the role of epiCaspase-1 in modulating immunogenic cell death, we genetically knocked out epiCaspase-1 in murine bladder cancer cells using CRISPR-Cas9 (G69Casp1KO) and knocked down epiCaspase-1 in breast cancer cells using shRNA (4T1shCasp1), respectively (Fig. 2a). Functional loss of epiCaspase-1 activity was confirmed by a marked reduction in extracellular LDH release in both models post-gemcitabine treatment (Fig. 2b, c; red vs blue bars). Next, we employed a gold-standard vaccination assay to assess the immunomodulatory role of epiCaspase-1 in eliciting immunogenic cell death in vivo. In brief, cancer cells were treated with IC50 dose of gemcitabine for 24 h and injected into the left hind flank of syngeneic, immunocompetent mice as “vaccines” (Fig. 2d). Seven days post-vaccination, mice were “rechallenged” with live wild type cells injected to the right flank. In the case when treated cancer cells had undergone immunogenic cell death and successfully conferred an immunogenic response, these mice would reject live tumor cell engraftment upon rechallenge (Fig. 2d).

a Representative of three western blot replicates confirming the efficiency of caspase-1 knockout by CRISPR-Cas9 in G69 bladder cancer cells and knockdown by shRNA in 4T1 breast cancer cells. b LDH release from wild-type (WT) and caspase-1 knockout (Casp1KO) G69 cells after gemcitabine treatment (3 biological replicates). c LDH release from wild-type (WT) and caspase-1 knockdown (shCasp1) 4T1 cells after gemcitabine treatment (3 biological replicates). d Schematic diagram illustrating the gold-standard vaccination assay for evaluating immunogenic cell death in vivo. Created in BioRender. Wong, S. (2026) https://BioRender.com/az159ga. e Kaplan-Meier plot showing tumor-free incidence of mice vaccinated with either vehicle, gemcitabine treated G69 wild-type (gemG69WT), or gemcitabine treated caspase-1 KO (gemG69Casp1KO) cancer cells as “vaccines” (n = 10 mice per group). f Corresponding changes in tumor burden from (e). g Kaplan-Meier plot showing tumor-free incidence of mice vaccinated with either vehicle (n = 10 mice per group), gemcitabine treated 4T1 wild-type (gem4T1WT, n = 10), or gemcitabine treated caspase-1 knockdown (gem4T1shCasp1, n = 8) cancer cells as “vaccines”. h Corresponding changes in tumor burden from (g). i Kaplan-Meier plot showing tumor-free incidence of mice vaccinated with either vehicle, gemG69WT, or gemG69Casp1KO cancer cell “vaccines” with prior treatment anti-CD8 mAb (αCD8) (n = 5 mice per group). j Kaplan-Meier plot showing tumor-free incidence of mice vaccinated with gemG69WT cancer cell as “vaccines” in mice depleted (αLy6G) and not depleted of neutrophils (n = 8 mice per group). k Schematic diagram depicting a modified vaccination assay in vivo. Red arrow: vaccination doses and treatment scheme. Created in BioRender. Wong, S. (2026) https://BioRender.com/6n2awul. l Flow cytometry gating strategy for the analysis of bone marrow progenitor cell subpopulations, following vaccination with vehicle [black], gemG69WT [red] or gemG69Casp1KO [blue] as “vaccines”. m Flow cytometry results showing the counts of granulocyte-monocyte progenitors (GMPs; Lin-Sca-cKit+CD16/32hiCD34hi) in mice vaccinated with gemG69WT, or gemG69Casp1KO (n = 6 mice per group). Statistics: Ordinary one-way ANOVA followed by Dunnett’s multiple comparison test (b, c, m), two-way ANOVA with Geisser-Greenhouse correction followed by Dunnett’s multiple comparison test (f, h), and log-rank (Mantel-Cox) test (e, g, i, j) were performed. Error bars represent standard error of mean (s.e.m.) unless otherwise stated. Source data are provided as a Source Data file.
To this end, mice vaccinated with gemcitabine-treated G69WT cells (gemG69WT) failed to reject tumor engraftment upon challenge (n = 10 mice per group) (Fig. 2e; red line), consistent with results from our previous report5. In contrast, a significant proportion of mice (6/10 mice) vaccinated with gemcitabine-treated G69Casp1KO (gemG69Casp1KO) cancer cells rejected tumor engraftment upon challenge (Fig. 2e; blue line), indicating that successful induction of immunogenic cell death is dependent on epiCaspase-1 deficiency. Moreover, in the few mice vaccinated with gemG69Casp1KO that exhibited tumor engraftment, their tumors displayed significantly restricted growth compared to control [4/10 gemG69Casp1KO (blue) versus 10/10 gemG69WT (red)] (Fig. 2f), indicating a partial but enhanced immune response during epiCaspase-1 deficiency. These observations were also generalized to the murine breast 4T1 cancer model where mice vaccinated with gemcitabine-treated 4T1shCasp1 (gem4T1shCasp1) cancer cells mounted a more effective immune response against the challenge as compared to those vaccinated with gemcitabine-treated 4T1WT (gem4T1WT) cancer cells (n = 8 mice per group) (Fig. 2g, h; red vs blue lines/bars). Taken together, these findings support the notion that epiCaspase-1 suppresses immunogenic cell death in the context of chemotherapy, highlighting that pyroptosis, while lytic and inflammatory, is not inherently immunogenic in this treatment context.
To investigate the mechanism by which epiCaspase-1 mediates its immunomodulatory effects, we evaluated the functional importance of cytotoxic CD8+ T cells, which are postulated as the key effector cells driving tumor rejection upon induction of immunogenic cell death3,24. This was achieved by depleting CD8+ T cells using a validated anti-CD8 monoclonal antibody (mAb)25. Mice were treated with anti-CD8 mAb on Days 4 and 6 post-vaccination before the challenge at Day 7 (Supplementary Fig. 3a). The efficiency of CD8+ T cells depletion was confirmed by flow cytometric analysis of peripheral blood (Supplementary Fig. 3b, c). In mice with CD8+ T cells depleted (αCD8), the previously observed immunogenic effects of gemG69Casp1KO vaccination were completely abolished, whereby all mice could no longer reject the live tumor cell challenge (n = 5 mice per group) (Fig. 2i) and tumors grew comprable to the control group (Supplementary Fig. 3d).
Given our clinical observation that CASCADEhigh tumor cells are associated with heavy neutrophil infiltration in chemo-resistant cancer patient (Fig. 1p–s), we next sought to investigate a potential immunomodulatory role of neutrophils using the same vaccination assay. To this end, we depleted neutrophils using a validated anti-Ly6G antibody following a previously established regimen, and evaluated how neutrophil depletion influences immunogenic cell death induced by vaccination of gemcitabine-treated G69WT (gemG69WT) cancer cells (n = 8 mice per group) (as in Fig. 2d). There was a significant increase in tumor-free survival for neutrophil-depleted mice when compared to vehicle-treated control mice (Fig. 2j); the efficacy of neutrophil depletion was confirmed by flow cytometry analysis of corresponding peripheral blood (Supplementary Fig 3e). Taken together, these findings support the notion that the immunomodulatory effects of epiCaspase-1 during chemotherapy is mediated, in part, by immunosuppressive neutrophils that dampen CD8⁺ T cell anti-tumoral activity.
epiCaspase-1 promotes granulocyte-monocyte progenitors distally in the bone marrow
While prior studies investigating immunogenic cell death have primarily focused on the immune landscape within the primary tumor26, to our knowledge, the role of bone marrow hematopoiesis--which is imperative for the rapid production of immune cells from early progenitor populations during acute infection27 or cancer development28--in modulating an immunogenic versus tolerogenic immune response remains unexplored. Therefore, this represent We, therefore, sought to first explore whether gemcitabine-treated cancer cells can remotely influence hematopoiesis in the bone marrow. To test this hypothesis, we modified the vaccination assay shown in Fig. 2d and used flow cytometry to characterize hematopoietic progenitor cell populations isolated from the bone marrow 48 h after the final vaccination dose (Fig. 2k, l). This modified vaccination assay allowed us to specifically examine how chemotherapy-induced cell death (i.e., immunogenic vs tolerogenic) might functionally regulate bone marrow hematopoiesis, while circumventing the direct systemic cytotoxic effects of chemotherapy on the host organism.
Intriguingly, mice vaccinated with gemG69WT exhibited a significant increase in the number of granulocyte-monocyte progenitors (GMPs; Lin-Sca-1-c-Kit+CD16/32hiCD34hi), whereas this increase was markedly diminished in gemG69Casp1KO vaccinated mice (Fig. 2m). Taken together, these findings revealed that gemcitabine-induced epiCaspase-1 can remotely influence bone marrow hematopoiesis by promoting a myeloid-biased skew, which are likely mediated by tumor-derived factors that are released upon chemotherapy treatment.
epiCaspase-1-driven myelopoiesis depends on NINJ1-mediated IL-1α release
To identify the effector molecules released from cancer cells that drive epiCaspase-1-dependent myeloid skew, we first targeted known signaling pathways associated with caspase-1—namely IL-33, HMGB1, Toll-Like receptor 4, IL-1β, IL-18—using corresponding inhibitors (ST2i, HMGB1i, TLR4i) and neutralizing antibodies (anti-IL-1β antibody, anti-IL-18 antibody) (Fig. 3a), using the same modified vaccination assay as Fig. 2k–l. Unexpectedly, mice vaccinated with gemcitabine-treated G69 cells, in the presence or absence of these inhibitors or neutralizing antibodies, showed no significant changes in GMP levels (Fig. 3b; red vs gray bars), suggesting the involvement of alternative mediators in modulating epiCaspase-1-dependent myeloid skew. To identify the alternative mediators, we performed an unbiased cytokine screen using supernatant collected from gemcitabine-treated G69 wild type (WT) and epiCaspase-1 knockout (Casp1KO) G69 cells (Fig. 3a, c). Notably, we observed a significant reduction in the alarmin, IL-1α, in gemcitabine-treated Casp1KO supernatant (Fig. 3d), alongside a concomitant increase in its antagonist, IL-1ra (Fig. 3d). This shift in the IL-1α axis suggests its potential role in epiCaspase-1 mediated hematopoietic regulation.

a Schematic diagram illustrating experimental approaches employed to identify the molecular mediator(s) crucial for facilitating gemcitabine-induced myelopoiesis. Created in BioRender. Wong, S. (2026) https://BioRender.com/8gnuj86. b Fold change in GMP levels in mice vaccinated with gemcitabine treated G69, in the presence or absence of inhibitors against the caspase-1 associated substrates (ST2i: IL-33 inhibitor, HMGBi: HMGB1 inhibitor, TLR4i: Toll-Like Receptor 4 inhibitor, α-IL-1β: anti-IL-1β antibody, α-IL-18: anti-IL-18 antibody) (n = 5 mice per group) c Cytokine array comprising to identify effector molecules released from gemcitabine-treated cancer cells (1 biological replicate). d Densitometry quantification of 1. IL-1α and 2. IL-1ra from the cytokine array (1 biological replicate). e ELISA quantifying the relative protein expression of IL-1α in supernatants of G69 wild-type (WT), Caspase-1 knockout (Casp1KO) and NINJ1 knockout (NINJ1KO) cancer cells treated with vehicle, gemcitabine (Gem; Gem + Casp1KO, Gem + NINJ1KO), or gemcitabine plus the caspase-1 inhibitor VRT-043198 (Gem + VRT-043198) (3 biological replicates). f Schematic diagram illustrating the identification and functional validation of signaling mediators downstream to epiCaspase-1 in modulating gemcitabine-induced myelopoiesis. Created in BioRender. Wong, S. (2026) https://BioRender.com/9dzk8x8. Absolute count of (g) GMP, (h) CMP, and (i) MEP, per 106 cells in the bone marrow of mice vaccinated with G69 cancer cells treated with vehicle (Vehicle, n = 11), gemcitabine-treated wild-type G69 (gemG69WT, n = 12), gemcitabine-treated IL-1α knockout G69 (gemG69IL-1αKO, n = 8), gemcitabine-treated NINJ1KO knockout G69 (gemG69NINJ1KO, n = 9) or gemG69WT followed by various agents targeting the IL-1α signaling, [anti-IL-1α antibody (α-IL-1α, n = 8), anti-IL-1R antibody (α-IL-1R, n = 10) or Anakinra (n = 7). j Kaplan-Meier plot showing tumor-free incidence of mice vaccinated with either vehicle, gemcitabine-treated G69 wild-type (gemG69WT) or gemcitabine-treated IL-1α KO (gemG69IL-1αKO) cancer cells as “vaccines” (n = 8 mice per group). k Kaplan-Meier plot showing tumor-free incidence of mice vaccinated with either vehicle, gemcitabine-treated G69 wild-type (gemG69WT) or gemcitabine-treated NINJ1KO (gemG69NINJ1KO) cancer cells as “vaccines” (n = 8 mice per group). l Expression of Il1r1 in HSC and distinct HSPCs in normal mouse BM. Data and images were generated from Gene Expression Commons. Statistics: Ordinary one-way ANOVA followed by Dunnetts’s multiple comparison test (b, e, g, h, i), and log-rank (Mantel-Cox) test (j, k) were performed. Error bars represent standard error of mean (s.e.m.) unless otherwise stated. Source data are provided as a Source Data file.
First, we confirmed using ELISA that the release of IL-1α during gemcitabine treatment was indeed caspase-1 dependent (Fig. 3e). Both Casp1KO G69 cells (Fig. 3e; blue) and WT cells co-treated with the caspase-1 inhibitor, VRT-043198 (Fig. 3e; light blue), exhibited significantly reduced IL-1α release upon gemcitabine treatment (Fig. 3e; red vs blue and light blue), demonstrating that gemcitabine-induced IL-1α release is dependent on epiCaspase-1. Intriguingly, knockout of NINJ1 (NINJ1KO), a key effector of lytic cell death and plasma membrane rupture29, also attenuated IL-1α release (Fig. 3e; red vs brown). The functional loss of NINJ1 was validated by the absence of LDH release following gemcitabine treatment (Supplementary Fig. 4a, red vs brown). To further investigate the functional contribution of this IL-1α/IL1r1 signaling axis and NINJ1 in cell death-induced myeloid skewing, we employed the previously described vaccination assay in Fig. 2k–l, in the presence or absence of various IL-1α pathway inhibitors (Fig. 3f). Specifically, we utilized a multimodal approach, including: (1) an anti-IL-1α antibody (direct), (2) an anti-IL-1R antibody (receptor) and (3) Anakinra (a peptide receptor antagonist) (Fig. 3f, Supplementary Fig. 4b; blue arrows). To rigorously examine whether cancer cell-derived IL-1α is the major driver for this observed myeloid progenitor skew, we further incorporated genetic knockouts of IL-1α (gemG69IL-1αKO) and NINJ1 (gemG69NNJ1KO) into this vaccination assay (Fig. 3f). Mice vaccinated with gemG69WT displayed increased number of GMPs as expected (Fig. 3g; black vs red bar). In addition, these mice exhibited a significant rise in CMPs (Fig. 3h; black vs red bar), but no notable significant changes in MEPs (Fig. 3i; black vs red bar). Since CMPs are progenitor cells that differentiate into either GMPs or MEPs30, the preferential expansion of GMPs (Fig. 3g; black vs red bar) over MEPs (Fig. 3i; black vs red bar) indicated a myeloid-biased hematopoiesis in gemG69WT-vaccinated mice.
Intriguingly, co-treatment with neutralizing antibodies or inhibitors significantly reduced the number of GMPs in the bone marrow (Fig. 3g; red vs dark blue bars). Consistent with this, similar inhibitory effects on GMPs were observed in mice vaccinated with gemG69IL-1αKO and gemG69NINJ1KO cancer cells (Fig. 3g–i; red vs light blue/ brown bars), definitively establishing epithelial cancer cells as the source of IL-1α in driving myeloid progenitor expansion in this setting. Notably, mice vaccinated with gemG69IL-1αKO and gemG69NINJ1KO cancer cells (Fig. 3g–i; red vs light blue/brown bars) cancer cells also showed significant alterations in CMP and MEP populations (Fig. 3h, i; red vs light blue/brown bars). However, we observed that co-treatment with neutralizing antibodies or inhibitors did not significantly affect CMPs or MEPs.
To further elucidate the role of IL-1α and NINJ1 in modulating immunogenic cell death, we employed the gold-standard vaccination assay as illustrated in Fig. 2d. Remarkably, mice vaccinated with either gemG69IL-1αKO (Fig. 3j; light blue line) or gemG69NINJ1KO (Fig. 3k; brown line) mounted a robust immunogenic response to the challenge, respectively, demonstrating that genetic ablation of IL-1α or NINJ1 in cancer cells was sufficient to restore immunogenic response in mice when compared with gemG69WT (Fig. 3j, k; red vs light blue/brown lines). These findings establish the epithelial caspase-1/IL-1α axis as a key suppressor of immunogenic cell death, with its regulatory function dependent on NINJ1.
To gain insight into how epiCaspase-1/IL-1α axis might impact myeloid skew, we analyze the expression pattern of the IL-1α receptor, i.e., Il1r1, across different hematopoietic progenitor populations within the bone marrow, by querying existing Gene Expression Commons dataset31. Interestingly, we discovered that the Il1r1 expression is highest in progenitor populations preceding GMPs, namely pGMPa (pre-Granulocyte/Macrophage Progenitor subset A) and pGMPb (pre-Granulocyte/Macrophage Progenitor subset B), suggesting a plausible role of IL-1α in promoting the formation of GMPs from these pre-GMP populations (Fig. 3l). Together, these results establish a role for the epiCaspase-1/IL-1α axis, originating from the primary tumor, in remotely skewing myelopoiesis at the distal bone marrow microenvironment. Importantly, our findings also demonstrate that this epiCaspase-1/IL-1α axis functionally suppresses immunogenic cell death, as validated by the classical vaccination assay. Given these insights, further preclinical evaluation is warranted to explore this phenomenon and the therapeutic implications of targeting this pathway.
Gemcitabine induces bone marrow myeloid progenitors in preclinical models in vivo
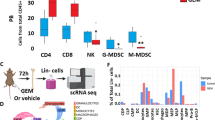
Building on our findings that chemotherapy-induced epiCaspase-1 can remotely modulate hematopoiesis, we next investigated its functional relevance while exploring the therapeutic implications of targeting this pathway in a preclinical treatment setting. To test this, immunocompetent mice bearing G69 tumors were treated with two cycles of gemcitabine-cisplatin chemotherapy (GC; Gemcitabine 120 mg /kg, Cisplatin 6 mg/kg) (Supplementary Fig. 4c). Hematopoietic cells from the tibia and femur were then analyzed at the molecular level. To achieve an unbiased characterization of the impact of chemotherapy treatment on bone marrow hematopoietic progenitor cells at a granular level, we employed single-cell RNA sequencing (scRNA-seq) to interrogate lineage-negative bone marrow hematopoietic progenitor cells from G69 tumor-bearing mice treated with GC or vehicle (n = 2 mice per group). Using immunomagnetic separation, we enriched bone marrow hematopoietic progenitor cells and subjected them to 10x Genomics scRNA-seq (Fig. 4a). Uniformed Manifold Approximation and Projection (UMAP) visualization revealed eight distinct hematopoietic progenitor cell clusters, annotated based on top differentially expressed genes and cross-referenced with established literature (Fig. 4b–d). These clusters included: 1. Hematopoietic stem and progenitor cells (HSPCs; Hlf/Cd34hi), 2. Pro-Erythrocytes (Pro-Ery; Gata1/Mt1hi) 3. Monocytic/dendritic progenitor cells (MDP; Irf8/Lgals1/Mpeg1hi), 4. Common lymphoid progenitor cells (CLP; Skap1hi), 5. Basophil/eosinophil/mast progenitor cells (Ba/Eo/MaP; Ms4a2/Cpa3hi), 6. Granulocyte-monocyte progenitor (GMP; Ms4a3hi), 7. Neutrophil progenitor (NeuP; S100a8/Elane/Mpohi), and 8. Pro-B cells (Pro-B cells; Ebf1/Pax5/Ighmhi) (Fig. 4b–d, Supplementary Fig. 4d, Supplementary Table 4). A representative gene marker for each progenitor cell type cluster was chosen in Fig. 4c and validated by expression specificity within the UMAP projection (Fig. 4d).

a Schematic illustrating the isolation and immunomagnetic enrichment of bone marrow hematopoietic progenitors for downstream single cell RNA sequencing (scRNAseq) analysis (Gemcitabine: 120 mg/kg, Cisplatin: 6 mg/kg, n = 2 mice per group). Created in BioRender. Wong, S. (2026) https://BioRender.com/6i8hwv9. b UMAP projection of eight distinct hematopoietic progenitor cell clusters with colored inference by specific cell type (n = 2 mice per group, 38,675 number of total cells analyzed). c Violin plot and d UMAP projection of hematopoietic progenitor cell clusters colored-based on their normalized expression of cluster-specific genes (Meis1, Gata1, Mt1, Mpeg1, Skap1, Ms4a2, Ms4a3, S100a8 and Ighm). e UMAP projection of hematopoietic progenitor cell clusters isolated from tumor-bearing mice treated with gemcitabine-cisplatin chemotherapy (Chemo) and vehicle (Veh) as a control. f Box plot illustrating the frequency of the GMP population (cluster 6) in tumor-bearing mice treated with gemcitabine-cisplatin chemotherapy and vehicle as a control (n = 2 mice per group). g Frequency of GMPs in the bone marrow of G69 tumor bearing mice treated with vehicle or chemotherapy (n = 5 mice per group) using flow cytometry. Abbreviations: hematopoietic stem and progenitor cell (HSPC), Pro-Erythrocytes (Pro-Ery), monocyte-dendritic progenitor (MDP), common lymphoid progenitor (CLP), basophil-eosinophil-mast cell progenitor (Ba/Eo/MaP), granulocyte-monocyte progenitors (GMP), neutrophil progenitors (NeuP) and pro-B cells (Pro-B cells). Statistics: Two-tailed unpaired t test with Welch’s correction was performed for g. Error bars represent standard error of mean (s.e.m.) unless otherwise stated. Box plots show the minimum and maximum values, and the center line represents the mean. Source data are provided as a Source Data file.
Leveraging these annotated clusters, we examined how chemotherapy induced alternations in each of these hematopoietic progenitor subpopulations in the bone marrow (Fig. 4e, Supplementary Fig. 4e). Notably, we observed an increase in GMPs (cluster 6) following chemotherapy (Fig. 4f). Quantitative flow cytometry analysis of the bone marrow in tumor bearing mice further validated the increase in GMPs after chemotherapy (Fig. 4g), reinforcing the paradigm that chemotherapy not only impacts the local immune TME, but also profoundly induces a preferential hematopoietic skew towards myeloid progenitors in a pre-clinical treatment setting. These findings set the stage to further evaluate whether tumor-driven epiCaspase-1 activation plays a key role in remotely modulating hematopoiesis in the bone marrow microenvironment in this preclinical model.
Caspase-1 inhibition enhances chemotherapeutic efficacy by normalizing hematopoiesis
Given the functional link between epiCapase-1 activation and myeloid skew, we next examined whether a clinical-grade caspase-1-specific inhibitor could mitigate myeloid skewing and provide therapeutic benefits in the preclinical setting. Mice received the caspase-1 inhibitor (Casp1i; Belnacasan/VX-765, 50 mg/kg) orally every 2 days, beginning 24 h before the first GC chemotherapy dose (Supplementary Fig. 5a). Bone marrow hematopoietic cells were subsequently collected for scRNA-seq and flow cytometry analysis at the endpoint to evaluate the effects of Casp1i in modulating myeloid progenitor skew in this preclinical model (n = 2 mice per group).
Using pseudotime trajectory analysis, coupled with gene expression mapping we annotated and validated thirteen different cellular clusters belonging to myeloid-lineage differentiation (Fig. 5a-c, Supplementary Fig. 5b, c): 1. HSPC (Msi2/Cd34/HlfHi), 2. Myelo-lymphoid lineage cells (LMPP; Dntt/Flt3Hi), 3. CMP (Sox4/Tmsb10/Gata2Hi), 4. GMP (Cd34/Ms4a3/ElaneHi), 5. Megakaryocyte-erythrocyte progenitors (MEP; Vamp5/Mt1/Tfgbr3Hi), 6. NeuP (Camp/ Ngp/ LtfHi), 7. Pre-Basophil/Mast progenitors (Pre-BMP; Lmo4/Cpa3/Gata2Hi), 8. Dendritic cell progenitors (DcP; Tcf4/Irf8/SiglechHi), 9. Monocyte progenitors (MoP; Pid1/Ccr2/S100a4Hi), 10. Megakaryocyte progenitors (MkP; Pf4/Pbx1/Prkg1Hi), 11. PreNeu (S100a9/Mmp8/RetnlgHi), 12. Basophil progenitors (BaP; Prss34/Mcpt8/Ms4a2Hi), 13. Mast cell progenitors (MaP; Itga1/Il18rap/Cd200r3Hi). Consistent with our previous findings, GC chemotherapy induces a pronounced expansion of GMPs, increasing from 4.0 to 7.2% (Fig. 5d, black vs red bars). Notably, the co-inhibition of epiCaspase-1 during chemotherapy (Chemo + Casp1i) reduced GMPs from 7.2 to 5.1% (Fig. 5d, red vs teal bars), confirming a role for caspase-1 in myeloid skew. These findings establish pharmacological caspase-1 inhibition as a viable strategy to counteract chemotherapy-induced myeloid skew in a preclinical setting.

a UMAP projection of pseudotime trajectory analysis, to infer cellular differentiation status of myeloid-lineage hematopoietic progenitor cell clusters, based on their differential gene expression using the Monocle3 pipeline (n = 2 mice per group, 16,130 number of total cells analyzed). b UMAP illustrating the hematopoietic and myeloid progenitor cell populations in various treatment conditions. c Stacked bar plot representing the relative distribution or frequency of each progenitor cell cluster, identified in the myeloid-lineage hematopoietic progenitor cell partitioned immune population. d Box plot illustrating the relative distribution of GMP amongst different treatment group (n = 2). e Schematic diagram illustrating GC chemotherapy treatment with or without caspase-1 inhibition, and endpoint collection of bone marrow, blood and tumor for further downstream analyses. Created in BioRender. Wong, S. (2026) https://BioRender.com/b3e0oiw. Flow cytometry analysis illustrating the percentage of GMP (f) and neutrophils (g), in the bone marrow of tumor-bearing mice treated with vehicle [black], gemcitabine-cisplatin chemotherapy (Chemo; Gemcitabine: 120 mg /kg, Cisplatin: 6 mg /kg, VX-765: 60 mg /kg) [red], VX-765 caspase-1 inhibitor (Casp1i) + Chemo combination [teal], and Casp1i [blue], respectively (n = 7 mice per group). h Flow cytometry analysis illustrating the percentage of neutrophils in the peripheral blood of tumor-bearing mice treated with vehicle [black], gemcitabine-cisplatin chemotherapy (Chemo; Gemcitabine: 120 mg/kg, Cisplatin: 6 mg/kg, VX-765: 60 mg/kg) [red], VX-765 caspase-1 inhibitor (Casp1i) + Chemo combination [teal], and Casp1i [blue], respectively (n = 5 per group). i Flow cytometry analysis illustrating the absolute count of intratumoral CD11b+Ly6G+ neutrophils, in tumor-bearing mice treated with vehicle [black], gemcitabine-cisplatin (Chemo; Gemcitabine: 120 mg/kg, Cisplatin: 6 mg/kg, VX-765: 60 mg/kg) [red], VX-765 caspase-1 inhibitor (Casp1i) + Chemo combination [teal], and Casp1i [blue], respectively (n = 7 mice per group). j Normalized absolute count of CD62L+CD11b+Ly6G+ neutrophils into tumors of mice treated with vehicle [black], gemcitabine-cisplatin (Chemo; Gemcitabine: 120 mg/kg, Cisplatin: 6 mg/kg, VX-765: 60 mg/kg) [red], VX-765 caspase-1 inhibitor (Casp1i) + Chemo combination [cyan], and Casp1i [blue], respectively (n = 7 mice per group). Tumor growth kinetics in mice harboring G69 (Gemcitabine: 120 mg/kg, Cisplatin: 6 mg/kg, VX-765: 60 mg/kg) (k) or 4T1 (Gemcitabine: 30 mg/kg, Cisplatin: 6 mg/kg, VX-765: 60 mg/kg) (l) xenografts treated with vehicle [black], gemcitabine-cisplatin (Chemo) [red], VX-765 caspase-1 inhibitor (Casp1i) + Chemo combination [teal], and Casp1i [blue], respectively (n = 7 mice per group). Statistics: Ordinary one-way ANOVA followed by Dunnett’s multiple comparisons test (f–j) and two-way ANOVA with Geisser-Greenhouse correction followed by Dunnett’s multiple comparison test (k, l) were performed. Error bars represent standard error of mean (s.e.m.) unless otherwise specified. Abbreviations: hematopoietic stem and progenitor cell (HSPC), lympho-myeloid progenitor (LMPP), granulocyte-monocyte progenitors (GMP), common lymphoid progenitors (CMP), megakaryocyte-erythrocyte progenitors (MEP), monocyte progenitors (MoP), dendritic cell progenitors (DcP), neutrophil progenitor (NeuP), pre-neutrophil (preNeu), Pre-Basophil/Mast progenitors (Pre-BMP), mast cell progenitors (MaP), basophil progenitors (BaP) and Megakaryocyte progenitors (MkP). Box plots show the minimum and maximum values, and the center line represents the mean. Bone Marrow, Peripheral Blood and Intra-Tumoral cartoon icons were Created in BioRender. Wong, S. (2026) https://BioRender.com/b3e0oiw. Source data are provided as a Source Data file.
We next conducted multi-color flow cytometry on the bone marrow, peripheral blood, and tumor of mice from each treatment arm to quantitatively validate the scRNA-seq observations (Fig. 5e). Consistent with results from Fig. 5d, mice treated with GC chemotherapy exhibited a significantly higher frequency of GMPs in the bone marrow (Fig. 5f; black vs red), which was significantly reduced by caspase-1 inhibition (Fig. 5f; red vs teal). Similarly, the frequency of CMPs increased following chemotherapy treatment (Supplementary Fig. 6a; black vs red) and was markedly decreased upon caspase-1 inhibition (Supplementary Fig. 6a; red vs teal). In contrast, MEP frequencies, which decreased after chemotherapy treatment (Supplementary Fig. 6b; black vs red), increased upon caspase-1 inhibition (Supplementary Fig. 6b; red vs teal). We also observed a significant increase in Ly6G-expressing CD11b+ neutrophils in the bone marrow, rising from 48 to 57% following GC chemotherapy treatment (Fig. 5g; black vs red), which was significantly reversed by caspase-1 inhibition (Fig. 5g; black vs teal). Collectively, these data demonstrate that in tumor-bearing mice, chemotherapy induces a myeloid skew and increases neutrophils within the bone marrow, but can be reversed by caspase-1 inhibition.
To assess whether these bone marrow perturbations extended to systemic immune cell alterations, we analyzed corresponding peripheral blood samples via flow cytometry (Fig. 5e). Consistent with bone marrow findings, chemotherapy induced a prominent and significant increase in circulating neutrophils from 18 to 30% (Fig. 5h; black vs red). Caspase-1 inhibition significantly reduced circulating neutrophil frequency (Fig. 5h; red vs teal).
Given the heightened neutrophil frequency in both the bone marrow and peripheral blood following chemotherapy treatment, we next examined whether this skew extended into the local tumor microenvironment (TME). Chemotherapy-treated tumors exhibited elevated neutrophil counts (Fig. 5i; red vs black) and a trending increase in frequencies (Supplementary Fig. 6c), that was significantly diminished upon caspase-1 inhibition (Fig. 5i; red vs. teal, Supplementary Fig. 6c). Additionally, analysis of CD62L expression—an adhesion marker32 that distinguishes newly infiltrated neutrophils (CD62L+) from aged neutrophils (CD62L−)—revealed a significant enrichment of CD62L+ neutrophils in chemotherapy-treated tumors, suggesting the influx of newly formed neutrophils after chemotherapy treatment (Fig. 5j, Supplementary Fig. 6d).
More importantly, we observed a significant therapeutic benefit in mice co-treated with caspase-1 inhibition alongside chemotherapy treatment. G69 tumor-bearing mice receiving concomitant chemotherapy and Casp1i administration exhibited significantly enhanced therapeutic response as compared to those in other treatment groups (Fig. 5k). Notably, pharmacological co-inhibition of caspase-1 also improved the efficacy of chemotherapy treatment in mice bearing 4T1 murine breast cancer tumors (Fig. 5l).
Collectively, our findings provide evidence that GC chemotherapy is able to rewire systemic hematopoiesis and promote a neutrophil-dominant TIME; this is characterized by a sustained myeloid skew originating from the bone marrow (Fig. 5f, g), followed by a systemic increase of neutrophils in both circulation (Fig. 5h) and TME (Fig. 5i, j), resulting in the lack of response to chemotherapy. Importantly, pharmacological inhibition of caspase-1 improves responses to chemotherapy, likely resulting from the normalization of bone marrow hematopoiesis.
Caspase-1/IL-1α inhibition recalibrates neutrophil-to-lymphocyte ratio and enhances T cell activation
Tumor-infiltrating neutrophils are well-documented as immunosuppressive mediators that hinder the tumor-eradicating effects of CD8+ T cells33. Clinically, the NLR is recognized as a non-invasive blood-based biomarker that is predictive of patient response to chemotherapy and immunotherapy33,34; however, the underlying molecular mechanism governing NLR is poorly understood. Given the dynamic changes within the TIME following chemotherapy and caspase-1 inhibition, we next examined whether targeting caspase-1 could modulate intratumoral NLR and restore anti-tumoral immunity. We analyzed tumors from various treatment groups: (i) vehicle, (ii) GC chemotherapy, (iii) Casp1i + GC chemotherapy, or (iv) Casp1i alone via multimodal methodologies, including multiplex immunofluorescence staining and flow cytometry (Supplementary Fig. 7a). Overall, chemotherapy treatment increased intratumoral neutrophils (Fig. 6a, Supplementary Figs. 7b, 8) while CD8+ T cell level remained relatively unchanged across vehicle- and GC chemotherapy-treated tumors (Fig. 6a, Supplementary Figs. 7c, 8). Consequently, the Ly6G:CD8 ratio (NLR) was significantly elevated in areas of interest (AOI) following chemotherapy treatment (Fig. 6b, black vs red, Supplementary Fig. 8). Intriguingly, Casp1i + GC chemotherapy co-treatment reshaped the immune cell composition, characterized by a concurrent increase in CD8+ T cells and a reduction in neutrophils (Fig. 6a, Supplementary Fig. 7b, c), resulting in a significantly lower Ly6G: CD8 ratio (NLR) compared with other groups (Fig. 6b, red vs teal, Supplementary Fig. 8). Flow cytometry independently confirmed the frequencies of CD8+ T cells in these tumors (Supplementary Fig. 7d). Notably, analysis of neutrophil (CD45+CD11b+Ly6G+) and CD8+ T cell (CD45+CD3+CD8+) frequencies across different treatment groups by both flow cytometry and immunofluorescence staining revealed a significant negative correlation, inferring a plausible inhibitory relationship between the two cell types (Supplementary Fig. 7e, f).

a Representative immunofluorescence staining of neutrophils (Ly6G; red) and T-cells (CD8; green) in tumors from mice treated with vehicle, gemcitabine-cisplatin chemotherapy (Chemo), VX-765 caspase-1 inhibitor (Casp1i) + Chemo combination, and Casp1i. b Quantification of neutrophil (Ly6G+) to lymphocyte (CD8+) ratio (NLR) in tumors from mice treated with vehicle (17 random AOI from 7 mice), chemo (14 random AOI from 7 mice), Casp1i + Chemo combination (14 random AOI from 7 mice), and Casp1i (8 random AOI from 7 mice), respectively. Flow cytometry results quantifying percent of CD107a and interferon-gamma (IFNγ) expressing CD8+ T cells (c), and percent of CD107a and granzyme B (Gzmb) expressing CD8 + T cells (d) in tumors of mice treated with vehicle [black], gemcitabine-cisplatin chemotherapy (Chemo) [red], VX-765 caspase-1 inhibitor (Casp1i) + Chemo combination [teal], and Casp1i [blue], respectively (n = 5 mice per group). e Expression of immunosuppressive genes CD27, CD47 and S100A9 within the neutrophil population from vehicle, chemotherapy and chemotherapy + Casp1i treated tumors (n = 2 mice per group, 31,362 number of cells analyzed). f Expression of immunosuppressive genes CD274 and S100A9 in neutrophils of responder (n = 1) and non-responder (n = 1) patient tissues analyzed with Xenium from Fig. 1o. g Tumor growth kinetics of mice harboring G69 tumors treated with vehicle [black], chemotherapy (Chemo; Gemcitabine: 60 mg/kg, Cisplatin: 6 mg/kg) [red], Anti-IL-1α + Chemo combination (Anti-IL-1α: 200 μg/mouse) [teal], and Anti-IL-1α [blue] (n = 5 mice per group). h Representative immunofluorescence staining of tumor sections for CD8+ T cells (CD8; green) and neutrophils (Ly6G; red) in vehicle, gemcitabine-cisplatin chemotherapy (Chemo), Anti-IL-1α + Chemo combination, and Anti-IL-1α. Statistics: Ordinary one-way ANOVA follow by Dunnett’s multiple comparison test (b–d) and two-way ANOVA with Geisser-Greenhouse correction followed by Dunnett’s multiple comparison test (g) were performed. Error bars represent standard error of mean (s.e.m.) unless otherwise stated. Source data are provided as a Source Data file.
To further assess the functional status of CD8+ T cells across treatment group, CD8+ T cells were enriched from tumors using immunomagnetic beads selection and activated overnight using an anti-CD3/-CD28 cocktail. Flow cytometry analyzing interferon-gamma (IFNγ), CD107a, and granzyme B (Gzmb)—complementary markers indicative of T cell activation state—revealed that GC chemotherapy induced a modest increase in activated T cells (CD107a⁺IFNγ⁺ and CD107a⁺Gzmb⁺) compared to vehicle-treated tumors (Fig. 6c, d, Supplementary Fig. 7g, h). Co-administration of Casp1i with GC chemotherapy significantly enhanced both CD107a+IFNγ+ (Fig. 6c, Supplementary Fig. 7g) and CD107a+Gzmb+ T cell frequencies relative to all other groups (Fig. 6d, Supplementary Fig. 7h). This increase in activated T cells in the Casp1i + GC chemotherapy group was accompanied by a significant reduction in tumor burden compared to other treatment groups. (i.e., vehicle, GC chemotherapy, and Casp1i treatment groups) (Fig. 5k). Importantly, co-staining of cleaved caspase-3 with CD8 indicated that chemotherapy and Casp1i co-treatment does not directly impact T cell viability in the local TIME (Supplementary Fig. 7i, j). These findings support that enhanced T cell activity, rather than viability, contributed to the overall improved treatment response.
To further characterize intratumoral neutrophils, we performed single-cell RNAseq (Supplementary Fig. 7k). Chemotherapy increased expression of immunosuppressive markers such as CD274 (PD-L1), S100A9, and the “don’t eat me” signal CD47 in neutrophils, which was diminished after Casp1i co-treatment (Fig. 6e). Consistently, these findings were recapitulated in patient tissues from Fig. 1o, showing an increased expression of these markers in non-responder versus responder patients (Fig. 6f).
Given that caspase-1-dependent release of IL-1α skews myelopoiesis (Fig. 3g–i), this prompted us to investigate the therapeutic effects of anti-IL-1α blockade. To this end, co-administration of anti-IL-1α antibody with chemotherapy significantly inhibited tumor growth in mice (Fig. 6g) and normalized intratumoral NLR (Fig. 6h) when compared with other treatment groups, highlighting IL-1α as a key mediator of caspase-1 mediated chemotherapy response.
These findings in Fig.6 demonstrate that chemotherapy induces neutrophil-dominant inflammation, whereas co-administration of a caspase-1 inhibitor or anti-IL-1α antibody with chemotherapy significantly reduces intratumoral NLR, enhances marker expression of T cell activation, and collectively improves chemotherapeutic response.
In summary, our study proposes that chemotherapy-induced epiCaspase-1 pyroptosis reshapes the systemic immune landscape by orchestrating a neutrophil-dominant program that originates from the bone marrow (Fig. 7). Tumor-derived IL-1α released from epiCaspase-1-dependent lytic cell death skews bone marrow myelopoiesis towards granulocyte-monocyte progenitor development and subsequent neutrophil production, which migrate into peripheral blood and primary tumors, resulting in elevated intratumoral NLR. Pharmaceutical inhibition of caspase-1 and IL-1α effectively reverses these effects, restoring immunogenic cell death by shifting NLR towards a CD8+ T cell-driven response, ultimately improving chemotherapy efficacy. Beyond these mechanistic insights, we developed CASCADE, an epiCaspase-1-driven gene signature that stratifies patients who are likely to benefit from combined caspase-1 inhibitor and chemotherapy co-treatment. These findings underscore that inflammatory pyroptosis is not inherently immunogenic,but rather, in the context of chemotherapy, reshapes the systemic immune landscape towards neutrophil-dominant inflammation.

The development of an epithelial caspase-1 (epiCaspase-1) signature, CASCADE, which associates with poor clinical outcomes in multiple cancers and neutrophil-dominant immune signature (Fig. 1k–r). Mechanistically, gemcitabine chemotherapy induces epiCaspase-1 expression in cancer cells (Fig. 1d-j), which suppresses immunogenic cell death (Fig. 2a–j) by promoting a shift toward myeloid progenitor expansion in the bone marrow via IL-1α signaling that is dependent on NINJ1 (Fig. 2m, Fig. 3). This leads to excessive neutrophil production in the bone marrow, driving their infiltration into primary tumors and skewing the intratumoral neutrophil-to-lymphocyte ratio (NLR) toward an immunosuppressive state (Figs. 5, 6). Notably, co-administration of a clinical-grade caspase-1 inhibitor significantly mitigated this chemotherapy-induced myeloid skewing in the bone marrow (Fig. 4), recalibrating NLR balance and enhancing T cell activation, thereby promoting an anti-tumor immune response (Figs. 5, 6). This study underscores the critical need to consider not only the local tumor microenvironment (TME) but also the distal bone marrow niche when evaluating the systemic effects of cytotoxic therapies. Created in BioRender. Wong, S. (2026) https://BioRender.com/it5hbww.
Discussion
Therapy-induced cell death and its far-reaching immunological ramifications are increasingly recognized as critical determinants of therapeutic response1,35. Recent investigations have explored whether specific chemotherapeutic agents can elicit immunogenic cell death to harness anti-tumoral immunity and enhance treatment efficacy36. In the current study, we propose a regulatory mechanism by which immunogenic cell death is attenuated by the immunosuppressive function of epiCaspase-1 in the primary tumor, whereby the distal bone marrow hematopoiesis is remotely skewed towards granulocyte-monocyte progenitor (GMP) production in the chemotherapy context. By shifting the focus of chemotherapy-induced inflammation from the local microenvironment to the bone marrow, our study expands the paradigm of immunomodulation in cancer. Additionally, by proposing a unique role for inflammatory epiCaspase-1 in remotely modulating hematopoiesis, our study not only challenges conventional view on caspase-1 biology but also underscores its dual-edged nature—beneficial during infection, yet deleterious in the context of chemotherapy-induced inflammation. Moresignificantly, we provide a clinically actionable strategy to stratify patients and to enhance chemotherapy efficacy by targeting an alternate axis of immune evasion. Our findings also illuminate a pivotal role of IL-1α as a non-canonical effector released from epiCaspase-1-induced pyroptotic cancer cells, instigating bone marrow hematopoietic skew, and orchestrating a systemic neutrophil-dominant immunosuppressive program. This program recalibrates the intratumoral NLR by fundamentally reshaping the TIME.
Intriguingly, recent studies have correlated elevated caspase-1 levels in breast cancer with an increase in pro-tumoral tumor-associated macrophages (TAMs) and the exclusion of CD8+ T cells from the TIME, resulting in enhanced breast tumor progression37. Independently, the pathological release of IL-1α during necroptosis—another form of lytic cell death—was shown to promote a myeloid-driven immunosuppressive microenvironment that constrains anti-tumor immunity9. Our study provides an alternative mechanism, unveiling how the epiCaspase-1/IL-1α axis orchestrates an immunosuppressive local TIME by skewing distal myelopoiesis in the bone marrow toward GMPs and fostering the influx of immunosuppressive neutrophils. Multiple studies have also corroborated our findings, highlighting the role of the IL-1 family in influencing hematopoietic dynamics in other contexts38,39. These insights not only redefine our understanding of chemotherapy effects and its immunological impact, but also offer a transformative perspective to therapeutically target the caspase-1/IL-1α axis for defusing this systemic immunosuppressive program.
The disappointing outcomes of recent Phase III clinical trials [KEYNOTE-36140 and IMvigor13041] combining conventional chemotherapy with immune checkpoint inhibitors (ICIs) underscore a critical knowledge gap in understanding how chemotherapy mechanistically modulates ICI efficacy beyond PD1/PDL1 regulation. Our study provides a mechanistic explanation for these failures by identifying an epiCaspase-1-driven immune evasion strategy. While chemotherapy-induced immunogenic cell death is known to influence ICI response, we provide compelling evidence that epiCaspase-1 attenuates immunogenic cell death in the chemotherapy context. Crucially, co-administration of a clinical-grade caspase-1 inhibitor significantly enhances chemotherapy efficacy, suggesting that combining caspase-1 inhibition with chemotherapy could synergize with ICIs. This discovery positions pharmaceutical caspase-1 co-inhibition as an alternative anti-cancer therapy—capable of reprogramming the tumor immune landscape but also preventing a systemic neutrophil-dominant immunosuppressive program from the bone marrow.
Previous studies42, including our own, have demonstrated that tumor-derived factors, such as PGE25,6 and CCL2043, reprogram various cell types within the TME to promote therapy resistance. For instance, extracellular release of PGE2 as an iDAMP during chemotherapy impairs CD103⁺ dendritic cell activation, dampening anti-tumor immunity5. In our most recent study, tumor-derived CCL20 and other CXCLs released during caspase-1-dependent pyroptosis converts αSMA⁺ cancer-associated fibroblasts (CAFs) into inflammatory CAFs (iCAFs), consequently promotes the expansion of chemo-resistant cancer stem cells and reduces chemotherapeutic response43. While these studies reveal parallel mechanisms of therapy resistance and immunosuppression within the local TME, our current work extends this paradigm by posing IL-1α as a second iDAMP in its class, which, in addition to its local effects within the TIME9, exerts immunomodulatory activity on the distal bone marrow site to reshape a systemic neutrophil dominant inflammation.
A limitation of our current study, however, includes the lack of understanding by which epiCaspase-1 influences the differentiation dynamics and maturation of GMPs into neutrophils in the bone marrow compartment. Future comprehensive analyses dissecting the dynamics of neutrophil differentiation from GMPs, such as that described by Kwok et al.44, will be essential for advancing our understanding on how tumoral epiCaspase-1 distally modulates neutrophil survival, maturation, and egress from the bone marrow.
The inherent heterogeneity of patient responses remains a major hurdle in the successful clinical translation of therapeutic strategies, particularly combination regimens. Overcoming this challenge requires not only a rational design of combination therapies but also the development of predictive biomarkers to guide patient stratification and inform clinical decision. In this regard, our CASCADE gene signature represents an important advancement in the field, offering a tool to prospectively identify patients predisposed to resistance against conventional treatments, upon validation. Through rigorous retrospective cohort analyses, we establish that CASCADE reliably stratifies patients with poor clinical outcomes and diminished therapeutic responses, laying the foundation for its future clinical integration as a cutting-edge prognostic biomarker. By prospectively validating this signature, we can move toward a more personalized treatment framework, optimizing patient-specific responses to caspase-1/IL-1α co-targeting therapies.
Finally, our study offers important insights amid the ongoing debate regarding the immunogenicity of distinct forms of lytic cell death, including ferroptosis, necroptosis, and pyroptosis. While some evidence suggests these cell death modalities are immunogenic and are capable of eliciting potent anti-tumor immunity45,46, other findings indicated a context-dependent and cell-type dependent nature for lack of immunogenicity8. Recent studies have elegantly shown that specific pyroptosis-inducing agents can sensitize breast tumors to immune checkpoint inhibitors47. Additionally, a selective agonist activating GSDMD pore formation and pyroptosis has been found to trigger immunogenic cell death and synergize with ICIs48. Our findings align with these recent studies while emphasizing the imperative need for rigorous molecular interrogations to delineate the distinct roles of lytic cell death modalities across different treatment contexts, such as chemotherapy and immunotherapy, respectively. As lytic cell death-inducing strategies advance toward clinical applications, rigorous mechanistic studies, such as ours, will be essential to guide their optimal integration and ensuring their maximal efficacy in oncology applications.
Methods
Cell culture
All cells are cultured under standard cell culture conditions (i.e., 37 °C, 5% CO2). Human T24 bladder cancer cell line was purchased from ATCC and maintained in DMEM high glucose media (Gibco, 11965092) supplemented with 10% fetal bovine serum (GenDEPOT, F0900-050) and 1x concentration of penicillin-streptomycin (Gibco, 15070063). The murine G69 bladder cancer cell line was generated as previously described5 and was maintained in DMEM/F12 media (Gibco, 11320033) supplemented with 10% fetal bovine serum, 1% 100 g/L D-glucose (Gibco, 15023021), 1% sodium pyruvate (Gibco, 160070), 1% NEAA (Gibco, 11140050), 1% penicillin-streptomycin and 18.5 μg/L of carrier-free recombinant mouse EGF (BioLegend, 585608). Murine breast cancer cell line 4T1 was maintained in RMPI-1640 media (Gibco, 11875093) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. ASC-knockout THP1 was purchased from InvivoGen (InvivoGen, thp-koascz) and RMPI-1640 media (Gibco, 11875093) supplemented with 10% heat-inactivated FBS (Corning, 35-011-CV), 1% penicillin-streptomycin, and 100 µg/mL of Normocin.
in vitro drug treatment and sample collection
All in vitro drug treatments were done in DMEM high glucose media supplemented 2% FBS, 1% penicillin-streptomycin and 1% sodium pyruvate. Drugs and compounds used include gemcitabine (G69: 120 µM, T24: 0.1 µM, 4T1: 0.1 µM, TCI, 501332958, CAS# 122111-03-9) and VRT-043198 (40 µM, MedChemExpress, HY-112226, CAS#244133-31-1). At the time of collection, cultured media were first centrifuged at 1200 × g for 5 min at 4 °C to pellet floating cells. Supernatants were then centrifuged at 10,000 × g for 3 min at 4 °C to pellet cellular debris. Debris-free supernatants were stored at −80 °C until usage in downstream assays. Adherent cells were dissociated using TrypLE express enzyme (Gibco, 12605028), combined with the floating cell pellets, and re-pelleted by centrifugation (1500 × g for 5 min at 4 °C) for downstream flow cytometric and western blot analyses.
LDH assay
LDH assay was performed using the LDH-Glo kit (Promega, J2381) follow manufacturer’s protocol. Briefly, cells were seeded in triplicates into 6-well plates at suitable cell densities and incubated overnight. Cells were then treated with vehicle or treatment of interests for 48 h. Supernatant were then collected for downstream LDH analysis. Maximum LDH release control was performed by adding 2 µL of 10% Triton-X per 100 µL of media to vehicle-only cells as per manufacturer protocol. % Maximal LDH release was calculated using the following formula: [(Experimental LDH Release − Medium Background)/(Maximum LDH Release Control − Medium Background)] × 100.
Western blotting
Western blot was performed using standard protocol. Briefly, cells were lysed in RIPA buffer (Thermo, 89901) that is supplemented with Protease Cocktail Inhibitor (Sigma, 4693116001) and PhosSTOP (Sigma, 4906837001). Lysates were cleared by centrifugation at 21,130 × g, 4 °C for 20 min. Protein concentration was determined using Pierce BCA Protein Assay Kit (Thermo, 23225) following manufacturer’s protocol. Equal amounts of proteins were loaded for SDS-PAGE, and wet transfer was done onto PVDF membrane (Millipore Sigma, IPVH85R). Membrane was blocked for 1 h with 5% BSA (Fisher, BP9703100) in TBST and subsequently incubated with primary antibodies overnight at 4 °C. Chemiluminescence imaging was done using ChemiDoc (Biorad) or iBright (Invitrogen) imaging systems. Primary human anti-Caspase-1 (CST, #2225 s, 1:1000), mouse anti-Caspase-1 (Adipogen, AG-20B-0044-C100, 1:1000), anti-Gasdermin D (Novus, NBP2-33422, 1:1000), anti-ASC (Adipogen, AG-25B-0006-C100, 1:1000), Anti-Beta-Actin (Santa Cruz, sc-47778, 1:5000), and corresponding secondary antibodies (CST, #7076 and #7074, 1:10000) were used.
Xenium sample processing and bioinformatics
All studies involving patient samples are approved by Houston Methodist Institutional Review Board under PRO00037670. Retrospective tissues are obtained under waiver of informed consent with IRB approval. FFPE blocks were processed for Xenium (10X Genomics) following manufacturer’s protocol. Raw Xenium spatial transcriptomic data were processed using the Seurat pipeline in R. Preprocessing steps included quality control filtering and normalization using the centered log ratio transformation method. High-quality cells were selected based on nFeature_Xenium and nCount_Xenium thresholds (from the 25th quartile to 75th quartile plus 1.5-fold of interquartile range). Dimensionality reduction was performed using principal component analysis (PCA), followed by Uniform Manifold Approximation and Projection (UMAP) for visualization. Unsupervised clustering was conducted with FindClusters, and cell types were annotated based on differentially expressed genes and marker-based classification. DimPlot and DotPlot were used to visualize the epithelial clusters with their corresponding CASCADE score. For spatial analysis, ImageDimPlot was used to visualize the distribution of different cell types of interest (i.e., epithelial cells with high or low CASCADE score, neutrophils and CD8 T cells) across the tissue architecture. All analyses were performed in R (v4.4.2). A total of 44,935 cells were analyzed.
Animals
All animal studies were performed in accordance with procedures approved by the IACUC under protocols #8777 (Cedars-Sinai) and IS00007158 (Houston Methodist Research Institute). The maximum allowable tumor size/ burden is 2000 mm³, or 2000 mm along any axis. No animals exceeded these limits. 6- to 10- weeks old male FVB mice (FVB/NJ) and female BALBc (BALB/cJ) purchased from The Jackson Laboratory were used in all animal studies. Animals were group housed in individually ventilated cages. Room lighting was set to a 12-h light-dark cycle as recommended by the National Advisory Committee for Laboratory Animal Research (NACLAR). Animals were provided with irradiated diet and autoclaved water, ad libitum. Animals were maintained in accordance with guidelines from the American Association of Laboratory Animal Care (AALAC).
In vivo studies
Gold standard vaccination assay
Cells were seeded at 4.7 × 104 cells per mm2 in a 10 cm2 dish and were treated with gemcitabine at IC50 dose (G69: 120 μM, 4T1: 0.1 μM) for 24 h in vitro. After 24 h, adherent cells were thoroughly washed with DPBS, dissociated using TrypLE express enzyme (Thermo, 12604013), and pelleted by centrifugation (1200 × r.p.m. for 5 min at room temperature). Cell pellets were washed with DPBS two more times to ensure clearance of residual enzyme and chemotherapy. 1 × 106 (G69) or 2 × 105 (4T1) cells were suspended in 50 μL of DPBS and injected subcutaneously into the left lower flank of mice. A week following vaccination, mice were challenged with 5 × 105 (G69) or 2 × 105 (4T1) cells suspended in 50 μL of DPBS on the right hind flank. Tumor incidence and growth were recorded twice a week using calipers and tumor volume was calculated using the formula (L × W2)/2, where L is the longest axis and W is the perpendicular axis.
Modified vaccination assay
A modified version of the gold-standard vaccination assay was implemented to study the effects of chemotherapy-induced cell death on the bone marrow without direct chemotherapy treatment. In the modified protocol, mice were vaccinated with gemcitabine treated G69 cells (180 μM) three times (on days 1, 2, and 8) and administered with or without compounds (on days 0, 1, 2, 4, 6, and 8) prior to harvesting the bone marrow for downstream analysis 48 h after the final injection. Compounds used for treatment were anti-IL-1α (BioXcell, BE0243, 100 µg per mouse), anti-IL-1R (BioXcell, BE0256, 200 µg per mouse), and Anakinra (MedChemExpress, HY-108841, CAS #143090-92-0, 500 µg per mouse), all administered intraperitonially.
In vivo drug treatments
Mice were injected with 1.5 × 106 G69 or 2 ×105 4T1 cells resuspended in DPBS in the left subcutaneous flank. Tumor growth was tracked weekly. Mice were randomized when the average cohort tumors reached approximately 40–50 mm3 in size. Mice were given two cycles of chemotherapy. One cycle of chemotherapy consists of Gemcitabine (G69: 120 mg/kg, 4T1: 30 mg/kg) AND cisplatin (G69/ 4T1: 6 mg/kg), i.p., on day 1, and Gemcitabine ALONE, i.p. at day 8. Mice were then given 1 week of drug holiday before the commencement of the second cycle. Vehicle group (veh) received 0.05% NaCl solution added with 10% DMSO and dosed at 0.1 ml per 10 g of body weight, i.p. Mice in the GC chemotherapy group (Chemo) were treated as per schedule described. GC chemotherapy plus caspase-1 inhibition group (Chemo + Casp1i) received chemotherapy together with caspase-1 inhibitor while Casp1i alone group received only the inhibitor. Casp1i (VX-765, MedChemExpress, HY-13205, CAS #273404-37-8), was administered by oral gavage at 50 mg/kg in 20% PEG and 2% Tween 80 solution, or corn oil, every other day until the completion of the experiment. Five days after the last dose of the second cycle, tissues (tumors, blood, and bone marrow) were harvested and processed for subsequent analyses.
Processing peripheral blood
Peripheral blood (~40–60 μL) via mouse tail-vein was collected into 1.5 mL BD Microtainer tubes (BD, BD365974). 40 μL from each sample was taken and centrifugation at 875 × g for 10 min at 4 °C to separate plasma and cells. Red blood cells were lysed using ACK buffer (Lonza, 10548E) and the remaining cell pellet was subjected to antibody staining for downstream flow cytometry analyses.
CD8+ depletion in vivo
Mice were injected subcutaneously with anti-CD8 monoclonal antibody (BioXcell, BE0004-1) at a concentration of 200 μg per 30 g of body weight. Mice were treated twice with anti-CD8 mAb 96- and 120-h post-vaccination prior to the live-cell challenge. The efficiency of CD8 depletion was determined by flow cytometry analysis on peripheral blood.
Neutrophil depletion in vivo
Mice were injected i.p. with anti-Ly6G (BioXcell, BE0075-1) at a concentration of 25 μg per mouse daily for the 1st week, and 50 μg per mouse daily for the 2nd week. Mice were also co-injected with anti-rat Kappa Immunoglobulin Light Chain (BioXcell; BE0122) every 2 days for the duration of anti-Ly6G injections. Depletion of neutrophils was determined by flow cytometry analysis on peripheral blood. IgG2a isotype control (BioXcell, BE0089) was used as a control injected at 25 μg per mouse daily for the 1st week, and 50 μg per mouse daily for the 2nd week. Treatment began 4 days after vaccination.
Flow cytometry/FACS
Flow cytometry panels are found in Supplementary Table 2. Antibodies used for flow cytometry are listed in Supplementary Table 3. Immune cell samples were suspended in 50 μL of anti-CD16/32 antibody (Biolegend, 101320) diluted to 1:200 in HBSS (Gibco, 14025076) supplemented with 2% heat-inactivated FBS (Thermo, A3840201) for 10 min on ice prior to immunophenotype staining (unless indicated otherwise). Gating strategies are found in Supplementary Fig. 9. BD Liquid Counting Beads (BD, 335925) or CountBright Absolute Counting Beads (Invitrogen, C36950) were used to quantify immune cells following the manufacturer’s protocol, where appropriate. For T-cell immunotyping, T-cell enrichment is done using the EasySep Mouse CD90.1 Positive Selection Kit (Stemcell, 18958) following manufacturer’s protocol. Live T-cells were incubated with anti-CD107a antibody an hour prior to subsequent staining procedures. All intracellular staining was performed using the BD Cytofix/Cytoperm (BD, 554714). In short, cells were washed with ice-cold PBS prior to being fixed/permed on ice with 150 μL of the BD Cytofix/Cytoperm buffer for 15 min. Live/Dead-NearIR stain (Thermo, L10119) was used to distinguish cell viability. Antibody-cell suspensions were incubated on ice for 20 min (unless indicated otherwise) and washed subsequently (2x) with ice-cold HBSS. For bone marrow samples, muscles were removed and bone marrow cells were collected by centrifugation at 13,000 × g for 15 s from the femur and tibia. Extracted bone marrow cells were treated with red blood cell removal buffer (Sigma, Cat#R7757) for 5 min at room temperature. Cells were then resuspended in FACS buffer (PBS, 0.8% BSA, 25 mM HEPES, 2 mM EDTA, 0.1% Sodium Azide) and counted before antibody staining. For antibody staining of bone marrow cells, incubation with CD34 was first done for 1 h on ice with mixing every 15 min before adding the rest of the antibody panel was added on top for an additional 20 min on ice. For staining of immune cells in the peripheral blood, RBC lysis was done for several rounds using red blood cell removal buffer (Sigma, Cat#R7757) for 5 min at room temperature per round. Flow cytometry was performed on BD Accuri™ C6, BD LSRFortessa™, or the Cytek™ Northern Lights cell analyzer. FACS was performed on the BD Aria™ II. All flow cytometry data were processed using FlowJo software v.10.10.0.
in vitro transfection of caspase-1 reporter
T24 and G69 cells were seeded at 3 × 104 cells per well in a 12-well plate on day 0 (a day prior to transfection). The Lipofectamine 3000 transfection reagent (Thermo, L3000015) was used following the manufacturer’s protocol. The DsRed2-Mito-7 plasmid (Addgene, 55838) was transfected at a concentration of 10 ng/well and the caspase-1 BIFC reporter plasmids, Pro-Casp1-Venus N-terminal and Pro-Casp1-Venus C-terminal17 (gifted by Dr. Lisa Bouchier-Hayes, Baylor College of Medicine), were transfected at a concentration of 20 ng/well. Images of cells were taken on the ECHO Revolve microscope.
Live cell imaging in vitro
The IncuCyte® Zoom system was used (10X: 1.22 μm/pixel) to track the caspase-1-specific bimolecular complementation (BiFC) reporter (green) and ds-Red2-mito-7 expression (red), which were transfected into cells as previously described. The system was housed in a culture incubator set at 37 °C and a humidified atmosphere containing 5% CO2. Images were taken hourly for up to 50 h. Analysis and quantification of green and red fluorescent cells were done on the IncuCyte software.
Sample preparation for scanning electron microscopy
T24 cells were cultured on a coverslip were treated with gemcitabine chemotherapy or vehicle in DMEM high culture medium for approximately 36 h at 37 °C with 5% CO2. For primary fixation, cells were fixed with 4% glutaraldehyde diluted in 0.1 M cacodylate buffer solution for 30 min at 37 °C. Cells were subsequently fixed with 1% OsO4 solution for 1 h at room temperature. Post-fixation, cells were dehydrated in 30%, 40%, 60%, 80%, 90%, and 100% ethanol for 15 min at each concentration. After the dehydration sequence, samples were immersed in 100% hexamethyldisilazane (Sigma, 440191) for 3 min. The coverslips were mounted on metal stubs and sputter coated with gold/palladium at 4 nm thickness using a sputter coater (EM ACE 600; Leica Microsystems, Vienna, Austria). Images were obtained using the Hitachi SU-8230 scanning electron microscope at 15 kV with a working distance of 5–7 mm and by using a secondary electron detector.
ELISA and AlphaLISA
ELISAs of IL-1β (R&D, MLB00C, and DLB50) and IL-1α (R&D, MLA00), and AlphaLISA of IL-18 (Revvity, AL3189HV) were performed according to the manufacturer’s protocol. Cultured media from in vitro experiments were processed by centrifugation (10,000 × g at 4 °C for 5 min) to rid of cell debris prior to assays.
Bone marrow sample preparation for scRNA-seq
Femurs and tibia were dissected from G69-tumor-bearing mice from various groups and kept on ice (n = 2 mice per group, total number of cells analyzed are reported in main text). Muscles were removed and bone marrow cells were collected by centrifugation at 13,000 × g for 15 s. Extracted bone marrow cells were treated with red blood cell removal buffer (Sigma, Cat#R7757) for 5 min at room temperature. Lysis buffer was neutralized by the addition of 10X volumes of PBS and centrifuged at 300 × g at 4 °C for 5 min. Centrifuged cells were resuspended in FACS buffer (0.5% BSA in PBS) and counted. Cells were then resuspended to a concentration of 1 × 107 cells/mL and hematopoietic progenitor cells were immuno-magnetically separated using the Hematopoietic Progenitor Isolation kit from following the manufacturer’s instructions (Stem Cell Technologies, 19856). Isolated hematopoietic progenitor cells were then counted and resuspended in FACS buffer to a concentration of 1000 cells/μL. Samples were finally processed according to the 10x Genomics protocol for the 3’ v3.1 assay and were loaded to target 10,000 cell recovery. Recovered single-cell beads were processed according to the 10x Genomics protocol (Cat#100424). Libraries were prepared according to the manufacturer’s instructions. Quality of cDNA and final libraries was performed on a BioAnalyzer 2100 (Agilent Technologies) using the High Sensitivity DNA analysis kit (Cat#5067-4626) and following the manufacturer’s instructions. Indexed complementary DNA (cDNA) libraries were sequenced with paired-end reads on an Illumina NovaSeq 6000 sequencer. Sequenced samples were processed with CellRanger (10x Genomics, v7.1) using a pre-mrna reference based on the mm10-2020-A-2.0.0 reference. Data from ambient RNA were trimmed based on nUMI-barcode saturation curve.
Single cell analyses
Single cell dimensionality reduction, clustering and annotation
The R package Seurat v4.3 was used under R v4.2.2 for data trimming, unsupervised clustering, and visualization49. Cells with poor sequence read quality, doublets, and stressed cells were excluded from downstream analysis. Cells that had less than 400 UMI, a gene number lower than 300 and were expressed in less than 10 cells were excluded from downstream analysis. Mitochondrial gene ratio was calculated to filter out cells with more than 5% mitochondrial genes50. Cell cycle score and cell phases were assigned by the function “CellCycleScoring” according to a gene list of cell cycle markers51. Next, the function “SCTransform” was applied to regress out confounders (i.e., UMI number, percentage of mitochondrial genes, and cell cycle scores), normalize for differences in sequencing depth across single cells, and define highly variable genes. Highly variable genes were then used as input for principal component analysis (PCA). Statistically significant PCs which explained the most variance were selected using the “Elbowplot” function. Based on significant PCs, “RunHarmony” function was used from the R package Harmony v0.1.1 to integrate scRNAseq data from the different treatment groups. Subsequently, “FindNeighbors” and “FindClusters” with a resolution of 0.2 were run to perform unsupervised, graph-based clustering. Cells were then projected to a 2-dimensional space for visualization by Uniform Manifold Approximation and Projection (UMAP, v0.1.14). Clusters were annotated using a panel of marker genes obtained from a literature search. Total number of cells analyzed are reported in main text.
Analysis of myeloid progenitor cells
To further analyze the myeloid lineage, we exclude the Erythrocyte Progenitor cluster, as well as the CLP and Pro-B cell clusters. Clusters were reanalyzed using “FindClusters” function with a resolution of 0.4 and projected to 2-dimenional space using “RunUMAP”. The resulting 13 clusters were annotated using a panel of marker genes obtained from the literature. Differentially expressed genes were obtained using “FindAllMarkers”. UMAP and Heatmap plots were generated using the R package ‘dittoSeq’ v.1.10.
Trajectory analysis of myeloid progenitor cells
Pseudotemporal ordering of myeloid progenitor cells to infer the potential lineage differentiation trajectory was done with Monocle 352. Cells were preprocessed, re-clustered, and the “learn_graph” function was used to order cells in pseudotime. The cluster with the highest expression of Hlf and Msi2 (i.e., HSPC) was used as a starting point to draw out inferred trajectories. Total number of cells analyzed are reported in main text.
CRISPR/Cas9-mediated genetic KO cell lines
To generate Caspase-1 knockout clones, G69 were transduced with a lentivirus carrying the TLCV2 plasmid (AddGene, 87360) and subsequently selected using puromycin (8 μg/mL). G69.TLCV2 were treated with 100 ng/mL of doxycycline (Sigma, D9891) in complete culture media on the day of transfection with sgRNAs. Caspase-1 sgRNA (Sigma, AGGGCAAGACGTGTACGAG) was transfected into cells using the Lipofectamine™ CRISPRMAX™ Cas9 Transfection Reagent (Thermo, CMAX00008); instructions were followed using the manufacturer’s protocol. Single cell clones were selected using limiting dilution and knockout efficiency was confirmed via western blot.
G69 IL-1αKO and NINJ1KO were generated and validated by EditCo Bio with the following guide RNA sequences:
IL-1α (CUUACCUGUAACAGUUCUUU); NINJ1 (CGGUUCCGCAAACCCCAGCG).
shRNA sequences
Sequences of shRNA used are as follows:
mouse Caspase-1
(CCGGGATTTCTTAACGGATGCAATTCTCGAG-AATTGCATCCGTTAAGAAATCTTTTT)
human PYCARD
(CCGGCCTGCACTTTATAGACCAGCACTCGAGTGCTGGTCTATAAAGTGCAGGTTTTTG).
Immunofluorescence staining
Slide sections were first incubated at 65 °C for 1 h and then incubated in Xylene (5 min) twice, followed by 50 dips in 100% Ethanol twice, 50 dips in 95% Ethanol twice, 50 dips in 70% Ethanol twice, and finally 50 dips in water once. Sections were fixed in 10% Formalin for 20 min and washed with TBST. For first staining, antigen retrieval was performed by steaming sections in 1x antigen retrieval buffer (Abcam, ab93684) for 20 min. Sections were then cooled and blocked with 1x animal free buffer (CST, 15019 L) for 10 min, and subsequently incubated with primary antibodies for either 2 h in room temperature or overnight at 4 °C. The sections were then washed in TBST (50 dips, 3 times) and incubated in the matched secondary antibodies for 30 min and again washed as per previously described in TBST. Sections were then incubated with Opal fluorophores diluted in Amplification buffer (Akoya, FP1498) for 10 min, and washed. For subsequent staining, antigen retrieval is done with 1x Citrate buffer (Sigma, C9999-1000ML) and staining was done as previously described. Sections were finally incubated after the last stain with DAPI in TBST (2 μg/mL) for 5 min, followed by 3 washes with TBST, and then mounted with ProLong Gold Antifade Reagent with DAPI (Invitrogen, P36935). Images were acquired using PhenoImager (Akoya) and analyzed using QuPath. Mouse MPO (R&D Systems, AF3667, 1:200), Ly6G (CST, #87048, 1:200), CD8a (CST, #98941, 1:200), Cleaved Caspase-3 (CST, #9661, 1:200), secondary HRP conjugated antibodies (abcam, ab214880 or Akoya, ARH1001EA), and Opal fluorophores were used (Akoya, 480: FP1500001KT, 520: FP1487001KT, 570: FP1488001KT, 620: FP1495001KT, 690: FP1497001KT).
Generation and validation of CASCADE signature
T24 cells were first primed with 1 μg/mL of ultrapure LPS (InvivoGen, tlrl-3pelps) for 3 h in DMEM supplemented with 2% FBS. 25 μM of nigericin (Sigma, N7143, CAS#28643-80-3) was then added and incubated for said number of hours for the induction of pyroptosis. Live cell imaging was done using Incucyte as previous described and Sytox green (Sartorius, 4633) was used as a nuclear dye. At specific timepoints described in text, cells were detached by scrapping and subjected to bulk RNA sequencing. The raw count data from bulk RNA sequencing were subject to differentially expressed genes (DEG) analyses using DEseq2 package. DEG that are significantly upregulated (log2 fold change >1 and p value less than 0.05) in nigericin-treated T24 cells compared to vehicle controls (n = 3 per group) were further refined with Cox proportional hazard regression analyses using the patient survival data and gene expression profile (GSE87304) as a training cohort with the threshold hazard ratio and p value of 1 and 0.05, respectively. The prognostic ability of the resulting 61-gene signature was evaluated using three independent bladder cancer cohorts (TCGA-BLCA, GSE32894, GSE48276), Breast (GSE25066) and other TCGA cancer cohorts. The patients in each cohort were split equally (50% percentile) into high- and low-signature score groups unless otherwise specified, and the overall survival of the two groups was compared using a log-rank test. The Kaplan-Meier curves were plotted using ggplot2, survival and survminer packages in R.
Statistics and reproducibility
All data were evaluated and graphed using Prism ver. 10 software (GraphPad). Statistics used are described in text. At least three independent biological replicates with at least technical triplicates per experiment were performed for all in vitro experiments. Data shown are represented as mean ± standard error of the mean (s.e.m.) unless otherwise specified. A p value of <0.05 is considered statistically significant. Numerical p values are reported wherever applicable. QQ plots of ANOVA analyses, wherever applicable, are provided in supplementary Fig. 10.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All experimentally generated sequencing data used in the manuscript are deposited into NCBI Gene Expression Omnibus and is publicly available under the following accession codes:T24 RNA-seq (GSE299107)[https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE299107],Single-cell RNAseq (GSE298319)[https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE298319],Xenium spatial transcriptomics of patient tissues (GSE305629)[https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE305629].Publicly available datasets used in the manuscripts are available in the NCBI Gene Expression Omnibus database under the following accession codes:GSE48277 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48277],GSE25066 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse25066],GSE32894 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE32894],GSE87304 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE87304],and GSE48276. Source data are provided with this paper. The remaining data are available within the Article, Supplementary Information or Source Data file. Source data are provided with this paper.
References
Green, D. R., Ferguson, T., Zitvogel, L. & Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 9, 353–363 (2009).
Kroemer, G., Galluzzi, L., Kepp, O. & Zitvogel, L. Immunogenic cell death in cancer therapy. Annu Rev. Immunol. 31, 51–72 (2013).
Galluzzi, L., Buque, A., Kepp, O., Zitvogel, L. & Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17, 97–111 (2017).
Krysko, D. V. et al. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 12, 860–875 (2012).
Hayashi, K. et al. Tipping the immunostimulatory and inhibitory DAMP balance to harness immunogenic cell death. Nat. Commun. 11, 6299 (2020).
Nikolos, F. et al. Cell death-induced immunogenicity enhances chemoimmunotherapeutic response by converting immune-excluded into T-cell inflamed bladder tumors. Nat. Commun. 13, 1487 (2022).
Ahmed, A. & Tait, S. W. G. Targeting immunogenic cell death in cancer. Mol. Oncol. 14, 2994–3006 (2020).
Wiernicki, B. et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat. Commun. 13, 3676 (2022).
Hänggi, K. et al. Interleukin-1α release during necrotic-like cell death generates myeloid-driven immunosuppression that restricts anti-tumor immunity. Cancer Cell 42, 2015–2031.e2011 (2024).
Bruchard, M. et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 19, 57–64 (2013).
Ghiringhelli, F. et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β–dependent adaptive immunity against tumors. Nat. Med. 15, 1170–1178 (2009).
Doulatov, S., Notta, F., Laurenti, E. & Dick, J. E. Hematopoiesis: a human perspective. Cell Stem Cell 10, 120–136 (2012).
Templeton, A. J. et al. Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: a systematic review and meta-analysis. J. Natl. Cancer Inst. 106, dju124 (2014).
Miao, E. A. et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142 (2010).
Choi, W. et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 25, 152–165 (2014).
Bergsbaken, T., Fink, S. L. & Cookson, B. T. Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109 (2009).
Parsons, M. J., Fassio, S. R. & Bouchier-Hayes, L. in Programmed Cell Death: Methods and Protocols (eds Hamsa Puthalakath & Christine J. Hawkins) 41–56 (Springer New York, 2016).
Atkin-Smith, G. K. et al. A novel mechanism of generating extracellular vesicles during apoptosis via a beads-on-a-string membrane structure. Nat. Commun. 6, 7439 (2015).
Mouasni, S. et al. The classical NLRP3 inflammasome controls FADD unconventional secretion through microvesicle shedding. Cell Death Dis. 10, 190 (2019).
Seiler, R. et al. Impact of molecular subtypes in muscle-invasive bladder cancer on predicting response and survival after neoadjuvant chemotherapy. Eur. Urol. 72, 544–554 (2017).
Seiler, R. et al. Divergent biological response to neoadjuvant chemotherapy in muscle-invasive bladder cancer. Clin. Cancer Res. 25, 5082–5093 (2019).
Aaron, M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nature Biotechnol. 37, 773–782 (2019).
Franchi, L., Eigenbrod, T., Munoz-Planillo, R. & Nunez, G. The inflammasome: a caspase−1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247 (2009).
Kroemer, G., Galassi, C., Zitvogel, L. & Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 23, 487–500 (2022).
Wang, W. et al. RIP1 kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell 34, 757–774 e757 (2018).
Legrand, A. J., Konstantinou, M., Goode, E. F. & Meier, P. The diversification of cell death and immunity: memento mori. Mol. Cell 76, 232–242 (2019).
Swann, J. W., Olson, O. C. & Passegué, E. Made to order: emergency myelopoiesis and demand-adapted innate immune cell production. Nat. Rev. Immunol. 24, 596–613 (2024).
Park, M. D. et al. Hematopoietic aging promotes cancer by fueling IL-1α–driven emergency myelopoiesis. Science 5, eadn0327 (2024).
Degen, M. et al. Structural basis of NINJ1-mediated plasma membrane rupture in cell death. Abstract Nature 618, 1065–1071 (2023)
Akashi, K., Traver, D., Miyamoto, T. & Weissman, I. L. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404, 193–197 (2000).
Seita, J. et al. Gene expression commons: an open platform for absolute gene expression profiling. PLoS ONE 7, e40321 (2012).
Zhang, D. et al. Neutrophil ageing is regulated by the microbiome. Nature 525, 528–532 (2015).
Jaillon, S. et al. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 20, 485–503 (2020).
Valero, C. et al. Pretreatment neutrophil-to-lymphocyte ratio and mutational burden as biomarkers of tumor response to immune checkpoint inhibitors. Nat. Commun. 12, 729 (2021).
Ichim, G. & Tait, S. W. A fate worse than death: apoptosis as an oncogenic process. Nat. Rev. Cancer 16, 539–548 (2016).
Hanggi, K. & Ruffell, B. Cell death, therapeutics, and the immune response in cancer. Trends Cancer 9, 381–396 (2023).
Zheng, W. et al. Caspase-1-dependent spatiality in triple-negative breast cancer and response to immunotherapy. Nat. Commun. 15, 8514 (2024).
Park, M. D. et al. Hematopoietic aging promotes cancer by fueling IL-1α–driven emergency myelopoiesis. Science 386, eadn0327 (2024).
Pietras, E. M. et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 18, 607–618 (2016).
Powles, T. et al. Pembrolizumab alone or combined with chemotherapy versus chemotherapy as first-line therapy for advanced urothelial carcinoma (KEYNOTE-361): a randomised, open-label, phase 3 trial. Lancet Oncol. 22, 931–945 (2021).
Galsky, M. D. et al. Atezolizumab with or without chemotherapy in metastatic urothelial cancer (IMvigor130): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 395, 1547–1557 (2020).
Lee, Y.-C. et al. The dynamic roles of the bladder tumour microenvironment. Nat. Rev. Urol. 19, 515–533 (2022).
Gao, H. et al. Caspase-1–dependent pyroptosis converts αSMA+ CAFs into collagen-IIIhigh iCAFs to fuel chemoresistant cancer stem cells. Sci. Adv. 11, eadt8697 (2025).
Kwok, I. et al. Combinatorial single-cell analyses of granulocyte-monocyte progenitor heterogeneity reveals an early uni-potent neutrophil progenitor. Immunity 53, 303–318.e305 (2020).
Wang, Y. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017).
Zhang, Z. et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579, 415–420 (2020).
Wang, Q. et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature 579, 421–426 (2020).
Fontana, P. et al. Small-molecule GSDMD agonism in tumors stimulates antitumor immunity without toxicity. Cell https://doi.org/10.1016/j.cell.2024.08.007 (2024).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529 (2021).
Ilicic, T. et al. Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 17, 29 (2016).
Kowalczyk, M. S. et al. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 25, 1860–1872 (2015).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Acknowledgements
We would like to thank Yu-Jung Tseng (Baylor College of Medicine) for scientific discussions about hematopoiesis. This study is funded in part by NIH F31CA247257 (K.H.), NIH R01CA255609 (K.S.C), CPRIT RR230010 (K.S.C.), and U.S. Department of Defense CA200750 (K.S.C., F.N.).
Author information
Authors and Affiliations
Contributions
S.Q.R.W., K.H., and E.J.S. contributed to the conceptualization, formal analysis, investigation, methodology, validation, visualization, writing of the original draft, as well as reviewing and editing the manuscript. K.H. made the initial discovery and established the project. H.G., S.J.P., H.E.G., X.P.H., M.D.A., A.K. and E.T. contributed to investigation and formal analysis. Y.H.H., M.K., Z.Y. and S.T.C.W. contributed to data curation, formal analysis, and software. D. Korentzelos, C.C., M.B., L.Z., Z.E.-E., D. Kaushik, R.S., C.S.S., L.B.-H., Y.G., Y.L., R.S.T. contributed to resources. F.N. contributed to conceptualization, data curation, formal analysis, software, funding acquisition, and resources. K.S.C. contributed to conceptualization, funding acquisition, resources, supervision, and writing—review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wong, S.Q., Hayashi, K., Subel, E.J. et al. Chemotherapy-induced activation of caspase-1 and IL-1α release by cancer cells remotely skews myelopoiesis to drive pro tumorigenic systemic neutrophil-dominant inflammation. Nat Commun 17, 4724 (2026). https://doi.org/10.1038/s41467-026-71471-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-71471-3
