
Abstract
The porcine gut microbiome is crucial for pig health and key to its production performance. However, genome-level analysis across multiple kingdoms remains limited. Here, we reconstructed the unified pig gastrointestinal genome (UPGG), including bacterial, archaeal, and annotated over 78 million non-redundant protein-coding genes using 5784 metagenome samples. We identified antibiotic resistance genes (ARGs) and mobile genetic elements (MGEs), and the distribution of 72,056 metabolic gene clusters within existing populations. We have constructed pan-genomes of 436 high-quality microbial species and, using these as references, discovered intraspecies genomic variations that revealed 23,350,975 single-nucleotide variants (SNVs). Finally, through comparative analysis of gut microbiome genomes conducted in this study, we observed that pigs may serve as a more suitable model than other animals for investigating human gut microbiota composition and functional patterns. In summary, we constructed a comprehensive reference catalog of the porcine gut microbiome and enhanced the understanding of the host-microbe coevolution.
Similar content being viewed by others
Introduction
Current perspectives suggest that the gut microbiome and the host genome constitute a holobiont1. The porcine gut harbors billions of bacteria2, which interact with the host genome in a symbiotic manner3, playing a crucial role in the host’s metabolism4, immunity5, and other activities6. In recent years, numerous studies have found that porcine gut microbiota are closely related to its production performances such as weaning stress7 and meat quality traits8. However, the majority of these studies have relied on existing microbial genomes and annotation information, primarily using 16S rRNA sequencing for analysis. In contrast, instead of merely delivering taxonomic insights, metagenomics facilitates a deeper exploration of the functional genomes9,10. Moreover, the metagenomic assembly and reconstruction of metagenome-assembled genomes (MAGs)11,12 is an effective strategy for addressing insufficient microbial diversity in incomplete reference datasets13.
To date, assembly catalogs of gut microbiomes have been publicly reported for various species, including human14,15,16,17, goat18,19, cattle20,21,22, mice23,24,25, chickens26,duck27, and horse28. With regards to pigs, the main datasets include PGC (pig gene catalog)29, PIGC (pig integrated gene catalog)30, weaned piglets31, and Chinese local pigs32. PGC constructed a reference gene catalog of the porcine gut microbiome, containing 7.7 million non-redundant genes29; PIGC utilized 500 metagenomic samples from four intestinal locations, eight breeds, and various populations, resulting in 6339 non-redundant gut microbiome-assembled genomes30. However, these catalogs are mainly characterized by medium-quality MAGs (completeness ≥ 50%, contamination ≤ 5%). The incomplete assembly quality not only affects subsequent functional analysis but also raises concerns about whether the microbial species represented by MAGs truly exist. Therefore, it is necessary and urgent to construct a complete and accurate catalog of pig MAGs. Moreover, we still need to expand the reference set across different dimensions to continually meet the research demands regarding the gut microbiota of pigs.
The construction of the MAG database significantly broadens our understanding of the interactions and co-evolution between gut microbiota and their host. Firstly, it facilitates the identification of uncultured microbial species, including bacteria, fungi, and viruses, allowing for a detailed examination of how microbiome distribution varies with diet, age, health status, and environmental conditions, as studied by Chen et al.30, who illustrated the effects of different farming environments on gut microbiota composition in pigs. Additionally, the MAG database aids in pinpointing crucial functional genes related to microbial resistance33 and metabolic pathways34, which also help unravel evolutionary narratives and phylogenetic connections among microorganism species. Incorporating information on microbial variation, such as single-nucleotide variants (SNVs), along with insights from the pan-genome, enhances our understanding of genetic diversity within microbial populations and can further illuminate these evolutionary trajectories35.
In this study, we constructed a relatively comprehensive catalog of the porcine gut genome. By analyzing 5784 metagenomic samples from a wide range of sources, including different ages, sexes, populations, and geographical regions, we obtained 42,710 MAGs that encompass 1431 bacterial species. The construction of the UPGG database has enabled a deeper exploration of the functional landscape within the pig gut microbiome. We identified ARGs and MGEs, and discovered horizontal gene transfer (HGT) phenomena. Additionally, we constructed 436 species-level microbial pangenomes using this resource, exploring the distribution and functions of core and accessory genes, as well as SNVs in different populations. At last, we presented a MAG database (https://alphaindex.zju.edu.cn/upgg/#/), which leverages customized database support for DNA-based taxonomic analysis, along with standardized processes for the quantification, classification, and functional annotation of MAGs, thereby contributing to the research community.
Results
Recovering 42,710 microbial genomes from over 5000 porcine gut metagenomes
To provide a comprehensive overview of the gut microbiota in the porcine gastrointestinal tract, we collected a total of luminal samples from five segments, i.e., rectum(n = 103), jejunum (n = 19), ileum (n = 35), cecum (n = 148), distal intestine (n = 17) and colon (n = 240), along with 5028 fecal samples, utilizing samples sequenced including 2063 metagenomes from the in-house dataset and 3721 from publicly available metagenomes, see details in Table S1. These samples encompass diverse age groups, sex, and geographical locations (Fig. 1a and Table S2). Metagenomes were recovered in samples from a total of eleven countries across four continents (Asia, Europe, North America, and Oceania), but the majority (45.09%) were sampled from China, Norway, Australia, and Denmark (Fig. 1b).

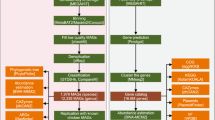
a The number and proportion of metagenomes stratified by sample source features, including age, sex, tissue. b Geographic distribution and breed classification of the number of samples retrieved from each country. c Quality metrics across high-quality, medium-quality MAGs. Boxes show the interquartile range (IQR), with the horizontal line as the median, the whiskers indicating the range of the data (up to 1.5×IQR), and points beyond the whiskers as outliers. Completeness and contamination scores for each of 38,786 genomes. d Overview of the computational pipeline to generate UPGG catalog. e The completeness of MAGs is consistent between the UPGG and PIGC databases. f Sparse curve analysis of sample size and number of high-quality MAGs. g Taxonomic classification of MAG according to GTDB-Tk. The annotations from inside to outside represent annotations of species level (different colors represent different phyla), unclassified genus (in orange), and unclassified species (in orange).
After removing adapter sequences, low-quality bases, short reads, and host genomic sequences, we obtained 42.8 TB of clean data, with an average of 4,178,444,134 reads per sample. Using these clean reads, we conducted metagenomic assembly and obtained a total of 42,710 bins, also known as MAGs. After performing redundancy removal on the MAGs with an average nucleotide identity (ANI) of 99% and filtering out MAGs with completeness less than or equal to 50% or contamination greater than or equal to 10%, we obtained a final set of 38,786 MAGs that were larger than 300 kb. The final ensemble, which serves as the foundation for the construction of the unified pig gastrointestinal genome (UPGG) catalog, was meticulously curated based on stringent criteria encompassing genome completeness, contamination levels, and assembly N50 metrics (Fig. S1, S2), ensuring the robustness and fidelity of the genomic inventory. According to the criteria of Bowers et al., we found that 24,802 (58.1%) of MAGs belonged to high-quality MAGs with completeness greater than 90% and contamination rate less than 5% (Fig. 1c and 1d). In addition, we identified 230 (0.54%) MAGs with 100% integrity (Table S3). 37,786 (97.42%) of the MAGs contained multiple tRNA genes (tRNA type number ≥ 10, Fig. 1c and Table S3). To enable amplicon-based metagenomic analysis, we sought to identify rRNA sequences for MAG within the UPGG catalog. Additionally, the presence of full-length 16S rRNA genes was detected in these MAGs, with only 5,210 (12.2%) MAGs containing one or more copies of these genes (Table S4). Conversely, a substantial number of MAGs, totaling 22,685 (53.1%), were found to encode partial 5S rRNA genes (Table S5).
For identical MAGs (Average Nucleotide Identity, ANI > 98%), the enhancement in genome assembly quality by UPGG is evident. We assessed 3,572 MAGs with 98% concordance between UPGG (9.2%) and PIGC (56.3%)30 (ANI > 98%) and found that 93.5% (3,342) of these MAGs had their genome completeness increased to 90% or higher in UPGG (Fig. 1e and Table S6). Additionally, rarefaction curve analysis indicated that UPGG, with a sample size ten times larger than PIGC, can obtain a sufficient number of high-quality MAGs (Completeness ≥ 90%, Contamination < 5%) (Fig. 1f). This highlights that our collection of porcine gut microbiome-assembled genomes already possesses a high degree of representativeness. The uniqueness of the UPGG catalog lies in its focus on the global porcine gut microbiome, enabling us to conduct an in-depth analysis of this specific ecological niche and expanding the knowledge base of microbial diversity represented by pigs as a mammalian model. Moreover, compared to other MAG resources, our study significantly broadens the geographical distribution, sample size, and breed of porcine, surpassing the number of samples and MAGs included in existing studies on pig metagenome assembly.
Taxonomic landscape of the porcine gut microbiome
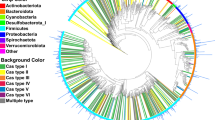
To explore taxonomic components in the UPGG catalog, we used GTDB-tk to annotate the taxonomical classification of MAGs, and 38,703 bacterial MAGs and 83 archaeal MAGs could be defined as species (Fig. 1g). We found that the UPGG catalog covered 26 known phylum (24 for bacteria and 2 for archaea) and 1,004 known genera (995 for bacteria and 9 for archaea) (Fig. 2a). Upon comparison, it was found that at the species level, there are 241 species common to both UPGG and PIGC (Fig. 2b). Additionally, UPGG encompasses 1,190 species not found in PIGC, while PIGC includes only 124 species that are not present in UPGG (Fig. 2b). Discrepancies in the results may be attributed to variations in the sample origins, the dimensions of the sample cohort, and the analytical pipeline applied. The 1,190 species unique to UPGG are distributed across the entire phylogenetic tree, highlighting the taxonomic breadth of new genomes (Fig. 1g). Subsequently, we further investigated the number of microbial expansions in MAGs at each taxonomic level with the highest counts. For instance, Firmicutes has the highest proportion in both UPGG and PIGC, but in terms of uniqueness, UPGG contains more species not found in PIGC (Fig. 2c), and there is a significant (p-value < 0.05) improvement in completeness (Fig. 2d). This phenomenon is observed at various taxonomic levels. Additionally, even within a species, different strains can exhibit varying functional capabilities36. Therefore, having a larger number of conspecific genomes for each species is advantageous. We assessed the genome counts for each of the 203 species common to both UPGG and PIGC and found that 99% of these species have a greater number of genomes in UPGG compared to PIGC (Fig. 2e). This increase in conspecific genomes is expected to expand the pan-genome size for each species. Utilizing Roary37, we generated the pan-genome for each species and observed significant expansion in the pan-genomes of many species, with two notable examples being UBA636_sp002299675 and Collinsella_sp002391315, which exhibited the greatest increase in the UPGG (Fig. 2e) Through rarefaction analysis, we confirmed that the pan-genome sizes for these two species are nearly saturated in UPGG (Fig. 2f).

a Taxonomic classification of 38,786 MAGs at different levels. b Consistency of species and genus level relationships between the PIGC and UPGG catalogs is assessed. c Bar graphs showing the number of species unique to UPGG and shared with PIGC across the highlighted lineages. d Box plots comparing estimated completeness of genomes for each lineage in PIGC and UPGG using CheckM. e A scatter plot compares the number of genomes for common species between UPGG and PIGC. The most abundant species marked in the UPGG. f Rarefaction and extrapolation curves for the pangenome size of two selected species, as determined using UPGG data. g The proportion of species detected within 26 prokaryotic phyla, categorized by their unclassified status, is presented. The numbers in parentheses denote the total species count within each respective phylum. h The number of MAGs in each kSGBs, and only the top 40 most represented kSGBs were displayed. The samples factor (i.e., sex, age) related to the MAGs per species is plotted.
However, a substantial subset of 11,705 MAGs from UPGG remained unidentifiable at the species level, hence categorized as unknown species-level genome bins (uSGBs). Moreover, 851 MAGs could not be classified even to the genus level, hence they were designated as unknown genus-level genome bins (uGGBs), as detailed in Table S7. The top genera of uGGBs classification are Prevotella (582), Eubacterium (571), and Faecousia (494). These generas have been consistently identified as part of the human core gut microbiome across large-scale metagenomic studies38,39. The proportion of uSGBs in the most dominant bacterial phyla is lower than that in rare phyla (Fig. 2g), highlighting the challenge of assembling rare microbial species. We categorized the MAGs within each species based on developmental stage, sex, age and the specific sampling site. It is noteworthy that the majority of MAGs were constructed from samples obtained from adult individuals. Sodaliphilus sp004557565 and Colinsella sp002391315, among others, exhibit equal proportions across two developmental stages, indicating their relatively stable presence in the porcine intestinal environment, potentially less influenced by factors such as environment, age, and feed composition (Fig. 2h). Analyzing the sample sources of MAGs enables the observation of specific patterns in the distribution of bacterial species. Despite the fact that MAGs are assembled from corresponding gut metagenome samples as dominant taxa, they are rare or exhibit differential abundance in the guts of most other pigs. These MAGs may offer a substantial reference genomic resource for elucidating the potential functions of specific microbes within the gut microbiome.
UPGG enables accurate taxonomic profiling
To analyze metagenomic sequencing data, Kraken2, which is frequently used in conjunction with Bracken and based on DNA sequences40, offers the convenience of integrating with a genome catalog as it can directly create customized databases of DNA sequences and their corresponding taxonomic information. Therefore, to enhance the universality of UPGG in taxonomic analysis, we created a custom Kraken2 database. Consequently, to further evaluate the accuracy of the custom UPGG in taxonomic analysis at the species level, we employed CAMISIM41 to create simulated metagenomic sequence samples that include MAGs. We then applied different taxonomic profiling pipelines: Kraken2+Bracken and MetaPhlAn4 with UPGG, PIGC, and GTDB as databases. It is worth noting that the “taxid” in the MetaPhlan4 classification results cannot be matched with other results, hence we assume that all genomes used for construction are present in this database, resulting in a “true positive” rate as described in Table S8. Ultimately, we observed that “Kraken2+UPGG” achieved the highest F1 score, demonstrating the best performance at the taxonomic level (Fig. 3a). Although PIGC achieved a 100% prediction rate in the statistical results, it actually did not yield good results in recall and accuracy due to its limited number of taxonomic categories; whereas GTDB, being the most comprehensive microbial database, generates redundant results in classification comparison, leading to a decreased prediction rate (Fig. 3a). Therefore, the above findings indicate that UPGG can provide more accurate taxonomic analysis of metagenomic data through Kraken2 custom database approach compared to the other pipelines.

a Using PIGC and UPGG genomes in conjunction with the Kraken2 method, boxplots illustrate F1 scores, recall, precision, and prediction rates for classifying metagenomic sequencing reads from 27 simulated datasets to their source species. b The boxplot illustrates the mapping rates across different databases for four independent studies and six breeds. c Beta diversity (Bray-Curtis) of metagenomic samples in the UPGG is represented, with different colors representing different breeds. d Factors significantly contributed to the overall microbial community compositions, as determined by multiple-factor and single-factor analysis results using bacteria. Horizontal bars represent the amount of inferred variance (adjusted R2) explained by each identified covariate. All factors were found to be significantly associated with gut microbial variations (FDR < 0.001). e The dominated genera are observed across 4497 metagenomes.
In order to evaluate the efficacy of UPGG in enhancing the mapping rate of metagenomic reads from the porcine gut microbiome, we conducted a comparative analysis using four independent datasets. These datasets comprise 162 fecal samples32,42,43,44 from distinct pig breeds and locations. Metagenomics reads from these datasets were aligned to the “Kraken2+UPGG”, “Kraken2+PIGC”, “Kraken2+GTDB” and “Metaphlan4” databases to facilitate taxonomic annotation and to ascertain the compositional diversity. Our findings revealed that the “Kraken2+UPGG” substantially improved the mapping rates of porcine gut microbial genomes (Fig. 3b). This enhancement in mapping rates was observed across both commercial and indigenous Chinese pig breeds (Fig. 3b), indicating a broader applicability of UPGG. In summary, we developed customized databases of UPGG genomes tailored of taxonomic profiling. Our results demonstrate that UPGG outperforms previous genomic catalogs in terms of classification ability, both on simulated data and independent real data.
UPGG reveals microbial distribution patterns by enhancing sample richness
The composition of microbial communities has been shown to vary with environmental, geographical, temporal, and biological factors. These patterns are primarily determined by focusing on one sample type and region at a time. To comprehensively assess the global distribution of porcine gut microbes, UPGG conducts large-scale surveys across regions, times, and hosts to ensure sample richness. In terms of microbial diversity, beta diversity (Fig. 3c and Fig. S3) and multivariate/single variance analyses indicate that host age has the most significant impact on microbial distribution. Additionally, breed, country, and sex also influence microbial distribution patterns (Fig. 3d). The alpha diversity indicators, including Shannon, species richness (SR), Pielou’s evenness index, and Simpson, for classified samples under factors such as age, breed, and country, show no significant bias and are fairly decent (Table S9 and Fig. S4). Specifically, samples spanning from birth to adulthood show high diversity; Most breeds exhibit uniform and high diversity levels, with the exception of Yunan and Wild breeds, which have different natural habitats and dietary patterns from controlled agricultural environments (Fig. S4); Geographically, sample diversity levels are uniform and high in most countries, except for Denmark, where the lower diversity values may be attributed to the inclusion of numerous fetal gut microbial samples during gestation (Fig. S4). These results suggest that UPGG, by enhancing the richness of sample sources, has to some extent ensured the consistency and diversity of microbes across various factors.
In this study, a total of 2,218 species were identified, among which 1479 had at least one representative genome in the UPGG catalog. s__Sodaliphilus sp04557565 exhibited the highest α-diversity across the sampled population, while an unclassified species belonging to o__Chitinophagales showed the lowest (Fig. S5). Notably, s__Treponema_D porcinum displayed the greatest variability in Shannon diversity, despite not ranking among the most diverse species on average (Fig. S6 and Table S10). β-diversity analysis revealed that the distribution of this species was strongly influenced by geographic origin, with minimal differentiation across host breed, age, or intestinal location (Fig. S7). Its relative abundance was higher in samples from China compared to those from Australia (https://alphaindex.zju.edu.cn/upgg/#/), suggesting a degree of geographic specificity in its distribution within the pig gut microbiota.
UPGG also suggests the impact of climatic conditions on the distribution of porcine gut microbiota. We conducted a subsequent study based on the climate indicators (https://worldclim.org/) of the 21 sampling sites involved in this study (Table S11). We assessed the correlation between the microbial abundance distance matrix and the climate indicator distance matrix of the respective sampling sites (Fig. S8). Although the analysis results showed that the explanatory power of climatic factors for the distribution of microbial communities was only 2.23%, significant associations with the microbial abundance distribution were still identified for five climatic indicators (climate stability and seasonal variation, and indicators of extreme temperature and humidity) through Mantel and RDA/CCA correlation analyses (Fig. S9 and S10).
Finally, examined the distribution of community composition of UPGG, the abundance composition distribution results at the genus level indicate that the microbiota showed a significantly high prevalence of Prevotella, Faecousia, Sodaliphilus, and Treponema_D (Fig. 3e). At the phylum and class taxonomic levels, our results align with those of prior investigations, confirming Firmicutes, Bacteroidota, Spirochaetota and Proteobacteria as the predominant phyla (Fig. S11). Furthermore, we observed variations in the ratios of Firmicutes to Bacteroidota, suggesting differences in adiposity levels across individuals45. Overall, UPGG has collected metagenomic data by considering various factors that influence microbial distribution, enhancing sample richness, and ensuring the diversity levels of microbes. This enhancement allows researchers to more comprehensively observe and analyze microbial communities, revealing patterns of co-evolution between microbes and their hosts.
UPGG expands the functional landscape of porcine gut bacterial proteins
We predicted and clustered protein sequences through UPGG genome sets, resulting in a total of 78,755,674 genes obtained, created a comprehensive catalog of porcine gut bacterial proteins. Subsequently, a functional annotation of the dataset was conducted, encompassing KEGG orthologys (KOs), Carbohydrate-active enzymes (CAZy), Virulence factor (VFDB), and Clusters of orthologous groups of proteins (COGs) (Fig. S12), as depicted in Fig. 4a. Among these, Carbohydrate Carbohydrate Esterases (53.9%) in the CAZy category accounted for the highest percentage (Fig. 4b). Together, 25.31% (n = 19,931,711) of the genes had no homologs in public protein databases and 49.67% (39,117,943) were not assigned to known functions by either database, suggesting that almost half of the proteins may code for novel functions.

a The non-redundant proteins were annotated based on four functional categories (COGs, KOs, and CAZy). Percentages of identified genes in the specified functional categories are shown. b The distribution shows carbohydrate enzyme classifications. c All ARGs were classified according to the antimicrobial for which they showed resistance. d The abundances and numbers of ARGs are presented for each sample. e The abundance of 22 core ARGs in each of pig populations, and their corresponding antimicrobial classes and resistance mechanisms. The ARGs existing in at least 95% of samples were defined as core ARGs in pig feces. Two color bars on the left of the heatmap represent the resistance mechanism and corresponding antimicrobial class of each resistance gene, respectively. f The identification shows the number of non-redundant proteins classified as various types of mobile elements. g We define and annotate ARG-MGE contigs, and subsequently integrate the analysis with the differential results between high-intensity and low-intensity farming communities. h MGCs of the UPGG are compared between kSGBs and uSGBs. All the MGCs were separated into eight classes. i The relative frequency of MGC types across dominant phyla shows that MGC genes are predominantly identified in Bacteroidota, Firmicutes, Proteobacteria and Actinobacteria. j The largest hybrid MGC predicted by gutSMASH harbored three MGC functional domains, potentially encoding different metabolic pathways. PDU pdu operon, NADHD Threonine to propionate; PROP, Threonine to propionate.
Furthermore, we identified antibiotic resistance genes (ARGs) and investigated their distribution. 10,769 genes (0.014%) were annotated to 546 different types of ARGs (Table S12). These were further categorized into 91 specific drug resistance classes (Table S12). Tetracycline resistance was most enriched in pig feces, followed by aminoglycoside and macrolide resistance classes (Fig. 4c). The predominant mechanisms underlying ARGs identified were the protection of antibiotic targets and the modification of antibiotic target (Fig. S13). The richness of ARGs was relatively consistent across countries and breeds (Fig. 4d). We identified 22 core ARGs (For detailed definitions, see the methods) that showing variable abundance across different pig breeds, especially in wild boar pig breed (Fig. 4e). Given the potential for ARGs to disseminate among commensal and pathogenic microbes, the presence of these core ARGs, particularly those enriched in wild or indigenous breeds, may indicate an increased risk for the development or transmission of antimicrobial-resistant infections. In addition, identifying mobile genetic elements (MGEs) is crucial for understanding horizontal gene transfer in microorganisms, such as the transmissibility of antibiotic resistance genes within the gut microbiome. The number of MGEs increases with the size of the sample (Fig. S14), and in our study, a total of 1,173,282 MGEs were obtained. The most abundant genes were those associated with integration/excision and replication/recombination repair (Fig. 4f). Our analysis focused on genes that harbor both ARGs and MGEs, leading to the identification of 20 contigs that met these criteria, designated as ARG-MGE46,47 (Fig. 4g and Table S13). These ARG-MGE contigs may serve as mobile reservoirs of resistance genes, raising concerns about their potential involvement in the emergence and spread of resistant pathogens, particularly under selective pressures such as disease outbreaks or antibiotic usage.
Anaerobic bacteria produce a variety of high-concentration primary metabolites, often encoded in metabolic gene clusters (MGCs), which can directly or indirectly affect the host. Given the vast microbial diversity in the porcine gut ecosystem, the UPGG catalog provides an important resource for mining metabolic gene clusters to elucidate host-microbe interactions. We identified metabolic gene clusters within each MAG as the unit, and a total of 72,056 MGC regions were identified in 27,362 MAGs. The identified MGCs were classified into 8 classes, with the majority being identified from uSGBs (Fig. 4h). Short-chain fatty acids (SCFA) at 50.13% and E-MGC (Energy-capturing-related gene clusters) at 25.56% are the most common classes. Subsequently, the distribution at the phylum level reveals differences in the distribution of MGC classifications (Fig. 4i). Among the identified MGC-encoding regions, PUB_DTU2018_MG_1469_bin.1 (Salmonella enterica) and PUB_29702_m1_bin.7 (Escherichia coli) contain the highest number of MGCs, and some of these regions carry multiple core functional domains, referred to as hybrid MGCs (Fig. 4j). These data indicate that the gut microbiome encodes a broad metabolic potential, and there is a significant variation in metabolic potential among different taxonomic groups in the pig gut. Overall, the porcine gut bacterial protein dataset we constructed has expanded the new landscape of functional proteins, providing an important resource for subsequent research on porcine gut microbiota functions and exploration of the metabolic potential of the pig gut.
Patterns of intraspecies genomic diversity
Core genes are shared by all species involved in fundamental biological processes, such as gene expression, energy production, and amino acid metabolism. Accessory genes are strain-specific genes48. To explore the intraspecific genomic diversity of the UPGG, we generated 436 pangenomes for SGBs with more than 10 high-quality MAGs by clustering protein sequences (Table S14). Investigating the correlation between pan-genome size and the number of conspecific genomes helps to clarify variations in gene richness among species. We observed that species originated from diverse gut microbial phyla, particularly those from distinct branches within the Firmicutes, demonstrated the most rapid rates of gene acquisition (Fig. 5a). Our stringent definition of core genes has resulted in a larger number of conspecific genomes contributing to a smaller proportion of core genes within the pan-genome, yet taxa from the Firmicutes and Bacteroidetes orders maintain a considerable proportion of these core genes. The median estimate of core genome proportion representing the percentage of core genes among all genes in the genome was calculated as 9.008% (Fig. 5b).

a The normalized pan-genome size as a function of the number of genomes from the same species was plotted, with regression curves drawn for each phylum. The coefficients of determination are displayed next to each curve, and the shaded areas represent the 95% confidence intervals. b Fraction of the core genome for each species is shown as a function of the number of genomes from the same species on the left, and as a histogram on the right, with colors indicating different phyla. The horizontal dashed line represents the median value across all species. c Proportion of core and accessory genes (n = 436 species) classified using various annotation schemes is displayed, along with the percentage of genes that lack functional annotation. The box lengths represent the interquartile range (IQR) of the data, and the whiskers extend to the minimum and maximum values within 1.5 times the IQR from the first and third quartiles, respectively. A two-tailed Wilcoxon rank-sum test was used to compare the classification between core and accessory genes, with significance indicated by ***p-value < 0.001. d Comparison of functional categories assigned to core and accessory genes highlights only statistically significant differences (FDR < 0.05). e The relationship between the number of single nucleotide variants (SNVs) and the number of participating species is shown. f The number of SNVs in the species-specific microbial genomes is presented.
To differentiate the functions encoded by core genes and accessory genes, we analyzed their associated functional annotations. Core genes exhibit better coverage compared to accessory genes, particularly in the COG and KEGG annotations, with median proportions reaching 153,928 (96%) and 156,494 (61%), respectively (Fig. 5c). Subsequently, we investigated the functions encoded by these genes based on COG functional classification. In comparison, the majority of gene functions remain concentrated in accessory genes, with transcription and unknown functions accounting for a larger proportion, exhibiting significant divergence from core genes (Fig. 5c). Key metabolic functions involving nucleotide, amino acid, and lipid metabolism, as well as other housekeeping functions, also show differences (Fig. 5d). Additionally, accessory genes account for a much larger proportion of COGs with no known function and genes involved in replication and recombination, which are often found in mobile elements. A significant number of accessory genes are related to defense mechanisms, including antibiotic resistance mechanisms, phage targeting, invasion modification, and mobile elements. These findings are consistent with the results of gene function exploration in the construction of the human UHGG pangenome14. Next, based on the core gene alignment of the pan-genome, we constructed phylogenetic trees for each species. We found that some species branches showed consistency with the population-assembled samples. For example, in the case of Lactobacillus delbrueckii_B (Fig. S16), where the Jinhua pig samples, sourced from disparate studies and encompassing varying gut segments and ages, exhibited a high degree of genomic similarity among samples within the Jinhua pig breed. This observation lends credence to the concept of co-evolution between hosts and their associated microorganisms. Our investigation revealed that this phenomenon at the genus level in our study was mainly concentrated in pathogenic microorganisms, Lactobacillus delbrueckii and Treponema_D sp016294035 (Fig. S17), and most frequently in unclassified MAGs.
We next investigated intraspecies single-nucleotide variants (SNVs) within the UPGG species. We generated a catalog consisting of 23,350,975 SNVs from 436 Sodaliphilus species with ten or more conspecific genomics (Fig. 5e and Table S15). Next, we assigned SNVs to different breeds, among which 0.4% are breed-specific SNVs. Notably, although the sample sizes for Jinhua pigs, Tibetan pigs, and Wild pigs were not the largest, these breeds had a higher number of breed-specific SNVs (Fig. 5f and Fig. S18). This phenomenon indicates that the gut microbiota of these Chinese local breeds exhibits higher uniqueness and diversity (Fig. S19). Specifically, these local breeds may possess unique genomic backgrounds and ecological environments, leading to distinctive changes in their gut microbiota. These findings contribute to the understanding of the genomic diversity and microbiome structure of different pig breeds.
Pigs as a potential model organism for studying the human gut microbiome in this study
We downloaded species-specific MAG databases constructed for cattle, sheep, human and mouse. Consistency comparisons have demonstrated that the UPGG exhibits the highest similarity to the representative assembled microbiota of humans (Fig. 6a), with both comprising a significant proportion of their own total MAG numbers. This suggests a closer taxonomic and functional resemblance between the pig and human gut microbiomes (Table S16). Furthermore, samples from Chinese local breeds made the greatest contribution to the assembly of MAGs that are closely aligned with human genomes. Despite their smaller numbers, they had a high proportion of samples, surpassing those from commercial breeds (Fig. 6b). This finding underscores the importance of local breeds in studying gut microbiota diversity and function.

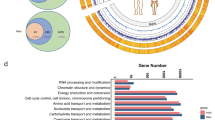
a The consistency ratio of UPGG with other species’ MAG databases is represented where green indicates the proportion of MAGs from different species’ MAG databases that are consistent with UPGG (ANI > 95%), and red represents the proportion of MAGs in UPGG that are consistent with those in other species’ databases (ANI > 95%). b The MAGs in the UPGG that are consistent with cattle, goat, mouse, and human are analyzed for the proportion of the population classification of the original metagenomic samples from which they were assembled. ‘Mixed breed’ refers only to hybrid varieties, while ‘commercial’ and ‘local’ refer only to pure breeds. c The distribution of metagenomic samples for the 5366 MAGs from Chinese human metagenome assemblies that are consistent with UPGG (ANI > 90%) is shown. The right pie chart illustrates these samples, with pink indicating local Chinese breeds. d The distribution of microbial diversity from humans and pigs, sourced from France and China, is shown. Colors represent countries, and symbol shapes denote species. e Differences in microbial abundance between two regions for the same species were compared, and the distribution of significantly different microbes was shown. Only microbes that were significantly upregulated in the groups are displayed. Those marked in red indicate microbes with consistent trends across different species at the same geographic location, with specific taxonomic levels shown in (f). g Overlap of core gut microbial species among humans, mice, and pigs. h Abundance distribution of core microbial species shared by pigs and humans across pig population samples (n = 4479).
Then, we focused on whether the gut microbiota of different species always exhibit similar preferences in a similar environment. Firstly, our study found that 5366 MAGs from UPGG could be identified as consistent with Chinese human population MAGs (ANI > 90%). Of these MAGs, 68.4% originated from samples assembled in China, with Chinese local breeds predominating these samples (Fig. 6c and Table S17). Secondly, we conducted a preliminary validation using both public and existing metagenomic data. The PCA results indicated that the gut microbiota of humans and pigs from the coastal regions of eastern China exhibited a higher degree of similarity to each other compared to those from France (Fig. 6d). Within the same geographical context, the affinity between the human and porcine microbiotas in China was more pronounced than that observed between their French counterparts, although a notable similarity in microbiota patterns was also detected between French humans and Chinese pigs (Fig. 6d). Subsequently, a comparative analysis of the differences in microbes between humans and pigs from two regions was conducted. The statistical analysis revealed that the number of MAGs showing consistent trends of change in both species was higher in China than that in France (Fig. 6e). Furthermore, in the France cohort, the majority of the microbial genera classification remains unassigned (Fig. 6f). In summary, our results revealed that the gut microbiota of humans and pigs in specific regions of China exhibit a certain degree of similarity. Therefore, Chinese local pigs have the potential to serve as model animals for human research.
Identifying shared core gut microbes across host species is critical for evaluating the suitability of model organisms. Our findings highlight that pigs share a substantially higher number of core gut microbial species with humans than mice do, underscoring their closer taxonomic similarity (Fig. 6g). Notably, several human core taxa such as Prevotella copri, Gemmiger qucibialis, and Roseburia inulinivorans are consistently abundant in pigs, suggesting functional convergence driven by similar dietary patterns (Fig. 6h and S20). In contrast, mice exhibit fewer overlapping core species and more divergent microbial compositions. Moreover, even within shared genera like Eubacterium, different host-specific species dominate in humans, pigs, and mice, reflecting host-adaptive specialization (Table S18). These results emphasize that pigs, by harboring a human-like core microbiota and possessing a similar hindgut structure49 (Fig. S21), serve as a more appropriate model than mice for studying human gut microbiome structure, function, and its region-specific variations.
Discussion
We have generated a unified sequence catalog representing 38,786 genomes and 78 million protein sequences of the porcine gut microbiome. Our study’s comprehensive sampling across developmental stages, geographic locations, breeds, and feeding methods has significantly expanded the pig genome database. The saturation curve analysis confirms that our extensive sample size is adequate for deriving representative MAGs. Customized databases leveraging UPGG genomes have outperformed previous catalogs in accuracy for taxonomic profiling. Mapping rates have significantly improved in independent studies using real metagenomic data. Additionally, compared to previous porcine gut microbiome databases, UPGG provides a comprehensive range of microbial gene sequences and functions, enhancing the application of genomic functional research for future researchers in the field of porcine gut microbiomes.
UPGG not only broadens the taxonomic diversity but also significantly enriches the functional landscape of the porcine gut microbiota. Pangenome and SNV analyses have revealed a plethora of unknown core genes that warrant further investigation36,48. Based on the proportion of core genes, we have identified differences in the openness of pangenomes between different phyla, indicating their disparate roles in the porcine gut ecosystem. Secondly, genetic variations in the host (such as breed), also lead to pronounced intraspecific differences within the gut microbial communities50. Additionally, breeds living in specific environmental regions, such as wild boars and Jinhua pigs, had a higher number of specific SNVs. Our data also provide insights into intra-species genomic variation. Even within the same microbial species, different intestinal niches may impose distinct selective pressures, leading to functional divergence. Using the cecum and colon as examples, we identified high-abundance MAGs specific to each region. Notably, some of these MAGs belong to the same species, yet their genomes exhibit differences in MGC composition. In particular, the relative proportions of classes such as SCFA, E-MGC, and Aliphatic_amine varied between the two gut regions (Fig. S15), suggesting that these functional differences may be associated with niche-specific physiological roles.
The UPGG catalog demonstrates a rich potential for discovering microbial functional diversity, making it an important resource for revealing interactions between microbial communities and their hosts, particularly ARGs and MGCs. ARGs significantly hinder the effective treatment of bacterial infections. More importantly, their impact extends beyond swine health, as ARGs can disseminate into the environment through MGEs, thereby posing a serious threat to human health and survival51. We found that the majority of identified MGCs originate from the genomes of uncultivated microbes, and there are differences in the potential metabolic gene clusters across various types, indicating a vast potential for mining previously unexplored microbial functional genes from uncultivated and unknown microbes in the pig gut. However, most MGCs identified from the UPGG catalog are fragmented, suggesting that short-read sequencing inhibits the recovery of full-length MGC sequences from uncultivated bacteria.
Our findings suggest that the pig may serve as an effective model organism for studying the human gut microbiome. The high similarity between the UPGG and human microbiota, as well as the significant contribution of local breed samples to MAGs aligned with humans, indicates a close taxonomic and functional relationship. These observations imply that environmental and geographical factors significantly influence the composition of the gut microbiota52, and the local breeds’ microbiota may offer a more relevant model for understanding human gut microbiota diversity and function.
Despite representing one of the most comprehensive efforts to compile porcine gut metagenomic data, the UPGG catalog has several inherent limitations that warrant discussion. First, although we integrated nearly all publicly available metagenomic samples, certain MAGs reported in previous studies could not be recovered after applying our standardized assembly, binning, and quality control workflows. This shortcoming largely stems from the limitations of short-read sequencing technologies, which struggle to accurately reconstruct genomes with highly conserved or repetitive regions53. As a result, some genomes may have been incompletely assembled or omitted, affecting the catalog’s overall representativeness54. Second, the dataset lacks detailed host phenotypic metadata, particularly regarding health status. While most samples were derived from healthy pigs, the absence of clinical diagnostics in public datasets, combined with the scarcity of large-scale phenotyped cohorts in livestock, restricts our ability to directly link microbial features to host health or disease. Finally, compared to human-focused microbial genome catalogs14, UPGG still lags in both scale and resolution. This underscores the need for continued efforts to build high-quality, longitudinal, and phenotype-rich porcine microbiome resources. We believe the UPGG resource, enriched with these future expansions, will support more comprehensive analyses of microbiome–host health interactions in pigs, and further validate their translational relevance as models for human microbiome research.
Together, our assembly and analyses of the UPGG catalog provided functional insights into the pig gastrointestinal microbiota that could potentially apply to pave the way to microbial interventions for better pig and eco-environmental qualities.
Methods
Sample collection
All animal-related work was strictly conducted in accordance with the “Regulations for the Administration of Affairs Concerning Experimental Animals issued by the Ministry of Agriculture and Rural Affairs of the People’s Republic of China. All the experimental procedures were approved by the Animal Welfare and Ethics Committee of Zhejiang University (approval number: ZJU20220224). In this study, a total of 5784 metagenomic samples were used, of which 3721 samples were derived from publicly available project data from NCBI (specific projects are listed in Table S1). The remaining 2063 samples were collected from previous studies conducted by our project group: 49 fecal samples were collected in 2020 from a pig breeding nucleus farm in Guigang, Guangxi, which included samples from 2 Yorkshire pigs, 24 Duroc pigs, and 23 Duroc2 pigs, all aged 21 days; 1661 fecal samples were collected in 2022 from the same pig farm in Guigang, Guangxi, including 5 breeds, 200 Duroc pigs, 39 Duroc2 pigs (another Duroc line), 300 Landrace pigs, 108 Pietrain pigs, and 1014 Yorkshire pigs, with ages ranging from 100 to 240 days; from 2021 to 2023, four batches of slaughtering were conducted in Jinhua, Zhejiang, collecting 6 jejunum samples, 73 cecum samples, 84 colon samples, and 176 fecal samples from Jinhua pigs; 35 Landrace × Yorkshire pigs from a farm in Jinhua, Zhejiang, yielding 30 cecum samples and 29 colon samples; 16 fecal samples from 8 Laiwu pigs and 8 Lulai pigs from a commercial farm in Laiwu, Shandong; 4 fecal samples from wild boars collected in Baishan, Jilin in 2022.
Fresh feces samples were collected from pigs and stored in liquid nitrogen. After cleaning the pigs’ anus, sterile swabs were carefully inserted into the rectum to stimulate defecation, and the fresh feces were immediately collected in sterile tubes; For pigs intended for slaughter, euthanasia is performed by placing them in a sealed chamber, introducing carbon dioxide at an appropriate rate until the pigs lose consciousness and cease breathing, confirming death before closing the gas valve and disposing of the carcasses. The luminal contents of the middle jejunum, middle ileum, cecum, and colon were collected within 30 minutes post-mortem and immediately placed into liquid nitrogen. All pigs involved in the experiments were healthy and had not been administered probiotics or antibiotics in the two months prior to sampling.
DNA extraction, library construction, and metagenomic sequencing
After sample collection, they were immediately placed in liquid nitrogen. After collecting all samples in each b−tch, they were transported to the laboratory and then transferred to a −80°C freezer for storage. The luminal DNA from each colon content sample was extracted using the QIAamp Fast DNA Stool Mini Kit (Qiagen, Germany) according to the manufacturer’s instructions. The amount and quality of DNA were measured using a NanoDrop 1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), and sterile water was used to adjust the DNA concentration to a final concentration of 1 ng/L. Libraries were generated using the Illumina TruSeq™ DNA Sample Prep Kit and subsequently sequenced on an Illumina HiSeq 2500 platform or MGI T7 platform by the sequencing companies Novogene (Beijing, China) and Wuhan Benagen Technology Co., Ltd. (Wuhan, China).
Metagenome assembly
Preprocess the raw sequencing data by using fastp (version 0.23.2)55 to filter adapters and low-quality sequences. Then, bowtie2 (v2.3.5.1)56 was applied to align the filtered sequences to the host genome. As suggested by Bush et al.57, we further employed snap (v2.0.1) to eliminate host contamination to obtain high-quality sequences (clean reads)58. Assemble the clean reads into contigs using megahit (v1.1.3) for single-sample assembly59. Set the assembly parameters as follows: ‘-min-count 2 -k-min 27 -k-max 87 -k-step 10 -min-contig-len 500’.
Genome reconstruction and annotation
We used some of the functional modules in MetaWRAP (v1.1.1) for the construction of microbial genomes (MAGs)60. The contigs of all samples were first binned using two binning tools, MetaBAT261 and Maxbin262. The minimum length of contigs considered for the construction of bins in MetaWRAP was set to 1000 bp and 1500 bp for Maxbin2 and MetaBAT2, respectively. The bins set AB is derived by merging bins from set A (produced by Maxbin2) with those from set B (produced by MetaBAT2). CheckM (v1.0.12)63 was then used to estimate the completeness and contamination rates of the three datasets (A, B, and AB), selecting the MAG with the highest bin score. To further enhance the quality of the bins, the metagenomic sequencing reads were remapped to each bin, and then reassembled using the metaSPAdes64 tool within the “reassemble_bins” module of MetaWRAP.
Redundancy of 41,769 MAGs was reduced using dRep’s two-step clustering method65. Initially, primary clustering was performed with Mash software66 using 90% MASH ANI. For each primary cluster, secondary clustering was conducted with 99% ANI and at least 25% overlap between genomes. The highest quality MAG from each secondary cluster was selected. The 24,802 representative non-redundant MAGs were divided into medium-quality MAGs (completeness ≥ 50%, contamination < 5%) and 16,967 high-quality MAGs (completeness ≥ 90%, contamination < 5%)67.
Taxonomic annotation of each species representative was performed with GTDB-tk v2.1.168 using the ‘classify_wf ’ function and default parameters. Names of taxonomic units above the genus level with suffixes indicate groups that are not monophyletic according to the GTDB reference tree. A maximum likelihood tree of the MAGs was constructed de novo using protein sequence alignments generated by GTDB-tk. The phylogenetic tree was visualized using Interactive Tree Of Life (iTOL v6.9.1)69. Annotation data for visualization were generated with the R package table2itol (https://github.com/mgoeker/table2itol). Genome-level annotation of the MAGs, including CDS, tRNA, and rRNA prediction, was performed using Prokka70 with the parameters ‘--metagenome --kingdom Bacteria --force --norrna --notrna --cpus 20’, effectively annotating each contig within the MAGs. Identification of 5S and 16S rRNA was conducted using Barrnap (v0.9) (https://github.com/tseemann/barrnap) with the parameters ‘--kingdom bac --threads 8 --quiet’. Primary metabolic gene clusters were predicted for each MAG genome using gutSmash71.
UPGG synthetic evaluation
To facilitate the comparison of different classification analysis methods, we utilized CAMISIM41 to generate a simulated dataset, setting the sequencing size for each sample to 5 Gbp while keeping all other parameters at their default settings. We constructed synthetic communities containing 50, 200, and 600 genomes, and based on the UPGG approach, we employed three strategies: (1) selecting only the representative genomes for each species; (2) randomly selecting one genome from among the conspecific genomes of each species; and (3) randomly selecting five genomes from each species, ensuring that each genome is evenly distributed across different samples. This resulted in a total of 27 simulated samples, and we aggregated the results of the simulated samples for each analysis method to evaluate the detection and quantification of genomes contained within the synthetic metagenome72. To this end, we defined (1) “true positives” as the detection of SGBs that correspond to genomes present in the synthetic metagenome; (2) “false positives” as the detection of SGBs that do not correspond to any genomes in the metagenome; and (3) “false negatives” as SGBs that were not detected but corresponded to genomes present in the synthetic metagenome. Detected SGBs representing overlapping SGBs within the community were also considered “true positives”. Based on these definitions, we calculated accuracy, precision, recall, and F1 score73.
Bioinformatic analysis of UPGG genomes
The Struo274 tool was employed to incorporate 38,786 MAGs into the construction of a Kraken2 customized database. Subsequently, the original metagenomic data were analyzed using Kraken240 and Bracken75 to perform quantitative analysis on MAGs within the UPGG. We performed sparse and extrapolated analyses of high-quality MAGs and sample numbers from PIGC and UPGG using the iNEXT76, with the data type set to “abundance”. In the context of this study, the metagenomic samples from CNP0002106 (from China National GeneBank DataBase)32, PRJNA984595 (from NCBI Sequence Read Archive)42, CRA010380 (from Genome Sequence Archive)43 and CNP003325 (from China National GeneBank DataBase)44 were analyzed using three distinct databases: PIGC30, Metaphlan4 (mpa_vOct22_CHOCOPhlAnSGB_202212)73 the GTDB database (release version r207)68, and the UPGG database constructed for this research.
Construction and function of gene catalog
Assembled contigs were used for gene prediction by Prodigal (v2.6)77 software. These contigs were subjected to gene prediction, and then incomplete genes were removed. Finally, the complete genes (with initiation codon and termination codon) were retained for subsequent analysis. All complete genes were clustered at the protein level using CD-HIT (v4.8.1)78 at 100%, 90%, and 50% similarity thresholds, guided by the UniRef database79. The longest sequence in each cluster served as the seed sequence, with each member in the 90% and 50% similarity clusters required to overlap with the seed sequence by at least 80%. The non-redundant gene sets were then compared to a database to filter out genes from eukaryotes. The final set of non-redundant microbial genes, with 90% sequence similarity, was obtained for subsequent analysis.
The amino acid sequences of the genes in the gene set were aligned to the database Uniprot TrEMBL using DIAMOND (v0.9.21.122)80. Genes that could not be aligned to any genes in the Uniprot TrEMBL database were defined as unknown genes. Annotation of KEGG functionality was performed by aligning the protein sequences of the genes to the KEGG database using kofam_scan (v1.3.0)81. Eggnog-mapper (v2.0.1)82 was employed to align the protein sequences of genes to the eggNOG 5.0 database for annotation83. The protein sequences of the genes were aligned to the dbCAN database84 for CAZy85 annotation using the hmmscan program of HMMER86. The annotation of resistance genes for antibiotics was performed using rgi (v6.0.3) to align sequences to the CARD database87, and the annotation of virulence factors was performed using BLAST (v2.15.0) to align sequences to the VFDB database88. The alignment with the smallest E value of each gene was selected for subsequent analysis. The gutSMASH tool89 was used to identify MGCs.
Salmon (v0.13.1)90 was used to estimate the coverage of genes (TPM). The abundance of microbial taxa, KEGG homologous protein clusters, KEGG metabolic pathways, eggNOG homologous protein clusters, ARGs, and virulence factors under each category item was summed as the abundance of this category item.
Antibiotic resistance gene and mobile genome element analysis
The criteria for selecting core resistance genes required that the calculated Transcripts Per Kilobase of exon model per Million mapped reads (TPM) values be greater than 30 and be present in 95% of the samples, resulting in a total of 22 core resistance genes identified. The sum of the abundances of all resistance genes in each sample was defined as the richness of ARGs91.
The number of MGEs in each sample was calculated by determining the TPM values of the contigs identified as MGEs. For instance, if there are 100 MGEs with a TPM greater than 0 in a sample, the number of non-redundant MGEs in the sample is considered to be 100. A random selection of a given number of samples was made from 3000 samples, and 1000 such random selections were made without replacement. The average value from these 1000 samples was used to plot the MGE rarefaction curve.
Analysis of microbial diversity and effects of host and environmental factors
To compute and visualize differences between groups, such as age, breeds, and geographical locations, we used the R package vegan (v2.6.4) to normalize the MAG quantitative abundance matrix92. The MAG quantitative abundance matrix was processed to compute diversity indices, including Shannon, Simpson, and Berger, as well as Fisher’s alpha diversity index. Additionally, beta diversity was calculated using Bray-Curtis dissimilarity measures. For strain-level diversity, we applied the same calculation method as described above, but focused on individual species within each sample. Species represented by only a single MAG were excluded from the analysis. In total, 1,479 species were assessed for their diversity, as summarized in Table S10. The permutational multivariate analysis of variance (PERMANOVA)93, implemented in the R package “vegan”, was used to determine the impacts of various host and environmental factors on the microbial diversities of the UPGG genomes.
The Redundancy Analysis/Canonical Correspondence Analysis method was employed to analyze the correlation between environmental factors and microbial distribution. A microbial matrix composed of samples containing information on environmental factors was filtered based on relative abundance (greater than 0.01%) and CLR transformation. A linear model of lm(MAG ~ climate factors + Age + Sex + Breed + Country) was used to evaluate the association between microbes and environmental factors. Only microbes that were significantly correlated with all five climate factors (FDR < 0.0001) were retained.
Comparing the UPGG genomes with the sequences in public datasets
Non-redundant MAGs were downloaded from the gut microbiome databases of mouse (MAG number = 1296), goat18 (MAG number = 4075), cattle20 (MAG Number=4941), and human14 (MAG number = 4746). FastANI was used to calculate the nucleotide identity between the MAGs in this study and those in the gut assembly genome databases of the aforementioned species. A threshold of 95% average nucleotide identity (ANI) was used to determine the similarity between two MAGs.
To explore whether gut microbiota across different hosts in the same environment exhibit consistency, we selected 81 human gut metagenomes (China Nucleotide Sequence Archive with the dataset identifier CNP0000175)94 from individuals with type 2 diabetes (T2D) in Suzhou, China, and 121 human gut metagenomes (European Nucleotide Archive with the dataset identifier PRJEB6070)95 from colorectal cancer patients in France. We also included 203 representative porcine gut metagenomes from the same geographical area in our study, with ages ranging from over 120 days to under 180 days. Using Kraken240 against the UPGG database developed in this study, alignment rates were approximately 60–70%. Subsequently, differential analyses were conducted separately in these two species groups. Initially, beta diversity (Bray-Curtis) was calculated to assess microbial community structures among different populations. Next, rank-sum tests were employed, and p-values were corrected using the Benjamini-Hochberg method. The R package UpSet96 was utilized to identify intersections and visualize them.
In another study, we utilized public data from the Mongolian population in China, specifically from the IMGG dataset in NCBI BioProject (PRJNA763692), which included individuals aged 18-38 years. From this dataset, 6729 MAGs were assembled using second and third-generation sequencing data17. These MAGs were then compared for nucleotide consistency with the MAGs in our study’s UPGG database using fastANI97.
Sources and identification of core bacterial strains for humans, pigs and mice
The human core species data were derived from two published studies encompassing a total of 24,342 human gut microbiome samples98,99. After merging and removing duplicates, 81 core species were identified. For mice, data were obtained from the Mouse Gastrointestinal Bacteria Catalog (MGBC)100, in which 102 core species were selected based on their presence in at least 80% of 2446 samples, as reported in the study. Similarly, for pigs, after excluding confounding samples such as fetal specimens, 160 core species were identified based on their presence in at least 80% of 4497 samples (Table S18).
Construction of strain pangenome
With the protein annotations and pan-genomes inferred for each of the 436 MAG species, we explored their intraspecies core and accessory gene repertoire. Only high-quality genomes (≥90% completeness) and species with at least ten independent conspecific genomes were analyzed14.
Protein-coding sequences (CDS) for each of the 41,769 genomes were predicted and annotated with Prokka (v1.14.6)70, using Prodigal (v2.6.3)77. Roary (v3.13)37 was used for pan-genome ortholog clustering analyses. COG, KEGG, CAZy, and viral annotations were derived from the eggNOG-mapper82 results. To assess the differences in annotation coverage and COG functional categories between core and accessory genes, we performed a two-tailed Wilcoxon rank-sum test using the ‘wilcox.test’ function in R (v4.2.3). The Benjamini-Hochberg method was employed to adjust the expected p-values for multiple testing. The effect size (Cohen’s d) was estimated using the ‘cohen.d’ function from the Effsize R package101.
Afterwards, we used core genes from each strain’s pan-genome and aligned them using FastTree102 for sequence comparison. Based on these alignments, we constructed an evolutionary tree using maximum likelihood methods. The resulting tree was visualized using the R package ggtree103.
Analysis of strain SNV
A pan-genome was built as a reference genome of 436 selected species, and snippy (v3.6.0)104 was employed to identify SNVs in the assembled genomes. Each SNV site was included in the catalog only if alternative alleles were detected in at least two genomes of the same species. The number of continent-specific SNVs was normalized per breed by the number of genomes, to estimate the contribution per genome to the breed-specific SNV discoveries.
UPGG database website construction
The website, UPGG, was deployed using the Nginx web server (https://nginx.org/en/) alongside Node.js (https://nodejs.org/en) to manage back-end operations. The front-end interface was developed with Vue3, a Progressive JavaScript Framework (https://vuejs.org/), delivering an intuitive and dynamic user experience. For web-based visualizations, Element Plus (https://element-plus.org/en-US/) and Apache ECharts (https://echarts.apache.org/en/index.html) were employed to facilitate comprehensive and interactive graphical displays. MySQL (https://www.mysql.com/) served as the database engine, ensuring reliable storage and management of all data within a MySQL database environment.
Data availability
Both the UPGG and microbial gene catalogs are available as part of a new genome layer within the UPGG website (https://alphaindex.zju.edu.cn/upgg/#/), where summary statistics of each species cluster and their functional annotations can be interactively explored and downloaded. The website encompasses a database in Kraken2 format, which facilitates the taxonomic quantification of metagenomic reads. The data used and generated in this study have been deposited in the Genome Sequence Archive (GSA) under project accession number PRJCA042329. Any other questions for data can be directed to the corresponding author (zhe_zhang@zju.edu.cn).
Code availability
The workflow used to generate the genome and protein catalogs, as well as the pan-genome and functional annotations, is described in a common workflow language pipeline. All scripts, software versions, and related information are available at https://github.com/Ls9917/UPGG. Additional code can be obtained from the corresponding author (zhe_zhang@zju.edu.cn) upon reasonable request.
References
Simon, J. C. et al. Host-microbiota interactions: from holobiont theory to analysis. Microbiome 7, 5 (2019).
Donaldson, G. P., Lee, S. M. & Mazmanian, S. K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol 14, 20–32 (2016).
Weissbrod, O. et al. Host genetics and microbiome associations through the lens of genome wide association studies. Curr. Opin. Microbiol 44, 9–19 (2018).
Tremaroli, V. & Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 489, 242–249 (2012).
Belkaid, Y. & Harrison, O. J. Homeostatic immunity and the microbiota. Immunity 46, 562–576 (2017).
Manor, O. et al. Health and disease markers correlate with gut microbiome composition across thousands of people. Nat. Commun. 11, 5206 (2020).
Xiong, X. et al. Nutritional Intervention for the Intestinal Development and Health of Weaned Pigs. Front Vet. Sci. 6, 46 (2019).
Luo, Z. B. et al. Fecal transplant from myostatin deletion pigs positively impacts the gut-muscle axis. Elife 12, e81858 (2023).
Zhou, Y. Y., Liu, M. & Yang, J. W. Recovering metagenome-assembled genomes from shotgun metagenomic sequencing data: Methods, applications, challenges, and opportunities. Microbiol. Res. 260, 127023 (2022).
Stothart, M. R., McLoughlin, P. D. & Poissant, J. Shallow shotgun sequencing of the microbiome recapitulates 16S amplicon results and provides functional insights. Mol. Ecol. Resour. 23, 549–564 (2023).
Ahn, T. H., Chai, J. & Pan, C. Sigma: strain-level inference of genomes from metagenomic analysis for biosurveillance. Bioinformatics 31, 170–177 (2015).
Segata, N. On the road to strain-resolved comparative metagenomics. mSystems 3, e00190–17 (2018).
Claesson, M. J. et al. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 38, e200 (2010).
Almeida, A. et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 39, 105–114 (2021).
Zeng, S. Q. et al. A compendium of 32,277 metagenome-assembled genomes and over 80 million genes from the early-life human gut microbiome. Nat. Commun. 13, 5139 (2022).
Huang, L. S. et al. A multi-kingdom collection of 33,804 reference genomes for the human vaginal microbiome. Nat. Microbiol. 9, 2185–2200 (2024).
Jin, H. et al. A high-quality genome compendium of the human gut microbiome of Inner Mongolians. Nat. Microbiol. 8, 150 (2023).
Cao, Y. et al. The multi-kingdom microbiome of the goat gastrointestinal tract. Microbiome 11, 219 (2023).
Jiao, J. Z. et al. Ecological niches and assembly dynamics of diverse microbial consortia in the gastrointestine of goat kids. ISME J. 18, wrae002 (2024).
Stewart, R. D. et al. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 37, 953–961 (2019).
Wilkinson, T. et al. 1200 high-quality metagenome-assembled genomes from the rumen of African cattle and their relevance in the context of sub-optimal feeding. Genome Biol. 21, 229 (2020).
Sato, Y. et al. Identification of 146 Metagenome-assembled Genomes from the Rumen Microbiome of Cattle in Japan. Microbes Environ. 37, ME22039 (2022).
Lesker, T. R. et al. An integrated metagenome catalog reveals new insights into the murine gut microbiome. Cell Rep. 30, 2909–2922.e6 (2020).
Rastegar, K., Kelley, S. T. & Thackray, V. G. Metagenome-assembled genomes from murine fecal microbiomes dominated by uncharacterized bacteria. Microbiol. Resour. Announc.12, e0116222 (2023).
Kieser, S., Zdobnov, E. M. & Trajkovski, M. Comprehensive mouse microbiota genome catalog reveals major difference to its human counterpart. Plos Comput. Biol. 18, e1009947 (2022).
Feng, Y. et al. Metagenome-assembled genomes and gene catalog from the chicken gut microbiome aid in deciphering antibiotic resistomes. Commun. Biol. 4, 1305 (2021).
Ma, L. Y. et al. Duck gut metagenome reveals the microbiome signatures linked to intestinal regional, temporal development, and rearing condition. Imeta 3, e198 (2024).
Li, C. et al. Expanded catalogue of metagenome-assembled genomes reveals resistome characteristics and athletic performance-associated microbes in horse. Microbiome 11, 7 (2023).
Xiao, L. et al. A reference gene catalogue of the pig gut microbiome. Nat. Microbiol 1, 16161 (2016).
Chen, C. Y. et al. Expanded catalog of microbial genes and metagenome-assembled genomes from the pig gut microbiome. Nat. Commun. 12, 1106 (2021).
Wang, W. et al. Metagenomic reconstructions of gut microbial metabolism in weanling pigs. Microbiome 7, 48 (2019).
Hu, J. et al. Characterizing the gut phageome and phage-borne antimicrobial resistance genes in pigs. Microbiome 12, 102 (2024).
Feng, Y. Q. et al. Metagenome-assembled genomes and gene catalog from the chicken gut microbiome aid in deciphering antibiotic resistomes. Commun. Biol. 5, 1289 (2022).
Cabral, L. et al. Gut microbiome of the largest living rodent harbors unprecedented enzymatic systems to degrade plant polysaccharides. Nat. Commun. 13, 629 (2022).
Vatanen, T. et al. Mobile genetic elements from the maternal microbiome shape infant gut microbial assembly and metabolism. Cell 185, 4921 (2022).
Van Rossum, T. et al. Diversity within species: interpreting strains in microbiomes. Nat. Rev. Microbiol. 18, 491–506 (2020).
Page, A. J. et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693 (2015).
Pasolli, E. et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176, 649–662.e20 (2019).
Falony, G. et al. Population-level analysis of gut microbiome variation. Science 352, 560–564 (2016).
Lu, J. et al. Metagenome analysis using the Kraken software suite. Nat. Protoc. 17, 2815–2839 (2022).
Fritz, A. et al. CAMISIM: simulating metagenomes and microbial communities. Microbiome 7, 17 (2019).
Sanjuán, J. M. O. et al. Effects of removing in-feed antibiotics and zinc oxide on the taxonomy and functionality of the microbiota in post weaning pigs. Anim. Microbiome 6, 18 (2024).
Chen, Q. et al. Tracking investigation of archaeal composition and methanogenesis function from parental to offspring pigs. Sci. Total Environ. 927, 172078 (2024).
Zhao, F. F. et al. Gut microbiome signatures of extreme environment adaption in Tibetan pig. Npj Biofilms Microbiomes 9, 27 (2023).
Magne, F. et al. The Firmicutes/Bacteroidetes ratio: a relevant marker of gut dysbiosis in obese patients? Nutrients 12, 1474 (2020).
Tian, L. et al. Metagenomic approach revealed the mobility and co-occurrence of antibiotic resistomes between non-intensive aquaculture environment and human. Microbiome 12, 107 (2024).
Gao, F. Z. et al. Swine farming shifted the gut antibiotic resistome of local people. J. Hazard. Mater. 465, 133082 (2024).
Golicz, A. A. et al. Pangenomics comes of age: from bacteria to plant and animal applications. Trends Genet. 36, 132–145 (2020).
Lunney, J. K. et al. Importance of the pig as a human biomedical model. Sci. Transl. Med. 13, eabd5758 (2021).
Zhernakova, D. V. et al. Host genetic regulation of human gut microbial structural variation. Nature 625, 813–821 (2024).
Pal, C. et al. The structure and diversity of human, animal and environmental resistomes. Microbiome 4, 54 (2016).
Rothschild, D. et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 555, 210 (2018).
Maghini, D. G. et al. Improved high-molecular-weight DNA extraction, nanopore sequencing and metagenomic assembly from the human gut microbiome. Nat. Protoc. 16, 458–471 (2021).
Chen, L. et al. Short- and long-read metagenomics expand individualized structural variations in gut microbiomes. Nat. Commun. 13, 3175 (2022).
Chen, S. F. et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890 (2018).
Langdon, W. B. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 8, 1 (2015).
Bush, S. J. et al. Evaluation of methods for detecting human reads in microbial sequencing datasets. Microb. Genomics 6, mgen000393 (2020).
Korf, I. Gene finding in novel genomes. BMC Bioinforma. 5, 59 (2004).
Li, D. H. et al. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11 (2016).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 158 (2018).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Wu, Y. W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Parks, D. H. et al. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Prjibelski, A. et al. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinforma. 70, e102 (2020).
Olm, M. R. et al. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. Isme j. 11, 2864–2868 (2017).
Wang, S. & Friedman, S. L. Found in translation-Fibrosis in metabolic dysfunction-associated steatohepatitis (MASH). Sci. Transl. Med. 15, eadi0759 (2023).
Bowers, R. M. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731 (2017).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785–D794 (2021).
Letunic, I. & Bork, P. Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–w82 (2024).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Pascal Andreu, V. et al. gutSMASH predicts specialized primary metabolic pathways from the human gut microbiota. Nat. Biotechnol. 41, 1416–1423 (2023).
Kim, N. et al. MRGM: an enhanced catalog of mouse gut microbial genomes substantially broadening taxonomic and functional landscapes. Gut Microbes 16, 2393791 (2024).
Blanco-Míguez, A. et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 41, 1633 (2023).
Youngblut, N. D. & Ley, R. E. Struo2: efficient metagenome profiling database construction for ever-expanding microbial genome datasets. PeerJ 9, e12198 (2021).
Lu, J. et al. Bracken: estimating species abundance in metagenomics data. Peerj Comput. Sci. 3, e104 (2017).
Hsieh, T. C., Ma, K. H. & Chao, A. iNEXT: an R package for rarefaction and extrapolation of species diversity (Hill numbers). Methods Ecol. Evol.7, 1451–1456 (2016).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinforma. 11, 119 (2010).
Fu, L. et al. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Suzek, B. E. et al. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Aramaki, T. et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252 (2020).
Cantalapiedra, C. P. et al. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Zhang, H. et al. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–w101 (2018).
Lombard, V. et al. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 (2014).
Finn, R. D. et al. HMMER web server: 2015 update. Nucleic Acids Res. 43, W30–W38 (2015).
Alcock, B. P. et al. CARD 2023: expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 51, D690–d699 (2023).
Liu, B. et al. VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–d917 (2022).
Andreu, V. P. et al. gutSMASH predicts specialized primary metabolic pathways from the human gut microbiota. Nat. Biotechnol. 41, 1416 (2023).
Patro, R. et al. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Bengtsson-Palme, J. The diversity of uncharacterized antibiotic resistance genes can be predicted from known gene variants-but not always. Microbiome 6, 125 (2018).
Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930 (2003).
Hu, Y. J. & Satten, G. A. A rarefaction-without-resampling extension of PERMANOVA for testing presence-absence associations in the microbiome. Bioinformatics 38, 3689–3697 (2022).
Zhong, H. et al. Distinct gut metagenomics and metaproteomics signatures in prediabetics and treatment-naïve type 2 diabetics. EBioMedicine 47, 373–383 (2019).
Ma, Y. et al. Metagenome analysis of intestinal bacteria in healthy people, patients with inflammatory bowel disease and colorectal cancer. Front Cell Infect. Microbiol 11, 599734 (2021).
Lex, A. et al. UpSet: visualization of intersecting sets. IEEE Trans. Vis. Comput Graph 20, 1983–1992 (2014).
Jain, C. et al. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114 (2018).
Hiseni, P. et al. HumGut: a comprehensive human gut prokaryotic genomes collection filtered by metagenome data. Microbiome 9, 165 (2021).
Goel, A. et al. Toward a health-associated core keystone index for the human gut microbiome. Cell Rep. 44, 115378 (2025).
Beresford-Jones, B. S. et al. The Mouse Gastrointestinal Bacteria Catalogue enables translation between the mouse and human gut microbiotas via functional mapping. Cell Host Microbe 30, 124–138.e8 (2022).
Torchiano, M. Effsize - a package for efficient effect size computation. (2016); Available from: https://doi.org/10.5281/ZENODO.1480624.
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490 (2010).
Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinforma. 69, e96 (2020).
T, S. snippy: fast bacterial variant calling from NGS reads. Available from: (2015). https://github.com/tseemann/snippy.
Acknowledgements
This work was supported by Zhejiang Provincial Natural Science Foundation of China (LZ23C170003), National Key Research and Development Program of China (2021YFD1200802, 2023YFF1001100), and National Natural Science Foundation of China (32272832).
Author information
Authors and Affiliations
Contributions
Z.Z. and Y.C. conceived and designed the experiments. S.L., X.L., Q.X., X.Y., C.C., J.G., W.Z., J.B,. and P.Y. collected samples. S.L., B.F., Z.Z.Y., and J.M. analyzed the data. S.L., B.F., Y.C. and Z.Z. guided the data mining. B.F. and S.L. constructed the website. S.L., B.F., Z.Z., Y.C., Q.W. and Z.W. wrote the manuscript. All authors have reviewed and agreed with the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, S., Feng, B., Zhang, Z. et al. UPGG: expanding the taxonomic and functional diversity of the pig gut microbiome with an enhanced genome catalog. npj Biofilms Microbiomes 11, 196 (2025). https://doi.org/10.1038/s41522-025-00828-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41522-025-00828-1
