
Abstract
An extracellular matrix (ECM) gene expression pattern (ECM3) distinguished truly aggressive grade III breast carcinomas (BCs). Here, we examined the biomechanical characteristics of the ECM in human BCs to identify the molecules that mediate the aggressiveness of ECM3/grade III (E3G3) tumors. By shotgun proteomics of decellularized human BCs, we found a significant enrichment in proteins involved in tumor-ECM interaction in E3G3 tumors. These tumors were characterized by high dense collagen deposition, a fibrillary cytoskeleton network and the highest stiffness. CLEC3A, a secreted C-type lectin domain family 3 member, was found unique of E3G3 tumors and was validated to be more expressed in these tumors by immunohistochemistry in 2 human BC cohorts, associating significantly with worse prognosis. Ectopic CLEC3A overexpression in MDA-MB-231, MDA-MB-361, and MDA-MB-468 BC cells increased intracellular mediators of tumor adhesion to the ECM, actin-stress fibers and YAP activation, and tumor migration. Accordingly, levels of the YAP/TAZ gene signature were higher in CLEC3A-positive ECM3-enriched tumors and correlated with tumor stiffness. These results implicate CLEC3A in mediating the ability of E3G3 BCs to sense cues in the surrounding ECM, accelerating tumor progression.
Similar content being viewed by others


Introduction
Breast cancer (BC) is the most common cancer in women and despite effective prevention strategies, early diagnostic screens, and innovative and personalized treatments, it remains a leading cause of cancer-related mortality worldwide1, necessitating further efforts to determine the complexity of tumor progression. Although BC is highly heterogeneous, based on the molecular features of tumor cells, and although these differences significantly affect treatment responses and patient outcomes2,3, the interactions of cancer cells with the surrounding cellular and noncellular microenvironment are proving to be crucial for tumor progression4,5.
The extracellular matrix (ECM)—the noncellular component of the tumor microenvironment (TME)—is biochemically distinct in BCs compared with normal breast, and the mechanical forces of the ECM collaborate with biochemical cues to modulate tumor cell behaviors through cell surface receptors that sense mechanical signals and activate specific signal transduction pathways6,7. In addition to being a complex network of structural proteins and carbohydrates that are essential for maintaining tumor structure, the ECM is a reservoir of molecules that are produced by tumor and host cells on which BCs rely to adapt to local conditions and advance8.
Analyses of genes that encode ECM molecules have shown that expression patterns vary between BCs and that these patterns are clinically significant, regardless of the intrinsic characteristics of neoplastic cells9,10,11. In this context, we identified an ECM gene expression pattern, which we termed ECM3, in approximately 40% of BCs. ECM3 features interact with grade in predicting BC progression, distinguishing tumors with accelerated metastatic potential only in the undifferentiated (grade III) phenotype whereas no significance was found in well-differentiated (grade I-II) carcinomas12,13,14,15. Notably, ECM3 grade III (E3G3) patients relapsed more frequently than did non-ECM3 grade III (E0G3) patients independently of tumor intrinsic subtype12,13,14,15, implicating unique crosstalk between undifferentiated tumor cells and the surrounding ECM3. Thus, a thorough biochemical analysis of proteins encoded by ECM3 genes and present in the tumor surrounding matrix represents a compulsory step to identify new promising targets to halt progression of aggressive grade III BCs.
Within the extensive extracellular network, C-type lectin domain family 3 member A (CLEC3A) has been reported to be a component of a filamentous extracellular system in BCs and be involved in tumor cell adhesion and invasion16,17,18. CLEC3A was first described as a cartilage-derived member of the C-type lectin superfamily19. It has heparin-binding activity, and its mRNA has been detected in human colon and BC cell lines, normal breast, and BC tissues16, but its function in cancer has not been examined extensively. On its overexpression in human cancer cell lines, CLEC3A enhances cell adhesion to integrin ligands on the ECM by binding to heparan sulfate proteoglycans on the cell surface16. High mRNA expression of CLEC3A is associated with poor prognosis in BCs18.
In this study, through a shotgun, label-free, proteomic analysis of decellularized matrices (dECMs) in human primary BCs, we observed an enrichment of cytoskeleton-related proteins among dECM-unique proteins in grade III BCs with the ECM3 gene expression pattern. These BCs harbored a peculiar cytoskeletal organization that correlated significantly with tumor stiffness. CLEC3A was among the matrisome proteins20 that were exclusive to the dECM proteome of E3G3 BCs, and its expression was associated with worse disease-free survival. In vitro, BC cell lines into which CLEC3A was knocked-in showed significant remodeling of the cytoskeleton, stiffness changes and increased focal adhesion signaling, migration, and invasiveness, indicating that this molecule is crucial in supporting E3G3 BC aggressiveness that is driven by tumor cell-ECM interactions.
Results
Protein composition of dECMs in BC by ECM gene expression pattern and tumor grade
To characterize the composition of the ECM at the protein level, dECMs were obtained from 29 BCs (proteomic cohort, Table 1) consisting of 6 ECM3/grade I-II (E3G0), 9 ECM3/grade III (E3G3), 5 non-ECM3/grade I-II (E0G0), and 9 non-ECM3/grade III (E0G3) BCs. Soluble extracts from the dECMs were analyzed using a shotgun, label-free, proteomic approach to characterize the corresponding proteomes, identifying 1158 proteins in E0G0, 2060 in E0G3, 1577 in E3G0, and 1626 in E3G3 (Fig. 1A, workflow in Supplementary Fig. 1).

A Venn diagram of proteins in dECMs of BCs (proteomic cohort) by ECM type and BC grade. Total numbers of proteins in each subgroup are also shown. B Heatmap of GO enrichment analysis by DAVID and Panther for differentially expressed proteins in E3G3 versus E3G0, E3G3 versus E0G3, and E0G3 versus E0G0. Only significant (p < 0.05) pathways are shown. Fold-enrichment is color-coded. C Principal component analysis for the total proteome of dECMs.
Proteins that were upregulated in or exclusive to E3G3 versus E3G0 samples (Supplementary Data 1) were significantly enriched in ribosomes, phagosome, focal adhesion, ECM-receptor interactions, and integrin pathways (Fig. 1B and Supplementary Data 2). These pathways do not overlap with those found comparing E0G3 and E0G0 samples (Fig. 1B, Supplementary Data 1 and Supplementary Data 2), supporting that molecules that are involved in ECM-tumor interactions are not a feature that characterizes all G3 tumors, but depend on E3 pattern. Compared with E0G3 samples (Supplementary Data 1), the ribosome, phagosome, and focal adhesion categories were enriched in E3G3 tumors, as in E3G3 versus E3G0 (Fig. 1B and Supplementary Data 2). The focal adhesion and ECM-receptor interaction pathways also appeared at the transcriptional level in E3G3 tumors versus the other groups as a whole (Supplementary Fig. 2), suggesting greater interaction between tumor cells and the ECM in these tumors.
Fatty acid metabolism pathways (e.g., propanoate metabolism, fatty acid elongation, unsaturated fatty acid biosynthesis, and fatty acid metabolism) decreased significantly in E3G3 compared with E3G0 tumors or were expressed exclusively in E3G0 tumors (Fig. 1B and Supplementary Data 2), whereas the TCA cycle pathway was upregulated. These characteristics were not specific to G3 tumors but depended on the ECM pattern, as none changed between E0G3 and E0G0 samples (Fig. 1B and Supplementary Data 2). In comparing E3G3 and E0G3 samples, lipid metabolism components declined in E3G3 samples or were expressed solely in E0G3 samples (Fig. 1B and Supplementary Data 2), arguing a sharp decrease in the synthesis of structural and functional lipids in E3G3 tumors. In addition, pathways that are associated with carbohydrate metabolism (e.g., pyruvate metabolism, pentose phosphate pathway, N-glycan biosynthesis, glyoxylate/dicarboxylate metabolism, and fructose/mannose metabolism) were negatively enriched only in E3G3 versus E0G3 samples, implicating the ECM in conditioning the glucose requirement. Molecules that are involved in antigen processing and presentation were overrepresented exclusively in E3G3 compared with E3G0 tumors (Fig. 1B).
Consistent with these analyses, E3G3 tumors behaved differently than other subtypes by principal component analysis (PCA) of the total dECM proteome (Fig. 1C). Among the 4 groups, we identified 51 proteins that were unique to E3G3 tumors (Fig. 1A). GO Term Mapper annotated 25% of these molecules to the cytoskeleton cellular component (Supplementary Data 3). In addition, manual classification of the 51 proteins, based on UniProt classification and the literature, classified 23 of the 51 proteins (45%) as cytoskeleton, cytoskeleton-associated, or cytoskeleton regulator proteins (Supplementary Data 3), suggesting peculiar cytoskeletal organization in E3G3 tumors as result of the tumor-ECM interaction. Notably, as observed in the entire cohort, PCA analysis of the total dECM proteome specifically in ER-positive tumors (n = 20 of the proteomic cohort) confirmed that E3G3 tumors clustered separately from other subtypes (Supplementary Fig. 3). This finding further supports the unique molecular profile of E3G3 tumors, independent of tumor-intrinsic features, consistent with our previous observations at the mRNA level13.
Morphological and biomechanical characterization of BC samples by ECM molecular signature and tumor grade
To corroborate the results of the proteomic analysis of dECMs, collagen, a contributor to the classification of ECM3 tumors, was examined by Masson’s trichrome staining in BC sections (stiffness cohort, n = 46, Table 1) selected from a consecutive BC cohort21 for which frozen samples were available (Fig. 2A). Significant differences between subgroups (p = 0.016) were observed, but the level of collagen did not characterize E3G3 tumors and did not differ in tumors with an ECM3 pattern, based on tumor grade. In contrast, the amount of dense collagen by Gomori trichrome staining was significantly higher in E3G3 versus E3G0 tumors (p = 0.044, Fig. 2B, C), suggesting aberrant collagen deposition in highly aggressive tumors.

A Box plot of pixel-based quantification of collagen in Masson’s thrichrome stains by ImageJ, normalized against total cell area, for BCs (stiffness cohort) by ECM and grade classifications. *p < 0.05, by Dunn’s test. B Box plot of pixel-based quantification of dense collagen in Gomori trichrome stains by ImageJ, normalized against total collagen (dense + interstitial), for BCs by ECM and grade classifications. # p = 0.0852 by Dunn’s test. C Representative images of Gomori trichrome staining in BCs by ECM and grade classifications. Images were obtained on an optical microscope at 10 × magnification; scale bar, 200 µm. Interstitial collagen is stained in light blue, and dense collagen is in purple, as clarified by the red and yellow arrows, respectively, in the E3G0 image.
By immunofluorescence, α-tubulin, a marker of the cytoskeleton that senses cell-microenvironment interactions22,23,24, was analyzed in 9 samples from each group, selected from the stiffness cohort. Due to the low frequency of E3G3, additional samples were also chosen from the IHC cohort to ensure adequate representation of this subset. All E3G3 tumors showed a fibrillary network that encompassed the entire tumor cytoplasm, whereas the other subgroups showed focal distribution that was restricted primarily to the external edges of the cytoplasm (Fig. 3A). The number of α-tubulin-positive pixels varied between subgroups, with significantly higher α-tubulin levels observed in E3G3 tumors compared to E3G0 tumors (p = 0.005; Fig. 3B).

A Representative images of α-tubulin staining (green) in E3G0, E3G3, E0G0, and E0G3 tumors. Nuclei are stained with DAPI (blue). Scale bar, 20 µm. B Box plot of α-tubulin-positive pixels, normalized against DAPI-positive pixels in BCs (n = 36, 9 for each group) by ECM and grade classifications. Two representative areas for each tumor sample were quantified. *p < 0.05, **p < 0.01 by Dunn’s test. C Histograms of elastic moduli (mean ± SEM) of BCs (stiffness cohort, n = 46) by ECM and grade classifications. p-values calculated by Kruskal-Wallis and Dunn’s test; *p < 0.05, **p < 0.01, ***p < 0.001.
Supporting the existence of a peculiar cooperation between tumor cells and the ECM in E3G3 tumors, these tumors showed the highest stiffness, based on high-strain elastic modulus (E-high) among the 46 BC samples of the stiffness cohort (Fig. 3C). Among BCs with the ECM3 gene pattern, E-high values and dense collagen levels correlated significantly (r = 0.63, p = 0.021), wherein E3G3 samples had high dense collagen content and E-high values (Supplementary Fig. 4). Among tumors that lacked the ECM3 pattern, E-high values did not correlate with dense collagen levels suggesting a distinct interplay between these BCs and ECM components, as indicated in our proteomic analysis.
Analysis of the prognostic value of CLEC3A, a unique protein to E3G3 dECMs
To investigate whether the aggressiveness of E3G3 BCs is linked to their interaction with ECM environment, we focused on CLEC3A, a matrix protein that is expressed exclusively in the E3G3 dECM proteome (Supplementary Data 3) and is reported to have a role in cell-ECM interaction16. Immunohistochemical analysis of CLEC3A in 55 BC tissues (IHC cohort, Table 1), by ECM pattern and grade, confirmed its significantly higher expression in E3G3 tumors (Fig. 4A). Whereas most non-E3G3 tumors were CLEC3A-negative (12/12 E0G0, 18/18 E0G3, 12/13 E3G0), 50% of E3G3 tumors showed strong CLEC3A immunoreactivity (6/12), predominantly at the tumor nodule edge (Fig. 4A). CLEC3A-positive BCs showed the worst prognosis of the entire cohort (p = 0.018, Fig. 4B), and even within E3G3 BCs of the same cohort, CLEC3A-positive cases trended toward worse disease-free survival (p = 0.073, Fig. 4C). The higher expression of CLEC3A in E3G3 tumors by IHC was confirmed in an independent cohort of G3 BCs (IHC validation cohort15, n = 45, Table 1) (p = 0.030) (Fig. 4D).

A Representative images of negative and positive CLEC3A stains. Scale bar, 50 µm. Right: CLEC3A expression in tumor tissue by ECM and grade classifications (IHC cohort: E0G0, n = 12; E0G3, n = 18; E3G0, n = 13; E3G3, n = 12). p-value by chi-square test. B, C Association between CLEC3A expression and disease-free survival in the entire cohort (n = 55, B) and in E3G3 patients (n = 12, C). p-values by log-rank test. D CLEC3A expression in G3 BC tissues by ECM classification in the IHC validation cohort (E0G3, n = 32; E3G3, n = 13). p-value by Fisher exact test.
Effect of CLEC3A expression in BC cells
Based on the low expression of CLEC3A in BC cell lines (CCLE dataset) in vitro (Supplementary Fig. 5A), the MDA-MB-361 HER2+ and MDA-MB-231 and MDA-MB-468 triple-negative (TN) BC cell lines, which express low levels of CLEC3A (Supplementary Fig. 5B), were transiently transfected with an expression vector that contained the CDS of human CLEC3A (Supplementary Fig. 6). As a result, CLEC3A localized predominantly to the ECM and dECM of transfected BC cells (Fig. 5A).

A Representative Western blot analysis of cell lysate (Lys), ECM, and dECM of MDA-MB-361, MDA-MB-231, and MDA-MB-468 cells seeded on collagen I, after transfection with pcDNA3 (Mock) or p3-CLEC3A plasmid (CLEC3A). Vinculin was used as control for cellular removal. B Representative stain of phalloidin (green) and tubulin (red) by immunofluorescence in cells grown on fibronectin. Nuclei are stained with DAPI (blue). Scale bar, 20 µm. C Representative WB analysis of mock and CLEC3A cells seeded on fibronectin. GAPDH was used as loading control.
To explore the function of CLEC3A in tumor cell interactions with the ECM, BC cells were transfected with CLEC3A or empty vector, seeded on fibronectin, and analyzed with regard to cytoskeletal organization and intracellular signaling. CLEC3A-overexpressing cells had a different cytoskeletal organization, with higher actin stress fibers, than mock cells (Fig. 5B). Intracellularly, pFak, RhoA, and Rac1 were upregulated in CLEC3A-transfected versus control cells, as evaluated by Western blot analysis (Fig. 5C). Moreover, YAP activation increased, based on the lower phosphorylation and/or the rise in YAP/TAZ expression (Fig. 5C), supporting YAP activity as a mechanosensor in CLEC3A-expressing compared with control cells.
Similar results were observed in cells that were seeded on collagen I (Supplementary Fig. 7). Consistently, the YAP/TAZ signature, which reflects the activation of the mechanosignaling cascade25,26, was higher in CLEC3A-positive ECM3 BCs and correlated with tumor stiffness (Supplementary Fig. 8). In addition, immunofluorescence analysis of CLEC3A and cortactin, an actin-binding and cytoskeletal protein used as a lamellipodia marker27, showed CLEC3A mainly localized at the perinuclear area and at lamellipodial protrusions in CLEC3A-expressing MDA-MB-231 cells (Supplementary Fig. 9A) and these cells exhibited an enhanced stiffness in the lamellipodial regions versus nuclei compared with mock cells, as measured by atomic force microscope (AFM, Supplementary Fig. 9B), sustaining a CLEC3A mechanical support for cell migration.
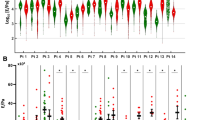
To go through CLEC3A-dependent modifications and BC aggressiveness, we analyzed the adhesion, proliferation, and migration/invasion of BC cells, based on CLEC3A expression. On overexpression of CLEC3A, BC cell lines exhibited enhanced adhesion to fibronectin (Fig. 6A), as expected17, comparable or reduced proliferation (observed only in MDA-MB-231 cells) (Fig. 6B) and increased migration in the wound healing assay (Fig. 6C) compared to control cells. Moreover, in MDA-MB-361 cells, which can generate spheroids, matrigel spheroid invasion was significantly higher in CLEC3A-expressing versus control cells (Supplementary Fig. 10).

A Adhesion rate of mock and CLEC3A-expressing MDA-MB-361, MDA-MB-231, and MDA-MB-468 cells on fibronectin. Data are presented as ratio of 1 h to 0 h values (mean ± SD, n = 6 and representative of 2–3 independent experiments). B Growth rate of mock and CLEC3A BC cells on fibronectin. Data are presented as ratio of values at various time points to 0 h values (mean ± SD, n = 4 and representative of 2–3 independent experiments). C Scratch test of mock and CLEC3A BC cells. Representative images are shown. Data are presented as percentage of wound closure at 5 days in MDA-MB-361 cells and at 6 h in MDA-MB-231 and MDA-MB-468 cells (mean ± SD, n = 6 and representative of 2 independent experiments). Representative images are shown. *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired Student’s t-test.
Discussion
We previously identified a robust cluster of genes (ECM3) that is upregulated in approximately 40% of BCs, encoding ECM proteins and identifying truly aggressive tumors among faster-growing BCs that are more likely to spread (i.e., grade III BCs13), regardless of the intrinsic BC subtype. In the current study, we characterized these BCs in depth—i.e., E3G3—focusing on their ECM composition and biomechanical properties.
Our proteomic analysis of dECMs identified proteins that are involved in ECM-receptor interactions, integrin signaling, and focal adhesion as being the most significant pathways enriched in E3G3 tumors. Thus, in addition to harboring primarily biochemical cues from the original tumor ECM, the dECMs of E3G3 BCs constitute a signature of the behavior of tumor cells that participate in tumor-ECM crosstalk. The highly significant enrichment of the ribosome pathway in these tumors is further evidence of a specific interaction between E3G3 BCs and the surrounding matrix that leads to a peculiar cytoskeleton organization in these tumors. Indeed, the cytoskeleton participates in the regulation of translation28 and high numbers of ribosomes are present at sites where local translation is required, such as focal adhesions in spreading cells29.
Focusing on collagen as the primary component of the ECM30 and consistent with the reproducible set of overexpressed collagen genes that define ECM3 tumors13, a high collagen content was observed in ECM3 tumors. Notably, only E3G3 tumors exhibited high dense collagen deposition in the ECM, which reportedly stiffens the ECM31. Accordingly, E3G3 samples had the highest rigidity among BC subtypes, significantly correlating with dense collagen levels and displaying a distinct α-tubulin organization, corroborating a peculiar dialogue between tumor cells and the ECM. The enrichment of cytoskeleton, cytoskeleton-associated, and cytoskeleton-regulating proteins among the unique proteins to E3G3 dECMs is further evidence of the interaction of tumor cells with a high stiffed ECM. Thus, it is conceivable that the external ECM3 mechanical forces that are sensed by G3 BC cells through the formation of focal adhesions mediate the cytoskeletal reorganization contributing to high rigidity of E3G3 tumors, consistent with the model that the cytoskeletal organization of tumor cells is the prime consequence of tissue rigidity32.
The unique expression of CLEC3A in E3G3 BCs is consistent with its ability to promote adhesion to ECM molecules in its putative function as an activator of integrins16 likely promoting tumor aggressiveness. Accordingly, CLEC3A levels were highest in E3G3 human BCs by IHC in various BC cohorts and were significantly associated with a worse prognosis. In this analysis, CLEC3A localized to the tumor nodule edge, supporting its function in ECM-tumor communication. In this context, we can also speculate that CLEC3A may contribute to the deposition of dense collagen in E3G3 tumors by activating integrins16. Indeed, upon binding to ECM molecules, integrins exert mechanical forces that lead to ECM fiber alignment31. Notably, YAP activity was higher in CLEC3A-positive BCs and correlated positively with tumor stiffness. The YAP and TAZ transcriptional activators are central mediators of mechanical forces from cell attachment to the surrounding ECM, shaping tumor cells and cytoskeletal organization33. Thus, we speculate that CLEC3A-mediated tumor-stroma interactions support YAP/TAZ activation with consequent sensing of ECM stiffness and induction of cancer aggressiveness. In addition, because YAP/TAZ activation in cancer cells, in turn, impacts the composition and physical properties of the ECM by regulating the secretion of ECM components and the recruitment of protumorigenic cells (e.g., cancer associated fibroblasts and immune cells), CLEC3A likely supports a vicious cycle of ECM signals and the production of matrix molecules, explaining why E3G3 BCs highly depend on the ECM to progress.
The results in human BC tissue samples were recapitulated in BC cell lines, wherein overexpression of CLEC3A increased their migration, invasion, and expression of intracellular adhesion mediators. Notably, ectopic CLEC3A reorganized the actin cytoskeleton and increased YAP activation, acting as a matrix mechanosensor of ECM rigidity. Interestingly, CLEC3A was found to localize at the nuclear area and cell boundaries contributing to cell movement and stiffness. Because the rigidity of fan-shaped lamellipodia protrusions is linked to the branched actin network, which, in turn, gives a relevant mechanical support for cell migration, it can be thought that CLEC3A contributes to the mechanical adaptability for cell migration.
Although matrisome proteins, as expected, were the most abundant in MS analysis, the sensitivity of this approach also allowed to detect cellular proteins in very low abundance that are peculiar of ECM-interacting cells. In this context, positive (TCA cycle) and negative (lipid and carbohydrate metabolism) enrichment of molecules in metabolic pathways emerged supporting the metabolic rewiring of these tumors. These proteomic data agree with our previous results on the whole proteome of BCs by ECM and grade34, which, in addition to ECM remodeling and signal mechanotransduction, demonstrated metabolic rewiring as a specific characteristic of E3G3 tumors.
Antigen-processing and presentation molecules were upregulated in E3G3 dECMs, likely reflecting greater antigen presentation on MHC-I in these tumors, which may stimulate an antitumor response.
These results may be explained by recent studies showing that the squeezing of tumor cells through a highly stiffened matrix, as seen in E3G3 tumors, deforms the nuclei. This deformation causes the migration of mobile nuclear factors away from DNA35, leading to impaired repair of DNA damage and increased expression of neoantigens36. However, recent molecular characterizations of E3G3 BCs have excluded the presence of immune-infiltrating cells in the TME13,15. This raises the possibility that, despite the potential activation of immune surveillance in these BCs, the tight tumor-ECM interaction prevents immune cell infiltration and the consequent stimulation of an immune response37.
A limitation of this study is the relatively small number of analyzed cases and the absence of a validation cohort in the proteomic analysis. However, this analysis was instrumental in identifying CLEC3A, which has been validated by IHC in other cohorts and preclinical models. Collectively, these results provide novel evidence that CLEC3A triggers a distinct tumor-ECM interaction in BCs. The concurrent action of CLEC3A at the edge of the tumor in response to ECM biomechanical signals constitutes the basis of the poor prognosis in high-grade tumors that spread aggressively. Further studies are needed to determine whether CLEC3A can be used as a surrogate marker of aggressive grade III tumors, regardless of their intrinsic subtype and as potential therapeutic targets. Indeed, CLEC3A itself may serve as a potential target to destabilize ECM-mediated signals and potentially influencing tumor metabolism and immunogenicity. Moreover, uncovering the molecular mechanism behind CLEC3A-driven tumor progression could reveal novel therapeutic targets, some of which may already have existing active drugs.
Methods
Human breast carcinoma samples
Human primary BC specimens were collected by our institutional biobank from women who underwent surgical treatment between 2004 to 2007 at Fondazione IRCCS Istituto Nazionale dei Tumori of Milan, Italy. To facilitate the mass spectrometry (MS) analyses, the proteomic cohort (n = 29) was specifically selected from cases for which a gene expression profile was available, to obtain a similar number of cases in each tumor subgroup, based on ECM and grade, and to balance the groups with regard to intrinsic BC subtype. The stiffness cohort (n = 46) included consecutive specimens of the ITA cohort21 for which frozen tissue was still available. The IHC cohort (n = 55) comprised BC cases from the proteomic and stiffness cohorts for which FFPE sections were available.
The IHC validation cohort consisted of 45 grade III primary BCs from women who underwent surgery at INT between 2008 and 2017, as described15. The clinicopathological characteristics of the BC specimens are summarized in Table 1. Institutional approval for this study (INT 144-14) was obtained from the Independent Ethics Committee (Comitato Etico Indipendente, Fondazione IRCCS, Istituto Nazionale dei Tumori). Patients provided informed consent for future use of their samples for research purposes. All data were analyzed anonymously, and all procedures were performed in accordance with the Declaration of Helsinki.
Bioinformatic analyses
To characterize BCs by ECM3 features, the gene expression profiles of BC tumors, previously examined on the Illumina (GSE5959521, GSE5534838) or Affymetrix platform (GSE8694539, GSE14747115), were analyzed by large average submatrix (LAS) bi-clustering method40, based on the expression levels of 738 ECM genes, as previously described13.
Gene-set enrichment analyses (GSEA) were performed using the GSEA v5.041 and c2.cp.kegg.v6.0 geneset. The YAP/TAZ activity signature, composed of validated direct targets of YAP/TAZ/TEAD25, was calculated as the standardized average expression of the genes in the signature, as described26. CLEC3A expression was analyzed in the BC cell lines of the Cancer Cell Line Encyclopedia (CCLE) dataset42.
BC sample decellularization and proteomic analysis
Twenty-micrometer slices of frozen tumor samples from the proteomic cohort (12 slices for each sample) were treated with hypotonic Tris-HCl solution, pH 8, for 72 h at 4 °C, followed by 24-h treatment with 4% sodium deoxycholate (SD) solution, prepared using distilled water and containing protease inhibitors (complete™ Mini, Protease inhibitor cocktail tablets, Roche), at room temperature to preserve protein content. This procedure allowed cell clearance as demonstrated by Hematoxylin and eosin (H&E) staining (Supplementary Fig. 11) and no glyceraldehyde 3-phosphate dehydrogenase (GAPDH) amplification by qPCR in RNA that was extracted from dECMs (data not shown). The 29 dECM samples were analyzed by mass spectrometry. The protein precipitate was resuspended in RapiGest (RG, Waters Corporation), reduced, derivatized, and digested with trypsin (protein:protease ratio of 20:1), as described43, before the MS/MS analysis. Nano-high-performance liquid chromatography (HPLC), coupled with MS/MS, was performed on a Dionex UltiMate 3000, which was connected directly to an Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific) using a nanoelectrospray ion source.
Peptide mixtures were enriched on a 75 μm ID × 150 mm Acclaim PepMap RSLC C18 column and separated on an LC gradient—4% acetonitrile (ACN) in 0.1% formic acid for 3 min, 4%–28% ACN in 0.1% formic acid for 130 min, 28%–40% ACN in 0.1% formic acid for 20 min, 40%–95% ACN in 0.1% formic for 2 min, and 95%–4% ACN in 0.1% formic acid for 3 min—at a flow rate of 0.3 μl/min.
The MS spectra of the eluted peptides were collected over an m/z range of 375–1500 using a resolution setting of 120,000 in data-dependent mode, to automatically alternate between Orbitrap-MS and Orbitrap-MS/MS acquisition. Higher-energy collisional dissociation (HCD) MS/MS spectra were collected for the 20 most abundant ions in each MS scan, using a normalized collision energy of 30% and an isolation window of 1.7 m/z. Rejection of +1 and unassigned charge states were enabled44.
Raw, label-free MS/MS files from Thermo Xcalibur (version 4.1)45 were analyzed using MaxQuant (version 1.6.0.1), with 6 ppm for the monoisotopic precursor. The initial maximum-allowed mass deviation was set to 6 ppm for monoisotopic precursor ions and 0.5 Da for MS/MS peaks. The enzyme specificity was set to trypsin, and a maximum of 2 missed cleavages was allowed. Carbamidomethyl cysteine was set as a fixed modification, and N-terminal acetylation, methionine oxidation, and asparagine/glutamine deamidation were set as variable modifications. The spectra were searched using the Andromeda search engine against the Homo sapiens Uniprot sequence database (release 27.06.2019). The reversed sequences of the target database were used as a decoy database. Protein identification required at least 1 unique or razor peptide per protein group. The quantification in MaxQuant was performed using the built-in XIC-based, label-free, quantification (LFQ) algorithm, using fast LFQ46. The required false-positive rate was set to a false-discovery rate (FDR) of 1% at the peptide level and 1% at the protein level, against a concatenated target decoy database, and the minimum required peptide length was set to 7 amino acids. Statistical analyses were performed using Perseus (version 1.5.5.3, www.biochem.mpg.de/mann/tools/). Only proteins that were present and quantified in at least 70% of repeats were considered to be positively identified in the four-group comparison, while at least 84% in the pairwise comparisons.
Bioinformatic analyses were performed in Panther (version 10.0) and DAVID (release 6.7) to cluster enriched annotation groups that were associated with gene ontology (GO) terms within a set of identified proteins. Homo sapiens genes were used as reference list in the enrichment analyses. The functional grouping was based on a p-value ≤ 0.05 and at least 3 counts.
The proteomic data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (project accession PXD019037).
Histochemistry
4-µm formalin-fixed, paraffin-embedded (FFPE) slices of BC samples of the stiffness cohort were stained using the Artisan Masson Trichrome Staining kit (Agilent) and Artisan Gomori’s Green Trichrome Stain kit (Agilent) according to the manufacturer’s instructions, on an automated Artisan Link Pro Instrument (Agilent). Images of the stained slices were then acquired on an Aperio ScanscopeXT® (Leica Biosystems Aperio) at 400x magnification for quantification.
Masson’s trichrome images were analyzed in ImageJ using color deconvolution, quantifying collagen as blue pixels and cells as red pixels. The entire dataset of the Gomori trichrome images was preprocessed to attenuate stochastic noise (which is unavoidable, due to the large size of the images) using the edge-preserving filter, as described47. To measure the percentages of pixels in each of the 3 classes (in false colors: denser collagen bundles in brown, interstitial collagen in green, and nuclei in red, excluding the acellular white areas), 25% of all available microphotographs were randomly chosen, to reduce decision bias, and quantified. Small round areas (diameter of 5 pixels) that were distributed uniformly throughout each image were labeled manually as belonging to any class. Subsequently, an equal number of areas per class was selected at random; the median values in the RBG color space of these zones were used to realize 3 palettes for the respective classes. The remaining 75% of the dataset was subsequently spanned, pixel by pixel, to compute the minimum Euclidean distance between the RBG triplet of the current pixel and all palette entries, assigning the pixel to its proper class. This approach avoids the definition of user-defined input parameters, and no postprocessing algorithm was used to further refine the segmentation.
Immunofluorescence
α-tubulin was analyzed in 4-μm FFPE slices using mouse monoclonal anti-human α-tubulin antibody (clone DM1A, 1:1000, Sigma-Aldrich) followed by anti-mouse AlexaFluor 488 secondary antibody (1:1000, Thermo-Fisher Scientific). Nuclei were stained using 4′,6-diamidino-2-phenylindole (DAPI) Prolong.
BC cell lines that had been grown for 24 h on coverslips that were coated with fibronectin were stained with α-tubulin antibody (Sigma-Aldrich), with anti-mouse AlexaFluor 555 (1:1000, Thermo-Fisher Scientific) as secondary antibody or with mouse anti-human CLEC3A antibody (1:50, Sigma-Aldrich) and rabbit anti-human cortactin (1:100, Abcam) with anti-mouse AlexaFluor 633 and anti-rabbit AlexaFluor 488 (1:1000, Thermo-Fisher Scientific) as secondary antibodies. Actin was stained with AlexaFluor 488 or Alexafluor 546 Phalloidin (1:1000, Thermo-Fisher Scientific) and nuclei using DAPI. Coverslips were mounted on glass slides using Prolong (Calbiochem).
Images were acquired on a Leica TCS SP8 X confocal laser scanning microscope (Leica Microsystems GmbH), as described48. DAPI was excited using the diode laser and detected from 410 to 458 nm; AlexaFluor 488 was excited using a 499-nm laser line and detected from 501 to 558 nm in BC specimens and from 509 to 543 in BC cell lines; AlexaFluor 555 and falloidin 546 were excited using a 554-nm laser line and detected from 564 to 643 nm and 564 to 616 nm, respectively; and AlexaFluor 633 was excited using a 629-nm laser line and detected from 639 to 738 nm. Images were acquired in scan format, at 512 × 512 pixels, in a Z-stack series (step size, 0.5 µm), using an HC PL APO 63X/1.40 CS2 oil-immersion objective and a pinhole set to 1 Airy unit. The data were analyzed in Leica LAS AF rel. 3.3 (Leica Microsystems GmbH). BC specimens were analyzed on images with an extended depth of field, consisting of 14 central frames in a stack (0.5-µm steps); a focus region was selected for maximum intensity.
Immunohistochemistry
CLEC3A was analyzed in FFPE specimens using a rabbit polyclonal anti-human CLEC3A antibody (1:5000, catalog number HPA051511, Sigma-Aldrich), after antigen retrieval in 5 mM citrate buffer, pH 6.0 for 4 min at 125 °C. Immunoreactions were enhanced by streptavidin-biotin-peroxidase, followed by counterstaining with Carazzi hematoxylin. CLEC3A positivity was evaluated independently by 2 of the authors, both of whom were blinded to the clinicopathological data. Discrepancies between observers were reviewed jointly to reach a consensus. Tumor cell membrane staining of any intensity was considered positive for CLEC3A expression.
Mechanical measurements
Frozen tumor samples of the stiffness cohort (n = 46) were thawed, and each was cut into a rectangular, slab-shaped piece, to obtain homogeneous and isotropic tissue samples. The specimens were placed in a Petri dish and subjected to 3 cycles of uniaxial compression on a Zwick/Roell material-testing machine (mod. Z005, Zwick), using a load cell of 10 N and a strain rate of 0.01 s−1. To prevent disruption of the specimens, the samples were stretched until 30% deformation. Elastic modules (E-high) were determined from the stress-strain curves. The stress-strain curves presented with nonlinear behaviors, with 2 linear zones (for small and large deformations) and a transition zone (toe region), near the 15% deformation point. For each sample, 2 elastic moduli were calculated: 1 for strain values less than 10% (E-low) and 1 for values higher than 25% (E-high). E values were determined according to the slopes of the linear regions of the stress-strain curve. E-low values, which correspond to the initial compression phase when fluids begin to escape tumors, were similar in all tumor tissues, whereas when fluids have been removed from the tumor, the differences in tumor tissue composition and organization determine the mechanical behaviors, classifying tumor tissue based on the resulting E-high values.
The Young’s modulus of elasticity of cells was measured by means of nanoindentation experiments performed using a Bioscope Catalyst atomic force microscope (AFM, Bruker) mounted on top of an inverted optical microscope (Olympus X71) and equipped with custom spherical tips49. The optical microscope was placed on an active antivibration base (DVIA-T45, Daeil Systems) and kept in an acoustic box (Schaefer, Italy) to isolate the system from outside noise. In the cell measurements, custom colloidal probes with spherical tips made of borosilicate glass beads mounted on tipless cantilevers (MikroMasch HQ:CSC38/Tipless/No Al) were used49. Reverse AFM imaging was used to calibrate the tip radius (r = 5460 nm), as described49. The cantilever spring constant (approx. 0.055 N/m) and deflection sensitivity (approx. 37 nm/V) were calibrated using the thermal noise and the SNAP methods, respectively, following procedures and guidelines detailed in refs. 49,50,51. The Hertz model (assuming a Poisson’s coefficient of 0.5) was fit to the force vs indentation curves (shortly force curves, FCs) to extract the Young’s modulus (YM), which characterizes cell elasticity51,52. In order to assess the YM of nuclear region and lamellopodia of cells, many cells were independently examined from 7–8 Petri plates. A minimum of 15 FCs on the surrounding substrate and 100 FCs on the selected region of interest (ROI) on the cell were typically acquired to obtain both the topographic and the YM maps of each cell. The local height of each individual cell was used to apply the bottom effect correction to the FCs, to account for the influence of the stiff substrate on the measured YM52. For every tested condition, the mean median value Ēmed of the YM values measured in different locations was calculated, with its corresponding standard deviation of the mean51.
Breast cancer cell lines and cell treatments
BC cells were purchased from ATCC (Rockville) and authenticated by short tandem repeat profiling method in 2021. BT474, MDA-MB-361, and MDA-MB-468 cells were maintained in DMEM medium, and MCF7, T47D, ZR75.1, HCC1954, and MDA-MB-231 cells were maintained in RPMI medium, supplemented with 10% (v/v) FBS (Thermo Fisher Scientific) and L-glutamine, in a 5% humidified CO2 chamber at 37 °C. The cells were transiently transfected with pcDNA3 (Promega) containing or not human CLEC3A CDS using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions; 24 h later, the cells were seeded on plastic that was coated with fibronectin (1 µg/cm2, F2006, Sigma-Aldrich) or collagen I (1 µg/cm2, C7661, Sigma-Aldrich) and used for further experiments. The p3-CLEC3A plasmid was generated by amplifying (forward 5’-TAAAGCTTATGGCAAAGAATGGACTTGTAATTTGCATCCTGGTG-3’; reverse 5’-TTGAATTCCTATTGAGGGATGGTGAACTCGCATATGTATCTC-3’) human CLEC3A CDS (NM_005752.6) from the cDNA of BT474 cells and cloning the PCR fragment into the HindIII and EcoRI sites of pCDNA3 (Promega). The resulting plasmid was sequenced to verify the correct sequence.
Relative cell growth and adhesion on fibronectin were determined by sulforhodamine B assay. Optical density was measured on an ELISA microplate reader (Bio-Rad Laboratories). The results were normalized to those of the control (seeding control at time 0). Cell migration was evaluated by scratch-wound migration test. Transfected cells were grown to confluence on human fibronectin-coated 24-well plates, and a scratch was made with a sterile pipette tip. The cells were photographed immediately after the scratch (0 h) and after 6–120 h. Self-assembled 3D tumor spheroids were obtained 2 days after seeding transfected BC cells (50,000 cells/well) on a solid layer of 1% agarose medium in a 24-well culture plate. Each spheroid was then positioned at the center of a well in a 24-well plate, embedded in Matrigel Matrix growth factor reduced, phenol red-free (356231, BD Biosciences) and monitored for spontaneous invasion into the matrix. Invasion was analyzed after 6 days. Matrigel-invading areas for each spheroid were selected manually and quantified in ImageJ.
RNA extraction and qRT-PCR analysis
RNA was extracted using QIAzol (Qiagen) according to the manufacturer’s instructions. The cDNA was reverse-transcribed using the High Capacity RNA-to-cDNA kit (Thermo Fisher Scientific). qRT-PCR was performed using TaqMan probe-based assays (CLEC3A: Hs.00194952_m1) and TaqMan Fast Universal PCR Master Mix on a StepOne PlusTM Real-time PCR system (Thermo Fisher Scientific). The relative abundance of transcripts was calculated by comparative Ct method using GAPDH (Hs.02758991_g1) as a reference gene.
Western blot analysis
Protein fractions were extracted from BC cells that were grown on fibronectin or collagen I for 24 h with TNTG lysis buffer, as described53. To evaluate the localization of CLEC3A, cells were seeded on collagen I and transfected, and 48 h later, the protein fraction was extracted with TNTG buffer. ECM and dECM were obtained by scraping the ECM on the plastic in boiled electrophoresis sample buffer after TNTG cell lysis or after treating the cell monolayer with 20 mM ammonium hydroxide (NH4OH) solution, respectively. The following primary antibodies were used: CLEC3A mouse mAb (SAB1411718, Sigma-Aldrich); pFAK mouse mAb (pY397, clone 14/FAK, catalog number 611722, BD Biosciences), FAK rabbit pAb (A-17, catalog number sc-557, Santa Cruz Biotecnology), RhoA rabbit mAb (clone 67B9, catalog number 2177, Cell Signaling), Rac1 rabbit mAb (clone 23A8, catalog number 05-389, Millipore), pYAP rabbit mAb (Ser127, clone D9W21, catalog number 13008, Cell Signaling), YAP rabbit mAb (clone D8H1X, catalog number 14074, Cell Signaling), TAZ rabbit pAb (HPA007415, Sigma-Aldrich), p53 mouse mAb (DO-1, catalog number sc-126, Santa Cruz Biotecnology), GAPDH mouse mAb (clone GAPDH-71.1, catalog number G8796, Sigma-Aldrich), and vinculin mouse mAb (clone hVIN-1, Sigma-Aldrich). Polyclonal anti-rabbit (NA9340V) or anti-mouse (NA931V) IgG horseradish peroxidase (HRP) (Amersham GE Healthcare) was used as a secondary antibody.
Statistical analysis
Differences among three or more groups were assessed using Kruskal-Wallis test, and then pairwise differences were evaluated by the post-hoc uncorrected Dunn’s test. Results of in vitro experiments were compared using two-tailed Student’s t-tests. Associations between categorical variables were analyzed by Fisher’s exact test or chi-square test and correlations between continuous variables were examined by Spearman correlation analysis. Survival curves were estimated using the Kaplan-Meier method, and the log-rank test was used for comparison. A p-value of less than 0.05 was considered statistically significant. All statistical analyses were conducted using GraphPad Prism version 5.01 (GraphPad).
Data availability
All data that were needed to evaluate the conclusions in this paper are included in the paper or in the Supplementary Materials. The proteomic data generated in this study have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (Project accession PXD019037). Gene expression profiles of the human BCs that were analyzed in this paper were previously deposited to the Gene Expression Omnibus database (Home - GEO - NCBI (nih.gov)), series GSE59595, GSE55348, GSE86945, and GSE147471.
Code availability
The data analysis was conducted without the use of any custom code.
References
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022).
Prat, A. & Perou, C. M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 5, 5–23 (2011).
Sims, A. H., Howell, A., Howell, S. J. & Clarke, R. B. Origins of breast cancer subtypes and therapeutic implications. Nat. Clin. Pr. Oncol. 4, 516–525 (2007).
Pupa, S. M., Ménard, S., Forti, S. & Tagliabue, E. New insights into the role of extracellular matrix during tumor onset and progression. J. Cell Physiol. 192, 259–267 (2002).
Pickup, M. W., Mouw, J. K. & Weaver, V. M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 15, 1243–1253 (2014).
Ungefroren, H., Sebens, S., Seidl, D., Lehnert, H. & Hass, R. Interaction of tumor cells with the microenvironment. Cell Commun. Signal 9, 9–18 (2011).
Bianchi, F. et al. Mechanical cues, E-cadherin expression and cell 'sociality' are crucial crossroads in determining pancreatic ductal adenocarcinoma cells behavior. Cells 11, 1318 (2022).
Frantz, C., Stewart, K. M. & Weaver, V. M. The extracellular matrix at a glance. J. Cell Sci. 123, 4195–4200 (2010).
West, R. B. et al. Determination of stromal signatures in breast carcinoma. PLoS Biol. 3, 1101–1110 (2005).
Farmer, P. et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med. 15, 68–74 (2009).
Giussani, M., Merlino, G., Cappelletti, V., Tagliabue, E. & Daidone, M. G. Tumor-extracellular matrix interactions: identification of tools associated with breast cancer progression. Semin Cancer Biol. 35, 3–10 (2015).
Bergamaschi, A. et al. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. J. Pathol. 214, 357–367 (2008).
Triulzi, T. et al. Neoplastic and stromal cells contribute to an extracellular matrix gene expression profile defining a breast cancer subtype likely to progress. PLoS ONE 8, e56761 (2013).
Tiwari, S. et al. Infrared spectroscopic imaging visualizes a prognostic extracellular matrix-related signature in breast cancer. Sci. Rep. 10, 5442 (2020).
Lecchi, M. et al. A combination of extracellular matrix- and interferon-associated signatures identifies high-grade breast cancers with poor prognosis. Mol. Oncol. 15, 1345–1357 (2021).
Tsunezumi, J., Higashi, S. & Miyazaki, K. Matrilysin (MMP-7) cleaves C-type lectin domain family 3 member A (CLEC3A) on tumor cell surface and modulates its cell adhesion activity. J. Cell Biochem. 106, 693–702 (2009).
Lau, D. et al. The cartilage-specific lectin C-type lectin domain family 3 member A (CLEC3A) enhances tissue plasminogen activator-mediated plasminogen activation. J. Biol. Chem. 293, 203–214 (2018).
Ni, J. et al. Overexpression of CLEC3A promotes tumor progression and poor prognosis in breast invasive ductal cancer. Onco Targets Ther. 11, 3303–3312 (2018).
Neame, P. J., Young, C. N. & Treep, J. T. Primary structure of a protein isolated from reef shark (Carcharhinus springeri) cartilage that is similar to the mammalian C-type lectin homolog, tetranectin. Protein Sci. 1, 161–168 (1992).
Naba, A. et al. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteomics 11, 1–18 (2012).
Huang, X. et al. Molecular portrait of breast cancer in China reveals comprehensive transcriptomic likeness to Caucasian breast cancer and low prevalence of luminal A subtype. Cancer Med. 4, 1016–1030 (2015).
Putnam, A. J., Schultz, K. & Mooney, D. J. Control of microtubule assembly by extracellular matrix and externally applied strain. Am. J. Physiol. Cell Physiol. 280, C556–C564 (2001).
Parker, A. L., Kavallaris, M. & McCarroll, J. A. Microtubules and their role in cellular stress in cancer. Front. Oncol. 4, 153 (2014).
Seetharaman, S. & Etienne-Manneville, S. Microtubules at focal adhesions—a double-edged sword. J. Cell Sci. 132, jcs232843 (2019).
Zanconato, F. et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 17, 1218–1227 (2015).
Ingallina, E. et al. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat. Cell Biol. 20, 28–35 (2018).
Jeannot, P., Besson, A. & Jeannot, G. P. Cortactin function in invadopodia. Small GTPases 11, 256–270 (2017).
Kim, S. & Coulombe, P. A. Emerging role for the cytoskeleton as an organizer and regulator of translation. Nat. Rev. Mol. Cell Biol. 11, 75–81 (2010).
Nilsson, J., Sengupta, J., Frank, J. & Nissen, P. Regulation of eukaryotic translation by the RACK1 protein: a platform for signalling molecules on the ribosome. EMBO Rep. 5, 1137–1141 (2004).
Bonnans, C., Chou, J. & Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 15, 786–801 (2014).
Winkler, J., Abisoye-Ogunniyan, A., Metcalf, K. J. & Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 11, 5120 (2020).
Yeung, T. et al. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil. Cytoskeleton 60, 24–34 (2005).
Zanconato, F., Cordenonsi, M. & Piccolo, S. YAP and TAZ: a signalling hub of the tumour microenvironment. Nat. Rev. Cancer 19, 454–464 (2019).
Moriggi, M. et al. ECM remodeling in breast cancer with different grade: contribution of 2D-DIGE proteomics. Proteomics 18, e1800278 (2018).
Irianto, J. et al. Nuclear constriction segregates mobile nuclear proteins away from chromatin. Mol. Biol. Cell 27, 4011–4020 (2016).
Pfeifer, C. R., Alvey, C. M., Irianto, J. & Discher, D. E. Genome variation across cancers scales with tissue stiffness—an invasion-mutation mechanism and implications for immune cell infiltration. Curr. Opin. Syst. Biol. 2, 103–114 (2017).
Yu, K. X., Yuan, W. J., Wang, H. Z. & Li, Y. X. Extracellular matrix stiffness and tumor-associated macrophage polarization: new fields affecting immune exclusion. Cancer Immunol. Immunother. 73, 115 (2024).
Triulzi, T. et al. Whole-transcriptome analysis links trastuzumab sensitivity of breast tumors to both HER2 dependence and immune cell infiltration. Oncotarget 6, 28173–20182 (2015).
Romero-Cordoba, S. et al. Decoding immune heterogeneity of triple negative breast cancer and its association with systemic inflammation. Cancers 11, 911 (2019).
Shabalin, A. A. et al. Finding large average submatricies in high dimensional data. Ann. Appl. Stat. 3, 985–1012 (2009).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Ghandi, M. et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019).
Mortarino, M. et al. Two-dimensional polyacrylamide gel electrophoresis map of bull seminal plasma proteins. Electrophoresis 19, 797–801 (1998).
Nicastro, R. et al. Snf1 phosphorylates adenylate cyclase and negatively regulates protein kinase A-dependent Transcription in Saccharomyces cerevisiae. J. Biol. Chem. 290, 24715–24726 (2015).
Schulte, C. et al. Scale invariant disordered nanotopography promotes hippocampal neuron development and maturation with involvement of mechanotransductive pathways. Front. Cell Neurosci. 10, 267 (2016).
Tamplenizza, M. et al. Nitric oxide synthase mediates PC12 differentiation induced by the surface topography of nanostructured TiO2. J. Nanobiotechnol. 11, 35 (2013).
Guastella, D. & Valenti, C. Cartoon filter via adaptive abstraction. J. Vis. Commun. Image Represent 36, 149–158 (2016).
Triulzi, T. et al. HER2 signaling regulates the tumor immune microenvironment and trastuzumab efficacy. Oncoimmunology 8, e1512942 (2019).
Indrieri, M., Podestà, A., Bongiorno, G., Marchesi, D. & Milani, P. Adhesive-free colloidal probes for nanoscale force measurements: production and characterization. Rev. Sci. Instrum. 82, 023708 (2011).
Schillers, H. et al. Standardized nanomechanical atomic force microscopy procedure (SNAP) for measuring soft and biological samples. Sci. Rep. 7, 5117 (2017).
Lorenc, E. et al. Correlation between biological and mechanical properties of extracellular matrix from colorectal peritoneal metastases in human tissues. Sci. Rep. 13, 5117 (2023).
Puricelli, L., Galluzzi, M., Schulte, C., Podestà, A. & Milani, P. Nanomechanical and topographical imaging of living cells by atomic force microscopy with colloidal probes. Rev. Sci. Instrum. 86, 033705 (2015).
Bianchi, F. et al. Fhit nuclear import following EGF stimulation sustains proliferation of breast cancer cells. J. Cell Physiol. 230, 2661–2670 (2015).
Acknowledgements
We thank Laura Mameli for excellent secretarial assistance. We also thank Dr. Vera Cappelletti and Dr. Andrea Vingiani for making the IHC validation cohort available to us and Prof. Nicoletta Gagliano for advising us on the functional analyses of CLEC3A knock-in cells. We also thank Dr. Claudio Tripodo for his supervision of the histochemical analysis and his insightful discussion; the OMIC facility of University of Milano for acquisition of the mass spectrometry data and Dr. Francesca Grassi Scalvini for assistance in the raw data acquisition and figure preparation. This study was supported by funding from Fondazione Associazione Italiana per la Ricerca sul Cancro (Fondazione AIRC) Special Program – 5 per mille 2011 No 12162 to ET and by “5 per 1000” funds financial support for healthcare research (Italian Ministry of Health). Marta Giussani was supported by a fellowship from Fondazione Italiana per la Ricerca sul Cancro (FIRC), No 16563.
Author information
Authors and Affiliations
Contributions
Conceptualization: T.T. and E.T.; methodology: T.T., M.G., V.R., E.J.L., E.M., F.B., and G.V.; data curation: T.T., M.G., E.M., E.J.L., V.C., C.V., M.S., G.V., and G.T.; investigation: T.T., M.G., E.M., V.R., E.J.L., V.A., S.P., M.V., V.C., and G.V.; funding acquisition: T.T. and E.T.; supervision: T.T., A.P., G.T., and E.T.; writing—original draft: T.T., G.T., and E.T.; writing—review & editing: E.J.L., F.B., V.C., A.P., L.S., and S.M.P.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Triulzi, T., Giussani, M., Maffioli, E. et al. Proteomic landscape of decellularized breast carcinomas identifies C-type lectin domain family 3 member A as a driver of cancer aggressiveness. npj Breast Cancer 11, 51 (2025). https://doi.org/10.1038/s41523-025-00769-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41523-025-00769-0
