
Abstract
In this study, we utilized a quantitative atomistic analysis approach to investigate the impact of chemical ordering structures on the diffusion behavior of interstitials and vacancies within the CrCoNi medium entropy alloy (MEA), employing an advanced neural network interatomic potential (NNP). We discovered that the degree of chemical ordering, which can be precisely controlled through annealing at elevated temperatures, significantly influences both interstitial and vacancy diffusion. This phenomenon contributes to the notable sluggish diffusion characteristic of CrCoNi, largely attributable to the restriction of diffusion pathways in regions with lower degree of chemical ordering. We also emphasized the crucial role of operating temperature on diffusion, which should be remained well below the annealing temperature to preserve the sluggish diffusion effect. Our research sheds light on the interplay between chemical ordering and defect diffusion in MEAs, and it proposes effective strategies for tailoring the diffusivity of MEAs by altering their chemical ordering. These insights are instrumental in the development of next-generation materials, which are optimized for use in challenging environments, such as high-temperature and irradiation conditions.
Similar content being viewed by others

Introduction
Multi-principal element alloys, which encompass both medium-entropy alloys (MEAs) and high-entropy alloys (HEAs), have garnered significant attention due to their outstanding mechanical properties1,2,3. Recently, their exceptional radiation resistance4,5,6,7 has also come into focus, as they exhibit the ability to withstand harsh environments, such as high temperatures, high pressures, and intense irradiation, for extended periods. Consequently, they are being considered as potential next-generation materials for nuclear structures8,9,10. During irradiation, the bombardment by high-energy particles creates numerous interstitials and vacancies11. The evolution of these defects through atomic diffusion leads to the formation of vacancy clusters, dislocations, and voids, ultimately resulting in materials failure12,13. The exceptional radiation resistance of MEAs and HEAs is believed to stem from their unique defect diffusion kinetics, characterized by sluggish diffusion dynamics14,15,16,17, which can impede the rapid formation of large defects, thereby preventing materials failure.
However, the precise mechanism behind this sluggish diffusion in MEAs and HEAs remains unclear and is a subject of debate. Consequently, the ability to manipulate and control sluggish diffusion has not yet been achieved. For instance, Tsai et al. reported that experimental results demonstrate a slower diffusion coefficient and higher activation energy in CoCrFeMnNi HEAs compared to reference metals18. In contrast, Vaidya et al. concluded that diffusion in CoCrFeNi and CoCrFeMnNi HEAs is not necessarily sluggish and can even be enhanced at a given temperature19. Additionally, recent theoretical work20 has suggested that there is no direct relationship between high configurational entropy and sluggish diffusion in MEAs and HEAs. These diverse findings underscore the need for further research to fully comprehend the phenomenon of sluggish diffusion in MEAs and HEAs.
Currently, some research has highlighted the formation of chemical ordering structures in MEAs and HEAs after annealing at elevated temperatures21,22,23, which could potentially influence the behavior of interstitial and vacancy diffusion as well as the radiation resistance24,25,26,27. Notably, an experimental study indicated that the local chemical order in CrCoNi can delay the formation and evolution of interstitial and vacancy, leading to improved radiation resistance at relatively low irradiation doses25. Atomistic simulations supported this by showing the local chemical order can reduce interstitial and vacancy diffusivity through increased diffusion energy barriers. Nevertheless, it is vital to recognize that these simulations are qualitative, primarily due to the difficulties in capturing the genuine chemical ordering structures and diffusion energy barriers in chemically ordered CrCoNi using the embedded atom method (EAM) potential. Some other vacancy diffusion simulation works have also pointed out that the formation of chemical short-range order can reduce vacancy diffusion by decreasing jump frequency and enhancing diffusion correlation28,29. However, the specific correlation between genuine chemical ordering structures and defect diffusion pathways has not been carefully explored. The precise nature of these chemical ordering structures is still debated, and a unanimous conclusion remains elusive. Not to mention the study how the modification of defect diffusion can be achieved by controlling the formation of these chemical ordering structures. The chemical ordering effect on defect diffusion deserves an in-depth understanding.
Recently, we successfully developed a precise neural-network potential (NNP) for CrCoMi MEA grounded in the density functional theory (DFT) produced training dataset. The NNP enabled us not only to specify realistic local chemical ordering structures but also to quantitatively predict their formation kinetics in CrCoNi MEA30,31. Moreover, the NNP accurately replicates the diffusion kinetics of interstitials and vacancies, largely because it well-reproduce the DFT activation energy for the interstitial and vacancy diffusion.
In the present study, utilizing the NNP-driven atomistic simulations, such as molecular dynamics (MD) simulations for interstitial diffusion and artificial-neural-network-accelerated diffusion-kinetic-Monte Carlo (ANN-kMC) simulations31 for vacancy jumps, we first precisely depict the genuine chemical ordering structures relative to annealing temperature and duration in CrCoNi MEA. Following this, we analyze its impact on interstitial and vacancy diffusion, revealing a clear link between the degree of chemical ordering and the diffusion kinetics. Our findings indicate that the genuine chemical ordering structures play a crucial role in defect sluggish diffusion by restricting interstitial and vacancy diffusion regions. In addition, the changes in diffusivity depending on the operational temperature of alloys was discussed, which is pivotal for understanding how alloys behave under practical operating conditions. Consequently, our results offer avenues for adjusting the diffusion kinetics of interstitials and vacancies by tuning chemical ordering through annealing conditions.
Results
Control of chemical ordering structures by thermal annealing
In our previous work31, we introduced a Time–Temperature–Chemical-ordering-degree (TTC) diagram for the equimolar CrCoNi MEA using ANN-kMC simulations combined with NNP. This diagram enables control over the degree of chemical ordering by adjusting the annealing time from a random solid solution (RSS) state at a fixed temperature. For the sake of clarity in the subsequent discussions, we have revisited this diagram in Fig. 1 utilizing the computational strategy previously published in ref. 31 with a modification of initial vacancy concentration by using experimental electric resistance change data32. It is important to note that the previous TTC diagram in reference31 was constructed under the assumption that the CrCoNi MEA had cooled down from 1500 K, and the equilibrium vacancy concentration at 1500 K represented the initial vacancy concentration just before annealing commenced. However, this assumption seems to have led to an overestimation of the initial vacancy concentration, especially when compared to the experimental Time–Temperature–Electric resistance relation32. In our current study, we took a different approach. We determine the initial vacancy concentration that best aligns with the experimental Time–Temperature–Electric resistance relation, which relies on electric resistance measurements of the CrCoNi MEA at various annealing times and temperatures (see Supplementary Fig. 1).

Star shows state of models RSS and Model_AN 1 ~ Model_AN 5 annealed at T = 673 K for various durations.
In Fig. 1, the degree of chemical ordering is quantified using the Warren–Cowley order parameters31 of the 1st neighbor shell, \({\alpha }_{1}^{{ij}}\), with both i and j representing Cr (refer to the “Method” section for details). Each C-shaped curve within the diagram stands for a specific chemical ordering degree. The plateau of each C-shape, in relation to time, indicates the equilibrium degree of chemical ordering reached at a given temperature. Significant changes in chemical ordering can be observed within a reasonable annealing time at T = 673 K as shown in Fig. 1. At higher temperatures than 673 K, chemical ordering is expected to be limited and not strongly affects vacancy and interstitial diffusion, despite rapid chemical ordering formation. In contrast, at temperatures lower than 673 K, the development of chemical ordering is considerably slower, though strong chemical ordering will eventually form. Therefore, our subsequent analysis of interstitial and vacancy diffusion focuses on models annealed at T = 673 K for various durations. Hereafter we referred to the RSS model and the annealed models as Model_RSS and Model_AN 1 ~ Model_AN 5, respectively.
Next we discerned the chemical ordering structures via a chemical ordering matching analysis31, yielding a chemical ordering degree, \({M}_{i}^{{{\rm{I}}}_{{\rm{d}}}}\) for specific ordering type \({{\rm{I}}}_{{\rm{d}}}\) (\({{\rm{I}}}_{{\rm{d}}}\) = types I, II, and III) around each atomic site i (see the “Method” section for the matching analysis details). The three chemical ordering types (\({{\rm{I}}}_{{\rm{d}}}\)= types I, II, and III, as depicted in Fig. 2a) were detailed in ref. 31. In these structures, Cr-rich and Co/Ni-rich layers are oriented along {110}, {100}, and {113} for types I, II, and III respectively. Specifically, type I displays an Immm symmetry, with a propensity for Cr atoms separation. Type II is reminiscent of the L10 structure, where Cr atoms are inclined to cluster in one layer and Co/Ni atoms in another. Type III is akin to the L11 structure, where Cr atoms locate at the eight vertices (see Supplementary Fig. 2 for the distribution of each chemical ordering structure in all models). The remaining structures all together such as other than the types I, II, and III are labeled “other” structures here. It should be noted that “other” structures also contain some weak chemical ordering, so they should be distinguished from the RSS structures (see Supplementary Fig. 3 for details). Figure 2b displays distribution of the chemical ordering sites i having \({\max }_{{{\rm{I}}}_{{\rm{d}}}={\rm{I}},{\rm{II}},{\rm{III}}}{M}_{i}^{{{\rm{I}}}_{{\rm{d}}}} > 0.85\) in an equimolar RSS and models annealed for 4.4, 13.2, 209, 293, and 407 h from the RSS state at T = 673 K. This figure illustrates the spatial and temporal evolution of chemical ordering. The chemical ordering increases with annealing time, with a noteworthy observation being that after 407 h of annealing, the volume fraction of chemical ordering approaches nearly 50% (see also Fig. 3b).

a Structure information of three representative chemical ordering (the red spheres represent Cr atoms, and the gray spheres represent Co/Ni atoms). b Distribution of chemical ordering structure after annealing at T = 673 K for different time (please refer to the text for details).

a Warren–Cowley order parameter changes during annealing simulation, dash lines represent Model_AN 1 ~ Model_AN 5 with different annealing time. b Volume fraction of types I, II, and III chemical ordering structures at different annealing time. The label “other” represents volume fraction of structure does not contain the types I, II, and III.
Figure 3a, b quantifies the evolution over time of the Warren–Cowley order parameters and the volume fraction of regions exhibiting type \({{\rm{I}}}_{{\rm{d}}}\) chemical ordering, represented as \({f}_{{\rm{v}}}^{{{\rm{I}}}_{{\rm{d}}}}={m}^{{{\rm{I}}}_{{\rm{d}}}}/N\). Here \({m}^{{{\rm{I}}}_{{\rm{d}}}}\) denotes the number of sites with \({M}_{i}^{{{\rm{I}}}_{{\rm{d}}}}\) > 0.85, and N represents the total atom count in the model. As depicted in Fig. 3a, the 407-h anneal resulted in the Cr-Cr and Co-Ni pairs exhibiting positive Warren–Cowley order parameters of \({\alpha }_{1}^{{\rm{CrCr}}}\) = 0.38 and \({\alpha }_{1}^{{\rm{CoNi}}}\) = 0.25, respectively, indicating repulsion tendencies between Cr-Cr and Co-Ni in the first nearest neighbor. Conversely, the Cr-Co and Cr-Ni pairs manifested negative parameters of \({\alpha }_{1}^{{\rm{CrCo}}}\) = −0.22 and \({\alpha }_{1}^{{\rm{CrNi}}}\) = −0.14, respectively, indicating attraction tendencies between Cr-Co and Cr-Ni in the first nearest neighbor. These observations align with the DFT results33. Figure 3b further reveals that the volume fractions of types I and II chemical ordering structures expand with annealing duration. Notably, after 407 h of annealing, the type I structure’s volume fraction approached 39.6%, and substantial chemical domain structures appeared in CrCoNi. In contrast, type III diminished, likely due to its inherent instability and gradual decomposition over extended annealing, as suggested by Zhou et al.34 and attributed to the energy penalty from increased Cr-Cr interactions31.
Impact of chemical ordering on interstitial and vacancy diffusion
Interstitials are usually formed in radiation damage due to the high formation energy. Materials will form a large number of point defects after radiation damage, including interstitials and vacancies. Since the high migration barrier of vacancy, interstitial diffusion plays an important role in the evolution of irradiation defects. Interstitials prefer to form dumbbells in CrCoNi and migrate along with the dumbbell in [100] direction and form new dumbbell to complete interstitial migration. Most of interstitials will combine with vacancies and annihilate, and the remaining interstitials will aggregate to form clusters after long-term diffusion, resulting in elements segregation within material17. For the interstitial diffusion simulation in this work, an atom was arbitrarily incorporated into models with a count of N = 2916 atoms. The diffusion temperature was established at T = 1200 K to ensure an adequate diffusion process during our 1 ns MD simulation time. Notably, no remarkable change in the existing chemical ordering were observed during this simulation time window, avoiding the potential influence of the segregation/ordering change on the diffusion results (please see Supplementary Fig. 4a, for details on chemical ordering variations throughout the diffusion simulation).
One the other hand, vacancy diffusion is activated at high temperature, and atoms will continue to exchange positions with vacancy through vacancy diffusion mechanism. These behaviors usually occur during the annealing process. Due to the different interaction between elements, a certain chemical ordering structures will be formed after long-term annealing26. Therefore, for the vacancy diffusion simulation, due to the higher migration energy barrier, we adopted the same simulation methodology as that used for the annealing simulation, namely ANN-kMC combined with NNP (refer to “Method” section for details). Here, a vacancy was generated by removing an atom from the model, and the simulation temperature was set to T = 1200 K. A total of 2000 accepted vacancy jumps were executed in ANN-kMC simulation, and similar to the interstitial diffusion simulation, no substantial change in the existing chemical ordering were detected within this MD simulation time window, thus avoiding the impact of the change in chemical ordering structures formed by vacancy diffusion (please see Supplementary Fig. 4b).
It is worth mentioning that both prolonged interstitial and vacancy diffusion lead to changes in element distribution, such as segregation and chemical ordering, but with different kinetic mechanisms. Since interstitial diffusion is much faster than vacancy, timescale required to change element distribution varies. Here, both interstitial diffusion MD simulations and vacancy jump kMC simulations were executed on the Model_RSS and Model_AN 1 ~ Model_AN 5, and chemical ordering impact on interstitial and vacancy diffusion was studied.
To quantify the diffusion behavior of both interstitials and vacancies, we calculated the average mean squared displacement (MSD) of all atoms for these models. The gradients of the MSD-time plots were employed to calculate diffusivity (for more details on MSD calculation, please see the “Method” section). It should be noted that the calculated diffusivity depends on the defect concentration in the simulation, and the concentration of interstitial and vacancy in this work are set to \(c=\,\frac{1}{N}\), where N = 2916 is the number of atoms in the defect free model. For self-interstitial defects, as mentioned before, they are usually formed under high energy irradiation environment due to the high formation energy of self-interstitial. Therefore, the relationship between the actual self-interstitial concentration and the calculated diffusivity will not be further discussed. For vacancy defects, the actual concentration depends on conditions such as the vacancy formation energy and the local temperature environment. If the actual vacancy concentration (cact) can be obtained, e.g., by fitting the experimental data as described in Supplementary Fig. 1, one can correct the actual vacancy diffusivity as \({D}^{{\rm{act}}}=\,\frac{{c}^{{\rm{act}}}}{{c}^{{\rm{cal}}}}{D}^{{\rm{cal}}}\) according to the calculated vacancy diffusivity (Dcal) and concentration (ccal).
Figure 4a, b displays the MSD results for interstitials and vacancies, respectively. The calculated diffusivities for both interstitials and vacancies across all models are shown in Fig. 4c, d. It is evident that all models after annealing (Model_AN 1~ Model_AN 5) exhibit lower MSD gradients compared to the Model_RSS. Additionally, these MSD slopes diminish with escalating chemical ordering or prolonged annealing. For instance, the Model_RSS yielded interstitial and vacancy diffusivities of 3.6 × 10−12 m2 s−1 and 3.6 × 10−13 m2 s−1, respectively. In contrast, the thoroughly annealed Model_AN 5 recorded diffusivities of just 2.0 × 10−12 m2 s−1 and 1.2 × 10−13 m2 s−1 for interstitials and vacancies, respectively (please see also Supplementary Table 1). It is discernible that diffusivity exhibits a monotonic decline with amplifying chemical ordering degree, leading to sluggish diffusion.

a Interstitial and b vacancy MSD - time curves in models with different degrees of chemical ordering. c Interstitial and d vacancy diffusivity in models with different degree of chemical ordering.
Correlation between chemical ordering region and diffuse region
To more precisely probe the relationship between the diffusive region of interstitial and vacancy and areas exhibiting chemical ordering, we undertook 50 independent diffusion simulations (both interstitial MD and vacancy ANN-kMC diffusion) for the Model_RSS and Model_AN 5 models. Each simulation had varied initial positions for the interstitials and vacancies.
For a finer understanding, we sectioned the model volume into a detailed mesh (see the “Method” section for the details). We then calculated the spatial distribution for both the chemical ordering degree and the residence densities of interstitials and vacancies during the simulation. Note that since interstitials evolve into a dumbbell pair structure as they diffuse, we utilized the coordinate centers of these dumbbell pairs to pinpoint the interstitial’s position.
As shown in Fig. 5a, a (010) plane was selected to display the chemical ordering degree and interstitial and vacancy residence density results for better understanding in the following discussions. Figure 5b, c renders the distribution of total chemical ordering degree of type I + type II + type III, \({P}_{k}=\mathop{\sum }\nolimits_{{{\rm{I}}}_{{\rm{d}}}={\rm{I}}}^{{\rm{III}}}{P}_{k}^{{{\rm{I}}}_{{\rm{d}}}}\) on the (010) plane and “other”, \({P}_{k}^{{\rm{o}}}=\) (\(1\,-\,{P}_{k})\) on the (010) plane, respectively, derived from Model_AN 5. \({P}_{k}^{{{\rm{I}}}_{{\rm{d}}}}\) is an average chemical ordering degree of type \({{\rm{I}}}_{{\rm{d}}}\) in a mesh k (please see also “Method” section). Concurrently, Fig. 5d, e renders interstitial and vacancy residence densities of mesh k\(,\,{Q}_{k}\) on the (010) plane (please see also “Method” section). The outcomes underscore a heterogeneous distribution pattern for interstitial and vacancy residence densities, with pronounced densities discerned in specific zones. This suggests a propensity for interstitials and vacancies to selectively diffuse within these zones. Contrarily, the Model_RSS exhibits a more homogeneous residence density, signaling a non-selective, random diffusion pattern across regions (see Supplementary Fig. 5, for details on diffusion results in the Model_RSS).

a A (010) plane was selected to display the chemical ordering degree and interstitial and vacancy residence density results. b, c Chemical ordering degree distribution of chemical ordering structures (type I + type II + type III) and “other” structures in the Model_AN 5 on the (010) plane, respectively. d, e Interstitial and vacancy residence density in the Model_AN 5 on the (010) plane, respectively.
Upon juxtaposing the residence density distribution of interstitials and vacancies with the chemical ordering degree distribution (type I + type II + type III chemical ordering structures and “other” structures), a prominent concurrence emerges between mesh regions boasting higher residence densities for interstitials and vacancies and those showing pronounced chemical ordering degrees. This infers that defects gravitate toward “other” regions devoid of higher degree of chemical ordering structures. Put differently, the inception of chemical ordering structures can delimit the diffusion pathways for defects. For additional confirmation, refer to Supplementary Figs. 6 and 7, which depict outcomes from two different models bearing the same chemical ordering degree Model_AN 5 but annealed from different RSS configurations, and Supplementary Figs. 8–10 for those from models with chemical ordering degree that are less pronounced than in Model_AN 5. These results also support above conclusion.
To quantify the interplay between diffusion regions for interstitials and vacancies and the spread of chemical ordering structures, we computed correlation factors between them. This was done using the relation between average chemical ordering degree \({P}_{k}\), and residence density \({Q}_{k}\), expressed as \(G=a\frac{1}{n}\mathop{\sum }\nolimits_{k=1}^{n}\mathop{\sum }\nolimits_{{{\rm{I}}}_{{\rm{d}}}={\rm{I}}}^{{\rm{III}}}{P}_{k}^{{{\rm{I}}}_{{\rm{d}}}}{Q}_{k}\), where \(n\) (=36 × 36 × 36) is the total number of mesh regions and a is a normalization factor (see “Method” section for the details).
Figure 6a–d presents distribution of the correlation for both interstitials and vacancies, differentiated by chemical ordering structures (type I + type II + type III) and “other” structures on the (010) plane. The total correlation factors for these groups were found to be: \(G\) = 0.41 for interstitials with chemical ordering structures, \(G\) = 0.38 for vacancies with chemical orderings structures, \(G\) = 0.59 for interstitials with “other” structures, \(G\) = 0.62 for vacancies with “other” structures. These results underline a stronger correlation of residence densities of both interstitials and vacancies with “other” structures. It reaffirms that the preferred diffusion regions for these defects are those with less chemical ordering degree.

a, b Correlation factor distribution of interstitial and vacancy for chemical ordering structures (type I + type II + type III) in the Model_AN 5 on the (010) plane, respectively. c, d Correlation factor distribution of interstitial and vacancy for “other” structures in the Model_AN 5 on the (010) plane, respectively. The total correlation factors between chemical ordering degree of chemical ordering structures and “other” structures and interstitial (vacancy) residence density were calculated as 0.41 (0.38) and 0.59 (0.62), respectively. e, f Distribution of interstitial and vacancy formation energy in the Model_AN 5 on the (010) plane, respectively.
To unravel why these diffusion-favorable regions emerge, we computed the formation energy distributions for interstitials and vacancies in the Model_AN 5 on the same (010) plane. As showcased in Fig. 6e, f (with more detailed histograms in Supplementary Fig. 11 for both Model_RSS and Model_AN 5), regions with pronounced chemical ordering structures exhibit elevated formation energies for interstitials and vacancies. This results in a tendency for these defects to migrate toward regions with less chemical ordering. Furthermore, the MSDs in both chemical ordering and “other” structures were evaluated, as illustrated in Fig. 7. This analysis reveals a slower diffusivity in the chemical ordering structures than in the “other” structures.

a Interstitial diffusion and b vacancy diffusion in chemical ordering (type I + type II + type III) and “other” structures in the Model_AN 5, respectively.
These results point to a significant conclusion: the main reason for the decreased diffusion of both interstitials and vacancies within chemical ordering structures can be attributed to constraints in regions characterized by low defect formation energy, such as the “other” structure regions with limited chemical ordering. Additionally, even when interstitials and vacancies transition from the “other” region to the chemical ordering region, their diffusivity significantly decreases, preventing quick diffusion. As a result, the overall diffusivity slows down in relation to the degree of chemical ordering.
The findings robustly indicate that by manipulating the chemical ordering degree, achieved by adjusting the annealing duration at a given annealing temperature, we can deftly regulate the diffusivity of both interstitials and vacancies. It is worth noting that as demonstrated in Fig. 1, to achieve a specific degree of chemical ordering and the corresponding target diffusivity, one can select the annealing temperature to match the nose-temperature of the iso-degree curve for the target degree in the TTC diagram. This approach is aimed at minimizing the annealing time.
Note that when we compare the MSD results of the “other” structures (Fig. 7) with those of the RSS structures (Fig. 4), we can find that the diffusivity of both vacancies and interstitials is certainly slower in the “other” structures. This suggests that even the “other” structures contain some degree of weak chemical ordering and its diffusivity is slower.
Impact of operating temperature on diffusion and chemical ordering
In our simulations, even though diffusion processes were modeled at high temperatures, they operated in short bursts. This was to ensure we analyzed diffusivity within a fixed state of chemical ordering. But when we use MEAs and HEAs over prolonged durations at elevated temperatures, diffusion can alter the existing chemical ordering structures.
The interplay of the annealing temperature and the operating temperature can be clearly depicted in the Time–Temperature–Chemical-ordering-degree diagram, illustrated in Fig. 8. The starting state of the MEA, post-annealing at a particular temperature and time, is denoted by a black dot. The blue arrows depict scenarios where \({T}^{{\rm{o}}}\) > \({T}^{{\rm{a}}}\). Here, when the MEA is suddenly subjected to a higher operating temperature, it rapidly achieves the thermal equilibrium for that temperature, driven by the diffusion of interstitials and vacancies. Conversely, the red arrows represent situations where \({T}^{{\rm{o}}}\) < \({T}^{{\rm{a}}}\). In such cases, upon initiation of operation, the MEA gravitates toward increasing its chemical ordering to match the equilibrium structure of the operating temperature. This transition is also facilitated by interstitial and vacancy diffusion but tends to be prolonged due to reduced diffusivity at lower temperatures and the notable disparity in chemical ordering degree between the initial and the equilibrium states.

One C-shaped curve stands for the same chemical ordering degree. The longer the annealing time or the lower the annealing temperature, the higher the chemical ordering degree.
In essence, if the operating temperature exceeds the annealing temperature, the sluggish diffusion effect progressively diminishes. On the other hand, when the operating temperature is below the annealing temperature, the sluggish diffusion effect becomes incrementally more pronounced.
Discussion
With the progress of MEAs and HEAs research, the impact of chemical ordering on their performances has gradually attracted attention. It is widely believed that the formation of chemical ordering can suppress defect diffusion, and its impact mainly lies in the diffusion barrier and correlation coefficient. For example, a MD simulation result concludes that chemical ordering caused diffusion barrier changes will lead to the vacancy trapping effect28. Meanwhile, DFT calculation shows that chemical ordered structure will reduce defects jump frequencies and correlation factors16. However, how chemical ordering structures directly affect defect diffusion behavior and how to control defect diffusion has not been carefully explored.
Here, we explored the influence of chemical ordering on interstitial and vacancy diffusion in CrCoNi MEA, illustrating the potential to modulate this diffusivity by adjusting the chemical ordering through controlling annealing conditions, such as temperature and duration. Our simulations show that distinct chemical ordering structures emerge in CrCoNi MEA when annealed at certain temperatures. These structures effectively repel interstitials and vacancies, resulting in a restricted diffusion region and, consequently, slower effective diffusion rates of entire system. Notably, diffusivity correlates directly with the degree of chemical ordering, which in turn is influenced by the annealing duration at a given temperature. This underscores the potential to regulate the diffusivity of interstitials and vacancies by modulating the degree of chemical ordering, achieved by fine-tuning the annealing temperature and duration. We conclude by highlighting the pivotal role of operating temperatures on diffusion, emphasizing that to preserve the slow diffusion effect, the operating temperature should remain well below the annealing temperature.
Methods
Kinetic Monte Carlo simulation
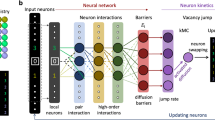
The annealing process in CrCoNi was modeled using the vacancy jump kinetic Monte Carlo (kMC) simulation. One vacancy was created in the model with FCC structure containing 2916 atoms. Vacancy will jump to one of its 12 nearest neighbor lattice sites, which is called as one kMC step. The jump frequency was calculated as \({v}_{i}={v}_{0}\exp \left(-\frac{\Delta {E}_{i}}{{k}_{{\rm{B}}}T}\right)\), where the pre-exponential factor \({v}_{0}\) = 4.16 × 1013 s−1 was adopted by fitting the MD vacancy diffusion simulation (details of the fitting are discussed below), \({k}_{{\rm{B}}}\) is Boltzmann’s constant, \(T\) is the absolute temperature, and \(\Delta {E}_{i}\) is the energy barrier for a vacancy jump to the nearest neighbor site \(i\), which is given by artificial neural network (ANN) predictions. The ANN predictions can significantly accelerate vacancy jump kMC simulations, especially when performing long kMC simulations31.
In order to determine the pre-exponential factor \({v}_{0}\) for the kMC vacancy diffusion simulation, long-term vacancy diffusion MD simulation was executed. The MD vacancy diffusion temperature was set to T = 1200 K, and total simulation time is 10 ns. For the MD simulations, vacancy diffusivity in Model_RSS and Model_AN 1 ~ 5 are calculated as 3.2 × 10−13 m2 s−1, 2.2 × 10−13 m2 s−1, 2.1 × 10−13 m2 s−1, 1.7 × 10−13 m2 s−1, 1.6 × 10−13 m2 s−1 and 1.1 × 10−13 m2 s−1, respectively. The modified pre-exponential factor for the vacancy kMC diffusion simulation was fitted by \({v}_{0}=\min \{\mathop{\sum }\nolimits_{k=1}^{k=6}{\left|{D}_{k}^{{\rm{MD}}}-{D}_{k}^{{\rm{kMC}}}\right|}^{2}\}\), where k is the index of models with different chemical ordering degree (k = 1 and k = 2 ~ 6 represent Model_RSS and Model_AN 1 ~ 5, respectively). This modified pre-exponential factor includes factors such as temperature depended vibration and enthalpy effects. Based on the MD simulation results, the modified pre-exponential factor is calculated as \({v}_{0}\) = 4.16 × 1013 s−1.
Figure 9 shows the MSD-time curve with modified pre-exponential factor and vacancy diffusivity in Model_RSS and Model_AN 1 ~ 5 are calculated as 3.6 × 10−13 m2 s−1, 2.1 × 10−13 m2 s−1, 1.8 × 10−13 m2 s−1, 1.4 × 10−13 m2 s−1, 1.3 × 10−13 m2 s−1 and 1.2 × 10−13 m2 s-1, respectively. In this way, the vacancy diffusivity calculated by kMC simulation shows good agreement with MD simulation results within at most 18% error in MSD.

Modified pre-exponential factor was applied for kMC simulation in Model_RSS and Model_AN 1 ~ 5, respectively.
The ANN predicts the vacancy jump barriers by the inputs of on-lattice atomic configuration of whole model, and then ANN can output 12 energy barriers of the vacancy jump to the 12 nearest neighbor sites of FCC. The energy barriers in the training dataset are computed using Nudged Elastic Band (NEB) method with the fully relaxed atomic configurations before and after the vacancy jump. The average error between the ANN predictions and NEB calculation in different vacancy configurations is only 0.05 eV and shows good accuracy of ANN predictions31. The average incubation time for a vacancy jump was calculated as \(\Delta {t}_{{\rm{kmc}}}=1/\mathop{\sum}\nolimits_{i=1}^{12}{v}_{i}\). The annealing time was calculated as \({t}_{{\rm{a}}}=\frac{{c}_{{\rm{v}}}}{{c}_{{\rm{v}}}^{{\rm{Exp}}.}\left(T\right)}\sum \left(\Delta {t}_{{\rm{kmc}}}\right)\), where \({c}_{{\rm{v}}}=\frac{1}{N}\) is the actual vacancy concentration during annealing simulation, \(N=2916\) is the total number of atoms, \({c}_{{\rm{v}}}^{{\rm{Exp}}.}\left(T\right)\) is the initial vacancy concentration at temperature \(T\) determined by experimental Time–Temperature–Electric resistance relation32.
It is crucial to understand that using a unified pre-exponential factor is an approximation. The local chemical structure (local chemical ordering) surrounding a vacancy significantly influences the vibrational entropy contributing to the free energy barrier of a vacancy jump. Specifically, this local chemical structure shapes the potential energy landscape and dictates the vibrational modes at the initial and saddle point configurations of the vacancy jump. Consequently, changes in the local chemical structure can alter the pre-exponential factor due to shifts in activation vibrational entropy.
As discussed in this paper, vacancy diffusion pathways vary according to the chemical ordering degree of the alloy. Therefore, during the vacancy diffusion process, the encountered local chemical structure statistics expected to vary based on the chemical ordering degree of the alloy. To address above discrepancy between KMC and MD simulations, we propose to incorporate activation vibrational entropy into the training dataset of energy barrier and to train the ANN using this dataset. This methodological enhancement will be further explored in our future research.
Interstitial diffusion molecular dynamics simulation
One interstitial atom was created in the model containing 2916 atoms to perform the interstitial diffusion MD simulation. The interstitial atom was randomly inserted in the model and migrated by constantly forming new dumbbell pairs with surrounding lattice atoms. The periodic boundary conditions were applied along X, Y and Z directions. Interstitial diffusion with a total time of 1 ns was performed, and the timestep was set to 0.001 ps. The simulation temperature was set to T = 1200 K to ensure an adequate diffusion process within the limited simulation time window. The NPT ensemble was adopted and temperature and pressure were controlled by Nosé-Hoover thermostat35. The MSD was calculated as \(<\, {R}^{2}\left(t\right)\, > \,=\frac{1}{N}\,{\sum }_{i=1}^{N}{\left|{{\boldsymbol{R}}}_{{\boldsymbol{i}}}\right.(t)-\left.{{\boldsymbol{R}}}_{{\boldsymbol{i}}}(0)\right|}^{2}\), where \({{\boldsymbol{R}}}_{{\boldsymbol{i}}}(t)\) and \({{\boldsymbol{R}}}_{{\boldsymbol{i}}}(0)\) are the position vectors of atom i at time t = t and t = 0, respectively, and N is the total number of atoms in the model with interstitial. Interstitial diffusivity can be assessed by calculating the gradients of the MSD-time plots. Diffusion trajectories were visualized by using the software OVITO, and interstitial positions were identified by using Wigner-Seitz method36,37,38.
Vacancy diffusion ANN-kMC simulation
Similar to the annealing simulation, ANN-kMC vacancy jump simulation was performed to study vacancy diffusion in CrCoNi, and ANN was used to predict the vacancy jump barrier in each kMC step. One vacancy was introduced in the model and periodic boundary conditions were applied in X, Y and Z directions. In each vacancy diffusion simulation, 2000 accepted vacancy jumps were performed. The vacancy diffusion temperature was set as T = 1200 K. Base on the kMC vacancy jump, the MSD of vacancy diffusion can be calculated, and the vacancy diffusivity can be evaluated by calculating the gradients of MSD-time plots. The software OVITO and Winger-Seitz method were used to visualize simulation results and identify vacancy position, respectively.
Chemical domain structure identifying
All chemical ordering structures were identified by using the chemical domain structure matching analysis. The matching degree of atom site i was defined as \({M}_{i}^{{{\rm{I}}}_{{\rm{d}}}}={\max }_{\left\{{{\rm{I}}}_{{\rm{d}}},{{\rm{T}}}_{{{\rm{I}}}_{{\rm{d}}}}\right\}}\frac{1}{{N}_{{{\rm{I}}}_{{\rm{d}}}}}\mathop{\sum }\nolimits_{K=1}^{{N}_{{{\rm{I}}}_{{\rm{d}}}}}{\delta }_{\alpha (K)\beta (K)}\), where \({{\rm{I}}}_{{\rm{d}}}\) is the chemical ordering type (type I, II, and III), \({{\rm{T}}}_{{{\rm{I}}}_{{\rm{d}}}}\) is the space group operation of chemical ordering type \({{\rm{I}}}_{{\rm{d}}}\) (e.g., rotating 90° around y axis for type I in Fig. 2a, so that the Cr-rich plane normal to [\(10\bar{1}\)] direction become to the normal [101] direction), \({N}_{{{\rm{I}}}_{{\rm{d}}}}\) is the total number of atom sites in type \({{\rm{I}}}_{{\rm{d}}}\) chemical ordering structure, \(\alpha (K)\) represents element type at K site in the chemical ordering, and \(\beta (K)\) represents the actual element type at K site in the model. \(\delta\) is Kronecker delta, which means that if \(\beta \left(K\right)=\alpha (K)\), then \({\delta }_{\alpha (K)\beta (K)}=1\), otherwise \({\delta }_{\alpha (K)\beta (K)}=0\). When \({M}_{i}^{{{\rm{I}}}_{{\rm{d}}}} > 0.85\), we define site i to belong to the chemical ordering type \({{\rm{I}}}_{{\rm{d}}}\). This matching analysis method has been successfully used to find the chemical ordering structures in our previous work31.
Residence density distribution and correlation with chemical ordering
Defect residence density and chemical ordering degree of each small mesh region was calculated using mesh region division. The model was divided into small mesh regions using a 36 × 36 × 36 mesh. The total sampling size of both MD and ANN-kMC simulations is 100,000. The average chemical ordering degree of each mesh region \({P}_{k}^{{{\rm{I}}}_{{\rm{d}}}}=\frac{{n}_{k}^{{{\rm{I}}}_{{\rm{d}}}}}{{N}_{k}}\) was assumed to be constant during diffusion, where \(k\) is the mesh region index, \({{\rm{I}}}_{{\rm{d}}}\) is the type of structure (type I, II, III, and “other”), \({n}_{k}^{{{\rm{I}}}_{{\rm{d}}}}\) is the total number of atoms of type \({{\rm{I}}}_{{\rm{d}}}\) around (1st nearest neighbor) mesh region \(k\), and \({N}_{k}\) is the total number of atoms around mesh region \(k\) (\(0\le {P}_{k}^{{{\rm{I}}}_{{\rm{d}}}}\le 1\), \(\sum _{{{\rm{I}}}_{{\rm{d}}}}{P}_{k}^{{{\rm{I}}}_{{\rm{d}}}}=1\)). We next defined a conditional function \({C}_{k}\left({\boldsymbol{r}}\right)=\left\{\begin{array}{c}1, {\boldsymbol{r}}(t)\in {\Omega }_{k}\\ 0, {\boldsymbol{r}}(t)\,\notin\, {\Omega }_{k}\end{array}\right.\) to calculate defect residence numbers in mesh region \(k\), where \({\Omega }_{k}\) is the mesh region \(k\), and \({\boldsymbol{r}}(t)\) is interstitial or vacancy defect position at time \(t\). When defects located in the mesh region \({\Omega }_{k}\) at sampling time t, mesh region \(k\) will be counted once. In this case, the normalized total defect residence density in mesh region \(k\) was calculated as \({Q}_{k}={a}{\int }_{0}^{{t}^{{\rm{F}}}}{C}_{k}\left({\boldsymbol{r}}(t)\right)\Delta {t\; dt}\), where \(a=\,\frac{n}{{t}^{{\rm{F}}}}\) is the normalization factor, \({t}^{{\rm{F}}}=\,\mathop{\sum }\nolimits_{k=1}^{n}{Q}_{k}\) is the total diffusion simulation time, \(n\) (=36 × 36 × 36) is the total number of mesh regions in the model, and \(\Delta t\) is the residence time for each sampling. For interstitial diffusion simulation, the residence time for each sampling is equal to the sampling interval, which is constant at 0.5 ps, while for vacancy diffusion simulation, the actual residence time for each sampling should be calculated based on the kMC jump time \(\Delta {t}_{{\rm{kmc}}}\). Finally, we defined the average correlation factor between the defect residence density and the chemical ordering degree of type \({{\rm{I}}}_{{\rm{d}}}\) as \({G}^{{{\rm{I}}}_{{\rm{d}}}}={a}{\frac{1}{n}\mathop{\sum }\nolimits_{k=1}^{n}{P}_{k}^{{{\rm{I}}}_{{\rm{d}}}}Q}_{k}\). A higher correlation factor \({G}^{{{\rm{I}}}_{{\rm{d}}}}\) indicates that defects prefer to diffuse in type \({{\rm{I}}}_{{\rm{d}}}\) region. In this work, adequate diffusion simulations were performed to capture the characteristic of interstitial and vacancy diffusion in different regions. See Supplementary Fig. 12 for convergence information on residence density and number of diffusion simulations.
Chemical ordering parameter
The Warren–Cowley order parameter \({\alpha }_{n}^{{ij}}=1-\frac{{P}_{n}^{{ij}}}{{C}_{j}}\) was used to descript the chemical ordering parameters for the element pairs i and j39, where \({P}_{n}^{{ij}}\) is the probability of occurrence of element j in the nth neighbor coordinate shell of element i, and \({C}_{j}\) is the concentration of element j in the system. A positive parameter indicates a tendency of repulsion pairs of each other (e.g., Cr-Cr pair has a positive \({\alpha }_{1}^{{\rm{CrCr}}}\) in this work), while negative parameters indicate a tendency of attraction with each other (e.g., the Cr-Co and Cr-Ni pairs have a negative \({\alpha }_{1}^{{\rm{CrCo}}}\) and \({\alpha }_{1}^{{\rm{CrNi}}}\) in this work). If the value equal to 0, it means that pairs of i and j elements are randomly distributed.
Data availability
The data used in this work are available from the authors upon request.
Code availability
The computer codes used in this work are available from the authors upon request.
References
Yeh, J.-W. et al. Nanostructured high‐entropy alloys with multiple principal elements: novel alloy design concepts and outcomes. Adv. Eng. Mater. 6, 299–303 (2004).
Cantor, B., Chang, I. T. H., Knight, P. & Vincent, A. J. B. Microstructural development in equiatomic multicomponent alloys. Mater. Sci. Eng. A 375–377, 213–218 (2004).
Enserink, M. Malaria treatment: ACT two (Science (560)). Science 318, 1866 (2007).
Lu, C. et al. Enhancing radiation tolerance by controlling defect mobility and migration pathways in multicomponent single-phase alloys. Nat. Commun. 7, 1–8 (2016).
Xu, Q., Guan, H. Q., Zhong, Z. H., Huang, S. S. & Zhao, J. J. Irradiation resistance mechanism of the CoCrFeMnNi equiatomic high-entropy alloy. Sci. Rep. 11, 1–8 (2021).
Luo, H., Li, Z., Mingers, A. M. & Raabe, D. Corrosion behavior of an equiatomic CoCrFeMnNi high-entropy alloy compared with 304 stainless steel in sulfuric acid solution. Corros. Sci. 134, 131–139 (2018).
Schuh, B. et al. Mechanical properties, microstructure and thermal stability of a nanocrystalline CoCrFeMnNi high-entropy alloy after severe plastic deformation. Acta Mater. 96, 258–268 (2015).
El-Atwani, O. et al. Outstanding radiation resistance of tungsten-based high-entropy alloys. Sci. Adv. 5, eaav2002 (2019).
Pickering, E. J. et al. High-entropy alloys for advanced nuclear applications. Entropy 23, 98 (2021).
Li, Y., Li, R., Peng, Q. & Ogata, S. Reduction of dislocation, mean free path, and migration barriers using high entropy alloy: Insights from the atomistic study of irradiation damage of CoNiCrFeMn. Nanotechnology 31, 8 (2020).
Derlet, P. M. & Dudarev, S. L. Microscopic structure of a heavily irradiated material. Phys. Rev. Mater. 4, 1–20 (2020).
Wirth, B. D. How does radiation damage materials? Science 318, 923–924 (2007).
Yang, T. et al. Irradiation responses and defect behavior of single-phase concentrated solid solution alloys. J. Mater. Res. 33, 3077–3091 (2018).
Huang, W. & Bai, X. M. Machine learning based on-the-fly kinetic Monte Carlo simulations of sluggish diffusion in Ni-Fe concentrated alloys. J. Alloy. Compd. 937, 168457 (2023).
Xu, B. et al. Mechanism of sluggish diffusion under rough energy landscape. Cell Rep. Phys. Sci. 4, 101337 (2023).
Zhao, S., Osetsky, Y. & Zhang, Y. Preferential diffusion in concentrated solid solution alloys: NiFe, NiCo and NiCoCr. Acta Mater. 128, 391–399 (2017).
Lu, C. et al. Radiation-induced segregation on defect clusters in single-phase concentrated solid-solution alloys. Acta Mater. 127, 98–107 (2017).
Tsai, K. Y., Tsai, M. H. & Yeh, J. W. Sluggish diffusion in Co-Cr-Fe-Mn-Ni high-entropy alloys. Acta Mater. 61, 4887–4897 (2013).
Vaidya, M., Trubel, S., Murty, B. S., Wilde, G. & Divinski, S. V. Ni tracer diffusion in CoCrFeNi and CoCrFeMnNi high entropy alloys. J. Alloy. Compd. 688, 994–1001 (2016).
Koyama, T., Tsukada, Y. & Abe, T. Simple approach for evaluating the possibility of sluggish diffusion in high-entropy alloys. J. Phase Equilib. Diffus. 43, 68–77 (2022).
Zhang, R. et al. Short-range order and its impact on the CrCoNi medium-entropy alloy. Nature 581, 283–287 (2020).
Inoue, K., Yoshida, S. & Tsuji, N. Direct observation of local chemical ordering in a few nanometer range in CoCrNi medium-entropy alloy by atom probe tomography and its impact on mechanical properties. Phys. Rev. Mater. 5, 85007 (2021).
Chen, X., Yuan, F., Zhou, H. & Wu, X. Structure motif of chemical short-range order in a medium-entropy alloy. Mater. Res. Lett. 10, 149–155 (2022).
Liu, L. et al. Local chemical ordering and its impact on radiation damage behavior of multi-principal element alloys. J. Mater. Sci. Technol. 135, 13–25 (2023).
Zhang, Z. et al. Effect of local chemical order on the irradiation-induced defect evolution in CrCoNi medium-entropy alloy. Proc. Natl Acad. Sci. 120, e2218673120 (2023).
Xu, B. et al. Influence of short-range order on diffusion in multiprincipal element alloys from long-time atomistic simulations. Phys. Rev. Mater. 7, 1–11 (2023).
Li, Y. et al. Chemical ordering effect on the radiation resistance of a CoNiCrFeMn high-entropy alloy. Comput. Mater. Sci. 214, 111764 (2022).
Xing, B., Wang, X., Bowman, W. J. & Cao, P. Short-range order localizing diffusion in multi-principal element alloys. Scr. Mater. 210, 114450 (2022).
Xing, B., Zou, W., Rupert, T. J. & Cao, P. Vacancy diffusion barrier spectrum and diffusion correlation in multicomponent alloys. Acta Mater. 266, 119653 (2024).
Yu, P., Du, J. P., Shinzato, S., Meng, F. S. & Ogata, S. Theory of history-dependent multi-layer generalized stacking fault energy—a modeling of the micro-substructure evolution kinetics in chemically ordered medium-entropy alloys. Acta Mater. 224, 117504 (2022).
Du, J. P. et al. Chemical domain structure and its formation kinetics in CrCoNi medium-entropy alloy. Acta Mater. 240, 118314 (2022).
Li, L. et al. Evolution of short-range order and its effects on the plastic deformation behavior of single crystals of the equiatomic Cr-Co-Ni medium-entropy alloy. Acta Mater. 243, 118537 (2023).
Ding, J., Yu, Q., Asta, M. & Ritchie, R. O. Tunable stacking fault energies by tailoring local chemical order in CrCoNi medium-entropy alloys. Proc. Natl Acad. Sci. USA 115, 8919–8924 (2018).
Zhou, L. et al. Atomic-scale evidence of chemical short-range order in CrCoNi medium-entropy alloy. Acta Mater. 224, 117490 (2022).
Nosé, S.A molecular dynamics method for simulations in the canonical ensemble A molecular dynamics method for simulations in the canonical ensemblet. Mol. Phys. 52, 255–268 (1984).
Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO-the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 18, 015012 (2010).
Stukowski, A. Structure identification methods for atomistic simulations of crystalline materials. Model. Simul. Mater. Sci. Eng. 20, 045021 (2012).
Stukowski, A., Bulatov, V. V. & Arsenlis, A. Automated identification and indexing of dislocations in crystal interfaces. Model. Simul. Mater. Sci. Eng. 20, 085007 (2012).
de Fontaine, D. The number of independent pair-correlation functions in multicomponent systems. J. Appl. Crystallogr. 4, 15–19 (1971).
Acknowledgements
Y.L., S.S., and S.O. were used computational resources of supercomputer Fugaku provided by the RIKEN Center for Computational Science (Project IDs: hp230205 and hp230212), the large-scale computer systems at the Cybermedia Center, Osaka University, the Large-scale parallel computing server at the Center for Computational Materials Science, Institute for Materials Research, Tohoku University, and Research Center for Computational Science, Okazaki, Japan (Project: 24-IMS-C503). S.S. and S.O. acknowledge the support by projects of MEXT of Japan (grant numbers JPMXP1122684766, JPMXP1020230325 and JPMXP1020230327). S.S. acknowledges the support by projects of JSPS KAKENHI (grant numbers JP21K14042, JP24K17170, and JP24H00994). S.O. acknowledges the support by projects of JSPS KAKENHI (grant numbers JP17H01238, JP21K18675, JP23H00161, and JP23K20037).
Author information
Authors and Affiliations
Contributions
Yangen Li: methodology, software, formal analysis, and original draft writing; Jun-Ping Du: software and methodology; Shuhei Shinzato: software and methodology; Shigenobu Ogata: conceptualization, writing—review & editing, supervision, and project management.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Du, JP., Shinzato, S. et al. Tunable interstitial and vacancy diffusivity by chemical ordering control in CrCoNi medium-entropy alloy. npj Comput Mater 10, 134 (2024). https://doi.org/10.1038/s41524-024-01322-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41524-024-01322-6
