
Abstract
Laminopathies are a group of rare disease due to mutations in the LMNA gene, which is crucial for nuclear integrity and cellular rigidity. Depending on the mutation, the disease manifests in striated muscles, adipose tissues, nerves, and the heart. Although many laminopathic patients exhibit accelerated aging syndromes, the connection as to why loss of LMNA drives aging remains unknown. Herein, we present evidence that cardiomyocytes from laminopathic heart sections exhibit shortened telomeres. Patient derived hiPSC-CMs we observed LMNA mutation results in myocardial enlargement and altered contractility in cardiomyocytes. Further, laminopathic murine cardiomyocytes recapitulates telomere attrition phenotype.
Similar content being viewed by others

Introduction
Within cells, the nuclear envelope is responsible for storing genomic material while allowing import and export of biomolecules, and its structure consists of nuclear membranes, nuclear lamina, and nuclear pore complexes. Lamin proteins are the major components of the nuclear lamina residing at the inner nuclear membrane facing chromatin. Laminopathy was first described in the late 1990s and coined in 2000 where mutations of the LMNA gene, that encodes lamin A and C, manifests in striated muscles, adipose tissues, nerves, and the heart. Clinically, laminopathies are classified as autosomal dominant Emery-Dreifuss muscular dystrophy (AD-EDMD)1, dilated cardiomyopathy with conduction defects (DCM-CD)2,3, and limb-girdle muscular dystrophy with cardiac conduction disturbances (LGMD1B)4. Cardiac laminopathy results in a wide spectrum of clinical manifestation, including electrical and mechanical alteration of cardiomyocytes contractility, cardiac fibrosis and arrythmias, leading towards life-threatening complication including sudden cardiac death and end-stage heart failure2,5. Besides heart transplants, currently there is no cure for this devastating disease.
Telomeres, composed of TTAGGG repeats located at the ends of chromosomes, maintain genomic stability and their shortening is a hallmark of aging6. It has been shown that cardiomyocytes remain largely non-proliferative post-birth7 and healthy myocardial telomere lengths remain relatively stable8. Previously we have demonstrated that telomere shortening occurs independent of proliferation in cardiomyopathies driven by sarcomeric mutations9,10,11 and it is now accepted that non-proliferative cells can also enter ‘senescence-like’ states12. Moreover, it has been shown that telomere localization is dependent on nuclear geometry13 Thus, we wondered whether perturbation to nuclear integrity due to LMNA mutation can result in accelerated telomere shortening due to increased contractile stress on the nucleus14. Here we show in laminopathic patient cardiac sections that loss of nuclear integrity correlates in telomere shortening. Further, using isogenic human induced pluripotent stem cell (hiPSC) and hiPSC-derived cardiomyocytes (hiPSC-CM) with or without mutation on one or two alleles, we demonstrate that telomere lengths are governed by lamin A/C levels and is cell type specific. We further observed that LMNA mutation results in increased cardiomyocyte surface area and increased contractile force.
Results
Laminopathic cardiomyocytes exhibit telomere shortening
To determine if mutation in LMNA gene would result in telomere shortening, we performed quantitative FISH (Q-FISH) and measured telomere fluorescence intensity per nucleus in cardiomyocytes (CMs) expressing the CM-specific marker Troponin-T (Fig. 1A). Cardiac sections from laminopathic patients who received heart transplant between 1997 and 2012 (Table 1) as well as healthy controls (Supplementary Table S1) were used. Hematoxylin and eosin staining showed presence of cardiac remodeling in all LMNA samples (Supplemental Figs. S1 & S2). Control biopsies of the grafted hearts were also included for telomere analysis. In accordance with our previous findings in hypertrophic and dilated cardiomyopathic cardiomyocytes11, the telomere levels of Troponin-T+ CMs in LMNA hearts were significantly reduced by 38% compared with healthy controls (LMNA, n = 13, 4.43 ± 0.48; control, n = 18, 7.13 ± 0.60; Fig. 1B). The trend is consistent when results were grouped based on clinical phenotype (DCM-CD, n = 2, 4.89 ± 1.28; EDMD, n = 4, 3.80 ± 0.80; LGMD1B, n = 6, 5.03 ± 0.64; Fig. 1C). Next, we examined the correlation between telomere level and age. Given the ages of the healthy donor hearts were not disclosed, we only compared telomere levels of LMNA and healthy controls. Healthy cardiomyocytes showed no telomere shortening across age which is in keep with previous observation15; LGMD1B patients exhibited faster telomere loss compared to EDMD patients (Fig. 1D). Together, these results show that loss of LMNA results in telomere loss in cardiomyocytes.

A Paraffin-embedded cardiac samples of laminopathy patients (N = 13 patients; n = 130–540 nuclei per patient tissue), control patients (healthy: N = 6 patients; n = 91-186 nuclei per patient tissue, and transplanted hearts: N = 12; n = 133–887 nuclei per patient tissue) were used for myocardial telomere Q-FISH quantification. Telomere intensities (a.u.) were scored in a blinded fashion within five to six regions of interest in two nonconsecutive sections. Created in BioRender. Wang, C. (2025) https://BioRender.com/8hmmn43. B Telomere levels per patient is or (C) per genotype are plotted. Data represent mean ± SEM. Student’s t-test for (B) and one-way ANOVA tests for (C) were used to calculate significance. D Average telomere levels and age of laminopathy patients and healthy controls are plotted and best-fit linear curves are shown. Extremely low telomere LGMD1B sample (blue triangle) was considered an outlier and was not included for determining best-fit linear curve.
LMNA copy number plays a role in hiPSC and hiPSC-CM telomere length homeostasis
To study the effect of LMNA copy number and its relationship with telomere length homeostasis, we generated human induced pluripotent stem cells from a LMNA+/mut patient with a frameshift mutations in exon 1 that results in expression of a truncated protein (Fig. 2A)16. Using TALEN, isogenic lines were generated17,18. Under aberrant nuclear integrity19, we first asked if LMNA copy number changes (2 copies, LMNA+/+; 1 copy, LMNA+/mut; 0 copy LMNAmut/mut and LMNAmut/null) can affect telomere levels in both hiPSC and hiPSC-CMs (Fig. 2B). In hiPSCs, restoration of LMNA+/+ copy number lengthened telomeres compared to LMNA+/mut cells (Fig. 2C). Interestingly, loss of both LMNA copies (LMNAmut/mut and LMNAmut/null) slightly increased telomere levels in hiPSCs compared to LMNA+/mut, albeit not to the extent of LMNA+/+ cells (Fig. 2C). However, after differentiation into hiPSC-CMs, both LMNAmut/mut and LMNAmut/null cardiomyocytes exhibited a further telomere loss compared to LMNA+/mut (Fig. 2D). Together, these data support the idea that telomere length homeostasis is regulated by the nuclear envelope integrity, which is determined by LMNA expression and the mutational status.

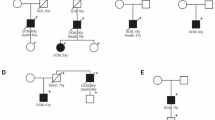
A Pt3, part of a three-generation family that had 15 affected members exhibiting autosomal dominant cardiac disease, used for the generation of hiPSC. Square, males; circles, females; slash, deceased. Pedigree originally reported in Heart Rhythm 2009 May; 6(5): 707–710. B Generation of hiPSC lines and subsequent hiPSC-CM differentiation. Representative micrographs of LMNA + / + , +/mut, mut/mut, and mut/null hiPSC-CMs stained with cardiac troponin cTnT (green), TelC telomeres (red), and nuclei (DAPI) are shown. C Telomere levels of hiPSC and (D) hiPSC-CMs were quantified. Data are shown as violin plots where blue median and gray quartiles are shown. One-way ANOVA tests by comparing the mean of each group with the mean of every other group, followed by Holm–Sídák multiple comparison test were used.
LMNA +/mut results in increased myocardial surface area and increased contractile force
To evaluate if changes in LMNA alters a cardiomyocyte’s ability to generate force when challenged with a fibrotic environment10, we subjected LMNA+/mut and LMNA+/+ hiPSC-CMs to traction force microscopy under electrical pacing at 1 Hz20. Single hiPSC-CMs were seeded on micropatterned hydrogel substrates to provide them a more physiological elongated shape and the hydrogel substrate stiffness was controlled to mimic healthy (10 kPa) and fibrotic (35 kPa) conditions. Interestingly, we observed that LMNA+/mut hiPSC-CMs displayed increased cell size (Fig. 3A) compared to LMNA+/+ hiPSC-CMs independently of substrate stiffness. Consequently, we measured that LMNA+/mut hiPSC-CMs produced overall more force compared to LMNA+/+ hiPSC-CMs (Fig. 3B), which was somewhat unexpected. However, while LMNA+/mut hiPSC-CMs also produced more strain energy under 10 kPa condition, (Fig. 3C); this was not the case when hiPSC-CMs were cultured under 35 kPa condition, which reveal an increased sensitivity to fibrotic stiffening in LMNA+/mut hiPSC-CMs. Since there was no difference in terms of average displacement (i.e., average hydrogel strain deformation) (Fig. 3D), the differences in terms of total force and strain energy between LMNA+/+ and LMNA+/mut can be attributed to the differences in cell size. Interestingly, cell size has been reported to correlate with nuclear size and inversely to lamin expression21. LMNA did not alter the contraction velocity (Fig. 3D, E) which suggests intact sarcomeric structure. However, under controlled electrical pacing conditions, we still observed small differences in average beating frequency (Fig. 3F), revealing potential arrhythmic contractions and deficient calcium handling capabilities. Together, these results suggest that expression of the LMNA truncated variant increases myocardial cell size and results in substrate-stiffness sensitive contractile force, strain energy generation, as well as in loss of beating rhythmicity.

Contractile assessment of LMNA+/mut and isogenic control LMNA+/+ hiPSC-CMs using traction force microscopy was performed on 10 kPa or 35 kPa hydrogels. (A) Force, (B) Displacement, (C) Contraction velocity, (D) Strain, (E) Area, and (F) beat rate of single LMNA+/+ and LMNA+/mut hiPSC-CMs subjected to 10 kPa and 35 kPa hydrogels under 1 Hz electrical pacing were measured (n = 3 independent experiments, 17–23 cells were analyzed). Data represent mean ± SEM. Student’s t-tests were used to calculate significance.
Cardiomyocytes from hLMNA-c.C1824T knockin mice also exhibit telomere shortening
To evaluate whether LMNA mutation or loss of expression can drive myocardial telomere shortening, we used heterozygous hLMNA-c.C1824T knockin mice to evaluate. Echocardiography assessment confirms an early onset of dilated cardiomyopathy in heterozygous hLMNA-c.C1824T knockin animals marked by decreased left ventricular ejection fraction (LVEF %) as well as fraction shortening (FS %) (Fig. 4A, B). In presence of hLMNA-c.C1824T mutant, myocardial telomere levels were significantly decreased in LMNA mice compared to controls (Fig. 4C). In Langendorff isolated cardiomyocytes, LMNA cardiomyocytes exhibit a significant decrease in cardiomyocyte shortening as well as sarcomeric baseline distance (Fig. 4D, E), indicative of contractile dysfunction. Together, these results suggest that LMNA mutation drives myocardial telomere attrition in heterozygous hLMNA-c.C1824T knockin mice.

Cardiac function evaluated by echocardiography. A Left ventricular ejection fraction (EF %) and (B) fractional shortening (FS %) at 6, 8, 10, 12, 14, 16, 18, and 20 weeks of age are shown. (n = 5 animals each). C Telomere intensities (a.u.) were scored in a blinded fashion within five regions of interest in two nonconsecutive sections per animal, five animals per group with a total of n = 126–138 nuclei per mouse scored. Quantification of (D) cardiomyocyte shortening and (E) sarcomeric baseline of Langendorff isolated cardiomyocytes are shown. Data are represented as mean ± SEM. Student’s t-tests were used to calculate significance.
Discussion
Here we demonstrate that laminopathic cardiomyocytes exhibit telomere loss and this phenomenon is recapitulated in patient hiPSCs. We observed that cardiomyocytes from patients classified with LGMD1B exhibit faster telomere shortening compared to EDMD patients while healthy controls exhibit very little telomere loss over chronological aging (Fig. 1C). Second, using TALENs17,18, we interrogated the relationship between LMNA copy number and telomere homeostasis. Using the four hiPSC lines, isogenic wildtype (LMNA+/+), patient-derived (LMNA+/mut), homozygous mutant (LMNAmut/mut) and knockout (LMNAmut/null), we demonstrate that telomere homeostasis is LMNA dependent and also cell-type specific. Telomeres are shorter in all LMNA mutated hiPSC lines compared to LMNA+/+ hiPSCs. Among the three LMNA mutational type, the homozygous LMNA+/mut mutation appeared to provides the least favorable nuclear integrity for telomere homeostasis in hiPSCs. However, upon differentiation into hiPSC-CMs, the high contractile demand in cardiomyocytes mostly impacted telomere level in LMNAmut/null and LMNAmut/mut, resulting in shorter telomeres in these lines compared to LMNA+/mut and control LMNA+/+ hiPSC-CMs. We also show on single cardiomyocyte level that LMNA+/mut hiPSC-CMs increase contractile force compared to LMNA+/+ hiPSC-CMs, an effect that appeared largely due to increased cell size. Myocardial telomere attrition was further validated in the hLMNA-c.C1824T heterozygous knockin mice. Overall, these results suggest that LMNA mutations alter telomere homeostasis and this phenomenon is cell-type and mutation-type specific, thereby correlating with the composition of the nuclear envelope and nuclear mechanotransduction. LMNA expression level has indeed been shown to affect the binding of the inner membrane protein emerin14 and of the structural Nesprins proteins22.
Compared to our previous studies where sarcomeric mutations drive telomere shortening in genetic DCM and HCM11 and that contraction of diseased cardiomyocytes promote telomere shortening10, it is clear from our results that disruption to LMNA expression, by various mutations, results in telomere attrition. Further, depending on mutation severity, the rate of telomere loss in laminopathic cardiomyocytes also differ. Although mice with extended telomeres exhibit less metabolic aging and have longer lifespans23, it remains elusive how telomere lengths are established and maintained. It has been demonstrated that regardless of starting cells, reprogramming resets the telomere length24 and this memory is kept in chimera mice derived from long telomere mESCs25. Furthermore, long telomeres, provided by C57BL/6 J mouse background, can override short telomeres, in CAST/Ei or SPRET/Ei background, and establish new telomere set points26 and it seems that telomere set points can be inherited27. These observations suggest that nuclear integrity, determined by composition of lamins, may determine telomere lengths in cells.
Second, it has been shown that telomeres are captured by the TERB1-TERB2-MAJIN complex and anchored onto nuclear envelope via SUN1 that regulates chromosomal rearrangements during meiosis28. Evidence from the Hockemeyer lab has demonstrated that TPP1 and POT1, two telomere binding proteins part of the shelterin complex, are required for telomere length homeostasis29,30,31 and TPP1 phosphorylation has been shown to initiate telomerase recruitment32. Whether these signaling pathways are actively present in non-dividing cardiomyocytes and whether these pathways are affected by LMNA disruption in the nuclear membrane integrity and nuclear mechanotransduction remains to be explored.
Third, unlike the heterozygous hLMNA-c.C1824T knockin mice, LMNA+/mut did not exhibit defective contractile force production, which raises the question of how do laminopathic cardiomyopathies develop? The C1824T mutation results in the production of progerin protein that is responsible for Hutchinson-Gilford progeria syndrome (HGPS), a rare genetic disorder mainly characterized by accelerated aging in children. Progerin, which lacks 50 amino acids on its carboxyl terminus, remain farnesylated at the cysteine of its CAAX sequence and prevents ZMPSTE24 protease cleavage to release it from the nuclear membrane33,34,35. To note, C1824T knockin mice36 and pigs37 have been shown to exhibit cardiac hypertrophy and diastolic dysfunction; whereas, LMNA deficient38 or myocardial LMNA knockout mice39 exhibit systolic dysfunction. It is known that isoflurane may suppress systolic function thus the systolic dysfunction we observed in our C1824T knockin animals warrant for further investigation of this new animal model. Transcriptionally, our group has previously demonstrated that LMNA loss can increase chromatin open state and non-sense mediated decay in laminopathic cardiomyocytes19. Enzymatically, it has been suggested that the leakiness of laminopathic nuclei can result in infiltration of matrix-metalloprotease-240. Moreover, overexpression of a dominant negative SUN1 protein was able to prevent laminopathic cardiomyopathy in mice39. However, whether all LMNA mutations can drive cardiac phenotype directly or compounded by endothelial dysfunction complications41 remains to be elucidated. To note, limited by tissue availability, we were unable to perform traditional telomeric repeat amplification protocol (TRAP) or RT-qPCR methods. The number of LMNA mutations examined are limited and further validations are warranted. In this study, we measured telomere signal using QFISH which captures relative telomeric length, but like all probe-based assays, are less sensitive to really short telomeres.
Together, this work provides evidence of accelerate telomere loss in human and murine laminopathic cardiomyocytes. We show that the level of telomere shortening may be correlated with disease severity. Using hiPSC and hiPSC-CMs as model, we demonstrate that truncated LMNA acts as a dominant negative in the pluripotent state, while the lack of full-length LMNA contributes to telomere attrition in differentiated cardiomyocytes. LMNA+/mut hiPSC-CMs exhibited larger cell size and produced more force when challenged with healthy and fibrotic-mimicking hydrogels, suggestive of altered cellular structure. Further, myocardial telomere attrition was further confirmed in the hLMNA-c.C1824T knockin mice. Our results provide the necessary tools in supporting the possibility of targeting telomeric ends in the treatment of laminopathic cardiomyopathies.
Methods
Human cardiac samples
The use of human samples was reviewed and approved by the Stanford Institutional Review Board (no. 13465). Deidentified laminopathic and engrafted heart sections were obtained from Normandie University, INSERM. Control hearts were isolated < 24 h post mortem from deidentified male patients who died of noncardiac disease at University of British Columbia. All tissue samples were formalin fixed, paraffin embedded, and 4 μm sections placed on ChemMate slides (Fisher Scientific) were used for telomere Q-FISH staining and telomere signal was quantified using Telometer as previously described in ref. 11. Histology was evaluated using hematoxylin and eosin staining (Fisher Scientific) per manufacturer’s instructions.
hiPSC-CM differentiation
All protocols using hiPSC were reviewed and approved by the Stanford Stem Cell Research Oversight committee (#602) as well as the Ethics Review committee at Ninth People’s Hospital, Shanghai Jiao.
Tong University School of Medicine (2018-207-K32). The LMNA patient hiPSC line was generated part of the Stanford Cardiovascular Institute Biobank where subsequent isogenic lines were generated as previously described17,18. hiPSC maintenance and hiPSC-CM differentiation were performed using the 2D matrigel protocol and traction force microscopy was used to evaluate contractile function as previously described10,11.
Animals
LMNA animals, C57BL/6 J hLMNA-c.C1824T knockin mice (Strain NO. T059801), were acquired from GemPharmatech (Nanjing, China). Pathogenic humanized LMNA point mutation at exon 11 (c.1824C>T [p.G608G]) was introduced via CRISPR-Cas9 construct according to supplier’s website (https://en.gempharmatech.com/product/details100035_4040548.html). Lethality was observed in homozygous C1824T animals; heterozygous animals were used in function and histological studies.
Animals were kept in an SPF barrier environment with a 12 h light/dark cycle and were provided with sufficient food and water. All animal studies complied with the Declaration of Helsinki, NIH guidelines, and were approved by the Animal Experiment Ethics Committee of Shanghai Ninth People’s Hospital (SH9H-2021-TK500-1). Mice were anaesthetized with intraperitoneal injection of 1.2% 2,2,2-tribromoethanol solution (0.2 mL/10 g body weight), monitoring respiratory and heart rates, and euthanized by CO2 overdose. Visual Sonics Vevo 3100 system (Visual Sonics, FUJIFILM) was used to perform echocardiographic assessment. During echocardiography, mice were anaesthetized with 2% isoflurane. Systolic functions were measured at the midventricular long-axis using M-mode scanning while maintaining the heart rate at the range of 425–475 beats per minute. 4 μm sections were used for telomere Q-FISH staining and telomere signal was quantified using Telometer as previously described in ref. 11. Murine cardiomyocytes were isolated using the Langendorff method as previously described in ref. 9 and an IonOptix HTC System (IonOptix) was used for function assessment. Briefly, Langendorff isolated AMCMs were seeded onto laminin (Sigma)-coated imaging dishes in DMEM supplemented with 10% FBS and incubated for 30 min at 37 °C. The cells were field stimulated (10 V) at a frequency of 1 Hz, and the changes of sarcomere length were simultaneously recorded and calculated.
Statistics
Statistical differences were analyzed using one-way ANOVA followed by Holm-Sidak’s multiple comparison test or by Student’s t-test as indicated. Image capture and quantification analyses were performed in a double-blinded fashion to avoid bias. All data are shown as the mean ± SEM. Significant differences were determined as p < 0.05.
Data availability
All data are available in the main text or the supplementary materials. The raw datasets used and/or analyzed during the current study are available from the corresponding author.
References
Bonne, G. et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet 21, 285–288 (1999).
Fatkin, D. et al. Missense Mutations in the Rod Domain of the Lamin A/C Gene as Causes of Dilated Cardiomyopathy and Conduction-System Disease. N. Engl. J. Med. 341, 1715–1724 (1999).
BÉCANE, H. et al. High Incidence of Sudden Death with Conduction System and Myocardial Disease Due to Lamins A and C Gene Mutation. Pacing Clin. Electrophysiol. 23, 1661–1666 (2000).
Muchir, A. et al. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum. Mol. Genet 9, 1453–1459 (2000).
Peretto, G. et al. Updated clinical overview on cardiac laminopathies: an electrical and mechanical disease. Nucleus 9, 380–391 (2018).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Bergmann, O. et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 161, 1566–1575 (2015).
Terai, M. et al. Association of telomere shortening in myocardium with heart weight gain and cause of death. Sci. Rep.-uk 3, 2401 (2013).
Chang, A. C. Y. et al. Telomere shortening and metabolic compromise underlie dystrophic cardiomyopathy. Proc. Natl. Acad. Sci. 113, 13120–13125 (2016).
Chang, A. C. Y. et al. Increased tissue stiffness triggers contractile dysfunction and telomere shortening in dystrophic cardiomyocytes. Stem Cell Rep. 16, 2169–2181 (2021).
Chang, A. C. Y. et al. Telomere shortening is a hallmark of genetic cardiomyopathies. Proc. Natl. Acad. Sci. 115, 201714538 (2018).
Gorgoulis, V. et al. Cellular Senescence: Defining a Path Forward. Cell 179, 813–827 (2019).
Makhija, E., Jokhun, D. S. & Shivashankar, G. V. Nuclear deformability and telomere dynamics are regulated by cell geometric constraints. Proc. Natl. Acad. Sci. 113, E32–E40 (2016).
Sullivan, T. et al. Loss of a-Type Lamin Expression Compromises Nuclear Envelope Integrity Leading to Muscular Dystrophy. J. Cell Biol. 147, 913–920 (1999).
Takubo, K. et al. Telomere lengths are characteristic in each human individual. Exp. Gerontol. 37, 523–531 (2002).
Pan, H. et al. A novel mutation in LAMIN A/C is associated with isolated early-onset atrial fibrillation and progressive atrioventricular block followed by cardiomyopathy and sudden cardiac death. Heart Rhythm 6, 707–710 (2009).
Termglinchan, V., Seeger, T., Chen, C., Wu, J. C. & Karakikes, I. Cardiac Gene Therapy, Methods and Protocols. Methods Mol. Biol. Clifton N. J. 1521, 55–68 (2016).
Karakikes, I. et al. A Comprehensive TALEN-Based Knockout Library for Generating Human-Induced Pluripotent Stem Cell–Based Models for Cardiovascular Diseases. Circ. Res 120, 1561–1571 (2017).
Lee, J. et al. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 572, 335–340 (2019).
Ribeiro, A. J. S. et al. Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proc. Natl. Acad. Sci. 112, 12705–12710 (2015).
Jevtić, P. et al. Concentration-dependent Effects of Nuclear Lamins on Nuclear Size in Xenopus and Mammalian Cells*. J. Biol. Chem. 290, 27557–27571 (2015).
Mounkes, L., Kozlov, S., Burke, B. & Stewart, C. L. The laminopathies: nuclear structure meets disease. Curr. Opin. Genet Dev. 13, 223–230 (2003).
Muñoz-Lorente, M. A., Cano-Martin, A. C. & Blasco, M. A. Mice with hyper-long telomeres show less metabolic aging and longer lifespans. Nat. Commun. 10, 4723 (2019).
Schaetzlein, S. et al. Telomere length is reset during early mammalian embryogenesis. Proc. Natl. Acad. Sci. 101, 8034–8038 (2004).
Varela, E., Muñoz-Lorente, M. A., Tejera, A. M., Ortega, S. & Blasco, M. A. Generation of mice with longer and better preserved telomeres in the absence of genetic manipulations. Nat. Commun. 7, 11739 (2016).
Hathcock, K. S. et al. Haploinsufficiency of mTR results in defects in telomere elongation. Proc. Natl. Acad. Sci. 99, 3591–3596 (2002).
Chiang, Y. J. et al. Telomere length is inherited with resetting of the telomere set-point. Proc. Natl. Acad. Sci. 107, 10148–10153 (2010).
Wang, Y. et al. The meiotic TERB1-TERB2-MAJIN complex tethers telomeres to the nuclear envelope. Nat. Commun. 10, 564 (2019).
Boyle, J. M. et al. Telomere length set point regulation in human pluripotent stem cells critically depends on the shelterin protein TPP1. Mol. Biol. Cell 31, 2583–2596 (2020).
Sexton, A. N. et al. Genetic and molecular identification of three human TPP1 functions in telomerase action: recruitment, activation, and homeostasis set point regulation. Gene Dev. 28, 1885–1899 (2014).
Kim, W. et al. Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. Embo J. 40, e107346 (2021).
Zhang, Y. et al. Phosphorylation of TPP1 regulates cell cycle-dependent telomerase recruitment. Proc. Natl. Acad. Sci. 110, 5457–5462 (2013).
Eriksson, M. et al. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature 423, 293–298 (2003).
Capell, B. C. et al. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. 102, 12879–12884 (2005).
Agarwal, A. K., Fryns, J.-P., Auchus, R. J. & Garg, A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum. Mol. Genet. 12, 1995–2001 (2003).
Subati, T. et al. Cardiomyocyte-Restricted Expression of Progerin Confers Cardiac Hypertrophy. Circ. Res. 137, 1374–1376 (2025).
Dorado, B. et al. Generation and characterization of a novel knockin minipig model of Hutchinson-Gilford progeria syndrome. Cell Discov. 5, 16 (2019).
Nikolova, V. et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C–deficient mice. J. Clin. Investig. 113, 357–369 (2004).
Chai, R. J. et al. Disrupting the LINC complex by AAV mediated gene transduction prevents progression of Lamin induced cardiomyopathy. Nat. Commun. 12, 4722 (2021).
Cho, S. et al. Mechanosensing by the Lamina Protects against Nuclear Rupture, DNA Damage, and Cell-Cycle Arrest. Dev. Cell 49, 920–935.e5 (2019).
Sayed, N. et al. Clinical trial in a dish using iPSCs shows lovastatin improves endothelial dysfunction and cellular cross-talk in LMNA cardiomyopathy. Sci. Transl. Med 12, eaax9276 (2020).
Acknowledgements
We thank Jon Mulholland and Cedric Espenel for expert microscopy assistance in the Nikon Spinning Disk Confocal microscope (funded jointly by the School of Engineering and the Beckman Center).This research was supported by National Natural Science Foundation of China (82070248 to A.C.Y.C.); Shanghai Pujiang Program (19PJ1407000 to A.C.Y.C.); The Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (0900000024 to A.C.Y.C.); Innovative Research Team of High-Level Local Universities in Shanghai (to A.C.Y.C.); the American Heart Association (13POST14480004 and 18CDA34110411 to A.C.Y.C.,18POST34080160 to G.P., 17MERIT33610009 to J.C.W., and 17CSA33590101 to H.M.B.); the Swiss National Science Foundation (SNSF) Early Postdoc Mobility Fellowship (#P2SKP2_164954 to G.P.); the National Institutes of Health (R01 AG044815 and R01 AR063963 to H.M.B.); the Baxter Foundation and the Li Ka Shing Foundation (to H.M.B).
Author information
Authors and Affiliations
Contributions
A.C.Y.C. and H.M.B. conceived and designed the research. A.C.Y.C., G.P., A.C.H.C., C.W., V.T., A.K., L.N., and F.B. performed the research. A.C.Y.C., G.P., A.C.H.C., and H.M.B. analyzed the data. A.C.Y.C., G.P., A.C.H.C., C.W., V.T., A.K., L.N., F.B., A.L., G.B., J.W. and H.M.B. contributed to the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
H.M.B. is an Associate Editor of npj Regenerative Medicine but is not part of a peer review process or decision making of the manuscript. The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, A.C.Y., Pardon, G., Chang, A.C.H. et al. Telomere shortening in laminopathic dilated cardiomyopathy. npj Regen Med 11, 16 (2026). https://doi.org/10.1038/s41536-026-00462-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41536-026-00462-1
