
Abstract
T-2 toxin is a typical mycotoxin that seriously threatens human and animal health. Liver is the major target organ of T-2 toxin. To elucidate the precise hepatotoxicity mechanism and discover a natural antagonist of T-2 toxin. T-2 toxin (0, 0.5, 1, 2 mg/kg BW)-induced liver injury model, Ferrostatin-1 (1 mg/kg·BW) interference model, Parkin−/− mice model, Nrf2-activating model (tBHQ, 20 mg/kg·BW) and lycopene (5 mg/kg·BW) treatment model were constructed. Proteomics revealed that ferroptosis is a critical hepatotoxicity mechanism of T-2 toxin. Blocking ferroptosis alleviated the liver damage and mitophagy under T-2 toxin threat. However, these processes were exacerbated in Parkin−/− mice. In vivo mouse model confirmed that Nrf2 activation increased PINK-Parkin mediated mitophagy and alleviated T-2 toxin-induced ferroptosis, suggesting that Nrf2/mitophagy axis was involved in T-2 toxin-induced hepatic ferroptosis. Further analysis revealed that lycopene promoted Nrf2 nuclear translocation and PINK-Parkin mediated mitophagy to mitigate T-2 toxin-induced hepatic ferroptosis.
Similar content being viewed by others


Introduction
T-2 toxin, an unavoidable contaminant, is a secondary metabolite of Fusarium in food and feedstuffs1. T-2 toxin is presented extensively in barley, wheat, maize and feeds based-on maize. The World Health Organization (WHO) and Food and Agriculture Organization (FAO) have classified T-2 toxin as a cause of foodborne diseases and an unavoidable food contaminant2. In a report from the FAO, T-2 toxin was listed as a mycotoxin that poses a great threat to food safety worldwide3. A global survey revealed a T-2 toxin-positive rate of 23% in 8721 agricultural commodities and the average T-2 toxin content was 25 mg/kg4. In feed samples from northern and eastern Europe, the detection rates of T-2 toxin were 30.3% and 48.2%, respectively4. In maize samples collected from New Zealand, the rate of T-2 toxin positivity reached 65%5. In 842 maize samples from Northeast China, 10% specimens were detected with T-2 toxin in which the highest concentration was up to 528.1 μg/kg6. T-2 toxin severely threatens human and animal health by poisoning the cardiovascular, digestive, skeletal, reproductive and nervous systems7,8,9,10,11. T-2 toxin has high structural stability and can maintain structural integrity and toxicity at various environments (such as high temperatures, strong acidic/alkaline conditions, ultraviolet irradiation and other conditions). Moreover, with the development of global warming, the threat of mycotoxins to public health is increasing12. Hence, illuminating the mechanisms of action and targets of T-2 toxin is critical for public health. The liver, the main site for energy metabolism, detoxification, and xenobiotic metabolism in body, become the target of mycotoxin toxicity13. In our previous review, we systematically summarized the structural and functional disorders induced by T-2 toxin hepatotoxicity and the recent advances in T-2 toxin hepatotoxicity14. Although oxidative imbalance, mitochondrial damage, and apoptosis are established modes of T-2 toxin hepatotoxicity15,16, the pivotal mechanism remains unclear.
As a regulated cell death process, ferroptosis is mechanistically unique compared to apoptosis, autophagy, and pyroptosis, which is only recently characterized with iron accumulation, lipid peroxidation, glutathione peroxidase 4 (GPx-4) depletion, and mitochondrial damage16. Ferric ions aggravate reactive oxygen species (ROS) accumulation through the Fenton reaction that initiates the ferroptotic cascade via induction of mitochondrial damage and lipid peroxidation17. When cells undergo ferroptosis, mitochondrial damage will be observed accompanied by mitochondrial cristae disintegrate, mitochondrial membrane rupture, and mitochondria shrink. Conversely, mitochondrial damage can exacerbate ROS production. Mitophagy is a selective autophagy pathway, through ubiquitin-mediated recognition (e.g., PTEN Induced Kinase 1 (PINK1)/E3 ubiquitin-protein ligase parkin (Parkin)) and lysosomal engulfment, maintaining cellular mitochondrial homeostasis, and intracellular ROS levels are decreased to ensure cell survival18,19. Hence, we speculated that imbalanced iron homeostasis and disordered mitophagy may be involved in the hepatotoxicity under T-2 toxin threat. NFE2 like bZIP transcription factor 2 (Nrf2) is an important molecule in cellular responses that regulates cellular redox homeostasis and exerts multiple cytoprotective mechanisms under stressful conditions by controlling multiple transcriptional targets20. Under physiological conditions, Nrf2 binds with kelch like ECH associated protein 1 (Keap1) in the cytoplasm, and the Nrf2 and Keap1 protein complex undergoes degradation via ubiquitination. Oxidative stress triggers Keap1 conformational changes which promotes Nrf2 dissociating from Keap1. Then, Nrf2 heterodimerizes with small MAF proteins in the nucleus to initiate target gene transcription. Nrf2 regulates multiple functions in the nucleus19. First, Nrf2 can increase antioxidant capacity to relieve oxidative stress by activating the transcriptional activity of antioxidant factors (such as heme oxygenase-1 (HO-1), NADPH:Quinone Oxidoreductase 1 (NQO1), catalase (CAT), and superoxide dismutase (SOD)). Second, Nrf2 controls cellular iron metabolism by transcriptionally coordinating various molecules (ferritin and solute carrier family 40 member 1 (SLC40A1)). Third, Nrf2 activation can increase mitophagy levels to maintain cellular mitochondrial homeostasis by increasing the expression of mitophagy-related molecules. As a transcription factor, Nrf2 may be the key upstream regulatory molecule that participates in T-2 toxin-induced hepatotoxicity via disturbing the cellular redox balance, ferroptosis and mitophagy.
Lycopene (LYC) is a lipophilic non-provitamin that is commonly seen in red-colored fruit and vegetables like apricots, peaches, watermelons, tomatoes, and carrots. LYC is constructed with 11 conjugated and 2 unconjugated double bonds that endow LYC with carotenoid properties and antioxidant activity21. As one of the most accepted antioxidants with no safety concerns, LYC has promise for inhibiting lipid peroxidation and promoting antioxidant capacity against liver injury. In addition, LYC has been shown to protect mitochondria from ROS damage and maintain a mitochondrial quality control system to maintain mitochondrial homeostasis22. LYC treatment can maintain mitochondrial homeostasis during mycotoxin exposure in the jejunum and intestinal cells23,24,25. Yang et al. reported that LYC alleviated hepatic mitochondrial stress induced by atrazine in the mouse26. LYC alleviated chicken hepatocyte injury induced by fumonisin B1 reported by Wang et al.27. Nevertheless, it is unknown whether LYC exerts a therapeutic impact on T-2 toxin hepatotoxicity and whether such an effect involves the Nrf2/mitophagy axis.
This article was designed to reveal the role and nuanced interplay of ferroptosis and Nrf2/mitophagy axis in the context of liver injury under T-2 toxin threat and to further explore the ameliorate impact of LYC on alleviating T-2 toxin hepatotoxicity. The results sought to elucidate the hepatotoxic mechanism of T-2 toxin and provide an in vivo reference for the application of LYC to resist mycotoxin-induced hepatotoxicity.
Results
T-2 toxin caused liver injury in mice
Weight gain significantly decreased (P < 0.05), while the liver coefficient and serum AST and ALT levels increased in a concentration-dependent manner with 28 d T-2 toxin gavage, compared with CG (Fig. 1A, B). During the autopsy, hypertrophic and yellow-stained livers were observed in groups exposed to T-2 toxin (Fig. 1C). HE staining of liver sections revealed varying degrees of apoptosis along the hepatic cord, disorganized arrangement of intrahepatic biliary epithelial cells around the central vein, and hepatocyte swelling with cytoplasmic vacuolation in T-2 toxin groups, and the Ishak score increased with T-2 toxin administration. Furthermore, sinusoid congestion and blurred hepatocyte boundaries in the HG indicated that pathological alterations were aggravated with increasing T-2 toxin doses. The Ishak score elevated in a concentration-dependent manner under T-2 toxin threat (Fig. 1B, C). T-2 toxin caused hepatic structural and functional disorder in a concentration-dependent manner. To further elucidate the T-2 toxin-induced hepatotoxic mechanism, we randomly selected liver samples from the CG and MG for proteomic sequencing and analysis. Principal component analysis (PCA) revealed sample clustering in the groups and scattering between the groups, distinguishing the MG from the CG (Fig. 1D). Through functional annotation and quantitative analysis, a total of 4752 proteins were identified; 175 proteins were significantly enriched and considered as DEPs (FC > 1.2, P < 0.05), among which 94 proteins were elevated, and 81 proteins were downregulated (Fig. 1E, F). In the Gene Ontology (GO) database (P < 0.05), the DEPs were enriched mostly in oxidation‒reduction processes, metabolic processes, and iron binding (Fig. 1H). Moreover, these DEPs were involved in glutathione metabolism, ferroptosis and unsaturated fatty acid metabolism according to KEGG annotation (P < 0.05) (Fig. 1G). Among the top 30 enriched KEGG pathways, ferroptosis was the only pathway that was directly linked to cell death. Moreover, other significantly enriched pathways, such as glutathione metabolism and lipid metabolism, were closely related to ferroptosis in the liver. The quantitative analysis also revealed that proteins related to ferroptosis, especially FTH and FTL, were significantly differentially expressed (Fig. 1I). Overall, the above analysis revealed that T-2 toxin cause liver damage through inducing ferroptosis.

A A schema diagram of C57 mice liver injury model. B Quantification of weight, liver coefficient, AST activity, ALT activity and Ishak score. C Representative images of liver autopsy photos and HE staining (Scale bar = 50 µm). Red triangle: inflammatory cell infiltration, Yellowe triangle: nuclear condensation, Blue triangle: cellular vacuolization. D–I The proteomic analysis of different treatment, CLP represents CG group, MLP represents MG group. (D) The PCA analysis of different treatment. E Quantification of protein expression expressed as Log2 Fold Charge present as volcano plot. The DEPs were statistically analyzed by T-test and P < 0.05 was considered significant, P < 0.01 was considered highly significant. F Cluster analysis of different groups presents as heatmap. G Enrichment analysis of DEPs using KEGG database to annotate show in bubble chart. H Enrichment analysis of DEPs using GO database to annotate show in bar chart. I Heatmap showing Ferroptosis related DEPs expression after Z-score normalization between CG and MG. Error bars are standard deviation (SD) (n = 3). Statistical analysis was done using one-way ANOVA; *, P ≤ 0.05; **, P ≤ 0.001 comparing to CG.
T-2 toxin induced hepatic ferroptosis by promoting iron metabolism disorder in mice
Based on the proteomic results, several ferroptosis indicators were evaluated. Compared with those in the CG, the iron content and MDA and 4-HNE levels were increased, whereas GPx-4 activity was decreased in the T-2 toxin groups (Fig. 2A). Based on the enriched KEGG pathways and DEPs in proteomic results, the molecular levels of related molecules were quantified (Fig. 2B). The protein expression levels of GPx-4 and SLC40A1 decreased, whereas those of FTH and FTL increased markedly in a concentration-dependent manner (P < 0.05). Compared with CG, the fluorescence intensity of FTH increased in a concentration-dependent manner in the T-2 toxin groups (Fig. 2C). Interestingly, T-2 toxin showed no markedly alteration in transferrin receptor (TFRC) expression (Fig. 2B). These results confirm that ferroptosis was triggered by the iron metabolism imbalance in the mouse liver under T-2 toxin threat.

A Quantification of liver iron content, GPx-4 activity, 4-HNE and MDA level. B Quantification of GPx-4, TFRC, SLC40A1, FTH and FTL expression normalized by GAPDH expression in liver injury model present as bar graph with representative images of western blot results. C Representative images of PB + DAB-stained liver section and immunofluorescence experiments of liver section stained for FTH and DAPI. D A schema diagram of ferroptosis inhibition model by intraperitoneal injection Fer-1 daily for 28 days using C57 mice. E Graph shows Ishak score in ferroptosis inhibition model. F Representative images of HE stained liver section in ferroptosis inhibition model (Scale bar = 50 µm). Red triangle: inflammatory cell infiltration, Yellowe triangle: nuclear condensation, Blue triangle: cellular vacuolization. G Quantification of liver iron content, GPx-4 activity, 4-HNE and MDA level in ferroptosis inhibition model show as bar graph. (H) Quantification of GPx-4, TFRC, SLC40A1, FTH and FTL expression normalized by GAPDH expression in ferroptosis inhibition model present as bar graph with representative images of western blot results. Error bars are standard deviation (SD) (n = 3). Statistical analysis was done using one-way ANOVA; *, P ≤ 0.05; **, P ≤ 0.001 comparing to CG group, #, P ≤ 0.05; ##, P ≤ 0.001 comparing to T-2 group.
Fer-1 is a ferroptosis inhibitor with antioxidant activity that can prevent oxidative membrane damage and inhibit ferroptosis. Thus, Fer-1 was examined to confirm the occurrence of ferroptosis in the mouse liver under T-2 toxin threat (Fig. 2D). HE staining revealed significant remission of hepatocyte swelling and cytoplasmic vacuolation and lower Ishak scores after the intraperitoneal injection of Fer-1 in the T-2 toxin groups (Fig. 2E, F). Fer-1 treatment partly reversed the elevation in 4-HNE and MDA levels and the reduction in GPx-4 activity (Fig. 2G). The iron content and expression of ferroptosis-involved proteins were subsequently quantified and showed that the injection of Fer-1 reversed the decreased expression of GPx-4 and SLC40A1 but increased FTH and FTL protein expression (Fig. 2H). These results further confirm the occurrence ferroptosis in the mice liver under T-2 toxin threat.
T-2 toxin induced mitochondrial damage and initiated PINK1-Parkin-mediated mitophagy
Mitochondria, as the centre of energy metabolism, also serve as targets of T-2 toxin upon exposure, which contributes immensely to ferroptosis. T-2 toxin exposure markedly increased ROS and cytochrome C levels in the cytoplasm and prominently decreased T-AOC and ATP content, indicating that T-2 toxin exposure severely impaired mitochondria (P < 0.05) (Fig. 3A, C). Zhang et al. reported that the mitophagy of PINK-Parkin-mediated pathway alleviated T-2 toxin-induced nephrotoxicity in mice1. In our research, the level of proteins in PINK-Parkin mediated pathway markedly escalated, suggesting that PINK-Parkin-mediated mitophagy was initiated under T-2 toxin threat (P < 0.05) (Fig. 3B). Parkin and LC3 colocalization also increased under T-2 toxin threat (Fig. 3C). We further evaluated ultrastructural damage by TEM. As shown in Fig. 3D, nuclear shrinkage and chromatin condensation were observed in the T-2 toxin groups. Moreover, compared with the normal structure of the rough endoplasmic reticulum and mitochondria in the CG, T-2 toxin exposure caused rough endoplasmic reticulum dilatation, the mitochondrial cristae disappearance and the mitochondrial membrane rupture. The occurrence of mitophagosomes in the T-2 toxin groups was in accordance with the expression patterns of mitophagy-related molecules. Overall, T-2 toxin damaged the mitochondria and activated PINK-Parkin-mediated mitophagy in the mouse liver.

A Liver ROS fluorescence, T-AOC activity, ATP activity and Cytochrome C level in liver injury model. B Mitophagy related protein expression (LC3, p62, PINK1 and Parkin), total protein normalized by GAPDH expression, mitochondrial protein normalized by COX IV with representative images of western blot results. C Representative images of ROS fluorescence results and LC3-Parkin immunofluorescence experiments using liver sections in liver injury model. D Representative images of liver TEM using liver samples in control group and T-2 toxin group. Error bars are standard deviation (SD) (n = 3). Statistical analysis was done using one-way ANOVA; *, P ≤ 0.05; **, P ≤ 0.001 comparing to CG. E A schema diagram of mitophagy inhibition model using wild type C57 mice and Parkin−/− C57 mice. F Charts of liver coefficient and Ishak score. G Representative images of HE stained and PB + DAB-stained liver section (Scale bar = 50 µm). Red triangle: inflammatory cell infiltration, Yellowe triangle: nuclear condensation, Blue triangle: cellular vacuolization. H Liver iron content, GPx-4 activity, 4-HNE and MDA level. I, J Mitophagy and ferroptosis related protein expression, total protein normalized by GAPDH expression, mitochondrial protein normalized by COX IV with representative images of western blot results. Error bars are standard deviation (SD) (n = 3). Statistical analysis was done using one-way ANOVA; *, P ≤ 0.05; **, P ≤ 0.001 comparing to CG group, #, P ≤ 0.05; ##, P ≤ 0.001 comparing to T-2 group.
Inhibiting PINK-Parkin mediated mitophagy exacerbated T-2 toxin-induced hepatic ferroptosis
Mitophagy is proven to be a double-edged sword. PINK–Parkin-mediated mitophagy regulates mitochondrial homeostasis. Parkin exhibits E3 ubiquitin ligase activity that can mark damaged mitochondria and initiate mitophagy. By knocking out Parkin, the ubiquitination of PINK1 on the mitochondrial outer membrane is blocked that further affects the sequestosome 1 (p62)-LC3 recruitment and inhibits the level of mitophagy in the mitochondria. To reveal how mitophagy affects hepatic ferroptosis under T-2 toxin threat, Parkin−/− mice were utilized in our study (Fig. 3E). The liver coefficient was significantly increased under T-2 toxin threat after blocking mitophagy (P < 0.05) (Fig. 3F). Histological alterations were observed through HE staining; compared with observations in the WT mice, after blocking mitophagy, the arrangement of the hepatic cord was more disorganized, vacuolation in the cytoplasm was more severe, and the Ishak score was increased in the PT mice (Fig. 3G, F). Iron accumulation was also increased after the inhibition of mitophagy, as shown in Fig. 3G, H. GPx-4 activity and 4-HNE and MDA levels were markedly different in PT mice than in WT mice (Fig. 3H). The protein expression of GPx-4 and SLC40A1 was consistent with the decreasing trend above. Interestingly, FTH and FTL expression were significantly decreased, possibly because severe damage to hepatocytes caused protein synthesis disorders (P < 0.05) (Fig. 3J). Overall, the inhibition of mitophagy by Parkin knockout exacerbated hepatic ferroptosis under T-2 toxin threat.
Nrf2 activation alleviated T-2 toxin-induced hepatic ferroptosis by inducing mitophagy
The above results exhibited that cellular iron accumulation led to ferroptosis in the liver under T-2 toxin exposure. Additionally, T-2 toxin exposure impaired mitochondria and activated mitophagy as a self-defense response. By blocking mitophagy, the protective function of mitophagy was confirmed under T-2 toxin exposure. Hence, identifying upstream regulators of mitophagy and ferroptosis is highly important. Using oxidative stress, iron metabolism, ferroptosis and mitophagy as key words in a GeneCards database search, 587 target molecules were selected (Fig. 4A). The molecules were found to be involved mainly in the response to oxidative stress and the regulation of ROS metabolic processes as shown in Fig. 4B (P < 0.05). The interactions between molecules were further analysed with a focus on oxidative homeostasis-related pathways in the GO database. As shown in Fig. 4C, TNF, EGFR, IL-6, RELA, Nrf2, transcription factor JUN and SRC were screened, revealing their significant roles in the pathways mentioned above. The TRRUST results revealed that Nrf2 had a transcriptional regulatory effect, which ranked first in terms of the p value (Fig. 4D). Nrf2, a transcription factor, contributes to redox balance, iron homeostasis, and mitophagy and has the potential to alleviate T-2 toxin-induced hepatotoxicity. Through Nrf2 immunofluorescence, we detected increased Nrf2 translocation and activation along with related genes, as shown in Fig. 4E, F, G. To elucidate the effect of Nrf2 on protecting hepatocytes from T-2 toxin-induced ferroptosis, the Nrf2 activator tBHQ was used (Fig. 4H). Compared with those in the T-2 groups, the histological alterations and Ishak scores in the tBHQ group were reversed (Fig. 4I). The iron content and 4-HNE and MDA levels were decreased, whereas GPx-4 activity was restored with tBHQ injection (Fig. 4L). Nrf2 protein expression was prominently elevated both in the plasma and in the nucleus, as mentioned above (P < 0.05) (Fig. 4J). In the downstream pathway of Nrf2, the molecular level of mitophagy-related genes and proteins significantly upregulated after tBHQ treatment (P < 0.05), which led to the alleviation of ferroptosis under T-2 toxin threat in the mice liver (Fig. 4K-N and Fig. S2). These results indicated that activating Nrf2 protected the mouse liver from ferroptosis by promoting mitophagy under T-2 toxin threat.

A Venn figure of oxidative stress, iron metabolism, ferroptosis and mitophagy related molecules from GeneCards database. B Pathway and process enrichment analysis network of related molecules. (P ≤ 0.05) C Molecular interaction network of related molecules. (P ≤ 0.05) D Top 20 results of TRRUST analysis. (P ≤ 0.05) E Quantification of Nrf2 protein expression normalized by GAPDH using total liver protein, and Nu-Nrf2 normalized by Histone using liver nuclear protein with representative images of western blot results. F Heatmap shows the quantification of Nrf2 related gene expression (Nrf2, Keap1, HO-1 and NQO1). G Representative images of immunofluorescence of liver section stained for Nrf2 and DAPI. H A schema diagram of Nrf2 activation model by intraperitoneal injection tBHQ daily for 28 days using C57 mice. I Representative images of HE stained liver section in and Ishak score in Nrf2 activation model (Scale bar = 100 µm). Red triangle: inflammatory cell infiltration, Yellowe triangle: nuclear condensation, Blue triangle: cellular vacuolization. J Quantification of Nrf2 protein expression normalized by GAPDH using total liver protein, and Nu-Nrf2 normalized by Histone using liver nuclear protein with representative images of western blot results from Nrf2 activation model. K Heatmap of the quantification of Nrf2 related gene expression in Nrf2 activation model. L Quantification of liver iron content, GPx-4 activity, MDA and 4-HNE level in Nrf2 activation model. M, N Mitophagy and ferroptosis related expression, total protein normalized by GAPDH expression, mitochondrial protein normalized by COX IV. Error bars are standard deviation (SD) (n = 3). Statistical analysis was done using one-way ANOVA; *, P ≤ 0.05; **, P ≤ 0.001 comparing to CG group. #, P ≤ 0.05; ##, P ≤ 0.001 comparing to T-2 group.
LYC alleviated T-2 toxin-induced hepatic ferroptosis by activating Nrf2
Above results indicated that activating the Nrf2 pathway could have a protective impact on alleviating T-2 toxin-induced hepatic ferroptosis in mice. Naturally, a substance that activates Nrf2 may be the ideal medication for T-2 toxin poisoning. In our previous research, LYC showed a favorable effect on relieving oxidative damage and restored mitochondrial homeostasis23. LYC has been reported to activate Nrf228. To further elucidate the potential remission impact and underlying mechanism of action of LYC in the context of T-2 toxin exposure, bioinformatic research was conducted. Simulated molecular docking revealed the binding of LYC and Nrf2 through hydrophobic interactions (−6.9 ± 0.5 kcal/mol). The binding results, affinity and pKi are shown in Table S5. LYC binds to the leucine zipper (514–521) of Nrf2 at positions 514, 517, 518 and 521, which may influence the dimerization of Nrf2 with small musculoaponeurotic fibrosarcomas (small MAFs) and reinforce its binding with AREs to activate the transcription of downstream genes (Fig. 5A, B; Table S4, S5). The molecular docking results also revealed that LYC can bind with Keap1 and Nrf2 by hydrophobic bonds, as shown in Fig. 5C and D. Keap1 is closely related to Nrf2 activation. Kelch repeats are involved in the connection of Keap1 and Nrf2 to prevent activation of the Nrf2 pathway. LYC interacts with multiple residues (329, 342, 355, 356, 369, 373, 374, and 376) in the Kelch1 (327–372) and Kelch2 (373–423) domains with a moderate affinity (−5.7 kcal/mol), which may competitively promote the releasing of Nrf2 from Keap1 and then further promote Nrf2 translocation (Fig. 5C, D; Table S6).

A 3D structure docking simulation of LYC-Nrf2. B 2D binding sites of LYC-Nrf2. C 3D structure docking simulation of LYC-Keap1. D 2D binding sites of LYC-Keap1. (E) A schema diagram of LYC treatment model by giving LYC daily for 28 days to C57 mice. F Mice weight, liver coefficient, AST activity, ALT activity and Ishak score. G Representative images of liver autopsy photos, HE stained liver section and ROS fluorescence results (Scale bar = 50 µm). Red triangle: inflammatory cell infiltration, Yellowe triangle: nuclear condensation, Blue triangle: cellular vacuolization. H Liver iron content, GPx-4 activity, MDA and 4-HNE level from 3 individual samples. I Nrf2 protein expression normalized by GAPDH using total liver protein, and Nu-Nrf2 normalized by Histone using liver nuclear protein present in bar graph with representative images of western blot results. J Heatmap shows the quantification of Nrf2 related gene expression in Nrf2 activation model. K Representative images of immunofluorescence experiments stained for FTH, Nrf2 and LC3-Parkin. L, M Mitophagy and ferroptosis related protein expression, total protein normalized by GAPDH, mitochondrial protein expression normalized by COX IV. Error bars are standard deviation (SD) (n = 3). Statistical analysis was done using one-way ANOVA; *, P ≤ 0.05; **, P ≤ 0.001 comparing to CG group, #, P ≤ 0.05; ##, P ≤ 0.001 comparing to T-2 group.
By treating C57 mice with 5 mg/kg LYC, the protective effect of LYC was further evaluated (Fig. 5E). During autopsy, the changes in the color of the liver and the Ishak score were reversed in the L + T group compared with the Con group, as shown in Fig. 5F, G. The body weight, liver coefficient, and AST and ALT activity levels improved markedly after LYC was administered (P < 0.05) (Fig. 5F). The histological alterations, especially cytoplasmic vacuolation and disorganized hepatic cord arrangement, were significantly alleviated (Fig. 5G). LYC reversed the hepatic structural and functional disorder under T-2 toxin threat in the liver. Moreover, LYC treatment significantly reduced ROS accumulation, as shown in Fig. 5G. The iron content and 4-HNE and MDA levels were reduced, while GPx-4 activity was restored, indicating alleviation of hepatic ferroptosis with LYC treatment (Fig. 5H). The Nrf2 translocation and increased expression of Nrf2 down-stream genes further confirmed the activation of Nrf2 (Fig. 5I, J, K). The molecular levels of FTH, FTL, SLC40A1 and GPx-4 were consistent with the results described above (Figs. 5L and S3A). The LC3, p62, PINK1 and Parkin expression levels and the colocalization of LC3 and Parkin were markedly increased in the T-2, L + T and LYC groups, as shown in Fig. 5K, M, indicating that LYC promoted mitophagy (Fig. S3B). These results showed that LYC treatment reversed the ferroptosis and mitochondrial damage under T-2 toxin threat and initiated Nrf2 and mitophagy to antagonize T-2 toxin-induced hepatotoxicity.
Discussion
T-2 toxin, listed as a common environmental and food contaminant, poses great threats to global health and food security, especially in the context of global warming12. The liver is the key metabolizing site of T-2 toxin and T-2 toxin targeted site. Despite extensive research in T-2 toxin hepatotoxicity the dominant toxic mechanism remains unclear14. To comprehensively elucidate the T-2 toxin toxic mechanism in its hepatotoxicity, proteomic analysis was conducted. The proteomics results indicated that ferroptosis may be the key final event in hepatotoxicity under T-2 toxin threat. Ferroptosis is a unique iron-reliant cell death program marked by three hallmarks: iron metabolism dysregulation, mitochondrial injury, and uncontrolled ROS generation29. He et al. confirmed that T-2 toxin caused testicular impairment through oxidative stress and ferroptosis30. A study reported the occurrence of ferroptosis through lipid ROS production and the reduction of SLC7A11 under T-2 toxin threat, which is the key molecule of system Xc- in vitro31. In-depth research revealed the inhibition on system Xc- from T-2 toxin, which leads to ferroptosis in articular chondrocytes32. Although these studies laid a solid foundation for studies on T-2 toxin-induced ferroptosis, they focused mainly on imbalances in system Xc-, whereas imbalances in iron metabolism have rarely been studied.
Iron metabolism is regulated by iron ion transportation and chelation. TFRC mediates the endocytosis of iron complex proteins to maintain iron transfer into cells. Redundant iron either binds with FTH and FTL to form ferritin and maintain cellular iron homeostasis or is transported out of the cell through SLC40A1. SLC40A1 is the only iron exporter protein in mammalian cells based on current research. A decrease in SLC40A1 expression impedes iron excretion and iron accumulation in cells. Hence, TFRC, ferritin and SLC40A1 are considered landmark indicators of iron metabolism. T-2 toxin exposure increased the liver iron content through damage of iron metabolism balance in this study. Interestingly, the expression of TFRC did not differ under T-2 toxin threat, whereas T-2 toxin decreased SLC40A1 expression, indicating that interfering with iron excretion instead of entry may be T-2 toxin’s toxic action. By inhibiting ferroptosis using Fer-1, T-2 toxin-induced liver damage was reversed functionally and histologically, confirming that ferroptosis mediated T-2 toxin hepatotoxicity via impaired iron efflux, resulting in pathological iron retention.
Mitochondrial damage is considered a feature of ferroptosis. Mitochondria constitute the main source of ROS since they are responsible for multiple physiological processes, such as energy metabolism, fatty acid synthesis and signal processing. Damage to mitochondria can cause leakage of cytochrome C and energy production disorders, eventually leading to ROS overproduction and contributing to ferroptosis. In this study, T-2 toxin disordered mitochondrial structure and function and further led to mitophagy activation. As a selective autophagic process, mitophagy maintains cellular mitochondrial integrity through the specifically removal of defective mitochondria33. The key pathway of mitochondrial homeostasis and functionality is PINK1-Parkin-mediated mitophagy. By recognizing PINK1 on the outer membrane of depolarized mitochondria, Parkin ubiquitinates then recruits the autophagy receptor p62 to engulf damaged mitochondria, which further promotes the progression of autophagy. Other studies have reported the important role of PINK-Parkin-mediated mitophagy in other organs; however, the regulatory mechanism remains unclear1. In this study, the mitophagy of PINK1-Parkin-mediated pathway was observed under T-2 toxin threat in the mouse livers. To further elucidate the function of mitophagy in liver ferroptosis under T-2 toxin threat, Parkin−/− mice were used. Parkin plays a crucial role in mitophagy progression for its E3 ubiquitin ligase property. By knocking out Parkin, the mitophagy activated by T-2 toxin was blocked, which caused even more damage to the liver and increased the levels of ferroptosis and mitochondrial injury. Overall, the activation of mitophagy via PINK1-Parkin mediated pathway was elevated against the ferroptosis under T-2 toxin threat.
In face of various dangerous substances and attack signals, cells have evolved exquisite regulatory mechanisms. In view of the close connections and crosstalk among mitophagy, iron metabolism and redox balance, these biological processes may be regulated by a mutual upstream molecule. Hence, uncovering the upstream comediator is important for revealing the hepatotoxic mechanism under T-2 toxin threat. Through bioinformatic analysis, Nrf2, Rela and Jun were selected as potential comediators, with Nrf2 ranked as the first transcriptional regulator. Nrf-2 is a classic self-defense transcription factor involved in antioxidant defense, mitophagy, and iron metabolism processes in cells34. T-2 toxin-induced ROS accumulation can activate cellular defense mechanisms, including Nrf2 activation, which was also observed in this study. Once the activation signal is received, Keap1 releases Nrf2 of its translocation into the nucleus to bind with AREs as a heterodimer form with small MAFs regulating target gene transcription. Nrf2 enhances cellular antioxidant capacity by transactivating antioxidant genes while promoting mitophagy via PINK1 and p62 upregulation. Although Nrf2 sensitivity differs across different organs, Peng et al. reported that Nrf2 promoted PINK1 expression in vitro, which was consistent with our findings35. The regulation of Nrf2 induced by T-2 toxin was distinctively reviewed in Wang’s review9, and Nrf2’s contributions to maintaining mitochondrial quality control (via mitophagy), iron homeostasis, and redox equilibrium in T-2 toxin hepatotoxicity remain incompletely characterized and warrants deeper investigation. By introducing the Nrf2 activator tBHQ into a T-2 toxin model, the contribution of Nrf2 activation on the hepatotoxicity under T-2 toxin threat was evaluated. The activation of Nrf2 reversed the cellular oxidative stress state and enhanced the transcription of iron homeostasis-related factors by restoring the excretion of iron and maintaining cellular iron homeostasis under T-2 toxin exposure. Additionally, Nrf2’s role in regulating mitophagy is accomplished via the PINK1-Parkin pathway. Studies showed that Nrf2 regulated PINK1 transcription in tubular cells in diabetic kidney disease36. tBHQ alleviated the mitochondrial damage in hepatocytes and activated mitophagy via PINK1-Parkin-mediated pathway under T-2 toxin exposure. However, mitophagy may be a result of multiple factors, and the specific role and level of Nrf2 influence in mitophagy still need more detailed evaluation.
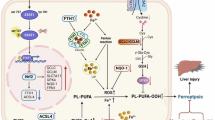
These findings indicate that T-2 toxin caused ferroptosis in mouse livers by inhibiting iron export and lipid peroxidation, leading to the retention of iron and ROS. As targets of iron and ROS, these accumulation severely injures mitochondria, and activated mitophagy via PINK1-Parkin pathway which contributed a protective role under T-2 toxin threat (Fig. 6). Bioinformatic analysis further revealed Nrf2 as the key transcriptional regulatory factor. The activation of Nrf2 and its protective effect on increasing mitophagy were verified through gain-of-function experiments using the Nrf2 agonist tBHQ. Therefore, the discovery of natural Nrf2 agonists would be beneficial for reducing T-2 toxin hepatotoxicity via practical production processes.

A schematic diagram of the mechanism of lycopene mitigates T-2 toxin induced hepatic ferroptosis by targeting Nrf2/mitophagy axis.
Lycopene, a natural antioxidant, has been shown to have a therapeutic effect on multiple organ damage37,38, and has confirmed as a potential activator for Nrf228. Hence, LYC is selected as a candidate to alleviate T-2 toxin induced hepatotoxicity. Hence, LYC was selected as a candidate to ameliorate the hepatotoxicity of T-2 toxin. However, the LYC-mediated mechanisms of Nrf2 activation is limited to the anti-inflammatory and antioxidant effects of LYC37. To further determine whether LYC activates Nrf2 through direct or indirect interactions, simulations of the molecular docking of LYC with Nrf2 and Keap1 were conducted. The activation of Nrf2 closely depends on the dissociation of Nrf2 and Keap1. Under normal physiological conditions, the Kelch repeat of Keap1 interacts with the DLG and ETGE segments of Nrf2. Through the action of CULLIN3 (CUL3), an E3 ubiquitin ligase, Nrf2 is subjected to proteasomal degradation. Blind molecular docking computationally probes binding affinities and modes of small molecules across a protein’s entire surface. These interactions include hydrogen bonds, hydrophobic bonds, halogen bonds and other intermolecular forces. Affinity is a measure of the binding ability between molecules. LYC binds with the Kelch 1 and Kelch 2 segments of Keap1 with medium affinity, which may contribute to the releasing of Nrf2 from Keap1 and promote the Nrf2 translocation. In addition, the leucine zipper of Nrf2 more closely binds with LYC, which may facilitate the dimerization of Nrf2 and small MAFs, further facilitating the binding of Nrf2 with ARE domains and activating the transcription of downstream genes. The Nrf2 activation induced by LYC is realized through the binding of Nrf2 or Keap1 structures. With the addition of LYC, liver damage was reversed, and ferroptosis levels were reduced in vivo with T-2 toxin threat. The activation of Nrf2 and the downstream pathway of PINK1-Parkin-mediated mitophagy were both elevated, which offers primary evidence that the protective effects of LYC may be realized through the Nrf2/mitophagy axis. The ability of LYC to promote mitophagy and act as an Nrf2 agonist have been verified in other studies23,37,39. The sophisticated mechanisms, however, remain inconclusive. Although the protective mechanism of LYC based on the Nrf2/mitophagy axis still needs further investigation, this preliminary research provides a new perspective on how LYC activates Nrf2 through a direct binding mechanism.
In summary, T-2 toxin elicited mitochondrial damage and iron export disorder, resulting in hepatic ferroptosis. The activation of the Nrf2/mitophagy axis was confirmed to resist ferroptosis under T-2 toxin exposure. We found that LYC likely had potential Nrf2 agonist-like activity. LYC could alleviate T-2 toxin-induced hepatotoxicity, possibly through activating Nrf2 to further promote mitophagy and maintain iron homeostasis (Fig. 6). This research uncovers the potential targets for developing hepatoprotective agents against T-2 toxin hepatotoxicity using natural plant extracts. However, the therapeutic effects of LYC in clinical trials need to be further investigated in the near future.
Methods
Animals and treatment
Wild-type C57 mice and Parkin−/− C57 mice, aged 6 to 8 weeks, were obtained from Henan Medical Laboratory Animal Center (Zhengzhou, China) and randomly divided into 18 groups (10 mice/group). Random numbers were generated using the standard = RAND() function in Microsoft Excel. The animals were maintained in pathogen-free conditions in the Laboratory Animal Center of Henan Agricultural University. The animals were kept in plastic cages (5 animals per cage) and maintained in an environmentally controlled room (temperature 24 °C, relative humidity 60%, and 12 h light/dark cycle) with free access to fresh solid pellet diet and water. The animal procedures were refer to ARRIVE guidelines and approved by the Animal Ethics Committee of the Henan Agricultural University (Approval number: HNND2021030801). At the end of the experiment, the mice were briefly anesthetized with 3–4% isoflurane (792632, Merck Ltd, Beijing, China) and then euthanized by gradual-fill CO2 (Qiangyuan Chemical Co., Ltd, Zhengzhou, China) asphyxiation (30–70% chamber volume per minute). Death was confirmed by the absence of cardiac and respiratory activity for ≥2 min. Subsequently, serum and liver tissue were immediately collected for further experiments. Serum was obtain by centrifuging (1200 × g, 15 min). The body and liver weights were recorded to calculate the liver coefficient. The sample storage condition is −80 °C.
For the T-2 toxin experiments, 40 wild-type C57 mice were randomly divided into 4 groups (CG, LG, MG, HG, n = 10) and treated daily with 0, 0.5, 1 or 2 mg/kg body weight (BW) of T-2 toxin (Table S1), respectively via oral gavage for 28 days. The dose of T-2 toxin in this research was designed according to the oral LD50 value (10 mg/kg BW) and preliminary experiments40. Wang et al.and our lab both verified that oral treatment with low dose of T-2 toxin (1 mg/kg BW) impaired the mice health and caused hepatic steatosis and oxidative stress in mice40,41. Thus, we chose 1 mg/kg as the medium dose. Namely, the low dose is 5% of the LD50, the medium dose is 10% of the LD50, and the high dose is 20% of the LD5042. T-2 toxin was purchased from Pribolab Biotech Co. (2C0B17).
For the ferroptosis and Nrf2 intervention experiments, 60 wild-type C57 mice (6 to 8 weeks) were randomly divided into 6 groups (n = 10): the control group (Con), the T-2 toxin group (T-2, 1 mg/kg BW), ferroptosis inhibitor group (Fer-1, 1 mg/kg BW), the tert-butylhydroquinone group (tBHQ, 20 mg/kg BW), the ferroptosis inhibitor + T-2 toxin group (F + T, 1 mg/kg BW Fer-1 + 1 mg/kg BW T-2 toxin), and tBHQ + T-2 toxin group (t + T, 20 mg/kg BW tBHQ + 1 mg/kg BW T-2 toxin). Fer-1 or tBHQ was injected to the mice intraperitoneally, then gavage with T-2 toxin after half an hour. For details on the Fer-1 and tBHQ treatment procedures, refer to a previous study43. Fer-1 (HPLC: 99.71%) and tBHQ (HPLC: 99.89%) were purchased from MedChemExpress LLC (HY-100579, HY-100489).
For the Parkin knockout mouse experiments, 20 wild-type C57 mice and 20 Parkin−/− C57 mice (6 to 8 weeks) were randomly divided into the following 4 groups (n = 10) and administered T-2 toxin through oral gavage for 28 days: wild-type C57 mice (WC, 0 mg/kg BW T-2 toxin), wild-type C57 mice (WT, 1 mg/kg BW T-2 toxin), Parkin−/− C57 mice (PC, 0 mg/kg BW T-2 toxin) and Parkin−/− C57 (PT, 1 mg/kg BW T-2 toxin).
For LYC treatment experiments, 40 wild-type C57 mice 96 to 8 weeks were randomly divided into 4 groups (n = 10): the control group (Con), the T-2 toxin group (T-2, 1 mg/kg BW T-2 toxin), LYC treatment group (L + Y, 5 mg/kg BW LYC + 1 mg/kg BW T-2 toxin), LYC group (LYC, 5 mg/kg BW LYC) by oral gavage for 28 days. LYC was purchased from Sichuan Vicky Biotechnology Co. (HPLC ≥ 98%, WP23040310). The dose of LYC used was selected based on other mouse model studies, which demonstrated that 5 mg/kg LYC has significant protective effects on the toxicity of various toxins18,44,45. Meanwhile, pharmacokinetic and targeting parameters of LYC in mouse tissues following oral administration showed that 8 mg/kg LYC have no adverse effect on mice health46. Therefore, we chose 5 mg/kg as LYC intervention dosage. The structures of the compounds mentioned above are presented in Fig. S1.
Histopathological and ultrastructural analysis
Liver samples were fixed in 4% paraformaldehyde for 24 h, then sent to Servicebio for haematoxylin‒eosin (HE) staining (Servicebio, Wuhan, China). The Ishak score was determined on the basis of fibrosis, necrosis, inflammation, and portal damage in liver sections stained with HE. Liver sections were also stained with Prussian blue using diaminobenzidine (PB + DAB, Servicebio, Wuhan, China), which binds ferric iron and appears brown in color. Histomorphometric analysis was performed by two independent pathologist to semi-quantitatively evaluate the index of liver damage and positive results of PB + DAB.
Ultrastructural observation of the liver was performed via transmission electron microscopy. Liver samples (1 mm3, fixed in 2.5% glutaraldehyde) were sent to RMC Boeckeler Instruments Inc. (China) for further staining and observation.
Immunofluorescence staining
Livers were fixed in 10% paraformaldehyde and then sent to XINLE Biotechnology (China) for further experiments. Liver sections were labelled with antibodies against ferritin heavy chain (FTH), Parkin, LC3 and Nrf2 (Table S2). Each section was scanned using a Pannoramic MIDI digital scanner (3DHISTECH Ltd., Hungary).
Serum AST and ALT detection
The level of serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were quantified by biochemical analyzer (AU680 Beckman Coulter, Inc., US).
Liver iron content detection
Using a tissue iron assay kit, hepatic iron content (Table S1) was determined in accordance with the manufacturer’s guidelines.
Redox status determination
The levels of ROS, T-AOC, malonaldehyde (MDA), 4-Hydroxynonenal (4-HNE) and GPx-4 activity in liver tissues were measured by commercial kits (Table S1). For ROS staining, collected liver samples were embedded in optimal cutting temperature compound, the prepared sections were stained following the experimental instructions of Servicebio.
Mitochondrial function determination
The levels of adenosine triphosphate (ATP) (S0026, Beyotime Institute of Biotechnology, China) and cytochrome C (JL13261, Jianglai Biology Co., Ltd. Shanghai, China) in liver tissue were measured using commercial kits accordance with the manufacturer’s guidelines.
Proteomic sequencing and analysis
Liver samples were sent to Novogene Biotech Co., Ltd., for further analysis. Three samples from the CG and MG were randomly selected for tandem mass tag (TMT) analysis and untargeted quantitative proteomics. The liver was extracted and prepared into 100-μL samples for TMT labelling and fraction separation. An EASY-nLCTM 1200 UHPLC system (Thermo Fisher) was used to construct the transition library for further proteomic analysis. For protein identification, raw data were analyzed using Proteome Discoverer 2.4 in the following database: 1056752-Mus_musculus.GRCm38.pep.all.fa_handled.fasta (66338 sequences). The differentially expressed proteins were statistically analysed by a t test. Thresholds for significance were set at P < 0.05 (significant) and P < 0.01 (highly significant). The GO and KEGG databases were used for annotation.
Bioinformatic analysis
Datasets were exported from Gene Cards under the key words “oxidative stress”, “iron metabolism”, “ferroptosis” and “mitophagy”. The molecules involved in all the biological processes were subjected to further enrichment analysis at https://www.bioinformatics.com.cn. and https://metascape.org. Pathway and process enrichment analyses used GO Biological Processes, TRRUST, KEGG Pathway, Comprehensive Resource of Mammalian protein complexes (CORUM), Reactome Gene Sets, Wiki Pathways, and Protein Analysis Through Evolutionary Relationships pathway (PANTHER). Enriched pathways were collected and grouped (P < 0.01, a minimum count of 3, and enrichment factor > 1.5). A subset of enriched terms was chosen and rendered as a network plot with a similarity > 0.3 are connected by edges. The molecules were also analyzed at https://cytoscape.org and were sorted by P < 0.01.
Western blotting
Liver cytoplasmic protein, nuclear protein and mitochondrial protein were extracted. The protein content was quantified with bicinchoninic acid (BCA) protein assay reagent. Protein lysates were resolved via SDS‒PAGE, electrotransferred onto PVDF membranes, and then probed with the primary antibodies (Table S2) at 4 °C for at least 12 h. An appropriate secondary antibody (GB23301/GB23303, Servicebio, China) was subsequently probed. Visualization was performed via an enhanced chemiluminescent reagent (BL520B, Biosharp, China). The relative band intensity of total proteins were normalized to the GAPDH level. Nuclear protein levels were normalized against Histone level. Mitochondrial protein levels were normalized to the COX IV level. The relative expression of LC3 was quantified by LC3 II/LC3 I (Table S2).
Quantitative RT‒PCR
Nrf2, Keap1, NQO1 and HO-1 mRNA expression levels were analysed by qRT‒PCR. Total liver mRNA was extracted with Trizon reagent (CW0580S, CWbio, Shanghai, China). cDNA synthesis and qRT‒PCR related regents were purchased from Servicebio (China) and performed on the qTOWER® (Analytik Jena, Germany) system. Each sample was analyzed for 3 times, and a mean value was calculated (normalized to β-actin). The primer pairs used are shown in Table S3.
Molecular docking
The 3D structures of Nrf2 (6QMK) and Keap1 (9F2Q) were obtained from the Protein Data Bank. The 3D structure of LYC was constructed using ChemDraw 22.0.0 (Revvity Signals Software, USA). The molecular binding docking was stimulated using AutoDock Vina 1.1.2 (CA, USA) for 3 times, and the mean binding energy and standard deviation were calculated. Visualization of the molecular docking results was performed with PyMOL (DeLano Scientific LLC., USA) and Ligplot 2.2 (European Molecular Biology Laboratory, GER).
Statistical analysis
Data are exhibited as the means ± SD of at least 3 independent experiments in each group, and statistical analyses were calculated using one-way analysis of variance (ANOVA) and Student’s t test (GraphPad Prism 9.5.0; GraphPad Software, Boston, MA, USA). * indicates a statistically significant difference between the experimental groups and the control group. # indicates a statistically significant difference between the T-2 group and the experimental groups. Statistical significance was defined as P < 0.05, with P < 0.01 denoting high significance.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Zhang, X. et al. PINK1/Parkin-mediated mitophagy mitigates T-2 toxin-induced nephrotoxicity. Food Chem. Toxicol. 164, 113078. https://doi.org/10.1016/j.fct.2022.113078 (2022).
Cancer, L. I. A. F. O., Humans, W. G. o. t. E. o. t. C. R. o. C. t., Lyon & Jun. IARC (International Agency for Research on Cancer) monographs on the evaluation of the carcinogenic risk of chemicals to humans. Food and Chemical Toxicology 25 (1980).
Eskola, M. et al. Worldwide contamination of food-crops with mycotoxins: validity of the widely cited ‘FAO estimate’ of 25. Crit. Rev. Food Sci. Nutr. 60, 2773–2789, https://doi.org/10.1080/10408398.2019.1658570 (2020).
Gruber-Dorninger, C., Jenkins, T. & Schatzmayr, G. Global Mycotoxin Occurrence in Feed: A Ten-Year Survey. Toxins 11. https://doi.org/10.3390/toxins11070375 (2019).
Shi, Y. et al. Involvement of TLRs/NF-κB/ESE-1 signaling pathway in T-2 toxin-induced cartilage matrix degradation. Environ. Pollut. 342, 123114. https://doi.org/10.1016/j.envpol.2023.123114 (2024).
Chen, J. et al. Comprehensive analysis of T-2 toxin contamination in maize from Northeast China using automated immunomagnetic beads system coupled with LC-MS/MS. Food Chem. 490, 145171. https://doi.org/10.1016/j.foodchem.2025.145171 (2025).
Janik, E. et al. T-2 toxin-the most toxic trichothecene mycotoxin: metabolism, toxicity, and decontamination strategies. Molecules 26, 6868, https://doi.org/10.3390/molecules26226868 (2021).
Dai, C. et al. T-2 toxin and its cardiotoxicity: New insights on the molecular mechanisms and therapeutic implications. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association 167, 113262. https://doi.org/10.1016/j.fct.2022.113262 (2022).
Wang, Y. et al. Review of neurotoxicity of T-2 toxin. Mycotoxin Res. 40, 85–95, https://doi.org/10.1007/s12550-024-00518-5 (2024).
Wang, Y. et al. T-2 toxin nephrotoxicity: toxic effects, mechanisms, mitigations, and future perspectives. J. Agric. Food Chem. 73, 2732–2744, https://doi.org/10.1021/acs.jafc.4c10015 (2025).
Yang, X. et al. p38 mediates T-2 toxin-induced Leydig cell testosterone synthesis disorder. Ecotoxicol. Environ. Saf. 253, 114695. https://doi.org/10.1016/j.ecoenv.2023.114695 (2023).
Hu, Z. Y. et al. AHR activation relieves deoxynivalenol-induced disruption of porcine intestinal epithelial barrier functions. J. Hazard. Mater. 480, 136095. https://doi.org/10.1016/j.jhazmat.2024.136095 (2024).
Adhikari, M. et al. T-2 mycotoxin: toxicological effects and decontamination strategies. Oncotarget 8, 33933–33952, https://doi.org/10.18632/oncotarget.15422 (2017).
Song, W. et al. T-2 toxin metabolism and its hepatotoxicity: New insights on the molecular mechanism and detoxification. Environ. Pollut. 330, 121784. https://doi.org/10.1016/j.envpol.2023.121784 (2023).
An, K., Shi, B., Lv, X., Liu, Y. & Xia, Z. T-2 toxin triggers lipid metabolism disorder and oxidative stress in liver of ducks. Ecotoxicol. Environ. Saf. 286, 117169. https://doi.org/10.1016/j.ecoenv.2024.117169 (2024).
Zhu, Y. et al. T-2 Toxin Exploits Gut-Derived Staphylococcus Saprophyticus to Disrupt Hepatic Macrophage Homeostasis. Adv. Sci. 12, e12828. https://doi.org/10.1002/advs.202512828 (2025).
Yang, X. et al. Deoxynivalenol induces testicular ferroptosis by regulating the Nrf2/System Xc(-)/GPX4 axis. Food Chem. Toxicol. Int. J. Published Br. Ind. Biol. Res. Assoc. 175, 113730. https://doi.org/10.1016/j.fct.2023.113730 (2023).
Chang, Y. H. et al. SIRT3 promotes mitophagy to attenuate fumonisin B1-induced chicken hepatocyte senescence and antagonism of lycopene. J. Hazard. Mater. 497, 139557. https://doi.org/10.1016/j.jhazmat.2025.139557 (2025).
Zhao, Y. et al. Inhibition of the p62-Nrf2-GPX4 pathway confers sensitivity to Butachlor-induced splenic macrophage ferroptosis. J. Agric. Food Chem. 72, 16998–17007, https://doi.org/10.1021/acs.jafc.4c01086 (2024).
Wang, Y. et al. Nrf2: A Main Responsive Element of the Toxicity Effect Caused by Trichothecene (T-2) Mycotoxin. Toxics 11. https://doi.org/10.3390/toxics11040393 (2023).
Zhu, R. et al. Lycopene in protection against obesity and diabetes: a mechanistic review. Pharmacol. Res. 159, 104966. https://doi.org/10.1016/j.phrs.2020.104966 (2020).
Assis, R. P. et al. Combined effects of curcumin and lycopene or bixin in yoghurt on inhibition of LDL oxidation and increases in HDL and paraoxonase levels in streptozotocin-diabetic rats. Int. J. Mol. Sci. 18, 332, https://doi.org/10.3390/ijms18040332 (2017).
Cai, Z. et al. Lycopene maintains mitochondrial homeostasis to counteract the enterotoxicity of deoxynivalenol. Antioxidants 12, 1958, https://doi.org/10.3390/antiox12111958 (2023).
Lin, J. et al. Lycopene alleviates multiple-mycotoxin-induced toxicity by inhibiting mitochondrial damage and ferroptosis in the mouse jejunum. Food Funct. 13, 11532–11542, https://doi.org/10.1039/d2fo02994d (2022).
Cai, Z. et al. Lycopene protects deoxynivalenol-induced intestinal barrier dysfunction and NLRP3 inflammasome activation by targeting the ERK pathway. Antioxidants 14, 1513 (2025).
Yang, T. N. et al. Holistic assessment based on hepatocyte mitochondria: lycopene repairs oxidized mtDNA to alleviate mitochondrial stress induced by atrazine. J. Agric. Food Chem. 71, 20325–20335, https://doi.org/10.1021/acs.jafc.3c05369 (2023).
Wang, X. Q. et al. SIRT1 Regulates Fumonisin B1-Induced LMH Cell PANoptosis and Antagonism of Lycopene. J. Agric. Food Chem. 73, 4923–4935, https://doi.org/10.1021/acs.jafc.4c11658 (2025).
Huang, W. et al. Lycopene ameliorates aflatoxin B(1)-induced testicular lesion by attenuating oxidative stress and mitochondrial damage with Nrf2 activation in mice. Ecotoxicol. Environ. Saf. 256, 114846. https://doi.org/10.1016/j.ecoenv.2023.114846 (2023).
Xue, Q. et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy 19, 1982–1996, https://doi.org/10.1080/15548627.2023.2165323 (2023).
He, J. et al. T-2 toxin-induced testicular impairment by triggering oxidative stress and ferroptosis. Ecotoxicol. Environ. Saf. 270, 115844. https://doi.org/10.1016/j.ecoenv.2023.115844 (2024).
Wang, G. et al. T-2 toxin induces ferroptosis by increasing lipid reactive oxygen species (ROS) and downregulating solute carrier family 7 member 11 (SLC7A11). J. Agric Food Chem. 69, 15716–15727, https://doi.org/10.1021/acs.jafc.1c05393 (2021).
Yang, Q. et al. Benchmark dose estimation from transcriptomics data for methylimidazolium ionic liquid hepatotoxicity: implications for health risk assessment of green solvents. Environ. Health, (2025).
Terešak, P. et al. Regulation of PRKN-independent mitophagy. Autophagy 18, 24–39, https://doi.org/10.1080/15548627.2021.1888244 (2022).
Bhattacharjee, A. et al. Loss of ubiquitinated protein autophagy is compensated by persistent cnc/NFE2L2/Nrf2 antioxidant responses. Autophagy 18, 2385–2396, https://doi.org/10.1080/15548627.2022.2037852 (2022).
Peng, C. et al. Mitochondrial ROS driven by NOX4 upregulation promotes hepatocellular carcinoma cell survival after incomplete radiofrequency ablation by inducing of mitophagy via Nrf2/PINK1. J. Transl. Med. 21, 218. https://doi.org/10.1186/s12967-023-04067-w (2023).
Xiao, L. et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 11, 297–311, https://doi.org/10.1016/j.redox.2016.12.022 (2017).
Wang, Y. et al. Lycopene attenuates the inflammation and apoptosis in aristolochic acid nephropathy by targeting the Nrf2 antioxidant system. Redox Biol. 57, 102494. https://doi.org/10.1016/j.redox.2022.102494 (2022).
Zhao, Y. et al. Lycopene prevents DEHP-induced hepatic oxidative stress damage by crosstalk between AHR-Nrf2 pathway. Environ. Pollut. 285, 117080. https://doi.org/10.1016/j.envpol.2021.117080 (2021).
Wang, Y. et al. LYC inhibits the AKT signaling pathway to activate autophagy and ameliorate TGFB-induced renal fibrosis. Autophagy 20, 1–20, https://doi.org/10.1080/15548627.2023.2287930 (2023).
Huang, T. et al. The role of gut microbiota in anorexia induced by T-2 toxin. Ecotoxicol. Environ. Saf. 281, 116612. https://doi.org/10.1016/j.ecoenv.2024.116612 (2024).
Wang, J. et al. Tannic acid ameliorates systemic glucose and lipid metabolic impairment induced by low-dose T-2 toxin exposure. J. Agric. Food Chem. 71, 12574–12586, https://doi.org/10.1021/acs.jafc.3c02934 (2023).
Li, Y. et al. T-2 toxin, a trichothecene mycotoxin: review of toxicity, metabolism, and analytical methods. J. Agric Food Chem. 59, 3441–3453, https://doi.org/10.1021/jf200767q (2011).
Xu, Y., Li, Y., Li, J. & Chen, W. Ethyl carbamate triggers ferroptosis in liver through inhibiting GSH synthesis and suppressing Nrf2 activation. Redox Biol. 53, 102349. https://doi.org/10.1016/j.redox.2022.102349 (2022).
Cui, J.-G. et al. Calcium homeostasis imbalance mediates DEHP induced mitochondrial damage in cerebellum and the antagonistic effect of lycopene. Sci. Total Environ. 954, 176351. https://doi.org/10.1016/j.scitotenv.2024.176351 (2024).
Chen, J. et al. Lycopene mitigates atrazine-induced hypothalamic neural stem cell senescence by modulating the integrated stress response pathway. Phytomedicine 135, 156114. https://doi.org/10.1016/j.phymed.2024.156114 (2024).
Guo, Y. et al. Oral delivery of lycopene-loaded microemulsion for brain-targeting: preparation, characterization, pharmacokinetic evaluation and tissue distribution. Drug Deliv. 26, 1191–1205, https://doi.org/10.1080/10717544.2019.1689312 (2019).
Acknowledgements
This work was supported by National Natural Science Foundation of China (32202877 and 32473108), Key Project of Henan Province Science and Technology Research and Development Plan Joint Fund (245200810018), Millions of Science and Education Service Action Fund (2024SFBQW37), the Key Scientific Research Projects of Colleges and Universities in Henan Province (26A230006) Special Funding from the China Postdoctoral Science Foundation (2024T170243) and the Outstanding Talents of Henan Agricultural University (30500996; 30500997). Parkin−/− C57 mice were gifted by Prof. Li Yanfei from Northeast Agricultural University.
Author information
Authors and Affiliations
Contributions
X.Y., W.S., and Z.L. conceived and designed the study, conducted the investigation, and wrote the original draft. Y.C. performed data curation and formal analysis. Y.W. contributed to visualization and validation. T.H. participated in conceptualization, methodology, and validation. Y.L. and Y.W. carried out software implementation and formal analysis. S.C. provided supervision. Y.L. acquired funding and supervised the project. W.B. contributed resources, project administration, and supervision. C.Z. supervised the study and reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, X., Song, W., Lu, Z. et al. Lycopene mitigates T-2 toxin-induced hepatic ferroptosis by targeting the Nrf2/mitophagy axis in mice. npj Sci Food 10, 94 (2026). https://doi.org/10.1038/s41538-026-00736-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41538-026-00736-4
