
Abstract
Tissue-scale architecture and mechanical properties instruct cell behaviour under physiological and diseased conditions, but our understanding of the underlying mechanisms remains fragmentary. Here we show that extracellular matrix stiffness, spatial confinements and applied forces, including stretching of mouse skin, regulate mitochondrial dynamics. Actomyosin tension promotes the phosphorylation of mitochondrial elongation factor 1 (MIEF1), limiting the recruitment of dynamin-related protein 1 (DRP1) at mitochondria, as well as peri-mitochondrial F-actin formation and mitochondrial fission. Strikingly, mitochondrial fission is also a general mechanotransduction mechanism. Indeed, we found that DRP1- and MIEF1/2-dependent fission is required and sufficient to regulate three transcription factors of broad relevance—YAP/TAZ, SREBP1/2 and NRF2—to control cell proliferation, lipogenesis, antioxidant metabolism, chemotherapy resistance and adipocyte differentiation in response to mechanical cues. This extends to the mouse liver, where DRP1 regulates hepatocyte proliferation and identity—hallmark YAP-dependent phenotypes. We propose that mitochondria fulfil a unifying signalling function by which the mechanical tissue microenvironment coordinates complementary cell functions.
Similar content being viewed by others


Main
Cells have the striking ability to measure the spatial and mechanical constraints imposed by their surroundings. Seemingly different constraints are sensed through a similar mechanism, based on the measurement of extracellular forces and deformations by active contraction of the actomyosin cytoskeleton1,2. In line with this model, any extrinsic or intrinsic condition associated with decreased actomyosin tension leads to similar biological responses. This mechanosensitive process, depending on the specific cell type, can regulate multiple cell behaviours in the context of tissue homeostasis, regeneration and disease3,4,5,6,7. In the face of a single force-sensing mechanism, multiple pathways and transcription factors can be regulated by extracellular forces8. It remains unclear whether any common regulatory mechanism exists downstream of actomyosin, which would coordinate distinct pathways into a unified and coherent mechanotransduction response.
An emerging theme is the potential for organelles, including mitochondria, to sense and transduce forces9. It was recently observed that cells cultured on a soft extracellular matrix (ECM) display increased mitochondrial fission10,11,12. Mitochondrial morphology is controlled by fission, which depends on the cytoplasmic GTPase Dynamin-related protein 1 (DRP1; also known as DNM1L), and by fusion, which depends on the mitochondrial GTPases Mitofusin 1 (MFN1), MFN2 and Optic atrophy 1 (refs. 13,14). Mitochondrial morphology is regulated to match the cellular metabolic demands or facilitate the structural rearrangements of mitochondria and the cell. Reflecting the importance of these processes, genes that regulate mitochondrial dynamics are found to be mutated in patients affected by neuropathies and neurodegenerative diseases15. In this Article, we sought to better explore the link between mitochondrial dynamics and the mechanical cell microenvironment.
Results
Extracellular forces induce mitochondrial elongation
We started this study by testing how general the regulation of mitochondrial morphology is in response to extracellular forces and constraints. Mitochondrial shortening was observed in different cell types in response to a soft ECM (Fig. 1a–c), direct inhibition of actomyosin players (Fig. 1d,e) and inducible knockout (KO) of Talin1 (Fig. 1f)16, in line with the role of integrin adhesions in mechanosensing, whereas there were no apparent effects upon depletion of Piezo1/2 mechanosensitive ion channels (Fig. 1g and Extended Data Fig. 1a). Mitochondrial shortening was also observed by culturing cells on small fibronectin microprinted islands and at high density (Fig. 1h–j)—conditions associated with a reduction of actomyosin tension17,18,19,20. These changes in mitochondrial morphology were reversible; when we enabled cell spreading by wounding a dense monolayer, this restored mitochondrial elongation (Fig. 1j). Moreover, the application of laminar fluid shear stresses of different magnitudes to endothelial cells induced mitochondrial shortening at a physiological wall shear stress (WSS) of 1.4 Pa (14 dyn cm−2) and elongation at a WSS of 4 Pa characteristic of areas prone to atherogenesis21 (Fig. 1k). This is in line with ref. 22 and with a shear stress set point typical of endothelial mechanotransduction responses23. Finally, stretching of dense immortalized keratinocytes also led to mitochondrial elongation (Fig. 1l), and coherent mitochondrial remodelling24,25 was observed in vivo upon mouse skin expansion (Fig. 1m), which activates the mechanosensitive Yes-associated protein 1 (YAP) factor for compensatory tissue growth26. These results were validated by an orthogonal quantification method (Extended Data Fig. 1b–g). Collectively, this indicates that extracellular forces broadly regulate mitochondrial dynamics in vitro and in vivo, with mitochondrial shortening occurring upon decreased actomyosin tension (soft or small ECM, dense cells and low WSS), as confirmed by monitoring Rho-associated protein kinase 1 and 2 (ROCK) activity (Extended Data Fig. 1h–o).

a, Mitochondrial length analysis in MCF10A-RAS cells cultured for 24 h on stiff (E = ~15 kPa) versus soft (E = ~0.5 kPa) fibronectin-coated polyacrylamide hydrogels (n = 71 (soft) and 95 cells (stiff) from four experiments). Scale bars, 10 μm. b,c, Mitochondrial length analysis in D2.0R cells (b) and MEFs (c) cultured as in a (n = 69 (stiff) and 48 cells (soft) in b; n = 57 (stiff) and 76 cells (soft) in c; both from three experiments). d, Mitochondrial length analysis in MCF10A-RAS cells transfected to express dominant-negative RHOA T19N or treated for 3 h with inhibitors of ROCK and MLCK (Y27632 and ML7; YM) or with the SMIFH2 inhibitor of the formin actin-nucleating proteins (n = 25 (GFP), 30 (RHOA T19N), 71 (DMSO), 89 (YM) and 93 cells (SMIFH2); from two experiments for GFP and RHOA T19N and from four experiments for the remainder). Scale bar, 10 μm. e, Mitochondrial length analysis in MEFs treated as in d (n = 86 (DMSO) and 71 cells (YM) from four experiments). Scale bar, 10 μm. f, Mitochondrial length analysis in mouse lung endothelial cells (MLECs) treated for 18 h with 4-OHT to trigger CreERT2-mediated Talin1 inactivation (Tln1 KO) (n = 26 (WT) and 25 cells (Tln1 KO) from two experiments). Scale bar, 10 μm. g, Mitochondrial length analysis in MCF10A-RAS cells transfected with control (siCO) or PIEZO1/2 siRNAs (n = 29 (siCO), 64 (siPIEZO1/2a) and 71 cells (siPIEZO1/2b) from three experiments). h, Mitochondrial length analysis in 3T3L1 cells cultured on large (10,000 μm2), medium (1,024 μm2) or small (300 μm2) micropatterned fibronectin islands for 24 h (n = 61 (large), 51 (medium) and 96 cells (small) from four experiments). Scale bars, 10 μm. i,j, Mitochondrial length analysis in 3T3L1 (i) and MCF10A-RAS cells (j) cultured under sparse (low-density (LD)) or dense (high-density (HD)) conditions for 72 h. Where indicated, cells were locally released from crowding by scratching the monolayer for 8 h (scratch) (n = 89 (LD) and 193 cells (HD) from four experiments in i; n = 69 (LD), 192 (HD without scratching) and 143 cells (HD with scratching) from three experiments in j). Scale bar, 10 μm. k, HUVECs forming mature monolayers were left untreated (static) or conditioned for 18 h with a flow yielding a nominal WSS of 1.4 or 4.0 Pa. Top left, analysis of cell orientation in response to flow (0° is the flow direction). Bottom left, representative TOMM20 immunofluorescence. Right, mitochondrial length analysis (n = 161 (static), 175 (1.4 Pa) and 202 cells (4.0 Pa) from three experiments). Scale bar, 30 μm. l, Mitochondrial length analysis in HaCaT cells cultured under dense conditions on a fibronectin-coated silicon membrane for 72 h then subjected to tonic uniaxial stretching for 3 h (stretch) (n = 128 (no stretch) and 153 cells (stretch) from three experiments). Bidirectional arrow represents the direction of stretching. Scale bar, 20 μm. m, Mitochondrial area analysis (right) from transmission electron microscopy images (left) of basal keratinocytes from normal mouse skin (CO) versus skin subjected to expansion for 6 d (EXP)26 (n = 88 (CO a), 78 (CO b), 68 (EXP c), 76 (EXP d) and 62 mitochondria (EXP e) in ≥15 cells per mouse). Scale bars, 2 μm (top images) and 500 nm (magnified images). The data are presented as means ± s.d. (a–l) or means and single points (m), with representative pictures. Statistical significance was determined by two-tailed Student’s t-test. In a–e and h–l, P values were calculated on punctate mitochondria. Source numerical data are available.
Mitochondrial fission regulates SREBP1/2 mechanotransduction
Prompted by such widespread connection between mitochondria and forces, we explored the possibility that mitochondrial fission regulates other downstream responses in addition to Nuclear factor erythroid 2-related factor 2 (NRF2)10. This possibility was also suggested by an analysis of gene expression in D2.0R cells, where we observed a substantial group of genes that were regulated on soft ECM in a DRP1-dependent but NRF2-independent manner (632/5,683 DRP1-dependent genes and 179/5,683 NRF2-dependent genes) (Supplementary Table 1). Hence, we analysed DRP1-dependent genes further.
Among the genes downregulated by DRP1 inhibition on soft ECM, we noticed the presence of enzymes related to lipid metabolism (Extended Data Fig. 2a). This suggested a link between mitochondrial fission and mechanical regulation of Sterol regulatory element-binding proteins 1 and 2 (SREBP1/2)27,28. Supporting this idea, DRP1 inactivation or its inhibition by Drpitor1a and MDIVI1 (mitochondrial division inhibitor 1) (refs. 29,30,31) prevented accumulation of the SREBP cleavage-activating protein (SCAP) chaperone to the Golgi apparatus and of the SREBP2 transcription factor to the nucleus in different cell types and in response to multiple experimental conditions associated with decreased actomyosin tension (Fig. 2a–g). Importantly, DRP1 inhibition was also sufficient to prevent activation of established SREBP1/2 target genes (Fig. 2h) and the accumulation of lipid droplets and cholesterol (Fig. 2,i,j), indicating effective inhibition of lipid metabolism. Inhibition of DRP1 was instead unable to prevent SREBP2 activation in response to non-mechanical inputs (Extended Data Fig. 2b–e)27,32,33, indicating that mitochondrial fission mediates the SREBP1/2 response to mechanical inputs.

a, MCF10A-RAS cells were treated with ROCK–MLCK inhibitors (YM) for 3 h. DRP1 was inhibited with Drpitor1a (DRP1A), Mitochondrial division inhibitor 1 (MDIVI1) or siRNA transfection. The percentages of cells displaying SREBP2 staining at the endoplasmic reticulum (ER) or in the nucleus (Nuc) were quantified (n = 95 (DMSO), 63 (YM), 87 (YM DRP1A), 88 (YM MDIVI1) and 75 cells (YM siDRP1) from three experiments). Scale bar, 10 μm. b, SREBP2 localization in MCF10A-RAS cells cultured on fibronectin hydrogels for 24 h (n = 80 (stiff), 64 (soft), 73 (soft DRP1A), 68 (soft MDIVI1) and 99 cells (soft siDRP1) from three experiments). Scale bar, 10 μm. c, MCF10A-RAS cells expressing Myc-tagged SCAP were treated as in b and the percentages of cells displaying SCAP immunofluorescence staining at the endoplasmic reticulum or concentrated at the Golgi apparatus were quantified (n = 40 (stiff), 51 (soft), 41 (soft DRP1A) and 43 cells (soft MDIVI1) from three experiments). Scale bar, 5 μm. d, SREBP2 localization in D2.0R cells with inhibition of ROCK–MLCK (YM) or DRP1 (DRP1A or shRNAa/b) (n = 101 (DMSO), 124 (YM), 106 (YM DRP1A), 98 (YM shDrp1a) and 102 cells (YM shDrp1b) from three experiments). Scale bar, 10 μm. e,f, SREBP2 localization in WT and Dnm1l KO (Drp1 KO) MEFs treated with YM (e) or cultured on fibronectin hydrogels for 24 h (f) (n = 103 (DMSO), 105 (YM), 92 (YM DRP1A), 69 (Drp1 KO) and 76 cells (Drp1 KO YM) in e; n = 56 (WT stiff), 71 (WT soft), 54 (KO stiff) and 61 cells (KO soft) in f; both from three experiments). Scale bars, 10 μm. g, SREBP2 localization in 3T3L1 cells cultured on large or small micropatterned islands for 24 h (n = 49 (large), 51 (small) and 63 cells (small DRP1A) from three experiments). Scale bar, 5 μm. h, Quantitative PCR (qPCR) in MCF10A-RAS cells cultured on stiff (E = ~50 kPa) or soft (E = ~0.2 kPa) collagen I-coated hydrogels for 24 h and treated with DRP1 inhibitors. The messenger RNA (mRNA) levels are relative to GAPDH levels and normalized to the stiff controls (n = 6 samples from three experiments). i,j, Quantification of lipid droplets by Oil Red O (ORO) staining (i) and of cholesterol by ultraviolet-fluorescent Filipin (j) in MCF10A-RAS cells treated with YM for 24 h and with DRP1 inhibitors (n = 50 cells in i and n = 53 cells in j; both from three experiments). Scale bars, 10 μm. The data are presented as means ± s.d. (a–g) or means and single points (h–j). Statistical significance was determined by two-tailed Student’s t-test. In a, b and d–g, this was calculated on nuclear SREBP2. Source numerical data are available.
Mitochondrial fission regulates YAP/TAZ mechanotransduction
We then turned our attention to genes upregulated by DRP1 inhibition on soft ECM. In this case, we observed some known target genes of YAP and Translational coactivator with PDZ-binding motif (TAZ; also known as WWTR1) (Extended Data Fig. 3a)34,35,36. Strikingly, inhibition of DRP1 prevented YAP inhibition in response to soft ECM and decreased actomyosin tension in different cell types (Fig. 3a–d and Extended Data Fig. 3b–d). Extending the relevance of these findings, inhibition of DRP1 prevented YAP inhibition in small and dense cells (Fig. 3e,f and Extended Data Fig. 3e) and had comparable effects to stretching in dense skin cells (Extended Data Fig. 3f), indicating that the response of YAP to mechanics and density occurs via the same mechanism20,37. Moreover, inhibition of DRP1 prevented YAP inhibition in endothelial cells subjected to physiological shear stress (Fig. 3g,h). In contrast, inhibition of DRP1 did not prevent YAP inhibition by the NF2 moesin-ezrin-radixin like tumour suppressor (Extended Data Fig. 3g,h)38,39, suggesting that mitochondrial fission broadly mediates YAP/TAZ regulation downstream of mechanical cues.

a, MCF10A-RAS cells were cultured on fibronectin hydrogels for 24 h and treated with DRP1 inhibitors. The percentages of cells displaying endogenous YAP immunofluorescence staining prevalently in the cytoplasm (Cyto), evenly distributed between the cytoplasm and nucleus (N = C) or prevalently in the nucleus (Nuc) were quantified (n = 176 (stiff), 135 (soft), 129 (soft DRP1A) and 67 cells (soft MDIVI1) from four experiments). Scale bar, 20 μm. b, qPCR in MCF10A-RAS cells cultured on collagen I-coated hydrogels for 24 h and treated with DRP1 inhibitors. The mRNA levels are relative to GAPDH levels and normalized to the stiff controls (n = 6 from three experiments). c, YAP localization in D2.0R cells stably expressing control (shCO) or Drp1 shRNAs and treated with ROCK–MLCK inhibitors (YM) for 3 h (n = 162 (DMSO shCO), 99 (YM shCO), 142 (shCO YM DRP1A), 162 (YM shDrp1a) and 142 cells (YM shDrp1b) from three experiments). Scale bar, 10 μm. d, YAP localization in WT and Drp1 KO MEFs cultured on fibronectin hydrogels for 24 h (n = 124 (WT stiff), 119 (WT soft), 137 (KO stiff) and 122 cells (KO soft) from three experiments). Scale bar, 10 μm. e, YAP localization in 3T3L1 cells cultured on large or small micropatterned islands and treated with Drpitor1a for 24 h (n = 38 (large), 61 (small) and 55 cells (small DRP1A) from three experiments). Scale bar, 10 μm. f, YAP localization in MCF10A-RAS cells cultured under sparse (LD) and dense (HD) conditions and treated with Drpitor1a. Cells within five cell diameters from the scratch were quantified for comparison (n = 80 (LD), 452 (HD), 231 (HD DRP1A) and 225 cells (HD scratch) from three experiments). Scale bar, 10 μm. g, YAP localization in HUVECs conditioned by flow, with or without Drpitor1a (n = 1,098 (static), 1,032 (1.4 Pa), 1,098 (1.4 Pa DRP1A) and 3,421 cells (4.0 Pa) from two experiments). Scale bar, 30 μm. h, qPCR in HUVECs treated as in g. The mRNA levels are relative to GAPDH levels and normalized to the static controls (n = 6 from three experiments). i, EdU incorporation assay in MCF10A-RAS cells cultured on fibronectin hydrogels and treated with Drpitor1a for 24 h (n = 276 (stiff), 253 (soft) and 156 cells (soft DRP1A) from three experiments). j, EdU incorporation assay in MCF10A-RAS cells cultured as in f for 72 h. Drpitor1a was added for the last 24 h (n = 147 (LD), 1,003 (HD) and 1,109 cells (HD DRP1A) from three experiments). k, EdU incorporation assay in 3T3L1 cells cultured on large or small micropatterned islands and treated with Drpitor1a for 24 h (n = 60 (large), 51 (small) and 54 cells (small DRP1A) from three experiments). l,m, Dominant-negative HA-DRP1 (DRP1 DN) and constitutively active FLAG-TAZ-4SA (TAZ4SA) induce cholangiocellular markers (l) and EdU incorporation (m) in mouse liver (n = 4 mice). Scale bars, 5 μm. The data are presented as means ± s.d. (a and c–g) or means and single points (b and h–m). Statistical significance was determined by two-tailed Student’s t-test. In a and c–f, P values were calculated on nuclear YAP levels. Source numerical data are available.
Strikingly, inhibition of DRP1 was also sufficient to resume DNA replication on soft ECM, in dense cells and small cells (Fig. 3i–k and Extended Data Fig. 3i,j), in line with rescued YAP–TAZ activity and with a mechanical checkpoint driving contact inhibition of proliferation20,37. As a final test, we challenged the function of DRP1 in mouse liver, where physiological low tissue tension acts as a gatekeeper of YAP–TAZ activity40,41. As shown in Fig. 3l,m, expression of dominant-negative DRP1 in mouse adult hepatocytes was sufficient to trigger ectopic expression of cholangiocyte markers and DNA replication, similar to the expression of constitutively active TAZ. DRP1 is thus broadly required for the regulation of YAP/TAZ responses by mechanical cues in vitro and in vivo.
In the above experiments, inhibition of DRP1 did not rescue ROCK activity or cell morphology (Extended Data Figs. 1k–m and 3k,l), placing DRP1 downstream of actomyosin. Hence, DRP1 mediates the regulation of SREBP1/2 and YAP/TAZ in response to mechanical cues. DRP1 also mediates the regulation of mitochondrial reactive oxygen species (mtROS) and NRF2 by mechanical cues10, but inhibition of ROS did not rescue YAP or SREBP2 (Extended Data Fig. 3m,o). This suggests that DRP1 regulates three distinct transcription factors by different mechanisms. For simplicity, we refer to this global process as mitochondrial mechanotransduction (MIME).
General workings of MIME
We next sought to better understand the nature and extent of MIME. We first focused on its temporal dynamics. Mitochondrial fission in response to ROCK–Myosin light chain kinase (MLCK) inhibitors was observed between 15 and 30 min (Fig. 4a), indicating an early response. This is coherent with fission acting upstream of the regulation of mtROS, YAP and SCAP–SREBP2, which occur within 1–2 h by the same treatment10,27,37. Moreover, adding or removing the DRP1 inhibitor at different time points indicated that fission continuously transduces a reversible signal downstream of actomyosin tension (Fig. 4b). This fast and reversible regulation also made it unlikely that MIME is mediated by a very general and indirect mechanism (for example, regulation of proliferation).

a, Mitochondrial length analysis in MCF10A-RAS cells treated with inhibitors of ROCK–MLCK (YM) for the indicated times (n = 22 (DMSO), 24 (15 min), 24 (30 min), 23 (60 min) and 25 cells (120 min) from two experiments). b, YAP localization in MCF10A-RAS cells with pulse–chase (+, washout) treatments with inhibitors of YM and DRP1 (DRP1A) (n = 66 (DMSO), 54 (YM), 53 (YM washout), 62 (DRP1A) and 87 cells (DRP1A washout) from two experiments). c, Representative immunostainings of deep ρ0 MCF10A-RAS cells largely lacking mitochondria. Scale bar, 10 μm. d–f, SREBP2 (d), SCAP (e) and YAP (f) localization in deep ρ0 MCF10A-RAS cells (n = 135 (stiff) and 141 cells (soft) in d; n = 81 (stiff) and 95 cells (soft) in e; n = 50 (stiff) and 45 cells (soft) in f; all from three experiments). Scale bars, 10 μm. g, YAP localization in MCF10A-RAS cells treated with FCCP (n = 78 (DMSO) and 80 cells (FCCP) from three experiments). Scale bar, 5 μm. h, YAP localization in MCF10A-RAS cells treated with ionomycin (Iono) (n = 63 (DSMO) and 72 cells (Iono) from two experiments). i,j, SREBP2 (i) and YAP (j) localization in MEFs upon activation of the ActuAtor system (n = 57 (−Act) and 58 cells (+Act) in i; n = 43 (−Act) and 42 cells (+Act) in j; both from two experiments). Scale bars, 10 μm. k,l, SREBP2 (k) and YAP (l) localization in MCF10A-RAS cells with MFN1 knockdown. Where indicated, cells were treated with Drpitor1a (n = 67 (siCO), 70 (siMFN1a), 66 (siMFN1a DRP1A), 65 (siMFN1b) and 71 (siMFN1b DRP1A) in k; n = 44 (siCO), 48 (siMFN1a), 56 (siMFN1a DRP1A), 49 (siMFN1b) and 67 (siMFN1b DRP1A) in l; both from three experiments). Scale bars, 10 μm. m, YAP localization in MCF10A-RAS cells with PLD6 knockdown (n = 52 (siCO), 45 (siPLD6a) and 39 cells (siPLD6b) from two experiments). Scale bar, 10 μm. The data are presented as means ± s.d. Statistical significance was determined by two-tailed Student’s t-test. This was calculated on nuclear SREBP2 (d and k) or nuclear YAP (f, g and l). Source numerical data are available.
By using dilutions of the Drpitor1a DRP1 inhibitor, we observed that the rescue of nuclear YAP closely mirrored the efficacy of the compound at elongating mitochondria, indicating a dose-dependent response (Extended Data Fig. 4a–d). On soft ECM, DRP1 inhibition mainly rescued intermediate mitochondria, suggesting that the number of punctate mitochondria or the fission process per se is associated with the regulation of YAP. Along the same idea, deep ρ0 cells devoid of mitochondrial DNA and largely missing mitochondria altogether (Fig. 4c and Extended Data Fig. 4e)42 displayed normal localization of YAP and SCAP–SREBP2 on stiff ECM but failed to regulate them on soft ECM (Fig. 4d–f). Overall, this suggests that mitochondria support fission-dependent signalling that inhibits YAP and activates SREBP2.
We then tested whether any alteration of mitochondrial morphology regulates MIME. Increased fission by uncoupling of the electron transport chain43 was not associated with inhibition of YAP (Fig. 4g and Extended Data Fig. 4f). Similarly, triggering fission by increasing cytoplasmic calcium44 or via the ActuAtor system45 was not sufficient to regulate YAP or SREBP2 (Fig. 4h–j and Extended Data Fig. 4g,h). In contrast, blocking mitochondrial fusion by inactivation of MFN1 (refs. 46,47) caused YAP inhibition and SREBP2 activation under stiff and sparse conditions (Fig. 4k,l and Extended Data Fig. 4i–l). Of note, these effects were rescued by DRP1 inhibition (Fig. 4k,l and Extended Data Fig. 4j) and we observed increased recruitment of DRP1 at mitochondria upon loss of MFN1 (Extended Data Fig. 4m,n), suggesting that loss of MFN1 leads to increased fission. Finally, inhibition of YAP was observed upon knockdown of the pro-fusion and anti-fission Phospholipase D family member 6 (PLD6) enzyme (also known as mitoPLD) (Fig. 4m and Extended Data Fig. 4o,p)48,49. This indicated that some fission events trigger MIME signalling, whereas others do not.
Collectively, these data suggest that activation of the DRP1 fission factor at mitochondria triggers a metabolic or signalling event that regulates YAP/TAZ, SREBP1/2 and NRF2. This event may be physically coincident with the formation of DRP1 fission foci or induced secondarily by shortened mitochondria.
MIME depends on MIEF1/2
The observation that MIME is only associated with some fission events indicated specificity, which we sought to understand at the molecular level. DRP1 can be recruited to mitochondria by the mitochondrial Fission 1 protein (FIS1), Mitochondrial fission factor (MFF) and Mitochondrial elongation factors 1 and 2 (MIEF1/2; also known as MID51 and MID49, respectively)13. We previously implicated MIEF1/2 in mtROS generation on soft ECM10, prompting us to test their role in MIME signalling. First, MIEF1/2 were required for increased fission on soft ECM (Fig. 5a). Second, MIEF1/2 were required for NRF2 target gene activation, cystine uptake and increased resistance of cells to oxidative stress (Fig. 5b–e and Extended Data Fig. 5a), indicating that they are required for the mechanical regulation of antioxidant metabolism. Third, we extended the MIEF1/2 requirement to the regulation of SREBP2 and YAP localization (Fig. 5f,g and Extended Data Fig. 5b,c). Of note, this included Mief1 Mief2 Mff KO mouse embryonic fibroblasts (MEFs)50 that were insensitive to the mechanical regulation of SREBP2 and YAP (Fig. 5h,i and Extended Data Fig. 5d). This indicates that MIME signalling depends on the MIEF1/2 factors. Moreover, as MIEF1/2 are only found at mitochondria13, this strengthens the idea that these regulations occur at or through mitochondria.

a, Mitochondrial length analysis in MCF10A-RAS cells depleted of MIEF1 and MIEF2 and cultured on fibronectin hydrogels for 24 h (n = 75 (siCO), 85 (siMIEF1/2a) and 91 cells (siMIEF1/2b) from four experiments). Scale bars, 10 μm. b, qPCR in MCF10A-RAS cells cultured on collagen I-coated hydrogels for 24 h. The mRNA levels are relative to GAPDH levels and normalized to the controls (n = 6 from three experiments). c, Uptake of FITC-labelled cystine quantified by flow cytometry in MCF10A-RAS cells depleted of MIEF1/2 and treated with ROCK–MLCK inhibitors (YM) for 24 h. The data are normalized to the mean intensity of the controls (n = 9 from three experiments). d,e, Survival rates of MCF10A-RAS cells depleted of MIEF1/2, plated on stiff and soft Matrigel and treated with cisplatin (Cis; d) or arsenic oxide (As2O3; e) for 48 h. The mean cell number of the controls was set to 100% (solid black) and all other sample values are relative to this (n = 21 per condition in d; n = 20 per condition in e; both from three experiments). f,g, SREBP2 (f) and YAP localization (g) in MCF10A-RAS cells depleted of MIEF1/2 and cultured on stiff and soft fibronectin-coated hydrogels for 24 h (n = 61 (stiff siCO), 68 (soft siCO), 72 (stiff siMIEF1/2a), 72 (soft siMIEF1/2a), 67 (stiff siMIEF1/2b) and 61 cells (soft siMIEF1/2b) in f; n = 88 (stiff siCO), 102 (soft siCO), 101 (stiff siMIEF1/2a), 103 (soft siMIEF1/2a), 86 (stiff siMIEF1/2b) and 98 cells (soft siMIEF1/2b) in g; both from three experiments). Scale bars, 10 μm. h,i, SREBP2 (h) and YAP localization (i) in WT and Mief1 Mief2 Mff KO MEFs cultured on fibronectin hydrogels for 24 h (n = 51 (stiff), 43 (soft), 44 (stiff Mief1/2 KO) and 48 cells (soft Mief1/2 KO) in h; n = 87 (stiff), 62 (soft), 76 (stiff Mief1/2 KO) and 71 cells (soft Mief1/2 KO) in i; both from two experiments). Scale bars, 10 μm. The data are presented as means ± s.d. (a and f–i) or means and single points (b–e). Statistical significance was determined by two-tailed Student’s t-test. This was calculated on punctate mitochondria (a), nuclear SREBP2 (f) or nuclear YAP (g). Source numerical data and unprocessed blots are available.
MIEF1/2 promote peri-mitochondrial F-actin and DRP1 activity
MIEF1/2 form heteromeric co-filaments together with DRP1 (ref. 51). These filaments constrict thanks to the GTPase activity of DRP1, leading to mitochondrial fission. Another player assisting fission is peri-mitochondrial F-actin52. We thus explored the relationship between these players in the context of mechanoregulated mitochondrial fission. MIEF1 forms discrete puncta along mitochondria that co-localize with DRP1 (Fig. 6a), in agreement with the co-filament model, and these puncta are enhanced on soft ECM (Fig. 6b). As for the requirements, MIEF1/2 knockdown reduces the number of DRP1 puncta on soft ECM (Fig. 6c). We also observed that DRP1 inhibition reduces the number of MIEF1 puncta (Fig. 6d), suggesting reciprocal stabilization during fission. We next monitored peri-mitochondrial F-actin, which co-localizes with DRP1 puncta10, and found that it depends on both MIEF1/2 and DRP1 activity (Fig. 6e). This is in line with the observation that MIEF1/2 overexpression can trigger peri-mitochondrial F-actin formation50,53. Conversely, inhibition of the actin-related protein 2/3 (ARP2/3) complex prevents the formation of peri-mitochondrial F-actin10,54 and decreases the number of both MIEF1 and DRP1 puncta (Fig. 6c–e). Overall, these results suggest a role for MIEF1/2 as both DRP1 receptors and promoters of peri-mitochondrial F-actin. Moreover, they suggest a mutually reinforcing role of MIEF1/2, DRP1 and F-actin in response to low actomyosin tension.

a, Co-localization analysis of HA-MIEF1 with endogenous DRP1 along mitochondria (Mito-RFP) in MCF10A-RAS cells upon ROCK–MLCK (YM) treatment for 3 h. Pearson’s indices (below) were calculated based on n = 10 cells per condition from one experiment. Scale bars, 2 μm and 5 μm. b, Quantification of MIEF1 puncta in MCF10A-RAS cells cultured on stiff and soft fibronectin-coated hydrogels (n = 36 cells from two experiments). c,d, Quantification of endogenous DRP1 (c) and MIEF1 (d) puncta in MCF10A-RAS cells with MIEF1/2 knockdown or treated with the CK869 Arp2/3 inhibitor (n = 36 cells in c; n = 21 cells in d; both from two experiments). e, Proximity ligation assay (PLA) between the TOMM20 mitochondrial outer membrane protein and F-actin in MCF10A-RAS cells with MIEF1/2 and DRP1 knockdown or treated with the CK869 Arp2/3 inhibitor (n = 20 cells from two experiments). Scale bar, 2 μm. The data are presented as means and single points. Source numerical data are available.
Actomyosin tension promotes MIEF1 phosphorylation
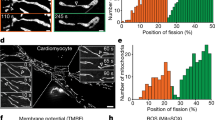
While developing this study, we started a parallel project by performing phosphoproteomics in response to ROCK–MLCK inhibition. As expected, we observed differential phosphorylation of many cytoskeletal and adhesion proteins (Supplementary Table 1). Phosphorylation of MIEF1 at a peptide encompassing serines 55 and 59, which was reduced upon ROCK–MCLK inhibition, immediately caught our attention (Fig. 7a). The same phosphopeptide was similarly regulated in fibroblasts by ECM stiffness and in hypercontractile hearts of patients with hypertrophic cardiomyopathy (Fig. 7b). Using immunoblotting, we observed an electrophoretic mobility shift of wild-type (WT) MIEF1 upon ROCK–MCLK inhibition and a similar shift was observed with a mutated variant of MIEF1 precluding phosphorylation (S55/59A; SA variant) (Fig. 7c), consistent with reduced phosphorylation. This was validated by developing affinity-purified antisera for the detection of reduced phosphorylation of endogenous MIEF1 upon ROCK–MLCK inhibition (Fig. 7d,e).

a, Volcano plot showing differentially phosphorylated peptides in MCF10A-RAS cells treated with ROCK–MLCK inhibitors (YM) for 2 h (n = 5 samples per condition in a single experiment). b, Heat maps showing the levels of phosphorylated S55/59 MIEF1 in fibroblasts cultured on hydrogels of differential stiffness77 (n = 3 per condition; P < 0.0001), of induced pluripotent stem cell (iPS)-derived cardiomyocytes (CMs) bearing the hyperactive MHY7 R403Q substitution (n = 6 per condition; P = 0.019) and of myectomy samples from patients with hypertrophic cardiomyopathy (HCM)78 (n = 5 per condition; P = 0.0015). c, Immunoblotting for HA-MIEF1 isoforms (WT, S55/59A (SA) and S55/59E (SE)) expressed in MCF10A-RAS cells. Where indicated, cells were treated with ROCK–MLCK inhibitors (YM). Asterisks denote electrophoretic shifts. GFP was co-transfected. Consistent results were obtained in n = 4 experiments. d, Immunoblotting with two anti-phospho-MIEF1 antisera on HA-MIEF1 variants immunopurified from MCF10A-RAS cell extracts. Where indicated, cells were treated with ROCK–MLCK inhibitors (YM). Consistent results were obtained in n = 3 experiments. unt, untransfected. e, Immunoblotting with two anti-phospho-MIEF1 antisera on endogenous MIEF1 after immunoprecipitation from MCF10A-RAS cell extracts. Consistent results were obtained in n = 3 experiments. f, Representative immunofluorescence staining for HA-MIEF1 variants in MCF10A-RAS cells treated with ROCK–MLCK inhibitors (YM). Scale bar, 2 μm. Consistent results were obtained in n = 3 experiments. g, Quantification of MIEF1 puncta in MCF10A-RAS cells as in f (n = 36 (MIEF1 WT DMSO), 36 (MIEF1 WT YM) and 43 cells (MIEF1 SA) from two experiments). h, Quantification of endogenous DRP1 puncta in MCF10A-RAS cells as in f (n = 30 (MIEF1 WT DMSO), 27 (MIEF1 WT YM) and 31 cells (MIEF1 SA) from two experiments). i, Mitochondrial length analysis in MCF10A-RAS cells depleted of MIEF1/2 (siMIEF1/2a) and transfected to reconstitute different levels of the WT, phosphorylation-mimicking (SE) and phosphorylation-null (SA) MIEF1 variants. Where indicated, cells were treated with ROCK–MLCK inhibitors (YM) (n = 39 (siCO DMSO), 41 (siCO YM), 39 (siMIEF1/2a DMSO), 40 (siMIEF1/2a YM), 43 (siMIEF1/2a MIEF1 WT low level YM), 46 (siMIEF1/2a MIEF1 WT medium level YM), 41 (siMIEF1/2a MIEF1 WT high level YM), 37 (siMIEF1/2a MIEF1 SA low level YM), 43 (siMIEF1/2a MIEF1 SA medium level YM), 38 (siMIEF1/2a MIEF1 SA high level YM), 21 (siMIEF1/2a MIEF1 SE low level YM), 23 (siMIEF1/2a MIEF1 SE medium level YM) and 25 cells (siMIEF1/2a MIEF1 SE high level YM) from three experiments). Scale bars, 10 μm. j, Mitochondrial length analysis in MCF10A-RAS cells with overexpression of WT and SA MIEF1 variants on stiff glass (n = 43 (untransfected), 34 (WT low level), 36 (WT high level), 35 (SA low level) and 42 cells (SA high level) from three experiments). Scale bars, 5 μm. The data are presented as means and single points (g and h) or means ± s.d (i and j). Statistical significance was determined by two-tailed Student’s t-test. In i and j, this was calculated on punctate mitochondria. Source numerical data and unprocessed blots are available (see source data and Supplementary Table 1). IP, immunoprecipitate.
Using immunofluorescence, we found that a mutated variant of MIEF1 mimicking constitutive phosphorylation (S55/59E; SE variant) remains evenly distributed on the mitochondrial surface and is unable to form foci, irrespective of actomyosin tension (Fig. 7f). The SA non-phosphorylated variant instead forms a higher number of DRP1-positive fission puncta under stiff and sparse conditions, approximating the effects of ROCK–MLCK inhibition on WT MIEF1 (Fig. 7f–h). This suggests that phosphorylated MIEF1 is deficient at forming fission foci. We then depleted MCF10A-RAS cells of MIEF1/2 and reconstituted the expression of small interfering RNA (siRNA)-insensitive MIEF1 variants to compare their activity in a null background. WT and SA MIEF1 restore mitochondrial fission in response to ROCK–MLCK inhibition, whereas the SE variant is less active (Fig. 7i and Extended Data Fig. 6a,b). Conversely, simple overexpression of the SA variant is sufficient to promote fission under stiff and sparse conditions (Fig. 7j). Overall, this indicates that high actomyosin tension limits fission by promoting MIEF1 phosphorylation.
MIEF1 phosphorylation mediates MIME signalling
We next tested the relevance of MIEF1 phosphorylation for downstream responses. In a striking parallel with the effects on mitochondrial morphology, the SE variant was less able to rescue the regulation of SREBP2 and YAP upon ROCK–MLCK inhibition (Fig. 8a,b and Extended Data Fig. 7a–d), whereas expression of the SA variant was sufficient to trigger SREBP2 activation and YAP inhibition (Fig. 8c,d and Extended Data Fig. 7e,f). Similar data were obtained with Mief1 Mief2 Mff KO MEFs (Extended Data Fig. 7g,h), indicating that this mechanism is conserved and excluding a role for Mff. Moreover, coherent results were obtained by monitoring the expression of NRF2, SREBP1/2 and YAP/TAZ target genes in response to ECM stiffness (Fig. 8e) and by monitoring cystine uptake (Fig. 8f)—a downstream readout of NRF2 activity10. These data indicate that MIEF1 phosphorylation is required and sufficient to regulate three transcriptional responses to mechanical cues.

a, SREBP2 localization in MCF10A-RAS cells depleted of MIEF1/2 and transfected to reconstitute the WT, SE and SA MIEF1 variants. Where indicated, cells were treated with ROCK–MLCK inhibitors (YM) (n = 62 (siCO DMSO), 71 (siCO YM), 51 (siMIEF1/2a DMSO), 56 (siMIEF1/2a YM), 58 (siMIEF1/2a MIEF1 WT low level YM), 59 (siMIEF1/2a MIEF1 WT medium level YM), 60 (siMIEF1/2a MIEF1 WT high level YM), 58 (siMIEF1/2a MIEF1 SA low level YM), 66 (siMIEF1/2a MIEF1 SA medium level YM), 68 (siMIEF1/2a MIEF1 SA high level YM), 69 (siMIEF1/2a MIEF1 SE low level YM), 63 (siMIEF1/2a MIEF1 SE medium level YM) and 70 cells (siMIEF1/2a MIEF1 SE high level YM) from three experiments). b, YAP localization in MCF10A-RAS cells reconstituted as in a (n = 91 (siCO DMSO), 102 (siCO YM), 87 (siMIEF1/2a DMSO), 99 (siMIEF1/2a YM), 104 (siMIEF1/2a MIEF1 WT low level YM), 89 (siMIEF1/2a MIEF1 WT medium level YM), 91 (siMIEF1/2a MIEF1 WT high level YM), 88 (siMIEF1/2a MIEF1 SA low level YM), 98 (siMIEF1/2a MIEF1 SA medium level YM), 101 (siMIEF1/2a MIEF1 SA high level YM), 81 (siMIEF1/2a MIEF1 SE low level YM), 73 (siMIEF1/2a MIEF1 SE medium level YM) and 75 cells (siMIEF1/2a MIEF1 SE high level YM) from three experiments). c, SREBP2 localization in MCF10A-RAS cells with overexpression of WT and SA MIEF1 variants on stiff glass (n = 66 (untrasfected), 59 (WT low level), 58 (WT high level), 61 (SA low level) and 69 cells (SA high level) from three experiments). d, YAP localization in MCF10A-RAS cells as in c (n = 104 (untrasfected), 63 (WT low level), 64 (WT high level), 65 (SA low level) and 67 cells (SA high level) from three experiments). e, qPCR in MCF10A-RAS cells stably expressing MIEF1 variants, transfected with MIEF1/2 siRNA and cultured on collagen I-coated hydrogels for 24 h. The heat maps indicate the mRNA levels of NRF2 targets (HMOX1 and NQO1), SREBP1/2 targets (DHCR7 and FASN) and YAP–TAZ targets (CYR61 and ANKRD1) in single samples relative to GAPDH levels and normalized to the mean control (n = 6 from three experiments). ind, doxycycline induction. f, Uptake of FITC-labelled cystine in MCF10A-RAS cells (n = 10 (siCO DMSO), 10 (siCO YM), 10 (siMIEF1/2a DMSO), 10 (siMIEF1/2a YM), 10 (siMIEF1/2a MIEF1 WT DMSO), 10 (siMIEF1/2a MIEF1 WT YM), 10 (siMIEF1/2a MIEF1 SA DMSO), 6 (siMIEF1/2a MIEF1 SA YM), 6 (siMIEF1/2a MIEF1 SE DMSO) and 6 replicates (siMIEF1/2a MIEF1 SE YM) from three experiments). g,h, YAP localization (g) and mitochondrial length analysis (h) in MCF10A-RAS cells cultured for 24 h on soft hydrogels and then subjected to 6 μm vertical confinement (compress) for 3 h (n = 70 (stiff), 84 (soft) and 78 (soft compress) in g; n = 52 (stiff), 68 (soft) and 71 (soft compress) in h; both from three experiments). i, YAP localization in MCF10A-RAS cells pre-treated for 1 h with inhibitors of ROCK–MLCK (YM) and then subjected to 6 μm vertical confinement (compress) with inhibitors for 2 h (n = 36 (DMSO), 41 (YM), 40 (YM compress), 39 (siMFN1a YM compress), 40 (siMFN1b YM compress) and 27 (MIEF1 SA YM compress) in i; n = 53 (DMSO), 70 (YM), 65 (YM compress), 56 (siMFN1a YM compress), 47 (siMFN1b YM compress) and 49 (MIEF1 SA YM compress) in j; both from two experiments). j, Adipogenic differentiation (FABP4 immunostaining) of 3T3L1 cells on micropatterned islands, treated with the Drpitor1a DRP1 inhibitor or expressing the indicated shRNAs (n = 69 (large), 79 (small), 61 (small DRP1A), 59 (small shDrp1a), 68 (small shDrp1b), 59 (small shMief1/2a) and 66 cells (small shMief1/2b) from three different experiments). Scale bars, 10 μm. k,l, Adipogenic differentiation of 3T3L1 cells at high density (HD), treated with the Drpitor1a DRP1 inhibitor or expressing the indicated shRNAs and scored by AdipoQ expression levels (k) or LipidTOX staining (l). The mRNA levels in k are relative to Gapdh levels and normalized to the low-density controls (n = 6 from three experiments). Scale bars in l, 10 μm. m, Adipogenic differentiation of 3T3L1 cells on micropatterned islands expressing the indicated shRNAs (Nrf2 and Scap) or the constitutively active TAZ4SA (n = 69 (large), 79 (small), 56 (small shNrf2a), 49 (small shNrf2b), 63 (small shScapa), 67 (small shScapb) and 57 cells (small TAZ4SA) from three different experiments). n, Adipogenic differentiation of 3T3L1 cells at high density (HD) expressing the indicated shRNAs or the constitutively active TAZ4SA. AdipoQ mRNA levels are relative to Gapdh levels and normalized to the low-density controls (n = 6 from three experiments). o, Adipogenic differentiation of 3T3L1 cells transfected with the WT and SA MIEF1 variants and cultured under low-density conditions without spatial confinement (n = 59 (LD), 67 (LD MIEF1 WT), 69 (LD MIEF1 SA) and 498 (HD) from three experiments). Scale bars, 5 μm. The data are presented as means ± s.d. (a–d and g–i) or means and single points (f and j–o). Statistical significance was determined by two-tailed Student’s t-test. This was calculated on nuclear YAP (b, d, g and i) or punctate mitochondria (h). P values for e and all source numerical data are available as source data.
The regulation of YAP by forces was previously linked to nuclear deformation and nuclear pore complexes55. In line with this, nuclear compression activates YAP on soft ECM (Fig. 8g and Extended Data Fig. 8a,b), as in ref. 56. However, inhibition of linker of nucleoskeleton and cytoskeleton (LINC) complexes—essential players in nuclear mechanotransduction57—and the proposed mediators of nuclear pore complex regulation in response to forces has a marginal effect on YAP (Extended Data Fig. 8c–f). Conversely, compression leads to mitochondrial elongation (Fig. 8h and Extended Data Fig. 8g) and is unable to activate YAP when mitochondrial elongation is prevented by expression of the SA variant or MFN1 knockdown (Fig. 8i and Extended Data Fig. 8h–k). This indicates that MIME also accounts for YAP regulation by compression and highlights an interesting difference in the way cells perceive spatial confinements: a confinement that limits actomyosin without gross nuclear deformation (small, soft or dense) activates fission, whereas a confinement that deforms the nucleus (compression) inhibits fission, which may have relevance for other responses58,59,60.
As a final test, we explored the role of MIME in the context of adipogenic differentiation—a process requiring the concerted regulation of proliferation, metabolism and cell fate—and an established response to mechanical cues17,37,61,62,63. As shown in Fig. 8j–l and Extended Data Fig. 9a–d, DRP1 and MIEF1/2 are required for the expression of adipogenic markers and the activation of lipogenic metabolism in response to a small cell geometry and cell density. Knockdown of NRF2 or the SCAP SREBP1/2 chaperone or activation of TAZ are individually sufficient to block differentiation (Fig. 8m,n and Extended Data Fig. 9e–h), suggesting a division of labour between these transcription factors downstream of fission. Finally, expression of the SA variant triggers differentiation in sparse and unconfined cells (Fig. 8o), indicating that MIEF1 phosphorylation mediates the coordinated regulation of three transcription factors and the differentiation of adipogenic cells in response to mechanical cues.
Discussion
These data unveil the backbone of a general pathway by which cells respond to forces, which we have termed MIME. Extracellular forces, tissue confinements, nuclear deformation and intracellular actomyosin tension converge on the regulation of mitochondrial dynamics. In turn, fission coordinates the activity of three transcription factors with complementary functions, including YAP/TAZ. This was observed in several cell types in vitro, is regulated in mice during the process of compensatory skin growth and in patients with heart disease and is relevant in vitro and in vivo for cell fate homeostasis. We thus propose that mitochondria fulfil a unifying mechanotransduction information-processing system role64 to match cell proliferation, cell metabolism and cell identity with tissue-scale organization and mechanical properties.
MIME is centred on phosphorylation of the MIEF1 fission factor, which limits fission when cells develop high actomyosin tension. MIEF1-mediated fission occurs in response to physiological tissue inputs and mostly regulates non-mitochondrial processes. MIME is thus different from other fission mechanisms in terms of upstream stimuli, molecular mechanism65,66,67,68 and downstream effects69,70. The quest is now open to identify the other players of this pathway, including the upstream MIEF1 kinase(s) and downstream molecular mechanism(s) by which mitochondrial fission regulates YAP/TAZ, SREBP1/2 and mtROS/NRF2. It also remains to be understood whether MIME regulates other mitochondrial functions.
This work opens avenues of research in many directions. For example, changes in mitochondrial morphology have been observed during the transition of mouse embryos from the blastocyst to the gastrula stage, or during induced pluripotent stem cell reprogramming71,72, and knockout of fission and fusion factors in mouse tissues revealed unexpected roles in stem cell self-renewal and differentiation73,74,75,76. Therefore, MIME might contribute to the physiological responses of tissues to environmental forces and could play a role in the development of pathologies associated with altered tissue mechanics, including fibrosis, atherosclerosis, cancer and ageing.
Methods
The research in this work complies with all of the relevant ethical regulations. Animal experiments were authorized by the Italian Ministry of Health.
Plasmids and reagents
Mito-RFP was kindly provided by L. Scorrano. Mito-Clover was kindly provided by D. De Stefani. Myc-tagged SCAP was kindly provided by Y. Chen. ActuAtor plasmids were kindly provided by T. Inoue and H. Nakamura. GFP-NESPRIN1-KASH and GFP-NESPRIN2-KASH plasmids were kindly provided by P. Roca-Cusachs. siRNA-insensitive human haemagglutinin (HA)-tagged human MIEF1 coding sequences (NM_019008.6; WT, S55/59A and S55/59E isoforms) were purchased from Integrated DNA Technologies and cloned into transient mammalian expression vectors. All plasmid preparations used for transfection were endotoxin free and sequence verified.
The compounds used (source and concentration) were: DRPITOR1a (AGVdiscovery; 2 µM)31, MDIVI1 (M0199; Sigma–Aldrich; 10 µM), Y27632 (1683; Axon; 20 µM); ML7 (I2764; Sigma–Aldrich; 20 µM), FASUDIL (HA-1077; Santa Cruz Biotechnology (STCB); 20 µM), BLEBBISTATIN (B0560; Sigma–Aldrich; 20 µM), SMIFH2 (S4826; Sigma–Aldrich; 10 µM), CK869 (C9124; Sigma–Aldrich; 30 µM), Rapamycin (553210; Sigma–Aldrich; 100 nM), Cisplatin (sc-200896; STCB; 5 µM); As2O3 (202673; Sigma–Aldrich; 5 µM), MitoTEMPO (SML0737; Sigma–Aldrich; 15 µM), NAC (A0737; Sigma–Aldrich; 5 mM), TROLOX (10011659; Cayman; 10 µM), Ionomycin (I0634; Sigma–Aldrich; 0.3 µM), FCCP (C2759; Sigma–Aldrich; 10 µM) and U18666A (sc-203306; SCBT; 3 µM).
Cell cultures
MCF10A and MCF10AneoT (MCF10A-RAS) human mammary epithelial cells were cultured in DMEM/F-12, 5% heat-inactivated horse serum, 10 µg ml−1 insulin (Sigma–Aldrich), 20 ng ml−1 human epidermal growth factor (PeproTech), 100 ng ml−1 cholera toxin (Sigma–Aldrich) and 0.5 µg ml−1 hydrocortisone (Sigma–Aldrich). Univocal identification was based on exon PCR sequencing to identify coding point mutations present in MCF10AneoT (MAP3K12 A662D), MCF10AT1 (also known as MCF10AT1k or MCF10A MII; PCSK5 M452I) and MCF10CA1a cells (also known as MCF10A MIV; PCSK5 M452I and PIK3CA H1047R)79. D2.0R mouse metastatic breast cancer cells, primary MEFs and mouse 3T3L1 preadipocyte cells were cultured in DMEM and 10% heat-inactivated foetal bovine serum. MEFs and 3T3L1 cells were expanded in a low-oxygen (5%) incubator. In experiments comparing KO and WT MEFs, both originated from the same laboratory. All other cells were kept at an ambient oxygen concentration with 5% CO2. ρ0 cells were obtained by culturing cells in 50 ng ml−1 ethidium bromide, 50 µg ml−1 uridine and 1 mM pyruvate for 3 weeks. Stable cell lines expressing Mito-Clover, TAZ4SA, WT and SE MIEF1 were obtained by retroviral infection followed by antibiotic selection. Stable short hairpin RNA (shRNA) expression was obtained by lentiviral infections. Stable doxycycline-inducible WT and SA MIEF1 expression was obtained by stably expressing pBCAG Tet3G and pBTRE-MIEF1 WT or pBTRE-MIEF1 SA in MCF10-RAS cells. To induce the expression of WT or SA MIEF1, cells were treated with 1 µg ml−1 doxycycline for 24 h. Primary human umbilical vein endothelial cells (HUVECs; neonatal, pooled donors; C-12203, PromoCell) were cultured in Endothelial Cell Growth Medium (C-22010; PromoCell) and used with three to four passages in vitro. Primary mouse lung endothelial cells were isolated, cultured and treated as in ref. 16. Adipogenic differentiation was induced by culturing 3T3L1 cells for 3 d in differentiation medium (insulin (I9278; Sigma–Aldrich; 10 µg ml−1), 3-isobutyl-1-methylxanthine (I5879; Sigma–Aldrich; 0.5 mM) and dexamethasone (D4902; Sigma–Aldrich; 1 µM) and then for 5 d in maintenance medium (insulin; 10 µg ml−1). Differentiated adipocytes were scored after 7 d from induction. Drpitor1a (0.5 µM) was added only with the differentiation medium, indicating an early requirement. Glutamine was added to a final concentration of 1 mM to all media. General media, serum and supplements were from Thermo Fisher Scientific. Cell lines were routinely cultured without antibiotics and tested with an N-GARDE Mycoplasma kit (EMK090020; Euroclone) to exclude contaminations.
Mechanical stimulations
Stiff (Young’s modulus, E = ~15 kPa) and soft (E = 0.5 ~kPa) fibronectin-coated polyacrylamide hydrogels were assembled in house using standard protocols40. For selected experiments, we used commercial collagen I-coated polyacrylamide hydrogels of E = ~50 kPa and E = ~0.2 kPa (Matrigen). Matrigel (BD Biosciences) was diluted 2% in 1× phosphate-buffered saline (PBS) to coat stiff tissue culture dishes (E = ~ 1 GPa), whereas it was used pure and gelled at 37 °C to obtain thick and soft substrata (E = ~250 Pa). Compression was obtained using the one-well static cell confiner (4Dcell) with 6 µm micropillars adapted on Softview collagen I-coated polyacrylamide hydrogels mounted on glass-bottom dishes (Matrigen). In all of these experiments, cells were completely isolated or contacted few neighbouring cells, and all cells were included in the analyses.
For cell density studies, MCF10A-RAS and 3T3L1 cells were seeded at 3,000 cells per cm2 (low density) and 100,000 cells per cm2 (high density) and HaCaT cells were seeded at 5,000 cells per cm2 and 150,000 cells per cm2 and cultured for 3 d to induce quantitative YAP inhibition. Tonic uniaxial stretching was achieved by plating dense cells onto a super clear 0.25 mm silicone sheet (HT6240GK; MVQ Silicones) mounted in a Brick Strex device80. The mounted apparatus was sterilized with ultraviolet light before coating of the silicone with fibronectin and plating. Cells and culture medium were contained by attaching a 30-mm-wide, 12-mm-high, 3-mm-thick three-dimensionally printed plastic ring (Protolabs) with high-vacuum silicone grease (Dow Corning). At 72 h, cells were slowly stretched to 200% that of the initial apparatus length. Cells were fixed for immunofluorescence analyses while stretched.
The fibronectin micropatterned glass slides were from Cytoo (PADO1-SQRS or PADO2-SQ17 custom masks; freely available to any customer upon request).
Flow experiments were performed in a custom-designed parallel plate flow chamber as previously described81. To fabricate the substrates that fit into the flow chamber, a 126 × 70 mm2 rectangle was cut from 120-μm-thick cyclic olefin copolymer foil (Ibidi) and patches with dimensions of 30 mm × 15 mm were cut from the centre of the substrate. The surfaces were cleaned with 70% ethanol, treated with oxygen plasma (80 W for 60 s; 1 ± 0.2 mbar) to increase their hydrophilicity and finally coated with glutaraldehyde cross-linked gelatin (104070; Merck Millipore) to increase cell adhesion82. HUVECs were seeded at a high initial density (450 cells per mm2) and grown under static conditions for 72 h to generate mature monolayers81. The flow rates were selected to reach the target WSS values, in accordance with the following equation: τ = 6Qμ/WH2, where τ is the WSS, Q is the volumetric flow rate, μ is the dynamic viscosity of water at 37 °C, W is the width of the chamber and H is the height of the chamber. Specifically, cells were conditioned for 18 h with flows yielding nominal WSSs of 1.4 and 4.0 Pa. At the end of each experiment, the cells were fixed and immunofluorescence staining was performed. Images of actin immunostainings were used to quantify cell orientation. Confocal Z-stacks were first projected with maximum intensity onto a single tiff image. From these images, the orientation distributions of actin were calculated using the ImageJ 1.54h plugin OrientationJ83. For each group (flow or static) in each independent experiment, ten fields of view were analysed and the results were pooled together to obtain the final orientation distribution. Finally, the orientation values were simplified to the range between 0 and 90° due to symmetry.
Transfections and RNA interference
Cell transfections were carried out with TransIT-LT1 (Mirus Bio), Lipofectamine 3000 (Thermo Fisher Scientific) or Lipofectamine RNAiMAX (Thermo Fisher Scientific). siRNAs were selected among FlexiTube GeneSolution (package of four) siRNA sets (Qiagen) and reordered after validation as dTdT-overhanging 19-nucleotide RNA duplexes (Thermo Fisher Scientific). The siRNA sequences were as follows: AACAAAGUCUCAGUAUUAC (hDRP1); CGGCTTTCCAAGCCTAATA (hMFN1 1); GGCGATTACTGCAATCTTT (hMFN1 2); GGUGCGUCGUGAGAAUCCA (hMIEF1 1); GUAUGAGCGUGACAAACAU (hMIEF1 2); GGUGCCUGAUGGUAGGUAU (hMIEF2 1); GCUGAGAGAAGGUGACUUA (hMIEF2 2); GCGUCUUCCUUAGCCAUUA (hPIEZO1 1); GCCUUGUAUGCACCGUCAA (hPIEZO1 2); CGACGGAAAUGGUUUGUUU (hPIEZO2 1); GCGCCGUGACUGACGUGUA (hPIEZO2 2); GCCUGUCCUUGGGUAGUAA (hPLD6 1); GCAACUACAUGGCCCUCAA (hPLD6 2); CGGUUAGAACAAGAGGUAA (hROCK1 1); AGCUCGAAUUACAUCUUUA (hROCK1 2); GCTACAUAUGGAGCUUAAA (hROCK2 1); and GCACUUGUAUAAAGCCAUA (hROCK2 2). AllStars Negative Control siRNA (Qiagen) was used. shRNAs were selected among five different pre-designed pLKO-1 vectors (Sigma–Aldrich) stably integrated in D2.0R cells by lentiviral infection and puromycin (50 µg ml−1) selection. The shRNA sequences were as follows: CGGUGGUGCUAGGAUUUGUUA (mDrp1 A (shDrp1a); TRCN0000321169); GGCAAUUGAGCUAGCGUAUAU (mDrp1 B (shDrp1b); TRCN0000012606); CCAAAGCUAGUAUAGCAAUAA (mNfe2l2 A (shNrf2a); TRCN0000054658); CUUGAAGUCUUCAGCAUGUUA (mNfe2l2 B (shNrf2b); TRCN0000054659); CAUCAUGAAUGUCCCUGGUUU (mMief1 A; TRCN0000192183); CCUGACACUAGAAGUUCAGUA (mMief1 B; TRC000192487); CCUCUUGGCUAAUGCUCGAUU (mMief2 A; TRCN0000201929); UCUCUAUGGGCCACGAAUUAC (mMief2 B; TRCN0000251424); GCGUACAUCCAACAGAUAUUU (mScap1 A (shScapa); TRCN0000341892); CCAUGGCGACAUTACCTTGTA (mScap1 B (shScapb); TRCN0000341892); and TTCTCCGAACGUGUCACGU (control (shCO)).
Flow cytometry
Staining for FITC-cystine uptake was carried out by incubating cells in a plate at the end of the experiments with the indicated reagents in full medium without phenol red (FITC-cystine; 5 µM; 30 min). After trypsinization and a brief wash in 1× Hanks’ Balanced Salt Solution, quantification was run on live cells in 1× Hanks’ Balanced Salt Solution (Ca and Mg free). Analysis was carried out on an LSRFortessa X-20 cell analyzer (BD Biosciences). Cells were gated based on side scatter pulse area versus forward scatter pulse area (to eliminate cell debris) and side scatter pulse width (to eliminate cell doublets). FITC-cystine was measured using a FITC-A filter. The median fluorescence intensity was measured for each independent biological sample, based on at least 3 × 104 gated events.
Survival assays
Cells were seeded in 96-well plates with the indicated coatings (5,000 MCF10A-RAS cells per well and 4,000 D2.0R cells per well), in parallel with standards in the linear range of detection (8,000, 4,000, 2,000, 1,000, 500 or 250 cells per well). Where pertinent, siRNA transfection was performed in bulk the day before seeding. At 24 h after seeding, culture medium was renewed by adding the indicated chemicals or corresponding amounts of vehicle. MCF10A-RAS cells were in glucose-free medium. After 48 h of treatment, cells were washed with 1× PBS and incubated with resazurin (R7017; Sigma–Aldrich; 100 µM) in culture medium, then incubated at 37 °C for 2 h. Fluorescence at 544/590 nm was measured on live cells by checking that the signal was in the temporal linear range. Absolute cell numbers were then calculated based on the standard curve after background subtraction (medium without cells). Relative survival was calculated as the percentage of remaining cells compared with control cells (100%).
EdU incorporation assays
Cells were incubated for 1 h with 5-ethynyl-2′-deoxyuridine (EdU; A10044; Invitrogen) before fixation with 4% formaldehyde for 15 min at room temperature, followed by a 0.5% Triton X-100 permeabilization step. Cells were then processed and analysed according to the manufacturer’s instructions for the Click-iT EdU Cell Proliferation Kit for Imaging (C10338; Invitrogen).
Quantitative PCR
Total RNA was isolated using commercial kits with DNAse treatment (Norgen Biotek). Complementary DNA (cDNA) synthesis was carried out with M-MLV Reverse Transcriptase (Thermo Fisher Scientific) and Oligo(dT) primers. Quantitative PCR reactions were assembled with FastStart SYBR Green Master mix (Roche) and run on a QuantStudio 6 thermal cycler (Thermo Fisher Scientific). GAPDH expression levels were used to normalize gene expression between samples based on eff(gene)−ΔCt(gene)/eff(GAPDH)−ΔCt(GAPDH), where eff is the primer amplification efficiency experimentally determined from three consecutive fivefold dilutions of one control sample of cDNA, centred on the dilution used for all of the other samples. Gene expression levels for each biological sample were quantified as the mean between three technical replicates.
The primer sequences for human genes were as follows: GTTGACTCCCCTTCCTGGTG and CTTCCAACTCTTCCCCGGAC (ACSS2); ACTTGGGCCGGTATTTCTTC and TGGGCTAGAAGTGTCTTCAGAT (ANKRD1); AGGAGTGGGTGTGTGACGA and CCAGGCAGTTGGCTCTAATC (CTGF (CCN2)); CCTTGTGGACAGCCAGTGTA and ACTTGGGCCGGTATTTCTTC (CYR61 (CCN1)); GGAGGAGTGTAAACAGCGCT and TTGGCAAACACACCCTCCTT (FASN); GCCAGTTTGTGCAGTTCCAG and GGTGGCCGAATCTTCCTTCA (FTH1); CTCCTGCACCACCAACTGCT and GGGCCATCCACAGTCTTCTG (GAPDH); TGCAGCAAACATTGTCACCG and CACCACCCACCGTTCCTATC (HMGCR); AGTCTTCGCCCCTGTCTACT and GCTGGTGTGTAGGGGATGAC (HMOX1); ACAGTCCAGGCACAGATGTC and AAGCCGCTCATTCACCTTGT (MFN1); GGTTCATTGACAGGGCCACT and GATCAGTTTCTGCAGGCCCT (MIEF1 (MID51)); ATTCGGGGAAAAGGAGCTGG and TCGAAGGTGGAGGAGTCTGT (MIEF2 (MID49)); GCACTGATCGTACTGGCTCA and CCACCACCTCCCATCCTTTC (NQO1); TTCCCCAAGTACATCCGTGC and CAGCTCGATGACCCACCATT (PIEZO1); TCTATGCCATGATCCACGCC and AGCTGGCACCCTTGAATCTC (PIEZO2); AGGGAAGATGTGAGTGTGCG and CAAGGACAGGCTGCTGAAGA (PLD6); GGTCGGTCGGTTGGGGTATTT and GCCCTAAACCTCACTTGGCTG (ROCK1); and GCCGCCGTTGCCATATTAAAG and ACCCACTTGTGCTGCTCTTTC (ROCK2).
The primer sequences for mouse genes were as follows: GCCAGTCATGCCGAAGATGA and AGTGCCATCTCTGCCATCAC (AdipoQ); CAGGTGGTGGGATTGGAGAC and CGGGCAGACAGTTTTCTTGC (Drp1); TGGGCGTGGAATTCGATGAA and ACATTCCACCACCAGCTTGT (Fabp4); ATCCTGCACCACCAACTGCT and GGGCCATCCACAGTCTTCT (Gapdh); CGAGCGCTCCATTCATCTCT and GCTGCTTCTCTTGCCTCTGA (Lpl); TCATGCATACCTGGGGAGGA and AGCCACCTACAACACATCGG (Mid49); TGCTACCCTGGCTGTAAAGC and CTCTCAGCAGGCTCAGTTCC (Mid51); AACAGAACGGCCCTAAAGCA and CCAGGACTCACGGGAACTTC (Nfe2l2); and GCTCGGCTTAATGGTTCCCT and CTGTGATGGGCTTCTGGTGT (Scap1).
Immunofluorescence and proximity ligation assay
Immunofluorescence was performed as in refs. 10,27 with minor modifications: 15 min with 1.5 mg ml−1 glycine in PBS before permeabilization to reduce the background; and blocking in 10% bovine serum albumin (BSA). Images were acquired sequentially with a Zeiss LSM 900 Airyscan, a Leica STELLARIS confocal microscope equipped with a charge-coupled device camera using ZEN 2 (version 3.9) or Leica LAS X (version 4.1.1) software. Typical acquisition settings for immunofluorescence were: image size = 1,024 × 1,024 pixels; acquisition mode = xyz; pixel size = 0.15 mm; image depth = 8 bits; acquisition speed = 5/10 (average = 2); and objective = Plan-Apochromat 40×/1.40 oil or 63×/1.40 oil DIC M27. For multichannel acquisitions, we used a 405/488/555/639 main beam splitter. Raw images (saved in .czi or .lif format) were opened in ImageJ for analysis.
For HUVEC assays, confocal microscopy was performed at room temperature using an automated Nikon Ti spinning disk confocal microscope (Nikon) equipped with an Andor DU-888 camera (Oxford Instruments) and a pE-100 LED illumination system (CoolLED) with violet (405 nm laser diode), blue (488 nm; Argon), yellow (561 nm; solid state) and red (633 nm; HeNe) excitation laser lines. For comparison purposes, different sample images were acquired under constant acquisition settings. Image acquisition was performed using a 40×/1.3 NA oil immersion objective (CFI Plan Fluor 40× Oil; Nikon). Fluorescent Z-stacks of the emitted signals were acquired by selecting the optical filters based on the respective emission.
For mitochondrial morphology analysis, cells were fixed for 10 min at 4 °C with 4% formic acid and washed with cold PBS before permeabilization, followed by immunofluorescence staining for the mitochondrial marker TOMM20. Images were acquired as above or with the Operetta automated imaging system. Two objective methods were used to quantify mitochondrial morphology: (1) a machine-learning approach measuring the percentage area of punctate, intermediate or filamentous mitochondria84; or (2) a mitochondrial fragmentation count85. Punctate, intermediate or filamentous mitochondria were classified manually by their morphology initially, followed by the application of a machine-learning algorithm (Columbus 2.9). The percentage area of these categories was calculated in each cell and the average distribution of the categories among different treatment groups was compared. The initial training was repeated for each cell type based on positive controls (DRP1 inhibition and FCCP treatment), due to basal differences in the mitochondrial network. The mitochondrial fragmentation count was calculated as the total number of mitochondria divided by the total area of mitochondria per cell.
For the proximity ligation assay (Duolink Proximity Ligation Assay) we followed the manufacturer’s instructions (DUO92102; Sigma–Aldrich) with minor modifications: cells were fixed in 4% formic acid for 10 min at 4 °C. After a brief wash, cells were incubated for 15 min with 1.5 mg ml−1 glycine in PBS before permeabilization and then blocked for 1 h at room temperature with Duolink Blocking Solution (Sigma–Aldrich). Primary antibodies were then incubated with 2% goat serum at 4 °C overnight. The primary antibodies used for the F-actin proximity ligation assay were TOMM20 (PA5-52843; Thermo Fisher Scientific) and biotin-labelled phalloidin (P8716; Sigma–Aldrich), together with an anti-biotin antibody (sc-53179; STCB).
Immunoblotting
Cells were harvested in HPO buffer (50 mM HEPES (pH 7.5), 100 mM NaCl, 50 mM KCl, 1% Triton X-100, 0.5% NP-40, 5% glycerol and 2 mM MgCl2) by sonication. Extracts were boiled in Laemli buffer (250 mM Tris-HCl (pH 6.8), 10% SDS, 50% glycerol and 0.5% BBP), loaded in NuPAGE Novex Bis-Tris acrylamide gels (Invitrogen) and transferred onto a nitrocellulose or polyvinylidene fluoride membrane in a wet transfer apparatus (Thermo Fisher Scientific) with buffer containing 50 mM Tris, 200 mM glycine, 1% SDS and 20% methanol). If possible, the membrane was cut horizontally to probe different antibodies in parallel on the same membrane. The membrane was washed in Tris-buffered saline (pH 7.5) (Tris with NaCl), 0.05% Tween-20 was added and then blocking was performed for 1 h at room temperature with 5% nonfat dry milk (Bio-Rad) or BSA (8076.4; Carl Roth). Primary antibodies were incubated overnight at 4 °C with 1% BSA blocking, except for Mid49 and Mid51, which were incubated for 1 h at 37 °C. Secondary antibodies were incubated for 1 h at room temperature. The horseradish peroxidase (HRP) signal was detected with Immobilon Crescendo Western HRP substrate (WBLVR0500; Merck Millipore) or Immobilon Forte Western HRP substrate (WBLUF0500; Merck Millipore) reagent in an ImageQuant 800 imaging system (Amersham).
Antibodies
The primary antibodies used were as follows: DRP1 (611113; BD Biosciences; 1:300; punctated immunofluorescence staining co-localizing with mitochondrial markers and disappearing upon Driptor1a treatment); FLAG (14793; Cell Signaling Technologies; 1:500); GAPDH (MAB374; Millipore; 1:20,000); green fluorescent protein (GFP; ab13970; Abcam; 1:500; immunofluorescence signal detected only in GFP-expressing cells); GM130 (610822; BD Biosciences; 1:100; immunofluorescence signal co-localizes with other Golgi markers); HA (ab9110; Abcam; 1:4,000; western blot signal detected only in HA-expressing cells); MIEF1 (20164-1-AP; Proteintech; 1:1,000; western blot signal lost in KO cells); MIEF2 (20718-1-AP; Proteintech; 1:1,000; western blot signal lost in KO cells); MFN1 (13798-1-AP; Proteintech; 1:1,000; western blot signal lost in KO cells); Myc (sc-789; STCB; 1:100); NF2 (sc-331; STCB; 1:200; western blot signal lost in KO cells); SREBP2 (10007663; CaymanChem; 1:200; immunofluorescence signal at the endoplasmic reticulum, Golgi or nucleus, depending on extracellular lipids); TOMM20 (PA5-52843 (Thermo Fisher Scientific) and sc-17764 (STCB); 1:500; immunofluorescence signal co-localizes with other mitochondrial markers); and YAP/TAZ (sc-101199; STCB; 1:200; western blot signal lost upon siRNA knockdown; immunofluorescence nucleo-cytoplasmic signals regulated by established inputs). The secondary antibodies used (1:1,000) were as follows: Alexa-488 anti-mouse (A21202; Thermo Fisher Scientific); Alexa-568 anti-rabbit (A11036; Thermo Fisher Scientific); Alexa-568 anti-mouse IgG1 (A21124; Thermo Fisher Scientific); Alexa-647 anti-mouse IgG2b (A21242; Thermo Fisher Scientific); anti-rabbit-HRP (A0545; Sigma–Aldrich); and anti-mouse-HRP (RPN4201V; GE Healthcare). Where indicated, cell extracts were subjected to immunoprecipitation with anti-HA (ab9110; Abcam) or anti-MIEF1 (sc-514135; STCB) antibodies. Custom-made rabbit (Innovagen; 1:1,000) and chick (Davids Biotechnologie; 1:2,000) polyclonal anti-human phosphorylated MIEF1 was developed by peptide immunization and subsequent positive (phosphopeptide) and negative (non-phosphopeptide) affinity purifications. Validation was based on enzyme-linked immunosorbent assay tests on peptides and on immunoblotting on immunoprecipitated WT and SA variant MIEF1 proteins.
For the HUVEC assays, the following primary antibodies were used: rabbit monoclonal anti-AIF antibody [E20] - mitochondrial marker (ab32516; Abcam 1:100) and mouse monoclonal anti-YAP (63.7) (sc-101199; Santa Cruz Biotechnology; 1:200). The secondary antibodies used were goat anti-mouse Alexa Fluor 555 (A28180; Thermo Fisher Scientific; 1:400) and goat anti-rabbit Alexa Fluor 647 (A27040; Thermo Fisher Scientific; 1:400). Cell nuclei were counterstained with DAPI (62248; Thermo Fisher Scientific; 1:1,000) and actin fibres with phalloidin (Alexa Fluor 555 Phalloidin; A34055; Thermo Fisher Scientific; 1 µM).
RNA sequencing
Library generation used the QuantSeq 3′ mRNA-Seq Library Prep kit (Lexogen) with Oligo(dT) priming. Sequencing was performed on NextSeq 500 Illumina instruments and based on at least 5 million reads (75 bp single end) per sample. The reads were trimmed using BBDuk (BBMap version 37.87) with the parameters indicated in the Lexogen data analysis protocol. After trimming, reads were aligned to the Mus musculus genome (GRCm38.p6) using STAR (version 2.7.6a). The gene expression levels were quantified using featureCounts (version 2.0.1). Genes were sorted to remove those that had a total number of counts below ten in at least three samples. All RNA sequencing analyses were carried out in the R environment (version 4.0.0) with Bioconductor (version 3.7). We computed differential expression analysis using the DESeq2 R package (version 1.28.1). Normalization to counts per million was performed exploiting the cpm function from the edgeR package (version 3.32.0).
Protein mass spectrometry
All of the experiments were performed in a labelling-free setting. Cells were lysed in 6 M guanidine and 100 mM Tris-HCl (pH 7.5) buffer supplemented with protease (11873580001; Roche) and phosphatase (P5726; Sigma–Aldrich) inhibitors. Samples were sonicated in a Bioruptor at 2 × 30 s, 4 °C and high amplitude. From each sample, 1 mg was treated with a final concentration of 10 mM tris(2-carboxyethyl)phosphine and 40 mM chloroacetamide at 95 °C for 10 min. Protein lysates were precipitated by methanol/chloroform using four volumes of ice-cold methanol, one volume of chloroform and three volumes of water. The mixture was centrifuged at 20,000g for 30 min and the upper aqueous phase was removed, then three volumes of ice-cold methanol were added. Proteins were pelleted by centrifugation, washed twice with two volumes of ice-cold methanol and air dried. Each pellet was resuspended in 50 ml 10 mM Tris-HCl (pH 8) and 8 M urea at room temperature. Proteins were digested with trypsin (ratio = 1:100) and Lys-C (ratio = 1:200) overnight at 37 °C on a mixer. The digestion reaction was acidified using 0.5% trifluoroacetate and peptides were purified using Sep-Pak tC18 cartridges (Waters; 50 mg), then lyophilized. Phosphopeptides were enriched using a High-Select TiO2 Phosphopeptide Enrichment Kit (A32993; Thermo Fisher Scientific).
Instruments for liquid chromatography with tandem mass spectrometry (MS/MS) analysis consisted of a NanoLC 1200 coupled via a nano-electrospray ionization source to a quadrupole-based Q Exactive HF benchtop mass spectrometer (Thermo Fisher Scientific). Peptide separation was carried out according to hydrophobicity on a home-made chromatographic column (inner diameter = 75 mm; tip = 8 µm; bed = 500 mm (packed with Reprosil-Pur C18-AQ); particle size = 1.9 mm; pore size = 120 Å) using a binary buffer system consisting of solutions A (0.1% formic acid) and B (80% acetonitrile and 0.1% formic acid). Runs of 140 min after loading were used for the proteome samples, with a constant flow rate of 250 nl min−1. After sample loading, runs started with a discontinuous gradient from 5–90% buffer B.
For Q Exactive HF, mass spectrometry spectra were acquired using 3 × 106 as an automatic gain control target, with a maximum injection time of 20 ms and 120,000 resolution at 200 m/z. The mass spectrometer operated in data-dependent Top20 mode with subsequent acquisition of higher-energy collisional dissociation fragmentation MS/MS spectra of the top 20 most intense peaks. The resolution for MS/MS spectra was set to 15,000 at 200 m/z, with an automatic gain control target of 1 × 105, a maximum injection time of 20 ms and an isolation window of 1.6 Th. The intensity threshold was set at 2.0 × 104 and dynamic exclusion at 30 s.
All of the experiments were performed in at least three independent biological replicates. All acquired raw files were processed using MaxQuant (version 1.6.2.10) and the implemented Andromeda search engine. For protein assignment, spectra were correlated with the UniProt human database (version 2021), including a list of common contaminants. Searches were performed with tryptic specifications and default settings for mass tolerances for mass spectrometry and MS/MS spectra. Carbamidomethyl at cysteine residues was set as a fixed modification, whereas oxidations at methionine, acetylation at the amino terminus and phosphorylation (at serines, threonines and tyrosines) were defined as variable modifications. The minimum peptide length was set to seven amino acids and the false discovery rate for proteins and peptide spectrum matches was set to 1%. The match-between-run feature with a time window of 1 min was used. For further analysis, Perseus software (version 1.6.2.3) was used and first filtered for contaminants and reverse entries, as well as proteins that were only identified by a modified peptide. The label-free quantification ratios were logarithmized, grouped and filtered for the minimum valid number (a minimum of three in at least one group). Missing values were replaced by random numbers that were drawn from a normal distribution. Two sample t-tests were performed using a false discovery rate of 0.05.
Mice
Hydrodynamic tail vein injection was used to transduce hepatocytes of 4- to 6-week-old BALB/C male mice with exogenous DNA. 50 μg piggyBac transposon plasmid DNA together with 10 μg hyperactive piggyBac transposase plasmid DNA (hyPBase) were diluted in sterile Ringer’s solution in a volume corresponding to 10% of the bodyweight (on average, 18–22 g) and injected via the tail vein for a maximum time of 8–10 s. piggyBac plasmids were obtained by subcloning the cDNAs of interest (HA-tagged DRP1 DN or FLAG-tagged mTAZ 4SA) in a piggyBac plasmid with a cytomegalovirus promoter. For EdU labelling, mice were injected with 12.5 mg kg−1 EdU (A10044; Molecular Probes) in sterile 1× PBS 16 h before tissue sampling. Mice were euthanized after 15 d and the abdominal contents were exposed. Transcardiac perfusion (29-gauge needle) with cold 1× PBS (10–20 ml) was performed to reduce blood contaminants. The liver was removed and placed in a clean petri dish with 1× PBS on ice. The liver tissue was either prefixed in 4% paraformaldehyde (A6 staining) or directly embedded in OCT (CK19 staining) and stored at −80 °C for subsequent immunofluorescence analyses. Every mouse was analysed by scoring six independent tissue sections. Mice were kept in standard cages with a limit of five mice per cage, with ad libitum feeding and drinking at an average temperature of 19–24 °C and with 12 h light/12 h dark cycles. Animal experiments were performed according to our institutional guidelines as approved by the University Animal Welfare Commission (OPBA) and authorized by the Ministry of Health (945/2015-PR and 54/2015-PR). Reporting was according to the ARRIVE guidelines.
Statistics and reproducibility
All of the data are based on multiple experiments with independent biological replicates, as specified in the figure captions, with the exception of RNA sequencing and proteomics, where independent biological replicates from a single experiment were used. Experiments were carried out by thawing a new aliquot of cells deriving from the same original stock. All values were pooled between experiments. No method was used to predetermine sample size, but our sample sizes are similar to those reported in previous publications10,20,27,28,37,40,41. No samples or experiments were excluded. The experiments were not blinded or randomized. Most results were qualitatively replicated across different cell lines and with independent reagents and techniques providing consistent results. Key results were replicated by another fellow in the laboratory.
Experimental data are presented as means and single points or means and s.d. Statistical significance was determined by unpaired two-tailed Student’s t-test or Dunnet’s test for multiple comparisons with a single control (Prism 9). The data distribution was assumed to be normal, but this was not formally tested.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Annotation of proteomics was based on UniProt human database (version 2021). RNA sequencing raw data have been deposited in the Gene Expression Omnibus with the accession code GSE189803. The results of DESeq2 analysis are provided in Supplementary Table 1. Proteomic raw data have been deposited in the PRIDE repository (PXD040502) and the results of the differential analysis are provided in Supplementary Table 1. Source data are provided with this paper. All of the other data supporting the findings of this study are available from the corresponding author upon reasonable request.
Change history
16 July 2025
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1038/s41556-025-01734-6
References
Iskratsch, T., Wolfenson, H. & Sheetz, M. P. Appreciating force and shape—the rise of mechanotransduction in cell biology. Nat. Rev. Mol. Cell Biol. 15, 825–833 (2014).
Janmey, P. A., Fletcher, D. A. & Reinhart-King, C. A. Stiffness sensing by cells. Physiol. Rev. 100, 695–724 (2020).
Discher, D. E. et al. Matrix mechanosensing: from scaling concepts in 'omics data to mechanisms in the nucleus, regeneration, and cancer. Annu. Rev. Biophys. 46, 295–315 (2017).
Mohammadi, H. & Sahai, E. Mechanisms and impact of altered tumour mechanics. Nat. Cell Biol. 20, 766–774 (2018).
Humphrey, J. D. & Schwartz, M. A. Vascular mechanobiology: homeostasis, adaptation, and disease. Annu. Rev. Biomed. Eng. 23, 1–27 (2021).
De Belly, H., Paluch, E. K. & Chalut, K. J. Interplay between mechanics and signalling in regulating cell fate. Nat. Rev. Mol. Cell Biol. 23, 465–480 (2022).
Romani, P., Valcarcel-Jimenez, L., Frezza, C. & Dupont, S. Crosstalk between mechanotransduction and metabolism. Nat. Rev. Mol. Cell Biol. 22, 22–38 (2021).
Dupont, S. & Wickström, S. A. Mechanical regulation of chromatin and transcription. Nat. Rev. Genet. 23, 624–643 (2022).
Phuyal, S., Romani, P., Dupont, S. & Farhan, H. Mechanobiology of organelles: illuminating their roles in mechanosensing and mechanotransduction.Trends Cell Biol. 33, 1049–1061 (2023).
Romani, P. et al. Mitochondrial fission links ECM mechanotransduction to metabolic redox homeostasis and metastatic chemotherapy resistance. Nat. Cell Biol. 24, 168–180 (2022).
Chen, K., Wang, Y., Deng, X., Guo, L. & Wu, C. Extracellular matrix stiffness regulates mitochondrial dynamics through PINCH-1- and kindlin-2-mediated signalling. Curr. Res. Cell Biol. 2, 100008 (2021).
Papalazarou, V. et al. The creatine–phosphagen system is mechanoresponsive in pancreatic adenocarcinoma and fuels invasion and metastasis. Nat. Metab. 2, 62–80 (2020).
Giacomello, M., Pyakurel, A., Glytsou, C. & Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 21, 204–224 (2020).
Fenton, A. R., Jongens, T. A. & Holzbaur, E. L. F. Mitochondrial dynamics: shaping and remodeling an organelle network. Curr. Opin. Cell Biol. 68, 28–36 (2021).
Chan, D. C. Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. Mech. Dis. 15, 235–259 (2020).
Nikolopoulou, P. et al. Talin regulates steady-state tensional homeostasis to drive vascular morphodynamics and cancer. Preprint at bioRxiv https://doi.org/10.1101/2022.08.03.502607 (2022).
McBeath, R., Pirone, D. M., Nelson, C. M., Bhadriraju, K. & Chen, C. S. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell 6, 483–495 (2004).
Chen, C. S., Alonso, J. L., Ostuni, E., Whitesides, G. M. & Ingber, D. E. Cell shape provides global control of focal adhesion assembly. Biochem. Biophys. Res. Commun. 307, 355–361 (2003).
Fu, J. et al. Mechanical regulation of cell function with geometrically modulated elastomeric substrates. Nat. Methods 7, 733–736 (2010).
Aragona, M. et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 154, 1047–1059 (2013).
Dhawan, S. S. et al. Shear stress and plaque development.Expert Rev. Cardiovasc. Ther. 8, 545–556 (2014).
Coon, B. G. et al. A mitochondrial contribution to anti-inflammatory shear stress signaling in vascular endothelial cells. J. Cell Biol. 221, e202109144 (2022).
Baeyens, N. et al. Vascular remodeling is governed by a VEGFR3-dependent fluid shear stress set point. eLife 4, e04645 (2015).
Song, M., Mihara, K., Chen, Y., Scorrano, L. & Dorn, G. W. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 21, 273–286 (2015).
Simpson, C. L. et al. NIX initiates mitochondrial fragmentation via DRP1 to drive epidermal differentiation. Cell Rep. 34, 108689 (2021).
Aragona, M. et al. Mechanisms of stretch-mediated skin expansion at single-cell resolution. Nature 584, 268–273 (2020).
Romani, P. et al. Extracellular matrix mechanical cues regulate lipid metabolism through Lipin-1 and SREBP. Nat. Cell Biol. 21, 338–347 (2019).
Bertolio, R. et al. Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat. Commun. 10, 1326 (2019).
Cassidy-Stone, A. et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14, 193–204 (2008).
Bordt, E. A. et al. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev. Cell 40, 583–594.e6 (2017).
Wu, D. et al. Identification of novel dynamin-related protein 1 (Drp1) GTPase inhibitors: therapeutic potential of Drpitor1 and Drpitor1a in cancer and cardiac ischemia-reperfusion injury. FASEB J. 34, 1447–1464 (2020).
Brown, M. S., Radhakrishnan, A. & Goldstein, J. L. Retrospective on cholesterol homeostasis: the central role of Scap. Annu. Rev. Biochem. 87, 783–807 (2018).
Takashima, K. et al. COPI-mediated retrieval of SCAP is crucial for regulating lipogenesis under basal and sterol-deficient conditions. J. Cell Sci. 128, 2805–2815 (2015).
Enzo, E. et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 34, 1349–1370 (2015).
Santinon, G., Enzo, E. & Dupont, S. The sweet side of YAP/TAZ. Cell Cycle 14, 2543–2544 (2015).
Santinon, G. et al. dNTP metabolism links mechanical cues and YAP/TAZ to cell growth and oncogene-induced senescence. EMBO J. 37, e97780 (2018).
Dupont, S. et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011).
Hamaratoglu, F. et al. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat. Cell Biol. 8, 27–36 (2006).
Kim, N.-G. & Gumbiner, B. M. Cell contact and Nf2/Merlin-dependent regulation of TEAD palmitoylation and activity. Proc. Natl Acad. Sci. USA 116, 9877–9882 (2019).
Pocaterra, A. et al. F-actin dynamics regulates mammalian organ growth and cell fate maintenance. J. Hepatol. 71, 130–142 (2019).
Pocaterra, A. et al. Fascin1 empowers YAP mechanotransduction and promotes cholangiocarcinoma development. Commun. Biol. 4, 763 (2021).
Chandel, N. S. & Schumacker, P. T. Cells depleted of mitochondrial DNA (ρ0) yield insight into physiological mechanisms. FEBS Lett. 454, 173–176 (1999).
Ishihara, N., Jofuku, A., Eura, Y. & Mihara, K. Regulation of mitochondrial morphology by membrane potential, and DRP1-dependent division and FZO1-dependent fusion reaction in mammalian cells. Biochem. Biophys. Res. Commun. 301, 891–898 (2003).
Chakrabarti, R. et al. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J. Cell Biol. 217, 251–268 (2018).
Nakamura, H. et al. ActuAtor, a Listeria-inspired molecular tool for physical manipulation of intracellular organizations through de novo actin polymerization. Cell Rep. 42, 113089 (2023).
Eura, Y., Ishihara, N., Yokota, S. & Mihara, K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J. Biochem. 134, 333–344 (2003).
Chen, H. et al. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200 (2003).
Choi, S.-Y. et al. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat. Cell Biol. 8, 1255–1262 (2006).
Adachi, Y. et al. Coincident phosphatidic acid interaction restrains Drp1 in mitochondrial division. Mol. Cell 63, 1034–1043 (2016).
Osellame, L. D. et al. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J. Cell Sci. 129, 2170–2181 (2016).
Kalia, R. et al. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 558, 401–405 (2018).
Fung, T. S., Chakrabarti, R. & Higgs, H. N. The multiple links between actin and mitochondria. Nat. Rev. Mol. Cell Biol. 24, 651–667 (2023).
Palmer, C. S. et al. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J. Biol. Chem. 288, 27584–27593 (2013).
Li, S. et al. Transient assembly of F-actin on the outer mitochondrial membrane contributes to mitochondrial fission. J. Cell Biol. 208, 109–123 (2015).
Elosegui-Artola, A. et al. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell 171, 1397–1410.e14 (2017).
Aureille, J. et al. Nuclear envelope deformation controls cell cycle progression in response to mechanical force. EMBO Rep. 20, e48084 (2019).
Guilluy, C. et al. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 16, 376–381 (2014).
Niethammer, P. Components and mechanisms of nuclear mechanotransduction. Annu. Rev. Cell Dev. Biol. 37, 233–256 (2021).
Miroshnikova, Y. A. & Wickström, S. A. Mechanical forces in nuclear organization.Cold Spring Harb. Perspect. Biol. 14, a039685 (2022).
Alraies, Z. et al. Cell shape sensing licenses dendritic cells for homeostatic migration to lymph nodes.Nat. Immunol. 25, 1193–1206 (2024).
Sordella, R., Jiang, W., Chen, G.-C., Curto, M. & Settleman, J. Modulation of Rho GTPase signaling regulates a switch between adipogenesis and myogenesis. Cell 113, 147–158 (2003).
Hong, J.-H. et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 309, 1074–1078 (2005).
Tang, Y. et al. MT1-MMP-dependent control of skeletal stem cell commitment via a β1-integrin/YAP/TAZ signaling axis. Dev. Cell 25, 402–416 (2013).
Picard, M. & Shirihai, O. S. Mitochondrial signal transduction. Cell Metab. 34, 1620–1653 (2022).
Cereghetti, G. M. et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl Acad. Sci. USA 105, 15803–15808 (2008).
Fung, T. S., Ji, W.-K., Higgs, H. N. & Chakrabarti, R. Two distinct actin filament populations have effects on mitochondria, with differences in stimuli and assembly factors. J. Cell Sci. 132, jcs234435 (2019).
Pangou, E. et al. A PKD–MFF signaling axis couples mitochondrial fission to mitotic progression. Cell Rep. 35, 109129 (2021).
Henschke, S. et al. Food perception promotes phosphorylation of MFFS131 and mitochondrial fragmentation in liver. Science 384, 438–446 (2024).
Ng, M. Y. W., Wai, T. & Simonsen, A. Quality control of the mitochondrion. Dev. Cell 56, 881–905 (2021).
Dayalan Naidu, S. & Dinkova-Kostova, A. T. Regulation of the mammalian heat shock factor 1. FEBS J. 284, 1606–1627 (2017).
Pernaute, B. et al. DRP1 levels determine the apoptotic threshold during embryonic differentiation through a mitophagy-dependent mechanism. Dev. Cell 57, 1316–1330.e7 (2022).
Bakare, A. B. et al. MitoCellPhe reveals mitochondrial morphologies in single fibroblasts and clustered stem cells. Am. J. Physiol. Cell Physiol. 321, C735–C748 (2021).
Khacho, M. et al. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell 19, 232–247 (2016).
Baker, N. et al. The mitochondrial protein OPA1 regulates the quiescent state of adult muscle stem cells. Cell Stem Cell 29, 1315–1332.e9 (2022).
Hong, X. et al. Mitochondrial dynamics maintain muscle stem cell regenerative competence throughout adult life by regulating metabolism and mitophagy. Cell Stem Cell 29, 1298–1314.e10 (2022).
Bean, C. et al. The mitochondrial protein Opa1 promotes adipocyte browning that is dependent on urea cycle metabolites. Nat. Metab. 3, 1633–1647 (2021).
Mattner, L. F. et al. Phosphoproteomics of cellular mechanosensing reveals NFATC4 as a regulator of myofibroblast activity. Preprint at bioRxiv https://doi.org/10.1101/2023.02.13.528335 (2023).
Moore, J. et al. Multi-omics profiling of hypertrophic cardiomyopathy reveals altered mechanisms in mitochondrial dynamics and excitation–contraction coupling. Int. J. Mol. Sci. 24, 4724 (2023).
Maguire, S. L. et al. Three-dimensional modelling identifies novel genetic dependencies associated with breast cancer progression in the isogenic MCF10 model. J. Pathol. 240, 315–328 (2016).
Mäntylä, E. & Ihalainen, T. O. Brick Strex: a robust device built of LEGO bricks for mechanical manipulation of cells. Sci. Rep. 11, 18520 (2021).
Wu, X. et al. A free-form patterning method enabling endothelialization under dynamic flow. Biomaterials 273, 120816 (2021).
Orsenigo, F. et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat. Commun. 3, 1208 (2012).
Rezakhaniha, R. et al. Experimental investigation of collagen waviness and orientation in the arterial adventitia using confocal laser scanning microscopy. Biomech. Model. Mechanobiol. 11, 461–473 (2012).
Chen, K.-H. et al. Epigenetic dysregulation of the dynamin-related protein 1 binding partners MiD49 and MiD51 increases mitotic mitochondrial fission and promotes pulmonary arterial hypertension: mechanistic and therapeutic implications. Circulation 138, 287–304 (2018).
Rehman, J. et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 26, 2175–2186 (2012).
Acknowledgements
We are deeply indebted to S. Archer for sharing the Drpitor1a compound. We thank staff at the Mass Spectrometry core at the Telethon Institute of Genetics and Medicine for proteomic analyses; D. De Stefani for the Mito-Clover plasmid; M. Ryan, S. Manley and H. Perreten for the Dnm1l KO and Mief1 Mief2 Mff KO MEFs; B. Gumbiner for the NF2 KO MCF10A cells; T. Inoue and H. Nakamura for the ActuAtor plasmids; L. Scorrano and A. Maracani for the Mfn1 KO MEFs; B. Burke and Y. Loon for the Sun1-2 KO MEFs; P. Roca-Cusachs for the NESPRIN-KASH plasmids; C. Blanpain for consenting to the use of mouse skin transmission electron microscopy; and Jean-David Durussel for lending bricks for the stretching apparatus. Funding was provided by Worldwide Cancer Research (grant 21-0156), the AIRC Foundation (Investigator Grants 21392 and 28940), the CARIPARO Foundation (Ricerca Scientifica di Eccellenza 2023; Mitochondrial drugs to cure mechanotransduction disease (MINDED)), the Italian Ministry of University and Research (grants 2022T9RM8A and P2022CE7SP) and the National Recovery and Resilience Plan (funded by NextGenerationEU Mission 4, Component 2, Investment 1.3; CUP B73C22001250006 MechanoMet) (to S.D.); 2020, 2021 and 2022 Veronesi Foundation fellowships and AIRC MFAG 27453 (to P.R.); 2023 and 2024 Veronesi Foundation fellowships (to M.C.); a Giovanni Armenise–Harvard Foundation Award and European Research Council Starting Grant MetEpiStem (to G.M.); Telethon Foundation grant TMPGMFU22TT and AIRC MFAG 24856 (to P.G.); and Swiss National Science Foundation grant 205321_188828 (to C.G.). M. Aragona acknowledges that the Novo Nordisk Foundation Center for Stem Cell Medicine is supported by a Novo Nordisk Foundation grant (NNF21CC0073729). The Hellenic Foundation for Research and Innovation grant HFRI-FM17-2644 was awarded to V.K.
Author information
Authors and Affiliations
Contributions
P.R. carried out most of the experiments and quantified and analysed the data with help from M.C. and G.B. M. Arboit performed RNA sequencing analysis with support from G.M. C.C. performed protein mass spectrometry analysis with support from P.G. X.W. performed the HUVEC flow experiments with support from C.G. G.R. performed the experiments with Talin1 KO mouse endothelial cells with support from V.K. M. Aragona provided the electron microscopy images of expanded mouse skin. S.D. conceptualized, coordinated and supervised the project and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cell Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article has been retracted. Please see the retraction notice for more detail: https://doi.org/10.1038/s41556-025-01734-6"
Extended data
Extended Data Fig. 1 Forces induce mitochondrial elongation.
a, qPCR in MCF10A-RAS cells transfected with PIEZO1-2 siRNA mixes. The mRNA levels are relative to the GAPDH levels, and normalized to control siRNAs (siCO) (n = 4 samples from two experiments). b-g, Quantification of mitochondrial morphology by mitochondrial fragmentation count (MFC) analysis in MCF10A-RAS cells cultured on hydrogels (b) or treated with YM (c), in MLECs with inducible Talin1 KO (d), in MCF10A-RAS cells cultured in sparse or dense conditions (e), in 3T3L1 cells cultured on micropatterns (f) and in HUVECs subjected to flows (g). The same set of pictures quantified in Fig. 1 were analyzed. h, MCF10A-RAS cells expressing a single-fluorophore excitation-ratiometric ROCK kinase Activity Reporter (ROCKAR-WT) or a version of the same reporter bearing a threonine to alanine mutation of the phosphorylation site (ROCKAR-TA) were transfected with ROCK1/2 siRNA. A lower ratio between the fluorescence emitted in response to excitation at 488 and the fluorescence emitted in response to excitation at 405 nm (Excitation ratio) indicates decreased phosphorylation of the reporter. (n = 59 (siCO), n = 59 (siROCK1/2a), n = 59 (siROCK1/2b) from two experiments). i, qPCR in MCF10A-RAS cells transfected with siRNA mixes targeting ROCK1 and ROCK2. The mRNA levels are relative to the GAPDH levels, and normalized to control siRNAs (siCO) (n = 4 from two experiments). j, MCF10A-RAS cells expressing the ROCKAR sensor were treated with ROCK inhibitors for 3 h (n = 30 (DMSO), n = 30 (Y27632) and n = 30 (FASUDIL) from two experiments). k, Excitation ratio of the ROCKAR sensors in MCF10A-RAS cells cultured on soft and stiff hydrogels for 24 h and treated with Drp1 inhibitor (DRP1A) (n = 30 (STIFF), n = 30 (SOFT), n = 30 (SOFT DRP1A) from two experiments). l, Excitation ratio of the ROCKAR sensors in 3T3L1 cells cultured in small and large micropatterned islands for 24 h (n = 25 (LARGE), n = 25 (SMALL), n = 25 (SMALL DRP1A) from two experiments). m, Excitation ratio of the ROCKAR sensors in MCF10A-RAS cells cultured in sparse and dense conditions for 48 h (n = 50 (LD), n = 50 (HD), n = 50 (HD DRP1A) from two experiments). n, Excitation ratio of the ROCKAR sensors in HaCaT cells cultured in sparse conditions, in dense conditions, and dense then subjected to tonic uniaxial stretching for 3 h (n = 30 (LD), n = 30 (HD), n = 30 (HD STRETCH) from two experiments). o, Excitation ratio of the ROCKAR sensors in MCF10A-RAS cells treated with SMIFH2 or with the non-muscle myosin type II inhibitor for 24 h (n = 30 (DMSO), n = 30 (SMIFH2), n = 30 (BLEBBI) from two experiments). Data are presented as mean values and single points (a,h-o) or box plots with median (centre), quartiles (25% and 75%) and extreme values (whiskers) (b-g). In b,c,e-g, p-values were calculated based on two-tailed Student’s t-tests. Source numerical data are available in the source data.
Extended Data Fig. 2 DRP1 is required for the mechano-regulation of SREBP1-2.
a, Expression levels (CPM, counts per million) of selected SREBP1-2 target genes from RNA sequencing of D2.0R cells, selected among the genes downregulated on a soft ECM upon DRP1 shRNA (n = 3 in a single experiment). b, SREBP2 localization in MCF10A-RAS cells deprived of serum and of exogenous lipids. DRP1 was inhibited with Drpitor1a (DRP1A) (n = 72 cells (SERUM) n = 120 (NO SERUM) and n = 131 (NO SERUM DRP1A) from three experiments). c, Excitation ratio of the ROCKAR sensor in MCF10A-RAS cells cultured as in (b) (n = 47 (FULL SERUM), n = 47 (NO SERUM), n = 47 (REFEED) from two experiments). d,e, SREBP2 localization in MCF10A-RAS cells treated with the U18666 NPC1 lysosomal cholesterol transporter inhibitor (d), or depleted of COPI27 (e) (d, n = 91 (ethanol), n = 103 (U18666A), n = 99 (U18666A DRP1A); e, n = 81 (siCO), n = 76 (siCO YM), n = 89 (siCO YM DRP1A), n = 98 (siCOPIa), n = 72 (siCOPIa DRP1A), n = 63 (siCOPIb), n = 81 (siCOPIb DRP1A) from three experiments). Scale bar 10 μm. Data are presented as mean values and single points (a,c) or mean ± s.d. In b,d,e, p-values were calculated based on two-tailed Student’s t-tests on nuclear SREBP2. Source numerical data are available in the source data and Supplementary Table 1.
Extended Data Fig. 3 DRP1 is required for the mechano-regulation of YAP-TAZ.
a, Expression levels (CPM, counts per million) of selected proliferation and YAP-TAZ target genes from RNA sequencing of D2.0R cells, selected among the genes upregulated on a soft ECM upon DRP1 shRNA (n = 3 in a single experiment). b, YAP localization in MCF10A-RAS cells treated with ROCK-MLCK inhibitors (YM) and Drpitor1a for 3 h (n = 91 cells (DMSO), n = 105 (YM) and n = 123 (YM DRP1A) from three experiments). Scale bar 10 μm. c, YAP localization in MCF10A-RAS cells treated with the SMIFH2 inhibitor of formin actin-nucleating proteins and with Drpitor1a for 3 h (n = 71 cells (DMSO), n = 78 (SMIFH2) and n = 81 (SMIFH2 DRP1A) from three experiments). Scale bar 10 μm. d, Immunoblotting of extracts from wt and Dnm1l KO MEFs (Drp1 KO). Consistent results were obtained in n = 3 experiments. e, YAP localization in 3T3L1 cells cultured in sparse (LD) and dense (HD) conditions, treated with Drpitor1a. (n = 60 (LD), n = 610 (HD) and n = 625 (HD DRP1A) from three experiments). f, YAP localization in HaCaT cells cultured at high density and either subjected to uniaxial stretching or treated with Drpitor1a for 3 h (n = 543 cells (HD), n = 621 (HD STRETCH) and n = 352 (HD DRP1A) from two experiments). Scale bar 20 μm. g, Immunoblotting of extracts from wt and NF2 KO MCF10A. Consistent results were obtained in n = 3 experiments. h, YAP localization in MCF10A cells (WT) treated with ROCK-MLCK inhibitors and in NF2-knockout MCF10A cells (NF2 KO) transiently transfected to reconstitute the expression of Nf2. Where indicated, cells were treated with Drpitor1a (n = 120 cells (wt DMSO), n = 114 (wt YM), n = 121 (wt YM DRP1A), n = 83 (NF2 KO), n = 51 (NF2 KO Nf2) and n = 72 (NF2 KO Nf2 DRP1A) from three experiments). i, EdU incorporation assay in 3T3L1 cells cultured on soft hydrogels and treated with Drpitor1a for 24 h (n = 134 cells (STIFF), n = 95 (SOFT) and n = 103 (SOFT DRP1A) from three experiments). j, EdU incorporation assay in 3T3L1 cells cultured at high density and treated with Drpitor1a for 24 h (n = 81 cells (LD), n = 343 (HD) and n = 391 (HD DRP1A) from three experiments). k,l, Cell area was measured in MCF10A-RAS cells cultured on a soft ECM for 24 h (k) or at high density for 48 h (l) and treated with Drpitor1a (k, n = 60 (STIFF), n = 60 (SOFT), n = 60 (SOFT DRP1A), n = 60 (SOFT MDIVI) from two experiments; l, n = 60 (LD), n = 60 (HD), n = 60 (HD DRP1A) from two experiments). m,n, SREBP2 (m) and YAP localization (n) in MCF10A-RAS cells treated with inhibitors of ROCK-MLCK (YM) or cultured on a soft ECM. Where indicated, cells were also treated with antioxidant compounds (NAC, TROLOX or MITOTEMPO) for 3 h (m, n = 93 cells (DMSO), n = 82 (YM), n = 56 (YM TROLOX), n = 65 (YM NAC), n = 87 (YM MITO), n = 56 (STIFF), n = 42 (SOFT),n = 62 (SOFT NAC), n = 53 (SOFT TROLOX), n = 41 (SOFT MITO); n, n = 78 (DMSO), n = 65 (YM), n = 41 (YM NAC), n = 67 (YM TROLOX), n = 73 (YM MITO), n = 55 (STIFF), n = 42 (SOFT), n = 39 (SOFT NAC), n = 43 (SOFT TROLOX), n = 39 (SOFT MITO) from two experiments). Scale bar 10 μm. Data are presented as mean values and single points (a,i,j), mean ± s.d. (b,c,e,f,h,m,n) or violin plots with median and quartiles (k,l). p-values were calculated based on two-tailed Student’s t-tests. In b,c,e,h, p-values were calculated on nuclear YAP levels. Source numerical data and unprocessed blots are available in the source data and Supplementary Table 1.
Extended Data Fig. 4 General workings of mitochondrial mechanotransduction.
a-d, Mitochondrial length analysis (a and c) and YAP localization (b and d) in MCF10A-RAS cells treated with inhibitors of ROCK-MLCK (YM; a and b) or cultured on soft hydrogels (c and d) and treated with the Drpitor1a inhibitor (DRP1A). (a n = 26 cells (YM), n = 21 (YM DRP1A 0.2microM), n = 22 (YM DRP1A 2microM) and n = 23 (YM DRP1A 10microM); b n = 63 cells (YM), n = 81 (YM DRP1A 0.2microM), n = 73 (YM DRP1A 2microM) and n = 53 (YM DRP1A 10microM); c n = 20 (SOFT), n = 20 (SOFT DRP1A 0.2microM), n = 20 (SOFT DRP1A 2microM) and n = 20 (SOFT DRP1A 10microM); d n = 56 cells (SOFT), n = 57 (SOFT DRP1A 0.2microM), n = 56 (SOFT DRP1A 2microM) and n = 56 (SOFT DRP1A 10microM) from two experiments). e, PCR for mitochondrial mtDNA loci in “deep” rho-zero (ρ0) MCF10A-RAS cells. The expression levels were normalized to nuclear Actin DNA (n = 4 from two experiments). f, Mitochondrial length analysis in MCF10A-RAS cells treated with the FCCP uncoupler of oxidative phosphorylation (n = 78 (DMSO) and n = 80 (FCCP) from three experiments). Scale bar 2 μm. g, Mitochondrial length analysis in MCF10A-RAS cells treated with Ionomycin (i n = 25 (DMSO) and n = 25 (IONO) from two experiments). Scale bar 2 μm. h, Mitochondrial length analysis in wt and Drp1 KO MEFs expressing the ActuAtor system45. Cells were left untreated or treated for 2 h with Rapamycin to activate the ActuAtor system (+Act) and induce fission (n = 26 cells (wt), n = 27 (wt Act), n = 26 (KO) and n = 26 (KO Act) from two experiments). Scale bar 2 μm. i, qPCR of MCF10A-RAS cells with MFN1 knockdown. The mRNA levels are relative to the GAPDH levels, and normalized to the stiff controls (n = 4 from two experiments). j, Mitochondrial length analysis in MCF10A-RAS cells with MFN1 knockdown. Where indicated, cells were treated with Drpitor1a (n = 41 cells (siCO), n = 56 (siMFN1a), n = 51 (siMFN1a DRP1A), n = 48 (siMFN1b), n = 49 (siMFN1b DRP1A) from three experiments). Scale bar, 2 μm. k, Immunoblotting of wt and Mfn1 KO MEFs. Consistent results were obtained in n = 2 experiments. l, YAP localization in Mfn1 KO MEFs (n = 55 cells (wt) and n = 65 (KO) from two experiments). m, Quantification of DRP1 mitochondrial puncta in MCF10A-RAS cells depleted of MFN1 and treated with Drpitor1a (n = 42 cells from two experiments). Scale bar 2 μm. n, Quantification of DRP1 mitochondrial puncta in Mfn1 KO MEFs (n = 43 from two experiments). o, qPCR of MCF10A-RAS cells with PLD6 knockdown. The mRNA levels are relative to the GAPDH levels, and normalized to the stiff controls (n = 4 from two experiments). p, Mitochondrial length analysis in MCF10A-RAS cells with PLD6 knockdown (n = 66 cells (siCO), n = 76 (siPLD6a) and n = 65 (siPLD6b) from two experiments). Data are presented as mean values ± s.d. (a-d,f-h,j,l,p) or mean and single points (e,i,m-o). p-values were calculated based on two-tailed Student’s t-tests. In f,j, this was calculated on punctate mitochondria. Source numerical data and unprocessed blots are available in the source data.
Extended Data Fig. 5 Mitochondrial mechanotransduction depends on the MIEF1-2 fission factors.
a, Representative gating strategy used to quantify FITC-Cystine uptake. Fluorescence was measured in the P2 gate. b,c, SREBP2 (b) and YAP localization (c) in MCF10A-RAS cells with MIEF1-2 knockdown treated with inhibitors of ROCK-MLCK (YM) for 6 h (b n = 67 (siCO DMSO), n = 81 (siCO YM), n = 89 (siMIEF1/2a DMSO), n = 84 (siMIEF1/2a YM), n = 84 (siMIEF1/2b DMSO) and n = 65 (siMIEF1/2b YM) from three experiments; c n = 131 (siCO DMSO), n = 141 (siCO YM), n = 112 (siMIEF1/2a DMSO), n = 119 (siMIEF1/2a YM), n = 123 (siMIEF1/2b DMSO) and n = 127 (siMIEF1/2b YM) from four experiments). d, Immunoblotting of wt and Mief1-Mief2-Mff KO MEFs. Consistent results were obtained in n = 2 experiments. Data are presented as mean values ± s.d. (b,c). p-values were calculated based on two-tailed Student’s t-tests. In b, this was calculated on nuclear SREBP2. In c, on nuclear YAP. Source numerical data and unprocessed blots are available in the source data.
Extended Data Fig. 6 Actomyosin tension inhibits fission by promoting MIEF1 phosphorylation.
a, Mitochondrial length analysis in MCF10A-RAS cells reconstituted as in Fig. 7i, but with an independent set of MIEF1-2 siRNAs (siMIEF1/2b) (siCO DMSO and siCO YM as in Fig. 7i, n = 49 (siMIEF1/2b DMSO), n = 44 (siMIEF1/2b YM), n = 45 (siMIEF1/2b WT low level YM), n = 39 (WT medium level), n = 43 (WT high level), n = 41 (SA low level), n = 39 (SA medium level), n = 44 (SA high level), n = 51 (SE low level), n = 39 (SE medium level) and n = 42 (SE high level) from three experiments). b, Immunoblotting of HA-MIEF1 isoforms reconstituted in MCF10A-RAS cells as in Fig. 7i. Consistent results were obtained in n = 2 experiments. Data are presented as mean values ± s.d. In a, p-values were calculated on punctate mitochondria based on two-tailed Student’s t-tests. Source numerical data and unprocessed blots are available in the source data.
Extended Data Fig. 7 MIEF1 phosphorylation mediates mitochondrial mechanotransduction.
a, Representative pictures of SREBP2 localization in MCF10A-RAS cells. Scale bar 10 μm. Refers to Fig. 8a. b, Representative pictures of YAP localization in MCF10A-RAS cells. Scale bar 10 μm. Refers to Fig. 8b. c, SREBP2 localization in MCF10A-RAS cells reconstituted as in Fig. 8a, but with an independent set of MIEF1-2 siRNAs (siMIEF1/2b) (siCO DMSO and siCO YM as in Fig. 8a, n = 57 (siMIEF1/2b DMSO), n = 65 (siMIEF1/2b YM), n = 56 (siMIEF1/2b WT low level YM), n = 61 (WT medium level), n = 58 (WT high level), n = 54 (SA low level), n = 66 (SA medium level), n = 60 (SA high level), n = 62 (SE low level), n = 64 (SE medium level) and n = 58 (SE high level) from three experiments). d, YAP localization in MCF10A-RAS cells reconstituted as in Fig. 8b, but with an independent set of MIEF1-2 siRNAs (siMIEF1/2b) (siCO DMSO and siCO YM as in Fig. 8b, n = 77 (siMIEF1/2b DMSO), n = 79 (siMIEF1/2b YM), n = 81 (siMIEF1/2b WT low level YM), n = 69 (WT medium level), n = 72 (WT high level), n = 73 (SA low level), n = 78 (SA medium level), n = 81 (SA high level), n = 84 (SE low level), n = 89 (SE medium level) and n = 88 (SE high level) from three experiments). e, Representative pictures of SREBP2 localization in MCF10A-RAS cells. Scale bar 10 μm. Refers to Fig. 8c. f, Representative pictures of YAP localization in MCF10A-RAS cells. Scale bar 10 μm. Refers to Fig. 8d. g, SREBP2 localization in Mief1-Mief2-Mff knockout MEFs transiently transfected to reconstitute different levels of the wild-type (WT), phosphorylation-mimicking (SE) and phosphorylation-null (SA) MIEF1 variants. Where indicated, cells were treated with ROCK-MLCK inhibitors (YM) (n = 71 cells (DMSO), n = 65 (YM), n = 67 (WT low level), n = 67 (WT medium level), n = 67 (WT high level), n = 63 (SA low level), n = 68 (SA medium level), n = 69 (SA high level), n = 68 (SE low level), n = 71 (SE medium level), n = 65 (SE high level) from three experiments). h, YAP localization in Mief1-Mief2-Mff knockout MEFs transiently transfected to reconstitute different levels of the wild-type (WT), phosphorylation-mimicking (SE) and phosphorylation-null (SA) MIEF1 variants. Where indicated, cells were treated with ROCK-MLCK inhibitors (YM) (n = 92 cells (DMSO), n = 89 (YM), n = 99 (WT low level), n = 97 (WT medium level), n = 102 (WT high level), n = 121 (SA low level), n = 99 (SA medium level), n = 105 (SA high level), n = 97 (SE low level), n = 92 (SE medium level), n = 104 (SE high level) from three experiments). Data are presented as mean values ± s.d. p-values were calculated based on two-tailed Student’s t-tests. In e,g, this was calculated on nuclear SREBP2. In f,h,this was calculated on nuclear YAP. Source numerical data are available in the source data.
Extended Data Fig. 8 Nuclear compression regulates mitochondrial mechanotransduction.
a, MCF10A-RAS were cultured for 24 h on soft hydrogels and then subjected to 6 μm vertical confinement (COMPRESS) for 3 h (n = 16 cells from one representative experiment; compression was evident upon visual inspection in all experiments). b, Representative pictures of YAP localization in MCF10A-RAS cells subjected to 6 μm vertical confinement. Scale bar 10 μm. Refers to Fig. 8g. c, Immunostainings for NESPRIN2 confirming defective assembly of the LINC complexes in Sun1-Sun2 KO MEFs. Scale bar 10 μm. d, YAP localization in Sun1-Sun2 KO MEFs (n = 195 (wt) and n = 179 (Sun1/2 KO) from three experiments). Scale bar 10 μm. e, YAP localization in MEFs transfected with increasing doses of dominant-negative GFP-NESPRIN1-KASH or GFP-NESPRIN2-KASH (n = 42 (EMPTY), n = 34 (NES1-KASH low level), n = 35 (NES1-KASH high level), n = 40 (NES2-KASH low level), n = 29 (NES2-KASH high level) from two experiments). Scale bar 10 μm. f, YAP localization in MCF10A-RAS cells transfected as in j (n = 56 (EMPTY), n = 64 (NES1-KASH low level), n = 45 (NES1-KASH high level), n = 43 (NES2-KASH low level), n = 55 (NES2-KASH high level) from two experiments). Scale bar 10 μm. g, Representative pictures of mitochondrial length in MCF10A-RAS cells subjected to 6 μm vertical confinement. Scale bar 10μm. Refers to Fig. 8h. h, Representative pictures of YAP localization in MCF10A-RAS cells subjected to 6 μm vertical confinement. Scale bar 10 μm. Refers to Fig. 8i. i, Mitochondrial length analysis in MCF10A-RAS cells pre-treated for 1 h with inhibitors of ROCK-MLCK (YM) and then subjected to 6 μm vertical confinement (COMPRESS) for 2 h with inhibitors. (n = 53 (DMSO), n = 70 (YM), n = 65 (YM COMPRESS), n = 56 (siMFN1a YM COMPRESS), n = 47 (siMFN1b YM COMPRESS) and n = 49 (MIEF1SA YM COMPRESS) from two experiments). j,k, YAP localization (j) and mitochondrial length analysis (k) and in MEFs cells cultured on soft hydrogel for 24 h and then subjected to 6 μm vertical confinement (COMPRESS) for 2 h. (j n = 43 (STIFF), n = 41 (SOFT), n = 39 (SOFT COMPRESS) and n = 25 (SOFT MIEF1SA COMPRESS); k n = 29 (STIFF), n = 31 (SOFT), n = 43 (SOFT COMPRESS), n = 33 (SOFT MIEF1SA COMPRESS) from two experiments). Data are presented as mean values ± s.d. (d-f, i-k) or mean and single points (a). p-values were calculated based on two-tailed Student’s t-tests. In d this was calculated on nuclear YAP. Source numerical data are available in the source data.
Extended Data Fig. 9 MIME mediates mechano-chemical adipogenic differentiation.
a,b, qPCR of 3T3L1 cells stably expressing Drp1 (a) or Mief1-2 shRNAs (b). The mRNA levels are relative to the Gapdh levels, and normalized to shRNA controls (n = 6 from three experiments). c,d, Adipogenic differentiation of 3T3L1 cells at high density (HD), treated with the Drpitor1a DRP1 inhibitor or expressing the indicated shRNAs or cDNAs, as scored by Lpl (c) or Fabp4 (d) expression levels. The mRNA levels are relative to the Gapdh levels, and normalized to the low density controls (n = 6 from three experiments). These are the same samples shown in Fig. 8k. e,f, qPCR of 3T3L1 cells stably expressing Nrf2 (e) or Scap shRNAs (f). The mRNA levels are relative to the Gapdh levels, and normalized to shRNA controls (n = 6 from three experiments). g,h, Adipogenic differentiation of 3T3L1 cells at high density (HD), treated with the Drpitor1a DRP1 inhibitor or expressing the indicated shRNAs or cDNAs, as scored by Lpl (g) or Fabp4 (h) expression levels. The mRNA levels are relative to the Gapdh levels, and normalized to the low density controls (n = 6 from three experiments). These are the same samples shown in Fig. 8n. Data are presented as mean values and single points. p-values were calculated based on two-tailed Student’s t-tests. Source numerical data are available in the source data.
Supplementary information
Supplementary Table 1 (download XLSX )
Raw data and differential analyses of RNA sequencing and phosphoproteomics.
Source data
Source Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Fig. 6 (download XLSX )
Statistical source data.
Source Data Fig. 7 (download XLSX )
Statistical source data.
Source Data Fig. 7 (download PDF )
Unprocessed western blots.
Source Data Fig. 8 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 3 (download PDF )
Unprocessed western blots.
Source Data Extended Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 4 (download PDF )
Unprocessed western blots.
Source Data Extended Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 5 (download PDF )
Unprocessed western blots.
Source Data Extended Data Fig. 6 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 6 (download PDF )
Unprocessed western blots.
Source Data Extended Data Fig. 7 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 8 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 9 (download XLSX )
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Romani, P., Benedetti, G., Cusan, M. et al. RETRACTED ARTICLE: Mitochondrial mechanotransduction through MIEF1 coordinates the nuclear response to forces. Nat Cell Biol 26, 2046–2060 (2024). https://doi.org/10.1038/s41556-024-01527-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41556-024-01527-3
This article is cited by
-
Mechanical regulation of metabolism, epigenetics, and their interplay
npj Biomedical Innovations (2026)
-
Protein networks are influenced by maternal BMI and differentiate preterm birth types
Communications Medicine (2026)
-
Biomechanics of the tumor extracellular matrix and regulatory T cells: regulatory mechanisms and potential therapeutic targets
Cell Communication and Signaling (2025)
-
Mechanical forces in the tumor microenvironment: roles, pathways, and therapeutic approaches
Journal of Translational Medicine (2025)
-
Extracellular matrix dynamics in tumor immunoregulation: from tumor microenvironment to immunotherapy
Journal of Hematology & Oncology (2025)
