
Abstract
Spatial domain detection methods often focus on high-variance structures, such as tumour-adjacent regions with sharp gene expression changes, while missing low-variance structures with subtle gene expression shifts, like those between adjacent normal and early adenoma regions. Here, to address this, we introduce ‘compare and contrast spatial transcriptomics’ (CoCo-ST), a graph contrastive feature representation framework. By comparing a target sample with a background sample, CoCo-ST detects both high-variance, broadly shared structures and low-variance, tissue-specific features. It offers technical advantages, including multisample integration, batch-effect correction and scalability across technologies from spot-level Visium data to single-cell Xenium Prime 5K and subcellular Visium HD data. We benchmarked CoCo-ST against ten state-of-the-art spatial-domain-detection algorithms using mouse lung precancerous samples, demonstrating its superior ability to identify low-variance spatial structures overlooked by other methods. CoCo-ST also effectively distinguishes cell clusters and niche structures in Visium HD and Xenium Prime 5K data. CoCo-ST is accessible at GitHub (https://github.com/WuLabMDA/CoCo-ST).
Similar content being viewed by others


Main
Analysing spatial transcriptomics (ST) data requires robust feature representation methods to effectively capture the intricate biological information or patterns enriched in these high-dimensional datasets. Although traditional dimension reduction techniques like principal component analysis (PCA)1 and non-negative matrix factorization (NMF)2 have been widely adopted as off-the-shelf approaches for ST data dimension reduction, they are primarily aimed at capturing global patterns and variations in the original high-dimensional ST datasets. More recently, the integration of spatial constraints into dimension reduction algorithms has led to the emergence of robust feature representation approaches such as non-negative spatial factorization3, spatial PCA4 and MEFISTO5. Moreover, advanced spatial domain detection methods such as GraphST6, SpaGCN7, Giotto8, STAGATE9 and BayesSpace10 have been introduced to better delineate spatially coherent domains.
While these methods represent considerable progress, they often prioritize identifying prominent global spatial patterns with high variability, potentially overlooking localized, low-variance spatial structures that are biologically important. This becomes particularly evident in studies of carcinogenesis, in which tumours progress from a few small isolated precancerous sites to invasive cancer across various tissue samples. The majority of these samples exhibit common global patterns representing normal tissue biology, which may not be of primary interest. Conversely, a small portion of samples contain unique, critical precancerous structures that require specific attention, patterns that traditional methods can fail to identify.
Moreover, most of these algorithms are designed to analyse a single sample at a time, making them less effective for studying dynamic processes, such as the evolution of the tumour microenvironment across multiple samples. Thus, they are vulnerable to batch effects, which can introduce noise and obscure meaningful biological signals when analysing multiple samples. Furthermore, the associated high computational costs make it difficult for these algorithms to analyse large-scale datasets or scale to single-cell or subcellular ST data.
To address these constraints, here we proposed a graph contrastive feature learning framework CoCo-ST (compare and contrast ST samples). CoCo-ST operates by taking two samples as inputs: one as the reference (background) and another as the target. These ST samples typically have certain common structures that are usually not the primary foci. The goal is to extract feature representations that emphasize the new and unique structures enriched in the target sample.
We used CoCo-ST to comprehensively investigate carcinogenesis using ST datasets from an in-house-curated mouse carcinogenesis model. We analysed a range of ST datasets, including Visium, Xenium Prime 5K and Visium HD, covering different spatial resolutions at spot, single-cell and subcellular scales. Furthermore, we benchmarked CoCo-ST against ten state-of-the-art algorithms, including GraphST, SpaGCN, Giotto, STAGATE and BayesSpace. We systematically evaluated CoCo-ST’s performance in handling diverse ST datasets, integrating multi-sample data, correcting batch effects and detecting biologically relevant spatial structures in both cancer and brain tissue samples.
Results
CoCo-ST algorithm
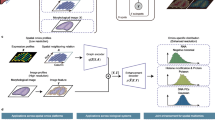
CoCo-ST was inspired by the recent successes of contrastive learning approaches11,12,13, which learn discriminative feature representations by contrasting positive pairs (similar samples) with negative pairs (dissimilar samples). In our CoCo-ST design workflow (Fig. 1a), we began by collecting tissue samples from mouse lungs and processing them using the Visium technology (10x Genomics) to obtain the ST data. We then organized the resulting gene expression data into a gene–spot matrix and further normalized the data to eliminate technical artifacts. CoCo-ST proceeded to construct two weighted graphs, one each for the background and target ST datasets, enabling us to capture the local structures within the datasets. We derived contrastive feature representations by comparing and contrasting the local variances of the background and target graphs. We achieved this by assessing the difference between their respective local total scatter matrices. In the case of a new target ST dataset, CoCo-ST simply uses the learned transformation to generate feature representations for the new data (Fig. 1a). These contrastive feature representations can be used as inputs for various other ST analysis tools for enhanced downstream analysis. We have illustrated the effectiveness of these contrastive feature representations across multiple downstream analysis tasks, including ST data visualization, spatial-domain identification, tissue-specific spatial trajectory inference, trajectory inference across multiple tissues and examination of cell–cell interaction. It is worth noting that CoCo-ST is generically applicable to broad ST data types that can be represented in form of a gene–spot matrix.

a, Overview of the CoCo-ST workflow. The diagram was created using BioRender. nCount_Spatial, total number of counts (UMIs or reads) captured per spot. b, A background tissue sample with mainly normal lung and a target ST tissue sample containing unique, interesting spatial structures annotated by a pathologist. c, The spatial domains and regions identified using the different feature representation methods. d, Quantitative comparisons of CoCo-ST and competing approaches in terms of ARI and NMI for hotspot domain detection. Genome-wide analysis (c and d) was performed on one independent biological sample (MLP-6).
CoCo-ST detects local structures in mouse carcinogenesis model
We first applied CoCo-ST to learn transformation by using a mouse normal lung tissue sample (MLP-1) as the background and an abnormal lung tissue sample (MLP-6) containing structures other than the normal spatial domain (Fig. 1b) as the target. We designated MLP-1 as the background ST data because its spatial structures belong to the normal lung spatial domain, which was also present in all the rest of the tissue samples. We then applied the learned transformation to the remaining tissue samples, resulting in contrastive feature representations that we subsequently used for spatial-domain identification and further downstream analysis. Note that CoCo-ST does not require much data to determine a good transformation compared with conventional machine learning approaches. Moreover, it has the potential to capture more specific local structures within individual samples. These properties make CoCo-ST a valuable complement to large-foundation-model-based approaches.
Uniform manifold approximation and projection (UMAP) embedding of the learned contrastive features in the target ST data (Extended Data Fig. 1a) illustrated CoCo-ST’s effectiveness in determining feature representations that provide robust discrimination of various spatial structures in the target tissue (Fig. 1c). Clustering the ST data based on the learned contrastive components led to the identification of six clusters, each corresponding to a unique spatial structure. These spatial structures detected using CoCo-ST’s contrastive components agreed well with the pathologist-annotated regions (Fig. 1b). Spatial clustering of spots on the basis of components determined using some of the compared methods, including BayeSpace, Giotto, GraphST, SpaGCN, STAGATE, NSF and MEFISTO, did not effectively detect the small hotspot region annotated as hyperplasia by the pathologist (Fig. 1c), owing to an inability to detect spatial structures of low variability. Contrastive PCA (cPCA) effectively captured the small hotspot region while NMF and Seurat (PCA) delineated the small hotspot region but annotated it as belonging to spatial domain 2 (SD 2). To provide a quantitative comparison, we evaluated all methods using an adjusted Rand index (ARI) and normalized mutual information (NMI) on the hotspot regions with the pathologist annotation as ground truth. CoCo-ST achieved the highest ARI value (0.88) and NMI value (0.73) among all of the methods tested (Fig. 1d), demonstrating its robustness in capturing hotspot regions and detecting the low-variance spatial domains aligning well with the pathologist-annotated hotspot regions.
We further annotated the detected spatial structures detected using CoCo-ST based on their differentially expressed marker genes (Extended Data Fig. 1b) and spatial locations. The distribution of these marker genes—including Epas1 for normal lung tissue (endothelial PAS domain), Slc26a4 for fibrotic/scarred tissue, Cybb for adjacent normal tissue, Hp for the bronchus/alveoli, Ctsh for the adenoma and Msln for the membrane—showed the expected high-expression patterns (Extended Data Fig. 1c). To further validate the adenoma region (hotspot) detected using CoCo-ST, we investigated the most differentially expressed marker genes for the detected adenoma regions and found 3,498 marker genes at a false-discovery rate of 5% (Extended Data Fig. 1d). The most differentially expressed marker genes were domain-specific metagenes for the adenoma region (including the small hotspot region). For example, a metagene consisting of Ctsh, Cxcl15 and Slc34a2 marked the hotspot regions clearly, as these genes exhibited high expression patterns in both the larger and smaller hotspot regions (Extended Data Fig. 1e). Cxcl15 and Slc34a2 were uniquely identified by CoCo-ST. The high expression of these genes at both the large and small hotspot adenoma regions indicates that these two spatial domains are biologically close. Moreover, Ctsh was reported to be differentially expressed in the adenoma region of patients with colorectal cancer14. On the other hand, the two methods Seurat and STUtility NMF, which detect the small adenoma region but misannotated it as SD 2, did not identify the important marker genes (Extended Data Fig. 1f), probably leading to the mis-annotation of this important spatial domain. Trf is the top marker gene differentiating the hotspot region (SD 5) for both Seurat (PCA) and NMF (Extended Data Fig. 1g). However, this gene had a high expression pattern in the larger adenoma region but not in the small hotspot region (Extended Data Fig. 1h). These results highlighted the limitations of the Seurat (PCA) and STUtility (NMF) methods, which emphasize identifying the large adenoma region with high variance but lack the ability to identify domain-specific metagenes that capture the smaller adenoma structure with significant biological relevance.
Examining the weights of the first five contrastive components revealed that CoCo-ST effectively identified major spatial domains (Extended Data Fig. 2a), indicating that it captured both global and local variations associated with the interesting spatial structures in the target data. For example, component 1 explained variation in multiple spatial domains, characterized by large positive weights around the adenoma and alveoli/bronchus and negative weights around the normal lung. For some of the compared approaches, such as Seurat (PCA) and NSF, the top five components of these methods predominantly focus on the normal lung structure with the largest variance (Extended Data Fig. 2b). For example, the first components of both Seurat PCA and NSF exhibited larger weights on the normal lung structures. As the first few components of these methods are expected to capture most of the information in the original data and are subsequently used as inputs for downstream analysis, relying solely on these components may result in overlooking crucial local biological insights. To gain deeper insights into the underlying biological processes associated with these components, we further investigated the top 20 genes with the largest weights on each of the CoCo-ST’s contrastive components (Extended Data Fig. 2c). These highlighted individual genes encoding domain-specific signatures such as Retnla, Cyp2f2, Ctsh, Ccl6 and Acta2 (Extended Data Fig. 2d), as well as gene sets linked with broader biological processes and pathways. Gene set enrichment analysis using the top 20 marker genes for each component revealed enriched Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways specific to each spatial domain. These included haem binding on component 1, lipoprotein and retinol metabolism on component 2, IgA immunoglobulin complex on component 3, lysosome on component 4 and extracellular matrix on component 5 (Extended Data Fig. 3).
CoCo-ST outperforms ten algorithms in detecting local structures
To evaluate CoCo-ST’s performance, we benchmarked it comprehensively against ten state-of-the-art approaches across multiple tissue samples (MLP-2–10). To ensure fair and optimal performance comparisons across all spatial-domain-detection methods, we systematically tuned key parameters for each algorithm using a randomized grid search strategy. We sampled a range of values for key parameters, evaluating each combination based on ARI and NMI against pathologist-annotated ground-truth labels. We then selected the parameter set yielding the highest average of ARI and NMI as the best-performing configuration. In all cases, we compared the performance of the author-recommended default parameters to those obtained through our tuning procedure and selected the configuration with the superior performance for inclusion in the comparative benchmarking. This strategy was applied consistently across methods to provide a rigorous and unbiased evaluation.
CoCo-ST consistently outperformed these methods in identifying biologically relevant spatial domains, particularly subtle hotspot regions that other methods missed (Fig. 2). Notably, in MLP-6 and MLP-9, CoCo-ST successfully detected hotspot regions such as the hyperplasia, early adenoma, late adenoma and adenocarcinoma, as annotated by pathologists. By contrast, other methods missed or only partially captured these spatial domains. Moreover, the hotspot regions delineated by CoCo-ST were more accurately aligned with expert annotations compared with those identified using other approaches. These results highlight CoCo-ST’s ability to detect both global and local spatial structures even in complex and heterogeneous tissue samples. By integrating contrastive learning framework, CoCo-ST can successfully detect spatial domains that were often masked by dominant high-variance structures. Its robust performance across multiple samples demonstrates its potential as a useful tool for spatial-domain identification and analysis in ST research.

Each panel displays the spatial domains identified using a different method. CoCo-ST consistently identifies biologically meaningful tissue structures, including subtle local regions relevant to early tumour evolution that are often missed or oversegmented by other methods. Genome-wide analysis was performed on nine independent biological samples (MLP-2–10).
CoCo-ST is robust to different graph construction approaches
To investigate how different graph construction methods (molecular versus spatial) would affect CoCo-ST’s performance, we constructed a similarity graph based on spatial coordinates, as opposed to the gene expression data used in our previous experiments. This approach has proven to be highly effective7 through assuming that neighbouring spots in the tissue share similar gene expression patterns and are probably part of the same spatial domain. Our findings demonstrated that the spatial graph based CoCo-ST effectively identified the major spatial domains across the target tissue samples (Extended Data Fig. 4), similar to analysis based on gene expression (Fig. 2). In summary, CoCo-ST demonstrates robust performance with similarity graphs constructed from both spatial coordinates and gene expression data.
CoCo-ST enables multiple sample integration
The analysis of ST data often requires integrating multiple batches or samples to investigate complex biological phenomena across diverse tissue types and conditions. However, technical variations between experiments, known as batch effects, can obscure meaningful biological patterns and hinder data interpretation. Through contrastive learning, CoCo-ST provides an effective framework to address these challenges and ensure the preservation of biologically relevant structures after integration. We used CoCo-ST to integrate multiple samples and align spatial domains across batches. The integration results are demonstrated in the UMAP embedding (Fig. 3a); spatial spots from different batches were well mixed and formed cohesive clusters without batch-specific separation. Quantitatively, CoCo-ST achieved a kBET15 value close to 1 (Fig. 3b), strongly indicating effective batch integration with no significant batch effect remaining. Moreover, spatial domains identified across batches were consistently mapped into coherent clusters (Fig. 3c), demonstrating CoCo-ST’s ability to harmonize datasets while maintaining spatial coherence and biological integrity.

a, UMAP embedding of multiple batches integrated using CoCo-ST, demonstrating well-mixed batches in the embedding space. b, kBET analysis to evaluate batch effects after integration with CoCo-ST. Higher observed kBET values indicate minimal batch effects after integration. c, Spatial clustering and domain identification based on the integrated batches. d, Spatial domains identified after multiple-sample integration with CoCo-ST. e, Spatial domains detected using competing approaches under multiple-sample integration settings. Genome-wide analysis (a–e) was performed on three independent biological samples.
Beyond batch correction, CoCo-ST excels in integrating multiple samples and identifying biologically relevant hotspot regions. For example, CoCo-ST distinguished early-adenoma regions (SD 6) in MLP-3 and MLP-4 from the late-adenoma region (SD 5) in MLP-7, even under challenging cross-sample integration scenarios (Fig. 3d). These early-adenoma regions maintained distinct spatial characteristics, highlighting CoCo-ST’s ability to capture subtle biological differences. By contrast, other methods such as STAGATE did not accurately identify these regions under similar conditions (Fig. 3e), further highlighting CoCo-ST’s superior performance in a multisample integration setting. CoCo-ST’s robust capabilities for batch effect correction and multiple-sample integration establish it as a versatile tool for large-scale ST studies. By harmonizing datasets and capturing biologically relevant spatial structures, CoCo-ST facilitates comprehensive analysis of tissue heterogeneity and spatial dynamics. These findings demonstrate its unique ability to identify subtle spatial structures and provide critical insights that are often overlooked by other methods.
CoCo-ST enables lung precancer evolution analysis
We next performed deconvolution analysis to infer the cell type composition at each of the spatial domains detected using CoCo-ST. For this analysis, we used matched single-cell RNA sequencing (scRNA-seq) data (Fig. 4a) obtained from the same MLP tissue samples as a reference. In the MLP-6 tissue sample (Fig. 4b), as expected, the normal lung spatial domain (endothelial PAS domain) showed a high concentration of endothelial cells (Fig. 4c,d and Extended Data Figs. 5 and 6). The fibrotic/scarred and bronchus/alveoli spatial domains were enriched with fibroblasts. In the adjacent normal spatial domain, there was an abundance of endothelial cells, whereas the adenoma spatial domain was enriched with macrophages and proliferating macrophages (Fig. 4c,d, and Extended Data Figs. 5 and 6). Notably, we observed tumour-associated macrophages (TAMs) in the adjacent normal spatial domain that exhibited significantly upregulated Ccl6. Notably, Ccl6 was closely associated with component 4 of CoCo-ST (Extended Data Fig. 2c,d). High expression of Ccl6 in a mouse model of lung cancer was reported to be associated with tumour growth and increased metastasis16. This evidence demonstrated the intricate cellular compositions within specific spatial domains, shedding light on potential implications for the progression of lung cancer.

a, UMAP embedding of the scRNA-seq dataset used as a reference for cell type deconvolution. cDC, conventional dendritic cells; NK, natural killer cells; pDC, plasmacytoid dendritic cells; prolif. mac., proliferating macrophages. b, Spatial domains identified in the MLP-6 tissue sample using CoCo-ST’s contrastive components. c, Cell type annotation on each of the spatial locations in MLP-6 tissue sample as inferred by the RCTD deconvolution algorithm. d, The percentage of different cell types (y axis) in the different spatial domains (x axis) detected using CoCo-ST. e, Cell–cell interaction weight plot for the MLP-6 tissue sample. The thicker the line, the stronger the interaction between the cell types. f, Chord plot of the cell–cell interactions through canonical WNT signalling. g, The communication probabilities (prob.) for WNT signalling from senders (sources) to receivers (targets). h, The network centrality scores for WNT signalling, highlighting the major signalling roles of the different cell groups. i, Spatial trajectory inference based on CoCo-ST’s determined contrastive components. The arrows indicate the direction of the trajectory, which points from the normal lung spatial domain to the adenoma spatial domain. j, Learned trajectory pseudotime, with red to green regions indicating tissue locations with low and high pseudotime. k, UMAP embedding of spots belonging to the combined normal, adenoma and adenocarcinoma spatial domains as determined using CoCo-ST. l, Trajectory inference of the cancer evolution from normal tissue to adenoma to adenocarcinoma coloured according to their corresponding pseudotimes. m, Gene modules containing differentially co-expressed genes that vary across the different stages of cancer as determined from the learned trajectory in l. n, The number of differentially co-expressed genes in each module in m. Genome-wide analysis in b–j was performed on one independent biological sample, and analysis in k–n was performed on nine independent biological samples.
After determining the composition of cell types in the various tissue samples through our deconvolution analysis, we next inferred their communication patterns. Initially, we identified cell–cell interactions by examining ligand–receptor patterns within the individual MLP tissue samples. Our analysis of the MLP-6 tissue sample revealed a strong pattern of communication between endothelial and epithelial cells as well as between endothelial cells and fibroblasts (Fig. 4e). Moreover, we observed strong communication initiating from both proliferating macrophages and B cells within the adenoma spatial domain, indicating an active immune response.
We observed that multiple signalling pathways, including PD-L1, GRN, ICOS, NECTIN, IL-6, WNT and CXCL, had pivotal roles in cell interactions across different spatial domains. Notably, we predominantly observed WNT ligand–receptor interactions in endothelial cells, epithelial cells, fibroblasts and macrophages (Fig. 4f). Moreover, we observed WNT signalling interactions between proliferating macrophages and B cells, which are enriched in the adenoma spatial domain. Meanwhile, we found self-interaction (among cells of the same group) to be the strongest in proliferating T cells, proliferating macrophages and endothelial cells (Fig. 4g). Network centrality analysis of the inferred WNT signalling network identified TAMs (macrophages and proliferating macrophages) as prominent mediators (gatekeepers) as well as influencers controlling the communication (Fig. 4h). Previous studies demonstrated that WNT signalling supports TAMs as drivers of tumour growth and that TAM-derived WNT ligands support tumorigenesis17.
We further examined the cell–cell interactions across the groups of tissue samples associated with the adenoma and adenocarcinoma spatial domains as determined using CoCo-ST. Specifically, we aggregated the communication weights of multiple tissue samples containing the adenoma (MLP-3, 4, 5, 6, 7 and 9) and adenocarcinoma (MLP-8 and 10) spatial domains to investigate the cell–cell interactions on a multitissue scale. Notably, we observed a bidirectional interaction between epithelial cells and proliferating macrophages in the adenocarcinoma group (Extended Data Fig. 7a,b) but did not see a similar interaction pattern in the adenoma group (Extended Data Fig. 7c,d). This is consistent with the established role of TAMs in promoting tumour growth and metastasis by engaging in an autocrine loop with cancer cells, thereby stimulating cancer cell progression18,19,20,21.
We next investigated how the normal endothelial, adjacent normal and tumour spatial domains are connected to each other during tumorigenesis. Specifically, we performed spatial trajectory inference with MLP-6 tissue based on the contrastive components derived from CoCo-ST. This analysis revealed a trajectory starting from the normal endothelial domain and moving toward the adjacent normal domain and further into the adenoma spatial domain (Fig. 4i,j). To gain a comprehensive view of the trajectory of precancer evolution across the entire population, we combined spots belonging to the adenoma and adenocarcinoma spatial domains as identified by our contrastive components. We then determine a UMAP embedding of the spots (Fig. 4k) with which the trajectories were reconstructed (Fig. 4l). As seen in Fig. 4k, the contrastive components effectively discriminated the three spatial domains and identified a trajectory starting from the normal lung, passing toward the adenoma, and ending at the adenocarcinoma cluster (Fig. 4l). These findings align with the biology of mouse tumorigenesis, consisting of a transition from normal tissue to hyperplasia, adenoma and, finally, adenocarcinoma. Furthermore, we identified modules of differentially expressed genes that were co-expressed across spots in the normal, adenoma and adenocarcinoma spatial domains as determined using CoCo-ST (Fig. 4m,n). Notably, these modules demonstrated high specificity for the different spatial domains, further indicating the effectiveness of CoCo-ST in determining feature representations that captured both the shared and unique spatial structures across the different tissues.
Scaling CoCo-ST to Xenium Prime 5K and Visium HD datasets
To evaluate the generalizability of CoCo-ST, we acquired Visium HD samples, a high-resolution ST platform and Xenium Prime 5K samples, providing single-cell-resolution ST data. These technologies enabled us to assess CoCo-ST’s performance across diverse resolutions. Using Visium HD, we evaluated CoCo-ST at resolutions of 16 μm, 8 μm and 2 μm to test its ability in detecting fine-grained spatial structures through scaling to subcellular data. Using dictionary learning22 and sketching, we sampled representative subsets from the two datasets, VHD1 and VHD2 (Fig. 5a), to create reference embeddings (Fig. 5b,d) based on CoCo-ST’s contrastive components. We then mapped the entire datasets onto the embedding spaces (Fig. 5c,e). CoCo-ST identified distinct spot clusters that aligned closely with pathologist annotations and captured the spatial domains within the tissues (Fig. 5f). Beyond spot clustering, CoCo-ST extended to identify spatial niches corresponding to pathologist-defined regions (Fig. 5g), demonstrating its ability to identify biologically important structures in subcellular high-resolution datasets.

a, The Visium HD tissue samples designated as the background and foreground datasets for CoCo-ST analysis. b, UMAP embedding of the CoCo-ST reference data from VHD1. c, Full embedding of the overall VHD1 samples onto the reference determined by CoCo-ST. d, UMAP embedding of the CoCo-ST reference data from VHD2. e, Full embedding of the overall VHD2 samples onto the reference determined by CoCo-ST. f, Spatial spot clusters detected by CoCo-ST on the VHD1 and VHD2 datasets. g, Spatial niches detected by CoCo-ST on the VHD1 and VHD2 datasets. Genome-wide analysis (b–g) was performed on two independent biological samples (VHD1 and VHD2).
In the Xenium Prime 5K datasets (Fig. 6a), we used similar approach as in the Visium HD analysis to construct reference sets using CoCo-ST’s contrastive components and then mapped the entire datasets onto these reference embedding spaces. The extracted contrastive components were then used for cell type clustering. CoCo-ST identified cell clusters within the tissue samples and was also able to distinguish tumour epithelial cells from bronchial epithelial cells (Fig. 6b,d and Extended Data Fig. 8a,c), a distinction that conventional methods of integrating scRNA-seq data with spatial single-cell Xenium data did not achieve (Fig. 6c,e and Extended Data Fig. 8b,d). By leveraging spatial and contrastive information, CoCo-ST effectively differentiates these distinct epithelial phenotypes. This highlights the power of its contrastive learning design to capture subtle differences within single-cell resolution datasets.

a, The Xenium samples designated as the background and target datasets for CoCo-ST analysis. b, Spatial clusters identified by CoCo-ST on the X5K1 sample. c, scRNA-seq integration results on the X5K1 sample. d, Spatial clusters identified by CoCo-ST on the X5K2 sample. e, scRNA-seq integration results on the X5K2 sample. Genome-wide analysis (b–e) was performed on two independent biological samples (X5K1 and X5K2).
These findings demonstrate CoCo-ST’s robust performance across advanced ST platforms, including high-resolution Visium HD and single-cell resolution Xenium Prime 5K. By leveraging its contrastive learning framework, CoCo-ST consistently captures spatial structures, including subtle domains and distinct cell phenotypes, across diverse spatial data modalities. This versatility further demonstrates CoCo-ST’s use as a scalable and adaptable tool for analysing complex ST datasets.
Extending CoCo-ST to brain tissues
Beyond cancer tissues, we analysed public Visium data generated from mouse brain (anterior and posterior) that profiles complex neurological structures. To examine the spatial-domain-identification performance of CoCo-ST, we used bidirectional contrastive learning, alternating the roles of the anterior and posterior slices as reference and target. The detected spatial domains (Fig. 7a) agree well with the Allen Institute for Brain Science reference atlas23 (Fig. 7b) and results reported in previous publications6,24,25. We further investigated the top five contrastive components as determined using CoCo-ST for both the anterior and posterior slices. These components captured spatial patterns highlighting specific major anatomical regions in the brain (Fig. 7c,f), such as the cerebral cortex (for anterior component 1) and cerebellum (for posterior component 1). Moreover, the top ranked genes for each component (Fig. 7d,g) displayed distinct spatial patterns and localized to specific brain regions (Fig. 7e,h).

a, Spatial domains/regions identified on anterior and posterior mouse brain tissue samples based on CoCo-ST’s contrastive components. b, Annotated mouse brain reference atlas from the Allen Institute23. c, Spatial patterns on the anterior mouse brain tissue sample captured by the first five contrastive components of CoCo-ST. d, The 20 genes with the largest weights on the first five contrastive components in c. The symbols to the right of the bars indicate the signs of the weights. e, Expression patterns for some representative genes in c. f, Spatial patterns on the posterior mouse brain tissue sample captured by the first five contrastive components of CoCo-ST. g, The 20 genes with the largest weights on the first five contrastive components in f. h, Expression patterns for some of the representative genes in g. Genome-wide analysis (a and c–h) was performed on two independent biological samples.
Scalability and sensitivity analysis of CoCo-ST
We performed a systematic parameter exploration by randomly sampling 20 values in the range [0,1] for the three parameters (η,μ1,μ2) and assessing how well CoCo-ST captured the hotspot regions. In each iteration, we fixed two parameters at a randomly chosen value and evaluated CoCo-ST’s performance under different settings of the third parameter. We repeated this procedure to cover all parameter combinations. CoCo-ST consistently achieved high and stable ARI and NMI scores across the tested configurations (Extended Data Fig. 9a), demonstrating its robust performance independent of specific parameter values. These results suggest that CoCo-ST requires minimal parameter tuning, enhancing its practicality for diverse datasets.
To evaluate scalability, we analysed its computational complexity using the Visium HD dataset. Experiments were carried out on a standard desktop computer, using 64-bit operating system with an Intel Xeon Silver 4210 CPU @ 2.20 GHz and 256 GB of RAM. The computation time scaled predictably with dataset size, requiring 518.8 s, 674.2 s and 4,258.4 s for resolutions of 16 μm (~0.14 million spots), 8 μm (~0.59 million spots) and 2 μm (~9.4 million spots), respectively (Extended Data Fig. 9b). Even for high-resolution datasets, CoCo-ST demonstrated computational efficiency, processing within reasonable timeframes. These results highlight CoCo-ST’s scalability and robustness, making it well suited for traditional and high-resolution ST, while meeting the computational demands of advanced technologies.
Discussion
Here we introduce CoCo-ST, a feature representation method leveraging graph contrastive learning for ST analysis. By incorporating a background dataset, CoCo-ST effectively identified unique low-variance spatial structures in target ST datasets while mitigating the influence of dominant high-variance spatial structures shared between target and background data. The contrastive design enables seamless integration of multiple samples.
We benchmarked CoCo-ST against ten state-of-the-art spatial-domain-detection methods, including GraphST, SpaGCN, STAGATE, Giotto and BayesSpace, demonstrating its superior performance across diverse tissue samples. CoCo-ST consistently outperformed these approaches, identifying biologically significant hotspot regions, such as early and late adenomas in a mouse carcinogenesis model, which were overlooked by other methods. Moreover, CoCo-ST can eliminate technical variability, enabling cross-sample identification of biologically relevant spatial domains and facilitating downstream analyses of cancer evolution, spatial heterogeneity and cell–cell interactions.
Another key strength of CoCo-ST is its scalability and versatility across multiple spatial resolutions, from spot-level Visium to single-cell Xenium Prime 5K and subcellular Visium HD. In Visium HD datasets, CoCo-ST identified distinct spot clusters and fine-grained spatial niches closely aligned with pathologist annotations. In Xenium Prime 5K datasets, it distinguished closely related cell types, such as tumour epithelial cells and bronchial epithelial cells, outperforming conventional scRNA-seq integration methods.
CoCo-ST also demonstrated robustness to parameter choices and effectively analysed brain tissue datasets. Its scalability and flexibility position CoCo-ST as a powerful tool for modern ST analysis, enabling researchers to identify intricate global and subtle local biological structures across diverse datasets and resolutions.
A notable limitation is the lack of universal or definitive markers to distinguish LUAD and its precursors, with diagnosis relying on pathology review based on morphological characteristics. We identified Ctsh as consistently highly expressed in hotspot regions annotated as hyperplasia, adenoma and adenocarcinoma (Extended Data Fig. 10). Previously linked to lung tumorigenesis and progression26,27,28,29,30, Ctsh supports the pathologist’s annotations despite not being a validated marker. These findings highlight its potential as an auxiliary marker, warranting further investigation.
In summary, CoCo-ST establishes a benchmark for ST analysis, combining robustness, scalability and versatility across diverse technologies. Its adaptability to various platforms and ability to identify intricate spatial structures make it an indispensable tool for advancing our understanding of tissue heterogeneity and spatial dynamics. These abilities position CoCo-ST to drive innovation in spatial biology research.
Methods
Ethics statement
All animal experiments were conducted according to MD Anderson Institutional Animal Care and Use Committee–approved protocols (00001217-RN03).
Problem definition and notation
We represented a spatially resolved ST slice from a spatial genomics technology as the set of pairs \({\left\{{\textbf x}_{i},{\textbf y}_{i}\right\}}_{i=1}^{n}\), with yi ∈ R2 denoting a vector of spatial coordinates and xi ∈ Rd denoting a vector of measured gene expression at a corresponding spatial location. We referred to a single spatial location xi as a spot and s ∈ {1, 2, …, Sl} as a slice containing ns spots. Let \({X}_{s}={\left[{\textbf x}_{1}^{s},{\textbf x}_{2}^{s},\ldots ,{\textbf x}_{{n}_{s}}^{s}\right]}^{T}\) denote the matrix containing the spot gene expression measurements and \({Y}_{s}={\left[{\textbf y}_{1}^{s},{\textbf y}_{2}^{s},\ldots ,{\textbf y}_{{n}_{s}}^{s}\right]}^{T}\) denote the corresponding spatial location matrix from slice s. Note that the number of spots can differ across different slices and that the slices may be from the same tissue sample or from two different tissue sample.
Our goal is to analyse these Sl slices by finding discriminative feature representations that capture the interesting spatial patterns within the different slices. To do this, we identified a background ST dataset containing dominant high-variance spatial structures that were present across all slices.
The background ST data have a crucial role in effectively contrasting dominant high-variance spatial structures, which was not the primary focus of this analysis, and in turn assists in detecting the intriguing unique spatial structures enriched in individual target slices. Three key advancements underlie the robust performance of our graph contrastive learning approach. First, we used paired slices to mitigate the impact of spatial structures that are not of primary interest, which subsequently aided the detection of unique spatial structures of particular interest in individual target slices. Second, we constructed local similarity graphs to capture the nuanced local structures in both the background and target ST datasets, thereby ensuring that important spatial structures are not lost. Third, we applied the concept of contrastive learning to compare and contrast the graph embedding of the background and target ST datasets, ensuring that similar spots are positioned close to each other and that dissimilar ones are distanced in the latent space. This collective methodology ensures the accurate identification and representation of distinctive spatial structures.
Graph representation learning
Recent advances in spatial molecular profiling made graph learning a focus of attention owing to the innate resemblance of spatial information to spatial graphs. Graph embedding techniques have great potential for various applications across spatially resolved transcriptomics. As ST datasets can be represented in a matrix format, we can identify spots as entities of interest and examine their interaction. This is equivalent to constructing gene or spot graphs based on suitable similarity measures. Here we describe the construction of such molecular similarity graphs. An essential task in ST data analysis is to find a lower dimensional manifold space that captures local neighbourhood information. Given an ST datum (slice), we can construct a weighted graph G = (V,E) representing complex, non-Euclidean structures, with edges eij ∈ E connecting nearby nodes i and j (i,j ∈ V) to each other if spots \({\textbf x}_{i}^{s}\) and \({\textbf x}_{j}^{s}\) are molecularly similar. A natural variation of this graph is to construct a graph of k-nearest neighbours in which similarity of nodes is usually quantified using the Euclidean metric (that is, nodes i and j are connected by an edge eij if \({\textbf x}_{i}^{s}\) is among the k-nearest neighbours of \({\textbf x}_{j}^{s}\) or \({\textbf x}_{j}^{s}\) is among the k-nearest neighbours of \({\textbf x}_{i}^{s}\)). The graph structure G = (V,E) is commonly encoded in an ns × ns affinity matrix S with entries in [0,1] and takes large values if \({\textbf x}_{i}^{s}\) and \({\textbf x}_{j}^{s}\) are close (or similar). Several approaches to computing the affinity matrix S are available31,32,33, one of which is the heat kernel weighting technique depicted by the equation
where \(N({\textbf x}_{j}^{s})\) denotes the set of k-nearest neighbours of \({\textbf x}_{j}^{s}\) and t is a user-specified parameter.
Based on the graph construction approach described above, the similarity among spots is quantified based on gene expression measurements at the corresponding spots. However, because gene expression measurements are captured alongside their spatial information in ST, these spatial locations can be used to construct similarity graphs. The spatial graphs constructed in this manner are similar to molecular similarity graphs in the sense that nodes correspond to spots. However, edges capture proximity of spots in the R2 coordinate space. The affinity matrix with the spatial locations can now be constructed as
Moreover, the spatial graph can be constructed using both the spatial locations and the molecular profiles treated as node features. Graph representation learning approaches are considered to determine biologically meaningful representations of these graphs by finding meaningful lower dimensional representations of nodes present in a complex graph, where local structures in the data are well captured. A widely used criterion for determining such a representation is to solve the objective function33
where \({\textbf z}_{i}^{s}={W}^{T}{\textbf x}_{i}^{\,s}\) denotes the lower dimensional representation of \({\textbf x}_{i}^{s}\). Solving equation (3) under appropriate constraints ensures that, if \({\textbf x}_{i}^{s}\) and \({\textbf x}_{j}^{\,s}\) are similar (or nodes i and j are connected in the graph), then \({\textbf z}_{i}^{s}\) and \({\textbf z}_{j}^{s}\) are similar (close), as well.
Contrastive representation learning
Contrastive learning has recently emerged as a successful method of unsupervised graph representation learning11,34,35. Contrastive learning methods first perform augmentation of the input data and enforce through a suitable objective function mapping of augmentation of the same data (positive pairs) close to each other in the representation (latent) space and augmentation of different data (negative pairs) far apart from each other. Arguably, a low-dimensional representation that is near optimal in the contrastive objective function is guaranteed to linearly separate similar data from dissimilar data. Such representations provide competitive performance in a host of downstream tasks. In early visual representation learning studies, researchers leveraged a pixel as local view to conduct local-to-local36 or local-to-global37 contrastive learning, whereas researchers recently found that randomly cropped image snippets help contrastive models better capture the relationships between image elements11. This motivated us to perform contrastive representation learning at the global image level.
Like several other machine-learning approaches, contrastive representation learning can be performed in an unsupervised (self-supervised) or supervised learning strategy. In self-supervised settings, contrastive learning methods learn discriminative feature representations based on some similarity measure defined according to the data. Consider the objective function defined by38
where m > 0 is a hyperparameter defining the lower bound distance between dissimilar samples, Y is a binary label with Y = 0 if xi and xj are similar, and Y = 1 if xi and xj are dissimilar. Minimizing the objective function is an attempt to determine a lower dimensional manifold subspace where similar input samples are mapped nearby and dissimilar samples are far apart. When sample labels are available, they can be integrated into the definition of similarity and dissimilarity to better guide the contrastive model to mapped samples belonging to the same class (same label) close to each other and samples of different classes farther apart. This approach is referred to as supervised contrastive representation learning. Both the self-supervised and fully supervised contrastive learning approaches are powerful methods of learning discriminative feature representations.
Graph contrastive feature representation using CoCo-ST
Most of the traditional feature representation approaches are designed to determine feature representations through maximization of data variance. These approaches can perform poorly if the ST data structures with maximal variances are not the structures of interest, as the local structures of interest are masked by the dominant high-variance structures. The feature representations determined using these approaches capture little to no useful information reflecting the unique low-variance local structures present in the ST data that are usually treated as noise. Furthermore, these traditional approaches are designed to explore one ST dataset at a time, which can hinder their performance in cases where there are multiple interconnected datasets that need to be explored.
To overcome these limitations, we propose CoCo-ST, which compares and contrasts the global and local variances in ST datasets to better capture discriminant and structural information. More generally, we use two ST datasets (background and target) and subsequently construct two similarity graph views: one for the background ST dataset and the other for the target ST dataset. We then design a contrastive objective function to learn feature representations that capture high global (and/or local) variances enriched in the target ST data while simultaneously attaining small global (and/or local) variances in the background ST data. Given a background ST dataset \({X}_{b}={\left[{\textbf x}_{1}^{b},{\textbf x}_{2}^{b},\ldots ,{\textbf x}_{{n}_{b}}^{b}\right]}^{T}\) containing spatial structures of no primary interest, such as a normal lung region, we can use the following two terms to measure the smoothness of the lower dimensional representation:
and
where tr(⋅) is the trace operator, D is a diagonal matrix of which the entries are the column (or row) sums of S, \({D}_{{ii}}={\sum }_{k}{S}_{{ik}}\) and L = D − S is the graph Laplacian matrix. We consider the symmetric normalized graph Laplacian matrix \(\bar{L}={D}^{-1/2}L{D}^{-1/2}\) in our later derivations. By minimizing \({{\mathscr{R}}}_{1}\), we aim to minimize the reconstruction error, whereas minimizing \({{\mathscr{R}}}_{2}\) is an attempt to preserve the local structure (that is, if two spots \({\textbf x}_{i}^{s}\) and \({\textbf x}_{j}^{s}\) are molecularly similar, their low-dimensional representations \({W}^{T}{\textbf x}_{i}^{b}\) and \({W}^{T}{\textbf x}_{j}^{b}\) are also similar). Combining equations (5) and (6), we can have the equivalent formulation
where \({H}_{b}=I-{\mu }_{1}{\bar{L}}^{b}\), I is an identity matrix, \({\bar{L}}^{b}\) is the normalized graph Laplacian for the background ST data and 0 ≤ μ1 ≤ 1 is a hyperparameter that controls the smoothness of the new representation. The matrix \({H}_{b}=I-{\mu }_{1}{\bar{L}}^{b}\) can be considered to be a graph Laplacian filter39 that helps to smooth the data while preserving underlying spatial structures in an ST slice. The first term in \({{\mathscr{O}}}_{1}\), \(\mathrm{tr}\left({W}^{T}{X}_{b}{X}_{b}^{T}W\right)\) aims to maximize the global variance of the data projected into the lower-dimensional space by extracting representations that span the principal directions of variance in the data. The second term, \({\mu }_{1}\mathrm{tr}\left({W}^{T}{X}_{b}{\bar{L}}^{b}{X}_{b}^{T}W\right)\), minimizes local variances based on the graph Laplacian \({\bar{L}}^{b}\), which captures the similarity structure among spots in the background dataset. By subtracting the smoothness term from the global variance term, the optimization ensures a balance between capturing global features (for example, principal variance directions) and preserving local structures in the lower-dimensional representation. Furthermore, the subtraction ensures that CoCo-ST avoids overfitting to irrelevant global variance by attenuating principal directions dominated by background noise. The parameter μ1 controls the weight of the smoothness term. This allows flexibility in adjusting the influence of local structures in the optimization process.
Similarly, for a target ST dataset \({X}_{t}={\left[{\textbf x}_{1}^{t},{\textbf x}_{2}^{t},\ldots ,{\textbf x}_{{n}_{t}}^{t}\right]}^{T}\) containing unique, interesting spatial structures, we can write the formulation
where \({H}_{t}=I-{\mu }_{2}{\bar{L}}^{t}\), \({\bar{L}}^{t}\) is the normalized graph Laplacian for the target ST data and 0 ≤ μ2 ≤ 1 is a hyperparameter.
Combining equations (7) and (8), CoCo-ST solves the following objective function
where η ≥ 0 is the contrastive parameter that determines the trade-off between high target global (and/or local) variance and low background global (and/or local) variance. The term \(\mathrm{tr}\left({W}^{T}{X}_{t}{H}_{t}{X}_{t}^{T}W\right)\) ensures that CoCo-ST focuses on extracting spatially coherent features unique to the target dataset. Whereas the term \(\eta \mathrm{tr}\left({W}^{T}{X}_{b}{H}_{b}{X}_{b}^{T}W\right)\) penalizes the representation of features that are common to the background dataset, effectively removing global noise and emphasizing local uniqueness. The constraint WTW = I ensures that the components are orthogonal to one another and capture distinct, non-overlapping sources of variation in the data. By maximizing spatially coherent features in the target dataset while minimizing those shared with the background dataset, the objective function enables CoCo-ST to prioritizes spatial domains unique to the target tissue sample.
We will first describe how to maximize the objective function \({{\mathscr{O}}}_{3}\). Let Λ be the Lagrange multiplier for the constraint WTW = I. The Lagrange \({\mathscr{L}}\) is
The partial derivative of \({\mathscr{L}}\) with respect to W is
The optimum solution to equation (10) satisfies \(\frac{\partial {\mathscr{L}}}{\partial W}=0\). We therefore have
Thus, the transformation matrix that maximizes the objective function \({{\mathscr{O}}}_{3}\) can be obtained by solving the eigenvalue problem (equation (12)). Let w1, w2, …, wp be the eigenvectors from equation (12) corresponding to the top p largest eigenvalues λ1 ≥ λ2 ≥ …, ≥ λp. The lower-dimensional representation can then be obtained as follows:
where \({\textbf z}_{i}^{t}\) is a p-dimensional representation of \({\textbf x}_{i}^{t}\), and W is a d × p matrix. This feature representation preserves the local structure of the ST datasets. A step-by-step description of the proposed CoCo-ST method is summarized in Algorithm 1.
Algorithm 1: CoCo-ST
Input: Background \({{{X}}}_{{{b}}}={\left[{{\textbf {x}}}_{{{1}}}^{{{b}}},{{\textbf {x}}}_{{{2}}}^{{{b}}},{\ldots },{{\textbf {x}}}_{{{{n}}}_{{{b}}}}^{{{b}}}\right]}^{{{T}}}\) and target \({{{X}}}_{{{t}}}={\left[{{\textbf {x}}}_{{{1}}}^{{{t}}},{{\textbf {x}}}_{{{2}}}^{{{t}}},{\ldots },{{\textbf{x}}}_{{{{n}}}_{{{t}}}}^{{{t}}}\right]}^{{{T}}}\) ST datasets, together with corresponding spatial locations \({{{Y}}}_{{{b}}}={\left[{{\textbf{y}}}_{{{1}}}^{{{b}}},{{\textbf{y}}}_{{{2}}}^{{{b}}},{\ldots },{{\textbf{y}}}_{{{{n}}}_{{{b}}}}^{{{b}}}\right]}^{{{T}}}\) and \({{{Y}}}_{{{t}}}={\left[{{\textbf{y}}}_{{{1}}}^{{{t}}},{{\textbf{y}}}_{{{2}}}^{{{t}}},{\ldots },{{\textbf{y}}}_{{{{n}}}_{{{t}}}}^{{{t}}}\right]}^{{{T}}}\), the number of nearest neighbours (k), and the hyperparameters μ1, μ2 and η.
Output: The low-dimensional contrastive feature representations for the target ST data Zt = WTXt
-
(1)
Construct the adjacency matrix for both the background and target ST datasets according to equations (1) or (2).
-
(2)
Construct the normalized graph Laplacian matrices \({\bar{{{L}}}}^{{{b}}}\) and \({\bar{{{L}}}}^{{{t}}}\) together with the graph Laplacian filters \({{{H}}}_{{{b}}}={{I}}-{{{\mu }}}_{{1}}{\bar{{{L}}}}^{{{b}}}\) and \({{{H}}}_{{{t}}}={{I}}-{{{\mu }}}_{{{2}}}{\bar{{{L}}}}^{{{t}}}\).
-
(3)
Compute the matrices \({X}_{b}{H}_{b}{X}_{b}^{T}\) and \({{{X}}}_{{{t}}}{{{H}}}_{{{t}}}{{{X}}}_{{{t}}}^{{{T}}}\).
-
(4)
Solve the eigenvalue problem in equation (12).
-
(5)
Compute the low-dimensional contrastive feature representations for the target ST data as Zt = WTXt.
We next investigate the computational complexity of the proposed CoCo-ST algorithm. Its complexity is dominated mainly by three parts: local similarity graph construction, matrix multiplication and solving an eigenvalue problem. Assuming we have nb and nt spots in d-dimensional spaces (d gene expression measurements) for the background and target ST datasets, to construct the similarity graphs, we first perform a k-nearest neighbour search for both datasets. The distance between any two spots in the background ST data can be computed in \(O\left({{dn}}_{b}^{2}\right)\), and the k-nearest neighbours can be found with \(O\left(k{n}_{b}^{2}\right)\). Thus, the k-nearest neighbour search for the background and target ST datasets has complexities \(O\left(\left(d+k\right){n}_{b}^{2}\right)\) and \(O\left(\left(d+k\right){n}_{t}^{2}\right)\), respectively. The complexities for computing the matrices \({X}_{b}{H}_{b}{X}_{b}^{T}\) and \({X}_{t}{H}_{t}{X}_{t}^{T}\) are \(O\left(\left({n}_{b}^{2}+{n}_{b}d\right)d\right)\) and \(O\left(\left({n}_{t}^{2}+{n}_{t}d\right)d\right)\), respectively. The last part is computing the eigenvectors corresponding to the top p eigenvalues of the eigenproblem in equation (12), of which the complexity is O(pd2). Thus, the time complexity of the CoCo-ST algorithm is \(O\left(\left(d+k\right)\left({n}_{b}^{2}+{n}_{t}^{2}\right)+\left(\left({n}_{b}+d\right){n}_{b}+\left({n}_{t}+d\right){n}_{t}+{pd}\right)d\right)\). Because \(k\ll {n}_{b}(\mathrm{or}{n}_{t})\) and \(p\ll d\), the overall complexity of CoCo-ST is determined by the number of spots nb (or nt) and the number of genes (d).
Several aspects of the proposed CoCo-ST approach are worth highlighting. Specifically:
-
(1)
If μ1 = μ2 = 0, the matrices \({X}_{t}{H}_{t}{X}_{t}^{T}\) and \({X}_{b}{H}_{b}{X}_{b}^{T}\) reduce to \({X}_{t}{X}_{t}^{T}\) and \({X}_{b}{X}_{b}^{T}\), respectively, so the objective function (\({{\mathscr{O}}}_{3}\)) reduces to that of cPCA35. Therefore, cPCA can be regarded as a variant of CoCo-ST.
-
(2)
Whereas cPCA and the majority of the traditional feature representation approaches focus on global geometrical structures, CoCo-ST can exploit the intrinsic geometric structures of ST datasets and incorporate them as additional regularization terms. Through construction of a graph to model local geometric structures, CoCo-ST can have more discriminating power than cPCA and the traditional feature representation approaches.
-
(3)
CoCo-ST simultaneously learns both global and local-level representations to complement tissue-wide representations, enabling it to distinguish different spatial areas in an ST tissue slice.
-
(4)
The graphs in our proposed CoCo-ST approach are solely unsupervised and constructed from molecular data or spatial location information. Other information, such as label information, can also be used to guide graph construction, leading to other extensions of CoCo-ST such as supervised or semisupervised CoCo-ST.
-
(5)
The proposed CoCo-ST approach differs from existing graph contrastive learning approaches that focus on graph neural network architectures for graph structured data. CoCo-ST considers the gene expression data and tries to learn local representations to better capture ST data structural information. As such, the objective functions of CoCo-ST and the conventional graph neural networks are different.
Why CoCo-ST excels for ST analysis
CoCo-ST excels in ST data analysis by preserving the geometric structure of ST datasets, ensuring that transcriptionally and spatially similar spots are close to each other in a lower-dimensional subspace while separating dissimilar ones. While many ST methods ensure that transcriptionally or spatially similar spots cluster together, CoCo-ST goes a step further. Its contrastive objective actively emphasizes differences between the background and target datasets, allowing disease-relevant or otherwise subtle patterns to stand out. This not only facilitates detection of large-scale spatial domains but also uncovers fine-grained interesting spatial structures, such as tumour–normal boundaries and disease-specific niches (hotspots), which often represent subtle low-variance spatial patterns that are overlooked by traditional methods focusing on dominant global variations. By leveraging both background and target datasets, CoCo-ST can provide even more discriminative feature representations than the traditional approaches that focus only on a single ST dataset. To explain this, we provide the following remarks and theorem.
Remark 1
When η = 0, CoCo-ST degenerates to a feature representation method that determines its discriminant vectors from the range space of the matrix \({X}_{t}{H}_{t}{X}_{t}^{T}\) associated with the target data alone. When η > 0, the matrix \({X}_{t}{H}_{t}{X}_{t}^{T}-\eta {X}_{b}{H}_{b}{X}_{b}^{T}\) is not guaranteed to be positive semidefinite even though \({X}_{t}{H}_{t}{X}_{t}^{T}\) and \({X}_{b}{H}_{b}{X}_{b}^{T}\) are both symmetric and positive semidefinite. Let w be the eigenvector of the matrix \({X}_{t}{H}_{t}{X}_{t}^{T}-\eta {X}_{b}{H}_{b}{X}_{b}^{T}\) corresponding to the eigenvalue λ < 0. We then have
Because both \({X}_{t}{H}_{t}{X}_{t}^{T}\) and \({X}_{b}{H}_{b}{X}_{b}^{T}\) are positive semidefinite, we can conclude that
Thus, the eigenvectors corresponding to the negative eigenvalues are derived from the range space of \({X}_{b}{H}_{b}{X}_{b}^{T}\) and contain some discriminant information.
Theorem 1
Suppose the matrix \({X}_{b}{H}_{b}{X}_{b}^{T}\) is singular and that w is an eigenvector of the matrix \({X}_{t}{H}_{t}{X}_{t}^{T}-\eta {X}_{b}{H}_{b}{X}_{b}^{T}\) corresponding to the eigenvalue λ > 0. The eigenvector w is then in the null space of \({X}_{b}{H}_{b}{X}_{b}^{T}\) when \(\eta \to \infty\).
Proof: because w is the eigenvector of the matrix \({X}_{t}{H}_{t}{X}_{t}^{T}-\eta {X}_{b}{H}_{b}{X}_{b}^{T}\) corresponding to the eigenvalue λ > 0, we have
Since λ > 0, we have the following:
Notably, both \({X}_{t}{H}_{t}{X}_{t}^{T}\) and \({X}_{b}{H}_{b}{X}_{b}^{T}\) are positive semidefinite (that is, \(w{X}_{t}{H}_{t}{X}_{t}^{T}w\ge 0\) and \(w{X}_{b}{H}_{b}{X}_{b}^{T}w\ge 0\)). As a result, we have
Thus, as \(\eta \to \infty\), the eigenvectors corresponding to the positive eigenvalues belong to the null space of \({X}_{b}{H}_{b}{X}_{b}^{T}\).
Remark 2
As \(\eta \to \infty\), the eigenvectors corresponding to the positive eigenvalues of the eigenproblem (equation (12)) contain the most discriminant information. We can rewrite the eigenvalue problem (equation (12)) as
Thus, as \(\eta \to \infty\), the eigenvectors corresponding to the positive eigenvalues contain the most discriminant information.
Remark 3
As \(\eta \to \infty\), the eigenvectors corresponding to the zero eigenvalues of the eigenproblem (equation (12)) contain no discriminant information. When λ = 0, the eigenvalue problem reduces to
Since \(w{X}_{t}{H}_{t}{X}_{t}^{T}w\) and \(w{X}_{b}{H}_{b}{X}_{b}^{T}w\) are finite and \(\eta \to \infty\), we have
Thus, the eigenvectors corresponding to the zero eigenvalues contain no discriminant information, as \(\eta \to \infty\). In general, we can conclude that CoCo-ST derives its discriminant feature vectors from the range spaces of both \({X}_{t}{H}_{t}{X}_{t}^{T}\) and \({X}_{b}{H}_{b}{X}_{b}^{T}\). The parameter η can be used to balance the contribution from the two spaces. Moreover, by extracting the eigenvectors of the eigenvalue problem in equation (12) corresponding to the largest positive eigenvalues, CoCo-ST can capture the most discriminant information in both the background and target ST datasets, enabling effective identification of the interesting spatial structures enriched in the target ST dataset.
Nonlinear extension of CoCo-ST
Thus far, we have focused on linear feature representation. However, biological data are well known to be complex and highly nonlinear40. We therefore extended CoCo-ST to perform nonlinear feature representation in a reproducing kernel Hilbert space \({\mathcal{H}}\), which gives rise to nonlinear CoCo-ST. We considered nonlinear mapping ϕ(⋅) of both the background Xb and target Xt ST datasets from the original input spaces to \({\mathcal{H}}\). Let Φb and Φt denote the background and target ST datasets in \({\mathcal{H}}\):
Denote by V the projection matrix in \({\mathcal{H}}\). The corresponding objective function (\({{\mathscr{O}}}_{3}\)) of CoCo-ST in \({\mathcal{H}}\) is
Let N = nb + nt, and define the data q1, q1, …, qN by
As the projection vectors v1, v2, …, vp (column vectors in V) are linear combinations41 of ϕ(q1), ϕ(q2), …, ϕ(qN), coefficients αi, i = 1, 2, …, N exist such that
where \(\alpha ={\left({\alpha }_{1},{\alpha }_{2},\ldots ,{\alpha }_{N}\right)}^{T}\in {R}^{N}\), \({\rm{{\rm A}}}=[{\alpha }^{1},{\alpha }^{2},\ldots ,{\alpha }^{p}]\). Following some algebraic formulations, we can rewrite the objective function (\({{\mathscr{O}}}_{4}\)) in the following equivalent form:
where \({K}_{{cc}}={\Phi}_{c}^{T}{\Phi}_{c},\) \({K}_{{ct}}={\Phi}_{c}^{T}{\Phi}_{t}\), \({K}_{{tc}}={\Phi}_{t}^{T}{\Phi}_{c}\), \({K}_{{cb}}={\Phi}_{c}^{T}{\Phi}_{b}\) and \({K}_{{bc}}={\Phi}_{b}^{T}{\Phi}_{c}\) are the kernel matrices. Several choices of the kernel functions are available, including the polynomial kernel \({\mathscr{K}}\left({\bf{x}}_{i}^{t},{\bf{x}}_{i}^{b}\right)={\left({\left({\bf{x}}_{i}^{t}\right)}^{T}{\bf{x}}_{i}^{b}+1\right)}^{d}\); Gaussian kernel \({\mathscr{K}}\left({\bf{x}}_{i}^{t},{\bf{x}}_{i}^{b}\right)=\exp (-\frac{{{|}{|}{\bf{x}}_{i}^{t}-{\bf{x}}_{i}^{b}{|}{|}}^{2}}{{\sigma}^{2}})\); and sigmoid kernel \({\mathscr{K}}\left({\bf{x}}_{i}^{t},{\bf{x}}_{i}^{b}\right)=({\left({\bf{x}}_{i}^{t}\right)}^{T}{\bf{x}}_{i}^{b}+\gamma)\).
Following approach similar to that in linear CoCo-ST, the projection vectors in equation (15) can be obtained as the eigenvectors corresponding to the top p largest eigenvalues of the generalized eigenvalue problem
To obtain a stable solution of the eigenvalue problem in equation (16), the kernel matrix Kcc must be nonsingular. When Kcc is singular, we can adopt the idea of regularization by adding a small constant value p to the diagonal of Kcc as Kcc + ρI for any ρ > 0. The matrix Kcc + ρI is nonsingular, and the projection vectors can be computed as the generalized eigenvectors of
Animal model
Wild-type mice (strain 009104) were purchased from The Jackson Laboratory and housed in colony cages under pathogen-free conditions at The University of Texas MD Anderson Cancer Center Research Animal Support Facility. The mice were housed at an ambient temperature of 20–26 °C and humidity range of 30–70% under a 12 h–12 h light–dark cycle. All of the animal experiments were conducted according to MD Anderson Institutional Animal Care and Use Committee–approved protocols (00001217-RN03). For carcinogen-induced mouse models, a urethane-induced mouse model was used. Specifically, the 129S4 wild-type mice described above received intraperitoneal injections of 1 mg per g (body weight) urethane three times over 8 days when they were 6 weeks old. A total of 50 mice (equally divided between males and females) were killed 7, 14, 20, 30 and 40 weeks after urethane administration, with a 0-week timepoint for mice that received no treatment. Randomization was used to assign mice to the treated or untreated group. Both normal lung and lung tumour tissue samples were collected from the mice for downstream analysis.
Single-cell sequencing and analysis
Fresh normal lung and lung tumour tissue samples collected from mice were immediately cut into pieces and placed into RPMI 1640 medium (Thermo Fisher Scientific) with 10% FBS (FBS; Gibco). The tissue samples were enzymatically digested using a tumour dissociation mixture composed of 1 mg ml−1 collagenase A (Sigma-Aldrich), 0.4 mg ml−1 hyaluronidase (Sigma-Aldrich) and 1:5 BSA fraction V (Thermo Fisher Scientific) according to the manufacturers’ instructions. Dissociation of tissue was carried out for 2 h on a rotary shaker at 37 °C until all large tissue fragments were digested. Next, the dissociated tissues were transferred to a conical tube and centrifuged at 350g for 5 min. The supernatant was removed, and 1–5 ml of prewarmed trypsin-EDTA was added to the collagenase/hyaluronidase-dissociated cells, resuspending them. Subsequently, 10 ml of cold RPMI 1640 without phenol red supplemented with 2% FBS was added and centrifuged at 350g for 5 min. As much of the supernatant as possible was collected, and 5 ml of prewarmed 5 U ml−1 dispase (StemCell Technologies) and 50 μl of DNase I solution (10 mg ml−1 in 0.15 M NaCl; StemCell Technologies) were added. The samples were pipetted for 1 min using a 1 ml micropipettor to further dissociate cell clumps. The cell suspension was diluted with an additional 10 ml of cold RPMI 1640 without phenol red supplemented with 2% FBS, and the cell suspension was filtered through a 40-μm Falcon cell strainer (Thermo Fisher Scientific) into a 50 ml tube. The cell suspension was further centrifuged at 450g for 5 min, and the supernatant was discarded. The pellet was resuspended in a 1:4 mixture of cold RPMI 1640 without phenol red supplemented with 2% FBS and an ammonium chloride solution (StemCell Technologies), which was followed by centrifugation at 450g for 5 min and discarding of the supernatant. Then, 10 μl of the cell suspension for each sample was analysed using an automated cell counter (Thermo Fisher Scientific) to determine the number of live cells. Throughout the dissociation procedure, cells were kept on ice when possible. The cells were then loaded onto a Chromium single-cell controller (10x Genomics) to create single-cell gel beads in an emulsion according to the manufacturer’s protocol. scRNA-seq libraries were constructed using a Single Cell 5′ Library and Gel Bead Kit v3.1 (10x Genomics) and sequenced using the NovaSeq 6000 sequencer (Illumina) at the Genomic and RNA Profiling Core at Baylor College of Medicine.
Tissue preparation and ST
Normal and tumour tissue samples from mouse lungs were fixed in 10% formalin at room temperature for 24-48 h using a fixative volume 5–10 times greater than that of the tissue volume. Fixed tissues were transferred to 70% ethanol for temporary storage at 4 °C. Paraffin embedding was conducted by the MD Anderson Research Histology Core Laboratory. Formalin-fixed, paraffin-embedded blocks were cut into 10-μm-thick sections (for traditional Visium) and 5-μm thick sections (for Visium HD) using a precooled RNase-free microtome. These sections were then transferred onto Visium Spatial Gene Expression slides (10x Genomics), which were pretreated by floating in a water bath at 43 °C. After sectioning, the slides were dried at 42 °C in a SimpliAmp Thermal Cycler (Thermo Fisher Scientific) for 3 h according to the manufacturer’s instructions. The slides were placed in a slide mailer, sealed with thermoplastic (Parafilm, Thermo Fisher Scientific) and stored overnight in a refrigerator at 4 °C. The slides were then deparaffinized, fixed, stained with haematoxylin and eosin and imaged at ×5 magnification using the DM5500 B microscope (Leica Microsystems). Tile scans of the entire array were acquired using Leica Application Suite X software and merged. Spatial gene expression libraries (Visium ST, 10x Genomics) were processed according to the manufacturer’s instructions and sequenced using the NovaSeq 6000 sequencer (Illumina). All haematoxylin and eosin staining, imaging, library preparation and sequencing processes were carried out at the Genomic and RNA Profiling Core at Baylor College of Medicine (for traditional Visium samples) and at the department of translational molecular pathology at MD Anderson Cancer Center (for Visium HD samples). For the Xenium Prime 5K assay, tissue preparation and sectioning are the same as the Visium ST assay.
Data processing
scRNA-seq data
Raw base call files were analysed using Cell Ranger v.3.0.2 software (10x Genomics). The mkfastq command was used to generate FASTQ files, and the count command was used to generate raw gene–barcode matrices aligned to the GRCh38 Ensembl 93 genome. The data were aggregated using the cellranger aggr command, and further downstream analysis was conducted in R v.4.1.0 using the Seurat package (v.4.1.1). To ensure that our analysis was performed using high-quality cells, filtering of cells was conducted by retaining cells that had unique feature counts greater than 200 or less than 5,000 and had mitochondrial content of less than 15%. After removing doublets, the total cell number was 70,698.
ST data
The ST datasets were processed using Space Ranger (v.2.0.1, 10x Genomics). The spatial sequencing data were aligned to mouse pre-mRNA genome reference version mm10 (downloaded from the 10x Genomics website) using Space Ranger, and mRNA count matrices were generated by adding intronic and exonic reads for each gene in each location. Paired histological haematoxylin- and eosin-stained images of tissues were processed using Space Ranger to select locations covered by tissue by aligning prerecorded spot locations with fiducial border spots in the images.
Data analysis
scRNA-seq analysis
The scRNA-seq data were first normalized, and the 2,000 most highly variable genes in the data were identified using variance-stabilizing transformation implemented in the Seurat package. Data were then scaled, and the first 30 principal components were extracted. The principal components were further transformed into the UMAP embedding space for which clustering analysis was conducted. The original Louvain algorithm was used for modularity optimization. The resulting 14 clusters were visualized in a 2D UMAP representation and annotated to known biological cell types using canonical marker genes. The following cell types were annotated (selected markers are listed in parentheses): endothelial cells (Pecam1, Vwf, Ets1, Ace, Eng, Cldn5 and Mcam), epithelial cells (Epcam, Muc1, Cdh1, Krt7 and Krt8), fibroblasts (Pdpn, Dcn, Col3a1, Mgp, Col1a1 and Col6a1), macrophages (Apoe, C1qa, C1qb, C1qc, Marco, Mrc1, Fabp4, Inhba, Ccl4, Cxcl10, Rsad2 and Herc6), conventional dendritic cells (cDC; H2-Aa, Ccr7, Flt3, Fscn1 and Clec9a), proliferating macrophages (Mki67, Tubb5 and Tuba1b), B cells (Cd19, Ms4a1, Cd79a, Cd79b and Blnk), T cells (Trbc2, Cd2, Cd3d, Cd3e, Cd3g, Cd4, Cd8a, Cd8b1, Il2ra and Foxp3), proliferating T cells (Mki67, Tubb5 and Tuba1b), plasmacytoid dendritic cells (pDC; Siglech, Ly6c2 and Cd209d), neutrophils (S100a8, S100a9 and Csf3r), plasma cells (Sdc1, Mzb1, Xbp1 and Jchain), monocytes (Cd14, Fcgr4, Lst1 and Vcan) and natural killer cells (Nkg7, Klrg1 and Ncr1).
ST analysis
The raw expression count matrices for both the background and target ST datasets were normalized using variance-stabilizing transformation implemented in the Seurat package. The normalized data were then standardized to have zero mean and unit s.d. The standardized expression data matrices with 3,000 genes were then used as inputs to our CoCo-ST method for low-dimensional feature representation. Clustering on the UMAP-embedded learned contrastive feature representations was then performed. Further differential gene expression analysis was conducted, and spatial domains were annotated based on the differentially expressed marker genes.
Pathway analysis
The most important genes (the 20 genes with the largest weights) on the top five contrastive components were identified, and the biological processes associated with these contrastive components were examined. Specifically, gene set enrichment analysis was performed with these 20 genes with the largest weights in the loading matrix using the g:GOSt function in the gprofiler2 package. In this analysis, all of the input 3,000 genes were used as the background, and the default options in the g:SCS method in gprofiler2 were used for multiple-testing correction. The gene sets were downloaded from the Molecular Signatures Database, including the KEGG, GO biological processes, GO cellular components and GO molecular functions.
Cell type deconvolution
Cell type deconvolution in ST enables estimation of cell type composition on each spatial location by leveraging a reference scRNA-seq dataset. Cell type deconvolution was performed using the RCTD42 method implemented in the spacexr R package. scRNA-seq data for the same mouse lung tumour samples (MLP samples) served as the reference data for deconvolution. The reference data contained 70,698 cells of multiple immune and malignant types as described in the ‘scRNA-seq analysis’ section. The RCTD method was run in doublet mode to estimate the reference cell type composition on each spatial location. Other parameters were set to the default settings.
Cell–cell interaction
Cell–cell interaction for the ST datasets was performed using CellChat43. The CellChatDB.mouse database of ligand–receptor interactions specifically curated for mice was used to identify overexpressed ligand–receptor interactions. The group-level communication probability or interaction weights were then computed using the truncated mean method with a 10% truncated mean. Subsequently, the communication probability at the signalling pathway level was computed by summarizing the communication probabilities of all ligand–receptor interactions associated with each signalling pathway. Finally, the cell–cell communication network was aggregated by summarizing the overall communication probabilities.
Trajectory inference analysis
For spatial trajectory analysis of individual tissue samples, the low-dimensional contrastive feature representations were used as inputs to the Slingshot algorithm44. Slingshot was applied to the contrastive feature representations so that nearby tissue spatial locations with similar gene expression would have similar pseudotimes. As Slingshot requires predefined cluster labels, the spatial domain labels from the spatial domain identification analysis were used for Slingshot. The normal lung spatial domain was set as the start cluster (beginning of the trajectory or pseudotime) with a focus on trajectory inference on tumour and tumour-adjacent spatial domains to determine how these locations are connected to one another during tumorigenesis.
For the trajectory analysis with combined tissue samples, spots belonging to normal lung, adenoma and adenocarcinoma spatial domains as determined using the contrastive feature representations were collected, and Monocle345 was used to infer the trajectory. First, the combined data (spots) were processed using the standard Seurat approach, including total count normalization, scaling and PCA analysis. Next, UMAP embedding was determined, which was used to learn the trajectory that fits the spots’ UMAP coordinates. A principal graph was then fit on the UMAP embedding, and the spots were ordered according to their progress along the learned trajectory. To identify genes that vary among spot clusters in the UMAP embedding space, spatial autocorrelation analysis (Moran’s I) was performed, and the obtained variable genes were grouped into modules by determining UMAP embedding of the genes followed by gene clustering based on Louvain community detection analysis.
Statistics and reproducibility
The pre-processing steps for the datasets used in this study are outlined in the relevant sections above. For all methods compared in this study, the default parameter settings were used unless otherwise specified in the ‘Results’ section. No statistical method was used to predetermine the sample size, and all data were included in the analysis. The normality of data distribution and the homogeneity of variances were assessed using the Kolmogorov–Smirnov test and Levene’s test, respectively; the data met the assumptions of the statistical tests used. Experiments were randomized, and data collection and analysis were not performed blind to the conditions of the experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The scRNA-seq and ST datasets analysed in this study have been deposited at Zenodo46 (https://doi.org/10.5281/zenodo.16891007). The mouse brain datasets, originally from 10x Genomics was downloaded using the SeuratData package at https://github.com/satijalab/seurat-data. Publicly available gene pathway databases used in this study include GO (https://geneontology.org/) and the KEGG (https://www.kegg.jp/). All other data supporting the findings of this study are available from the corresponding author on reasonable request. Source data are provided with this paper.
Code availability
Installation instructions and tutorials, together with the code46 used for data analysis and generating figures, can be found at GitHub (https://github.com/WuLabMDA/CoCo-ST) and Zenodo (https://doi.org/10.5281/zenodo.16891007). A detailed protocol47 for using CoCo-ST is provided at Protocols.io (https://doi.org/10.17504/protocols.io.x54v95m6pl3e/v1).
References
Liu, W., et al. Joint dimension reduction and clustering analysis of single-cell RNA-seq and spatial transcriptomics data. Nucleic Acids Res. 50, e72 (2022).
Bergenstråhle, J., Larsson, L. & Lundeberg, J. Seamless integration of image and molecular analysis for spatial transcriptomics workflows. BMC Genom. 21, 482 (2020).
Townes, F. W. & Engelhardt, B. E. Nonnegative spatial factorization applied to spatial genomics. Nat. Methods 20, 229–238 (2023).
Shang, L. & Zhou, X. Spatially aware dimension reduction for spatial transcriptomics. Nat. Commun. 13, 7203 (2022).
Velten, B. et al. Identifying temporal and spatial patterns of variation from multimodal data using MEFISTO. Nat. Methods 19, 179–186 (2022).
Long, Y. et al. Spatially informed clustering, integration, and deconvolution of spatial transcriptomics with GraphST. Nat. Commun. 14, 1155 (2023).
Hu, J. et al. SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 18, 1342–1351 (2021).
Dries, R., et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 22, 78 (2021).
Dong, K. & Zhang, S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat. Commun. 13, 1739 (2022).
Zhao, E. et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 39, 1375–1384 (2021).
Chen, T., Kornblith, S., Norouzi, M. & Hinton, G. A simple framework for contrastive learning of visual representations. In Proc. International Conference on Machine Learning 1597–1607 (PMLR, 2020).
You, Y. et al. Graph contrastive learning with augmentations. Adv. Neural Inf. Process. Syst. 33, 5812–5823 (2020).
Wang, Y., Wang, J., Cao, Z. & Barati Farimani, A. Molecular contrastive learning of representations via graph neural networks. Nat. Mach. Intell. 4, 279–287 (2022).
Dang, H. et al. Cancer-associated fibroblasts are key determinants of cancer cell invasion in the earliest stage of colorectal cancer. Cell. Mol. Gastroenterol. Hepatol. 16, 107–131 (2023).
Büttner, M., Miao, Z., Wolf, F. A., Teichmann, S. A. & Theis, F. J. A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods 16, 43–49 (2019).
Yi, F., Jaffe, R. & Prochownik, E. V. The CCL6 chemokine is differentially regulated by c-Myc and L-Myc, and promotes tumorigenesis and metastasis. Cancer Res. 63, 2923–2932 (2003).
Tigue, M. L. et al. Wnt signaling in the phenotype and function of tumor-associated macrophages. Cancer Res. 83, 3–11 (2023).
Schmall, A. et al. Macrophage and cancer cell cross-talk via CCR2 and CX3CR1 is a fundamental mechanism driving lung cancer. Am. J. Respir. Crit. Care Med. 191, 437–447 (2015).
Garrido-Navas, C. et al. Cooperative and escaping mechanisms between circulating tumor cells and blood constituents. Cells 8, 1382 (2019).
Sarode, P., Schaefer, M. B., Grimminger, F., Seeger, W. & Savai, R. Macrophage and tumor cell cross-talk is fundamental for lung tumor progression: we need to talk. Front. Oncol. 10, 324 (2020).
Ge, Z. & Ding, S. The crosstalk between tumor-associated macrophages (TAMs) and tumor cells and the corresponding targeted therapy. Front. Oncol. 10, 590941 (2020).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Allen Reference Atlas—Mouse Brain (Allen Institute for Brain Science, 2008); atlas.brain-map.org
Shi, H. et al. Spatial atlas of the mouse central nervous system at molecular resolution. Nature 622, 552–561 (2023).
Zhang, M. et al. Molecularly defined and spatially resolved cell atlas of the whole mouse brain. Nature 624, 343–354 (2023).
Deng, T. et al. Cathepsins and cancer risk: a Mendelian randomization study. Front. Endocrinol. 15, 1428433 (2024).
Olson, O. C. & Joyce, J. A. Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 15, 712–729 (2015).
Yang, Y. et al. Integrating genomics and proteomics data to identify candidate plasma biomarkers for lung cancer risk among European descendants. Br. J. Cancer 129, 1510–1515 (2023).
Li, J., Tang, M., Gao, X., Tian, S. & Liu, W. Mendelian randomization analyses explore the relationship between cathepsins and lung cancer. Commun. Biol. 6, 1019 (2023).
Luyapan, J. et al. Candidate pathway analysis of surfactant proteins identifies CTSH and SFTA2 that influences lung cancer risk. Hum. Mol. Genet. 32, 2842–2855 (2023).
Aminu, M. & Ahmad, N. A. Complex chemical data classification and discrimination using locality preserving partial least squares discriminant analysis. ACS Omega 5, 26601–26610 (2020).
Aminu, M. & Ahmad, N. A. Locality preserving partial least squares discriminant analysis for face recognition. J. King Saud. Univ. Comput. Inf. Sci. 34, 153–164 (2022).
He, X. & Niyogi, P. Locality preserving projections. In Proc. 17th International Conference on Neural Information Processing Systems (eds Thrun, S. et al.) 153–160 (ACM, 2003).
Khosla, P. et al. Supervised contrastive learning. Adv. Neural Inf. Process. Syst. 33, 18661–18673 (2020).
Abid, A., Zhang, M. J., Bagaria, V. K. & Zou, J. Exploring patterns enriched in a dataset with contrastive principal component analysis. Nat. Commun. 9, 2134 (2018).
Wang, W. et al. Exploring cross-image pixel contrast for semantic segmentation. In Proc. IEEE/CVF International Conference on Computer Vision 7303–7313 (IEEE, 2021).
Miao, J., Yang, Z., Fan, L. & Yang, Y. FedSeg: class-heterogeneous federated learning for semantic segmentation. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition 8042–8052 (IEEE, 2023).
Hadsell, R., Chopra, S. & LeCun, Y. Dimensionality reduction by learning an invariant mapping. In Proc. IEEE Computer Society Conference on Computer Vision and Pattern Recognition 1735–1742 (IEEE, 2006).
Liu, Y. et al. Simple contrastive graph clustering. In Proc. IEEE Transactions on Neural Networks and Learning Systems 13789–13800 (IEEE, 2023).
Almeida, J. S. Predictive non-linear modeling of complex data by artificial neural networks. Curr. Opin. Biotechnol. 13, 72–76 (2002).
Rosipal, R. & Trejo, L. J. Kernel partial least squares regression in reproducing kernel hilbert space. J. Mach. Learn. Res. 2, 97–123 (2001).
Cable, D. M. et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 40, 517–526 (2022).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Street, K., et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 19, 477 (2018).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Aminu, M. et al. Datasets for ‘CoCo-ST detects global and local biological structures in spatial transcriptomics datasets’. Zenodo https://doi.org/10.5281/zenodo.16891007 (2025).
Aminu, M., Zhu, B., Vokes, N., Wu, J. & Zhang, J. Protocol for ‘CoCo-ST detects global and local biological structures in spatial transcriptomics datasets’. protocol.io https://doi.org/10.17504/protocols.io.x54v95m6pl3e/v1 (2025).
Acknowledgements
This work was supported by contributions to The University of Texas MD Anderson Lung Moon Shot Program, the MD Anderson Cancer Center Support Grant P30CA016672. This research was partially funded by the National Institutes of Health (NIH) grants R00CA218667, R01CA262425, R01CA276178 and 5R50CA265307, as well as CPRIT RP240117 and RP250399. This work was sponsored by contributions from A. Mugnaini and E. L. C. Smith, as well as the Rexanna’s Foundation for Fighting Lung Cancer, QIAC Partnership in Research (QPR) funding and Permanent Health Funds. We thank D. Norwood for editing this article.
Author information
Authors and Affiliations
Contributions
M.A. and J.W. formulated and applied the method. B.Z. and J.Z. acquired the data. M.A. developed the software. M.A., B.Z., N.V., C.C., K.C., J.Z. and J.W. designed the experiments. M.A., B.Z., H.C, N.V. and L.H. analysed the data. J.L., J.F., M.C., Y.Y., B.W., A.P., M.B.N., X.L., T.C., D.J., N.N., T.W., L.A.B., D.L.G. and J.H. provided feedback on the software, case studies and/or the paper draft. All of the authors contributed to the interpretation of the data. M.A., B.Z. and H.C. prepared the first draft of the paper. L.H., N.V., C.C., K.C., J.Z. and J.W. revised the paper. J.Z. and J.W. supervised the project. All of the authors read and approved the final version of the paper. All of the authors were responsible for the final decision to submit the paper for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cell Biology thanks Qing Nie and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Differential gene expression analysis of detected spatial domains by CoCo-ST.
a, UMAP embedding of the contrastive components determined using CoCo-ST on the target ST tissue sample. b, UMAP embedding of spots showing the expression of some of the most differentially expressed genes in different clusters identified using the contrastive feature representations from CoCo-ST. c, Violin plots of the expression levels for the most differentially expressed genes for the different spatial domains identified using CoCo-ST. d, Volcano plot of the most differentially expressed genes for the adenoma spatial domain identified by CoCo-ST. e, Spatial expression patterns for the most differentially expressed genes (Ctsh, Cxcl15, and Slc34a2) for the adenoma spatial domain identified using CoCo-ST. These genes had high expression patterns in both the larger and smaller (hotspot) adenoma spatial domains. f, Volcano plot of the most differentially expressed genes for the adenoma spatial domain identified using the Seurat (PCA) and STUtility (NMF) approaches. g, Violin plots of the expression levels for the most differentially expressed genes for the different spatial domains identified using the Seurat (PCA) and STUtility (NMF) approaches. h, Spatial expression pattern for the most differentially expressed gene (Trf) for the adenoma spatial domain identified using the compared approaches. This gene had high expression patterns only within the larger adenoma spatial domain, with no such pattern observed in the smaller (hotspot) region. Genome-wide analyses (a–g) were performed on one independent biological sample.
Extended Data Fig. 2 CoCo-ST’s contrastive components marked interesting spatial structures enriched in ST data sets.
a, Spatial patterns captured by the first five contrastive components of CoCo-ST. b, Spatial patterns captured by the first five components of the Seurat (PCA) and NSF. c, The top 20 genes with the largest weights on the corresponding first five contrastive components. Symbols to the right of the bars indicate the signs of the weights. d, Expression patterns for the top representative genes for each of the first five contrastive components. Genome-wide analysis (a–d) were performed on one independent biological sample.
Extended Data Fig. 3 Biological processes and pathways associated with CoCo-ST’s contrastive components.
Pathway enrichment analysis was performed using the top 50 genes most strongly associated with each CoCo-ST contrastive component. Genome-wide analysis was performed on one independent biological sample.
Extended Data Fig. 4 Spatial domains identified on all MLP tissue samples using CoCo-ST’s contrastive components.
The similarity graphs for both the background and target ST data sets were constructed based on spatial locations. Genome-wide analysis were performed on nine independent biological samples.
Extended Data Fig. 5 Predicted spatial distributions of major cell types in the MLP-6 tissue sample.
The figures shows the inferred spatial localization patterns of major cell types across the MLP-6 sample, as predicted by RCTD. Genome-wide analysis was performed on one independent biological sample.
Extended Data Fig. 6 Distribution of different cell types in each spatial domain on the MLP-6 tissue sample determined using CoCo-ST.
The cell type percentages in each spatial domain add up to 100%. Genome-wide analysis was performed on one independent biological sample.
Extended Data Fig. 7 Aggregated cell-cell interaction plots on the combined MLP tissue samples containing the adenoma and adenocarcinoma spatial domains.
a, Cell-cell interaction weight plot for the adenocarcinoma-related MLP tissue samples. b, Simplified cell-cell interaction plots for a showing signalling sent from each cell group. The thicker the line, the stronger the communication. c, Cell-cell interaction weight plot for the adenoma-related MLP tissue samples. d, Simplified cell-cell interaction plots for c showing signalling sent from each cell group. The thicker the line, the stronger the communication. Genome-wide analyses (a–d) were performed on nine independent biological samples.
Extended Data Fig. 8 CoCo-ST analysis on Xenium Prime 5 K samples.
a, Spatial clusters identified by CoCo-ST on the X5K1 sample. b, scRNA-seq integration results on the X5K1 sample. c, Spatial clusters identified by CoCo-ST on the X5K2 sample. d, scRNA-seq integration results on the X5K2 sample. Genome-wide analysis (a–d) were performed on two independent biological sample.
Extended Data Fig. 9 Parameter sensitivity and performance analysis of CoCo-ST.
a, Performance comparison of CoCo-ST and cPCA for hotspot detection, demonstrating CoCo-ST’s superior performance. b, Time complexity analysis of CoCo-ST across various Visium HD assay sizes, showcasing its scalability and computational efficiency for spatial transcriptomics data.
Extended Data Fig. 10 High expression of Ctsh in pathologist-annotated regions of hyperplasia, early adenoma, late adenoma and adenocarcinoma.
The figure illustrates pathologist-defined annotations aligned with biologically meaningful patterns, as evidenced by elevated Ctsh expression in regions representing distinct tumour stages. Genome-wide analyses were performed on nine independent biological samples.
Supplementary information
Source data
Source Data Fig. 1 (download XLSX )
Pathologist annotations and comparison of ARI and NMI across different domain detection algorithms.
Source Data Fig. 3 (download XLSX )
a, CoCo-ST embedding of multiple. c, Spatial clustering on the integrated batches. d, Spatial domains identified after multiple sample integration.
Source Data Fig. 4 (download XLSX )
Results from scRNA-seq integration, cell-cell interaction analysis on tissue samples, and trajectory analysis performed both on tissue and across multiple precancer stages. Cell-cell interaction data focuses on molecular interactions between cell types within the tissue. Trajectory analysis results capture cellular transitions within tissue samples and across various stages of precancer evolution.
Source Data Extended Data Fig. 1 (download XLSX )
a, UMAP embeddings of the contrastive components determined using CoCo-ST on target ST tissue sample.
Source Data Extended Data Fig. 2 (download XLSX )
c, The 20 genes with the largest weights across the first five contrastive components. d, Gene expression patterns for the most representative genes in CoCo-ST components.
Source Data Extended Data Fig. 3 (download XLSX )
Biological processes associated with CoCo-ST’s contrastive components.
Source Data Extended Data Fig. 5 (download XLSX )
Spatially resolved enrichment of cell types is mapped onto the tissue sample following scRNA-seq integration.
Source Data Extended Data Fig. 6 (download XLSX )
Proportions of cell types after scRNA-seq integration. Cell type distributions are represented as percentages, illustrating the composition of the tissue sample after integration.
Source Data Extended Data Fig. 7 (download XLSX )
a, Cell-cell interaction weights for the adenocarcinoma tissue samples. c, Cell-cell interaction weights for the adenoma tissue samples.
Source Data Extended Data Fig. 9 (download XLSX )
Parameter sensitivity results showing the robustness of CoCo-ST under different parameter settings.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Aminu, M., Zhu, B., Vokes, N. et al. CoCo-ST detects global and local biological structures in spatial transcriptomics datasets. Nat Cell Biol 27, 2019–2031 (2025). https://doi.org/10.1038/s41556-025-01781-z
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41556-025-01781-z
