
Abstract
Human Schlafen proteins restrict viral replication by cleaving tRNA, thereby suppressing protein synthesis. Although the ribonuclease domain of Schlafen proteins is conserved across all domains of life, its function in prokaryotes has remained unclear. Here we demonstrate that prokaryotic Schlafen nucleases are widespread antiviral effectors that protect bacteria from bacteriophages and are fused to a diverse array of phage-sensing domains. We expressed seven Enterobacterales Schlafen systems in Escherichia coli, identifying two that confer defence against coliphages. We focused on a system where Schlafen nuclease is fused to a previously unknown immunoglobulin-like sensor domain and demonstrated that it recognizes tail assembly chaperones of T5-like phages. Upon activation, the Schlafen nuclease cleaves both E. coli and phage-encoded tRNAs and restricts T5 phage by reducing its burst size. Our findings redefine Schlafens as an ancient, mechanistically conserved family of immune effectors, revealing the deep evolutionary origin of tRNA-targeting antiviral immunity in humans.
Similar content being viewed by others

Main
The mammalian Schlafen (Slfn) protein family was identified in 1998 and named for its ability to induce growth arrest in murine thymocytes (‘Schlafen’ is German for ‘sleep’)1. Later work found that mammalian Slfn proteins share a conserved N-terminal endoribonuclease domain that suppresses protein synthesis by cleaving transfer RNA (tRNA) and rRNA2,3,4,5,6 and that several Slfn members act as interferon-inducible antiviral factors7,8. In particular, human SLFN11 restricts diverse viruses, including human immunodeficiency virus 1 (ref. 5), human cytomegalovirus9 and positive-strand RNA flaviviruses, such as Zika and Dengue viruses10. Human sterile alpha motif domain-containing protein 9 (SAMD9) and its paralogue, SAMD9-like protein (SAMD9L), similarly use an Slfn-like tRNase domain to inhibit translation during poxviral and lentiviral infections11,12.
The Slfn ribonuclease domain is highly conserved in animals and, in addition to antiviral defence, has been linked to suppressing transposable elements in roundworms13. This recurring recruitment of the Slfn ribonuclease domain for immune functions suggests deep evolutionary roots of its role in restricting genetic parasites8,13,14. Slfn domains are found in prokaryotic genomes and a few instances have been reported to associate with anti-phage genes, hinting at a potential immune function15,16,17,18. However, direct experimental evidence for the antiviral role of prokaryotic Schlafens (pSlfns) has been lacking and their molecular mechanisms remained unknown.
Here, we use a combination of computational, functional and biochemical assays to demonstrate that pSlfn domains are anti-phage effectors fused to a variety of potential phage sensors. We then determine the molecular mechanism of phage defence mediated by pSlfn nuclease fused to an immunoglobulin-like domain. We show that pSlfn-mediated tRNA cleavage triggers growth arrest in response to phage infection, revealing evolutionary, functional and mechanistic conservation with human Slfn-mediated antiviral immunity.
Results
Bacterial Schlafen proteins mediate phage defence
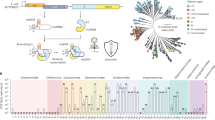
The human SLFN11 protein has an N-terminal ribonuclease (RNase) domain, a linker domain and a C-terminal DNA/RNA helicase domain4. The N-terminal domain consists of two lobes (N- and C-lobe), where the C-lobe contains the ribonuclease active site with a catalytic Ex4ExK motif that is essential for tRNA cleavage4 (Fig. 1a). To identify prokaryotic Slfn homologues, we searched for Slfn nuclease domains in reference prokaryotic genomes and identified 5,930 unique (9,937 total) protein sequences across 4 archaeal and 34 bacterial phyla (Fig. 1a, Extended Data Fig. 1a and Supplementary Table 1). Subsequent multiple sequence alignments show a highly conserved Ex4(E/D)xK sequence motif in the predicted active site, which matches the catalytic residues of SLFN11, suggesting a conserved enzymatic mechanism (Fig. 1a and Extended Data Fig. 1b).

a, The computational approach to identify prokaryotic homologues of Schlafen nucleases (Slfn) with predicted roles in phage defence. This search found 5,930 unique Slfn protein sequences (100% CD-HIT cut off). The Weblogo plot shows the conserved Ex4(E/D)xK motif in prokaryotic Slfn domains. b, Annotation of domains fused to Slfn in the 5,930 identified proteins. c, Experimental approach to test for immune activities of Slfn proteins. d, Immune activities of tested Slfn proteins (top to bottom) against a panel of phages that infect E. coli (left to right). Cfr, Citrobacter freundii; Eco, Escherichia coli; Eho, Enterobacter hormaechei; Elu, Enterobacter ludwigii; Pag, Pantoea agglomerans; Ror, Raoultella ornithinolytica; dsDNA, double-stranded DNA; ssDNA, single-stranded DNA; ssRNA, single-stranded RNA; λvir, virulent mutant of phage λ.
Prokaryotic defence genes frequently cluster in the host genome, forming genetic neighbourhoods known as defence islands15,19. To determine whether pSlfn proteins reside in defence islands, we performed a genetic neighbourhood analysis and found that 69.6% of identified pSlfn genes are located within a ±10-kb distance to at least one known defence gene (Extended Data Fig. 1a and Supplementary Table 1). On average, identified pSlfn genes co-localize with 1.9 defence genes, which is comparable to AriB (2.74), the nuclease effector of the phage anti-restriction-induced system (PARIS)20,21, and approximately tenfold higher than the housekeeping genes rpL3 (0.13) and L1p (0.19) (Extended Data Fig. 1c). This genetic association suggested that pSlfns may function in phage defence. Notably, 30.4% of identified pSlfn genes were not associated with defence genes, suggesting that in some instances, pSlfn domains may have non-immune functions or that these pSlfn genes are in defence hot spots that have not been annotated yet21,22.
Phage defence systems often function through sensors that detect infection and effectors that execute the immune response. These functional modules can be encoded within a single gene or split across multiple genes23,24. We found that 97.1% of identified pSlfn domains are fused to other protein domains, where pSlfns most probably act as effectors, while the identified fused domains may function as sensors (Fig. 1b). To classify the diversity of domain architectures among pSlfn proteins, we used a combination of sequence and structural homology-based computational methods, identifying 55 distinct domain compositions, which we designated pSlfn1 through pSlfn55 (Supplementary Table 2). The most frequent domain compositions include fusions of pSlfn to histidine kinase (HisKinase, 34.0%), HisKinase and helix-turn-helix domains (HisKinase–HTH, 19.2%), HTH domains (8.8%), DUF5929 (5.1%), domains with immunoglobulin-like fold (Ig-like, 3.9%), DUF262 (1.8%) and TBP-like domains (1.5%) (Fig. 1b). Of 5,930 identified pSlfns, 1,020 (17.2%) are fused to protein domains that show no substantial homology to the domains annotated in the Pfam and Evolutionary Classification of protein Domains (ECOD) databases25,26. We further grouped these proteins into 558 sequence similarity clusters (Supplementary Table 2). This diversity of domain compositions in pSlfn proteins probably represents combinations of the core Slfn effector module with various sensors that respond to different cues and regulate its nuclease activity.
To test whether pSlfn genes confer phage defence, we focused on four domain architectures found in Enterobacterales, comprising fusions with HisKinase, Ig-like, DUF262 and unannotated domains. We manually picked two systems from each group, prioritizing those with the most neighbouring defence genes and greatest amino acid sequence divergence (Extended Data Fig. 1d and Supplementary Table 3). One construct failed during synthesis, leaving seven systems. We expressed these in the E. coli K-12 strain MG1655 under their native promoters and challenged the cells with a panel of diverse E. coli phages (Fig. 1c,d and Supplementary Table 4). As a positive control, we used a plasmid encoding the E. coli B185 PARIS-2 immune system21, while an empty vector (EV) served as a negative control. In the tested subset, two pSlfn genes provided defence against T phages (Fig. 1d and Extended Data Fig. 1e,f). We found that the DUF262–Slfn fusion from E. coli UC4224 (EcoSlfn7) significantly reduced the efficiency of plating (EOP) of a broad range of phages, with the most robust activity against the T7 phage (538.5 ± 194.4-fold EOP reduction, P = 6.9 × 10−4). By contrast, the Slfn fusion with Ig-like domain from Raoultella ornithinolytica (RorSlfn5) protected E. coli only against T5 phage, reducing the EOP by 22.3 ± 1.0-fold (P = 3.0 × 10−11) (Extended Data Fig. 1f). Overall, these data demonstrate that prokaryotic Schlafen proteins protect bacteria from phage infection and exhibit distinct phage specificities that are probably determined by their fused domains.
RorSlfn5 reduces the productivity of the T5 phage
To elucidate the mechanisms that underpin the immune function of pSlfn proteins, we focused on RorSlfn5 defence, which features an uncharacterized Ig-like domain, suggesting a mode of phage sensing that was not previously described. We challenged MG1655 cells with T5 phage at different multiplicities of infection (MOIs) and monitored cell growth over time. At low MOIs (0.01 and 0.1), cells expressing RorSlfn5 survived the infection compared with cells without the defence system, whereas infections at high MOIs (5 and 10) resulted in all cultures collapsing approximately 60 min post infection, which agrees with the reported lysis time for T5 phage27 (Fig. 2a,b). Further, at a MOI of 10, the phage titre in supernatants of RorSlfn5-expressing cells at 60 min post infection was reduced by 59.3 ± 8.6% compared with the no defence control (P = 7.6 × 10−3; Extended Data Fig. 2a). Alanine substitutions in the catalytic Ex₄DxK motif (E15A, D20A) of the Slfn domain abolished defence at low MOIs and rescued phage titre at a high MOI, indicating that the nuclease activity of RorSlfn5 is required for the phage defence (Fig. 2c and Extended Data Fig. 2a).

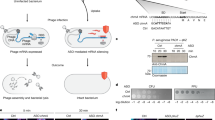
a–c, The growth kinetics of E. coli K-12 MG1655 cells without the defence (a), with RorSlfn5 (b) or the nuclease-dead RorSlfn5E15A, D20A mutant (c) following T5 bacteriophage infection at various MOIs. Each line shows a biological replicate (n = 4). d,e, The ECOI (d) and average burst size (e) of T5 phage in RorSlfn5-expressing E. coli MG1655 cells compared with the EV control. The data are shown as the mean of three biological replicates ± s.d. A two-sided Welch’s t-test was used to compare the experimental and control groups. f, An experimental approach to identify viral triggers of RorSlfn5. g, Top: plaque assays with RorSlfn5-escaping T5 phages (esc1–esc4). Bottom: mapping of mutations in T5 phage escapers compared with a reference phage genome. The vertical lines indicate mutations found in the laboratory T5 phage stock compared with the reference NCBI sequence. Vertical orange box highlights mutations present in escaper phages but not in the original viral stock. h, A toxicity assay in MG1655 cells co-transformed with the RorSlfn5 plasmid and a plasmid for arabinose-inducible expression of T5.142 protein. Tenfold serial dilutions were spotted on agar plates with 0.2% glucose (−) or 0.2% arabinose (ara) (+). pBAD-GFP was used as a control for arabinose-dependent induction.
The collapse of RorSlfn5-expressing cultures at high MOIs and reduced phage titres suggested that the phage defence may act by triggering abortive infection, killing the host cell before phage replication is completed28,29. To test this hypothesis, we performed the efficiency of centre of infection (ECOI) assay, which quantifies the number of productive infections that release at least one infectious progeny phage30. However, we found no significant difference in the number of infective centres between RorSlfn5-expressing cells and the EV control, indicating that RorSlfn5 does not abort T5 phage infection (P = 0.94; Fig. 2d). Reduced EOP (Fig. 1d) and lower phage titres with no changes in ECOI suggest that RorSlfn5 reduces T5 phage productivity without completely eliminating the phage31. Consistent with this, the average burst size of T5 was reduced by 81.2 ± 3.0% in RorSlfn5-expressing cells compared with the control (P = 3.5 × 10−4; Fig. 2e). Taken together, these assays indicate that RorSlfn5 restricts T5 phage through a non-abortive mechanism that acts downstream of infection, limiting infectious phage progeny.
T5 tail assembly chaperone triggers RorSlfn5 phage defence
To determine the viral trigger for the RorSlfn5 defence, we isolated T5 phages that escaped the immunity and sequenced their genomes (Fig. 2f). Four viral clones that we sequenced contained three distinct missense mutations (that is, D18G, D68G and I72T) in the T5.142 gene, which encodes for the phage tail assembly chaperone (NCBI accession: YP_006970.1)32 (Fig. 2g and Supplementary Table 5). To test whether the T5.142 protein triggers RorSlfn5 defence, we cloned the viral gene and its escape variants into high-copy plasmid vectors under the control of the arabinose-inducible promoter (pBAD; Fig. 2h). Expression of T5.142 in cells co-transformed with the RorSlfn5 plasmid resulted in toxicity, demonstrating that this viral protein is sufficient to trigger the defence. By contrast, cells expressing a catalytically inactive mutant of RorSlfn5 (RorSlfn5E15A, D20A) showed no growth defect compared with uninduced cells, indicating that the toxicity is mediated by the T5.142-triggered nuclease activity of RorSlfn5. In addition, escape mutations in the viral T5.142 protein rescued toxicity. Notably, while the T5.142D18G variant reduced toxicity, it did not completely abolish it, suggesting that, in our experimental set up, it is less effective at evading RorSlfn5 defence than D68G and I72T mutations.
The immunoglobulin-like domain of pSlfn5 is a phage sensor
Homologues of pSlfn5 from Pantoea agglomerans (PagSlfn5; 39.6% sequence identity to RorSlfn5) and Serratia fonticola (SfoSlfn5; 79.6% sequence identity to RorSlfn5) did not protect E. coli from T5 phage or any other phage tested (Extended Data Figs. 1f and 2b). The sequence divergence between these proteins is primarily driven by their C-terminal domains, while the N-terminal Slfn nuclease domains are more conserved (Extended Data Fig. 3a). The AlphaFold-predicted structure of RorSlfn5 suggests that its C-terminal domain has an Ig-like β-sandwich fold with structural similarity to human integrin ectodomains (Extended Data Fig. 3b,c and Supplementary Table 6). These observations led us to hypothesize that the Ig-like domains of pSlfn5 proteins function as sensor modules that recognize viral proteins, and thereby PagSlfn5 and SfoSlfn5 might recognize T5.142 homologues from other phages.
To test this, we synthesized genes encoding six representative homologues of the T5 phage tail assembly chaperone (Fig. 3a). In addition to T5.142, RorSlfn5 was triggered by expression of homologues from Salmonella phage S114, Pantoea phage vB_PagS_AAS21 and an unclassified phage from a human gut metagenome (Fig. 3b and Extended Data Fig. 2c). Consistent with the lack of defence against the T5 phage, PagSlfn5 and SfoSlfn5 were not activated by expression of T5.142, while the tail assembly chaperone from the human gut metagenome triggered all tested pSlfn5 homologues (Fig. 3b and Extended Data Fig. 2d,e). None of the identified triggers caused toxicity when expressed alone or co-expressed with a nuclease-dead RorSlfn5E15A, D20A mutant, confirming that toxicity is mediated by the nuclease activity of pSlfn5 (Extended Data Fig. 2f,g). Together, these findings demonstrate that pSlfn5 proteins recognize tail assembly chaperones from phages in the Demerecviridae family, with different pSlfn5 homologues exhibiting distinct trigger specificities.

a, A maximum-likelihood phylogenetic tree of T5.142 homologues. Selected representatives are listed for each homologue cluster and indicated with black dots. b, A summary of toxicity assays in MG1655 cells co-transformed with pSlfn5 (top to bottom) and T5.142 homologues from various phages (left to right) shown in Extended Data Fig. 2. The experiment was performed in three biological replicates and the results for one representative replicate are shown. The numbers correspond to the homologue clusters in a. c,d, Toxicity (c) and EOP assay (d) with chimeric pSlfn5 proteins with swapped Ig-like domains. The toxicity assays (Extended Data Fig. 4) were repeated independently three times with similar results. The data are shown for one replicate. EOP assay results are shown as mean ± s.d. of three biological replicates. The white dots show the average of three technical replicates for each biological replicate. A one-way ANOVA with post-hoc Tukey HSD test was used to compare the experimental groups. Sfo, Serratia fonticola.
To confirm the sensor function of Ig-like domains in pSlfn5 defence, we created chimeric proteins by swapping the Ig-like domains of RorSlfn5 and SfoSlfn5. Cellular toxicity assays showed that phage trigger specificity was transferred along with the sensor domain (Fig. 3c and Extended Data Fig. 4). Furthermore, the chimeric protein containing the nuclease domain of SfoSlfn5 and the RorSlfn5 sensor (Sfo–Ror chimera) gained protection against T5 phage, whereas changing the Ig-like domain of RorSlfn5 for that of SfoSlfn5 (Ror–Sfo chimera) abolished defence against T5 (Fig. 3d). Wild-type (WT) RorSlfn5 reduced T5 phage EOP by 18.9 ± 2.1-fold (P = 3.5 × 10−9), while the Sfo–Ror chimera was ~3.6-fold less efficient and reduced EOP by 5.2 ± 0.1-fold (P = 2.4 × 10−3).
Overall, these results demonstrate that the Ig-like domain determines phage specificity, indicating that it functions as a sensor module within the pSlfn5 defence system. The acquired defence in the Sfo–Ror chimera shows that these domains are modular and can be exchanged between orthologues. However, the reduced phage defence of the chimera compared with RorSlfn5 suggests that effective immunity requires co-evolution between sensor and effector domains.
RorSlfn5 is a phage-activated tRNase
After identifying the viral cue that triggers pSlfn5 defence, we then investigated its mechanism. Co-expression of RorSlfn5 with its viral trigger led to growth arrest ~30 min after inducing T5.142 expression (Fig. 4a). This timing mirrors the kinetics of GFP expression under the same promoter, which became detectable 30 min post-induction (Fig. 4b). Control cells co-expressing RorSlfn5 and GFP or RorSlfn5E15A, D20A and T5.142 showed no growth arrest, demonstrating that it is mediated by T5.142-triggered nuclease activity of the pSlfn domain (Extended Data Fig. 5a,b).

a, Growth kinetics of MG1655 cells constitutively expressing RorSlfn5 with (0.2% arabinose (ara)) and without (0.2% glucose (gluc)) induction of T5.142 expression. b, Kinetics of GFP expression after induction of the pBAD promoter with arabinose. c, Schematics of the reporter assay for detecting SOS response upon DNA damage. d,e, Growth kinetics (d) and SOS-response reporter assay (e) of MG1655 treated with 100 nM mitomycin C or PBS. f, SOS-response reporter assay with RorSlfn5-expressing cells after induction of T5.142 expression. The kinetic assays in a–f were performed in four biological replicates. The data are shown as mean (centre line) ± s.d. (ribbon). g, Urea–PAGE of total RNA extracted from RorSlfn or RorSlfnE15A, D20A-expressing MG1655 at various timepoints after induction of T5.142 expression. Black arrows highlight RNA cleavage fragments. h, Schematics of the 3′-end RNA-seq approach used to map RorSlfn5-mediated RNA cleavage in T5 phage-infected cells. i, Total abundance of non-native (internal) 3′ ends in different RNA types in T5 phage-infected cells expressing RorSlfn5 or its nuclease-dead version (E15A and D20A mutation). The data are shown as the mean of three biological replicates ± s.d. Means were compared using a two-sided Welch’s t-test. The resulting P values were adjusted using the Holm method to account for multiple comparisons. j, The total abundance of internal 3′ ends in bacterial and viral tRNAs. The data are shown as the mean of three replicates.
To test whether RorSlfn5 targets DNA, we assessed bacterial DNA damage following T5.142-triggered RorSlfn5 nuclease activity. We used a fluorescent reporter plasmid (pSOS-GFP) that encoded a gfp gene under the control of a recA promoter, which is upregulated in the bacterial SOS response to DNA damage (Fig. 4c). Treatment of MG1655 with mitomycin C, a DNA-damaging agent, induced a strong fluorescent signal, which was absent in PBS-treated cells (Fig. 4d,e). By contrast, cells co-transformed with RorSlfn5, T5.142 and pSOS-GFP did not show increased fluorescence, suggesting that RorSlfn5 activation does not cause DNA damage and the SOS response (Fig. 4f and Extended Data Fig. 5c,d). These results indicate that RorSlfn5 does not act on DNA and probably retains RNase activity as its primary mechanism of toxicity, consistent with the function of human SLFN proteins3,4.
To test whether RorSlfn5 targets RNA, we co-transformed MG1655 cells with plasmids encoding the T5.142 trigger and either WT RorSlfn5 or a catalytically inactive mutant RorSlfn5E15A, D20A. We extracted total RNA from cells at 0, 30 and 60 min post-T5.142 induction. Urea–PAGE analysis of extracted RNA identified RNA fragments between 25 and 50 nt in size that appeared at 30- and 60-min post-induction in cells expressing RorSlfn5 but not the catalytically inactivated mutant, demonstrating that T5.142 expression triggers RorSlfn5-mediated RNA cleavage (Fig. 4g).
To map RNA cleavage induced by RorSlfn5, we performed RNA 3′-end sequencing (Fig. 4h). To validate this approach, we analysed RNA from E. coli expressing the PARIS-2 immune system. RNA sequencing (RNA-seq) revealed a significant enrichment of internal 3′ ends in tRNALys(UUU) (false discovery rate (FDR) of 0.002) in T5-infected cells expressing the active PARIS-2 defence compared with cells expressing the inactivated PARIS-2 system (AriBE26A mutant; Extended Data Fig. 6a–c). This result agrees with previous work demonstrating that activation of PARIS-2 immunity triggers AriB-mediated cleavage of tRNALys(UUU)20,33.
Next, we applied the same RNA-seq strategy to T5 phage-infected MG1655 cells expressing RorSlfn5 or its nuclease-dead mutant (Fig. 4h and Extended Data Fig. 6g). This analysis found no significant difference in the abundance of internal 3′ ends in messenger RNA (mRNA), non-coding RNA (ncRNA) or ribosomal RNA (rRNA), while internal tRNA 3′ ends were significantly enriched in cells expressing WT RorSlfn5 (Padj = 3.4 × 10−3; Fig. 4i). Analysis of tRNA mapping reads identified significant enrichment of total internal 3′-end counts in tRNALys(UUU) (194.1-fold, FDR of 2.4 × 10−4), T5 phage-encoded tRNAAla(UGC) (34.8-fold, FDR of 5.2 × 10−3) and tRNAMet(CAU) (145.4-fold, FDR of 4.4 × 10−5) in cells expressing WT RorSlfn5 compared with the nuclease-dead mutant (Slfn5dead; Fig. 4j and Supplementary Table 7). Position-specific analysis confirmed these enrichments and identified additional tRNAs with significant cleavage signatures, including tRNAVal(UAC), tRNAPhe(GAA) and tRNAIle(CAU) (FDR <0.05; Extended Data Fig. 6h). Together, these results indicated that RorSlfn5 preferentially cleaves tRNA upon phage infection, with tRNALys(UUU) fragments being most abundant in the cell.
Phage-activated RorSlfn5 cleaves the tRNA anticodon arm
We next sought to map RorSlfn5-mediated tRNA cleavage sites at single-nucleotide resolution. Mapping of 3′ ends in tRNALys(UUU) identified three highly enriched positions within the anticodon arm (29, 30 and 32), while the 5′-end mapping showed peaks at positions 39 and 41 (Fig. 5a,b). Correspondingly, sequence coverage across the entire anticodon loop of tRNALys(UUU) was absent, indicating complete removal of this region (Fig. 5c). A similar sequence coverage gap was observed between the 3′ and 5′ ends of tRNAMet(CAU) fragments (Extended Data Fig. 6i–k,m). This cleavage pattern suggests that RorSlfn5 may either excise the anticodon loop through two precise cuts or perform a single cut followed by trimming of the resulting fragments by host ribonucleases. Supporting the latter model, RNA-seq of cells expressing the PARIS immune system revealed a similar 11-nucleotide gap in the anticodon loop of tRNALys(UUU) spanning nucleotides 29 and 41, the latter position matching the AriB cut site identified in vitro20,33. These findings support a mechanism in which cleavage is followed by trimming of the anticodon loop (Extended Data Fig. 6d–f,l).

a,b, Position-specific mapping of internal 3′ ends (a) and 5′ ends (b) in tRNALys(UUU) in T5 phage-infected cells expressing RorSlfn5. The data are shown as the mean of three biological replicates. c, Alignment of sequencing reads to the lysW gene of MG1655 cells. One representative replicate from three biological replicates is shown. d, SEC of affinity-purified complex of RorSlfn5 and T5.142 (left). The elution fraction highlighted with a light blue rectangle was analysed with SDS–PAGE (right). The full SEC profile can be found in Extended Data Fig. 7. e, tRNA cleavage assays with 100 nM tRNALys(UUU) and 100 nM RorSlfn5 in the presence of T5.142 trigger (2 µM) and Mn2+ (2 mM). f, The same as in e but with 2 mM Mg2+, 2 mM Mn2+ or no metal and 10 mM EDTA. All shown assays were reproduced independently three times with similar results. g, Potential RorSlfn5 cut sites (triangles) in the anticodon arm of tRNALys(UUU), according to RNA-seq data in a and b. The red triangle shows the cut site supported by cleavage assays in e and f. h, A proposed model for RorSlfn5-mediated tRNA cleavage in vivo and subsequent host exoribonuclease (exoRNase) trimming of the anticodon loop.
To reconstitute tRNA cleavage in vitro, we expressed and purified RorSlfn5 and T5.142 proteins. The size-exclusion chromatography (SEC) profile of the RorSlfn5 (43.1 kDa) indicates that it assembles in higher-order oligomers, while T5.142 (13.8 kDa) eluted at a size consistent with a dimer or a trimer, based on gel filtration standards (Extended Data Fig. 7a,b). SEC with multi-angle light scattering (SEC–MALS) analysis confirmed this oligomerization, estimating an average mass of 261.0 ± 0.4 kDa, indicating that RorSlfn5 predominantly forms a hexamer at the tested conditions (Extended Data Fig. 7c). As Ig-like domains often mediate protein–protein interactions34, we hypothesized that the Ig-like domain of RorSlfn5 may detect phage by directly binding T5.142. To test this hypothesis, we co-expressed RorSlfn5dead with the T5.142 and performed a reciprocal affinity co-purification using a Twin-Strep-tag (Extended Data Fig. 7d). Both proteins co-purified from cell lysates regardless of which had the affinity tag and, after cleaving off the tags, RorSlfn5dead and T5.142 co-eluted in a single SEC peak, confirming a direct interaction and suggesting that binding of T5.142 triggers the tRNase activity of RorSlfn5 (Fig. 5d and Extended Data Fig. 7e).
Human SLFN nucleases catalyse tRNA cleavage using a manganese-dependent mechanism3,4. To test whether RorSlfn5 shares a similar mechanism, we incubated synthetic tRNALys(UUU) with purified RorSlfn5 and T5.142 in the presence of manganese, which yielded two major cleavage fragments sized between 25 and 50 nucleotides (Fig. 5e). Neither protein alone produced detectable cleavage and no activity was observed in the absence of metal or in the presence of EDTA (Fig. 5e,f). The detected tRNase activity was manganese specific as neither magnesium nor any other tested divalent metal supported cleavage in the tested conditions (Fig. 5f and Extended Data Fig. 8a). Mutations in the pSlfn active site (E15A, D20A) abolished cleavage activity, demonstrating that the Slfn nuclease domain of RorSlfn5 catalyses the cleavage reaction (Extended Data Fig. 8b). Upon titration of the trigger, tRNA cleavage was detectable with 1 µM T5.142 (100 nM RorSlfn5, 100 nM tRNALys(UUU)), while concentrations of T5.142 higher than 2 µM did not further enhance activity, suggesting that this trigger concentration saturates RorSlfn5 under the tested conditions (Extended Data Fig. 8c). Together, these cleavage assays demonstrate that RorSlfn5 functions as a manganese-dependent tRNase, activated by the phage tail assembly chaperone.
Testing additional tRNA substrates showed that bacterial tRNALys(UUU) and T5-encoded tRNAAla(UGC) were cleaved most efficiently, consistent with these tRNAs being the top hits in our RNA-seq data (Fig. 4j and Extended Data Fig. 8d). In vitro cleavage of tRNALys(UUU) produced two major products, consistent with a single cut (Fig. 5e,f), while the in vivo RNA-seq data identified two candidate RorSlfn5 cut sites at nucleotides 30–31 (site 1) and 39–40 (site 2) in tRNALys(UUU) (Fig. 5g). The fragment sizes observed in vitro support cleavage at predicted site 1 (30- and 46-nt products) over site 2 (37- and 39-nt products) (Fig. 5e,f). We therefore propose that in vivo RorSlfn5 cleaves the anticodon arm between nucleotides 30 and 31 (site 1), which is then followed by trimming of the anticodon loop by host ribonucleases (Fig. 5h).
Discussion
tRNA cleavage as an antiviral strategy was first demonstrated with the discovery of the bacterial PrrC anticodon nuclease35,36. Since then, several other examples of anti-phage tRNases have been described, such as AriB from the PARIS system20, PtuAB from Retron Ec7837 and ribonucleases in prophage competition elements38. More recently, virus-induced tRNA cleavage has also been recognized as a component of antiviral defence in mammals3,4,12. Although mechanistically similar, these shared antiviral strategies were thought to have emerged independently through convergent evolution12. Our work establishes a direct evolutionary link by demonstrating that homologues of human Schlafen tRNA ribonucleases function in bacterial phage defence through a tRNA cleavage mechanism conserved across the tree of life.
The evolutionary and functional conservation of the Schlafen ribonuclease domain suggests that it belongs to an ancient ancestral immune system that predates the divergence of prokaryotes and eukaryotes39 (Fig. 6). In prokaryotes, this antiviral effector is fused to a variety of accessory domains that probably act as phage sensors, regulating the nuclease activity in response to distinct viral cues. This modular architecture of Schlafen proteins appears to be a deeply conserved feature, with the core ribonuclease domain fused to diverse regulatory modules in both prokaryotes and mammals17,40. Binding of single-stranded DNA (ssDNA) to the helicase domain of human SLFN11 activates its tRNase activity, whereas recent work found that SAMD9, which has a complex multidomain architecture, is activated by cytosolic double-stranded nucleic acids6,12,41,42. This recurrent fusion of the Schlafen nuclease with diverse sensory domains highlights its versatility as an antiviral effector, co-opted by immune systems across the domains of life.

A model for Schlafen-mediated phage defence (right). This mechanism parallels antiviral Schlafen function in humans (left), revealing functional and mechanistic conservation.
Many domains associated with prokaryotic Schlafen have not been previously linked to phage defence, revealing a previously unknown repertoire of phage-sensing mechanisms. In particular, we have identified an uncharacterized C-terminal immunoglobulin-like sensor domain in pSlfn5 that detects tail assembly chaperones of T5-like phages, dictating the phage specificity. We found that phages with substitutions in T5.142 can evade pSlfn5 defence without manifesting any apparent fitness defects. Selection of such escape variants, in turn, might drive adaptations in the sensor domain to regain the defence. Consistent with this co-evolution model, the Slfn nuclease domain is highly conserved across pSlfn5 orthologues, whereas the sensor domain shows greater sequence variation.
Functional, genetic and biochemical assays in our work demonstrate that the T5.142 phage tail assembly chaperone activates RorSlfn5, which results in cleavage of the anticodon arm of host- and phage-encoded tRNAs. In this work, we expressed RorSlfn5 under its native promoter in a medium-copy plasmid vector (p15A origin), maintained at ~10–20 copies per cell43,44, which may result in expression levels higher than physiological. The elevated levels of RorSlfn5 may expand its target RNA specificity beyond that observed under physiological expression. While our data establish trigger-dependent tRNA cleavage by RorSlfn5, additional work would be needed for a more nuanced understanding of its preferred cleavage targets in the native context.
Phage tRNAs often accumulate mutations that make them resistant to cleavage by immune anticodon nucleases45. Our work indicates that RorSlfn5 might cleave phage tRNAAla(UGC) more efficiently than its host equivalent, suggesting that these mutations can backfire, rendering phage tRNAs susceptible. Moreover, phage tRNAs can supplement the cellular tRNA pools, fine-tuning them to the codon usage of late phage genes46. Therefore, targeting phage tRNA may be effective for subverting the late stages of infection and limiting phage progeny.
The modular architecture of prokaryotic Schlafen immunity parallels that of recently described single-gene Shedu systems, which pair a shared nuclease core with diverse sensory domains47,48,49. In Shedu, the nuclease forms a central oligomeric core with variable sensors projecting outwards41,42. While RorSlfn5 also purifies as a higher-order oligomer, whether oligomerization is driven by the nuclease or the sensor remains unknown. Structural and biochemical studies will be essential to determine the architecture of this complex, how phage components activate the nuclease, what is the molecular basis for tRNA specificity and whether distinct domain fusions of prokaryotic Schlafens converge on a shared activation mechanism or have evolved unique mechanistic solutions.
Methods
Plasmids, bacterial strains and bacteriophages
The oligonucleotide primers and plasmid DNA sequences are listed in Supplementary Table 4. Candidate phage defence genes with native promoters (100–150 upstream sequence) were synthesized and cloned into a medium-copy plasmid vector (pTwist_Amp_MC) by Twist Bioscience. The plasmid expressing the PARIS-2 immune system of E. coli B185 was a gift from Dr. Blake Wiedenheft20. Site-directed mutagenesis was used to make nucleotide substitutions in plasmid pAN248 to create the nuclease-dead variant of RorSlfn5 (E15A, D20A mutations) and in plasmid pRF85 to inactivate AriB nuclease (E26A mutation) in the PARIS-2 system. The genes encoding the T5 tail assembly chaperone (T5.142; NCBI accession: YP_006970.1) and its variants (D18G, D68G and I72T) were PCR amplified from genomic DNA of the T5 phage and its escape variants. Amplified genes were cloned into the pBAD vector. Homologues of T5.142 gene were synthesized by Twist Bioscience and cloned into pBAD. To perform the cell toxicity assays, the plasmid backbones of pAN248 (RorSlfn5), pAN254 (PagSlfn5) and pAN294 (SfoSlfn5) were modified to replace the ampicillin resistance (AmpR) gene with the kanamycin resistance (KanR) gene. Plasmids pAN309 and pAN310 expressing chimeras of RorSlfn and SfoIg-like and SfoSlfn and RorIg-like were assembled using Hi-Fi (NEB, E2621). Gene fragments for pAN301 (pSOS-GFP) were synthesized by Twist Biosciences and assembled using NEBridge Golden Gate Assembly Kit (BsmBI-v2) (NEB, E1602S). For protein expression, the genes encoding for RorSlfn5 and T5.142 were cloned into the pDF0118 (Addgene #172503) or pRSF-1b vector backbones in frame with a 6xHis-TwinStrep-SUMO affinity tag. All plasmid sequences were confirmed with whole-plasmid sequencing at Plasmidsaurus (https://www.plasmidsaurus.com/). All bacterial strains and phages used in this work are listed in Supplementary Table 4.
Computational search for prokaryotic Slfn
The initial search was done using jackhmmer to find homologues of the human SLFN11 nuclease domain (amino acids 1–354; NCBI: NP_001098057.1) in a database of reference proteomes50. The hidden Markov model (HMM) generated by jackhmmer was used to perform an HMMsearch (from HMMER 3.1b2) on a database of bacterial and archaeal amino acid sequences, created by translating RefSeq complete genomes, downloaded in July 2023, using prodigal51. The non-redundant hits from the first HMMsearch (e-value <0.001; CD-HIT with -c 0.9) were aligned using MAFFT v7.526. A new HMM was created from the alignment using HMMbuild and HMMsearch was repeated on the same database. The second round found 9,937 potential homologues (e-value <0.001) with full-length Slfn domains (>100 amino acids; PF04326). After redundancy (CD-HIT with -c 1) was removed, a total of 5,930 protein sequences were left.
Prokaryotic Slfn domains were mapped in the HMMsearch hits using HMMscan with the PF04326.19 HMM profile from the Pfam database25. Prokaryotic Slfn domains (5,930) were extracted and aligned using MAFFT v7.526 with 596 unique mammalian Slfn domain sequences (CD-HIT with -c 1) found with jackhmmer. The resulting alignment was used to build a phylogenetic tree using FastTree v2.1.11 with -pseudo -wag -gamma options. For visualization, the tree was pruned to remove prokaryotic sequences with >50% sequence similarity (CD-HIT -c 0.5) and mammalian sequences with >70% sequence similarity (CD-HIT -c 0.7). The resulting tree was visualized and annotated using iTOL52.
For every identified pSlfn, genes within a ±10-kbp distance were extracted and annotated using HMMscan (i-E-value <0.001) against HMM profiles downloaded from the DefenseFinder database in March 202553. The number of defence genes identified in the genetic proximity was then calculated for every pSlfn gene (Supplementary Table 1). Defence scores were calculated for pSlfn clusters as the number of pSlfn homologues with at least one annotated defence gene in the genetic neighbourhood divided by the total number of pSlfn homologues within the cluster.
Classification of pSlfn proteins
Sequences of 5,930 pSlfn proteins were searched against the Pfam database25 (v37.2) using HMMER’s hmmscan50, and against the ECOD database26 (v292) using BLAST54. Pfam hits (i-E-value <0.0001) were filtered to remove overlapping or nested annotations by selecting predictions with the highest confidence. Proteins with significant sequence homology to multiple Pfam families, that is, fusions of Slfn (PF04326) to other domains, were further classified by assignment to their respective Pfam clan, representing a higher-order grouping of related domain families. Proteins with Slfn domains and less than 40 amino acids of remaining sequence were annotated as ‘pSlfn only’. Proteins with Slfn domains and more than 40 amino acids of remaining sequence without confident hits to Pfam or ECOD (2,134 total) were further annotated using structural homology as described before55. Briefly, 2,134 proteins were grouped in 903 clusters using MMseqs with 50% sequence identity and 0.8 coverage cut offs56. AlphaFold 3 (AF3) was used to predict protein structure models for each cluster representative57. AF3 models were processed by Domain Parser for AlphaFold Models (DPAM) to identify domain architectures and map domains to the ECOD database classification58. The resulting ECOD domain classifications were assigned to cluster members, converted into corresponding Pfam clans (where possible) and integrated with the sequence-based Pfam domain annotation. For 1,020 pSlfn proteins, a confident annotation could not be assigned. These proteins were designated as unannotated fusions of pSlfn domains and grouped into 558 sequence similarity groups using MMseqs. The domain annotations can be found in Supplementary Table 2.
Computational search for T5.142 homologues
Homologues of T5.142 were identified in the database of non-redundant protein sequences (NCBI) using PSI-BLAST. Identified homologues were clustered using CD-HIT with -c 0.9, and cluster representatives were used for experimental testing. Protein sequences of T5.142 homologues were aligned with MAFFT v7.526. The phylogenetic tree was constructed using FastTree v2.1.11 with -pseudo -wag -gamma options and visualized using iTOL.
Bacterial growth assays
E. coli K-12 MG1655 cells expressing RorSlfn5, RorSlfn5E15A, D20A or carrying no defence system were grown with shaking at 37 °C to an OD600 of 0.3. Then, 180 µl of the cell culture was transferred to a 96-well plate and mixed with 20 µl of T5 phage dilution at varying MOIs. The 96-wells were shaken and the OD600 was measured every 60 s for 6 h at 37 °C using a SpectraMax M5e reader. To measure bacterial growth upon viral trigger expression, E. coli K-12 MG1655 cells were co-transformed with RorSlfn5 or RorSlfn5E15A,D20A and a plasmid with an inducible trigger T5.142 or GFP gene. Double-transformed cells were plated on LB agar plates with dual antibiotics and 0.2% glucose to suppress basal expression. Night cultures of double-transformed cells were used to inoculate liquid cultures and cells were grown to an OD600 of 0.3. After reaching optical density, 180 µl of bacterial cultures were mixed with 20 µl of d-glucose or L-arabinose to a final concentration of 0.2% in a 96-well plate. Optical density and GFP signal were measured every 60 s for 6 h at 37 °C using a SpectraMax M5e reader. All experiments were performed in three or four biological replicates.
EOP assay
E. coli K-12 MG1655 or E. coli NovaBlue (for testing phages M13, MS2 and Qbeta) expressing candidate phage defence genes, the PARIS-2 system or an EV (negative control) were grown in LB medium with shaking at 37 °C to an OD600 of 0.3. Cultures were supplemented with antibiotics, 10 mM MgCl2 and 10 mM CaCl2. Day cultures were then mixed with soft agar containing the same supplements and antibiotics and overlaid onto LB agar plates. The plates were allowed to dry for 1 h at room temperature. Serial dilutions of phages were prepared and 10 μl of each dilution was spotted onto the plates and left to dry until the drops were no longer visible. Plates were incubated overnight at 37 °C. The following day, the phage titre (PFU ml−1) was determined from the dilution that produced countable plaques. For T2, T4 with PARIS-2, M13, MS2 and Qbeta phages, the least concentrated dilution that yielded full lysis was considered to be 100 plaques following previously published methods59. The EOP was calculated as the ratio of the phage titre with cells expressing the immune system to that of the EV control. All experiments were performed in three biological replicates.
ECOI assays
Day cultures of E. coli K-12 MG1655 cells expressing either an EV or RorSlfn5 defence system were grown as previously described. Once OD600 reached 0.3, 1 ml of cells was infected with T5 bacteriophage at 0.1 MOI and incubated at 37 °C for 10 min to allow phage adsorption. Immediately after, samples were centrifuged at 4 °C, 3,000g for 5 min, washed twice with SM buffer (50 mM Tris–HCl, pH 7.5, 100 mM NaCl, 8 mM MgSO4 and 0.01% gelatin) and serially diluted. Ten microlitres of diluted cells was spotted onto plates containing bottom agar and soft agar mixed with naive E. coli K-12 MG1655 cells (OD600 of 0.3) supplemented with 10 mM MgCl2 and 10 mM CaCl2. Plates were dried at room temperature and incubated at 37 °C overnight. The next day, the infection centres were counted and the ECOI was calculated as the ratio of the phage titre on cells expressing the RorSlfn5 candidate defences to that on cells expressing the EV control. All experiments were performed in three biological replicates.
Burst size
Average burst sizes were quantified as previously described with modifications60. E. coli K-12 MG1655 cells expressing either an EV or RorSlfn5 defence system were grown to an OD600 of 0.3. Cells (2 ml) were infected with T5 phage at an MOI of 0.1 and incubated with shaking (180 rpm) for 0, 20 or 60 min. At each time point, 200 μl of cells were mixed with 800 μl of SM buffer with chloroform (2% final). Samples were serially diluted and 10 μl was spotted onto plates containing bottom agar and soft agar mixed with naive E. coli K-12 MG1655 cells (OD600 of 0.3) and supplements (10 mM MgCl2 and 10 mM CaCl2). Plates were dried at room temperature and incubated overnight at 37 °C. Phage titres (PFU ml−1) were determined the following day and burst size was calculated using the following formula: burst size = (PFU ml−1 at 60 min) – (PFU ml−1 at 20 min)/(PFU ml−1 at 0 min) – (PFU ml−1 at 20 min). The experiment was performed in three biological replicates.
Phage escapers isolation
The T5 phage was serially diluted and 100 µl of each dilution was mixed with 500 µl of E. coli K-12 MG1655 cells expressing RorSlfn5 at an OD600 of 0.3. This mixture was added to 8 ml of soft agar supplemented with ampicillin (100 µg ml−1), 10 mM MgCl2 and 10 mM CaCl2 and overlaid onto LB agar plates containing the same supplements. Plates were incubated overnight at 37 °C. The following day, several individual plaques were picked to inoculate E. coli K-12 MG1655 expressing RorSlfn5 (OD600 of 0.3). The cultures were incubated overnight in LB media supplemented with ampicillin (100 µg ml−1) at 37 °C with shaking at 180 rpm. The next day, cultures were centrifuged at 5,000g for 10 min at 4 °C and the supernatants containing phages were transferred to fresh tubes. The phages were serially diluted and spotted on the lawns of E. coli K-12 MG1655 cells expressing RorSlfn5 or no system to assess their ability to escape the immune system.
Phage genomic DNA isolation and sequencing
Genomic DNA of T5 phage and T5 phage escapers was isolated from 1 ml of phage supernatant using Norgen Biotek Phage DNA Isolation Spin Column kit (SKU 46800). Then 500 ng of each genomic DNA sample was used to prepare a sequencing library as described in the SQK-LSK114 protocol using the Native Barcoding kit 24 V14 (SQK-NBD114.24). The flow cell was primed and the barcoded sequencing library (300 ng) was loaded according to the Oxford Nanopore protocol (SQK-LSK114). Raw sequencing data (POD5 files) were base called in the super-accuracy mode (dna_r10.4.1_e8.2_400bps_sup@v5.0.0) and demultiplexed using Dorado basecaller v0.8.3 (Oxford Nanopore). Base-called reads were aligned to the reference (NCBI: NC_005859.1) using minimap2 (v2.28-r1209) with Nanopore preset (-ax map-ont setting). Resulting alignments (BAM files) were used to call consensus sequences using samtools v1.21 and call sequence variants using bcftools v1.21.
Cell toxicity assay
E. coli K-12 MG1655 cells were co-transformed with plasmids encoding for defence systems and inducible pBAD vectors with viral trigger genes or GFP. Double-transformed cells were selected by plating on LB agar supplemented with ampicillin (100 µg ml−1), kanamycin (100 µg ml−1) and D-glucose (0.2%), followed by overnight incubation at 37 °C. Individual colonies were then used to inoculate overnight cultures in LB medium containing the same antibiotics. The following day, cultures were adjusted to OD600 of 0.6, tenfold serially diluted and 3 µl of each dilution was spotted onto LB agar plates supplemented with ampicillin (100 µg ml−1), kanamycin (100 µg ml−1) and either 0.2% D-glucose or 0.2% L-arabinose. The resulting plates were incubated overnight at 37 °C. All experiments were performed in three biological replicates.
SOS response reporter assay
E. coli K-12 MG1655 cells were co-transformed with RorSlfn5 or RorSlfn5E15A, D20A plasmid, pBAD plasmid with T5.142 or GFP gene and pSOS-GFP reporter plasmid. Triple-transformed cells were grown to an OD600 of 0.3 and 180 µl of these cultures were mixed with 20 µl of D-glucose or L-arabinose to a final concentration of 0.2% in a 96-well plate. Bacterial growth (OD600) and GFP signal were measured every 60 s for 6 h at 37 °C using a SpectraMax M5e reader. Mitomycin C (RPI #M92010) was added to the bacterial culture to a final concentration of 100 nM as a positive control to induce the SOS response. PBS was used as a negative control. All experiments were performed in three biological replicates.
Total RNA extraction
E. coli K-12 MG1655 cells were double transformed with plasmids expressing either RorSlfn5 or RorSlfn5E15A, D20A, along with a plasmid encoding the viral trigger T5.142. Cultures were grown to an OD600 of 0.3 and trigger expression was induced by adding L-arabinose to 0.2% final concentration. Cells were collected at 0, 30 and 60 min post-induction for total RNA extraction using the Direct-zol RNA Miniprep kit (Zymo Research, R2052). Briefly, the cells were spun down at 3,000g for 5 min at 4 °C. The supernatants were removed and the cell pellets were resuspended in 900 µl of TRI-reagent (Sigma-Aldrich). Then, the samples in TRI-reagent were loaded on columns and total RNA purification was performed according to the kit instructions with a DNAse I on-column treatment step. Then ~500 ng of total RNA was used to run a 12% urea–PAGE. The E. coli K-12 MG1655 cell cultures expressing RorSlfn5, RorSlfn5E15A, D20A, PARIS or PARISAriB(E26A) (OD600 of 0.3) were infected with T5 phage at an MOI of 10. Culture samples (5 ml) were taken right before the infection (0 min) and 30 min after for total RNA extraction using the Direct-zol RNA Miniprep kit from Zymo Research (R2052) as described above. The quality of RNA samples was assessed using the Agilent 2100 Bioanalyzer at the University of Florida ICBR Gene Expression and Genotyping Core Facility (RRID: SCR_019145).
RNA-seq
Total RNA samples (~1,200 ng each) were treated with T4 PNK (NEB, M0201) in a reaction buffer (50 mM Tris–HCL, pH 7.5, 10 mM MgCl2, 1 mM DTT, 1 mM ATP and murine RNase inhibitor 1 U µl−1 (NEB)) for 1 h at 37 °C. Next, RNA was purified with the Monarch RNA Cleanup kit (NEB, T2030L). PNK-treated RNA samples were polyadenylated using E. coli Poly(A) polymerase (NEB, M0276S) in a reaction buffer (50 mM Tris–HCl, pH 8.0, 250 mM NaCl, 10 mM MgCl2, 1 mM ATP and murine RNase inhibitor 1 U µl−1) for 30 min at 37 °C. The RNA samples were purified with the Monarch RNA Cleanup kit (NEB, T2030L) and sonicated with a Bioruptor Pico sonication device. For shearing, RNA samples were diluted in TE buffer (10 mM Tris–HCl, pH 8.0, and 1 mM EDTA) to 50 µl and sonicated for 30 cycles, with 30 s of ON time and 30 s of OFF time (15 min total sonication time). RNA was purified with RNAClean XP beads (Beckman Coulter, A63987) using a 2:1 ratio of beads to RNA (v/v) and used to prepare a sequencing library as described in the SQK-PCB114.24 protocol using Barcoding kit CDNA-PCR 24 V14 (Oxford Nanopore). Briefly, the RNA was reverse transcribed and the resulting cDNA was amplified with barcoded PCR primers. Barcoded PCR products were purified using Mag-Bind TotalPure NGS magnetic beads (Omega Bio-tek) and pooled together for sequencing library preparation. The flow cell was primed and the barcoded sequencing library (~10 ng) was loaded according to the Oxford Nanopore protocol (SQK-PCB114.24). Raw sequencing data (POD5 files) were base called using the super-accuracy model (dna_r10.4.1_e8.2_400bps_sup@v5.0.0) and demultiplexed with Dorado basecaller, v0.9.1, with the --no-trim option (Oxford Nanopore). Primer sequences and polyA (sense strand reads) or polyT (anti-sense strand reads) tails were removed using cutadapt v5.0. Bowtie 2 (v2.5.4) was used to align trimmed reads to a reference sequence created by concatenating sequences of the E. coli K-12 MG1655 genome (NCBI: NC_000913.3), the T5 phage genome (NCBI: NC_005859.1) and the pAN248 plasmid (this work). To map RNA ends, start and end coordinates for every read were extracted using bedtools bamtobed (v2.31.1). RNA end coordinates were mapped to genetic features, quantified and normalized using total mapped read counts to produce count per million mapped reads (CPM) values. RNA ends were classified as internal if the RNA end position was >10 nucleotides from the transcript start and transcript end. Read alignments were plotted using Gviz v1.44.2 R package.
Protein expression and purification
For individual protein expression and purification, the vectors pAN261 (6xHisTwinStrepSUMO-RorSlfn5) and pAN302 (6xHisTwinStrepSUMO-T5.142) were transformed into T7 Express lysY cells (NEB, C3010I). Cells were grown in LB broth (Lennox) supplemented with ampicillin (100 µg ml−1) at 37 °C to an OD600 of 0.5–0.7, incubated on ice for 30 min and then induced with 0.5 mM IPTG for overnight expression at 16 °C. Cells were lysed with sonication in lysis buffer (20 mM Tris–HCl pH 8.0, 500 mM NaCl, 1 mM TCEP and protease inhibitor (PI78430, Thermo Fisher)). Lysates were clarified by sequential centrifugation: first at 10,000g for 20 min at 4 °C and then the supernatant was re-centrifuged at 10,000g for an additional 10 min at 4 °C. The affinity-tagged proteins were bound to StrepTrap XT-columns (Cytiva) and eluted with elution buffer (20 mM Tris–HCl pH 8.0, 500 mM NaCl, 1 mM TCEP and 50 mM Biotin). Proteins were concentrated using 10-kDa spin concentrators. Affinity tags were removed by overnight dialysis at 4 °C against SUMO digest buffer (30 mM Tris–HCl pH 8.0, 500 mM NaCl, 1 mM TCEP and 0.15% Igepal) with His-tagged SUMO protease made in house. The affinity tag and protease were removed using a HisTrap HP column (Cytiva) and the flow-through containing untagged RorSlfn5 or T5.142 was concentrated using Corning Spin-X concentrators at 4 °C. Finally, the proteins were purified using a Superdex 200 10/300 size-exclusion column (Cytiva) in a buffer (20 mM Tris–HCl, pH 7.5, 500 mM NaCl and 1 mM TCEP). Fractions containing the target protein were pooled, concentrated, aliquoted, flash-frozen in liquid nitrogen and stored at −80 °C.
For SEC–MALS analysis of RorSlfn5, the protein was first purified as described above; then 100 µl of pooled SEC peak fractions was loaded again on the Superdex 200 10/300 column equilibrated with SEC buffer and passed through a miniDAWN MALS detector and an Optilab refractive index detector (Wyatt Technology). Data collection and analysis were performed using the ASTRA v8.1.2.1 software (Wyatt Technology).
For the purification of the RorSlfn5-T5.142 complex, two co-expression schemes were used. 6xHisTwinStrepSUMO-tagged RorSlfn5E15A,D20A (pAN372) was co-expressed with untagged T5.142 (pAN316), or untagged RorSlfn5 (pAN338) was co-expressed with 6xHisTwinStrepSUMO-tagged T5.142 (pAN302) in T7 Express lysY E. coli. The cells were grown to an OD600 of 0.5 and induced with 0.5 mM IPTG at 16 °C. Cell pellets were lysed with sonication in Lysis buffer (20 mM Tris–HCl, pH 8.0, 500 mM NaCl, 1 mM TCEP and protease inhibitor (PI78430, Thermo Fisher)) and lysates were clarified by centrifugation as described above. The Strep-tagged complex was affinity purified from the lysates using a StrepTrap XT column. For SEC, the affinity tag was removed and the complex was loaded on the Superdex 200 10/300 gel filtration column (Cytiva), as described above.
tRNA synthesis
Bacterial and T5 phage tRNA were synthesized using in vitro transcription (IVT) as previously described33. Briefly, single-stranded DNA oligos encoding tRNA with an upstream T7 promoter sequence were ordered from Eurofins Genomics (Supplementary Table 4) and used as templates for PCR. Each 25 µl PCR reaction contained 1× Q5 Reaction Buffer (NEB), 200 µM dNTPs, 0.5 µM forward primer, 0.5 µM reverse primer, 0.2 U µl−1 Q5 High-Fidelity DNA Polymerase (NEB) and 0.04 µM of DNA oligo template. PCR products were purified using the DNA Clean and Concentrator-5 kit (Zymo Research, D4014). To synthesize tRNA, 0.8–1 μg of PCR product was used as a template for an IVT reaction using the HiScribe T7 High Yield RNA Synthesis kit (NEB, E2040L). The 10 μl reactions were prepared following the manufacturer’s instructions and incubated for 16–18 h at 37 °C. IVT products were treated with DNase I (NEB, M0303L) and then purified using the Monarch RNA Cleanup kit (NEB, T2050S).
tRNA cleavage assay
To test RorSlfn5 nuclease activity in vitro, 100 nM of RorSlfn5 or RorSlfn5E15A, D20A was incubated with 100 nM of tRNA with or without the purified T5.142 trigger in the reaction buffer (50 mM Tris–HCl, pH 7.5, 2 mM MnCl2, 100 mM NaCl and 1 mM DTT). To test other divalent metals, MgCl2, CaCl2, ZnCl2 or NiSO4 were used instead of MnCl2 in the same buffer conditions. All reactions were incubated for 1 h at 37 °C, and 50 mM EDTA was added to stop the reaction. Samples were incubated for 10 min at 70 °C with 2× RNA loading dye before loading into a preheated 15% urea–PAGE. The gel was run at 20 W, stained with SYBR Gold (Invitrogen, S11494) for 30 min and imaged on a Bio-Rad Chemidoc MP Imaging System.
Statistics and reproducibility
Statistical analyses were performed using R version 4.3.0 with functions from the stats package. Experiments that compare three or more groups were analysed using a one-way analysis of variance (ANOVA). Post-hoc pairwise mean comparisons were performed using Tukey’s honestly significant difference (HSD) or Dunnett’s test. Means in experiments with two groups were compared using a two-sided Welch’s t-test. For RNA-seq data, normalized (CPM) and log2-transformed RNA end counts were compared using a two-sided Welch’s t-test with unequal variances and the resulting P values were adjusted using the Holm method or Benjamini–Hochberg correction to calculate the FDR. The statistical test, the level of statistical significance and the sample size (n) for each experiment are provided in the figure legends. Results of all statistical tests and exact P values are provided in the Source Data. Data were plotted using the ggplot2 and pheatmap packages in R.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw sequencing data were deposited in the NCBI Sequence Read Archive and are publicly available under Bioproject accession numbers PRJNA1272589 (phage genome sequencing) and PRJNA1272590 (3′-end RNA sequencing). Source data are provided with this paper.
References
Schwarz, D. A., Katayama, C. D. & Hedrick, S. M. Schlafen, a new family of growth regulatory genes that affect thymocyte development. Immunity 9, 657–668 (1998).
Li, M. et al. DNA damage-induced cell death relies on SLFN11-dependent cleavage of distinct type II tRNAs. Nat. Struct. Mol. Biol. 25, 1047–1058 (2018).
Yang, J.-Y. et al. Structure of Schlafen13 reveals a new class of tRNA/rRNA-targeting RNase engaged in translational control. Nat. Commun. 9, 1165 (2018).
Metzner, F. J. et al. Mechanistic understanding of human SLFN11. Nat. Commun. 13, 5464 (2022).
Li, M. et al. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 491, 125–128 (2012).
Zhang, P. et al. Schlafen 11 triggers innate immune responses through its ribonuclease activity upon detection of single-stranded DNA. Sci. Immunol. 9, eadj5465 (2024).
Jo, U. & Pommier, Y. Structural, molecular, and functional insights into Schlafen proteins. Exp. Mol. Med. 54, 730–738 (2022).
Kim, E. T. & Weitzman, M. D. Schlafens can put viruses to sleep. Viruses 14, 442 (2022).
Nightingale, K. et al. Human cytomegalovirus protein RL1 degrades the antiviral factor SLFN11 via recruitment of the CRL4 E3 ubiquitin ligase complex. Proc. Natl Acad. Sci. USA 119, e2108173119 (2022).
Valdez, F. et al. Schlafen 11 restricts flavivirus replication. J. Virol. 93, e00104–e00119 (2019).
Xie, X. et al. SAMD9L acts as an antiviral factor against HIV-1 and primate lentiviruses by restricting viral and cellular translation. PLoS Biol. 22, e3002696 (2024).
Zhang, F. et al. Human SAMD9 is a poxvirus-activatable anticodon nuclease inhibiting codon-specific protein synthesis. Sci. Adv. 9, eadh8502 (2023).
Podvalnaya, N. et al. piRNA processing by a trimeric Schlafen-domain nuclease. Nature 622, 402–409 (2023).
Bustos, O. et al. Evolution of the Schlafen genes, a gene family associated with embryonic lethality, meiotic drive, immune processes and orthopoxvirus virulence. Gene 447, 1–11 (2009).
Makarova, K. S., Wolf, Y. I., Snir, S. & Koonin, E. V. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J. Bacteriol. 193, 6039–6056 (2011).
Aravind, L., Nicastro, G. G., Iyer, L. M. & Burroughs, A. M. The prokaryotic roots of eukaryotic immune systems. Annu. Rev. Genet. 58, 365–389 (2024).
Burroughs, A. M., Aravind, L. & Stock, A. M. Identification of uncharacterized components of prokaryotic immune systems and their diverse eukaryotic reformulations. J. Bacteriol. 202, e00365–20 (2020).
Kibby, E. M. et al. Bacterial NLR-related proteins protect against phage. Cell 186, 2410–2424 (2023).
Makarova, K. S., Wolf, Y. I. & Koonin, E. V. Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res. 41, 4360–4377 (2013).
Burman, N. et al. A virally encoded tRNA neutralizes the PARIS antiviral defence system. Nature 634, 424–431 (2024).
Rousset, F. et al. Phages and their satellites encode hotspots of antiviral systems. Cell Host Microbe 30, 740–753 (2022).
Fillol-Salom, A. et al. Bacteriophages benefit from mobilizing pathogenicity islands encoding immune systems against competitors. Cell 185, 3248–3262 (2022).
Georjon, H. & Bernheim, A. The highly diverse antiphage defence systems of bacteria. Nat. Rev. Microbiol. 21, 686–700 (2023).
Rousset, F. & Sorek, R. The evolutionary success of regulated cell death in bacterial immunity. Curr. Opin. Microbiol. 74, 102312 (2023).
Paysan-Lafosse, T. et al. The Pfam protein families database: embracing AI/ML. Nucleic Acids Res. 53, D523–D534 (2025).
Schaeffer, R. D. et al. ECOD: integrating classifications of protein domains from experimental and predicted structures. Nucleic Acids Res. 53, D411–D418 (2025).
Zweig, M., Rosenkranz, H. S. & Morgan, C. Development of coliphage T5: ultrastructural and biochemical studies. J. Virol. 9, 526–543 (1972).
Lopatina, A., Tal, N. & Sorek, R. Abortive infection: bacterial suicide as an antiviral immune strategy. Annu. Rev. Virol. 7, 371–384 (2020).
Aframian, N. & Eldar, A. Abortive infection antiphage defense systems: separating mechanism and phenotype. Trends Microbiol. 31, 1003–1012 (2023).
Moineau, S., Durmaz, E., Pandian, S. & Klaenhammer, T. R. Differentiation of two abortive mechanisms by using monoclonal antibodies directed toward lactococcal bacteriophage capsid proteins. Appl. Environ. Microbiol. 59, 208–212 (1993).
Hyman, P. & Abedon, S. T. Bacteriophage host range and bacterial resistance. Adv. Appl. Microbiol. 70, 217–248 (2010).
Zivanovic, Y. et al. Insights into bacteriophage T5 structure from analysis of its morphogenesis genes and protein components. J. Virol. 88, 1162–1174 (2014).
Belukhina, S. et al. Specificity and mechanism of tRNA cleavage by the AriB Toprim nuclease of the PARIS bacterial immune system. Philos. Trans. R Soc. Lond. B Biol. Sci. 380, 20240074 (2025).
Chatterjee, S., Basak, A. J., Nair, A. V., Duraivelan, K. & Samanta, D. Immunoglobulin-fold containing bacterial adhesins: molecular and structural perspectives in host tissue colonization and infection. FEMS Microbiol. Lett. 368, fnaa220 (2021).
David, M., Borasio, G. D. & Kaufmann, G. Bacteriophage T4-induced anticodon-loop nuclease detected in a host strain restrictive to RNA ligase mutants. Proc. Natl Acad. Sci. USA 79, 7097–7101 (1982).
Levitz, R. et al. The optional E. coli prr locus encodes a latent form of phage T4-induced anticodon nuclease. EMBO J. 9, 1383–1389 (1990).
Azam, A. H. et al. Evasion of antiviral bacterial immunity by phage tRNAs. Nat. Commun. 15, 9586 (2024).
Sargen, M. R. & Helaine, S. A prophage competition element protects Salmonella from lysis. Cell Host Microbe 32, 2063–2079 (2024).
Bernheim, A., Cury, J. & Poirier, E. Z. The immune modules conserved across the tree of life: towards a definition of ancestral immunity. PLoS Biol. 22, e3002717 (2024).
Mekhedov, S. L., Makarova, K. S. & Koonin, E. V. The complex domain architecture of SAMD9 family proteins, predicted STAND-like NTPases, suggests new links to inflammation and apoptosis. Biol. Direct 12, 13 (2017).
Peng, S. et al. Structure and function of an effector domain in antiviral factors and tumor suppressors SAMD9 and SAMD9L. Proc. Natl Acad. Sci. USA 119, e2116550119 (2022).
Hou, G. et al. SAMD9 senses cytosolic double-stranded nucleic acids in epithelial and mesenchymal cells to induce antiviral immunity. Nat. Commun. 16, 3756 (2025).
Jahn, M., Vorpahl, C., Hübschmann, T., Harms, H. & Müller, S. Copy number variability of expression plasmids determined by cell sorting and droplet digital PCR. Microb. Cell Fact. 15, 211 (2016).
Shao, B. et al. Single-cell measurement of plasmid copy number and promoter activity. Nat. Commun. 12, 1475 (2021).
van den Berg, D. F., van der Steen, B. A., Costa, A. R. & Brouns, S. J. J. Phage tRNAs evade tRNA-targeting host defenses through anticodon loop mutations. eLife 12, e85183 (2023).
van den Berg, D. F. & Brouns, S. J. J. Phage tRNAs: decoding the enigma. Trends Microbiol. 33, 1121–1131 (2025).
Doron, S. et al. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359, eaar4120 (2018).
Gu, Y. et al. Bacterial Shedu immune nucleases share a common enzymatic core regulated by diverse sensor domains. Mol. Cell 85, 523–536.e6 (2025).
Loeff, L., Walter, A., Rosalen, G. T. & Jinek, M. DNA end sensing and cleavage by the Shedu anti-phage defense system. Cell 188, 721–733 (2025).
Potter, S. C. et al. HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204 (2018).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Letunic, I. & Bork, P. Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82 (2024).
Tesson, F. et al. Systematic and quantitative view of the antiviral arsenal of prokaryotes. Nat. Commun. 13, 2561 (2022).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Medvedev, K. E. et al. Structure classification of the proteins from Salmonella enterica pangenome revealed novel potential pathogenicity islands. Sci. Rep. 14, 12260 (2024).
Steinegger, M. & Soding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Zhang, J., Schaeffer, R. D., Durham, J., Cong, Q. & Grishin, N. V. DPAM: a domain parser for AlphaFold models. Protein Sci. 32, e4548 (2023).
Ofir, G. et al. Antiviral activity of bacterial TIR domains via immune signalling molecules. Nature 600, 116–120 (2021).
Watson, B. N. J. et al. Type I-F CRISPR–Cas resistance against virulent phages results in abortive infection and provides population-level immunity. Nat. Commun. 10, 5526 (2019).
Acknowledgements
We thank B. Wiedenheft and R. Wilkinson for generous discussions and for sharing E. coli strains and bacteriophages; M. Jinek for insightful discussions and valuable advice throughout the course of this work; M. Kladde and M. Gauthier for Bioruptor Pico sonicator access; M. Buyukyoruk for advice on computational analyses; and University of Florida ICBR Gene Expression and Genotyping Core Facility for RNA analysis using the Agilent 2100 Bioanalyzer (RRID: SCR_019145). A. Nemudryi was supported by a start-up package from the University of Florida College of Medicine, University of Florida Office of the Vice President for Research, and University of Florida Emerging Pathogens Institute. This work was supported by the National Institutes of Health grant R00AI171893 (A. Nemudryi) and 1T32GM156737-01 (V.P.T.). L.L. was supported by the LUMC Junior Principal Investigator grant 2025.
Author information
Authors and Affiliations
Contributions
A. Nemudryi and A. Nemudraia conceived, designed and supervised the project. A. Nemudryi performed the initial bioinformatic search and neighbourhood analysis. K.E.M. performed classification of pSlfn domain architectures. A. Nemudryi, A. Nemudraia, R.C. and V.P.T. designed and generated the plasmids. A. Nemudraia, V.P.T. and R.C. performed phage assays. A. Nemudryi performed a bacterial growth assay. A. Nemudraia and A. Nemudryi performed phage escaper isolation and sequencing. A. Nemudryi and V.P.T. performed the cell toxicity assays. A. Nemudryi and A. Nemudraia performed RNA extractions and sequencing experiments. A. Nemudryi, A. Nemudraia, Y.W. and L.L. performed protein expression and purification. V.P.T. performed tRNA synthesis and tRNA cleavage assays. A. Nemudryi and A. Nemudraia wrote the original draft, with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
A. Nemudryi and A. Nemudraia are inventors of patents and patent applications related to CRISPR–Cas systems and applications thereof. The other authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Prokaryotic Schlafen proteins have anti-phage functions.
a, phylogenetic tree of bacterial, archaeal, and mammalian Slfn domains. Tree branches are colored according to the taxonomic group. To improve visualization, the phylogenetic tree was pruned to keep representative sequences identified by CD-HIT from the original dataset. A 50% CD-HIT threshold was used for prokaryotic Slfn domains, while mammalian Slfn domains were pruned with a 70% threshold. Blue bars show the number of phage defense genes in the ±10 kb neighborhood of prokaryotic Slfn genes. b, alignment of the AlphaFold predicted structure of the bacterial Slfn domain (light blue) and the C-lobe of the human SLFN11 nuclease domain. The inset shows the ribonuclease active site with amino acid residues required for the catalytic activity. c, Scatter plot showing results of genetic neighborhood analysis for AriB (known anti-phage gene), pSlfn, L1p, and rpL3 genes. L1p and rpL3 were used as controls with established non-immune functions. Each dot represents the number of known phage defense genes within a 10 kb neighborhood of AriB, pSlfn, L1p, or rpL3 homologs in our database of prokaryotic genomes. The red dot shows the median value. Vertical red lines show interquartile ranges. d, pSlfn proteins were clustered based on domain architectures, annotated using sequence and structural homology methods. Each point represents a pSlfn protein group. Y-axis: number of unique sequences in the group; x-axis: defense score, which was calculated as the ratio of group members that are found within ± 10 kbp distance from at least one known defense gene to the total number of members in the group (see Methods). Clusters with more than ten representatives (horizontal dashed line) and a defense score > 0.5 (vertical dashed line) were further considered as candidate defense systems (red dots). Black circles show domain architectures experimentally tested in Fig. 1. e, A representative phage spot assay showing that EcoDUF262-Slfn (1) and RorSlfn5 (2) protect from phages. f, Efficiency of plating (EOP) of a panel of Escherichia coli phages on MG1655 cells expressing prokaryotic Schlafen proteins. EOP was calculated by dividing plaque-forming units (PFU mL−1) in pSlfn-expressing cells over empty vector control. Data is shown as mean ± SD of three biological replicates. White dots show the average of three technical replicates for each biological replicate. Data were analyzed using one-way Analysis of Variance (ANOVA), and means were compared to the empty vector control using one-sided post-hoc Dunnett’s test. The p values for statistically significant differences are shown in the plot.
Extended Data Fig. 2 T5 phage tail assembly protein triggers pSlfn5-mediated phage defense.
a, T5 phage titer in supernatants of cells with no defense, RorSlfn5, or catalytic mutant of RorSlfn5 (RorSlfn5dead) was quantified at 50 min post infection with T5 phage at MOI = 10. Phage titers were normalized to the mean of the no-defense control. Data is shown as the mean of three biological replicates ± S.D. One-way ANOVA with post hoc Tukey HSD test was used to compare the experimental groups. The resulting p values are shown in the plot. b, Antiviral activities of SfoSlfn5 ortholog against a panel of phages. EOP – efficiency of plating. Data is shown as the mean of three biological replicates ± S.D. White dots show the average of three technical replicates for each biological replicate. c-g, Cellular toxicity assay in E. coli K-12 MG1655 cells co-transformed with a plasmid expressing RorSlfn5 (c), PagSlfn5 (d), or SfoSlfn5 (e) and a plasmid for arabinose-inducible expression of T5.142 homologs (T5.142 – T5 phage, S114 – Salmonella phage S114, DU_PP_V - Pectobacterium phage DU_PP_V, My1 - Pectobacterium phage My1, KKP_3711 - Enterobacter phage KKP_3711, vB_PagS_AAS21 - Pantoea phage vB_PagS_AAS21). Assay was performed in three biological replicates, with one representative replicate shown in the figure. Asterisk (*) indicates toxicity phenotypes. pBAD-GFP was used as a positive control for inducible protein expression. f-g, Cellular toxicity assay in MG1655 transformed only with T5.142 homologs (f) or co-transformed with a plasmid expressing RorSlfn5E15A,D20A and T5.142 homologs (g).
Extended Data Fig. 3 The C-terminus of pSlfn5 contains an Ig-like phage sensor domain.
a, Multiple sequence alignment of pSlfn5 homologs. Red asterisks (*) indicate the conserved catalytic motif of the Slfn ribonuclease domain. b, AlphaFold-predicted structure of RorSlfn5. c, Comparison of AlphaFold-predicted structures of the C-terminal domain of RorSlfn5 and the Ig-like domain of human Complement 5.
Extended Data Fig. 4 Ig-like domain of pSlfn5 defense confers phage trigger specificity.
Cellular toxicity assay in E. coli K-12 MG1655 cells co-transformed with a plasmid expressing RorSlfn5, SfoSlfn5, or chimeric proteins with swapped Ig-like domains and a plasmid for arabinose-inducible expression of T5.142 homologs (T5.142 – T5 phage, S114 – Salmonella phage S114, DU_PP_V - Pectobacterium phage DU_PP_V, My1 - Pectobacterium phage My1, KKP_3711 - Enterobacter phage KKP_3711, vB_PagS_AAS21 - Pantoea phage vB_PagS_AAS21). Asterisk (*) indicates toxicity phenotypes. pBAD-GFP was used as a positive control for inducible protein expression. Toxicity assays were repeated independently three times with similar results, with data shown for one representative replicate.
Extended Data Fig. 5 RorSlfn5 activation does not trigger SOS response.
a, growth kinetics of MG1655 cells co-transformed with RorSlfn5 and inducible T5.142 plasmid after addition of L-arabinose or D-glucose. b, Same for MG1655 co-transformed with RorSlfn5 plasmid and pBAD-GFP. c, SOS response reporter assay for MG1655 cells co-transformed with plasmid encoding inactivated RorSlfn5 defense (E15A, D20A mutation). d, Schematics of different plasmids used in the SOS response and cell toxicity assays.
Extended Data Fig. 6 Phage-activated RorSlfn5 cleaves tRNA anticodon arm.
a, Agilent 2100 Bioanalyzer gel image for total RNA extracted 30 min post T5 phage infection of MG1655 expressing active PARIS defense or its inactive mutant [PARISdead; AriB(E26A) mutation]. 1,2,3 – biological replicates. Red asterisks mark phage-associated transcripts absent in the uninfected (uninf) control. b, quantification of total 3′-end abundance in tRNA from phage-infected cells expressing PARIS defense vs. PARISdead control. Data is shown as the mean of three biological replicates ± S.D. Means were compared using a two-sided Welch’s t-test. **p < 0.01.c, quantification of total 3′-ends counts in specific tRNAs. Data is shown as the mean of three biological replicates. d-e, position-specific mapping of 3′-ends (d) and 5′-ends (e) in tRNALys(UUU) in cells with active (left) or inactive (right) PARIS defense. Data is shown as the mean of three biological replicates. f, alignment of sequencing reads to the lysW gene of MG1655 cells in cells with active (left) or inactive (right) PARIS defense. One representative replicate of three biological replicates is shown. g, Agilent 2100 Bioanalyzer gel image for total RNA extracted 30 min post T5 phage infection of MG1655 expressing active RorSlfn5 defense or its inactive mutant (RorSlfnE15A, D20A). 1,2,3 – biological replicates. Red asterisks mark phage-associated transcripts absent in the uninfected (uninf) control. h, position-specific quantification of internal tRNA 3′-ends in cells expressing active vs. inactive RorSlfn5 defense. FDR – false discovery rate. i-j, position-specific mapping of 3′-ends (i) and 5′-ends (j) in tRNAMet(CAU) in cells with active (left) or inactive (right) RorSlfn5 defense. Data is shown as the mean of three biological replicates. k, alignment of sequencing reads to the metU gene of MG1655 cells in cells with active (left) or inactive (right) RorSlfn5 defense. One representative replicate of three biological replicates is shown. l, position of PARIS nuclease-dependent RNA ends in tRNALys(UUU). The red triangle shows the position of mapped internal 5′-ends; the gray triangle shows the position of mapped internal 3′-ends. m, same for RorSlfn5 nuclease-dependent RNA ends in tRNAMet(CAU).
Extended Data Fig. 7 RorSlfn5 and T5.142 form a stable complex.
Size-exclusion chromatography (SEC) profiles of RorSlfn5 (a) and T5.142 (b) proteins expressed and purified separately. All SEC runs were performed on a Superdex 200 10/300 GL column. Vertical blue rectangles highlight elution fractions used for SDS-PAGE on the right. c, SEC with Multi-Angle Static Light Scattering (SEC-MALS) analysis of purified RorSlfn5 protein. d, Reciprocal affinity co-purification of RorSlfn5dead and T5.142. RorSlfn5dead (left) or T5.142 (right) protein was tagged with a TwinStrep-6xHis-SUMO tag at the N-terminus (black circle). Proteins were co-expressed, and lysates were applied to the Strep-Tactin XT column. Eluted proteins were visualized with SDS-PAGE followed by protein staining. e, Same as in (a) and (b), but RorSlfn5dead and T5.142 that were co-expressed and co-purified. Experiments shown in (a), (b), (d), and (e) were reproduced independently three or more times with similar results.
Extended Data Fig. 8 T5.142 triggers RorSlfn5-mediated tRNA cleavage.
a, tRNA cleavage assay with 100 nM tRNALys(UUU) and 100 nM RorSlfn5 in the presence of trigger T5.142 (2 µM) and 2 mM divalent metals or 2 mM EDTA. RNA fragments were resolved with 15% Urea-PAGE. b, tRNA-Lys(UUU) cleavage assay with RorSlfn5 or RorSlfn5E15A, D20A (“dead”) in the presence of trigger T5.142 and 2 mM Mn2+. Concentrations of the nuclease, tRNA, and T5.142 are same as in (a). c, tRNA-Lys(UUU) (100 nM) cleavage assay with RorSlfn5 (100 nM) and varying concentrations of T5.142. Reactions were supplemented with 2 mM Mn2+. d, Cleavage assay with varying synthetic tRNAs (100 nM) with 100 nM RorSlfn5 and 2 µM T5.142 in reaction buffer supplemented with 2 mM Mn2+. All shown cleavage assays were reproduced independently three times with similar results.
Supplementary information
Supplementary Table 1 (download XLSX )
Taxonomy and gene neighbourhood analysis of prokaryotic Schlafen proteins.
Supplementary Table 2 (download XLSX )
Domain annotation of prokaryotic Schlafen proteins.
Supplementary Table 3 (download XLSX )
Candidate Schlafen systems tested in the study.
Supplementary Table 4 (download XLSX )
Phages, plasmids, bacterial strains and oligonucleotides used in the study.
Supplementary Table 5 (download XLSX )
Nucleotide variant data in T5 escaper phages.
Supplementary Table 6 (download XLSX )
Foldseek analysis of Ig-like domain in RorSlfn5.
Supplementary Table 7 (download XLSX )
RNA 3′-end counts.
Source data
Source Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Fig. 2 (download PDF )
Uncropped plate images.
Source Data Fig. 3 (download XLSX )
Tree data and statistical source data.
Source Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Fig. 4 (download PDF )
Uncropped gel image.
Source Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Fig. 5 (download PDF )
Uncropped gel images.
Source Data Extended Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 4 (download PDF )
Uncropped plate images.
Source Data Extended Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 6 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 6 (download PDF )
Bioanalyser data.
Source Data Extended Data Fig. 7 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 7 (download PDF )
Uncropped gel images.
Source Data Extended Data Fig. 8 (download PDF )
Uncropped gel images.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Perez Taboada, V., Wu, Y., Cassidy, R. et al. Bacterial Schlafen proteins mediate phage defence. Nat Microbiol (2026). https://doi.org/10.1038/s41564-026-02277-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41564-026-02277-8
