
Abstract
Homarine (N-methylpicolinic acid) is a ubiquitous marine metabolite produced by phytoplankton and noted for its infochemical properties among marine animals, yet its microbial degradation pathways are uncharacterized. Here we identify a conserved operon (homABCDER) that mediates homarine catabolism in bacteria using comparative transcriptomics, mutagenesis and targeted knockouts. Phylogenetic and genomic analyses show that this operon is distributed across abundant bacterial clades, including coastal copiotrophs (for example, Rhodobacterales) and open-ocean oligotrophs (for example, SAR11, SAR116), and in genomes from non-marine environments. High-resolution mass spectrometry revealed N-methylglutamic acid and glutamic acid as key metabolic products of homarine in both model and natural systems, with N-methyl-glutamate dehydrogenase catalysing their conversion. Metatranscriptomics showed responsive and in situ expression of hom genes aligned with homarine availability. These findings uncover the genetic and metabolic basis of homarine degradation, establish its ecological relevance and highlight homarine as a versatile growth substrate that feeds into central metabolism via glutamic acid in diverse marine bacteria.
Similar content being viewed by others


Main
Microbial metabolites are central to ocean biogeochemistry, facilitating rapid cycling of labile organic carbon through the microbial loop and driving the annual turnover of tens of petagrams of carbon1. Many metabolites also have compound-specific functions such as enabling micronutrient acquisition2, serving as chemoattractants3 or acting as toxins4. Advances in environmental metabolomics have uncovered abundant metabolites whose biogeochemical and ecological roles are unknown, partly because their metabolic pathways are unknown5,6,7. These unknown pathways hinder genomics, transcriptomics and proteomics approaches for studying microbial function. Identifying the genes and transformations linked to metabolites enhances our understanding of microbial metabolism and better integrates metabolites into a collective biological, ecological and biogeochemical framework.
Among marine metabolites, homarine (N-methylpicolinic acid) stands out for both its widespread distribution and multifaceted bioactivity. Homarine, a polar alkaloid with a methylated pyridine ring, was first discovered in lobsters nearly a century ago8 and is now recognized as ubiquitous in surface seawater6,7, with turnover times ranging from days to weeks9. While heterotrophic bacteria are assumed to consume homarine, the metabolic pathway, molecular intermediates and ecological distribution of its degradation remain uncharacterized. Homarine is produced by abundant phytoplankton groups such as diatoms and Synechococcus at millimolar intracellular concentrations, while other groups such as dinoflagellates and prasinophytes have not been observed to produce it6. Homarine’s pyridine ring suggests specialized enzymatic machinery for its degradation, as seen with other pyridine-containing compounds10. This, along with its production patterns, may contribute to niche partitioning among phytoplankton and heterotrophs. Homarine also exhibits bioactivity in marine animals, influencing oyster shell morphology11, deterring feeding in butterfly fish and sea stars12, altering larval development in hydroids13 and serving as a crab predation deterrent14.
This study investigates the heterotrophic bacterial degradation of homarine across biochemical, genetic and ecological dimensions through an integrative approach. Comparative transcriptomics identifies a conserved operon for homarine degradation, while mass spectrometry reveals its biochemical transformation into metabolic products. Comparative genomics maps the global distribution of homologous operons, and metatranscriptomic analyses in natural and amended communities link gene expression to homarine availability, emphasizing its ecological relevance.
Results
Identification of homarine operon
Motivated by homarine’s abundance in marine environments, we sought to understand its bacterial degradation. We isolated a bacterium from seawater at Owen Beach, Tacoma, Washington, USA, and demonstrated its ability to use homarine as a primary carbon and nitrogen source (Fig. 1a). Genome sequencing identified this Gammaproteobacterium as Cobetia sp. within the order Oceanospirillales, family Halomonadaceae, with high similarity to Cobetia marina (98.51% average nucleotide identity (ANI)) and Cobetia pacifica (98.3% ANI, Supplementary Table 1)15. We named this isolate Cobetia sp. OBi1.

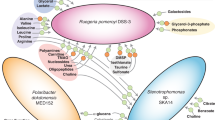
a, Cobetia sp. OBi1 growth on glucose (12 mM C), glucose (12 mM C) with homarine (1 mM C) or homarine (12 mM C) for comparative transcriptomics (n = 3). Data are presented as mean ± s.d. of 2 different cultures. Grey vertical lines indicate RNA sampling times (13 h for cultures with glucose and cultures with glucose+homarine and 16.5 h for cultures with homarine). b, Differential gene expression fold change (FC) of Cobetia sp. OBi1 in homarine vs glucose. c, Differential gene expression of Cobetia sp. OBi1 in glucose+homarine vs glucose. P values were adjusted for multiple comparisons using Benjamini–Hochberg (BH) correction (two-sided Wald test). Black vertical lines indicate significantly upregulated genes; positive values represent upregulation in the homarine condition. d, Cobetia sp. OBi1 genes (arrows) upregulated in homarine relative to glucose and homologous gene clusters in R. pomeroyi DSS-3. Arrow fill colour corresponds to the type of function performed by each gene. Arrow outline colour indicates the results of growth tests with in-frame deletion mutants of Cobetia sp. OBi1 or transposon insertion KanR mutants of R. pomeroyi DSS-3. GBT, glycine betaine transporter. Grey stars (*) indicate genes upregulated in glucose+homarine compared to glucose alone; black stars (*) indicate genes upregulated in homarine compared to glucose. Full descriptions of genes found in Supplementary Table 3. e, Proposed homarine catabolic pathway inferred from omics data.
To uncover genes involved in homarine degradation, we grew Cobetia sp. OBi1 in minimal media with homarine as the sole carbon source and conducted comparative transcriptomics and metabolomics experiments. We compared three conditions: glucose (12 mM C, 0.8 mM NH4, control), homarine (12 mM C, no additional NH4), and glucose+homarine (12 m C and 1 mM C, respectively, with 0.8 mM NH4). In both conditions with homarine, 22 genes were enriched compared to the glucose-only condition (s-value < 0.05; Fig. 1b,c and Supplementary Table 2), including three gene clusters. The first cluster encoded a transcriptional regulator, a predicted glycine betaine transporter (GBT) and five putative catabolic genes with unclear function (ACFLL8_11490–ACFLL8_11520; Fig. 1d and Supplementary Table 2). The second cluster contains a putative sarcosine oxidase complex and the methylenetetrahydrofolate dehydrogenase folD (ACFLL8_07290–ACFLL8_07310; Fig. 1d and Supplementary Table 2). The third cluster included motility-associated genes fliG and flgD (Fig. 1d and Supplementary Table 2). In the homarine-only treatment, we observed broader upregulation of the flagellar and chemotaxis genes, including fliM, fliN, motB, aer, cheR and cheY16 (Fig. 1d and Supplementary Table 2). These gene enrichments suggest that homarine may induce motility and chemotaxis.
To target functional assessments, we identified orthologues of the homarine-induced genes from Cobetia sp. OBi1 in the model marine heterotrophic bacteria Ruegeria pomeroyi DSS-3 using reciprocal best-hit searches (e-value < 10−5). Of the seven genes in Cobetia sp. OBi1’s homarine-induced operon with unknown function (ACFLL8_11490–ACFLL8_11520), five were found in a single operon in R. pomeroyi DSS-3 (SPO3188–SPO3192; Fig. 1d and Supplementary Table 3). Orthologues of ACFLL8_11490 (the first catabolic gene) and ACFLL8_11505 (the transporter) were absent. R. pomeroyi DSS-3 was capable of growth on homarine as the sole carbon and nitrogen source (Extended Data Fig. 1a). These results support a homarine catabolic operon, here termed homABCDER (Fig. 1d and Supplementary Table 3), consisting of four core catabolic genes (homABCD), a transcriptional regulator (homR) and variably present auxiliary gene (homE).
To investigate the functions and structures of the proteins encoded by the homABCDER operon, we combined domain annotation17,18, structural modelling19,20 and functional prediction21. In both Cobetia sp. OBi1 and R. pomeroyi DSS-3, HomA contains conserved motifs for a flavin adenine dinucleotide (FAD) binding domain (Supplementary Table 3). Functional characterization of HomB suggests that it binds cyclic compounds and exhibits catalytic activity (confidence score of the predicted Gene Ontology terms, CscoreGo > 0.65; Supplementary Table 4). AlphaFold modelling yielded high-confidence structures for HomA and HomB (interface predicted template modelling + predicted template modelling: 0.9471 for R. pomeroyi DSS-3; 0.9246 for Cobetia sp. OBi1). Visualization in ChimeraX22 revealed hydrogen bonds and electrostatic interactions consistent with heterodimer formation. This resembles the trigonelline catabolic enzymes TgnA and TgnB, which operate as a flavin-supplying and oxygen-activating pair to open the pyridine ring of trigonelline23, a structural isomer of homarine. We hypothesize a similar functional relationship for HomA and HomB. HomC is predicted to function as a peptidase (CscoreGO > 0.65; Supplementary Table 4), while HomD is probably an aldehyde dehydrogenase-like oxidoreductase (CscoreGO > 0.65; Supplementary Tables 3 and 4). These findings are consistent with a model in which the operon encodes enzymes involved in distinct but coordinated steps of homarine degradation.
Phenotypic analysis of homarine-associated genes
We phenotypically characterized the proposed homarine catabolic operon using recombinant techniques and a transposon barcoded mutant library of our model organisms. In Cobetia sp. OBi1, homologous recombination was used to generate a ΔhomD mutant (ACFLL8_11515), which retained normal growth on glucose but was unable to grow with homarine as a sole source of carbon and nitrogen (Extended Data Fig. 1b). Complementation with the wild-type homD gene restored growth on homarine, confirming its role in homarine catabolism (Extended Data Fig. 1b). Similarly, mutants from a Tn5 transposon barcoded mutant library in R. pomeroyi DSS-3 (ref. 24) were used to assess gene phenotypes (Extended Data Fig. 1a and Supplementary Tables 3 and 5). Knockouts of three catabolic genes (homB, SPO3189; homC, SPO3190; homD, SPO3191) and the transcriptional regulator (homR, SPO3192) resulted in the loss of growth on homarine at 4 mM C (Extended Data Fig. 1b for homD; Supplementary Tables 3 and 5 for others). These results confirm the role of the homarine operon (homABCDER) in the degradation of homarine in our model organisms.
A knockout strain targeting the transporter gene (ACFLL8_11505) within the hom operon of Cobetia sp. OBi1 (ΔhomT) exhibited reduced growth on homarine compared to the wild-type strain (Extended Data Fig. 2a). Plasmid-based complementation with homT restored growth relative to a control expressing red fluorescent protein (RFP) (Extended Data Fig. 2). Given its location in the hom operon, its upregulation in response to homarine and the observed growth effects, this gene probably encodes a homarine transporter, which we designate as HomT. However, ΔhomT mutants eventually reached wild-type yields, suggesting the presence of functionally redundant transport proteins in the cell that can also facilitate homarine uptake. Such redundancy is common for betaines in marine bacteria25 and competitive inhibition of homarine uptake by other betaines and sulfoniums in microbial communities supports this9. In R. pomeroyi DSS-3, the absence of a mutant strain precluded testing of the function of any transporter near the hom operon (SPO3186). Further characterization of homarine transporters was not pursued.
Identification of homarine degradation products using mass spectrometry
To investigate the catabolism of homarine, we analysed the metabolomes of Cobetia sp. OBi1 grown under the glucose, homarine and glucose+homarine conditions. Using a comparative approach, we identified intracellular and extracellular metabolites that were enriched in homarine treatments (Fig. 2a and Extended Data Fig. 3). Enrichment was determined relative to previously identified ‘core’ metabolites, or compounds that remain relatively constant in abundance across diverse marine organisms and environmental regimes, serving as a baseline for comparison. Homarine, glutamic acid and several unidentified compounds were enriched (Fig. 2a, Extended Data Fig. 3 and Supplementary Tables 6 and 7). Two compounds were subsequently identified: N-methylglutamic acid (via comparison to standard) and N-methylglutamine (via MS1/MS2 spectra, Extended Data Figs. 4 and 5). To explore conserved degradation products of homarine, we analysed the metabolomes of R. pomeroyi DSS-3 grown under similar glucose and homarine conditions. We found the enrichment of N-methylglutamic acid, N-methylglutamine and compounds with molecular formulas of C6H9NO3, C6H9NO4 and C7H9NO5, when grown with homarine, consistent with Cobetia sp. OBi1 (Fig. 2a–c and Supplementary Tables 8 and 9).

a, Enrichment factors of select metabolites in Cobetia sp. OBi1 and R. pomeroyi DSS-3 during growth on homarine and on glucose+homarine as compared to glucose-only growth (glucose-only treatment serves as the denominator for enrichment calculations and is not shown independently). ‘Core metabs’ show the maximum (max), median (med) and minimum (min) calculated enrichment factor for the subset of metabolites used as normalizing factor for enrichment calculations for comparison. Molecular formulas are reported for identified compounds. Compounds without identification are described with the putative molecular formula, ionization mode (− or +) and retention time (in minutes), assuming [M + H]+ or [M−H]− forms. Asterisks designate enrichments found to be significant (FDR-adjusted p < 0.05). b, Extracted ion chromatograms of N-methylglutamic acid in Cobetia sp. OBi1 and R. pomeroyi DSS-3 during growth on homarine and glucose. c, Same as b but for N-methylglutamine. d, Map of stable isotope experiments. e, Structure of homarine, N-methylglutamic acid and N-methylglutamine, with atoms subjected to stable isotope probing highlighted. f, Enrichment factors of select metabolites in stable isotope experiment for TN397 where 2H3-labelled homarine was added (violet Hs in e). The selected metabolites were all detected with 2H3 stable isotope. Core metabolites, scale and labels are the same as in a. g, Enrichment factors of select metabolites in stable isotope experiment for RC104 and TN412 where 13C7,15N-homarine was added (blue C and orange N in e). The selected metabolites were all detected with all Cs as 13C and 1 N as 15N isotope. Core metabolites, scale and labels are the same as in a. Detailed data in Supplementary Tables 6, 8 and 9.
Stable-isotope probing was performed to track homarine degradation products in natural marine microbial communities from five locations across three research cruises (cruises TN412, RC104 and TN397; Fig. 2d). Seawater incubated with isotopically labelled homarine (2H3-homarine for TN412 experiments; 13C7,15N-homarine for RC104 and TN412 experiments; Fig. 2e) was analysed for the compounds enriched in our model organisms, with the isotopic labels. In the TN397 experiments, 3H3-labelled N-methylglutamic acid and N-methylglutamine were enriched, alongside the previously observed unidentified compounds with formulas of C6H9NO3, C6H9NO4 and C7H9NO5 (Fig. 2f and Supplementary Table 9). Consistent with the removal of a 2H3-methyl group from N-methylglutamic acid, we did not find 2H3-glutamic acid (Supplementary Table 9). In the RC104 and TN412 experiments, we identified enrichment of 13C5–7,15N1-N-methylglutamic acid, glutamic acid, N-methylglutamine, and C6H9NO3, C6H9NO4 and C7H9NO5 features, confirming them as direct degradation products of homarine (Fig. 2g and Supplementary Table 9).
The consistency in the enriched metabolites in R. pomeroyi DSS-3, Cobetia sp. OBi1 and natural microbial communities support a conserved degradation of homarine to N-methylglutamic acid and subsequently to glutamic acid. Complete isotopic labelling of N-methylglutamic acid and glutamic acid confirms their origins from homarine, while the isotopic labelling of 2H3-N-methylglutamic acid highlights that homarine’s methyl group is preserved through ring opening, transferred into N-methylglutamic acid and subsequently removed to form glutamic acid. Untargeted analysis of the 2H3-homarine experiments found no evidence for 2H3-containing compounds beyond those displayed in Fig. 2f (Supplementary Table 10), confirming that homarine’s pyridine ring is opened before demethylation. While earlier studies imply that homarine functions as a methyl donor to form picolinic acid26, in these stable isotope experiments we only observed the methylated pyridine ring opening intact.
Role of N-methyl-glutamate dehydrogenase in homarine degradation
The discovery of N-methylglutamic acid as a product of homarine degradation prompted us to reexamine upregulated genes in Cobetia sp. OBi1 for involvement with this N-methylglutamic acid. Two gene clusters originally annotated as coding for the heterotetrameric sarcosine oxidase (TSOX) were identified: ACFLL8_02220–02235 and ACFLL8_07290–07305, with the latter showing increased transcription during growth on homarine (Fig. 1d and Supplementary Table 2). Genes encoding TSOX (soxBDAG) and N-methyl-glutamate dehydrogenase (NMGDH; mgdABCD) often share high sequence similarity, leading to misannotation by automated pipelines27,28. Both enzymes act on a methylated secondary amine, with similar CH3NHCH(R)COOH backbones (R = H in sarcosine, R = propanoic acid in N-methylglutamic acid). Because of this similarity, we compared the Cobetia sp. OBi1 gene sets to reference sox and mgd clusters from organisms with experimentally verified activities. Phylogenetic analysis of each subunit revealed that sox-like genes in Cobetia sp. OBi1 and R. pomeroyi DSS-3 cluster with mgd genes from bacteria with demonstrated NMGDH activity rather than with genes that encode TSOX (Supplementary Figs. 1–4). Furthermore, growth of mgdA::Tn5 and mgdD::Tn5 mutants of R. pomeroyi DSS-3 was significantly reduced when grown on homarine or N-methylglutamic acid compared to the wild type (Extended Data Fig. 1a and Supplementary Table 5). Together, these results suggest that the sox-like genes upregulated in Cobetia sp. OBi1 correspond to the mgd gene cluster.
Previously, the only known route for the formation of N-methylglutamic acid was as an intermediate in glutamate-assisted methylamine utilization, where a methyl group is transferred from methylamines onto glutamic acid, followed by NMGDH-catalysed demethylation to glutamic acid and formaldehyde or 5,10-methylene-H4folate28. Our field experiments revealed accumulation of 13C5,15N-glutamic acid and 13C6,15N-N-methyglutamic acid from 13C7,15N-homarine (Fig. 2g), demonstrating a new pathway for N-methylglutamic acid formation during the conversion from homarine to glutamic acid as displayed in Fig. 1c. In 2H3-homarine incubations, a few 2H2-labelled metabolites were detected (Supplementary Table 10), consistent with methyl group transfer into the folate cycle. Combined with mgd upregulation in Cobetia sp. OBi1, these results support the role of NMGDH in catalysing the conversion of homarine-derived N-methylglutamic acid to glutamic acid natural communities (Fig. 1e).
Homarine-degrading bacteria with conserved homarine catabolic genes
To evaluate the distribution of the genetic mechanisms for homarine catabolism, we isolated 26 bacteria strains from coastal and pelagic environments using homarine-enriched agar plates and sequenced their genomes. After sequencing, we found that many isolates were highly similar to one another (>99% ANI), so we selected one representative from each group, resulting in 16 isolates for further analysis. These were then screened for axenic growth in oligotrophic seawater supplemented with homarine and phosphate; 9 of these were capable of growing on homarine as a sole carbon and nitrogen source, all of which were Alphaproteobacteria or Gammaproteobacteria. All homarine-utilizing isolates encoded homABCD, homR and sometimes homE in a conserved operon (Fig. 3a and Supplementary Table 11). An mgd operon was identified in 8 of 9 genomes with the hom operon, and all mgd-positive isolates also carried soxGADB (Fig. 3a, Supplementary Table 11 and Figs. 1–4). Notably, Vibrio isolate PS01 contained the hom operon but lacked mgdABCD, yet still grew on homarine, suggesting genome incompleteness or an alternative pathway for processing N-methylglutamic acid. One isolate, Nitratireductor sp. G4i25, encoded mgdABCD but lacked the hom operon and could not grow on homarine, reinforcing the need of the homABCD operon for homarine catabolism (Fig. 3a and Supplementary Table 11).

a, Isolates tested in this study, with ‘+’ showing growth on homarine as primary source of carbon and nitrogen, from 3 independent replicates, based on optical density (OD600); checkmarks note the presence of hom genes, mgd genes and sox genes (more details in Supplementary Figs. 1–4). The class and family of each isolate are colour and symbol coded. b, Sunburst plot highlighting taxonomy of genomes with identified hom operon in NCBI genomes. c, Geographic distribution and environment type of NCBI genomes from environmental samples (circles), with stars showing location of additional marine isolates from a (see Supplementary Tables 11 and 12 for details).
Widespread distribution and conservation of hom operon
To explore the taxonomic and geographic distribution of bacteria capable of homarine degradation, we searched complete genomes in the NCBI database for operons encoding the homarine catabolic genes (homABCD and optionally homE). Due to sequence similarity between sox and mgd genes, we did not attempt the same search for the mgd operon27 (Supplementary Figs. 1–4). Across 11,755 genomes containing the hom operon, we identified primarily Alphaproteobacteria (3,148 genomes, 26.8%) and Gammaproteobacteria (8,094 genomes, 68.9%) lineages (Fig. 3b and Supplementary Table 12). Operon organization was highly conserved; homA and homB were adjacent in 97.7% of genomes, while homC and homD were typically nearby (Extended Data Fig. 6a and Supplementary Table 12). In contrast, the presence and positioning of homE was variable (Extended Data Fig. 6a and Supplementary Table 12). Approximately one-third of the genomes containing homABCD do not include homE. In genomes with homE, it is positioned upstream of homA in 49.9% (such as Cobetia sp. OBi1) and downstream of homD in 50.1% (Extended Data Fig. 6a and Supplementary Table 12). This variability suggests that homE functions as an auxiliary module in the hom operon but is not a core component. Gammaproteobacteria retained homE in 93% of the genomes containing the hom operon (Supplementary Table 12), suggesting that it may extend or enhance catabolic potential. By contrast, 94% of the Alphaproteobacteria genomes lacked homE, including all 120 genomes of the highly abundant Pelagibacter clade (Extended Data Fig. 6a and Supplementary Table 12). Bacteria with homABCD without homE had smaller genomes overall (t-test, p = 2.2 × 10−16, n = 11,755; Extended Data Fig. 6b), consistent with broader patterns of gene loss in streamlined lineages. These observations support a model where homE serves a taxon-specific auxiliary role for homarine metabolism.
We identified bacteria encoding the hom operon from a range of settings, including soil, freshwater, marine, sediments, food (for example, cheese, oysters) and human clinical samples (for example, urine, faecal; Fig. 3c and Supplementary Table 12). Although gene presence does not guarantee functionality, detecting the operon in non-aquatic environments suggests that homarine may play roles in microbial metabolism beyond marine systems. Among genomes encoding the operon were members of ecologically important marine heterotrophic clades, including SAR11 (Pelagibacterales)29, SAR116 (Puniceispirillales)30, SAR92 (Cellvibrionales)31 and Rhodobacterales including members of the Roseobacter clade32 (Fig. 3c and Supplementary Table 12). Notably, SAR11 accounts for ~25% of microbial cells in the ocean29, SAR92 represents up to 10% of cells in nearshore waters31, and Roseobacters comprise up to 20% of bacterial cells in coastal ecosystems33. SAR11’s streamlined genome retains the hom operon, underscoring its importance to Pelagibacterales and suggesting a key role for homarine in their ecological success. The widespread occurrence of the hom operon across diverse regimes and its presence in dominant marine microbes highlights homarine’s broad ecological relevance and its pervasive role in microbial metabolism.
Expression of hom catabolic genes in marine microbes
To determine whether the genomic potential of homarine catabolism translates to metabolic activity in situ, we evaluated gene expression under experimental and environmental conditions. In homarine-amended incubations in Puget Sound, significant transcriptional enrichment was detected for 13 bacterial genes relative to unamended controls (Supplementary Table 13). Transcripts of homC and homD were enriched in members of the Porticoccaceae (Gammaproteobacteria), and homB was enriched in Oceanospirillaceae (Alphaproteobacteria) (Supplementary Table 13). These taxa were identified in our genome search of the genetic capacity for homarine catabolism (Fig. 3b and Supplementary Table 12). In addition, mgdA, the gene encoding the α-subunit of NMGDH was upregulated in Porticoccaceae (Supplementary Table 13), reinforcing the role of NMGDH in converting homarine-derived N-methylglutamic acid to glutamic acid and linking genomic capacity to catabolic activity in natural microbial assemblages.
We examined the in situ expression of the hom genes using quantitative metatranscriptomics along a surface ocean transect from the subtropical gyre into the North Pacific Transition Zone (25.87° N to 40.88° N on 158° W). Spatially structured expression of homB and homA was observed, with elevated transcript abundance north of 35° N (Fig. 4 and Extended Data Fig. 7, respectively). Differences in expression between homA (~700 nt) and homB (~1,700 nt) probably stem from length-dependent biases in messenger (m)RNA recovery, with shorter transcripts being less efficiently captured during metatranscriptomic library preparation34. Expression of homB was primarily attributed to Gammaproteobacteria (for example, Vibrionales, Porticoccaceae) and Alphaproteobacteria (for example, Pelagibacter, SAR116, Rhodobacterales; Fig. 4 and Supplementary Table 14). Across stations, homarine concentrations (sum of dissolved and particulate measurements) were significantly correlated with hom gene transcript abundance (R2 = 0.518, p = 0.0052, Spearman’s correlation), supporting coupling between substrate availability and gene expression. Normalizing hom gene transcripts by cell abundance using 10 core housekeeping genes as a proxy35 supported increased per-cell expression at higher substrate concentrations (Extended Data Fig. 8). Taxa expressing hom genes in situ overlapped with those identified in our perturbation experiments and predicted to encode homarine catabolic pathways from genome-based analyses (Supplementary Tables 12–14). These taxa spanned both copiotrophic and oligotrophic lineages, such as Porticoccaceae, Vibrionales, Pelagibacteraceae, SAR116, Oceanospirillaceae and Rhodobacterales (Supplementary Table 14).

Discussion
This study defines a core microbial pathway for the degradation of homarine, a ubiquitous yet previously enigmatic metabolite in marine systems6,7. Using comparative transcriptomics, transposon mutagenesis and targeted gene deletions, we identified a conserved operon (homABCDER) that mediates homarine catabolism in two model organisms, a newly isolated Gammaproteobacteria Cobetia sp. OBi1 and the genetically tractable model marine Alphaproteobacteria R. pomeroyi DSS-3. Metabolomic analyses in both model organisms and natural communities reveal that homarine degradation produces N-methylglutamic acid, which is subsequently converted into glutamic acid by N-methyl-glutamate dehydrogenase (NMGDH). This refines the understanding of NMGDH, a gene that is frequently misannotated27,28, and expands its ecological importance in marine microbial metabolism. Glutamic acid can be converted into alpha-keto glutarate for entry into the Krebs cycle; plays a central role in nitrogen assimilation via glutamine oxoglutarate aminotransferase (GOGAT); contributes to the biosynthesis of metabolites, peptides and proteins; and functions as an osmolyte. This positions homarine as a versatile growth substrate for bacteria equipped with genes to convert homarine into glutamic acid. The hom operon is broadly distributed across bacterial genomes from diverse environments, including coastal and open-ocean systems, as well as non-aquatic sources. Within bacteria with demonstrated homarine-degradation capabilities, there is a frequent co-occurrence of the hom operon with the mgd gene cluster, which encodes NMGDH and facilitates the conversion of N-methylglutamic acid to glutamic acid. This genomic association underscores the interdependence of the hom and mgd pathways, highlighting their integrated role in nutrient cycling. Genomic mining further revealed that the hom operon is conserved in globally dominant bacterial clades that are key drivers of carbon and nitrogen cycling in marine ecosystems, including SAR11 (ref. 36), SAR116 (ref. 30) and Rhodobacterales32. Furthermore, variability in the auxiliary homE gene reveals operon modularity and its adaptive tailoring to ecological or genomic constraints.
Through perturbation experiments and in situ analyses of gene expression, we show the coupling of homarine availability to homB gene expression in natural marine environments. The responsiveness of these genes to substrate availability suggests that homarine degradation actively shapes microbial community structure, gene expression and metabolic dynamics in situ. Co-expression of chemotaxis and motility genes alongside the hom operon in cultures exposed to homarine indicates that bacteria not only sense and metabolize homarine but may actively seek it out in the environment, potentially leading to organismal interactions. The detection of transcripts from both fast-growing coastal taxa and genome-streamlined open-ocean bacteria suggests that homarine catabolism operates across diverse ecological strategies and niches. These findings highlight homarine degradation as a responsive and ecologically widespread metabolic capacity, with gene expression linked to substrate availability in marine environments.
This study provides a foundational framework to link metabolic gene function to ecological activity, bridging molecular mechanisms with environmental processes. By elucidating the genetic basis, biochemical transformations and ecological distribution of homarine catabolism, we expand our understanding of how microbial communities integrate metabolite turnover into broader oceanic biogeochemical cycles. These findings open opportunities for further exploration into how homarine interacts with co-occurring metabolites, impacts organismal interactions and supports ecosystem functioning in the ocean and beyond.
Methods
Isolation, growth and sequencing of Cobetia sp. OBi1
Owen Beach isolate 1 (Cobetia sp. OBi1) was obtained by enrichment culturing of seawater collected in Owen Beach (Tacoma, Washington). Seawater was collected in sterile polycarbonate bottles in February 2020. We amended 100 ml seawater samples with 12 mM C homarine and 0.1 mM sodium phosphate and incubated in aerated flasks at 22 °C. Culture optical density (OD600) was measured daily to monitor growth. After 3 days, 50 μl samples were streaked on agar plates made with marine broth (Difco 2216) to isolate single colonies. Single colonies were picked and restreaked twice in the same agar media. The isolated colonies were tested for growth on homarine in seawater from Owen Beach sterilized by filtering through a 0.22 μm PES membrane and autoclaving. We supplied the medium with homarine and phosphate as before. Cobetia sp. OBi1 colonies regrew on homarine, and the isolate was selected for further analysis.
We grew Cobetia sp. OBi1 in seawater medium containing homarine and phosphate in aerated flasks at 25 °C to obtain DNA for genome sequencing. Genomic DNA was purified with the NEB Monarch tissue lysis kit using a modified lysis solution containing lysozyme, Proteinase K and RNase A. For whole-genome sequencing, we generated a set of Illumina paired-end reads with a depth of 400 Mbp on a NextSeq 2000 and a set of Oxford Nanopore reads at the Microbial Genome Sequencing Center (Pittsburgh, Pennsylvania). Quality control and adapter trimming were performed with bcl2fastq37 and porechop38 for Illumina and Nanopore reads, respectively. A hybrid assembly with both sets of reads was performed with Unicycler using default parameters39. Assembly statistics were recorded with QUAST40. Assembly annotation was performed using Prokka41. The genome was analysed with the NCBI prokaryotic annotation and TypeMat tools from the Microbial Genomes Atlas (MiGA) web server15 to resolve its taxonomic classification and completeness.
Transcriptomics of Cobetia sp. OBi1 grown on homarine
We generated and compared transcriptomes of Cobetia sp. OBi1 cultures grown in three conditions with (1) glucose, (2) homarine or (3) glucose+homarine to identify genes involved in homarine catabolism. The growth media for transcriptomics experiments were prepared with sterile-filtered and autoclaved oligotrophic surface seawater from Hawaii to reduce background carbon and nutrients. The medium was amended with PRO99 trace metals and 0.1 mM sodium phosphate. Growth medium (1) was amended with 12 mM carbon as glucose and 0.8 mM nitrogen as ammonium. Growth medium (2) contained 12 mM carbon as homarine and no additional nitrogen source. Growth medium (3) was the same as medium (1) but was spiked with 1 mM carbon as homarine 30 min before RNA extraction. Before the transcriptomics experiment, we grew 5-ml cultures of Cobetia sp. OBi1 in each medium (1–3) to determine the OD600 of cultures in the mid-exponential phase. For the transcriptomic experiment, we pre-grew Cobetia sp. OBi1 in 5 ml of glucose medium (1), of which 4 ml was pelleted and washed three times with 1 ml of sterile oligotrophic Hawaii seawater and resuspended back in 4 ml of seawater. We used 0.5 ml of washed cells to inoculate 75 ml of each growth medium (1–3) in duplicate for transcriptomics. Cultures were incubated in aerated flasks at 25 °C and 200 r.p.m. for a period of 29–30 h during which we subsampled 0.5–1 ml from each replicate to track growth by OD600. Absorbance measurements were obtained with a Genesys 20 spectrophotometer (Spectronic Instruments). Cells were collected from 40 ml per replicate when cultures were in mid-exponential phase. Samples were split before RNA extraction with a Zymo Research Quick-RNA Fungal Bacterial MiniPrep kit. DNA was digested with DNase I from NEB. RNA samples were repurified with a Zymo Research RNA Clean & Concentrator-25 kit. The samples obtained were measured in a NanoDrop (ThermoFisher) and those with >50 ng μl−1 concentration were selected for RNA sequencing. RNA samples were sent to the Microbial Genome Sequencing Center for RNA-seq. DNA samples were treated with RNase-free DNase (Invitrogen). Library preparation was performed using Illumina’s Stranded Total RNA Prep Ligation with Ribo-Zero Plus kit and 10 bp integrated DNA technologies (IDT) for Illumina indices. Sequencing was done on a NextSeq 2000 system giving 2 × 51 bp reads. Demultiplexing, quality control and adapter trimming were performed with bcl2fastq (v.20.20.0.445). RNA-seq generated >12 M paired-end reads per sample.
RNA-seq paired-end reads were aligned to the Cobetia sp. OBi1 genome assembly to quantify the number of reads mapped to each coding gene using the Rsubread R package42. DESeq2 was used to normalize gene counts and resolve differentially expressed genes43 between each pair of Cobetia sp. OBi1 growth conditions. Significant results were resolved with a log-fold change threshold >(±)1.99 and p < 0.05 (Wald test) following adjustment for false discovery using the Benjamini–Hochberg (BH) procedure.
Functions and structures of the homABCDER operon
To functionally annotate proteins of the homABCDER operon, we used domain and pathway annotation tools including InterProScan18, KEGG17 and COFACTOR21. Gene Ontology (GO) terms and enzyme classifications were retrieved from InterPro and COFACTOR, and annotations were retained when COFACTOR CscoreGO values exceeded 0.65. KEGG annotations were obtained by querying the KEGG REST API using gene identifiers for R. pomeroyi DSS-3 (SPO3188–SPO3192). Structural models of HomA and HomB were generated using AlphaFold19, and the highest-scoring model for each protein was selected on the basis of the combined iptm+ptm confidence score. Predicted structures were visualized in ChimeraX22 to assess domain organization and potential protein–protein interaction interfaces.
Generation of mutants of homarine operon in Cobetia sp. OBi1
All Cobetia sp. experiments were conducted using the Cobetia sp. OBi1 strain. Plasmids were conjugated into Cobetia sp. OBi1 using the donor bacterial strain Escherichia coli WM3064 [a derivative of B2155 (ref. 44) provided by W. Metcalf, University of Illinois, Urbana], which can grow only in the presence of diaminopimelate (DAP). WM3064 carrying plasmids was grown overnight at 30–37 °C, shaking in LB media containing 30–50 µg ml−1 kanamycin final concentration and 0.3 mM DAP final concentration. Cobetia sp. OBi1 was grown overnight at 25–30 °C, shaking in Marine Broth 2216 (MB) media (either Difco or NutriSelect Plus). To set up conjugations, overnight cultures were mixed at a 1:1 volume ratio, pelleted for 1 min at 14,000 r.p.m., resuspended in 1/30th of its initial mixed volume and spotted onto an MB agar plate containing 0.3 mM DAP. Conjugations were then incubated at room temperature for at least 24 h. Transconjugant selection was performed by dilution streaking for single colonies from overnight spotted mating on MB agar plates without DAP. The kanamycin concentration of MB agar plates for Cobetia sp. OBi1 transconjugants is 5 µg ml−1 for integrating plasmids and 25 µg ml−1 for replicating plasmids.
Strains harbouring replicating plasmids for empty vector, fluorescence or genetic complementation used the pBVMCS-2 plasmid45 or the pGingerBK-LacUV5 plasmid46. Both plasmid transformations used the conjugation protocol outlined in the previous paragraph. Gene deletion strains were generated using the integrating plasmid pNPTS138 (ref. 47) for homD (protein accession ACFLL8_11515) and a modified pNPTS138 for homT (protein accession ACFLL8_11505). pNPTS138 transformation and integration occurs at a chromosomal site homologous to the insertion sequence in pNPTS138. The modified pNPTS138 replaced the aph(3’)-Ia kanamycin resistance gene with aph(3’)-IIa, added the lacI gene and replaced the sacB gene promoter with the lac operator. Resistance cassette, lacI and lac-operator were sourced from ref. 46; henceforth, the modified pNPTS138 plasmid is called pMZT1.
Single-colony transconjugants were inoculated into liquid MB media at 25–30 °C, shaking for 48–72 h for non-selective growth. Non-selective liquid growth allows for a second recombination event to occur, which either restores the native locus or replaces the native locus with the insertion sequence that was engineered into pNPTS138. Counterselection for the second recombination of pNPTS138 excision was carried out by passing non-selective cultures (1:100 dilution) into new liquid MB media with 10–20% (w/v) final sucrose concentration for 6–8 h. For counterselections with pMZT1, a final concentration of 0.5 mM isopropyl-β-D-thiogalactoside (IPTG) was added. Serial dilutions of counterselection cultures were plated on MB agar with 10–20% sucrose, and for counterselection with the pMZT1 plasmid, the sucrose agar contained 0.5 mM IPTG.
Colonies were subjected to PCR genotyping and sequencing to confirm allele replacement. For homD deletion analysis, primers homD_KO check F/R were utilized, while primers homT_comp F and homT_KO check R validated the deletion of homT (Supplementary Table 16). Plasmid construction involved various combinations of restriction enzyme digestion, PCR amplification and Gibson assembly. The plasmid pNPTS138-homD-KO was generated by double digestion of pNPTS138 with NheI and HindIII, followed by assembly with a synthesized in-frame knockout (KO) allele of homD from GeneWiz, then transformed into chemically competent WM3064 cells. Similarly, pMZT1-homT-KO was constructed by double digestion of pMZT1 with NheI and HindIII, assembly with a synthesized KO allele of homT from GeneWiz, followed by Gibson assembly and transformation into WM3064 cells. The pMZT1 plasmid itself was created using two PCR fragments: one amplified from pGinger-LacUV (2.6 kb) and another from pNPTS138 (4 kb) assembled via Gibson assembly and transformed into Zymo Mix and Go DH5⍺ chemically competent cells. For pBVMCS-homD, pBVMCS-2 was linearized via double digestion with EcoRI and NdeI, and assembled with a PCR fragment containing the native homD promoter and coding sequence amplified from Cobetia sp. OBi1 genomic DNA (gDNA), followed by transformation into WM3064 cells. Construction of pLacUV-homT involved linearization of pGinger-LacUV with EcoRI and BamHI, assembly with a PCR fragment containing the homT coding sequence amplified from Cobetia sp. OBi1 gDNA, and placement of homT under an IPTG-inducible promoter. The final construct was assembled with Gibson assembly and transformed into WM3064 cells.
Phenotype experiments with Cobetia sp. OBi1 knockout mutants
To determine whether Cobetia sp. OBi1 requires homD to grow on homarine, we grew the wild-type and ΔhomD strains in oligotrophic Hawaii surface seawater medium containing homarine at 12 mM carbon (test condition) or 12 mM carbon glucose with 1.7 mM ammonium (positive control). The base seawater medium contained PRO99 trace metals and 0.2 mM phosphate to support growth; we did not measure background nitrogen concentrations. All strains, including the wild type, ΔhomD, the ΔhomD complementation strain (ΔhomD + pBVMCS-homD) and its empty-vector control (ΔhomD + pBVMCS) were first grown only for the overnight starter cultures in ½ YTSS medium from a single colony (with kanamycin included at 50 µg ml−1 for the plasmid-bearing strains). Once strains reached an OD600 of 0.8, 1 ml aliquots were pelleted at 3,000 × g to collect cells. Cells were washed five times with base seawater medium and starved overnight to reduce background growth. Washed cells were diluted 100-fold in seawater and 10 μl were arrayed into 96-well plates containing 0.3 ml per well of the homarine or glucose test media. The plates were incubated on a shaker at 22 °C removed from light. Discrete OD600 measurements were obtained on a SpectraMax Plus microplate reader (Molecular Devices). The background seawater absorbance was subtracted from all measurements to quantify growth.
To compare the growth of wild-type Cobetia sp. OBi1 and ΔhomT, cultures were pre-grown in ½ YTSS incubated overnight at 200 r.p.m. and 25 °C. Overnight cultures of ΔhomT plasmid complementation strains were incubated with 0.5 mM IPTG to induce RFP or HomT expression. Cell pellets of 1 ml of culture were washed five times in base seawater medium. Washed cells were diluted 20-fold in seawater and 10 μl were arrayed into 96-well plates containing 0.3 ml per well of the homarine or glucose test media. Plasmid complementation strains were incubated with varying concentrations of IPTG (0–0.5 mM). Because ΔhomT was expected to exhibit slower growth, plates were incubated directly in the microplate reader to obtain high-frequency measurements over several days. This required sealing of plates with breathable film to prevent evaporation during the extended incubation. Plates were incubated at 25 °C in the microplate reader to record absorbance and RFP fluorescence (excitation and emission). A logistic growth model was implemented in R with the logit and nonlinear least squares (nls) function in the ‘car’ package to estimate the growth rate parameter. A one-way analysis of variance (ANOVA) and the median test for multiple comparisons from the ‘agricolae’ package were implemented in R to test for significant differences in growth rates.
Phenotype experiments with mutants of R. pomeryoi DSS-3
Gene clusters for Cobetia sp. OBi1: ACFLL8_11490 (homE), ACFLL8_11495 (homA), ACFLL8_11500 (homB), ACFLL8_11510 (homC), ACFLL8_11515 (homD) and ACFLL8_11520 (homR) were pairwise aligned to R. pomeroyi DSS-3 using blastp with a BLOSUM62 matrix48 (Supplementary Table 3). To confirm the involvement of homarine catabolic genes, we utilized RB-TnSeq mutants of R. pomeroyi DSS-3. Detailed methods for generating and arraying the R. pomeroyi DSS-3 barcoded mutant library are provided and summarized in previous work24,49. Mutant cultures were pre-grown overnight in ½ YTSS broth containing 50 μg ml−1 kanamycin. Screens were performed in L1 minimal medium amended with Basal Medium vitamins50, 0.8 mM ammonium, 50 μg ml−1 kanamycin and 100 μM phosphorus as PO43−. Washed (3×) overnight cultures of individual mutants were inoculated at an OD600 of 0.01 into modified L1 medium with a single substrate as the sole carbon source, either homarine or glucose at 4 mM carbon (n = 6). Plates were incubated at 25 °C with shaking at 425 r.p.m. for 72 h, and OD600 was measured at 5-min intervals using a Synergy H1 microplate reader (BioTek), corrected to a pathlength of 1 cm, assuming a volume of 200 μl. As a positive control, the same medium was inoculated with washed overnight cultures of wild-type R. pomeroyi DSS-3 (n = 3). Wild-type R. pomeroyi DSS-3 was also grown in kanamycin to test the effectiveness of the antibiotic (n = 3). For media control, wells filled with 200 μl of medium without inoculum were incubated (n = 3).
Preparation, mass spectrometry and analysis of metabolomics experiment with Cobetia sp. OBi1
Cobetia sp. OBi1 was grown in three conditions, as described for comparative transcriptomic analyses: homarine, glucose and glucose+homarine. Overnight cultures of Cobetia sp. OBi1 (100 ml) were grown at 25 °C grown in glucose-amended seawater media as described for Cobetia sp. OBi1 transcriptomics experiments. Next, 10 ml subsamples were taken from overnight culture, centrifuged for 15 min at 2,800 g, and resuspended in the experimental growth media homarine, glucose or glucose+homarine, in triplicate (again, as described for Cobetia sp. OBi1 transcriptomics experiments). These samples were incubated for 1 h at 25 °C in a dark shaker before collection. Cells were collected by centrifugation for 15 min at 2,800 g, and supernatant was collected and filtered through a 0.22 μm PES membrane filter. Cell pellets and supernatant samples were stored at −80 °C and −20 °C, respectively.
For particulate metabolomics, cell pellets were extracted using a combination of mechanical and chemical disruption techniques as described in previous work51. Metabolites from the supernatant were extracted using a cation-exchange-based solid-phase extraction technique as described previously7, with 1 ml of supernatant diluted into 10 ml of HPLC grade water. Isotopically labelled internal standards were added for normalization purposes, as reported in Supplementary Table 17.
Metabolomics data were acquired by liquid chromatography paired with high-resolution mass spectrometry (LC–MS) on a ThermoOrbitrap Q-Exactive HF mass spectrometer (QE). Particulate analyses were performed as reported51 with modifications reported previously52. Briefly, samples were introduced via hydrophilic interaction liquid chromatography (HILIC) in both positive and negative modes using polarity switching. All samples were introduced in full scan mode. In addition to the full scan analyses, samples were pooled and monitored in data-dependent acquisition (DDA) mode to acquire MS2 data. These data were acquired at three different collision energies (20, 35 and 50 V), with separate injections for each collision energy and ionization mode (positive or negative). Pooled samples were injected at full strength and also diluted 1:1 with water to aid in normalization, as previously described51.
Data from the QE (.raw files) were converted to the open source .mzML file format using MSConvert52. For targeted analyses, peaks were integrated in Skyline53 using a template of known compounds for which we have authentic standards. Peaks were identified by comparing to standards run in reconstitution solvent as well as spiked into a pooled sample. Further quality control was performed to remove small and low-quality peaks using the quality control procedure as described51. Quality control parameters for dissolved and particulate data from HILIC chromatography included a signal to noise ratio of at least 3, a mass within 6 ppm of the standards’ calculated mass, a retention time within 2.5 min of the standards’ retention time, a minimum area of 40,000 and a ratio of signal over signal from blank of at least 3. Finally, peaks were normalized to reduce variability introduced during data acquisition using best-matched internal standard (B-MIS)51.
For untargeted analyses, we used MS-Dial (v.4.9)54 to extract mass features using the parameters supplied in Supplementary Table 18. This resulted in a list of mass features (m/z and retention time features) that could be compared quantitatively between samples, regardless of the ability to identify these compounds. We dereplicated mass features from MS-Dial by identifying and annotating adducts (including between positive and negative polarity) and isotopes on the basis of retention time matching and calculated m/z differences and consolidated mass features for downstream statistical analyses. Mass features that correspond to our targeted compounds were annotated by comparing to authentic standards in the same run and matching m/z (within 5 ppm) and retention time (within 0.3 min). For instances where a mass feature was not detected in all samples of an experiment, we replaced missing values with 0.02 times the minimum detected value for that mass feature within the same experiment for all calculations, ensuring that all features have a baseline value to facilitate comparisons in downstream analyses.
Next, we performed an enrichment analysis to calculate the relative enrichment of individual metabolites or mass features between treatments when compared to core metabolites. This is similar to normalizing mass feature area to biomass, a common normalization technique, but does not rely on a separate biomass measurement. Instead, the enrichment analysis calculates a scaled area for each compound or mass feature relative to each ‘core metabolite’ to assess its enrichment under different treatment conditions. Core metabolites were decided on the basis of previous work6 that identified metabolites that follow biomass trends by depth and latitudinally, and are observable in most phytoplankton when analysed under our analytical conditions (15 total), reported in Supplementary Table 9. These scaled areas were used to compute fold changes and perform statistical analyses. To account for the fact that some of the ‘core metabolites’ might also be affected by treatments, we performed an outlier detection step, where Grubbs’ test55 was applied to identify extreme values in fold changes for core metabolites, and those core metabolites detected as outliers were excluded from further analysis. Final core metabolites per sample set ranged from 11 to 13 and are reported in Supplementary Table 9. After filtering, the final enrichment results were summarized by calculating the median p values (after applying a Benjamini–Hochberg false discovery rate correction56) and fold changes for each metabolite across sample treatments.
Preparation, mass spectrometry and analysis of metabolomics experiment with R. pomeryoi DSS-3
Cultures of R. pomeroyi DSS-3 were revived from cryostocks onto ½ YTSS agar plates and incubated at 30 °C for 6 days. Single colonies were inoculated into 11 ml of glucose minimal media (GMM) and grown overnight at 30 °C with shaking at 200 r.p.m. GMM was prepared using a modified L1 minimal medium with glucose 12 mM C as the sole carbon source. All cultures were maintained in sterile 15 ml assay tubes. Overnight cultures were diluted to an OD600 of 0.1 in fresh GMM, incubated for 11–12 h and amended with glucose (4 mM C, 0.8 mM NH4, control), homarine (500 nM C, no additional NH4) or glucose+homarine (1 mM C and 2 mM C, respectively with 0.8 mM NH4). Homarine additions were staggered across timepoints to ensure consistent incubation durations. At each time point, samples were collected for cell counts and particulate metabolites. For cell counts, 1 ml of culture was fixed with glutaraldehyde (final concentration 1%) in labelled cryovials, held at 4 °C for 20 min and then stored at −80 °C. Particulate metabolites were collected by filtering cultures through combusted glass fibre filters using a vacuum manifold set to 8 psi; filters were wrapped in combusted foil and flash frozen in liquid nitrogen. The experiment cultures were sampled after 2 h in biological triplicate, as well as the three control conditions: glucose-only controls, glucose+homarine controls (uninoculated) and a filter blank.
For metabolite extractions, a one-phase extraction was performed with 40:40:20:0.01 methanol:acetonitrile:water:formic acid solution as the extraction solvent57. Filters were placed in 15 ml Teflon tubes with pre-chilled extraction solvent, incubated at −20 °C for 10 min, bead beaten with silica beads and centrifuged. The solvent was then collected, transferred into glass tubes, and the procedure was repeated three times while keeping samples cold as much as possible. This extraction method was selected because comparisons between the one-phase and two-phase methods showed minimal difference in recovery of hydrophilic compounds, and the quicker one-phase method was used since the non-polar fractions were not of interest and quantification was not a goal. Samples were dried down under nitrogen gas, reconstituted in 400 μl of H2O and stored at −80 °C until analysis by LC–MS. Isotopically labelled internal standards were added for normalization purposes, as reported in Supplementary Table 17.
Samples were filtered, extracted and run on the instrument as reported for the metabolomics experiment for Cobetia sp. OBi1, but only particulate samples were acquired and run with the HILIC method for LC–MS, as these data were the most promising for identification of intermediates of homarine based on the Cobetia sp. Obi1 experiment. We used Skyline53 to mine the R. pomeroyi DSS-3 metabolomics data for features that showed enrichment in Cobetia sp. OBi1 under homarine conditions, and all the core metabolites, as reported in Supplementary Table 9. These features underwent the same normalization and calculations for enrichment as the Cobetia sp. OBi1 data as reported above.
Field experiments using isotopically labelled homarine
Experiments using 2H3-homarine were performed on research cruise TN397 in the Fall of 2021 at two different stations in the North Pacific (described in Supplementary Table 8 and displayed in Fig. 2d). 2H3-homarine was purchased from Toronto Research Chemicals and was injected onto a Q-Exactive HF Orbitrap mass spectrometer (QE-HF) to confirm the mass of the deuterium label (141.0743 m/z) and the retention time (6.4 min, same as non-labelled homarine). Seawater was collected into 21 acid-washed 10-l polycarbonate carboys from a trace metal clean stayfish system suspended at a depth of 8 m and prefiltered through 100 µm nylon mesh. Three unamended samples were collected immediately after seawater collection to provide samples of the starting community. Nine treatment bottles were spiked with 500 nM 2H3-homarine, with 9 control bottles receiving no additions. The concentration was selected to align with the observed range of dissolved homarine concentrations in the surface ocean (60 pM to >13 nM (refs. 7,9)) while being sufficiently elevated to track its metabolic products’ isotopic signature without overwhelming the microbial system. Bottles were incubated in blue-shaded (~50% shading as measured using a LICOR-180 spectrometer) temperature and light-controlled incubators designed to mimic mixed-layer conditions of the sampling location. Triplicate bottles with and without homarine addition were collected at 2, 24 and 48 h. All particulate samples (4 l) were collected using peristaltic pumps onto Durapore 0.22 μm, 47 mm, hydrophilic PVDF membrane filters, flash frozen in liquid nitrogen and stored at −80 °C. Additional stable isotope experiments with 13C7-15N-labelled homarine, prepared as described below, were performed on two different cruises: TN412 (Winter 2023) in the North Pacific, and RC104 (Summer 2023) in Puget Sound (described in Supplementary Table 8 and displayed in Fig. 2d). For TN412, the experiments were performed at two different stations; for RC104, the experiment was performed at one station. For all TN412 and RC104 experiments, seawater was collected through a trace metal clean stayfish system suspended at a depth of 8 m and prefiltered through 100 µm nylon mesh. For TN412, incubations were performed in blue-shaded (~50% shading as measured using a LICOR-180) temperature and light-controlled incubators designed to mimic mixed-layer conditions of the sampling location. For RC104, incubations were performed in mesh-shaded (~50% shading as measured using a LICOR-180) flow-through incubators. Triplicate samples were collected into acid-washed 2-l polycarbonate bottles, spiked with 90 nM of 13C7-15N-labelled homarine, and incubated in temperature and light-controlled incubators for 5 different timepoints (6, 12, 24, 48 and 96 h). The concentration of homarine for these incubations was selected to remain within the observed range of dissolved homarine concentrations in the surface ocean (60 pM to >13 nM (refs. 7,9)) while being low enough to avoid overwhelming the microbial system. A lower concentration of homarine was used for 13C7-15N-labelled homarine experiments than for the 2H3-homarine incubations after observations from the TN397 cruise showed sufficient signal. Triplicates of spiked and unspiked samples were filtered as quickly as possible, no more than 30 min (T0 and unamended control samples, respectively). Particulate samples were collected identically as in the TN397 experiment described above. To prepare 13C and 15N-labelled homarine, Synechococcus sp. WH8102 was grown in Pro99 media58 prepared with 15N sodium nitrate and 13C sodium bicarbonate and no HEPES buffer. Synechococcus sp. WH8102 has been previously shown to produce a large amount of homarine6. Cultures were grown in 250 ml glass media bottles with minimal headspace. Cells were collected by filtration onto 0.2 µm Durapore filters using combusted borosilicate filter towers. All filters were stored at −80 °C until extraction. Filters were extracted using 40:40:20:0.01 methanol:acetonitrile:water:formic acid solution as the extraction solvent in 15 ml Teflon tubes. The solvent was dried under N2 gas, reconstituted with H2O and injected onto a QE-HF Orbitrap mass spectrometer to confirm the production of 3C-15N-labelled homarine (146.0754 m/z). After confirming the 13C-15N-labelling of homarine, Synechococcus sp. WH8102 was grown in ~10 l Pro99 media58, collected after 7 days and extracted as previously described. Next, this extract was purified for spiking into environmental samples for stable isotope experiments. Purification was done using cation exchange chromatography and a Supelcosil LC-SCX column (25 cm × 4.6 mm, 5 µm particle size) with a Thermo Scientific Vanquish UHPLC, fraction collector and diode array detector. Solvent A was water with 2% formic acid and solvent B was water with 1% formic acid and 100 mM ammonium formate. The column was held at 0% B for 2 min, ramped to 20% B over 10 min, ramped to 100% B over 8 min, held at 100% B for 5 min and equilibrated back to 0% B for 5 min (total run time was 30 min). The column temperature was maintained at 25 °C and the flow rate was 1.0 ml min−1. Purification of labelled homarine was achieved by UV-based collection of a peak at 270 nm wavelength and a retention time of 4.3 min, which was confirmed by a homarine standard. Fractions containing labelled homarine were pooled together, dried under N2 gas and injected into the QE-HF to confirm the purity of the labelled homarine and quantify its concentration by comparison to a non-labelled standard. Its purity was measured by determining the concentration of interfering compounds relative to homarine concentration by comparison to standards. Combined interfering compounds (glycine betaine and alanine) were found to comprise less than 10%, based on signal intensity compared to standards. Samples were extracted and data were acquired as reported for the metabolomics experiment for Cobetia sp. OBi1 above. To prevent confusion in using the isotope labels, we used a subset of isotopically labelled internal standards, as reported in Supplementary Table 17. Using the approach described above for the R. pomeroyi DSS-3 metabolomics data, we mined these data for the core metabolites as well as the features that showed enrichment in Cobetia sp. OBi1 under homarine conditions, both monoisotopic and expected isotopologues, as reported in Supplementary Table 9. Again, these features underwent the same normalization and calculations for enrichment as the Cobetia sp. OBi1 data as reported above, using unamended control samples for comparison.
For the TN397 with 2H3-homarine amendments, we also performed an untargeted analysis in an attempt to find additional features that would correspond to a methyl transfer. This approach is analogous to previous studies that utilized natural abundance isotopes to track sulfur and iron-containing metabolites59. We used CoreMS60 to identify mass features using a persistent homology approach with liberal thresholds, yielding thousands of features per sample (of which many were expected to be false positives or very low abundance peaks). For each sample’s features, we identified pairs of features that could correspond to 2H3, 2H2 and 13C isotopologues; these pairs needed to be within 0.1 min of one another, with a mass error of expected isotopologues of no more than 4 ppm. From there, we collected features with putative 2H3, 2H2 isotopologues within all the samples into retention time and m/z groups and filtered out any features that were present in control samples or a single sample. We visually inspected these mass feature pairs and discarded pairs that did not have consistent peak shapes, resulting in 7 quality mass features (Supplementary Table 10). Note that this approach only yields features that have an observable level of the monoisotopic version of a compound in the samples.
Isolation, preparation and genome sequencing of additional marine isolates
Additional homarine-degrading bacteria were obtained by culturing bacteria in seawater samples on solid media made with sterile oligotrophic seawater, 1.5% ultrapure agarose (SeaKem), 12 mM carbon as homarine and 0.1 mM sodium phosphate. Seawater samples were collected at various nearshore locations in Puget Sound near Tacoma, Washington, and in the equatorial North Pacific during Gradients IV cruise TN397. Bacteria were concentrated by vacuum filtration onto a 0.22 µm Supor membrane, resuspended in 1 ml of sterile oligotrophic seawater, plated on homarine plates prepared with sterile oligotrophic seawater, 1.5% ultrapure agarose (SeaKem), 12 mM carbon as homarine and 0.1 mM sodium phosphate, and incubated for 1–2 weeks at 25 °C. Single colonies were restreaked in marine broth agar plates to obtain axenic strains. The isolate collection was rescreened in oligotrophic seawater media containing homarine and phosphate to confirm growth on homarine. Two colonies per isolate were tested in 24-well plates containing 2 ml per well of media containing homarine or control media lacking homarine. Growth (OD600) was measured on a SpectraMax Plus microplate reader (Molecular Devices) after 4 and 8 days of incubation. Isolates with confirmed growth on homarine were regrown in 5 ml of marine broth medium for 1–2 days. Cultures were pelleted and stored at −20 °C before DNA extraction. Bacterial cell pellets were rinsed in 5 ml of 0.9% saline solution by vortexing, 1 ml was transferred to sterile microtubes and centrifuged at 3,500 × g for 5 min, and the supernatant was discarded. DNA was extracted with a Monarch Genomic DNA Purification kit using the tissue lysis solution supplemented with lysozyme, followed by treatment with Proteinase K and RNase A. Purified DNA was analysed on a Nanodrop spectrophotometer. Samples with >20 ng µl−1 were sent to SeqCenter (Pittsburgh, Pennsylvania) for Illumina whole-genome sequencing. Sample libraries were prepared using the Illumina DNA Prep kit and IDT 10 bp unique dual indexes (UDI) indices and sequenced on an Illumina NextSeq 2000, producing 2 × 151 bp reads. Illumina reads were trimmed and assembled with Trim Galore61 (https://doi.org/10.5281/zenodo.5127898; https://doi.org/10.14806/ej.17.1.200) and the Unicycler SPAdes-optimizer39, respectively. We annotated assembly contigs with Prokka41.
Identification of the homarine operon and sox and mgd genes in isolates
To identify genes involved in homarine catabolism, we conducted reciprocal best BLAST hit (RBH) analyses across isolate genomes. Protein sequences from all genomes were used to generate BLASTP databases (NCBI BLAST+), and an all-vs-all BLASTP search was performed using a parallelized script. Gene pairs were considered RBHs if they were each other’s top-scoring match by bitscore.
RBH results were screened for homologues of the homABCDER operon from R. pomeroyi DSS-3 (for example, SPO3186–SPO3192) and Cobetia sp. OBi1 (ACFLL8_11490–ACFLL8_11520), as well as for subunits of sarcosine oxidase (sox) and N-methyl-glutamate dehydrogenase (mgd). Reference sequences for sox subunits were obtained from Stenotrophomonas maltophilia (AAY34150.1 (ref. 62)) and Corynebacterium sp. (P40875.2 (ref. 63)) and mgd subunits from Cobetia sp. OBi1 (ACFLL8_07290–07305), R. pomeroyi DSS-3 (SPO1585–1588), Methylocella silvestris (WP_008064110.1 (ref. 27)) and Methyloversatilis universalis (WP_012591629 (ref. 28)).
Filter hits were concatenated into a non-redundant set, and corresponding protein sequences were extracted from genome-wide protein files and annotated by gene identity and genome of origin. To guide alignment strategies, sequence variability was first assessed by calculating the average pairwise identity, with average identities as follows: soxA/mgdC 39.64%, soxG/mgdD 31.02%, soxB/mgdA 46.18% and soxD/mgdB 34.62%. Due to the high sequence divergence observed across gene pairs, alignments were performed using MAFFT64 with the E-INS-i algorithm and a BLOSUM62 matrix, to optimize for sequences with multiple conserved domains and long gaps. The alignments were subjected to model selection using ProtTest (v.3)65. Best-fit substitution models were chosen on the basis of Akaike information criterion and Bayesian information criterion and applied per gene pair as follows: soxB/mgdA and soxA/mgdC: LG + I + G, soxD/mgdB: WAG + I + G, and soxG/mgdD: WAG + G. Maximum likelihood phylogenetic trees were generated using RAxML with bootstrapping (1,000 replicates) on each gene pair alignment, using the chosen substitution models, automated maximum likelihood inference, bootstrap generation and support value mapping. Tree diagrams were generated using iTol66.
Identification of the homarine operon in NCBI genomes
We conducted a broad survey to identify and characterize homarine operon orthologues. First, we downloaded and processed the NCBI ClusteredNR BLAST database67. We ran BLAST searches to identify homologues of the homarine catabolic genes (homA, homB, homC, homD and homE). We retrieved cluster members of relevant hits using the ClusteredNR scripts68 and downloaded their corresponding nucleotide coordinates from NCBI with Entrez Direct69 to identify candidate genomic loci with at least three of the six conserved genes co-located within 10,000 bp. To ensure completeness of our initial survey, we downloaded the genomes in which we found candidate homarine catabolic operons to perform a second round of BLAST searches and selected loci that contained at least one orthologue of homA, homB, homC and homD co-located within 10,000 bp. The resulting genomic loci data are reported in Supplementary Table 12.
We retrieved BioSample metadata from NCBI for each of the genomes where we found at least homA, homB, homC and homD co-located within 10,000 bp. Metadata were parsed and mined to retrieve key biosample attributes such as isolation source, environmental context, geographic location and additional identifiers. Next, we cleaned and organized the BioSample metadata for comparison. This involved categorizing isolation sources into environmental and non-environmental types and further classifying environmental sources by specific habitats; latitude and longitude data were parsed directly from metadata or inferred from reported geographic location. Note that many of the biosample records accompanying the NCBI genomes lacked sufficient metadata to parse the environmental context or geographical location of the source. Resulting mapping data are reported in Supplementary Table 12.
Environmental transcriptomics perturbation experiment
To identify microorganisms and genes responsive to homarine in Puget Sound, we conducted short shipboard incubations with seawater amended with homarine and collected total RNA for metatranscriptomics. Seawater was sampled with the RV Rachel Carson conductivity–temperature–depth rosette system from 4 m depth during RC0078 (Station 2) in the Salish Sea near the Strait of Juan de Fuca on 5 June 2022. Seawater was transferred to acid-clean, blue-tinted, 5-l polycarbonate carboys for acclimation in a deck incubator with flow-through surface seawater. A set of three carboys were supplied with 0.1 mM carbon as homarine and three other unamended carboys served as a control group. We incubated samples for 3.5 h and proceeded to concentrate the microbial fraction by filtering each through a 47-mm-diameter, 0.22-µm-pore-size polycarbonate membrane with a peristaltic pump. The filters were flash frozen in liquid nitrogen at sea and were transferred to −80 °C for storage in the laboratory. Total RNA was extracted from filters using a ZymoBIOMICS RNA miniprep kit following manufacturer protocol. RNA was quantified on a NanoDrop spectrophotometer. Samples with >50 ng μl−1 were sent to SeqCenter (Pittsburgh, Pennsylvania) for RNA-seq. RNA samples were DNAse treated with Invitrogen DNAse (RNAse free). Library preparation was performed using Illumina’s Stranded Total RNA Prep Ligation with Ribo-Zero Plus kit and 10 bp IDT for Illumina indices. Sequencing was done on a NovaSeq 6000 giving 2 × 51 bp reads. Demultiplexing, quality control and adapter trimming were performed with bcl-convert (v.4.0.3). For metatranscriptomic analysis, we first parsed ribosomal (r)RNA from non-rRNA reads with SortMeRNA. Non-rRNA reads (mRNA fragments) were assembled with PLASS70 and mapped back to the 9,574,265 assembled proteins of length >100 amino acids with MMSeqs2 (ref. 71) with the sensitivity parameter and maximum accepted targets set to 1. To quantify mapped reads, we retained only the best alignment for each unique read and reads with 100% identity to an assembled protein, resulting in 42,155,143 reads and 1,367,930 proteins. We used DESeq2 (ref. 43) and the tabulated read count data to detect differentially expressed genes between seawater samples supplied with homarine and unamended seawater. DESeq2 was implemented with a log2 fold change threshold of 1, the alternative hypothesis statement set to greater Abs, the significance level set to 0.05 and the default false discovery rate adjustment. We annotated 49 differentially expressed genes with the NCBI non-redundant database using BLASTP (excluding environmental samples)48 and the eggNOG 5.0.2 protein database using eggNOG-mapper (v.2.1.9)72 in ultra-sensitive mode.
In situ homarine concentration
Samples for particulate and dissolved in situ homarine concentrations were taken on the Gradients 3 cruise (KM1906) in the North Pacific in 2019. Samples were collected and measured as described in previous work6,7. Briefly, water was collected from the shipboard flow-through underway sampling system and particulate metabolites were sampled by filtering the seawater using peristaltic pumps onto 142-mm-diameter, 0.2-μm-pore-size PTFE Omnipore filters, flash frozen in liquid nitrogen and stored at −80 °C until analysis. Dissolved metabolites were sampled by collecting the filtrate in 50-ml acid-washed polypropylene Falcon tubes. Particulate metabolites were extracted using modified Bligh and Dyer extraction as previously described51. Dissolved metabolites were extracted using cation-exchange solid-phase extraction as reported7. Following extraction, metabolites were dried down under N2 gas, reconstituted in water with isotope-labelled internal standards and analysed using LC–MS with the same acquisition approach as described for Cobetia sp. OBi1 samples. Homarine concentrations were quantified by comparison to a 2H3-homarine internal standard.
In situ metatranscriptomes
Metatranscriptome samples were collected during the Gradients 3 cruise (KM1906) in the North Pacific in 2019 (same cruise as in situ homarine concentration). Samples were collected from the ship’s underway system by subsampling ~5–10 l of seawater into 10-l polycarbonate carboys, prefiltered through a 100 μm mesh filter, and sequentially filtered through 3 μm and 0.2 μm polycarbonate filters. Filters were flash frozen in liquid nitrogen and stored at −80 °C before processing. Metatranscriptomes were generated by extracting total RNA from the filters using the Direct-zol MiniPrep Plus kit (Zymo Research R2720). During extraction, a set of 14 synthetic spike-in mRNA standards was added as previously described34,73 to enable quantification of absolute transcript abundances across samples. The extracted RNA was quantified with Qubit fluorometer 1.0 (ThermoFisher) and quality controlled using a Bioanalyzer 2100 instrument (Agilent). Total community mRNA was generated by rRNA depletion using the ThermoFisher RiboMinus (Yeast+Bacteria; K15503, K15504) kit. Samples were sequenced on the Illumina NovaSeq 6000 platform using an S2 flow cell with 300 cycles. Library preparation and sequencing was done at the Northwest Genomics Center (University of Washington, Seattle).
Raw Illumina reads from rRNA depleted samples were quality controlled with Trimmomatic v.0.36 (MAXINFO:135:0.5, LEADING:3, TRAILING:3, MINLEN:60 and AVGQUAL:20)74. Short reads were assembled into longer contigs using Trinity de novo assembler v.2.3.2 (–normalize_reads –min_kmer_cov 2 –min_contig_length 300)75. All assemblies were performed on short reads from combined replicates for each sample. Assembled contigs were translated into amino acid space in six frames with transeq76 vEMBOSS:6.6.0.059 using Standard Genetic Code, and the longest coding frame was retained for future analysis. The best frame translated assemblies were clustered at 99% amino acid identity with MMseqs2 (ref. 71). To quantify assembled contig abundances and obtain raw transcript counts, unassembled short reads were mapped to the assembled contigs using kallisto (v.0.46.1)77. The raw transcript counts for each assembled contig were normalized to total transcripts per litre using normalization factors based on the recovery of 14 synthetic internal mRNA34,59. Standard counts were recovered with Bowtie 2 aligner v.2.5.2 using the standard pipeline for paired-end reads73. Taxonomic annotation of the assembled contigs was done with DIAMOND alignment to a combined reference database containing MarFeRReT (v.1.1)78 and MARMICRODB (.1.0)79 reference databases. Functional annotation of the assembled contigs was done using HMMER (3.3)80 against the Kyoto Encyclopedia of Genes and Genomes (KEGG) KOfam Hidden Markov Models (HMMs) database17, with best KOfam predictions with a threshold score >30 retained for each contig, and against Pfam (35.0)81 HMMs where the ‘trusteed cut-off’ assigned by Pfam to each hmm profile was used as a minimum score threshold. Resulting unclassified hits for hom genes were aligned against the NCBI nr database using BLASTP and assigned taxonomy at the class level.
To estimate abundances of selected HMM profiles in total community metatranscriptomes, assembled and annotated contigs of non-selected transcripts from prokaryotes and eukaryotes from the 3 μm and 0.2 m fractions of each sample replicate were combined. Only contigs taxonomically annotated as ‘Bacteria’ at the kingdom level were retained during the analysis.
Single-copy gene normalization of homA and homB transcript profiles
To quantify changes in homA and homB transcription across the transect, metatranscriptomic read counts for each gene were normalized on a per-cell basis by dividing transcript abundances by the median transcript abundance of ten universal single-copy housekeeping genes (K06942, K01889, K01887, K01875, K01883, K01869, K01873, K01409, K03106 and K03110; Supplementary Table 19). This procedure follows the per-cell normalization framework described previously35, in which expression of marker genes provides an estimate of relative bacterial cell numbers within each sample. Normalized transcript abundances for each metatranscriptome replicate were transformed into log2 space and averaged per station, with standard deviations representing among-replicate variability. These values were used to resolve fold-change variation in homA and homB transcription across the latitudinal gradient.
Hidden Markov model profiles for genes in the hom operon
Homarine catabolic protein sequences for each gene were aligned using MAFFT with a BLOSUM62 substitution matrix. Alignments were manually curated to remove poorly conserved N- and C-terminal regions and internal gaps. Hidden Markov Models (HMMs) were then built from the curated alignments using HMMER (v.3.3.2)82. HMM profile specificity was tested against a curated genome database of isolates capable of growing in homarine with co-located hom catabolic genes. Profiles for homA and homB showed strong specificity with minimal off-target hits. However, homC returned several false positives, and homD produced numerous non-specific matches, rendering these profiles unsuitable for further analysis.
Each HMM profile was used to search for homologues in environmental metatranscriptomes collected on the Gradients 3 cruise using hmmsearch (HMMER v.3.3.2) with the following parameters: –cpu 4 -T 30 –incT 30. The results were parsed and filtered, retaining only high-confidence hits based on alignment coverage, score and contig completeness. Hits were manually curated against the reference HMM profile, and sequences that did not align properly or did not match the expected profile length were discarded. Finally, filtered gene hits were cross-referenced with sample metadata. We focused our analysis on the homB results, which showed the highest significance (e-values ranging from 3.8 × 10−43 to 3.9 × 10−240), whereas homA, while still significant, had comparatively weaker support (e-values ranging from 1.5 × 10−06 to 9.5 × 10−57).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All datasets generated or analysed during this study are publicly available or will be made available upon publication. Genome assemblies for Cobetia sp. OBi1 and additional homarine-degrading isolates, and transcriptomic data from Cobetia sp. OBi1 grown under homarine, glucose-only, and glucose + homarine conditions are available in NCBI BioProject PRJNA862506. Metabolomic datasets for Cobetia sp. OBi1 and R. pomeroyi DSS-3 are deposited in the Metabolomics Workbench repository under study IDs ST004324 and ST004337, respectively. Metabolomics data from environmental seawater incubations with 2H3- or 13C7-15N-labelled homarine are deposited in the Metabolomics Workbench repository under study IDs ST004352, ST004353 and ST004354. Environmental data from the Gradients North Pacific transect are available through Simons CMAP (https://simonscmap.com/catalog/datasets/Gradients3_KM1906_Optics_LISST_ACS_ECO). Environmental metatranscriptome data from the Gradients 3 North Pacific transect are available through NCBI SRA under BioProject PRJNA1091352. Environmental metabolomics data from the Gradients 3 North Pacific transect are available through the Metabolomics Workbench repository under study ID ST004347. Contact corresponding authors to request any of the environmental isolates (including Cobetia sp. OBi1). All data for generating figures available in supplementary tables.
Code availability
Custom analysis scripts used in this study are available on GitHub at https://github.com/IngallsLabUW/Homarine_Catabolism_MS (ref. 83).
References
Moran, M. A. et al. The Ocean’s labile DOC supply chain. Limnol. Oceanogr. 67, 1007–1021 (2022).
Schalk, I. J. Bacterial siderophores: diversity, uptake pathways and applications. Nat. Rev. Microbiol. 23, 24–40 (2025).
Seymour, J. R., Simó, R., Ahmed, T. & Stocker, R. Chemoattraction to dimethylsulfoniopropionate throughout the marine microbial food web. Science 329, 342–345 (2010).
Harvey, E. L. et al. A bacterial quorum-sensing precursor induces mortality in the marine coccolithophore, Emiliania huxleyi. Front. Microbiol. 7, 59 (2016).
Boysen, A. K. et al. Particulate metabolites and transcripts reflect diel oscillations of microbial activity in the surface ocean. mSystems 6, e00896-20 (2021).
Heal, K. R. et al. Marine community metabolomes carry fingerprints of phytoplankton community composition. mSystems 6, e01334-20 (2021).
Sacks, J. S., Heal, K. R., Boysen, A. K., Carlson, L. T. & Ingalls, A. E. Quantification of dissolved metabolites in environmental samples through cation-exchange solid-phase extraction paired with liquid chromatography–mass spectrometry. Limnol. Oceanogr. Methods 20, 683–700 (2022).
Hoppe-Seyler, F. A. Über das Homarin, eine bisher unbekannte tierische Base. Z. Für Physiol. Chem. 222, 105–115 (1933).
Sacks, J. S. et al. The cycling of glycine betaine and homarine in marine microbial communities: quantitative flux measurements and the role of competitive uptake inhibition. Limnol. Oceanogr. Lett. 11, e70069 (2026).
Zhang, Y. et al. Biodegradation of picolinic acid by Rhodococcus sp. PA18. Appl. Sci. 9, 1006 (2019).
Roney, S. H. et al. Common fear molecules induce defensive responses in marine prey across trophic levels. Oecologia 202, 655–667 (2023).
Maldonado, A. et al. Hard coral (Porites lobata) extracts and homarine on cytochrome P450 expression in Hawaiian butterflyfishes with different feeding strategies. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 179, 57–63 (2016).
Berking, S. Homarine (N-methylpicolinic acid) and trigonelline (N-methylnicotinic acid) appear to be involved in pattern control in a marine hydroid. Development 99, 211–220 (1987).
Poulin, R. X. et al. Chemical encoding of risk perception and predator detection among estuarine invertebrates. Proc. Natl Acad. Sci. USA 115, 662–667 (2018).
Rodriguez-R, L. M. et al. The Microbial Genomes Atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 46, W282–W288 (2018).
Sarkar, M. K., Paul, K. & Blair, D. Chemotaxis signaling protein CheY binds to the rotor protein FliN to control the direction of flagellar rotation in Escherichia coli. Proc. Natl Acad. Sci. USA 107, 9370–9375 (2010).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677 (2025).
Blum, M. et al. InterPro: the protein sequence classification resource in 2025. Nucleic Acids Res. 53, D444–D456 (2025).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Cianfrocco, M. A., Wong-Barnum, M., Youn, C., Wagner, R. & Leschziner, A. COSMIC2: a science gateway for cryo-electron microscopy structure determination. In Practice and Experience in Advanced Research Computing 2017: Sustainability, Success and Impact 1–5 (ACM, 2017).
Zhang, C., Freddolino, L. & Zhang, Y. COFACTOR: improved protein function prediction by combining structure, sequence and protein–protein interaction information. Nucleic Acids Res. 45, W291–W299 (2017).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Perchat, N. et al. Elucidation of the trigonelline degradation pathway reveals previously undescribed enzymes and metabolites. Proc. Natl. Acad. Sci. USA 115, E4358–E4367 (2018).
Mejia, C. et al. An arrayed transposon library of Ruegeria pomeroyi DSS-3. Preprint at bioRxiv https://doi.org/10.1101/2022.09.11.507510 (2022).
Noell, S. E. & Giovannoni, S. J. SAR11 bacteria have a high affinity and multifunctional glycine betaine transporter. Environ. Microbiol. 21, 2559–2575 (2019).
Netherton, J. C. III & Gurin, S. Biosynthesis and physiological role of homarine in marine shrimp. J. Biol. Chem. 257, 11971–11975 (1982).
Chen, Y. et al. γ-Glutamylmethylamide is an essential intermediate in the metabolism of methylamine by Methylocella silvestris. Appl. Environ. Microbiol. 76, 4530–4537 (2010).
Latypova, E. et al. Genetics of the glutamate-mediated methylamine utilization pathway in the facultative methylotrophic beta-proteobacterium Methyloversatilis universalis FAM5. Mol. Microbiol. 75, 426–439 (2010).
Giovannoni, S. J. et al. Genome streamlining in a cosmopolitan oceanic bacterium. Science 309, 1242–1245 (2005).
Rappé, M. S., Kemp, P. F. & Giovannoni, S. J. Phylogenetic diversity of marine coastal picoplankton 16S rRNA genes cloned from the continental shelf off Cape Hatteras, North Carolina. Limnol. Oceanogr. 42, 811–826 (1997).
Stingl, U., Desiderio, R. A., Cho, J.-C., Vergin, K. L. & Giovannoni, S. J. The SAR92 Clade: an abundant coastal clade of culturable marine bacteria possessing proteorhodopsin. Appl. Environ. Microbiol. 73, 2290–2296 (2007).
Luo, H. & Moran, M. A. Evolutionary ecology of the marine Roseobacter clade. Microbiol. Mol. Biol. Rev. 78, 573–587 (2014).
Moran, M. A. et al. Ecological genomics of marine roseobacters. Appl. Environ. Microbiol. 73, 4559–4569 (2007).
Satinsky, B. M., Gifford, S. M., Crump, B. C. & Moran, M. A. Use of internal standards for quantitative metatranscriptome and metagenome analysis. Methods Enzymol. 531, 237–250 (2013).
Salazar, G. et al. Gene expression changes and community turnover differentially shape the global ocean metatranscriptome. Cell 179, 1068–1083.e21 (2019).
Giovannoni, S. J. SAR11 bacteria: the most abundant plankton in the oceans. Annu. Rev. Mar. Sci. 9, 231–255 (2017).
Holtgrewe, M., Messerschmidt, C., Nieminen, M. & Beule, D. DigestiFlow: from BCL to FASTQ with ease. Bioinformatics 36, 1983–1985 (2020).
Bonenfant, Q., Noé, L. & Touzet, H. Porechop_ABI: discovering unknown adapters in Oxford Nanopore Technology sequencing reads for downstream trimming. Bioinform. Adv. 3, vbac085 (2023).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595 (2017).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Liao, Y., Smyth, G. K. & Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47 (2019).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Dehio, C. & Meyer, M. Maintenance of broad-host-range incompatibility group P and group Q plasmids and transposition of Tn5 in Bartonella henselae following conjugal plasmid transfer from Escherichia coli. J. Bacteriol. 179, 538–540 (1997).
Thanbichler, M., Iniesta, A. A. & Shapiro, L. A comprehensive set of plasmids for vanillate- and xylose-inducible gene expression in Caulobacter crescentus. Nucleic Acids Res. 35, e137 (2007).
Pearson, A. N. et al. The pGinger family of expression plasmids. Microbiol. Spectr. 11, e00373-23 (2023).
Ried, J. L. & Collmer, A. An nptI-sacR cartridge for constructing directed, unmarked mutations in Gram negative bacteria by marker exchange eviction mutagenesis. Gene 57, 239–246 (1987).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Schroer, W. F. et al. Functional annotation and importance of marine bacterial transporters of plankton exometabolites. ISME Commun. 3, 37 (2023).
Guillard, R. R. L. & Hargraves, P. E. Stichochrysis immobilis is a diatom, not a chrysophyte. Phycologia 32, 234–236 (1992).
Boysen, A. K., Heal, K. R., Carlson, L. T. & Ingalls, A. E. Best-matched internal standard normalization in liquid chromatography–mass spectrometry metabolomics applied to environmental samples. Anal. Chem. 90, 1363–1369 (2018).
Boysen, A. K. et al. Glycine betaine uptake and metabolism in marine microbial communities. Environ. Microbiol. 24, 2380–2403 (2022).
Adams, K. J. et al. Skyline for small molecules: a unifying software package for quantitative metabolomics. J. Proteome Res. 19, 1447–1458 (2020).
Tsugawa, H. et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 38, 1159–1163 (2020).
Grubbs, F. E. Procedures for detecting outlying observations in samples. Technometrics 11, 1–21 (1969).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Methodol. 57, 289–300 (1995).
Canelas, A. B. et al. Quantitative evaluation of intracellular metabolite extraction techniques for yeast metabolomics. Anal. Chem. 81, 7379–7389 (2009).
Durham, B. P. et al. Chemotaxonomic patterns in intracellular metabolites of marine microbial plankton. Front. Mar. Sci. 9, 864796 (2022).
Durham, B. P. et al. Sulfonate-based networks between eukaryotic phytoplankton and heterotrophic bacteria in the surface ocean. Nat. Microbiol. 4, 1706–1715 (2019).
Corilo, Y. E., Kew, W. R., McCue, L. A., Heal, K. & Carr, J. C. EMSL-computing/CoreMS: CoreMS 3.0.0. Zenodo https://doi.org/10.5281/zenodo.14009575 (2024).
Krueger, F. Trim Galore!: A Wrapper Around Cutadapt and FastQC to Consistently Apply Adapter and Quality Trimming to FastQ files, with Extra Functionality for RRBS Data (Babraham Institute, 2015).
Hassan-Abdallah, A. et al. Cloning, expression and crystallization of heterotetrameric sarcosine oxidase from Pseudomonas maltophilia. Protein Expr. Purif. 43, 33–43 (2005).
Suzuki, M. Purification and some properties of sarcosine oxidase from Corynebacterium sp. U-96. J. Biochem. 89, 599–607 (1981).
Katoh, K. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Abascal, F., Zardoya, R. & Posada, D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21, 2104–2105 (2005).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Sayers, E. W. et al. Database resources of the National Center for Biotechnology Information in 2023. Nucleic Acids Res. 51, D29–D38 (2023).
Celebi, F. M., Eisen, J. A., Hochstrasser, M. L. & Reiter, T. Clustering the NCBI NR Database to Reduce Database Size and Enable Faster BLAST Searches (Arcadia Science, 2023).
Kans, J. Entrez Direct: E-utilities on the UNIX Command Line (National Center for Biotechnology Information, 2025).
Steinegger, M., Mirdita, M. & Söding, J. Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold. Nat. Methods 16, 603–606 (2019).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Groussman, R. D., Coesel, S. N., Durham, B. P., Schatz, M. J. & Armbrust, E. V. The North Pacific Eukaryotic Gene Catalog of metatranscriptome assemblies and annotations. Sci. Data 11, 1161 (2024).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European molecular biology open software suite. Trends Genet. 16, 276–277 (2000).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Groussman, R. D., Blaskowski, S., Coesel, S. N. & Armbrust, E. V. MarFERReT, an open-source, version-controlled reference library of marine microbial eukaryote functional genes. Sci. Data 10, 926 (2023).
Hogle, S. L. MARMICRODB database for taxonomic classification of (marine) metagenomes (1.0.0). Zenodo https://doi.org/10.5281/zenodo.3520509 (2019).
Eddy, S. R. Accelerated profile HMM searches. PLoS Comput. Biol. 7, e1002195 (2011).
Mistry, J. et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021).
Potter, S. C. et al. HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204 (2018).
Heal, K. R., Ferrer-González F. X., Sacks, J. S. & Finch, A. H. Data analysis for homarine catabolism manuscript. GitHub https://github.com/IngallsLabUW/Homarine_Catabolism_MS (2026).
Schymanski, E. L. et al. Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ. Sci. Technol. 48, 2097–2098 (2014).
Acknowledgements
We thank the captains, crews and science parties of the RV Kilo Moana on cruise KM1906, of the RV Thomas G. Thompson on cruises TN397 and TN412, and RV Rachel Carson on cruises RC078 and RC104. We thank L. T. Rodriguez and S. Garcia for assistance with laboratory analyses; S. N. Coesel, S. Graff Van Creveld, S. Blaskowski, D. Weidner, B. Grodner and J. E. Schreier for valuable conversations and assistance with data analysis; M. A. Moran and C. R. Reisch for access to the R. pomeroyi DSS-3 arrayed BarSeq library. This work was supported by grants from the Simons Foundation (LS award ID: 385428, A.E.I.; SCOPE award ID 329108, A.E.I.; award ID 916384, B.P.D. and 721244 to E.V.A.), the National Science Foundation (OCE-2125886 to A.E.I. and K.R.H.; OCE-2124712 to O.A.S.), the University of Florida (Research Opportunity Seed Fund award ID 00131632 to B.P.D.), and the Center for Chemical Currencies of a Microbial Planet (C-CoMP; award ID 2019589 to F.X.F-G).
Author information
Authors and Affiliations
Contributions
K.R.H., O.A.S. and A.E.I. designed the study. F.X.F.-G., K.R.H., J.S.S., A.H.F., Z.B., C.B.L., M.P.G., E.V.A., O.A.S. and A.E.I. performed field work. FXF-G, K.R.H., J.S.S., A.H.F., L.T.C., Z.B., Y.R.-M., S.K.A., C.B.L., M.P.G., S.E.F., C.I.B.G., J.M.L.D. and O.A.S. performed data and/or laboratory analyses. L.S.Y.C. and B.P.D. provided and validated R. pomeroyi mutant strains. F.X.F.-G., K.R.H., J.S.S., A.H.F., Z.B. and O.A.S. analysed and visualized data. M.Z.T., C.I.B.G., J.M.L.D., K.J.B. and L.A.Y. collected data and built mutant strains. F.X.F.-G., K.R.H., O.A.S. and A.E.I. wrote the manuscript. All authors edited and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Growth phenotypes of transposon and deletion mutants affecting homarine catabolism.
(a) A transposon insertion in R. pomeroyi DSS-3 gene SPO3191 (aldehyde dehydrogenase) prevents growth on homarine. Insertion in mgdA (SPO1585) and mgdD (SPO1588) disables growth on N-methylglutamic acid (NMGA). (b) Deletion of homD (ACFLL8_11515) (ΔhomD) in Cobetia sp. OBi1 disables growth on homarine. Complementation with the wild-type (WT) allele (p-homD) rescues growth on homarine. Empty complementation vector (p-null) shows no growth on homarine.
Extended Data Fig. 2 Deletion of the GBT gene (ΔhomT) in the proposed homarine catabolic operon impairs growth of Cobetia sp. OBi1 on homarine.
(a) Growth of wild-type and ΔhomT on glucose or homarine. (b) Growth of ΔhomT plasmid complementation strains expressing wild-type HomT or RFP induced with 50 µM IPTG. (c) HomT complementation increases the growth rate of Cobetia sp. OBi1 ΔhomT on homarine relative to a control strain expressing RFP. Letter labels (b, c, and d) denote significantly different growth rates between sample groups (median test, p < 0.9). The y-axis position of letters labels corresponds to the median growth rate of each group. (d) RFP fluorescence of Cobetia sp. OBi1 ΔhomT cultures expressing RFP normalized to sample absorbance. The RFP data corresponds to the experiment in (b) and (c).
Extended Data Fig. 3 Summarized results from metabolomics samples in Cobetia sp. OBi1.
Each marker is a mass feature, shaped by detection in the particulate or dissolved fraction of the metabolome. Highlighted mass features were significantly enriched in both the homarine vs glucose and glucose+homarine vs homarine comparisons. Highlighted mass features are annotated with identifications when available, or their retention time (RT), m/z, and polarity (- or +) when not identified. Note that only mass features where more than one ion type (that is adduct or polarity) was detected are shown (with the derived [M + H]+ or [M-H]- ion shown). Full results are given in Tables S14 and S15 for particulate and dissolved results, respectively.
Extended Data Fig. 4 Support for the identification of N-methylglutamic acid.
Extracted ion chromatograms at 5 ppm tolerance at 162.0759 m/z (predicted m/z for [M + H]+ ion of N-methylglutamic acid) show that retention time matches from sample of Cobetia sp. OBi1 to commercially acquired standard. This sample was re-run to ensure similar chromatography. MS2 spectra of isolated ions with m/z of 162 show near-identical fragments in samples of Cobetia sp. OBi1 and commercially acquired standard.
Extended Data Fig. 5 Support for the identification of N-methylglutamine.
We were not able to obtain a commercial standard for this compound. Shown is MS2 spectra of isolated ions with m/z of 161 (matching retention time shown in Fig. 2c) from Cobetia sp. OBi1 (top, in green). Inverted spectra is MS2 spectra of isolated 147 ions of a glutamine standard (in black), and those same fragments shown with mass difference corresponding to an additional methyl group (in red, corresponding to putative fragments associated with N-methylglutamine associated with glutamine fragments by arrows). All the major fragments in the Cobetia sp. OBi1 spectra (green, top) are associated with either fragments from glutamine (black) or fragments corresponding to an additional methyl group on fragments (red), as one would expect for N-methylglutamine. Combining this spectral interpretation with the matched m/z (green dashed line) to the predicted m/z for [M + H]+ ion of N-methylglutamine (red dashed line) leads us to grant the identification of the mass feature as a highly likely N-methylglutamine (level 2b confidence according to guidance84).
Extended Data Fig. 6 Conservation of hom operon arrangement.
(a) Distance (in base pairs) of homBCDE genes from homA in NCBI genomes, with families with > 120 genomes highlighted separately. Each subpanel indicates if the family is Alphaproteobacteria or Gammaproteobacteria, number of genomes, and percentage of genomes with homE. (b) There is a statistically significant difference (p < 0.001 by t-test) in genome size between groups with and without homE.
Extended Data Fig. 7 In-situ expression of homA in the environment.
Transcript abundance of homA in heterotrophic bacteria, alongside chlorophyll a (μg/L) concentrations.
Extended Data Fig. 8 Cell abundance normalized transcript expression across the Pacific transect.
Mean log2-normalized transcript counts for homA and homB are shown for each station after per-cell normalization using the median abundance of ten universal single-copy housekeeping genes. Points represent mean transcript abundance across biological replicates and horizontal error bars show one standard deviation. These fold-change profiles provide a higher-resolution view of per-cell variation in homarine catabolic gene transcription across the latitudinal gradient and complement the absolute transcript abundance patterns shown in Fig. 4 and Extended Data Fig. 7.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–4.
Supplementary Tables (download XLSX )
Supplementary Tables 1–19.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ferrer-González, F.X., Heal, K.R., Sacks, J.S. et al. Conserved pathway for homarine catabolism in environmental bacteria. Nat Microbiol (2026). https://doi.org/10.1038/s41564-026-02313-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41564-026-02313-7
