
Abstract
Cancer immunotherapies with antibodies blocking immune checkpoint molecules are clinically active across multiple cancer entities and have markedly improved cancer treatment1. Yet, response rates are still limited, and tumour progression commonly occurs2. Soluble and cell-bound factors in the tumour microenvironment negatively affect cancer immunity. Recently, growth differentiation factor 15 (GDF-15), a cytokine that is abundantly produced by many cancer types, was shown to interfere with antitumour immune response. In preclinical cancer models, GDF-15 blockade synergistically enhanced the efficacy of anti-PD-1-mediated checkpoint inhibition3. In a first-in-human phase 1–2a study (GDFATHER-1/2a trial, NCT04725474), patients with advanced cancers refractory to anti-PD-1 or anti-PD-L1 therapy (termed generally as anti-PD-1/PD-L1 refractoriness) were treated with the neutralizing anti-GDF-15 antibody visugromab (CTL-002) in combination with the anti-PD-1 antibody nivolumab. Here we show that durable and deep responses were achieved in some patients with non-squamous non-small cell lung cancer and urothelial cancer, two cancer entities identified as frequently immunosuppressed by GDF-15 in an in silico screening of approximately 10,000 tumour samples in The Cancer Genome Atlas database. Increased levels of tumour infiltration, proliferation, interferon-γ-related signalling and granzyme B expression by cytotoxic T cells were observed in response to treatment. Neutralizing GDF-15 holds promise in overcoming resistance to immune checkpoint inhibition in cancer.
Similar content being viewed by others


Main
Empowering the immune system to fight cancer has revolutionized tumour treatment in the past two decades1. Anti-PD-1 and anti-PD-L1 antibodies are now established as a standard of care for first-line treatment of cancer. However, response rates of just 15–50% in many solid tumour types and high progression and relapse rates suggest that resistance mechanisms often prevent successful tumour control. Tolerogenic factors such as transforming growth factor-β (TGFβ) are often co-opted by cancer cells for immune escape4,5. Recently, we demonstrated that the divergent TGFβ superfamily member GDF-15 (ref. 6), which is expressed at high levels in placenta and solid tumours, stands out as a prominent molecule used by tumours to keep the immune system at bay3. This mirrors the physiological role of GDF-15 in feto-maternal tolerance towards placental alloantigens7,8. Here we report results from a clinical trial of the GDF-15-blocking antibody visugromab (CTL-002) co-administered with anti-PD-1 to patients in late- or last-line treatment with relapsed or refractory cancer under ongoing prior anti-PD-1 or anti-PD-L1 treatment.
GDFATHER clinical trial phase 1
The first-in-human exploration of the GDF-15 neutralizing antibody visugromab was a combined phase 1 and 2a trial (CTL-002-001; NCT04725474). Preclinical toxicology testing of visugromab in cynomolgus monkeys showed no toxicologic findings up to the maximum feasible dose of 100 mg kg−1 per week. Phase 1 (part A) was a classic ‘3 + 3’ dose escalation in advanced-stage, metastatic mixed solid tumours. To investigate safety and tolerability, patients in last-line therapy who had relapsed or were refractory to prior checkpoint-inhibitor (CPI) treatment received visugromab in combination with the CPI nivolumab. In phase 2a (part B), antitumoural activity of the combination of visugromab and nivolumab was explored in various defined advanced-stage, metastatic solid tumour types. Again, patients had relapsed or were refractory to prior CPI treatment.
In phase 1 (December 2020 to November 2021), 25 patients were enrolled to receive 5 predefined dose levels (DLs) of visugromab (0.3, 1, 3, 10 and 20 mg kg−1; once every other week) following a 3 + 3 dose-escalation design. The first three patients for each DL received visugromab as a monotherapy for one cycle (14 days) as a safety run-in, followed by a combination of visugromab with nivolumab (second and all subsequent cycles; once every other week). Treatment was continued until disease progression, unacceptable toxicity or withdrawal of informed consent. After the respective dose-limiting toxicity period for each cohort had been cleared, the dose was escalated. For the highest three DLs, additional backfill cohorts of three patients each were enrolled, providing additional information on the combination of visugromab and nivolumab for the respective DL. Patients in the backfill cohorts received the combination treatment starting from the first cycle.
The patients with mixed solid tumours who were enrolled had been heavily pretreated, entering the trial with a median of 4.4 prior systemic lines of treatment, and no available established treatment options were left for their malignant disease. All patients had to have experienced relapse or progression to at least one prior anti-PD-1/PD-L1 treatment by strict criteria, either in monotherapy or in combination. Sequential tumour biopsies were performed at the baseline (n = 22/25), on day 14 (before cycle 2, monotherapy phase; n = 13/16) and on day 28 (after having received one cycle of visugromab and nivolumab; n = 17/25). Patient baseline characteristics are provided in Supplementary Table 1.
Visugromab in combination with nivolumab was overall well tolerated at all DLs with no dose-limiting toxicity and just one treatment-emergent adverse event (TEAE) ≥ grade 4 occurring (acute respiratory failure), not considered by the investigator as treatment related. Grade 3 TEAEs occurred in 12 patients (48%) reporting at least 1 TEAE. The median duration of treatment across all DLs was 10 weeks, and that in the highest three DLs was 14 weeks. For five individuals in this population of heavily pretreated patients with tumours in last-line therapy, indications of clinical benefit were observed (Fig. 1a), all at DLs 3−5. Three patients experienced a confirmed partial response (PR) (1 × mesothelioma, 1 × liver hepatocellular cancer (HCC), 1 × cancer of unknown primary origin; Supplementary Table 2). The patient with cancer of unknown primary origin underwent radiofrequency ablation of a remaining, sole small positron emission tomography-positive liver lesion (at 8 months on treatment, with subsequent staging as complete response (CR) by positron emission tomography with computed tomography) and had a prolonged CR until occurrence of a single new lung lesion that had to be irradiated. The mean duration of response (DOR, from start of treatment9) was 12.9 months (median 7.1 months; Supplementary Table 2). Two additional patients experienced prolonged disease stabilization longer than 6 months (1 × non-small cell lung cancer (NSCLC) and 1 × ocular melanoma). A sixth patient with mucosal melanoma achieved 5.5 months of disease stabilization, and then had local progression in a single liver lesion compressing a vessel. Local palliative irradiation was conducted. Consecutive computed tomography staging revealed a PR (−34% by response evaluation criteria in solid tumours (RECIST) 1.1 criteria) with notable abscopal antitumoural effects in liver lesions not included in the irradiation field, all under continued treatment with visugromab and nivolumab.

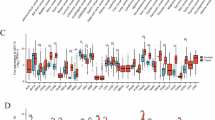
a, Swimmer plot for 25 patients with mixed solid tumours treated with escalating doses (DLs) of visugromab (0.3, 1.0, 3.0, 10 and 20 mg kg−1, once every other week) and nivolumab in last-line treatment, with relapsed or refractory cancer under prior anti-PD-1 or anti-PD-L1 treatment. Data show time course and remission status, with tumour regression (blue; greater than −5%) and PR (light green) as per RECIST 1.1. Broken lines indicate not evaluable (NE). MES, mesothelioma; UCEC, uterine corpus endometrial carcinoma; OMEL, ocular melanoma; CCA, cholangiocarcinoma; MEL, melanoma; RCC, renal cell carcinoma; CUP, cancer of unknown primary origin; OVA, ovarian cancer; TNBR, triple-negative breast cancer; CER, cervical cancer; H&N, head and neck squamous cell carcinoma; CRC, colorectal cancer; BF, backfill; PD, progressive disease; SD, stable disease; RT, radiotherapy; RFA, radiofrequency ablation. b, Fold change of CD4+FOXP3−, CD8+, CD3+Ki67+ and CD3+GZMB+ T cells in evaluable, sequential biopsies of the above patients at day 14 (visugromab monotherapy) and day 28 (combination) compared to baseline (Bsl). Patients with >2-fold increase are depicted in black and patients with <2-fold increase are depicted in light grey. c, Representative immunohistochemical and immunofluorescence images of tumour area (green square 400 × 400 µm) demonstrating baseline (day 0; upper panel) versus monotherapy (day 14; middle panel) and combination therapy impact (day 28; lower panel), showing increase in CD8+ (purple label; n = 8), CD4+ (yellow label; n = 7), in the left panel, and CD3+Ki67+ (red label; n = 8) and increase in GZMB synthesis (pink label; n = 5), in the right panel.
Pharmacodynamic observations in phase 1
As previously reported, tumour-derived GDF-15 inhibits the LFA-1–ICAM-1 axis in T cells3, impairing T cell migration into tumour tissue. Neutralization of GDF-15 enhances T cell infiltration and efficacy of immune checkpoint blockade in mouse GDF-15-expressing tumour models3. Sequential biopsies were taken at the baseline and day 14 and day 28 of treatment for immunohistochemical T cell quantification and characterization. Neutralization of GDF-15 led to a tumour-selective increase (>2-fold) in the numbers of CD4+FOXP3− T cells (7/14 patients) and CD8+ T cells (8/15 patients) and induction of CD3+Ki67+ proliferating T cells (8/15 patients), as well as increases in CD3+GZMB+ cytotoxic T cell numbers (5/10) across all DLs (Fig. 1b,c). The median fold change at day 14 was 2.13 for CD4+FOXP3− T cells, 1.78 for CD8+ T cells, 1.46 for CD3+Ki67+ T cells and 1.6 for CD3+GZMB+ T cells, with much higher increases observed in tumour tissue of individual patients. At day 28, the median fold change decreased for CD4+FOXP3− and CD8+ T cells to 1.92 and 1.22, respectively, whereas it further increased for proliferating T cells (2.32) and remained stable for CD3+GZMB+ T cells (1.52). Notably, the increase in T cell number, proliferation induction and increase in CD3+GZMB+ cell number were observed under initial visugromab monotherapy (Fig. 1b,c), possibly setting the stage for successful antitumour immune responses by T cells during anti-PD-1 treatment. The data are indicative of immunosuppressive effects of GDF-15 on intratumoural T cell abundance and activation, which can be reversed by treatment with visugromab. As the number of on-treatment biopsies from responding patients is still limited, it is not possible to correlate these observations with clinical response.
Phase 2a indication selection
To identify the tumour types that are affected by GDF-15, integrative high-throughput pan-cancer immune-transcriptomic analyses were performed. Clinical data from 33 cancer types (Fig. 2a and Extended Data Tables 1 and 2), comprising approximately 10,000 patient-derived tumours, were retrieved from The Cancer Genome Atlas (TCGA). In detail, GDF15 expression patterns in tumour (primary, checkpoint naive) and normal tissues were analysed, segregated by molecular subgroups and genotypes and correlated with clinical parameters (including outcome), and immune signatures in the tumour microenvironment (TME). As a result, in at least 10 of 13 selected immune-related analyses, an inverse relation between GDF15 mRNA expression and T cell transcriptomic signatures could be revealed for colorectal cancer (microsatellite stable (MSS) and microsatellite instability-high), urothelial cancer (UC) or, breast cancer, pancreatic adenocarcinoma, lung adenocarcinoma (LUAD), kidney renal clear cell carcinoma, testicular germ cell tumours, and cervical squamous cell carcinoma and endocervical adenocarcinoma. This is consistent with the postulated GDF-15-related T cell exclusion mechanism and hence may predict promising indications for related therapy (Fig. 2a). Stratification for different molecular subtypes of NSCLC revealed a significantly higher level of GDF15 expression in lung adenocarcinoma when compared with lung squamous carcinoma (Fig. 2b). Correlation analyses also showed a strong negative correlation between GDF15 expression and immune markers in lung adenocarcinoma, not found in lung squamous carcinoma (Fig. 2c and Extended Data Table 2). To estimate the average distribution of immune-cell populations from the TCGA bulk RNA-sequencing data, a quanTIseq-based deconvolution was performed (Fig. 2d). A statistically significant decrease in the number of infiltrating CD8+ T cells was observed in LUAD with higher compared to lower levels of GDF15 expression, and the same subset clearly decreased in number in basal squamous UC with increasing GDF15 expression level. On the basis of these transcriptomic analyses, non-squamous NSCLC and UC were selected for phase 2a investigation.

a, Immune-related analyses and relation to GDF15 mRNA expression ranked by a consensus score. IFNγ, interferon-γ. Two-sided Spearman’s rank correlation (coefficient ρ) test was performed. P values were adjusted for multiple testing for each indication across tested signature scores on the basis of the false discovery rate (FDR) according to the Benjamini–Hochberg method. Significant positive (FDR < 0.1 and ρ > 0.1) and negative (FDR < 0.1 and ρ < 0.1) relationships between GDF15 expression and immune signature score are indicated in red and blue, respectively. For inflamed to T cell exclusion (excl.), an absolute value of the difference in z-scores (|∆z|) > 0.2 was used as the cutoff (z-scores were calculated using Fisher’s z-transformation on ρ). ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; DLBC, diffuse large B-cell lymphoma; ESCA, oesophageal carcinoma; GBMLGG, glioblastoma and low-grade gliomas; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumours; THCA, thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma. b, Normalized GDF15 mRNA expression in LUAD (n = 515), lung squamous cell carcinoma (n = 501; left panel), luminal UC subtypes (luminal, luminal infiltrated, luminal papillary, n = 246) and basal squamous UC (n = 142, right panel). TPM, transcripts per million. Lower and upper hinges, first and third quartile (interquartile range (IQR)); thick line, median. The whiskers extend to the smallest and largest values no further than 1.5 × IQR. P values are from two-sided Wilcoxon rank sum test. c, Correlation analyses of normalized GDF15 expression in lung and bladder cancer subtypes with immune-related signatures; Spearman rank-based correlation coefficient rho (ρ) is indicated according to the legend. P values are from two-sided Spearman’s rank correlation test and not adjusted for multiple comparisons. IMPRES, immuno-predictive score. CTL, cytotoxic T lymphocytes. d, Distribution of averaged cell fractions according to GDF15 expression level (low, mid, high). NK, natural killer.
Phase 2a dose selection
Pharmacokinetics (PK) demonstrated a dose-proportional increase in visugromab exposure across the dose range from 0.3 to 20 mg kg−1 in phase 1, with little accumulation and no obvious loss of exposure after repeated treatment cycles, indicative of no anti-drug antibody response over the observation period. To ensure neutralization of elevated and variable GDF-15 levels in serum and tumour microvasculature of patients with cancer, a population PK–pharmacodynamics (PD) modelling approach was developed on the basis of serum analyses from the phase 1 study (Extended Data Fig. 1a,b). By combining this modelling of visugromab and total GDF-15 levels with estimated population parameters, and with a previously developed model for the distribution of GDF-15 in tumours, free GDF-15 concentration in serum and the tumour microvasculature could be predicted for 3, 10 and 20 mg kg−1 dosing once every second, third or fourth week. This PK–PD model was fully consistent with the observed total visugromab and total GDF-15 levels in the phase 1 study. Higher GDF-15 baseline levels (>2 ng ml−1) require more visugromab for neutralization. These calculations support a dosing regimen of ≥10 mg kg−1 every 2 weeks, or ≥20 mg kg−1 every 3 or 4 weeks, to constantly maintain free GDF-15 in the tumour below the nominal average physiologic level of 0.5 ng ml−1 (ref. 10) for the whole duration of 2- to 4-weekly cycles. For the subsequent phase 2a dose-expansion part of the study, 10 mg kg−1 visugromab every second week was thus selected as the recommended phase 2 dose in combination with nivolumab at the standard dose. In phase 2a, visugromab continued to show linear and cycle-independent PK behaviour.
Phase 2a clinical activity
Sample size calculation followed a single-arm, exploratory Simon two-stage design, with 14 participants in stage 1 and 13 participants in stage 2. All participants were in late or last line of treatment with a median of three prior systemic cancer therapies. To rule out substantial visugromab-independent antitumoural activity of nivolumab, patients were included only when their cancer was relapsed or refractory to prior, approved anti-PD-1- or anti-PD-L1-containing treatment as per defined, strict criteria. Patients were enrolled only if they had a minimum of 12 weeks of continuous prior exposure to anti-PD-1 or anti-PD-L1, to rule out inclusion of pseudoprogressors, and if their relapse or progression had occurred on ongoing anti-PD-1- or anti-PD-L1-containing treatment. Based on data in the literature, the expected rate of patients responding to monotherapy retreatment with an approved anti-PD-1 or anti-PD-L1 agent such as nivolumab in such strictly defined relapsed or refractory populations is ≤5% for both NSCLC11,12,13,14,15 and UC16. Response assessment was performed locally throughout phase 2a (Supplementary Clinical Trial Protocol).
NSCLC cohort
As of May 2024, 20 male and 7 female patients had been enrolled in the NSCLC cohort. Baseline characteristics are provided as Supplementary Table 3. Histologic subtypes were non-squamous in 21 (77.8%) and squamous in 6 (22.2%) participants; 1 participant had a mixed, dominantly neuroendocrine tumour histology and was assigned to the non-squamous group. The mean duration of study treatment was 3.9 months with two patients still ongoing on treatment. Regarding efficacy, the overall response rate (ORR) is 4/27 (14.8%) as per RECIST 1.1, with 2 PR and 2 CR (1 initially as a lasting PR on trial treatment, maturing to a confirmed CR post treatment discontinuation due to a TEAE and prior focal irradiation of a progression-suspected lymph node area). Of note, all responses were observed in non-squamous NSCLC, resulting in an ORR of 19.0% (4/21) in this subgroup (Fig. 3a, Extended Data Fig. 2a and Supplementary Table 4). Responding patients had experienced clinical benefit on prior anti-PD-1- or anti-PD-L1-containing treatment (two in combination with poly-chemotherapy; Supplementary Table 5). This may be due to a general bias for trials in patients in late-line treatment based on the fact that approximately only 8% of all patients with NSCLC reach third-line treatment (retrospective study of 3,995 patients with NSCLC17, for which most surviving patients had to have experienced some prior benefit from earlier lines of treatment). The 4 responding patients had received prior anti-PD-1 or anti-PD-L1 treatment for a median of 19.7 months (versus 23 non-responding patients with a median of 5.8 months). Responses occurred in both PD-L1+ and PD-L1− tumours. The current DOR is 15.3 and 16.6 months (mean and median), respectively, with 3 of 4 responses ongoing.

a,b, Swimmer plot for patients with NSCLC (a) and UC (b) illustrating response assessment according to RECIST 1.1 and time on study. Patients with PR or CR are highlighted in light and dark green, respectively; patients with SD but decrease in target lesion size (>5%; tumour shrinkage) are highlighted in blue. Light blue indicates SD, and PD is depicted in grey (X indicates detection of PD). Patients who received radiotherapy on treatment are not evaluable (NE (post RT)) anymore, which is indicated by a hatched fill. Scans were performed approximately every 8 weeks.
Regarding safety, at least one TEAE with a grade of ≥3 was reported in 16 (59.3%) participants. Among those were 15 (55.6%) participants with at least 1 grade 3 TEAE, 1 (3.7%) participant with at least 1 grade 4 TEAE, and 3 (11.1%) participants with at least 1 grade 5 TEAE (Supplementary Table 6). At least one treatment-related adverse event (TRAE) with a grade of ≥3 was reported in two (7.4%) participants. Among those, two (7.4%) participants had at least one TRAE of grade 3, one (3.7%) had at least one grade 4 TRAE, and one (3.7%) had at least one grade 5 TRAE. The grade 4 (hepatic failure and acute kidney injury) TRAEs, which led to the grade 5 (multiple organ dysfunction syndrome) TRAE, were observed in an individual with NSCLC, who had (low-grade) persisting prior kidney damage at study entry from an experimental antibody–drug conjugate treatment that had ended 1 month before study entry. Kidney and liver tissue examination indicated acute interstitial nephritis, compatible with drug-toxic renal injury and potential drug-induced liver injury, with no indication of chronic hepatopathy (Supplementary Note 1). The grade 3 TRAEs consisted of gastrointestinal disorder (diarrhoea (n = 1)) and laboratory abnormalities (elevated aspartate aminotransferase levels and hypokalaemia; one individual).
UC cohort
As of May 2024, 27 efficacy-evaluable participants had been enrolled in the UC cohort. Baseline characteristics are provided as Supplementary Table 7. The median for prior lines of therapy was 3 (mean 3.3). The mean duration of treatment was 5.4 months and 5 participants were still on study. The observed ORR was 18.5% (5/27) as per RECIST 1.1 (Fig. 3b, Extended Data Fig. 2b and Supplementary Table 8). The current mean DOR is 16.4 months (median 14.4 months; Supplementary Table 5). One patient continues experiencing a confirmed CR, ongoing for over 20 months. Three of five responding patients had experienced clinical benefit on prior anti-PD-1- or anti-PD-L1-containing therapy (Supplementary Table 5). All responding patients had received prior anti-PD-1- or anti-PD-L1-containing therapy for a median of 4.9 months (versus 22 non-responding patients with a median of 6.9 months).
Regarding safety, at least 1 TEAE with a grade of ≥3 was reported in 14 (51.8%) patients. Among those, 14 (51.8%) patients had at least 1 grade 3 TEAE, no patients had at least 1 grade 4 TEAE, and 1 patient (3.7%) had at least 1 grade 5 TEAE (urinary tract infection). In the UC expansion cohort, at least one TRAE with a grade of ≥3 was reported in two (7.4%) patients (Supplementary Table 9). Within those, two (7.4%) patients had at least one TRAE of grade 3, and no patients had at least one TRAE of grade 4 or 5. The grade 3 TRAEs consisted of immune-mediated pneumonitis in one patient and hypertension and platelet count decrease in one patient.
Notably, more than half of all responding patients in the NSCLC and UC cohorts experienced a response depth level on study treatment as per RECIST 1.1 criteria that had not been reached on initial, prior anti-PD-1 or anti-PD-L1 treatment that was mostly administered as first-line treatment and in combination with (poly-)chemotherapy (Supplementary Table 5). In addition, 3 of the 4 patients with a CR had not achieved a CR on any prior line of systemic treatment, including their initial anti-PD-1 or anti-PD-L1 treatment. Seven of nine responses in these two cohorts are ongoing at the time of this report. Representative scans of tumour regressions in phase 1 and phase 2a are provided as Extended Data Fig. 7.
Further cohorts
Additional tumour types that were also investigated in phase 2a included melanoma, HCC (after seeing a short-lived but confirmed PR in the sole patient with HCC in phase 1) and MSS colorectal cancer (Supplementary Tables 10–12). Antitumoural activity comparable to that seen in the non-squamous NSCLC and UC cohorts is being detected in individuals with HCC (3 PRs, 1 CR and 1 patient with durable tumour shrinkage in 20 currently evaluable HCC participants of the trial, with the HCC expansion cohort ongoing), reflective of an overall interim ORR of 20.0% as per RECIST 1.1 for this tumour type (Supplementary Table 13). The dedicated phase 2a HCC cohort has been accordingly expanded to n = 27 patients and data will further mature. No relevant activity was seen in melanoma and colorectal cancer (Supplementary Tables 14 and 15). TEAEs of grade 3 or greater are summarized for melanoma, HCC and colorectal cancer in Supplementary Tables 16–18.
Chemokines and interferon signalling
As T cell density increase and proliferation were observed under visugromab monotherapy in phase 1, in-depth RNA analysis using the nCounter PanCancer IO 360 panel for immuno-oncology was performed. The analysis of differentially expressed genes revealed the induction of inflammatory genes such as CXCL9, CXCL10, CCL8, CCR5 and IRF7 and pathways related to interferon and cytokine signalling in tumour tissue from participants with an increased level of T cell influx (>2-fold) on day 14 of therapy (Fig. 4a,b and Extended Data Table 3). The induction of an interferon-γ signature is indicative of the inflammatory properties and functionality of infiltrating immune cells. It was not observed in biopsies from participants with <2-fold T cell density increase (Extended Data Fig. 3b). In addition, the exhaustion markers PDCD1, LAG3 and HAVCR2 were not induced (Extended Data Fig. 3a). The serum levels of the interferon-γ-inducible chemokines CXC motif chemokine ligand 9 (CXCL9) and 10 (CXCL10) were significantly increased following treatment and remained elevated over the monitored period of 6 weeks (Fig. 4c for CXCL9 and Extended Data Fig. 4a for CXCL10). In addition, a comparison between different response groups (progressive disease and stable disease versus PR and CR) showed higher exposure (area under the curve) of CXCL9 (progressive disease and stable disease: 631.7, s.e. ±267.3; PR and CR: 864.0, s.e. ±345.8) and of CXCL10 (progressive disease and stable disease: 322.4, s.e. ±99.26; PR and CR: 414.4, s.e. ±94.19) during therapy in participants who developed PR or CR (Fig. 4d for CXCL9 and Extended Data Fig. 4b for CXCL10). Of note, baseline levels of CXCL9 and CXCL10 did not differ between response groups (Fig. 4e for CXCL9 and Extended Data Fig. 4c for CXCL10).

a, Volcano plot presenting differentially expressed genes in biopsies on day 14 of visugromab monotherapy compared to pretreatment biopsies in participants with >2-fold increase in CD8+ and CD4+ T cells (n = 5). P values are from two-sided moderated t-test using limma. Dashed vertical lines, −1.5 and +1.5 fold change. Dashed horizontal line, P = 0.05. b, Reactome pathway analysis shows that significantly upregulated genes from the PanCancer IO 360 panel in a are part of important inflammation-related pathways. P values are from two-sided Fisher’s exact test and adjusted according to the Benjamini–Hochberg method (Padj). Numbers of PanCancer IO 360 panel genes in the respective pathway (second number to right of each bar) and of significantly upregulated genes in the respective pathway (first number to right of each bar) are indicated. Grey vertical line, Padj = 0.1. OAS, oligoadenylate synthetases. c, Significant induction and maintenance of CXCL9 following treatment for 6 weeks in the blood of participants with NSCLC, HCC and UC in the phase 2a study. P values are reported from repeated-measures one-way analysis of variance (ANOVA) with Geisser–Greenhouse correction, corrected for multiple comparisons by Dunnett’s test. d, Baseline CXCL9 levels of participants with best overall response according to RECIST 1.1. P values are reported from two-tailed Mann–Whitney test. e, Heat map representing higher mean values (pg ml−1) of CXCL9 following treatment in participants with best overall response of PR and CR (lower row) compared to participants with best overall response of PD and SD (upper row). For the box plots in c,d: centre line, median; box, IQR; whiskers, 1.5 × IQR; outliers are depicted individually. C1D1, cycle 1 day 1; C1D2, cycle 1 day 2; C2D1, cycle 2 day 1; C3D1, cycle 3 day 1.
GDF-15 correlates with immune infiltrates
Patients with a baseline serum GDF-15 level of >1.5 ng ml−1 showed a statistically significant reduction in CD4+FOXP3+ regulatory T cell density, cytotoxic CD3+GZMB+ T cell density and proliferating CD3+Ki67+ T cell density in the tumour, indicative of a GDF-15 suppressive effect on the abundance of T cell subsets, T cell proliferation and functionality (Fig. 5a). Effects on other analysed immune subsets did not reach statistical significance. The negative correlation of GDF-15 in serum with distinct immune-cell subsets in the TME was supported by a translational research study of a cohort of patients in early-line treatment, most newly diagnosed with UC. Patients with elevated GDF-15 levels showed a reduced percentage of PD-L1+ tumour cells, CD8+ T cells and proliferating CD45+Ki67+ immune cells (Extended Data Fig. 5a). Owing to the small cohort size, the results were not statistically significant. Serum GDF-15 levels of phase 2a participants at baseline were significantly higher in patients with UC as compared to patients with NSCLC but elevated in both indications when compared to physiological levels in healthy individuals10 (Extended Data Fig. 5b). Of note, correlation analyses of GDF15 expression with immune markers and signatures in UC (Extended Data Table 2) revealed a stronger negative correlation in the basal squamous subtype compared to the luminal subtype (Fig. 2c), even though the latter showed a significantly higher level of GDF15 expression (Fig. 2b). Notably, in both molecular subtypes of BLCA, distinct immune-cell fractions change depending on the level of GDF15 expression, with the basal squamous subtype showing a more pronounced negative correlation suggestive of the immunosuppressive impact of GDF-15 on the TME (Fig. 2d).

a, CD4+FOXP3+ T cell density, CD4+FOXP3− T cell density and CD3+Ki67+ T cell density are significantly reduced in tumour biopsies from patients in phase 1 and 2 with serum GDF-15 (sGDF-15) of >1.5 ng ml−1. P values are from two-sided Mann–Whitney test. b, In baseline tumour tissue with very high CD8+ T cell infiltration from patients in phase 1 and 2, low (pro-)GDF-15 expression is observed. P value is reported from two-sided Mann–Whitney test. c, TME characterization by PD-L1 tumour proportion score (TPS) and CD8+ T cell density (cells mm−2) in biopsies from patients in phase 1 and 2 before treatment (n = 97); PR and CR are highlighted in green.
Notably, baseline tumour tissue from patients with a very high level of CD8+ T cell infiltrates showed a consistently low level of pro-GDF-15 staining (H score; Fig. 5b) across all evaluable participants in phase 1 and 2a, supporting the suppressive role of GDF-15 on T cell extravasation into the TME3. This could be confirmed for NSCLC and UC in indication-specific analyses, separately (Extended Data Fig. 5c).
Immunohistochemical characterization of biopsies before treatment revealed that five out of nine PR/CR showed a positive PD-L1 tumour proportion score (>5) and elevated CD8+ T cell density (>300 cells mm−2; Fig. 5c), suggesting that most of the responding tumours may still be intrinsically immunogenic, but capable of limiting antitumoural immune responses. Further exploration is needed to assess whether PD-L1 expression in combination with elevated CD8+ T cell counts and/or other similar markers may serve as biomarker(s) for enrichment of responding patients.
Discussion
Although immune checkpoint inhibition has become a mainstay of cancer treatment, deep and lasting responses are achieved only in a minority of individuals with metastatic cancer, and cures are mostly limited to very small subpopulations, if any, perhaps with the exception of individuals with cutaneous melanoma18.
Most metastatic solid tumours in patients are refractory to current immunotherapy from the start of anti-PD-1 or anti-PD-L1 treatment. For most tumour types treated with CPI, 50–85% of patients fail to respond to initial anti-PD-1 or anti-PD-L1 monotherapy as per RECIST 1.1 criteria and in most tumour progression or relapse and death occurs19.
So far, blocking inhibitory signals or adding activating stimuli on top of PD-1-based immune checkpoint blockade has still mostly failed to improve response rates or survival20,21,22. Thus, there is a clear need to better understand the reasons for primary immune checkpoint resistance in non-responding patients and for secondary refractoriness in initially responding patients.
GDF-15 was originally discovered as a macrophage inhibitory cytokine (MIC-1)23 and found to be potentially important for feto-maternal tolerance, with high miscarriage rates in pregnancy if the GDF-15 level in serum does not reach the levels observed in healthy pregnancy7,8. Metabolically, GDF-15 was shown to induce anorexia and cachexia through the GFRAL receptor expressed by neurons in the brainstem24. In tumours, GDF-15 is often expressed by cancer cells already at diagnosis, and its expression is further induced following tumour treatment and progression25, making it the most prominently overexpressed cytokine in cancer26, but its role as a tumour-derived potent local immunosuppressant has only recently been recognized3.
No cognate receptor has been discovered on leukocytes yet. However, GDF-15 has been shown to suppress tumour surveillance by macrophages27 and to inhibit inflammatory signalling in macrophages in the liver28. Tumoural GDF-15 expression prevents effector T cell adhesion and transmigration into the TME3. This mechanism was found to correlate with a disruption of LFA-1 β2-integrin-dependent actin engagement, which may affect not only diapedesis but also the immune synapse, which it is likely to destabilize29,30. In our investigations, high intratumoural and serum GDF-15 levels correlated with reduced levels of immune infiltration in the TME, reduced levels of T cell proliferation and reduced levels of granzyme B (GZMB) expression in both patients with cancer in last-line treatment and those newly diagnosed with cancer. This further supports the immunosuppressive role of GDF-15 in cancer.
Preclinical research has shown that intratumoural expression of GDF-15 renders tumours unresponsive to PD-1 blockade3. In addition, GDF-15 expression in CPI-treatment-naive patients was correlated with lack of response to immunotherapy. In the present study, T cell infiltration, proliferation and activation, accompanied by induction of interferon-related genes and pathways in the TME, were observed after 14 days of anti-GDF-15 monotherapy (Figs. 1b,c and 4a,b), whereas expression of T cell exhaustion markers was not affected (Extended Data Fig. 3a). As blocking GDF-15 is not expected to substantially alter immunogenicity of the tumour or to overcome exhaustion of antitumoural T cells, combination therapy with CPIs is considered the best therapeutic option to achieve and sustain maximum antitumoural efficacy. Data from immune monitoring of interferon-γ-inducible chemokines during co-therapy in this study (Fig. 4c–e and Extended Data Fig. 4a–c) support this combination treatment concept. In addition, the median fold change in intratumoural T cell numbers under visugromab monotherapy further increased for proliferating T cells and remained stable for CD3+GZMB+ T cells following PD-1 blockade. This supports the hypothesis that neutralization of intratumoural GDF-15 enables T cells to infiltrate irrespective of their antigen specificity, but combination therapy with a CPI enhances survival and proliferation of stimulated T cells within the TME.
The first-in-human trial of visugromab (CTL-002-001; NCT04725474) was conducted in individuals with anti-PD-1 or anti-PD-L1 relapsed or refractory tumours, most having exhausted all treatment options and received visugromab–nivolumab combination therapy as the fourth line (median) of treatment (range second–seventh line).
Overall, the combination treatment was very well tolerated. However, an aspect to consider is that trial participants included could not have experienced serious side effects during previous exposure to CPI treatment as a minimum of 12 weeks continuous treatment had to have been tolerated (to exclude pseudoprogressors). Therefore, those patients prone to such side effects could have been filtered out, and this could, at least in part, explain the very benign adverse event profile. Of note, and in line with the known metabolic and nutritional effect of GDF-15 (ref. 31), the subpopulation of patients with high serum GDF-15 levels showed an increase in body weight following GDF-15 blockade.
Remarkably, the treatment induced a substantial number of deep and long-lasting objective tumour regressions up to confirmed CRs in non-squamous NSCLC and UC. This provides evidence for long-term antitumoural activity and effective tumour mass removal by the immune system in a distinct subset of patients. At this early stage of visugromab exploration, the response rate and durability of responses in these mostly third- to fourth-line-treated patients compare already favourably with the standard of care in these indications (for example, docetaxel in second-line NSCLC has an ORR of 9–12% and a DOR of just 5–8 months).
Notably, more than half of all responding patients in the NSCLC and UC cohorts of the trial experienced a response depth on study treatment as per RECIST 1.1 criteria that had not been reached on initial, prior anti-PD-1 or anti-PD-L1 treatment. In addition, 3 of the 4 patients with a CR for NSCLC, UC and HCC had not achieved a CR on any prior line of treatment, including their initial anti-PD-1 or anti-PD-L1 treatment.
These results warrant further investigation of visugromab in combination with CPIs and other treatment modalities in randomized clinical trials, including trials recruiting patients at earlier lines of treatment and stages of their disease for which most immunotherapies do unfold their full potential.
Two other anti-GDF-15 neutralizing monoclonal antibodies have entered clinical exploration in patients with cancer. Ponsegromab was well tolerated in a phase 1b study and demonstrated preliminary evidence for efficacy against cachexia by inducing weight gain in patients with cancer with elevated serum GDF-15 (ref. 32) and cachexia. AZD8853 has completed a first-in-human trial (NCT05397171) but achieved only transient GDF-15 neutralization and did not result in any objective clinical response when treating mainly patients with MSS colorectal cancer33.
Rechallenge of patients with anti-PD-1 or anti-PD-L1 relapsed or refractory disease with nivolumab or pembrolizumab in monotherapy is rarely successful. In patients meeting the strict inclusion criteria for anti-PD-1 or anti-PD-L1 relapsed or refractory disease of our study, reported ORRs for monotherapy retreatment attempts are 0–5% (refs. 11,12,13,14,15,16). Likewise, a large retrospective analysis on treatment beyond progression (another form of retreatment in monotherapy for patients that progressed on anti-PD-1 treatment with pembrolizumab) revealed a response rate as per RECIST 1.1 of just 2.8% in a patient population not too different from that of our trial34. Addition of visugromab, however, restored anti-PD-1 activity to levels nearly reaching response rates reported in populations with checkpoint-naive NSCLC and UC (reported to be at 18–21% for anti-PD-1-naive NSCLC and UC in second-line treatment35,36). Therefore, it can be inferred that GDF-15 is a druggable roadblock for successful immune checkpoint inhibition in a distinct target population.
Responses occurred in both PD-L1+ and PD-L1− tumours, with a tendency for enrichment of responses in patients positive for PD-L1 with pre-existing T cell infiltration. GDF-15 serum levels were not predictive for response, suggesting that even low GDF-15 levels can already be suppressive of antitumour immune responses (Extended Data Fig. 6a), and that serum levels of GDF-15 may not be a direct reflection of intratumoural levels, but influenced by tumour size and production rate per volume. At present, the datasets available are too small and do not allow one to assess whether serum GDF-15 can ultimately be used to enrich for responding patients. Weight gain, however, seemed to be higher in patients with elevated serum GDF-15 levels during visugromab treatment (pooled data from patients with NSCLC, UC and HCC; Extended Data Fig. 6b). Overall, it seems reasonable to assume that local GDF-15 levels in the TME suppress antitumour immune responses, whereas systemic GDF-15 serum levels determine weight loss due to anorexia.
Taken together, the provided and published data3,27,28,37 suggest that GDF-15 may be a new resistance factor for cancer immunotherapy in a distinct subpopulation of patients and tumour types. GDF-15 seems to have a potentially significant role as a T cell repellent and T cell suppressant in the TME. This implies that GDF-15 inhibition can improve success rates of anti-PD-1-based cancer immunotherapy not only in late- to last-line treatment of non-squamous NSCLC and UC and possibly HCC, as shown here, but also in earlier stages of these diseases. Future clinical trials with anti-GDF-15 in early and advanced cancer are thus warranted, to further elucidate the role of GDF-15, discover potentially response-predictive biomarkers and fully elucidate the mechanisms of action and ultimate clinical potential of GDF-15 inhibition.
Methods
Patients and treatment
The trial was termed GDFATHER, for GDF-15 antibody-mediated human effector cell relocation (ClinicalTrials.gov NCT04725474). This study was conducted in compliance with the International Council for Harmonisation E6 guideline for Good Clinical Practice and the Declaration of Helsinki. Complete and signed written informed consent was obtained from patients for inclusion in the study. The protocol was approved by the regulatory authorities in Germany (Paul-Ehrlich-Institut), Spain (Agencia Española de Medicamentos y Productos Sanitarios) and Switzerland (Swissmedic) and the local ethics committees in charge of the clinical trial site: Comité de Ética de la Investigación con Medicamentos, Pamplona, Spain; Swissethics, Cantonal Ethics Committee, Zurich, Switzerland; and the Ethics Committee of the University of Würzburg, Würzburg, Germany. The redacted protocol is available in the Supplementary Information and publicly available at the Clinical Trials Information System, a database set up and maintained by the European Commission.
Patients were eligible for enrolment if they had advanced-stage, relapsed or refractory solid tumours; had exhausted available approved standard treatments, including being relapsed or refractory to prior anti-PD-1 or anti-PD-L1 treatment; (for phase 1; part A and selected phase 2a cohorts) presented with biopsy-accessible tumour for serial biopsy taking; were 18 years or older; and had signed the informed consent form.
The study consisted of two parts, part A (phase 1 (dose escalation)) being a classic ‘3 + 3’ dose-escalation study and part B (phase 2a (expansion)) to explore the antitumoural activity of the combination. In part A of the study, a total of 25 patients were enrolled to receive five predefined DLs and received escalating doses of visugromab intravenous infusion (0.3, 1, 3, 10 and 20 mg kg−1) every 2 weeks. The first three patients for each DL received visugromab as monotherapy for one cycle (14 days) followed by the combination of visugromab and nivolumab. Nivolumab was also administered as an intravenous infusion at 240 mg every 2 weeks. Triple tumour biopsies were taken at baseline, day 14 and day 28.
In part B of the study, up to 5 cohorts with up to 27 participants per cohort with defined tumour entities expected to be GDF-15 dependent were treated with a recommended phase 2 dose, and safety and preliminary efficacy of CTL-002 monotherapy and the combination were evaluated further. To rule out significant visugromab-independent antitumoural activity of nivolumab, patients were included only if they were relapsed or refractory to prior, approved anti-PD-1- or anti-PD-L1-containing treatment as per defined, strict criteria. Patients were enrolled only if they had a minimum of 12 weeks of continuous prior exposure to anti-PD-1 or anti-PD-L1, and their relapse or progression on prior approved anti-PD-1- or anti-PD-L1-containing treatment had occurred while this anti-PD-1 or anti-PD-L1 treatment was ongoing. Based on data in the literature, the expected rate of patients responding to retreatment with an approved PD-1 or PD-L1 agent such as nivolumab in monotherapy in these populations is ≤5% for NSCLC10,11,12,13,14,15.
The study was initiated in December 2020 and the first patient was enrolled on 9 December 2020. As of October 2023, phase 1 of the study has been completed and phase 2a is ongoing with a total of 174 patients enrolled overall in the study.
Endpoints
The main endpoints were safety of visugromab (CTL-002) in combination with nivolumab and antitumoural activity. Safety parameters evaluated for this purpose were the number of participants with adverse events, including serious adverse events; clinical laboratory data; vital signs; electrocardiograms; physical examination (including neurological assessment); and Eastern Cooperative Oncology Group performance status. For phase 1 (part A) dose-limiting toxicities and maximum tolerated dose were also evaluated using National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) Version 5.0. Investigator-assessed evaluation of the antitumour activity was performed according to RECIST V1.1 including the assessment of the proportion of participants with tumour shrinkage (declared if RECIST V1.1-defined reduction in target lesions was ≥5% or more and <30%), a confirmed PR (≥30% reduction in target lesions) and/or CR, and ORR and various related parameters such as time to response, time to progression and DOR (measured from the time point of signing the informed consent). Secondary and exploratory endpoints included PK, pharmacodynamics (for example, degree of GDF-15 neutralization achieved and change in immune-cell number and composition in the tumour tissue) and cachexia-related parameters such as change in weight.
TCGA data and correlation analysis
The analysis consisted of 30 different cancer types with a varying number of solid tumours analysed. Material included in the TCGA database is derived from primary tumours of untreated (meaning immune checkpoint blockade naive) patients. The full list of abbreviations used, study names and number of samples for solid tumours is available in Extended Data Table 1.
For these indications, gene expression data (RNAseq-v2 raw counts and TPM) were downloaded from http://firebrowse.org on 1 August 2019 (Broad Institute of MIT and Harvard). Duplicated and ambiguous genes were removed, and normalized data were log2-transformed (log2[TPM + 1]). Correlation analyses of normalized GDF15 expression with immune-related genes and signatures (Extended Data Table 2) were performed using Spearman’s rank correlation (test) with the normalized enrichment score from single-sample gene set enrichment analyses or averaged expression levels and visualized as heat maps. GDF15 expression was analysed in different molecular subtypes and in association with clinical parameters in primary tumours of selected indications. Differences in expression between levels of molecular cancer subtypes were tested using a two-sided Wilcoxon rank sum test. Subsequently, P values were adjusted for multiple testing with the Benjamini–Hochberg method.
Change of estimated immune infiltrates between GDF15 expression groups
Fractions of immune infiltrates and other cell types (including tumour and stromal cells) were estimated through quanTIseq38,39 using the immunodeconv R package40 on RNA-sequencing data (TPM) corrected for purity as determined by ESTIMATE41. Average distribution within a subgroup of patients was computed according to GDF15 expression log2[TPM + 1] (low, medium and high terciles) within the respective tumour types. The distribution of immune-cell fractions was averaged over patients in these groups and visualized as a stacked bar plot (including a fraction with other cell types such as tumour and stromal cells).
Software and resources
All calculations, correlations and visualization analyses were performed using the statistical software environment R as well as the resources outlined in Extended Data Table 4.
Measurement of chemokine levels in GDFATHER patient samples
Serum samples for assessment of chemokine levels were taken at screening and each scheduled visit day from cycle 1 until cycle 3, and at the end-of-treatment visit. At dosing days (day 1), the samples were taken within 30 min before infusion. The serum was isolated using standard procedures and cryopreserved at −80 °C until use. Concentrations of the CXCL9 (MIG) and CXCL10 (IP10) chemokines were determined using validated solid-phase sandwich enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s instructions (R&D Systems; human CXCL9/MIG Quantikine ELISA kit, catalogue no. DCX900; human CXCL10/IP10 Quantikine ELISA kit, catalogue no. DIP100). Both assays were conducted in compliance with the Principles of Good Laboratory Practices regulations.
Measurement of serum visugromab levels in GDFATHER patient samples
Serum samples for PK assessment of total visugromab were taken at screening and every treatment cycle within 30 min before dosing and just at the end of each infusion. The serum was isolated using standard procedures and cryopreserved at −80 °C until use. Concentrations of visugromab were determined using a validated electrochemiluminescence assay method. The PK assay was conducted in compliance with the Principles of Good Laboratory Practices regulations.
Measurement of serum GDF-15 levels in GDFATHER patient samples
Serum samples for GDF-15 assessment were taken at screening and every treatment cycle within 30 min before dosing, isolated using standard procedures and cryopreserved at −80 °C until use. Samples from patients at screening were analysed for baseline GDF-15 levels using a validated quantitative solid-phase sandwich ELISA kit according to the manufacturer’s instructions (R&D Systems; human GDF-15 DuoSet ELISA Kit, catalogue no. DY957). Total GDF-15 levels (free GDF-15 plus visugromab-GDF-15 complex) during treatment were determined by a validated ECL method, using a custom visugromab non-competitive anti-GDF-15 nanobody as a capture reagent and, following saturation with visugromab, a custom non-competitive anti-visugromab antibody as a detection reagent. Both GDF-15 assays were conducted in compliance with the Principles of Good Laboratory Practices regulations.
Measurement of serum GDF-15 levels in translational patient samples
Tumour, serum samples and patient data used in the translational part of the study were provided by the University Cancer Center Frankfurt (UCT). The study was approved by the institutional review boards of the UCT and the responsible ethical committees at the Goethe University Frankfurt (project number SUG-2-2022).
For analysis of serum GDF-15, archived samples, which were taken within 1–89 days before surgery for patients with invasive bladder cancer (n = 34) or invasive upper urinary tract carcinoma (n = 3), as well as before treatment start with systemic therapy and within 1 year before–after biopsy for patients with metastatic urothelial cancer (n = 13), were provided. The serum was analysed for GDF-15 levels using a quantitative solid-phase sandwich ELISA kit according to the manufacturer’s instructions (R&D Systems; human GDF-15 DuoSet ELISA Kit, catalogue no. DY957).
Multiplex histological analyses of GDFATHER patient biopsies
Immunohistochemical (IHC) staining and immunofluorescence staining were performed on 4-µm formalin-fixed and paraffin-embedded (FFPE) sections of tumour tissue from consenting patients. Sections were deparaffinized and pretreated by protease digestion or heat-mediated treatment before antibody incubation. Before conducting any IHC or immunofluorescence analysis, a histology assessment was performed by a board-certified medical pathologist on slides stained with haematoxylin–eosin. The slides were stained using the Ventana HE 600 automated staining system (Roche Diagnostics) and scanned using bright-field imaging on the Leica Aperio AT2 platform (Leica Biosystems) using Scanscope software (console, v102.0.7.5; controller, v102.0.8.60) and a UPlanSapo 20×/0.75 objective (plus Doppler lens for ×40 images). The assessment consisted of confirmation of tumour type, assessment of histological features, presence of invasive margin, and determination of percentage of necrotic area on the whole slide and in the malignant lesion area.
Evaluation of intratumoural pro-GDF-15 expression levels in human FFPE samples
Determination of the intratumoural pro-GDF-15 expression levels was carried out applying a rabbit polyclonal anti-GDF-15 antibody (Sigma Aldrich; product no. HPA011191) as the primary antibody for automated staining using the Ventana BenchMark Ultra platform (Roche; software version no. 12.3.1 and 12.5.4). The binding of the anti-GDF-15 antibody was visualized using a secondary horseradish peroxidase (HRP) enzyme-conjugated antibody (Roche Diagnostics; Ventana optiView Universal DAB Detection Kit, catalogue no. 760-700). This specific antibody–enzyme complex was then visualized with a precipitating enzyme reaction product. Evaluation was carried out by a board-certified pathologist. Evaluation considered cytoplasmic staining (and in cases in which it was applicable, also membranous staining) of tumour cells. Cytoplasmic staining was assessed in four staining intensity categories, ranging from 0 (no staining) to 3+ (intensive staining). The percentage of stained cells per staining intensity category (0 to 3+) was recorded. This classification provided the basis for the calculation of the H score, which describes the GDF-15 protein levels in the tumour. The H score was determined by adding the results of multiplication of the percentage of cells with their respective intensity values as follows:
IHC evaluation of PD-L1 protein expression in human FFPE samples
Determination of the PD-L1 protein expression level was performed equivalent to the pro-GDF-15 assessment using a rabbit monoclonal anti-PD-L1 (SP263) antibody (Roche Diagnostics; catalogue no. 790-4905) as a primary antibody for automated staining following the manufacturer’s protocol. A rabbit IgG monoclonal antibody (Roche Diagnostics; catalogue no. 790-4795) was used as an isotype control. PD-L1 IHC was evaluated by a board-certified pathologist applying TPS and combined positive score as previously described42.
Multiplex IHC evaluation of CD4, CD8 and FOXP3 expression
CD8, CD4 and FOXP3 IHC staining was performed as a triplex IHC assay on one FFPE tissue slide. CD8 staining was carried out using a monoclonal mouse anti-human CD8 antibody (clone C8/144B; Agilent Technologies; product no. M710301-2) as a primary antibody. CD4 staining was carried out using a monoclonal rabbit anti-human CD4 antibody (clone SP35; CellMarque; product no. 104R), and FOXP3 was stained using a monoclonal rabbit anti-human FOXP3 antibody (clone SP97; Abcam; product no. Ab99963).
Depending on the primary antibody, rabbit- or mouse-specific hydroxyquinoxaline (HQ)-conjugated secondary antibodies (Roche Diagnostics; anti-mouse HQ, product no. 760-4814, anti-rabbit HQ, product no. 760-4815) or rabbit-specific nitropyrazole (NP)-conjugated secondary antibodies (Roche Diagnostics; anti-rabbit NP, product no. 760-4817) were used to allow signal amplification. The binding of the specific primary antibody was visualized using a secondary HRP-conjugated antibody or alkaline phosphatase-conjugated antibody (Roche Diagnostics; anti-HQ HRP, product no. 760-4820; anti-NP alkaline phosphatase, product no. 760-4827).
The enzyme coupled to the secondary antibody catalysed a chromogenic reaction at the binding site of the actual primary antibody resulting in teal-, yellow- and purple-coloured precipitation (Roche Diagnostics; DISCOVERY Teal HRP Kit, product no. 760-247; DISCOVERY Yellow Kit, product no. 760-239; DISCOVERY Purple Kit, product no. 760-229). Triplex IHC staining was carried out on a Ventana Discovery Ultra stainer (Roche; software version 12.5.4). For the identification of tumour epithelium, additional staining for pan-cytokeratin IHC analysis was performed on a separate FFPE tissue slide using a mouse anti-human pan-keratin antibody (clone AE1/AE3/PCK26; Roche Diagnostics; product no. 05267145001). For melanoma samples, an additional SOX10 IHC staining was performed on a separate FFPE tissue slide using a monoclonal rabbit anti-human SOX10 antibody (clone SP267; Roche Diagnostics; product no. 760-4968) to support the tumour-stroma separation process in the digital image analysis. Stained slides were scanned on a Leica Aperio AT2 scanner using Scanscope software (console, v102.0.7.5; controller, v102.0.8.60) and a UPlanSapo 20×/0.75 objective (Olympus; plus Doppler lens for ×40 images).
Evaluation of the triplex IHC staining was carried out by pathologist-assisted digital image analysis of representative areas using Visiopharm software (Visiopharm; version no. 2020.01.1 or higher). Visiopharm software uses undisclosed, linear display lookup tables. Visiopharm analysis output gave a readout on the density (positively stained cells per square millimetre) of cells positive for CD4, CD8, FOXP3 and FOXP3 plus CD4 in four different annotated regions of interest (‘tumour’, ‘tumour stroma’, ‘peritumoural stroma’ and adjacent non-neoplastic ‘normal tissue’).
Multiplex immunofluorescence evaluation of CD3, GZMB, Ki67, panCK and SOX10
For the evaluation of CD3, GZMB, Ki67, panCK and SOX10 expression and a nuclear counterstain in human FFPE patient samples, the semi-quantitative UltiMapper I/O T-act kit (Ultivue; product no. ULT20104 or ULT20110) was used. The slides were stained on a Leica Bond RX (Leica Biosystems; software version Bond 6.0.0.431 or higher), and scanned on the Zeiss Axio Scan Z1 fluorescence scanner (Hamamatsu Orca Flash, v4.0, camera (Hamamatsu Photonics); Colibri7 LED light source (Carl Zeiss Microscopy)) using Zen Blue (v3.1) software and a Plan-Apochromat 20×/0.8 M27 (Carl Zeiss Microscopy) objective. The slides were subsequently analysed by pathologist-assisted digital image analysis using Visiopharm software (Visiopharm; version no. 2020.01.1 or higher). Visiopharm software uses undisclosed, linear display lookup tables. The output of the digital analysis gave information on cell densities (positively stained cells per square millimetre) in four different annotated regions of interest (‘tumour cells’, ‘tumour stroma’, ‘tumour area’ (combining ‘tumour cells’ + ‘tumour stroma’), and in adjacent non-neoplastic ‘normal tissue’, in cases in which it was applicable).
Multiplex histological analyses of biopsies from patients with UC under early-line therapy
Slides were stained with haematoxylin and eosin (Sakura Finetek). IHC staining of GDF-15 (HPA011191, polyclonal, 1:100) was performed manually on 4-µm fresh FFPE slides. Semi-quantitative evaluation of IHC results was performed by a pathologist blinded to clinical data using a semi-quantitative approach using the H score.
The multiplex immunofluorescence analysis on whole-slide images was described previously43. Slides were stained with Opal 7‐Color Automation Kits (Akoya Biosciences). We stained a tumour microenvironment panel: panCK (C-11, Abcam), CD45 (polyclonal, Abcam), PD-L1 (SP142, Abcam), αSMA (1A4, Sigma), Ki67 (SP6, Abcam) and vimentin (EPR3776, Abcam); and an immune-cell panel: CD3 (D7A6E, Cell Signaling), CD8 (C8/144B, Dako/Agilent), CD4 (EPR6855, Abcam), FOXP3 (236A/E7, Abcam). The dye 4′,6‐diamidino‐2‐phenylindole (DAPI; SouthernBiotech) was used for staining of nuclei. Corresponding Opal fluorophore antibodies were used for visualization, and images were taken with the PhenoImager HT imaging system (Akoya Biosciences). Representative areas with urothelial cancer were selected and analysed with the inForm software (Akoya Biosciences). Cells were segmented and a machine learning algorithm in the inForm software was trained to identify cell populations.
Gene expression analysis of GDFATHER patient biopsies
Gene expression was measured using the NanoString nCounter PanCancer IO 360 Panel (NanoString Technologies). The PanCancer IO 360 Panel consists of 770 genes, including 20 housekeeping genes. Tissue samples were placed on glass slides as 4-μm-thick FFPE sections and five slides were subjected to RNA extraction using the RNeasy FFPE kit (QIAGEN; catalogue no. 73504) and quality control by NanoDrop (Thermo Scientific). The analysis of gene expression was conducted on the nCounter PanCancer IO 360 Panel and NanoString (NanoString Technologies) platform. A quality check had been performed using NanoStringQCpro v1.14.0 (NanoString Technologies). Raw data normalization using the R package NanoStringNorm resulted in very similar relative log expression distributions compared to normalization using nSolver v4.0 (NanoString Technologies) with standard settings. No batch effect between different runs or cartridges was observed by principal component analyses. Normalized expression data were log2-transformed, and housekeeping genes were filtered. Differential gene expression analyses were performed using the R/Bioconductor package limma (linear models for microarray and RNA-sequencing data) between visugromab treatment (day 14) and pretreatment (baseline) by applying a paired moderated t-test. These analyses were performed for two groups of participants, one group of participants with immune-cell influx (IMM) indicated by a more than twofold increase in the numbers of CD8+ and CD4+ T cells, and another group of participants with less than twofold increases in the same T cell subsets (NOIMM). Differential expression of individual genes between IMM and NOIMM was tested using two-sided Wilcoxon rank sum test and visualized as box plots. Volcano plots were generated using the R package EnhancedVolcano. Over-represented Reactome pathways for significantly upregulated genes on visugromab treatment (day 14) versus pretreatment (baseline) in the IMM group were determined using pathway information from ConsensusPathDB (http://cpdb.molgen.mpg.de/CPDB) and a Fisher’s exact test adjusted for the PanCancer IO 360 gene panel in the statistical software environment R v4.3.1 (R Development Core Team). P values were adjusted for the number of Reactome pathways with at least two matching genes using the Benjamini–Hochberg method.
PK–PD modelling of the distribution, elimination and interaction of visugromab and GDF-15
The PK–PD model was derived from non-human primates and describes the distribution, elimination and interaction of visugromab and GDF-15 in the serum compartment and the peripheral compartment. The distribution between the two compartments was modelled with an inter-compartmental clearance. Visugromab clearance was modelled as a first-order elimination from the serum compartment. Mass action kinetics was used to describe the binding of visugromab to GDF-15, forming a complex that was assumed to be eliminated at the same rate as free visugromab from the serum compartment.
Model parameters for non-human primates were scaled to humans using allometric scaling with a 3 kg body weight for non-human primates and 70 kg body weight for human. The allometric scaling coefficients were 1 for volumes of distribution, 0.75 for clearance, 0.667 for the inter-compartmental clearance, and −0.25 for rates.
The scaled human PK–PD model was coupled to a tumour model to predict free tumour GDF-15 levels for the first-in-human dose selection, reflecting visugromab’s mechanism of action and GDF-15 biology. The tumour model included a tumour microvasculature compartment with a blood flow rate of 0.2 ml−1 min−1 g−1, assuming a tumour size of 36 g. Visugromab was entering from the serum side, whereas free GDF-15 was released from the tumour, resulting in reported serum GDF-15 levels in patients with cancer of 0.5, 2 and 10 ng ml−1 (low, medium and severe scenarios). The calculation of free GDF-15 used the duration it takes the blood to flow through the tumour.
The parameters of this PK–PD model were estimated using total visugromab and total GDF-15 concentration measurements from the dose-range-finding and Good Laboratory Practice toxicology studies. Once the clinical phase 1 study data were available, the parameters were re-estimated. Both estimations were performed using the SEAM algorithm with Monolix software version 2019R1.
Statistical analysis
This phase 1 and 2a trial was largely evaluated on a descriptive basis as antitumoural activity was unknown for this first-in-human trial.
For the phase 2a cohorts, n = 14 response-evaluable participants were to be initially recruited for each tumour indication. Assuming a true response rate of 20%, the probability of observing at least 2 responding patients out of 14 participants was 80%. For an assumed true response of 10%, the probability of observing at least 1/14 responses in the cohort was 77%.
If at least one response was observed, cohort expansion was warranted per the design (5% one-sided α-level, 80% power, 5% maximum response probability of a ‘poor drug’, 20% response probability of a ‘good drug’). An additional n = 13 participants were then to be added to a cohort. Observing at least 4/27 responses would confirm that the drug warrants further investigation in that indication.
To assess the statistical significance between two independent groups, a two-tailed Mann–Whitney test was performed with a P value of ≤0.05 deemed as statistically significant. The statistical significance between three or more groups was determined by repeated-measures ANOVA with Geisser–Greenhouse correction followed by Dunnett’s test corrected for multiple comparisons with a P value of ≤0.05 deemed as statistically significant. The correlation between two parameters was computed using a non-parametric Spearman’s rank correlation with a 95% confidence interval. All statistical analysis was performed using GraphPad Prism software version no. 10.1.2.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Normalized RNA-sequencing data (TPM) from pan-cancer analyses of TCGA data were obtained from http://firebrowse.org and together with related immune signatures, estimated immune-cell fractions, and detailed information for selected indications (LUAD, LUSC, BLCA phenotypes), are available via Zenodo at https://doi.org/10.5281/zenodo.13716226 (ref. 44). Pathway analysis of gene expression data in patients is based on (Reactome) pathway information from ConsensusPathDB (http://cpdb.molgen.mpg.de/CPDB). Source data are provided with this paper.
Change history
25 February 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41586-025-08827-0
References
Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 11, 3801 (2020).
Santiago-Sánchez, G. S., Fabian, K. P. & Hodge, J. W. A landscape of checkpoint blockade resistance in cancer: underlying mechanisms and current strategies to overcome resistance. Cancer Biol. Ther. 25, 2308097 (2024).
Haake, M. et al. Tumor-derived GDF-15 blocks LFA-1 dependent T cell recruitment and suppresses responses to anti-PD-1 treatment. Nat. Commun. 14, 4253 (2023).
Batlle, E. & Massagué, J. Transforming growth factor-β signaling in immunity and cancer. Immunity. 50, 924–940 (2019).
Lodyga, M. & Hinz, B. TGF-β1 - a truly transforming growth factor in fibrosis and immunity. Semin. Cell Dev. Biol. 101, 123–139 (2020).
Wischhusen, J., Melero, I. & Fridman, W. H. Growth/differentiation factor-15 (GDF-15): from biomarker to novel targetable immune checkpoint. Front. Immunol. 11, 951 (2020).
Tong, S. et al. Serum concentrations of macrophage inhibitory cytokine 1 (MIC 1) as a predictor of miscarriage. Lancet 363, 129–130 (2004).
Kaitu’u-Lino, T. J. et al. Plasma MIC-1 and PAPP-a levels are decreased among women presenting to an early pregnancy assessment unit, have fetal viability confirmed but later miscarry. PLoS ONE 8, e72437 (2013).
Delgado, A. & Guddati, A. K. Clinical endpoints in oncology - a primer. Am. J. Cancer Res. 11, 1121–1131 (2021).
Tavenier, J. et al. Longitudinal course of GDF15 levels before acute hospitalization and death in the general population. Geroscience 43, 1835–1849 (2021).
Fujita, K. et al. Retreatment with anti-PD-L1 antibody in advanced non-small cell lung cancer previously treated with anti-PD-1 antibodies. Anticancer Res. 39, 3917–3921 (2019).
Fujita, K. et al. Retreatment with anti-PD-1 antibody in non-small cell lung cancer patients previously treated with anti-PD-L1 antibody. Thorac. Cancer 11, 15–18 (2020).
Watanabe, H. et al. The effect and safety of immune checkpoint inhibitor rechallenge in non-small cell lung cancer. Jpn. J. Clin. Oncol. 49, 762–765 (2019).
Katayama, Y. et al. Retrospective efficacy analysis of immune checkpoint inhibitor rechallenge in patients with non-small cell lung cancer. J. Clin. Med. 9, 102 (2019).
Fujita, K. et al. Retreatment with pembrolizumab in advanced non-small cell lung cancer patients previously treated with nivolumab: emerging reports of 12 cases. Cancer Chemother. Pharmacol. 81, 1105–1109 (2018).
Makrakis, D. et al. Treatment rechallenge with immune checkpoint inhibitors in advanced urothelial carcinoma. Clin. Genitourin. Cancer 21, 286–294 (2023).
Stenehjem, D. et al. Treatment patterns in patients with metastatic non-small-cell lung cancer in the era of immunotherapy. Future Oncol. 17, 2940–2949 (2021).
Sun, L. et al. Clinical efficacy and safety of anti-PD-1/PD-L1 inhibitors for the treatment of advanced or metastatic cancer: a systematic review and meta-analysis. Sci. Rep. 10, 2083 (2020).
Fitzsimmons, T. S. et al. Immune checkpoint inhibitors efficacy across solid cancers and the utility of PD-L1 as a biomarker of response: a systematic review and meta-analysis. Front. Med. 10, 1192762 (2023).
Long, G. V. et al. Epacadostat (E) plus pembrolizumab (P) versus pembrolizumab alone in patients (pts) with unresectable or metastatic melanoma: results of the phase 3 ECHO-301/KEYNOTE-252 study. J. Clin. Oncol. https://doi.org/10.1200/JCO.2018.36.15_suppl.108 (2018).
Ribas, A. et al. MASTERKEY-265: a phase III, randomized, placebo (Pbo) controlled study of talimogene laherparepvec (T)plus pembrolizumab (P) for unresectable stage IIIB–IVM1c melanoma (MEL). Ann. Oncol. 32, S868–S869 (2021).
Diab, A. et al. Bempegaldesleukin plus nivolumab in untreated advanced melanoma: the open-label, phase III PIVOT IO 001 trial results. J. Clin. Oncol. 41, 4756–4767 (2023).
Bootcov, M. R. et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc. Natl Acad. Sci. USA 94, 11514–11519 (1997).
Borner, T. et al. GDF15 induces anorexia through nausea and emesis. Cell Metab. 31, 351–362 (2020).
Breen, D. M. et al. GDF-15 neutralization alleviates platinum-based chemotherapy-induced emesis, anorexia, and weight loss in mice and nonhuman primates. Cell Metab. 32, 938–950 (2020).
Welsh, J. B. et al. Large-scale delineation of secreted protein biomarkers overexpressed in cancer tissue and serum. Proc. Natl Acad. Sci. USA 100, 3410–3415 (2003).
Ratnam, N. M. et al. NF-κB regulates GDF-15 to suppress macrophage surveillance during early tumor development. J. Clin. Invest. 127, 3796–3809 (2017).
Weng, J. H. et al. Colchicine acts selectively in the liver to induce hepatokines that inhibit myeloid cell activation. Nat. Metab. 3, 513–522 (2021).
Dustin, M. L. The immunological synapse. Cancer Immunol. Res. 2, 1023–1033 (2014).
Wurzer, H. et al. Actin cytoskeleton straddling the immunological synapse between cytotoxic lymphocytes and cancer cells. Cells. 8, 463 (2019).
Yang, L. et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat. Med. 23, 1158–1166 (2017).
Crawford, J. et al. A phase Ib first-in-patient study assessing the safety, tolerability, pharmacokinetics, and pharmacodynamics of ponsegromab in participants with cancer and cachexia. Clin. Cancer Res. 30, 489–497 (2024).
Carneiro, B. A. et al. Safety and efficacy of AZD8853, an anti-growth and differentiation factor 15 (GDF15) antibody, in patients (pts) with advanced/metastatic solid tumors: first-in-human study. Cancer Res. 84, abstr. CT100 (2024).
Topp, B. G. et al. Tumor dynamics in patients with solid tumors treated with pembrolizumab beyond disease progression. Cancer Cell 41, 1680–1688 (2023).
Herbst, R. S. et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387, 1540–1550 (2016).
Sharma, P. et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 18, 312–322 (2017).
Wang, Z. et al. GDF15 induces immunosuppression via CD48 on regulatory T cells in hepatocellular carcinoma. J. Immunother. Cancer 9, e002787 (2021).
Finotello, F. et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 11, 34 (2019).
Finotello, F. et al. Next-generation computational tools for interrogating cancer immunity. Nat. Rev. Genet. 20, 724–746 (2019).
Sturm, G. et al. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics. 35, i436–i445 (2019).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from 884 expression data. Nat. Commun. 4, 2612 (2013).
De Ruiter, E. J. et al. Comparison of three PD-L1 immunohistochemical assays in head and neck squamous cell carcinoma (HNSCC). Mod. Pathol. 34, 1125–1132 (2021).
Koll, F. J. et al. Tumor-associated macrophages and Tregs influence and represent immune cell 889 infiltration of muscle-invasive bladder cancer and predict prognosis. J. Transl. Med. 21, 124 (2023).
Hubert, H., Falk, G. & Zlatko, T. Association of GDF-15 expression with immune parameter in a pan-cancer analysis. Zenodo https://doi.org/10.5281/zenodo.13716226 (2024).
Acknowledgements
We thank all of the participants in the trial and their families. The results shown regarding the indication selection are in part based on data generated by the TCGA Research Network (https://www.cancer.gov/tcga). Manuscript editing support was provided by M. Hartmann. Study funding was provided by CatalYm, serving also legally as the trial sponsor of the GDFATHER-1/2a trial.
Author information
Authors and Affiliations
Contributions
Study conceptualization: I. Melero, A.H.-C., P.L., M.M., J.W., T.F.G., W.H.F., A.d.S., C.Z., M. Rüdiger, M.A., C.S.-W., P.F., K.K. and E.L. Methodology: I. Melero, R.B., A.d.S., C.Z., M. Rüdiger, P.F., K.K. and E.L. Clinical investigation: I. Melero, M.d.M.L., G.d.V., E.G., J.M.-L., M.J., G.A., M.-E.G., M.S., D.K., R.D., M. Reig, M.-E.R.R., E.C., J.E.-V., A.O., P.S., J.J.S.-C., K.-L.K., O.S., C.S., T.G., H.L., E.R., M.F., A.L.-M., I. Moreno, J.T.-J., A.H.-C., D.H., F.R., H. Richly, A.M.S., C.E., M.S.-Z., A.V.-L., J.T., C.K., P.R.G. and F.F. Biomarker validation: Z.T., H.H., F.G., S.S., J.R., A.T., M.A. and K.K. PK–PD analyses: I. Melero, P.L., M.M., A.G., M.A., M.L., K.K. and E.L. Formal analysis: I. Melero, P.L., M.M., A.A., A.G., C.Z., B.R., F.H., M.A., M.L., P.F., K.K. and E.L. Resources: M. Rüdiger, P.L’H. and E.L. Data curation: I. Melero, M.d.M.L., G.d.V., E.G., J.M.-L., M.J., G.A., M.-E.G., M.S., D.K., R.D., M. Reig, M.-E.R.R., E.C., J.E.-V., A.O., P.S., J.J.S.-C., K.-L.K., O.S., C.S., T.G., H.L., E.R., M.F., A.L.-M., I. Moreno, J.T.-J., A.H.-C., D.H., F.R., H. Richly, A.M.S., C.E., M.S.-Z., A.V.-L., J.T., C.K., P.R.G., F.F., Z.T., H.H., F.G., F.J.K., P.W., F.K.H.C., H. Reis, M.M., S.S., J.R., A.T., A.d.S., F.S.L., J.A., A.D., A.A., A.G., B.R., F.H., M.A., M.L., D.R., P.F., K.K. and E.L. Writing (original draft): I. Melero, F.H., M.A., K.K. and E.L. Writing (review and editing): all authors. Visualization: A.G., C.Z., B.R., M.A., M.L., D.R., P.F., K.K. and E.L. Project administration: A.A., A.G., B.R., F.H., M.A., M.L., D.R., P.F., K.K. and E.L. Supervision: I. Melero, P.F., K.K. and E.L. Funding acquisition: M. Rüdiger, P.L’H. and E.L.
Corresponding authors
Ethics declarations
Competing interests
M.H. and J.W. are co-founders and stock owners of CatalYm. A.d.S., M. Rüdiger, P.L’H., M.H., J.A., A.D., A.A., A.G., F.H., M.A., M.L., D.R., C.S.-W., F.S.L., P.F., K.K., F.H., B.R. and E.L. are current or former employees of and/or hold stock options in Catalym. R.D., M.H., J.W., K.K., E.L., F.H., M. Rüdiger and C.S.-W. are inventors on patents related to visugromab and anti-GDF-15 treatment (WO2014049087, WO2015144855, WO2017055613, WO2022101263, WO2024052532, WO2024126808 and unpublished). I. Melero, H.H., R.B., R.D., J.W., F.J.K., A.M.M.E., W.H.F., T.F.G., P.L., M.M., S.S., A.T. and J.R. have received research funding or consulting or advisory fees from CatalYm. Several authors and investigators declare potentially competing interests (grants or contracts and/or royalties or licences, stock or stock options, consulting fees, honoria, travel or meeting support, or advisory or data safety monitoring board participation) apart from their relationship with CatalYm: I. Melero (Agenus, Alligator, Allmiral, AstraZeneca, Biontech, BMS, Boehringer Ingelheim, Bright Peak, Crescendo Biologics, Curon, Dynamicure, F-Star, Genmab, Highlight Therapeutics, HotSpot, Merck Serono, MSD, Merus, Mestag, Numab, PharmaMar, Pieris, Pierre Fabre, Pioneering Medicines, Roche, Sanofi and Servier); G.d.V. (Astellas, AstraZeneca, Bayer, BMS, Ipsen, Janssen MSD, Merck Serono, Pfizer and Roche); E.G. (Anaveon, BeiGene, Boehringer Ingelheim, Ellipses Pharma, F-Star Therapeutics, Hengrui, Incyte, Janssen Global Services, MabDiscovery, Medscape, MSD, Novartis, Seattle Genetics, Sanofi, Roche, SeaGen, Taiho and Thermo Fisher); J.M.-L. (Astellas, BMS, Highlight Therapeutics, Merck Serono, MSD, Novartis, Ipsen, Roche, Sanofi and Pierre Fabre); M.J. (Adoram, AstraZeneca, Debiopharm, Novartis, Roche, Sanofi and Takeda); M.-E.G. (BMS, Janssen, Novartis and Roche); M.S. (Amgen, AstraZeneca, Blueprint Medicines, Boehringer Ingelheim, Bristol Myers Squibb, GlaxoSmithKline, Janssen, MSD, Novartis, Roche, Sanofi and Tacalyx); D.K. (AstraZeneca, Amgen, BMS, Merck Serono, MSD, Mirati, PharmaMar, Sanofi and Swiss Oncology in Motion); R.D. (Alligator, Amgen, BMS, MSD, Novartis, Pierre Fabre, Roche, Sun Pharma, Takeda, Sanofi, Second-Genome, Regeneron, T3 Pharma, MaviVax, Pfizer, Simcere and touchIME); M. Reig (AstraZeneca, Bayer, BMS, Boston Scientific, Biotoscana Pharma, Engitix Therapeutics, Eli Lilly, Geneos, Gilead, Ipsen, Merck, Roche and Universal DC); M.-E.R.R. (BMS, Imcore-Roche, Roche and Highlight Therapeutics); E.C. (Achilles, Adcendo, Alkermes, Amunix, Anaveon, Amcure, AstraZeneca, BeiGene, BMS, Boehringer, Chugai, Debiopharm, Diaccurate, Ellipses, Incyte, iTeos, Janssen, Merus, MonTa, MSD, Nanobiotix, Nouscom, Novartis, OncoDNA, PharmaMar, PsiOxus Therapeutics, Roche/Genentech, Sanofi, Servier, Shionogi, Syneos Health, TargImmune, T-knife and Tolremo); J.E.-V. (BMS, MSD and Pfizer); J.J.S.-C. (Eisai, Novartis, Pfizer and Seagen); K.-L.K. (Janssen and Takeda); O.S. (Affimed); C.S. (BMS, Eli Lilly and Boehringer Ingelheim); E.R. (BMS, Eli Lilly, Kyowa Kirin, Leo Pharma, MSD, Pierre Fabre and Sanofi); A.L.-M. (Incyte, Merck, PharmaMar, Pfizer, Roche, Rovi and Sanofi); I. Moreno (Digicore Initiative, Fondacion Intheos, Exafield, Guidepoint and Ellipses Pharma); A.H.-C. (BMS, Gilead, Kyowa Kirin, Merus and MSD); D.H. (Novartis and Roche); M.S.-Z. (Bayer, Instituto de Salud Carlos III (grant FI19/00222), BTG, MSD and Roche); A.V.-L. (AstraZeneca, BMS, Health in Code and Roche); J.T. (Amgen, AstraZeneca, Bristol Myers Squibb, Eisai, EXACT Therapeutics, Institut für Qualitätssicherung und Transparenz im Gesundheitswesen, Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen, Ipsen, Lilly ImClone, medupdate, Merck Serono, Merck Sharp & Dome, Oncolytics Biotech, onkowissen.de, Roche, Servier and streamedup!); C.K. (AstraZenca, BMS, DGVS, Incyte, MCI Deutschland, MSD, Servier, Ipsen, Merck Serono and Roche); P.R.G. (AstraZeneca, Bayer, BMS, Boston Scientific, Eisai, Guerbet, Ipsen, MSD, Lilly, Roche and Sirtex); F.F. (AstraZeneca, BMS, Eisai, Merck Serono, Servier and Roche); Z.T. (Boehringer Ingelheim); H.H. (Secarna); F.J.K. (German Cancer Aid (Deutsche Krebshilfe)); H. Reis (AstraZeneca, BMS, Boehringer Ingelheim, Chop GmbH, Diaceutics, GSK, HUeG, Janssen, MCI, Merck, Novartis, Roche Pharma and Sanofi); M.M. (LYO-X AG and TATAA Biocenter); T.G. (Allogene, Bicara, BMS, Merck, Pyxis and Samyang); W.H.F. (Anaveon, Atreca, Incendia, Ichnos Sciences, Genenta, OSE Immunotherapeutics, Oxford Biotherapeutics, Mestag, Novartis, Roche and Tabby); A.M.M.E. (Acetra, Agenus, BMS, Boehringer Ingelheim, BioInvent, BioNTech, Ellipses, Galecto, GSK, IO Biotech, IQVIA, Isa Pharmaceuticals, Merck & Co, MSD, Pfizer, Pierre Fabre, Sairopa, Sellas, SkylineDX, TigaTx and Trained Immunity); R.B. (Amgen, Avencell); J.R. (Astellas, AstraZeneca, BMS, Daiichi Sankyo, GSK, MSD, Merck and Sanofi); F.H. (Thermosome); P.F. (Curevac); J.W. (Bayrische Forschungsstiftung (FORTiTher) and the German Ministry for Education & Research (Transcan-3)). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 PK/PD data observed in phase 1 is consistent with population-based model and predicts neutralization of free GDF-15 in the tumour.
(a) Observed concentrations of total visugromab and total GDF-15 (free and drug-bound GDF-15) determined in serum of phase 1 participants across all tested dose levels within 8 weeks of treatment and at end of treatment. Mean and individual data points. (b) A population PK/PD model was derived from NHP studies and updated with clinical phase 1 data to describe GDF-15 neutralization in tumour micro-vasculature. Free GDF-15 is predicted for serum GDF-15 baseline levels of 0.5 ng/mL (left panel), 2 ng/mL (middle panel) and 10 ng/mL (right panel) and dosing schemes (upper panel = q2wk, middle panel = q3wk, lower panel = q4wk) as well as different concentrations (red, solid line = 3 mg/kg, green dashed line = 10 mg/kg and blue dashed line = 20 mg/kg) of visugromab. The threshold indicates the average physiological serum GDF-15 level (0.5 ng/mL).
Extended Data Fig. 2 Visugromab in combination with nivolumab induces remissions in a fraction of anti-PD-1/-L1 relapsed/refractory last-line NSCLC and urothelial cancer.
(a, b) Spider plots of NSCLC (a) and UC (b) patients illustrating changes in tumour size, response assessment was done according to investigator-assessed RECIST1.1, and time on study. Patients with partial or complete response (PR and CR) are highlighted in light and dark green respectively, arrow indicates ongoing treatment.
Extended Data Fig. 3 Pharmacodynamic effects of visugromab monotherapy in phase 1.
(a) Exhaustion markers PDCD1, HAVCR2 and LAG3 are not differentially expressed in pre-(baseline) (n = 6) and on-treatment (D14) (n = 5) biopsies following visugromab therapy in trial participants with >2-fold (IMM) and participants with <2-fold (NOIMM) increase in CD8+ and CD4+ T cells. Boxplots black line, median; red line, mean; box, IQR; ns, not significant, (P > 0.5 from two-sided Wilcoxon rank sum test). (b) Volcano plot presenting differentially expressed genes (NS = no significant change) in biopsies on day 14 of visugromab monotherapy compared to pre-treatment biopsies in participants with <2-fold (NOIMM) increase in CD8+ and CD4 + T cells. P values are from moderated t-test using limma.
Extended Data Fig. 4 Impact of induction of Interferon-γ signalling in a fraction of anti-PD-1/PD-L1 relapsed/refractory last-line cancer on CXCL10 levels.
(a) Significant induction and maintenance of chemokine CXCL-10 following treatment for six weeks in the blood of phase 2a NSCLC, HCC and UC study participants. P values are reported from repeated measures ANOVA with Geisser-Greenhouse correction followed by Dunnett’s test corrected for multiple comparisons. (b) Baseline CXCL-10 levels between participant with best overall response (BOR) according to RECIST 1.1 of PD/SD (grey) and PR/CR (green). P values are reported from two-tailed Mann-Whitney test. (c) Heatmap representing higher mean values [pg/ml] of CXCL-10 following treatment in participants with BOR of PR or CR (lower row) compared to participants with BOR of PD or SD (upper row). (a-c) Boxplots centre line, median; box, IQR; whiskers, 1.5 x IQR; outliers are depicted individually.
Extended Data Fig. 5 Serum and intra-tumoural (pro-)GDF-15 inversely correlate with immune cell numbers in UC and NSCLC.
(a) Early-line, treatment-naïve UC (MIBC, NMIBC, mUC and UTUC) patients with high (n = 9) vs. low (n = 31) serum GDF-15 levels show reduced CD8+ T cell density, CD45+Ki67+ cell density and PD-L1+ tumour cell density in the tumour. P values were calculated by two-sided Mann-Whitney test. (b) Serum GDF-15 levels at baseline in phase 2a NSCLC and UC patients. The box bounds the inter-quartile range (IQR) divided by the median, and Tukey-style whiskers extend to a maximum of 1.5 x IQR beyond the box. Individual data points represent outliers. P value is reported from two-sided Mann-Whitney test. (c) Association of pro-GDF-15 H score and CD8+ T cell density in tumour tissue in NSCLC patients (left panel, n = 30) and UC patients (right panel, n = 19) of phase 2a. PR/CR with available baseline biopsies of the respective indications are highlighted in green. Best-fit of simple linear regression is depicted as red line with 95% confidence interval as light grey shaded area.
Extended Data Fig. 6 Serum GDF-15 at baseline is not predictive for response, but positively associated with weight gain in phase 2a patients.
(a) Serum GDF-15 levels at baseline of phase 2a NSCLC, UC and HCC patients according to RECIST 1.1 response assessment. (b) In NSCLC, UC and HCC patients with elevated GDF-15 (n = 17) serum levels at baseline weight gain is significantly higher than in patients with low GDF-15 (n = 21). Boxplots center line, median; box, IQR; whiskers, 1.5 x IQR; outliers are depicted individually. P values are reported from two-sided Mann-Whitney test.
Extended Data Fig. 7 CT and PET-CT scans of responding tumours.
(a-d) Participant with mesothelioma and large, bulky disease (phase 1 part). RECIST 1.1 response under treatment with partial response (a) and (c) show baseline tumour assessment with bulky disease in upper mediastinum at two levels. (b) and (d) show respective significant tumour reduction at corresponding scan level at Cycle 8, Day 1 (week 16). (e, f) Participant with metastatic non-squamous NSCLC, Baseline (e) vs. Cycle 12 (f) demonstrating partial response as per RECIST 1.1. (g, h) Participant with metastatic non-squamous NSCLC (phase 2a part). Baseline (g) vs. Cycle 35 with partial response (h) as per RECIST 1.1. (i-l) Participant with metastatic urothelial cancer (phase 2a part), shown is large retroperitoneal lesion. RECIST 1.1 response with −100% reduction (complete response). (i) Baseline assessment at Screening with lesion marked by red arrow and red line. (j) Cycle 5 Day 1 assessment: Partial response. (k) PET-CT at Cycle 28 Day 1 demonstrating complete metabolic response. (l) CT scan at Cycle 45 Day 1 with continued complete response.
Supplementary information
Supplementary Information (download PDF )
Supplementary Note 1 and Tables 1–18.
Clinical Trial Protocol (download PDF )
Clinical trial protocol number CTL-002-001 (version 6.0).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Melero, I., de Miguel Luken, M., de Velasco, G. et al. Neutralizing GDF-15 can overcome anti-PD-1 and anti-PD-L1 resistance in solid tumours. Nature 637, 1218–1227 (2025). https://doi.org/10.1038/s41586-024-08305-z
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-024-08305-z
This article is cited by
-
Targeting mitochondrial homeostasis as a cancer treatment strategy: current status and future prospects
Molecular Cancer (2026)
-
Growth differentiation factor 15: from stress response to clinical utility in chronic liver diseases
Journal of Gastroenterology (2026)
-
A GDF-15–GFRAL axis controls autoimmune T cell responses during neuroinflammation
Nature Immunology (2026)
-
Research progress of CD73-adenosine signaling regulating hepatocellular carcinoma through tumor microenvironment
Journal of Experimental & Clinical Cancer Research (2025)
-
GDF15 blockade can reduce immunotherapy resistance in urothelial cancer
Nature Reviews Urology (2025)
