
Abstract
Lentiviral vector (LV)-mediated ex vivo gene therapy for haematopoietic stem and progenitor cells (HSPCs) has delivered on the promise of a ‘one-and-done’ treatment for several genetic diseases1. However, ex vivo manipulation and patient conditioning before transplantation are major hurdles that could be overcome by an in vivo approach. Here we demonstrate that in vivo gene delivery to HSPCs after systemic LV administration is enabled by the substantial trafficking of these cells from the liver to the bone marrow in newborn mice. We improved gene-transfer efficiency using a phagocytosis-shielded LV, successfully reaching bona fide HSPCs capable of long-term multilineage output and engraftment after serial transplantation, as confirmed by clonal tracking. HSPC mobilization further increased gene transfer, extending the window of intervention, although permissiveness to LV transduction declined with age. We successfully tested this in vivo strategy in mouse models of adenosine deaminase deficiency, autosomal recessive osteopetrosis and Fanconi anaemia. Interestingly, in vivo gene transfer provided a selective advantage to corrected HSPCs in Fanconi anaemia, leading to near-complete haematopoietic reconstitution and prevention of bone marrow failure. Given that circulating HSPCs in humans are also most abundant shortly after birth, in vivo HSPC gene transfer holds strong translational potential across multiple diseases.
Similar content being viewed by others


Main
Ex vivo gene transfer using LVs in HSPCs has demonstrated both safety and efficacy in treating haematological and metabolic disorders. Several gene-therapy products are now in advanced clinical development or available on the market1. However, certain challenges hinder their broader application to all eligible patients2,3, such as the need to collect, manipulate and preserve enough functional HSPCs to ensure effective haematopoietic reconstitution, as well as the need for patient conditioning before cell infusion. Moreover, ex vivo gene therapy enables correction only in HSPCs, whereas many genetic diseases have systemic manifestations that would benefit from broader correction. In vivo gene therapy presents a potential solution to these limitations, but targeting HSPCs in their niche remains a major challenge4,5,6.
Here we identified a unique window of opportunity in early postnatal life, when high numbers of circulating HSPCs (cHSPCs) are trafficking from residual haematopoietic niches to the bone marrow (BM). Building on this observation and our previous development of engineered LVs for in vivo administration and tissue-targeted expression7, we report successful gene transfer into bona fide long-term HSPCs in newborn mice. To assess the translational potential of this approach, we carried out tests in models of disease in mice in which distinct haematopoietic lineages and/or extra-haematopoietic tissues are involved. We selected: (1) adenosine deaminase (ADA) deficiency, which causes severe combined immunodeficiency (ADA-SCID) through the accumulation of toxic adenosine metabolites, impairing T cell, B cell and natural killer cell differentiation and function; (2) autosomal recessive osteopetrosis (ARO), a severe inherited bone disease in humans caused by mutations in the T cell immune regulator 1 (TCIRG1) gene, leading to defective osteoclast function and impaired bone resorption; and (3) Fanconi anaemia, a rare genetic disorder classified as a DNA repair deficiency syndrome, caused by mutations in the Fanconi anaemia pathway, resulting in severe HSPC depletion and leading to BM failure and increased cancer susceptibility.
High haematopoietic stem cell trafficking in the blood of newborn mice
To assess the feasibility of in vivo LV gene transfer into HSPCs, we first examined the spatiotemporal evolution of the haematopoietic system in C57BL/6 mice. We characterized the composition and absolute counts of haematopoietic cells in the BM, peripheral blood (PB), spleen and liver in newborn (postnatal day 0–1), 1-, 2- and 8-week-old (adult) mice (Fig. 1a and Extended Data Fig. 1a–d).

a, Haematopoietic cells were collected from the BM, spleen, liver and PB of newborn and 1-, 2- and 8-week-old C57BL/6 mice and analysed by FACS. FSC, forward scatter; SSC, side scatter. b–i, Mean and s.e.m. of the percentage of different populations between Lin−KIT+ HSPCs (b–e) or single values and mean and s.e.m of the absolute count of HSCs (f–i) in the BM (b,f), PB (c,g), spleen (d,h) and liver (e,i) at the indicated ages. n = 5 mice per group. Newborn PB: 10 pools from 50 mice. Spleen: n = 10. Kruskal–Wallis test with Dunn’s multiple-comparisons test (compared with newborn). EP, erythroid progenitor cell (PC); GMLP, granulocyte–monocyte–lymphoid PC; GMP, granulocyte–monocyte PC; KSL, KIT+SCA-1+Lin− cell; LK CD150, Lin−KIT+CD150+ cell; MEP, megakaryocyte–erythroid PC; MPP, multipotent PC. j, Experimental scheme: LV was administered i.v. to newborn C57BL/6 mice (510 transducing units, TU kg−1). GFP expression was monitored monthly in PB of treated mice. After 17 weeks, GFP+ cells were FACS-sorted from the BM and transplanted into busulfan-conditioned adult C57BL/6 mice. k,l, Mean and s.e.m. of GFP+ cells measured over time in the PB of treated mice (n = 19, k) and in the indicated populations 17 weeks after LV administration (l). Prog, progenitor cells. m–q, Single values of GFP+ cells measured over time in PB of individual surviving transplanted mice (m–p) and in the indicated populations 20 weeks after transplant (q). DN, double negative cells; DP, double positive cells. r, Heat map showing integration sites (ISs) (columns) shared between LV-treated newborn mice (pooled, n = 19) 17 weeks after administration and transplanted mice (pooled, n = 4) 20 weeks after transplant (shown in k,l). TPX: indicated organs of transplanted mice shown in m–q. Sample count indicates the number of samples (rows) sharing a given IS. A complete list of the ISs is provided in Supplementary Table 2. Schematics in a and j created with BioRender. Milani, M. (2025): a, https://BioRender.com/hvji2y3; j, https://BioRender.com/5z7zf6u.
Analysis of the HSPC compartment revealed a consistent proportion of stem cells and different progenitor subtypes in the BM across all ages (Fig. 1b). By contrast, haematopoietic stem cells (HSCs) were more prevalent in the PB and spleen in newborns compared with older mice (Fig. 1c,d). The composition of HSPCs in the newborn liver closely resembled that of the BM (Fig. 1e).
Further analysis specifically focusing on the HSC distribution confirmed the highest concentrations in the PB of newborns, which declined progressively over time and was paralleled by an increase in the BM (Fig. 1f,g), indicating ongoing HSC homing to the maturing BM niche. Furthermore, HSCs expanded significantly in the spleens of 1- and 2-week-old mice but were absent in the adult liver (Fig. 1h,i). To confirm active haematopoiesis in the livers of young mice, we detected cycling cells in haematopoietic foci that also included megakaryocytes (Extended Data Fig. 1e).
Together, these findings highlight a unique window of opportunity for the genetic engineering of HSC in newborns, marked by the persistence of fetal haematopoietic niches shortly after birth and extensive HSC trafficking in the blood.
LV in vivo gene transfer into HSCs
To evaluate the potential of in vivo LV gene transfer into HSCs, we intravenously (i.v.) administered GFP-expressing LVs to newborn C57BL/6 mice. This procedure resulted in stable GFP expression in approximately 0.5% of all blood lineages (Fig. 1j,k). We observed comparable GFP marking across different lineages in the spleen and BM, including KIT+SCA-1+Lin− cells, in most analysed mice (Fig. 1l). To confirm gene transfer into long-term HSCs, we transplanted GFP+ cells collected from the BM into busulfan-conditioned adult mice. As GFP-marking levels were low, we pooled CD45+GFP+ cells from the BM sorted using fluorescence-activated cell sorting (FACS) at 16 weeks after LV administration from four to five mice treated as newborns and transplanted each pool into busulfan-conditioned adult mice (Fig. 1j). Molecular analysis of sorted HSPCs showed an average of one vector copy per diploid genome (VCN; Extended Data Fig. 1f). We observed long-term engraftment of transplanted cells in four out of five mice (Fig. 1m–p). One transplanted mouse died owing to engraftment failure, like the non-transplanted control mouse. Notably, this mouse had received the lowest number of CD45+GFP+ cells (Extended Data Fig. 1g and Supplementary Table 1). Engraftment levels varied between the four surviving mice, which was expected given the low number of transplanted cells and the heterogeneous composition of GFP+ cells in each pool (Fig. 1m–p). However, we detected GFP+ cells across all analysed lineages in the PB and haematopoietic organs at comparable levels in each mouse, with a corresponding VCN (Fig. 1q and Extended Data Fig. 1h).
To confirm whether the GFP+ haematopoietic output in the transplanted mice originated from bona fide HSCs, we performed clonal tracking using LV integration site analysis. Examination of the GFP+ cells collected for transplantation from the BM of LV-treated mice, and of sorted GFP+ splenic myeloid, B cells and T cells revealed hundreds of clones that contributed homogeneously to the haematopoietic output across multiple lineages, without dominant clones. Analysis of total BM, spleen and thymus cells 20 weeks after transplant in recipient mice revealed heterogeneous contributions from a subset of clones originally identified in donor mice (Fig. 1r and Extended Data Fig. 1i–l). None of the clones that expanded in transplanted mice was predominant in donor mice or had integrations near oncogenes (Extended Data Fig. 1m). These findings show that LV administration in newborn mice successfully targeted multipotent, self-renewing HSPCs that contributed to long-term haematopoiesis in both donor and recipient mice.
To further assess long-term transduction, we monitored newborn-treated mice for more than 1 year after LV administration and observed stable GFP expression across all analysed PB lineages (Extended Data Fig. 1n). Next, to compare HSPC transduction efficiency between newborn and adult mice, we administered i.v. LVs to adult C57BL/6 mice at a dose matched to the body weight. GFP marking in the circulation was initially detected but rapidly declined by 4 weeks after LV administration (Extended Data Fig. 1o). This delayed loss was probably due to an emerging anti-GFP immune response in mice treated as adults. As i.v.-administered LVs predominantly transduce liver cells7,8, we examined the liver at the study end point and found few, if any, GFP+ cells (Extended Data Fig. 1p), suggesting clearance by cytotoxic T cells. Overall, these findings demonstrate successful in vivo gene transfer into bona fide HSCs, facilitated by the unique features of newborn haematopoiesis.
Enhancement of HSPC gene transfer
Our initial analyses showed that HSC content was highest in the liver and spleen of 2-week-old mice (Fig. 1h,i). We therefore tested the efficiency of in vivo HSPC gene transfer at this age but found that it was significantly lower than in newborns (Extended Data Fig. 2a–c). These data suggest that HSPCs are targeted mostly when circulating in newborns. To increase the amount of HSPC accessible to LV transduction, we administered 2-week-old mice a granulocyte-colony stimulating factor (G-CSF) and plerixafor mobilization regimen, modelling its clinical use. This treatment sharply increased the number of circulating HSCs (cHSCs) and enhanced in vivo gene transfer, reaching a mean of 8.5% GFP+CD45+ cells—fivefold higher than in control mice (Fig. 2a–d and Extended Data Fig. 2d). At 20 weeks after LV administration, GFP expression and VCN in the BM, thymus and spleen remained higher in mobilized mice than in controls (Fig. 2e,f and Extended Data Fig. 2e,f). We then performed total BM transplantation into busulfan-conditioned CD45.1 recipient mice. Cells derived from LV-treated donor mice successfully engrafted in all transplanted mice, with GFP-marking levels matching those of the corresponding donor, in PB and all analysed organs at 16 weeks after transplant (Fig. 2g–j and Extended Data Fig. 2g–l). We then applied a shorter mobilization regimen to newborn C57BL/6 mice that resulted in a 15-fold increase in gene transfer efficiency, reaching 11% GFP+CD45+ circulating cells (Fig. 2k,l). These findings indicate that in vivo LV gene-transfer efficiency is highest in newborn mice, reflecting a window of HSC accessibility that can be extended to juvenile mice through mobilization.

a,b, Single values and mean and s.e.m. of HSC absolute count in BM (a) and PB (b) of 2-week-old mobilized (n = 6) or control (n = 7) C57BL/6 mice. Mann–Whitney U-test. c–f, Mean and s.e.m. (c,d) with single values (e,f) of GFP+ cells (c,e) and VCN (d,f) in PB (c,d), BM (e) and whole organs (f) of 2-week-old mobilized (n = 6) or control (n = 6) C57BL/6 mice over time (PB, 1.1 × 1011 TU kg−1) and 20 weeks after LV administration (e,f). DC, denditric cells; Gran, granulocytes; LSK DP, Lin−SCA-1+KIT+ double positive cells; Mono, monocytes; NK: natural killer cells. Mann–Whitney U-test. g–j, Mean and s.e.m. (g–i) with single values (j) of GFP+ cells (g,h,j) and VCN (i) in PB (g–i) or BM (j) of mice transplanted with BM cells from control (g, n = 5) or mobilized (h, n = 6) mice over time (PB, 15 million CD45+ cells per recipient) and 16 weeks after transplant (j). Mann–Whitney U-test. k,l, Mean and s.e.m. of GFP+ cells (k) and VCN (l) in PB of mobilized (n = 7) or control (n = 6) newborn C57BL/6 mice (9 × 1010 TU kg−1). Mann–Whitney U-test. m,n, Mean and s.e.m. (m) with single values (n) of GFP+ cells in PB over time (m) and BM (n) 20 weeks after LV administration (5 × 1010 TU kg−1) in NSG mice (CD47high-LV newborns, n = 12; CD47free-LV newborns, n = 7; CD47high-LV adults, n = 6). Kruskal–Wallis test with Dunn’s multiple-comparisons test. o, Mean and s.e.m. of GFP+ cells in PB of mobilized (n = 8) and control (n = 7) 2-week-old and adult NSG mice mobilized (n = 4) or not (n = 8) (4.5 × 1010 TU kg−1). Kruskal–Wallis test with Dunn’s multiple-comparisons test (compared with 2-week-old controls). p, Single values and mean and s.e.m. of GFP+ cells in PB 16 weeks after LV administration in indicated mice. NSG, CD47high-LV 6 × 1010 TU kg−1. The same data reported in m and o. Kruskal–Wallis test with Dunn’s multiple-comparisons test. q,r, Mean and s.e.m. of GFP+ cells in PB of indicated mice treated at the indicated LV doses and ages (newborn, q; 2-week-old or adult mice, r).
In addition to mobilization, we tested other approaches to enhance gene-transfer efficiency. These included administration of an anti-interferon-α (IFNα) receptor-blocking antibody (MAR-1), the use of a phagocytosis-shielded LV (CD47high-LV) with high surface content of the ‘don’t-eat-me’ signal7 CD47 and higher LV doses. MAR-1 was administered i.v. 3 h before LV injection in newborn C57BL/6 mice, resulting in a 2.5-fold increase in GFP+ cells compared with IgG-treated controls (Extended Data Fig. 3a,b). We confirmed this increase across haematopoietic subpopulations in the spleen, thymus and BM at 16 weeks after LV administration (Extended Data Fig. 3c–f). These data suggest that IFNα restricts in vivo LV transduction of HSPCs, a limitation that MAR-1 can mitigate.
We next evaluated the effect of the CD47high-LV, using non-obese diabetic/SCID/IL2Rγ (NSG) mice bearing a CD47-receptor polymorphism that enables it to interact with human CD47 displayed on the LV surface9. A spatiotemporal analysis of haematopoiesis in these mice showed that, similar to C57BL/6 mice, cHSC numbers declined over time whereas BM HSC content modestly increased (Extended Data Fig. 4a–g). In contrast to C57BL/6 mice, however, NSG mice had fewer cHSCs as newborns but an increased HSC presence in the liver and spleen (Extended Data Fig. 4h,i), suggesting that haematopoiesis in NSG mice occurs mainly in these organs rather than in the BM. When we administered CD47high-LV expressing GFP to newborn and adult NSG mice, we observed a fourfold increase in GFP marking in NSG mice treated as newborns compared with both C57BL/6 mice treated as newborns and NSG mice treated as adults (Figs. 1k and 2m). By contrast, administration of CD47free-LV (produced in CD47-deficient cells) to newborn NSG mice resulted in a marked reduction of GFP+ cells in the PB. This finding demonstrates that phagocytosis shielding enhances LV gene transfer into HSPCs (Fig. 2m). We confirmed similar GFP expression in HSPCs in the BM of treated mice 20 weeks after LV administration (Fig. 2n). To further assess clonogenic capacity, we FACS-sorted GFP+ or GFP−Lin− cells from the BM of LV-treated NSG mice, performed colony-forming unit assays and found comparable clonogenic potential (Extended Data Fig. 4j). We also confirmed a higher VCN in Lin−GFP+ cells FACS-sorted from the BM of mice treated as newborns compared with those treated as adults (Extended Data Fig. 4k). To evaluate the range of VCN in HSPCs transduced in vivo, we analysed single colonies from colony-forming unit assays and measured a VCN of 1 in most colonies (Extended Data Fig. 4l). We also performed a biodistribution analysis of GFP+ cells using immunohistochemistry, 20 weeks after LV administration to newborn or adult NSG mice. Immunohistochemistry revealed GFP+ cells in the liver and spleen, with scattered cells in the lung, kidney and brain only in NSG mice treated as newborns. These cells were predominantly haematopoietic and endothelial, with liver parenchymal cell transduction also observed (Extended Data Fig. 4m,n). This distribution probably reflects the tissue-specific accessibility of LVs through the bloodstream.
We next combined HSC mobilization with CD47high-LV administration in 2-week-old or adult NSG mice. Although mobilization was more effective in adults than in 2-week-old mice (Extended Data Fig. 5a–c), GFP marking was sevenfold higher in 2-week-old mice (Fig. 2o and Extended Data Fig. 5d–f). Mobilization increased gene-transfer efficiency in both age groups, although cHSCs in mobilized NSG mice remained lower than in C57BL/6 mice (Fig. 2b and Extended Data Fig. 5b). Furthermore, LV doses were 2.5-fold lower in NSG compared with C57BL/6 mice as they had reduced tolerance to extensive manipulation by the mobilization regimen.
Analysed together, these data show significantly fewer GFP+ cells in the circulation of mice treated at increasing ages, with the highest permissiveness observed in newborn mice (Fig. 2p). Moreover, despite mobilization increasing gene transfer in both young and adult mice, transduction was twofold higher in the former, suggesting that cell-intrinsic factors influence LV transduction efficiency (Fig. 2p).
Finally, we assessed long-term HSPC gene transfer after increasing LV doses. We tested LV gene-transfer efficiency in newborn and 2-week-old C57BL/6 mice versus CD47high-LV in newborn, 2-week-old and adult NSG mice (Fig. 2q,r), confirming that HSPC accessibility to LV transduction was highest in newborns, with CD47high-LV enhancing gene-transfer efficiency in NSG mice. All mice treated as newborn or at 2 weeks old tolerated all LV doses without toxicity, whereas NSG adults had dose-limiting toxicity, preventing higher-dose administration (Fig. 2q,r).
Overall, these findings establish that the newborn period is the optimal window for in vivo HSC gene transfer, although HSC mobilization and CD47high-LV could enhance transduction efficiency in young and adult mice.
In vivo LV gene therapy corrects ADA-SCID
To evaluate the therapeutic potential of in vivo LV gene transfer, we selected ADA-SCID as our initial disease model for two key reasons. First, gene-corrected lymphocytes possess a selective survival advantage over uncorrected cells10. Second, the ADA enzyme is ubiquitously expressed, enabling gene-corrected hepatocytes to serve as a permanent enzyme replacement therapy by detoxifying the entire organism11.
We tested three LV constructs with distinct expression patterns: PGK.ADA, which drives ubiquitous ADA expression under the phosphoglycerate kinase (PGK) promoter; PGK.ADA.122mirT, which excludes hepatocyte expression using complementary target sequences for the liver-specific miR-122; and ET.ADA.142mirT, which restricts ADA expression to hepatocytes using the enhanced transthyretin (ET) promoter along with miR-142 target sequences to suppress expression in haematopoietic cells12 (Fig. 3a). We administered these LVs to newborn ADA-SCID mice and monitored weight and survival over time up to 20 weeks after LV administration. Whereas all untreated ADA-SCID mice died before weaning, we observed longer survival in LV-treated mice, with healthy growth comparable with wild-type (WT) littermates (Fig. 3b and Extended Data Fig. 6a). PGK.ADA-LV achieved the highest rate of long-term survival (5 out of 7 mice), whereas PGK.ADA.122mirT-LV was less effective (6 out of 14 mice), highlighting the importance of hepatocyte-mediated detoxification. Meanwhile, 4 out of 5 ET.ADA.142mirT-LV mice also survived up to 20 weeks after LV administration, consistent with previous reports showing enzyme replacement therapy efficacy in this model and robust LV expression in hepatocytes13,14 (Fig. 3b).

a, Newborn ADA-SCID mice or WT littermates were treated with 2.5 × 1010 TU kg−1 of the indicated LV. At 20 weeks after LV administration, 10–13 million total BM cells were transplanted into irradiated NSG mice. b, Survival of ADA-SCID mice treated as newborns with PGK.ADA-LV (n = 7), PGK.ADA.122mirT-LV (n = 14) or ET.ADA.142mirT-LV (n = 5), untreated (knockout (KO), n = 17) and LV-treated WT (WT, n = 30) littermates. WT littermates were treated in parallel with ADA-SCID mice; a pooled dataset of all LV-treated WT mice is reported, regardless of the administered LV construct. log-rank Mantel–Cox test with Bonferroni’s correction (compared with the PGK.ADA group). c–e, Mean and s.e.m. of the VCN (c), T lymphocytes measured by FACS (d) and lymphocyte counts measured by haemocytometer (e) in the PB of ADA-SCID mice treated with the indicated LV as newborns. Sample sizes (n) at each time point are reported below the graphs. Linear mixed-effects (LME) model (compared with WT; Supplementary Tables 3–5). f,g, Mean and s.e.m. of the percentage of the indicated populations in the spleen (f) and thymus (g) of ADA-SCID mice treated as newborns with the indicated LV or left untreated, 20 weeks after LV administration. Sample sizes (n) are reported above the bars. h, Survival of NSG mice transplanted with total BM cells isolated from ADA-SCID mice shown in b–g. log-rank Mantel–Cox test with Bonferroni’s correction (compared with WT). i–m, Mean and s.e.m. of lymphocyte counts in PB (8 weeks (i) or 12 weeks (j) after transplant), percentage of the indicated populations in the spleen (k) and thymus (l) or single values and mean and s.e.m. of VCN measured in the indicated organs (m) 12 weeks after transplant. Kruskal–Wallis test followed by post hoc analysis. Sample sizes (n) are reported above the bars (k,l) or in the graphs. Schematics in a created with BioRender. Milani, M. (2025) https://BioRender.com/7wda8gx.
To assess the selective advantage of the corrected cells, we measured the VCN in the PB of LV-treated ADA-SCID and WT littermates as a proxy. We observed a marked selective advantage only in PGK.ADA-LV-treated mice, in which 7% of circulating cells were transduced compared with 2–4% in LV-treated WT littermates (Fig. 3c). Immune reconstitution in the PB was fastest in PGK.ADA-LV-treated mice, whereas animals treated with the other LVs had a delayed immune recovery (Fig. 3d,e and Extended Data Fig. 6b,c). Analysis of spleen and thymus composition at 20 weeks after LV administration showed no differences between all surviving LV-treated mice and WT littermates, further confirming immune system reconstitution (Fig. 3f,g).
We measured ADA enzyme activity in red blood cells in the PB and found threefold higher activity in PGK.ADA-LV-treated mice compared with all other LV groups, although it remained below WT levels. Plasma ADA activity, however, was similar across all LV-treated mice and with compared WT levels (Extended Data Fig. 6d,e). We also confirmed the expected pattern of expression dictated by each LV design (Extended Data Fig. 6f).
To determine whether gene-corrected HSCs retained engraftment potential, we transplanted total BM cells from ADA-SCID mice treated as newborns into irradiated adult NSG mice (Fig. 3a), enabling the monitoring of engraftment owing to an H-2Dq mismatch. Notably, despite NSG mice having normal ADA expression, only recipients transplanted with BM cells from PGK.ADA-LV-treated mice showed long term survival, similar to mice transplanted with WT cells (Fig. 3h). Mice that received BM cells from ET.ADA.142mirT-LV-treated donors had the lowest survival (four out of eight), as expected, as these cells lacked ADA transgene expression. The low survival of mice that received BM cells from PGK.ADA.122mirT-LV-treated donors suggests that these cells were less fit, despite expressing ADA in a fraction of them. This effect probably resulted from poor donor detoxification, similar to cells isolated from ET.ADA.142mirT-LV-treated donors (Fig. 3h). There were no significant differences in body weight and engraftment of transplanted mice between groups (Extended Data Fig. 6g,h). However, lymphocyte counts were restored only in mice that received BM cells collected from PGK.ADA-LV-treated donors (Fig. 3i,j and Extended Data Fig. 6i,j). Similarly, spleen and thymus compositions were indistinguishable from WT-transplanted controls (Fig. 3k,l). In line with donor marking levels, mice that received BM cells from PGK.ADA-LV-treated donors and WT mice had similar VCNs, with an average 0.04 in PB and BM, confirming stable transduction of long-term repopulating HSCs (Fig. 3m and Extended Data Fig. 6k).
LV integration site analysis in the spleen, thymus and BM of LV-treated ADA-SCID mice and NSG transplant recipients confirmed integration site sharing across different organs, supporting that i.v. LV administration in newborns successfully targets bona fide HSCs (Extended Data Fig. 6l).
Finally, we compared in vivo LV gene therapy efficacy in newborn versus young mice by administering PGK.ADA-LV to 2-week-old ADA-SCID mice at a weight-matched dose. Only one out of five mice treated at 2 weeks old survived long term, compared with five out of seven mice treated as newborns (Fig. 3h and Extended Data Fig. 6m,n). Together, these findings demonstrate the therapeutic potential of in vivo ADA-LV administration in newborn ADA-SCID mice and highlight the advantage of vector designs that allow expression in both haematopoietic and liver parenchymal cells.
Restoration of bone remodelling in ARO
As a second model for LV-mediated early postnatal in vivo gene transfer to HSPCs, we selected ARO. This condition requires early intervention to prevent disease progression, as BM failure in patients with osteopetrosis severely limits treatment options. The underlying genetic defect impairs osteoclast resorption capacity, preventing the formation of a properly structured bone matrix. This disruption hinders HSPC homing to the BM, resulting in an increased concentration of cHSPCs15,16.
In this setting, we showed that in vivo LV gene transfer to HSPCs in osteopetrotic (OC) newborn mice extended postnatal survival and enabled the differentiation of gene-corrected cells into functional osteoclasts capable of bone resorption in vivo. However, given the severe and rapidly progressing phenotype of this model, complete rescue could not be achieved under the current experimental conditions (Extended Data Fig. 7). For a detailed description of gene therapy outcomes in this model, see the Supplementary Notes.
Full rescue of BM failure in Fanconi anaemia
Last, we selected Fanconi anaemia, a rare genetic disorder belonging to the DNA repair deficiency syndromes. In Fanconi anaemia, the HSC compartment is directly affected, posing additional constraints to the collection and subsequent infusion of sufficient numbers of genetically corrected HSCs in patients with this disorder. Nevertheless, clinical trials have shown that even a small population of corrected HSCs can expand in vivo and repopulate the BM progressively, even without conditioning17,18.
First, we analysed PB from Fanca knockout (Fanca−/−) mice over time, focusing on cHSC numbers (Extended Data Fig. 8a,b). As observed in the other analysed mouse strains, cHSC numbers were higher in newborns than older mice; however, the absolute number of cHSCs in newborn Fanca−/− mice was very low, mirroring patients with Fanconi anaemia (Fig. 1g and Extended Data Fig. 8b).
We then administered PGK.FANCA-LV i.v. to Fanca−/− newborn mice and tracked the VCN over time in PB cells. The VCN progressively increased in all treated mice during physiological growth and homeostatic haematopoietic renewal up to 1 year after LV administration, demonstrating that gene-corrected Fanca−/− cells have a selective advantage over uncorrected Fanca−/− cells in vivo (Fig. 4a,b). Parallel analysis of PB parameters revealed that counts of white blood cells and lymphocytes normalized progressively starting at 30 weeks after LV administration. This suggests that the expansion of corrected HSCs eventually reached the threshold needed to restore PB parameters (Fig. 4c,d and Extended Data Fig. 8c–e).

a,b, Single VCN values (a) and VCN fold expansion (b; over 5 weeks) in PB of Fanca−/− mice treated as newborns (1.5 × 1010 TU kg−1, FANCA, LV, n = 5) or untreated (FANCA, untreated, n = 5). LME model (Supplementary Table 6). P value: Fanca−/−, LV 5 weeks compared to 45 weeks. c,d, Mean and s.e.m. of white blood cell (WBC; c) and lymphocyte (d) counts in the PB of mice in a and from WT untreated mice. Kruskal–Wallis test with Dunnett’s multiple-comparisons test (compared with WT, untreated). e, Single values and mean and s.e.m. of VCN in the indicated organs 1 year after LV administration in mice in a. f,g, Single VCN values (f) and VCN fold expansion (g; over 4 weeks) in PB of Fanca−/− mice transplanted when 8 weeks old with BM cells from FANCA, LV (n = 7) or FANCA, untreated (n = 7) mice in a. LME model (Supplementary Table 7). P value: Fanca−/−, LV 4 weeks compared to 28 weeks. h, Single values and mean and s.e.m. of VCN in indicated organs 1 year after transplant in mice in f. i,j, Single VCN values (i) and VCN fold expansion (j; over 5 weeks) in PB of mice treated as newborns (1.5 × 1010 TU kg−1, n = 29) or untreated (n = 14). A subset of FANCA, LV (n = 15) or FANCA, untreated (n = 7) mice received two doses of MMC (arrows). LME model (Supplementary Table 8). P values: Fanca−/−, LV and Fanca−/−, LV, MMC 5 weeks compared to 52 weeks. Two independent experiments. k–n, Mean and s.e.m. of neutrophil count (k), red blood cell (RBC) count (l), haemoglobin concentration (HGB, m) and platelet count (n) in PB of mice from i. Kruskal–Wallis test followed by post hoc analysis (compared with FANCA, LV). o, Single values and mean and s.e.m. of VCN in indicated organs 1 year after LV administration in mice from i. Mann–Whitney U-test. p,q, Mean and s.e.m. of VCN (p) and VCN fold expansion (q; over 5 weeks) in PB of mice treated as adults (8-week-old controls n = 6; mobilized n = 5) or at 2 weeks old (controls n = 5; mobilized n = 6) (1.5 × 1010 TU kg−1). LME model (Supplementary Table 9). P values: 2 weeks, mobilized and adults, mobilized 5 weeks compared to 15 weeks.
At the end of the 1-year follow-up, we detected a high VCN in the spleen, thymus and BM of LV-treated Fanca−/− mice, except for one mouse with low PB marking (Fig. 4e). Sorting of PB subpopulations and KIT+SCA-1+Lin− cells from the BM confirmed a VCN consistent with PB measurements (Extended Data Fig. 8f). LV integration site analysis in these sorted PB and BM samples and whole BM, spleen and thymus confirmed transduction of bona fide HSCs that expanded owing to their selective advantage (Extended Data Fig. 8g,h). We then transplanted BM cells from LV-treated or untreated Fanca−/− mice into irradiated adult Fanca−/− recipients. Gene-corrected cells expanded over non-corrected cells in this stringent setting, again confirming the ability of transduced HSCs to outcompete native cells (Fig. 4f,g). VCN measurements in the organs of transplanted mice 1 year after transplant matched those observed in the PB, further supporting the stability of gene correction (Fig. 4h).
As Fanca−/− mice have a milder disease phenotype compared with humans, we applied a mitomycin C (MMC) challenge to model Fanconi-anaemia-associated BM failure. MMC is a DNA cross-linking agent, which is toxic to Fanca−/− cells owing to their defective DNA damage response19,20. Interestingly, LV-treated Fanca−/− mice exposed to two MMC doses exhibited substantial expansion of corrected cells, with the highest measured VCN reaching 1 (Fig. 4i,j). By contrast, untreated Fanca−/− mice exposed to MMC experienced severe haematopoietic decline, mimicking the BM failure observed in patients with Fanconi anaemia. By contrast, LV-treated Fanca−/− mice underwent a rapid recovery of all these parameters (Fig. 4k–n). One year after LV administration, we measured the VCN in the BM, spleen and thymus of LV-treated Fanca−/− mice. We found that values again mirrored those measured in the PB and were the highest in MMC-challenged LV-treated Fanca−/− mice, reinforcing the idea that corrected HSCs expanded after selective pressure from MMC treatment (Fig. 4o).
Finally, to explore strategies to further increase the number of corrected HSCs, we applied an HSPC mobilization regimen to 2-week-old or adult Fanca−/− mice (Fig. 4p,q). As expected, HSC gene-transfer efficiency in young and adult mice was much lower than in newborns. However, mobilization improved gene-transfer efficiency in both groups, with the highest VCN observed in 2-week-old mice (Fig. 4p), highlighting the potential of using mobilization in Fanconi anaemia to enhance gene-transfer efficiency and extend the intervention window.
Overall, we achieved full rescue of the haematopoietic defects observed in Fanca−/− mice, leveraging the selective advantage of corrected cells and progressive expansion of the initial pool of in vivo transduced HSCs.
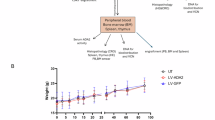
High numbers of cHSPCs in human neonates
Given that in vivo gene transfer to HSCs in newborn mice was facilitated by high levels of cHSPCs, we next examined whether a similar window of accessibility exists in humans. Using a previously published multiparametric flow-cytometry analysis21, we assessed PB samples from healthy participants at birth, 0–2 months, 3–8 months and when adults (>18 years). We observed a peak in phenotypically defined cHSCs and multipotent progenitor cells during the neonatal and early infantile periods, both in absolute numbers and in percentage of total HSPCs, with amounts progressively declining around weaning and becoming nearly undetectable in adults (Fig. 5). These findings suggest that, as in mice, early postnatal life in humans may represent a unique and time-sensitive window for in vivo HSC gene therapy. By leveraging this stage, future therapeutic strategies could bypass ex vivo manipulations and enable direct, clinically relevant HSC correction in paediatric patients.

a, Mean and s.e.m. of the percentage of different cHSPC populations in the PB of healthy donors of the indicated ages. CMP, common myeloid progenitor cell; ETP, early T cell precursor cell; MKP, megakaryocyte progenitor cell; MLP, multi-lymphoid progenitor cell; pre-BNK, B cell and natural killer cell progenitor cell. Neonates (P0), n = 63; 0–2 months, n = 4; 3–8 months, n = 6; >18 years, n = 21. b,c, Single values and mean and s.e.m. of the absolute count of cHSCs (b) and MPPs (c) in the PB of healthy donors of the indicated ages. Neonates, n = 63; 0–2 months, n = 4; 3–8 months, n = 6; >18 years, n = 21. Kruskal–Wallis test with Dunn’s multiple-comparisons test (compared with the neonate group). HSC and MPP counts from some donors are also reported in a separate study providing a detailed biological characterization of cHSPCs24.
Discussion
Here we show that the early postnatal period provides a unique opportunity to access HSCs in vivo for LV gene transfer in mice, bypassing the hurdles associated with ex vivo engineering. Although the transduction rate and extent of genetically modified haematopoiesis remained below what can be achieved by ex vivo transduction and infusion after conditioning, we provide proof of concept of substantial therapeutic benefit achieved in three models of severe genetic diseases.
Bona fide HSC gene transfer was proven by long-term multilineage output, self-renewal after serial transplantation and clonal tracking of in vivo transduced HSCs. Notably, the levels of genetically modified LV-positive cells in the haematopoietic compartment remained stable across all observation time points. Our investigation of the spatiotemporal dynamics of haematopoiesis in mice during postnatal development has provided insight into the biological mechanisms that underlie in vivo HSC gene transfer in newborns, with a substantial proportion of HSCs persisting in the liver and spleen of young mice, consistent with recent reports22,23. However, we also observed that the high numbers of cHSPCs seen in newborns, probably trafficking to newly forming definitive niches, rapidly declined in parallel with the efficiency of gene transfer. This suggests that LV access to HSCs is improved when they are in circulation. Supporting this hypothesis, we found that HSPC mobilization at 2 weeks of age, when significant numbers of HSPCs remain in tissue reservoirs, rescued effective gene transfer to levels exceeding those in non-mobilized newborns. These findings underscore the translational potential of this strategy, extending the window of efficacy to pre-weaning, a time point that may be more favourable for therapeutic intervention in humans.
HSPC mobilization with a G-CSF and plerixafor regimen markedly increased gene-transfer efficiency not only in WT mice but also in a mouse model of Fanconi anaemia. However, physical accessibility alone may not fully account for LV gene-transfer efficiency. Cell-intrinsic factors, including LV receptor expression levels, metabolic activity and the proliferative state of circulating cells during early life, may also contribute to productive transduction. Even after mobilization, in vivo HSPC gene-transfer efficiency declined at increasing ages. It is well known that HSPCs collected from the circulation of mobilized individuals are poorly permissive to LV gene transfer and require ex vivo activation for robust transduction. Similarly, quiescent HSCs in their BM niche impose notable barriers to genetic engineering, whether mediated by gene transfer or gene editing. These obstacles include limited vector entry, restricted reverse transcription owing to unbalanced cellular dNTP pools, inefficient nuclear translocation and constraints in DNA accessibility, repair pathways and replication competence. The permissiveness to in vivo HSC gene transfer observed in early postnatal life might, therefore, extend beyond LV transduction, potentially informing strategies to enhance genetic-engineering approaches. Notably, the few previous reports of in vivo genetic modification of HSCs have relied on either HSPC mobilization followed by chemotherapy-based selection of the limited number of cells engineered using adenoviral vectors or on targeted lipid nanoparticles delivering limiting amounts of RNA cargo5,6.
Our findings indicate that a permissive condition for LV gene transfer also exists in neonatal humans, probably extending into early infantile life, given the high numbers of cHSPCs, including the most primitive subsets24, and the well-established notion that human HSPCs isolated from cord blood exhibit greater permissiveness to LV-mediated transduction compared with those obtained from adult BM.
Systemic LV administration has not yet been reported in humans, but we have previously demonstrated its safety and tolerability in non-human primates7,25. Notably, non-human primates and humans exhibit greater permissiveness to LV gene transfer and respond more effectively to phagocytosis-shielded CD47high-LV than do mice7,25. In a mouse model engineered to be sensitive to human CD47 signalling, we achieved up to 9% stable haematopoietic engineering without transduction enhancers or mobilization protocols. Moreover, the LV doses used in this study fall in the range of doses already applied in preclinical liver-directed LV gene therapy for haemophilia in mouse models25. Interestingly, the estimated initial LV concentration in the PB after infusion also aligns with levels typically used for ex vivo HSPC transduction. Further increases in in vivo HSC gene transfer may be achieved through early-life mobilization and co-administration of clinically compatible transduction enhancers26,27,28, as shown here with IFNα receptor blockade.
In Fanconi anaemia, we successfully exploited the selective advantage conferred by correction of the genetic defect in DNA repair, which specifically affects HSCs. This approach led to a remarkably high proportion of genetic correction in the haematopoietic compartment over the long term, albeit sustained by only a small number of expanded HSC clones. Importantly, ex vivo Fanconi anaemia gene therapy has demonstrated the importance of treating patients at early stages of BM failure to facilitate the collection of clinically relevant numbers of HSCs17,18. An in vivo gene therapy approach would extend the application of treatment to patients with limited HSC content while avoiding ex vivo manipulation of these fragile cells. For ADA-SCID and ARO outcomes see Supplementary Discussion.
In conclusion, our findings support further preclinical studies to assess the translational potential of in vivo HSC gene transfer in early infants. The well-established clinical use of LVs, the advantages of the vector design used here for in vivo delivery and the increasing availability of newborn screening for monogenic diseases provide a strong rationale for progressing towards the first clinical testing of this approach. By bypassing the major barriers associated with ex vivo HSC gene therapy, this strategy stands to offer broader accessibility to life-changing treatments.
Methods
Study design
Sample size in experiments with mice was chosen according to previous experience with experimental models and assays. No sample or animal was excluded from the analyses. Mice were randomly assigned to each experimental group. Investigators were blinded at the moment of LV administration to newborn ADA-SCID or OC mice. In all the other experimental settings investigators were not blinded.
Plasmid construction
Plasmids pCCLsin.cPPT.PGK.GFP, pCCLsin.cPPT.PGK.ADA, pCCLsin.cPPT.PGK.TCIRG1 and pCCLsin.cPPT.PGK.FANCA were previously described14,19,29,30. Plasmid pCCLsin.cPPT.ET.ADA.142T was constructed by standard cloning techniques, by exchanging the human FIX cDNA7 with human ADA cDNA (SpeI–SpeI) into the previously described pCCLsin.cPPT.ET.coFIX-R338L.142T (NheI–SalI) after blunting the ends. Plasmid pCCLsin.cPPT.PGK.ADA.122T was constructed by standard cloning techniques, by inserting the miRNA-122 target sequence into the pCCLsin.cPPT.PGK.ADA plasmid (SalI–ScaI).
Vector production
Third-generation self-inactivating LVs were produced by calcium phosphate transient transfection into 293T cells. 293T cells were transfected with a solution containing a mix of the selected LV genome-transfer plasmid, the packaging plasmids pMDLg-pRRE and pCMV.REV, pMD2.G and pAdVantage (Promega), as previously described7. In brief, the medium was changed 14–16 h after transfection and the supernatant was collected 30 h after medium change. LV-containing supernatants were sterilized through a 0.22-μm filter (Millipore), pre-concentrated four times with a Vivaflow 200 Tangential Flow Filtration Cassette 100 kDa (Sartorius), according to manufacturing instructions, transferred into sterile polyallomer tubes (Beckman) and centrifuged at 20,000g for 120 min at 20 °C (Beckman Optima XL-100K Ultracentrifuge). The LV pellet was dissolved in the appropriate volume of PBS to allow 500–1,000× concentration.
LV titration
For LV titration, 1 × 105 293T cells were transduced with serial LV dilutions in the presence of polybrene (8 μg ml−1). For PGK.GFP-LV, cells were analysed by flow cytometry using a FACSCanto analyser (BD Biosciences), equipped with DIVA software, 5–7 days after transduction and the infectious titre, expressed as transducing units (TU) ml−1, was calculated using the formula TU ml−1 = ((% GFP+ cells/100) × 100,000×(1/dilution factor)). For all other LVs, genomic DNA (gDNA) was extracted 14 days after transduction, using the Maxwell 48 Cell DNA Purification Kit (Promega), following the manufacturer’s instructions. The VCN was determined by ddPCR, starting from 5–20 ng of template gDNA using primers (HIV FW: 5′-TACTGACGCTCTCGCACC-3′; HIV REV: 5′-TCTCGACGCAGGACTCG-3′) and a probe (FAM 5′-ATCTCTCTCCTTCTAGCCTC-3′) designed against the primer-binding region of LV. The amount of endogenous DNA was quantified by a primers–probe set designed against the human telomerase gene (Telo FW: 5′-GGCACACGTGGCTTTTCG-3′; Telo REV: 5′-GGTGAACCTCGTAAGTTTATGCAA-3′; Telo probe: VIC 5′-TCAGGACGTCGAGTGGACACGGTG-3′ TAMRA) or the human GAPDH gene (Applied Biosystems HS00483111_cm). The PCR reaction was performed with each primer (900 nM) and the probe (250 nM; 500 nM for Telo) following the manufacturer’s instructions (Bio-Rad), read with a QX200 reader and analysed with QuantaSoft software (Bio-Rad). The infectious titre, expressed as TU ml−1, was calculated using the formula TU ml−1 = (VCN × 100,000×(1/dilution factor). LV physical particles were measured by HIV-1 Gag p24 antigen immunocapture assay (Perkin Elmer) following the manufacturer’s instructions. LV-specific infectivity was calculated as the ratio between infectious titre and physical particles.
VCN determination
For mouse experiments, DNA was extracted from the whole liver using the Maxwell 48 Tissue DNA Purification Kit (Promega), from a cell suspension obtained from the thymus, spleen or BM using the Maxwell 48 Cell DNA Purification Kit (Promega) or from whole PB using the Maxwell 48 Whole Blood DNA Purification Kit (Promega). DNA was extracted from lineage-negative (Lin−) cultured cells using the Maxwell 48 Cell DNA Purification Kit (Promega) and from single-picked colonies using Quick extract DNA (Biosearch Technologies), according to cell number. The VCN in mouse DNA was determined by ddPCR, starting from 5–20 ng of template gDNA using a primers–probe set designed against the primer-binding region of LVs (see ‘LV titration’ section). In some cases, the VCN was determined using an ad hoc ddPCR (QX200 EvaGreen Digital PCR Supermix, Bio-Rad), which selectively amplifies the reverse-transcribed vector genome (both integrated and non-integrated), discriminating it from possible plasmid contamination29 (RT-LV; ΔU3 FW: 5′-TCACTCCCAACGAAGACAAGATC-3′, gag REV: 5′-GAGTCCTGCGTCGAGAGAG-3′). The amount of endogenous mouse DNA was quantified by a primers–probe set designed against the mouse Sema3a gene (Sema3a FW: 5′-ACCGATTCCAGATGATTGGC-3′; Sema3a REV: 5′-TCCATATTAATGCAGTGCTTGC-3′; Sema3a probe: HEX 5′-AGAGGCCTGTCCTGCAGCTCATGG-3′ BHQ1). The PCR reaction was performed with each primer (900 nM; 150 nM for RT-LV primers) and the probe (250 nM) following the manufacturer’s instructions (Bio-Rad), read with a QX200 reader and analysed with QuantaSoft software (Bio-Rad).
RNA extraction and ddPCR
RNA extraction was performed using a Maxwell 48 SimplyRNA Blood extraction kit (Promega, AS1380), according to the manufacturer’s instructions and reverse transcribed using the SuperScript IV VILO kit (11766050; ThermoFisher Scientific). LV gene expression was assessed by ddPCR, starting from 10–25 ng of template cDNA using a primers–probe set designed against the Woodchuck hepatitis virus post-transcriptional regulatory element (Wpre) sequence of the LV genome (Wpre FW: 5′-GGCTGTTGGGCACTGACAAT-3′; Wpre REV: 5′-ACGTCCCGCGCAGAATC-3′; Wpre probe: FAM 5′-TTTCCATGGCTGCTCGCCTGTGT-3′ MGB). Hprt was used as a reference gene for mouse samples (dMmuCPE5095493, Bio-Rad). The PCR reaction was performed with each primer (900 nM) and the probe (250 nM) following the manufacturer’s instructions (Bio-Rad), read with a QX200 reader and analysed with QuantaSoft software (Bio-Rad).
Cell cultures and in vitro experiments
The 293T cell line was maintained in Iscove’s modified Dulbecco’s medium (IMDM, Sigma) supplemented with 10% fetal bovine serum (FBS; FetalClone II, HyClone, Euroclone), 4 mM glutamine (Lonza), 100 international units (IU) ml−1 penicillin and streptomycin (Lonza). Primary mouse Lin− cells were FACS-sorted from cell suspensions obtained from the BM or the liver of LV-treated mice and cultured in StemSpan supplemented with 10% FBS, 4 mM glutamine, 100 IU ml−1 penicillin and streptomycin, 100 ng ml−1 mouse stem-cell factor (mSCF, Peprotech), 50 ng ml−1 mouse thrombopoietin (mTPO, Peprotech), 100 ng ml−1 mouse fms-like tyrosine kinase 3 ligand (mFLT3L, Peprotech) and 20 ng ml−1 mouse interleukin-3 (mIL-3, Peprotech) for 24 h. The medium was then exchanged with IMDM supplemented with 10% FBS, 100 IU ml−1 penicillin and streptomycin, 100 ng ml−1 mSCF (Peprotech) and mFLT3L 100 ng ml−1 (Peprotech) for 48 h. Finally, GFP expression was determined using a FACSCanto analyser (BD Biosciences), equipped with DIVA software. Some of the sorted Lin− cells were plated (800 cells per plate) in classic Methocult (STEMCELL Technologies) supplemented with 100 ng ml−1 mSCF (Peprotech), 50 ng ml−1 mTPO (Peprotech), 100 ng ml−1 mFLT3L (Peprotech) and 20 ng ml−1 mIL-3 (Peprotech). All cells were maintained in a 5% CO2 humidified atmosphere at 37 °C. Cell lines were routinely tested for mycoplasma contamination.
Mice experiments
Founder FVB,129-Adatm1Mw Tg(PLADA)4118Rkmb/J mice (referred to as ADA-SCID mice) were obtained from The Jackson Laboratory31 (stock 003265). Founder B6C3Fe a/a-Tcirg1oc/J mice (referred to as OC mice) were obtained from The Jackson Laboratory16 (stock 00230) and fully backcrossed on the C57BL/6 background. Founder mice with a targeted disruption in the Fanca gene32 were provided by H. J. van de Vrugt (Free University Medical Center) and backcrossed with FVB mice to obtain the Fanca−/− FVB strain (referred to as Fanca−/− mice). C57BL/6 and NSG mice were purchased from Charles River Laboratories. All mice were maintained in specific-pathogen-free conditions. Genotyping of ADA-SCID and OC mice was performed at 2–3 weeks of age according to the protocol available on The Jackson Laboratory website. Vector administration was carried out in adult female C57BL/6 or NSG (7–10-week-old) mice by tail-vein injection (250–300 µl per mouse), in newborns by temporal-vein injection (30 µl per mouse) and in 2-week-old mice by retro-orbital injection (100–150 µl per mouse). Mice were bled from the retro-orbital plexus using capillary tubes and blood was collected in 0.38% sodium citrate buffer, pH 7.4 or in an EDTA-coated tube (Microvette 20.1341). For transplantation experiments, 7-week-old female C57BL/6 recipient mice were conditioned with a lethal regimen of busulfan (6 mg ml−1), consisting of four daily intraperitoneal (i.p.) administrations of 27 µg g−1 of busulfan (prediluted 1:2 in 0.9% NaCl injectable solution). The transplant was performed by i.v. injection of total BM cells or total CD45+ cells FACS-sorted (BD FACSAria Fusion Cell Sorter, BD Biosciences or MoFlo Astrios EQ Cell Sorter, Beckman Coulter) from the liver or the BM of donor mice the day after the last dose of busulfan. Cells were resuspended in 200 µl of PBS. The type and number of transplanted cells are indicated in the experimental schemes. The weight of recipient mice was monitored daily during busulfan administration and weekly for 1 month after transplant. In case of transplantation experiments in NSG mice, conditioning was performed by sublethal irradiation of recipient mice (200 rad) and transplantation 3–4 h after irradiation. For the transplantation experiments in Fanca−/− mice, conditioning was performed by lethal irradiation of recipient mice using two doses of 4.5 Gy 24 h apart. A total of 3–4 h after conditioning, 4–5 × 106 BM cells from LV-treated or untreated Fanca−/− mice were transplanted by i.v. injection in the tail vein of recipient mice. HSPC mobilization was performed in 2-week-old or adult mice by administering five i.p. doses of 250 ng g−1 G-CSF per mouse 12 h apart. Then 8 h after the last dose of G-CSF, 2 µg g−1 plerixafor per mouse was administered i.p. to mobilized mice. LVs were administered i.v. 2 h after plerixafor and, at the same time, haematopoietic cells were collected from the BM, PB or spleen for FACS analysis. HSPC mobilization was performed in newborn mice following the same protocol used in 2-week-old or adult mice, except for a reduced G-CSF regimen that consisted of three instead of five doses. Anti-IFNα receptor-blocking antibody (MAR-1, InVivoMAb, MAR1-5A3) or IgG control (InVivoMAb, mouse IgG1 Isotype control, MOPC-21) were administered i.v. to newborn mice 3 h before LV administration at 50 mg kg−1 dose. BM failure was induced in Fanca−/− mice by i.p. administration of two doses of 0.3 mg kg−1 of mitomycin C (MilliporeSigma), spaced 7 days apart, 6 weeks after LV injection20. Mice were euthanized by CO2 inhalation at the scheduled times. Experimental procedures in Fanca−/− mice were conducted according to European and Spanish regulations (European convention ETS 123, regarding the use and protection of vertebrate mammals used in experimentation and other scientific purposes, Directive 2010/63/UE, Spanish Law 6/2013 and Real Decreto (R.D.) 53/2013 regarding the protection and use of animals in scientific research). All animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee.
Fractionation of liver, thymus, spleen and BM
The mouse liver was perfused (15 ml min−1) through the inferior vena cava with 12.5 ml of the following solutions at subsequent steps: (1) PBS EDTA (0.5 mM); (2) HBSS (Gibco) and HEPES (10 mM); and (3) HBSS and HEPES containing 0.03% collagenase IV (Sigma). The digested mouse liver tissue was collected, passed through a 100-μm cell strainer (BD Biosciences) and processed into a single-cell suspension. This suspension was subsequently centrifuged three times (30g, 25g and 20g, for 3 min, at room temperature) to remove hepatocyte-containing pellets. The supernatant containing non-parenchymal cells was centrifuged (650g, 7 min, at room temperature) and recovered cells were subsequently incubated with the monoclonal antibodies indicated in the ‘Flow cytometry’ section. To obtain a single-cell suspension of spleen or thymus, organs were smashed on a 40-µm filter using PBS–2% FBS (FetalClone II, HyClone, Euroclone), red blood cells (RBCs) were lysed with 0.5 or 1 ml of ACK Lysing buffer (GIBCO) for 5 or 10 min on ice, respectively. The lysis was stopped by adding 10 ml of PBS–2% FBS. The cell suspension was then filtered through a 40-µm filter and cells were counted and used for gDNA extraction or flow-cytometry analysis (see ‘Flow cytometry’ section). BM cells were flushed from the tibia and femur of one or two legs, as needed, or extracted by bone crushing in the case of newborn or OC mice and then processed as described for cells isolated from the spleen.
Flow cytometry
Flow-cytometry analyses using cell suspensions obtained from BM, spleen, thymus, PB or liver were performed using a FACSCanto analyser (BD Biosciences), equipped with DIVA software. Between 500,000 and 1,000,000 cells were processed, washed with PBS–2% FBS (FetalClone II, HyClone, Euroclone) or MACS buffer (PBS pH 7.2, 0.5% bovine serum albumin (BSA), 2 mM EDTA), treated with fragment crystallizable (Fc) Receptor-Block (Miltenyi Biotec) when antibody-stained and then resuspended in the buffer used for washing. Staining was performed in MACS buffer, incubating cells with antibodies (in the proportion indicated in Supplementary Table 15) for 20 min at 4 °C in the dark. Right before analysis, 3 µl of 7-AAD (Biolegend 420404) was added to each tube to exclude dead cells. Rainbow beads (BD Biosciences) were used to calibrate the instrument detectors. Gating strategies are provided in Supplementary Figs. 1–4. A BD FACSAria Fusion Cell Sorter (BD Biosciences), equipped with DIVA software (BD Biosciences), or a MoFlo Astrios EQ Cell Sorter (Beckman Coulter), equipped with Summit software (Beckman Coulter), was used for cell sorting.
For PB samples collected from healthy human participants of different ages and mouse samples shown in Fig. 1b–i and Extended Data Fig. 1a–d, samples were analysed using two standardized multiparametric flow-cytometry assays21,33 (through whole blood dissection). In brief, after RBC lysis with ACK lysing buffer (STEM CELL Technologies), samples were labelled with the fluorescent antibodies reported in Supplementary Table 16. Titration assays were performed to assess the best antibody concentration. Samples from mice were pre-incubated with a rat anti-mouse FcR blocking reagent (BD, dilution 1:100). After surface marking, cells were incubated with propidium iodide (PI; BioLegend) to stain dead cells. Absolute cell quantification was performed by adding Precision count beads (BioLegend) to PB samples before the staining procedure. Images of all stained samples were acquired through a BD Symphony A5 (BD Bioscience) cytofluorimeter after calibration with Rainbow beads (Spherotech) and raw data were collected using the DIVA software (BD Biosciences). Data were subsequently analysed with FlowJo software v.10.5.3 (BD Biosciences). A list of antibodies used for mouse or human whole-blood-dissection panels is reported in Supplementary Table 16.
PB samples from paediatric and adult healthy donors were collected and analysed after written informed consent was given, using protocols approved by the ethics committees of IRCCS Ospedale San Raffaele (Protocol_TIGET09 for both paediatric and adult participants). Written consent for paediatric participants was given by their parents.
Immunohistochemistry imaging
Mice were perfused through the left ventricle with PBS containing 0.5 mM EDTA to remove blood, after which organs were collected and fixed in zinc-formalin for at least 24 h. Embedding, sectioning, slide preparation, staining and image acquisition were performed at the Centro di Imaging Sperimentale facility at San Raffaele Hospital (Italy). Formalin-fixed, paraffin-embedded consecutive sections (4 µm) were dewaxed and rehydrated through a graded alcohol series before histological or immunohistochemical staining. Immunostaining was performed using the Automatic Leica BOND RX system (Leica Microsystems). Tissues were first deparaffinized and subjected to epitope retrieval using Epitope Retrieval Solution 1 (ER1 Citrate Buffer). Slides were incubated with either an anti-GFP primary antibody (Invitrogen, A11122, 1:1,000) or an anti-Ki-67 primary antibody (D3B5, Cell Signaling Technology, 1:200) for 30 min at room temperature, followed by detection using the Bond Polymer Refine Detection Kit (Leica, DS9800). Images of stained slides were acquired using an Aperio AT2 digital scanner at 20× magnification (Leica Biosystems) and analysed with ImageScope software (Leica Biosystems).
Integration-site analysis
Integration sites were retrieved by Sonication Linker Mediated (SLiM)-PCR, as previously described34, with minor modifications. In brief, for each sample up to 300 ng of gDNA was processed by DNA shearing using the Covaris E220 Ultrasonicator, generating fragments with an average size of 1,000 bp. The fragmented DNA was subjected to end repair and 3′ adenylation, and then ligated to linker cassettes containing an eight-nucleotide sequence barcode used for sample identification and all the sequences required for Read 2 paired-end sequencing. The ligation products were split in three technical replicates and subjected to 35 cycles of exponential PCR using primers specific for the LV LTR and the linker cassette. A subsequent amplification with an additional ten PCR cycles was performed using a primer specific for the linker cassette and the LTR. These primers contain an 8-nucleotide barcode used for sample identification (coupled with the barcode on the linker cassette) and the sequences needed for Read 1 sequencing, plus a random 12-nucleotide sequence, enabling easier cluster recognition in the first sequencing cycles on the next-generation sequencing system. The generated SLiM-PCR products are therefore associated with a unique pair of barcodes, assembled into libraries and subjected to Illumina next-generation sequencing.
Bioinformatic analysis
Sequencing reads were processed using a custom bioinformatics pipeline35 (VISPA2) that isolates genomic sequences flanking the vector LTR and maps them to the mouse genome (mm9). Because vector integration in the same genomic position in different cells is a very low probability event, identical integration sites in independent samples were considered contamination or amplification artefacts, which may occur during the technical procedure. Datasets were pruned of potential contamination and false positives for each primary mouse and of integration sites derived from unrelated secondary mice. Identical integration sites shared between mice belonging to different experimental groups were reassigned by identifying the insertion site in at least two SLiM technical replicates and through sequence counts. Downstream analyses of vector-integration sites, such as relative abundance, sharing and CIS analysis, were performed using ISAnalytics36. Clonal abundance, estimated as the relative percentage of genome numbers, was determined by the R package sonicLength37. Common insertion sites were calculated using the Grubbs test for outliers38.
Haemocytometric analysis
Haemocytometric analysis of PB was performed using a Procyte DX Analyser (IDEXX) at the time points indicated in the experimental design schemes. Approximately 30 µl of PB was collected into EDTA-coated tubes (Microvette 20.1341) and analysed. The measured parameters were: haemoglobin, total RBC count, differential count (lymphocyte, monocyte and neutrophil (absolute count and percentage)), platelet count and total leukocyte count (white blood cell).
ADA activity
ADA activity was measured in plasma or the cellular fraction of PB as previously described39. In brief, 20 μl of packed RBCs were lysed in 20 μl of lysis buffer (0.25 mol l−1 HEPES, 0.025 mol l−1 CHAPS and 0.025 mol l−1 dithiothreitol). The cell lysate was centrifuged at 12,000g for 5 min. The enzyme activity was evaluated in a mixture containing 50 mmol l−1 Tris (pH 7.2) and 0.4 mmol l−1 adenosine by adding 10 μl of lysed RBC supernatant or 20 μl of plasma. The reaction was stopped by adding PCA (final concentration, 0.21 mmol l−1) and neutralized with KOH (final concentration, 0.22 mmol l−1). The reaction progression was analysed using aliquots at two time points with a Bio-Rad Biofocus 3000 instrument (Bio-Rad) after inosine plus hypoxanthine formation at 0 and 10 min for the cellular fraction or at 0 and 30 min for plasma. The enzyme activity was measured as mol h−1 per (ml packed RBCs or ml plasma).
Osteoclast differentiation, CTX and parathyroid hormone ELISA and dentine assay
Osteoclasts were differentiated from BM cells as previously described40 and assayed for resorption functions on dentine slides (AE8050). Dentine discs were stained with toluidine blue as previously described40. The concentration of released CTX in the culture supernatant was measured by enzyme-linked immunosorbent assay (ELISA; CrossLaps, AC-07F1, Immunodiagnostic System) following the manufacturer’s protocol. The concentration of parathyroid hormone in the serum of LV-treated OC or WT mice was measured by ELISA (MicroVue Mouse parathyroid hormone 1-84 EIA) following the manufacturer’s protocol.
Micro-computed tomography
For micro-computed tomography analyses, vertebral columns of OC mice and their WT littermates were dissected, cleaned from connective tissues and fixed in 4% paraformaldehyde. Bones were scanned at a nominal image resolution of 7 µm using a SkyScan 1276 (Bruker) at 55 kV and 72 µA, using a 0.25-mm aluminium filter. Images were captured every 0.4° in a rotation to 360° with a 2 × 2 camera pixel binning. The reconstruction was carried out with the SkyScan NRecon 2.0.0.5 software using the InstaRecon reconstruction engine, ring artefact reduction and beam-hardening correction. Image analysis of the L5 vertebra was performed using the CT Analyser software (CTAn 1.18.8.0+). An in-house-developed semiautomatic approach was used to segment the trabecular region of the vertebral body in the coronal plane. The bone mineral density of the trabecular region was calculated by calibrating a linear extrapolation against the mean attenuation coefficients of selected regions of a 2-mm phantom rod pair containing 0.25 and 0.75 g cm−3 CaHA (Skyscan). A three-dimensional morphometric analysis of the trabecular region was performed after binarization of the trabeculae by an adaptive thresholding algorithm. The ratio between the bone volume and total volume (%) and the trabecular separation (mm) were assessed.
Statistical analyses
Statistical analyses were performed after consulting with professional statisticians at the San Raffaele University Center for Statistics in the Biomedical Sciences (CUSSB). When normality assumptions were not met, nonparametric statistical tests were performed. A two-tailed Mann–Whitney U-test was performed to compare two independent groups and for more than two independent groups, a Kruskal–Wallis test was used, followed by post hoc analysis (Dunn’s test for multiple comparisons against the reference control group along with Bonferroni’s correction). For paired observations, a Wilcoxon matched-pairs test was performed. Data were analysed using GraphPad (v.10). To model the dynamics of biological processes, properly accounting for dependencies induced by the repeated-measures design, LME models were estimated, specifying random-effect terms for experimental units or session. To fulfil underlying model assumptions, various transformations of the outcome variable, including logarithmic, square/cubic root and ordered quantile normalization, were considered. After the estimation of LME models, a post hoc analysis was conducted, enabling the pairwise comparison of treatment groups at a fixed time point. To account for multiplicity issues, P values were adjusted using Bonferroni’s approach. LME were estimated using R software (v.4.3.1). In all the analyses, the significance threshold was set at 0.05. Inferential techniques were used for adequate sample sizes (n ≥ 5), otherwise only descriptive statistics are reported.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data associated with this study are available in the Source data files. Source data are provided with this paper.
References
Ferrari, G., Thrasher, A. J. & Aiuti, A. Gene therapy using haematopoietic stem and progenitor cells. Nat. Rev. Genet. 22, 216–234 (2021).
Naldini, L. Genetic engineering of hematopoiesis: current stage of clinical translation and future perspectives. EMBO Mol. Med. 11, e9958 (2019).
Ferrari, S. et al. Genetic engineering meets hematopoietic stem cell biology for next-generation gene therapy. Cell Stem Cell 30, 549–570 (2023).
Ferrari, S. & Naldini, L. A step toward stem cell engineering in vivo. Science 381, 378–379 (2023).
Richter, M. et al. In vivo transduction of primitive mobilized hematopoietic stem cells after intravenous injection of integrating adenovirus vectors. Blood 128, 2206–2217 (2016).
Breda, L. et al. In vivo hematopoietic stem cell modification by mRNA delivery. Science 381, 436–443 (2023).
Milani, M. et al. Phagocytosis-shielded lentiviral vectors improve liver gene therapy in nonhuman primates. Sci. Transl. Med. 11, eaav7325 (2019).
Milani, M. et al. GP64-pseudotyped lentiviral vectors target liver endothelial cells and correct hemophilia A mice. EMBO Mol. Med. 16, 1427–1450 (2024).
Takenaka, K. et al. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat. Immunol. 8, 1313–1323 (2007).
Aiuti, A. et al. Multilineage hematopoietic reconstitution without clonal selection in ADA-SCID patients treated with stem cell gene therapy. J. Clin. Invest. 117, 2233–2240 (2007).
Ferrua, F. & Aiuti, A. Twenty-five years of gene therapy for ADA-SCID: from bubble babies to an approved drug. Hum. Gene Ther. 28, 972–981 (2017).
Cantore, A. et al. Hyperfunctional coagulation factor IX improves the efficacy of gene therapy in hemophilic mice. Blood 120, 4517–4520 (2012).
Carbonaro, D. A. et al. Gene therapy/bone marrow transplantation in ADA-deficient mice: roles of enzyme-replacement therapy and cytoreduction. Blood 120, 3677–3687 (2012).
Mortellaro, A. et al. Ex vivo gene therapy with lentiviral vectors rescues adenosine deaminase (ADA)-deficient mice and corrects their immune and metabolic defects. Blood 108, 2979–2988 (2006).
Capo, V., Abinun, M. & Villa, A. Osteoclast rich osteopetrosis due to defects in the TCIRG1 gene. Bone 165, 116519 (2022).
Scimeca, J. C. et al. The gene encoding the mouse homologue of the human osteoclast-specific 116-kDa V-ATPase subunit bears a deletion in osteosclerotic (oc/oc) mutants. Bone 26, 207–213 (2000).
Rio, P. et al. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat. Med. 25, 1396–1401 (2019).
Rio, P. et al. Haematopoietic gene therapy of non-conditioned patients with Fanconi anaemia-A: results from open-label phase 1/2 (FANCOLEN-1) and long-term clinical trials. Lancet 404, 2584–2592 (2025).
Rio, P. et al. In vitro phenotypic correction of hematopoietic progenitors from Fanconi anemia group A knockout mice. Blood 100, 2032–2039 (2002).
Casado, J. A. et al. Upregulation of NKG2D ligands impairs hematopoietic stem cell function in Fanconi anemia. J. Clin. Invest. 132, e142842 (2022).
Basso-Ricci, L. et al. Multiparametric whole blood dissection: a one-shot comprehensive picture of the human hematopoietic system. Cytometry A 91, 952–965 (2017).
Li, Y. et al. Single-cell analysis of neonatal HSC ontogeny reveals gradual and uncoordinated transcriptional reprogramming that begins before birth. Cell Stem Cell 27, 732–747 (2020).
Collins, A. et al. Maternal inflammation regulates fetal emergency myelopoiesis. Cell 187, 1402–1421 (2024).
Quaranta, P. et al. Circulating hematopoietic stem/progenitor cells subsets contribute to human hematopoietic homeostasis. Blood 143, 1937–1952 (2024).
Milani, M. et al. Liver-directed lentiviral gene therapy corrects hemophilia A mice and achieves normal-range factor VIII activity in non-human primates. Nat. Commun. 13, 2454 (2022).
Santoni de Sio, F. R., Cascio, P., Zingale, A., Gasparini, M. & Naldini, L. Proteasome activity restricts lentiviral gene transfer into hematopoietic stem cells and is down-regulated by cytokines that enhance transduction. Blood 107, 4257–4265 (2006).
Santoni de Sio, F. R. et al. Lentiviral vector gene transfer is limited by the proteasome at postentry steps in various types of stem cells. Stem Cells 26, 2142–2152 (2008).
Brown, B. D. et al. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood 109, 2797–2805 (2007).
Matrai, J. et al. Hepatocyte-targeted expression by integrase-defective lentiviral vectors induces antigen-specific tolerance in mice with low genotoxic risk. Hepatology 53, 1696–1707 (2011).
Capo, V. et al. Expanded circulating hematopoietic stem/progenitor cells as novel cell source for the treatment of TCIRG1 osteopetrosis. Haematologica 106, 74–86 (2021).
Blackburn, M. R., Datta, S. K. & Kellems, R. E. Adenosine deaminase-deficient mice generated using a two-stage genetic engineering strategy exhibit a combined immunodeficiency. J. Biol. Chem. 273, 5093–5100 (1998).
Cheng, N. C. et al. Mice with a targeted disruption of the Fanconi anemia homolog Fanca. Hum. Mol. Genet. 9, 1805–1811 (2000).
Colantuoni, M. et al. Constitutive IL-1RA production by modified immune cells protects against IL-1-mediated inflammatory disorders. Sci. Transl. Med. 15, eade3856 (2023).
Cesana, D. et al. Retrieval of vector integration sites from cell-free DNA. Nat. Med. 27, 1458–1470 (2021).
Spinozzi, G. et al. VISPA2: a scalable pipeline for high-throughput identification and annotation of vector integration sites. BMC Bioinformatics 18, 520 (2017).
Pais, G. et al. ISAnalytics enables longitudinal and high-throughput clonal tracking studies in hematopoietic stem cell gene therapy applications. Brief. Bioinform. 24, bbac551 (2023).
Berry, C. C. et al. Estimating abundances of retroviral insertion sites from DNA fragment length data. Bioinformatics 28, 755–762 (2012).
Biffi, A. et al. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood 117, 5332–5339 (2011).
Carlucci, F. et al. Capillary electrophoresis in diagnosis and monitoring of adenosine deaminase deficiency. Clin. Chem. 49, 1830–1838 (2003).
Palagano, E. et al. Generation of an immunodeficient mouse model of tcirg1-deficient autosomal recessive osteopetrosis. Bone Rep. 12, 100242 (2020).
Acknowledgements
We thank A. Fiocchi and the Experimental Imaging Center (San Raffaele Scientific Institute) for imaging techniques and analysis, all the members of the Cantore laboratory for helpful discussions and L. Martínez de la Cal, L. García and M. Angel Martín for their technical support in the experiments performed with the Fanca−/− mouse model. L.S. conducted this study as partial fulfilment of his International PhD Course in Molecular Medicine, Cellular and Molecular Biology Programme at the San Raffaele University. This work was supported by Fondazione Telethon SR-Tiget Core Grant (TTACC0422TT) to A.C., Fondo Italiano per la Scienza (FIS 2021 Advanced Grant FIS00002157) to L.N., Fondazione Telethon SR-Tiget Core Grant E2 (TTAVC0522TT) to A.V., Fondazione Telethon SR-Tiget Core Grant (Tgt2016) to A.A. and S.S. and by Fondazione CARIPLO Giovani Ricercatori (GR-2019-12369499) to S.S. A.A. is the recipient of the Else Kröner Fresenius Prize for Medical Research 2020. J.A.B., M.P.-R. and P.R. are supported by Instituto de Salud Carlos III (ISCIII)/Red Española de Terapias Avanzadas RICORS/TERAV and by the European Union NextGenerationEU (RD21/0017/0027) Plan de Recuperación Transformación y Resiliencia. P.R. receives funding and M.P.-R. is supported by the PID2021-125077OB-C21 grant from Ministry of Science, Innovation and Universities.
Author information
Authors and Affiliations
Contributions
M.M. designed and performed experiments, analysed and interpreted data, coordinated research and wrote the manuscript. A. Fabiano designed and performed experiments and analysed data. M.P.-R. performed experiments and analysed data related to Fanconi anaemia. R.J.H. designed and performed experiments and analysed data related to ADA-SCID. A.Z. and V.I. designed and performed experiments and analysed data related to ARO. E.Z. designed experiments, provided intellectual input and edited the manuscript. M.V. analysed integration-site analysis data. L.O. performed experimental work related to integration-site analysis. S.O., L.B.-R., C.C., P.Q., L.S. and M.B. performed experiments and analysed data. F.R. performed experiments. C.B. performed statistical analysis. F.C. performed ADA activity quantification. M.F. performed micro-computed tomography scans and image analysis. A. Forlino analysed micro-computed tomography images and interpreted data. E.M. supervised work related to integration-site analysis. S.S supervised work related to cHSPCs, provided intellectual input and edited the manuscript. V.C. and A.V. supervised ARO work, provided intellectual input and edited the manuscript. P.R. and J.A.B. supervised Fanconi anaemia work, provided intellectual input and edited the manuscript. A.A. supervised ADA-SCID work, provided intellectual input and edited the manuscript. A.C. supervised research, interpreted data and wrote the manuscript. L.N. supervised research, interpreted data and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
P.R. and J.A.B. receive funding and have licensed the PGK.FANCA-LV to Rocket Pharmaceuticals and are inventors on patents filed by CIEMAT, CIBERER and Fundación Jiménez Díaz, and may be entitled to receive financial benefits from the licensing of such patents. J.A.B. is consultant of Rocket Pharmaceuticals, A.A. is the principal investigator of clinical trials with LV-transduced haematopoietic stem cells sponsored by Orchard Therapeutics or Fondazione Telethon. L.N. and A.C. are inventors on patent applications submitted by Fondazione Telethon or the San Raffaele Scientific Institute on LV technology for in vivo use related to the work presented in this paper. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Jose-Carlos Segovia and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Characterization of the hematopoietic system in mice of different ages and in vivo HSPC gene transfer by LV.
a-d, Mean with SEM of the percentage of different populations among CD45-positive cells in the BM (a), PB (b), spleen (c), or liver (d) of newborn, 1-week-old, 2-week-old, or 8-week-old (adult) C57Bl/6 mice, as indicated. n = 5 mice per group. Newborn PB samples represent 10 pools derived from 50 mice. e, Representative immunohistochemistry image (out of 5 collected) of a liver section from a 1-week-old C57Bl/6 mouse stained with an anti-Ki67 (brown) antibody. Black arrowheads indicate proliferating hematopoietic cells; the red arrowhead indicates a representative megakaryocyte. f, Single VCN values measured in the indicated spleen populations or whole BM from the pooled donor mice shown in Fig. 1k,l. BM cells from 19 LV-treated newborn mice were pooled into five groups (each containing 4–5 mice) for transplantation into five recipient mice after sorting. g, Body weights of individual busulfan-conditioned adult mice that were transplanted (TPX n = 5) or not transplanted (not-TPX, n = 1). The gray area indicates the period of busulfan treatment. Black crosses mark deceased mice. h, Single values of CD45-positive GFP-positive cells in the indicated organs of recipient mice 20 wks post-transplant. VCN values are indicated above the bars. i-l, LV integration site (IS) analysis of GFP-positive cells from LV-treated donor mice and transplanted recipient mice at the study endpoint. Shared IS are color-coded. Clones with an abundance of <1% are shown in gray. The total number of IS detected is indicated above each bar. m, Common integration site analysis of LV-treated and transplanted mice, showing the distribution of integration events relative to known oncogenes. n, Mean with SEM of GFP-positive cells in PBMCs of LV-treated mice as newborns (n = 4) and monitored for 1 year after LV administration (5e10 TU/kg). o, Mean with SEM of GFP-positive cells in PBMCs of LV-treated mice as adults (n = 5, 8-week-old) via i.v. LV administration (8e10 TU/kg). p, Representative immunofluorescence image of the liver from a mouse shown in (o), revealing few-to-no GFP-positive cells. Scale bar: 500 µm. Blue: Hoechst; Green: GFP.
Extended Data Fig. 2 Gene transfer efficiency into HSPC in 2-week-old C57Bl/6 mice after in vivo LV administration.
a,b, Mean with SEM of GFP-positive cells (a) or VCN (b) measured in the PB of C57Bl/6 mice administered LV (LV, n = 15; 3e10 TU/kg) at 2 weeks-of-age or untreated (Unt, n = 1). c, Single values and mean with SEM of the VCN measured in the indicated organs in Unt and LV mice, 16 weeks after LV administration. d, Single values and mean with SEM of the absolute count of HSCs in the spleen of 2-week-old mobilized (n = 6) or untreated control (n = 7) mice, as indicated. Mann-Whitney test. e,f, Single values and mean with SEM of GFP-positive cells in the thymus (e) or spleen (f) of mobilized (n = 6) or control (n = 7) mice, 20 wks after LV administration. Mann-Whitney test. g,h, Mean with SEM of the engraftment of CD45.2 cells in the PB of CD45.1 recipient mice over time. Controls: n = 5 recipient; Mobilized: n = 6 recipient. Mice were transplanted with 15x106 total BM cells harvested from donor mice, 20 wks after LV administration. i,j, Single values and mean with SEM of the VCN (i) or engraftment of CD45.2 cells (j) measured in CD45.1 recipient mice, 16 wks after transplant in the indicated organs. Mann-Whitney test in (i). k,l, Single values and mean with SEM of GFP-positive cells in the thymus (k), or spleen (l) of CD45.1 recipient mice, 16 wks after transplant. Mann-Whitney test.
Extended Data Fig. 3 Administration of anti-interferon-α receptor blocking antibody (MAR-1) increase in vivo gene transfer to HSPCs.
a,b, Mean with SEM of GFP-positive cells (a) or the VCN (b) measured over time in the PB of C57Bl/6 mice treated as newborns with MAR-1 (i.v., 50 mg/kg, n = 13) or an IgG control (i.v., 50 mg/kg, n = 13) 3 h before LV administration (8e10 TU/kg). Mann-Whitney test on the last time point analyzed. In (a), comparisons were made among CD45-positive cells. Data from a pool of two independent experiments are shown. c-e, Single values and mean with SEM of GFP-positive cells in the spleen (c), thymus (d), and BM (e) of the mice shown in (a,b) 16 wks after LV administration. Mann-Whitney test. f, Single values and mean with SEM of the VCN measured in the indicated organs, 16 wks after LV administration in the mice shown in (a-e). Mann-Whitney test.
Extended Data Fig. 4 Characterization of the HSPC compartment in NSG mice of different ages and in vivo HSPC gene transfer by LV.
a, Experimental scheme: hematopoietic cells were collected from the bone marrow (BM), spleen, liver, or peripheral blood (PB) of newborn, 1-week-old, 2-week-old, or adult NSG mice and analyzed by FACS to evaluate the composition of total CD45-positive cells. n = 5 mice per group. Newborn PB samples from 40 mice were pooled into eight groups. b-e, Mean with SEM of the percentage of different populations among lineage-negative (Lin-) and cKit-positive (cKit+) cells in the BM (b), PB (c), spleen (d), or liver (e) of the indicated NSG mice. HSC: hematopoietic stem cells; MPP: multipotent progenitor cells; GMLP: granulocyte-monocyte-lymphoid progenitor cells; KSL: cKit+ Sca1+ Lin- cells; LK CD150: Lin- cKit+ CD150+ cells; EP: erythroid progenitor cells; MEP: megakaryocyte-erythroid progenitor cells; GMP: granulocyte-monocyte progenitor cells. n = 5 mice per group. Newborn PB samples from 40 mice were pooled into eight groups. f-i, Single values and mean with SEM of the absolute count of HSCs in the BM (f), PB (g), spleen (h), or liver (i) of the indicated NSG mice. n = 5 mice per group. Newborn PB samples from 40 mice were pooled into eight groups. Kruskal-Wallis test with Dunn’s multiple comparisons test (comparison to the newborn group). j, Mean with SEM of the count of HSPC red or white colonies grown from GFP-positive (GFPpos) or GFP-negative (GFPneg) Lin- cells isolated from the BM of the mice in Fig. 2k–m, 20 wks after LV administration. k, Single VCN values measured in FACS-sorted GFPpos or GFPneg Lin- cells isolated from mice shown in Fig. 2m,n and cultured for 72 h. l, Single VCN values measured from single-picked red (RFC) or white (WFC) colonies grown in MethoCult from GFPpos or GFPneg FACS-sorted Lin- cells isolated from the mice shown in Fig. 2m,n. m, Single values and mean with SEM of the GFP-positive area measured by immunohistochemistry in the indicated tissues, 20 wks after LV administration in NSG mice treated as newborns (n = 6) or adults (n = 4) with CD47hi-LV (shown in Fig. 2m,n), or untreated (Unt, n = 3). n, Representative immunohistochemistry images of the indicated tissues taken from the mice shown in (m). GFP-positive cells are shown in brown (black arrowheads). Scale bar: 300 µm. Schematics in a created with BioRender (https://biorender.com).
Extended Data Fig. 5 Gene transfer efficiency into HSPC in 2-week-old mice after in vivo LV administration.
a-c, Single values and mean with SEM of the absolute HSC counts in the BM (a), PB (b), or spleen (c) of 2-week-old or adult mobilized (n = 5 per group) or untreated (n = 2 per group) NSG mice, as indicated. Mann-Whitney test. d,e, Single values and mean with SEM of GFP-positive cells measured in the indicated populations in the spleen (d) or BM (e) of the mice shown in Fig. 2o, 16 wks after LV administration. Kruskal-Wallis test with Dunn’s multiple comparisons (comparison to the 2-week-old control group). f, Single values and mean with SEM of the VCN measured in the indicated organs of the mice shown in Fig. 2o, 16 wks after LV administration. Kruskal-Wallis test with Dunn’s multiple comparisons (comparison to the respective 2-week-old control groups).
Extended Data Fig. 6 In vivo HSPC gene transfer in ADA-SCID mice by ADA-LV.
a, Mean with SEM of the weight over time of mice shown in Fig. 3b. b,c, Mean with SEM of neutrophil (b) or monocyte (c) counts measured by hemocytometer in the PB of ADA-SCID mice treated with the indicated LV as newborns. The number of mice at each time point is reported below the graphs. LME model (only comparisons vs. WT and p-values < 0.05 are reported; complete analyses in Supplementary Tables 10, 11). d,e, Single values and mean with SEM of ADA activity measured over time in the PB cellular fraction (d) or plasma (e) from ADA-SCID or wild-type (WT) mice treated with the indicated LV or left untreated. LME model (only comparisons vs. WT and p-values < 0.05 are reported; complete analyses in Supplementary Tables 12, 13). f, Single values and mean with SEM of LV-RNA expression in the indicated organs or liver subpopulations 8 wks after administration of the indicated LV to newborn ADA-SCID mice (n = 5 per group, 2e10 TU/kg). Wpre: Woodchuck hepatitis virus post-transcriptional regulatory element, used to measure LV genome expression. Kruskal-Wallis test with Dunn’s multiple comparisons test (comparison to the PGK.ADA group). g, Mean with SEM of the weights of the mice shown in Fig. 3h. h, Single values and mean with SEM of the engraftment of cells isolated from ADA-SCID or WT mice treated with the indicated LV as newborns and used as donors for irradiated NSG mice. LV-treated WT mice have been pooled, irrespective of the administered LV. i,j, Mean with SEM of the percentage of the indicated populations among CD45-positive cells in the PB of transplanted NSG mice, 8 (i) or 12 wks (j) after transplant. k, Mean with SEM of the VCN measured in the PB of NSG-transplanted or untreated (Unt) mice over time. LME model (only comparisons vs. WT and p-values < 0.05 are reported; complete analyses in Supplementary Table 14). l, Representative LV integration site analysis of cells from one LV-treated newborn ADA-SCID mouse, 20 wks after PGK.ADA-LV administration (see Fig. 3b, left panel) and the corresponding NSG recipient mouse, 12 wks after transplant (see Fig. 3h, right panel). Shared integrations are color-coded. Clones with abundance <1% are shown in grey. The total number of integrations is reported on top of the bars. m,n, Survival (m) and individual body weights (n) of ADA-SCID mice treated at 2 weeks-of-age with PGK.ADA-LV (n = 5, 2.5e10 TU/kg) compared to WT (WT, n = 2) littermates.
Extended Data Fig. 7 In vivo HSPC gene transfer in osteopetrotic newborn mice.
a, Survival of LV-treated osteopetrotic (OC-LV, n = 11) or WT mice (WT-LV, n = 27) treated as newborns with PGK.TCIRG1-LV (1-2e10 TU/kg) compared to untreated OC mice (OC-unt, n = 8) mice. b, Single values of the weight of OC-LV or WT-LV mice over time post LV treatment. c,d, Mean with SEM of white blood cell (WBC, c) or lymphocyte (d) counts measured by hemocytometer in the PB of OC-LV (n = 10) or, WT-LV (n = 16) mice, or untreated WT (WT-unt) mice (n = 3) over time. Sample sizes (n) at each time point are reported below the graphs. e, Mean with SEM of the VCN measured in the PB of OC-LV (n = 9), WT-LV mice (n = 15), or WT-unt mice (n = 3) over time. Sample sizes (n) at each time point are reported below the graphs. Šídák’s multiple comparisons test. f, Single values and mean with SEM of the total cell count isolated at necropsy from the BM or spleen of OC-LV (n = 7) or WT-LV (n = 15) mice. Mann-Whitney test. g,h, Single values and mean with SEM of the indicated populations among CD45-positive cells analyzed at necropsy in the spleen (g) or BM (h) of OC-LV (n = 7) or WT-LV (n = 15) mice. i, Single values and mean with SEM of the VCN measured in the indicated organs at necropsy in OC-LV (n = 9), WT-LV mice (n = 14), or WT-unt mice (n = 4). Mann-Whitney test. j, Representative images of toluidine blue staining of dentine discs after a resorption assay of osteoclasts differentiated from the BM of LV-treated WT or OC mice, as indicated. The purple area represents resorbed dentine. Scale bar: 200 µm. k, Single values and mean with SEM of C-terminal telopeptide (CTX) measured in the medium of osteoclast cultures from OC-LV (n = 5) or WT-LV (n = 4) mice following dentine resorption. l-o, Representative micro-tomography scans (l) or single values and mean with SEM of the quantification of bone mineral density (m), bone volume fraction (n), and trabecular separation (o) of WT-LV (n = 7), OC-LV-treated (n = 8), or WT-unt (n = 1) mice. Asterisks indicate the longest-surviving OC-LV mice, one of which is also shown in the bottom-left panel in (l). Scale bar: 500 µm. p, Single values and mean of parathyroid hormone (PTH) concentration in the serum of OC-LV (n = 10) or WT-LV (n = 7) mice at the indicated time points. Sample sizes (n) at each time point are reported below the graphs.
Extended Data Fig. 8 In vivo HSPC gene transfer in Fanca-/- mice by PGK.FANCA-LV.
a, Mean with SEM of the percentage of different populations among lineage-negative (Lin-) and cKit-positive (cKit + ) cells (HSPCs) in the PB of newborn, 2-week-old, or 8-week-old (adult) Fanca-/- mice, as indicated. HSC: hematopoietic stem cells; MPP: multipotent progenitor cells; GMLP: granulocyte-monocyte-lymphoid progenitor cells; KSL: cKit+ Sca1+ Lin- cells; LK CD150: Lin- cKit+ CD150+ cells; EP: erythroid progenitor cells; MEP: megakaryocyte-erythroid progenitor cells; GMP: granulocyte-monocyte progenitor cells. b, Single values and mean with SEM of the absolute count of HSCs in the PB of newborn, 2-week-old, or 8-week-old Fanca-/- mice, as indicated. n = 5 mice per group. Newborn PB samples represent six pools from 30 mice. Kruskal-Wallis test with Dunn’s multiple comparisons test (comparison to the newborn group). c-e, Mean with SEM of neutrophil (c), monocyte (d), or platelet (e) counts measured by hemocytometer in the PB of LV-treated (FANCA-LV) or untreated (FANCA-unt) Fanca-/- mice (n = 5) or untreated wild-type (WT-unt) mice (n = 5). Kruskal-Wallis test with Dunnett’s multiple comparisons test (comparison to WT). f, Single values and mean with SEM of the VCN measured after FACS-sorting cell populations from PB or BM of the mice shown in Fig. 5a–e, 1year after LV administration. g, Representative LV integration site (IS) analysis of cells from one LV-treated newborn Fanca-/- mouse, 20 wks after PGK.FANCA-LV administration (shown in Fig. 5a–e). Shared integrations among the indicated organs or FACS-sorted populations from PB or BM are color-coded. Clones with an abundance <1% are shown in grey. The total number of integrations is reported on top of the bars. h, Representative heatmap showing shared integrations (columns) in the indicated organs or FACS-sorted populations isolated from a PGK.FANCA-LV-treated mouse (shown in Fig. 5a–e). “Sample count” refers to the number of samples (rows) that share a given IS.
Supplementary information
Supplementary Information
Supplementary Figs. 1–4, Supplementary Tables 1–16, Supplementary Notes, Supplementary Discussion and Supplementary references.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Milani, M., Fabiano, A., Perez-Rodriguez, M. et al. In vivo haemopoietic stem cell gene therapy enabled by postnatal trafficking. Nature 643, 1097–1106 (2025). https://doi.org/10.1038/s41586-025-09070-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09070-3
This article is cited by
-
A neonatal window for in vivo gene therapy in haematopoietic stem cells
Nature Reviews Drug Discovery (2025)
