
Abstract
Calcium (Ca2+) is an essential mineral that must be strictly regulated to support numerous physiological activities1,2. Extracellular fluid Ca2+ is regulated in vertebrates through endocrine systems that manage the vast Ca2+ reservoir in the bones3,4,5,6, but the Ca2+ regulatory mechanisms used by invertebrates, which lack bones, remain largely unclear. Here we identified a potent peptide hormone, Capa, which is responsible for regulating extracellular fluid Ca2+ levels in the fruit fly Drosophila melanogaster. Capa-deficient larvae exhibit low haemolymph Ca2+ levels, resulting in reduced locomotion and induced elongated pupae that mimic those of animals reared on a Ca2+-free diet. Capa secreted from specific neurosecretory cells acts on specialized Ca2+ storage compartments in the anterior Malpighian tubules, elevating haemolymph Ca2+. This endocrine mechanism governing Ca2+ regulation in terrestrial invertebrates resembles the parathyroid hormone system in vertebrates.
Similar content being viewed by others

Main
Ca2+ is an essential mineral that is strictly regulated in support of numerous physiological activities across all eukaryotes1,2. Extracellular fluid Ca2+ levels must be accurately maintained within a tight range to ensure a proper supply of Ca2+ to the cells3. Particularly in terrestrial vertebrates, which acquire Ca2+ solely from their diet, evolution has honed bone remodelling and endocrine systems related to Ca2+ metabolism to facilitate precise homeostatic Ca2+ regulation4. Among the endocrine factors regulating Ca2+ homeostasis, parathyroid hormone (PTH) is crucial for maintaining serum Ca2+ homeostasis during Ca2+ deficiency5,6. When serum Ca2+ levels drop, PTH secreted from the parathyroid glands acts primarily on the bones and kidneys to elevate serum Ca2+ levels. PTH deficiency leads to hypocalcaemia, which in turn causes neuronal and muscle dysfunction7. By contrast, most invertebrates, including terrestrial arthropods, lack bones for Ca2+ storage or PTH orthologues in their genomes. Yet somehow these boneless PTH-lacking terrestrial invertebrates still thrive, carrying out numerous Ca2+-dependent physiological activities that are evolutionarily conserved in vertebrates. This implies the existence of unknown but presumably endocrine mechanisms in terrestrial invertebrates that maintain homeostatic extracellular fluid Ca2+ levels.
The impact of hypocalcaemia on fly larvae
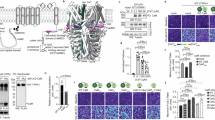
To investigate the endocrine mechanisms regulating Ca2+ homeostasis in terrestrial invertebrates, we used the fruit fly D. melanogaster as a model system. First, we examined the developmental and physiological effects of rearing larvae on food lacking dietary Ca2+. For this purpose, we modified a previously reported chemically defined diet for D. melanogaster, known as a holidic medium (HM)8, to create a Ca2+-free HM (CFHM) (Extended Data Fig. 1a and Supplementary Table 1). While standard fly food contains approximately 2.5 mM Ca2+ and standard HM (SHM) contains approximately 1.2 mM Ca2+, we could not detect any Ca2+ in CFHM (Extended Data Fig. 1a). Wandering third instar (L3; that is, the final instar) larvae reared on CFHM from the beginning of L3 exhibited approximately half the haemolymph Ca2+ of those reared on SHM (Fig. 1a,b), suggesting hypocalcaemia. CFHM-reared larvae also showed reduced locomotor activity (Fig. 1c and Supplementary Video 1). Moreover, larvae reared on CFHM from L3 showed a reduction in the intensity and frequency of skeletal muscle contraction characterized by changes in fluorescent signals from the Ca2+ probe GCaMP just before puparium formation (PF) (Extended Data Fig. 1b–d). This resulted in the formation of elongated pupae with an increased pupal axis ratio (that is, the ratio of pupal length to width) (Fig. 1d–f). Such elongated pupae are characteristic of mutants with impaired muscle function9,10, suggesting that the phenotype arises from Ca2+-deficiency-induced muscle dysfunction. In support of this interpretation, we observed similar elongated pupal phenotypes in animals with motor neuron-specific knockdown of voltage-gated Ca2+ channels (Extended Data Fig. 1e,f). Furthermore, we were able to induce elongated pupae by injecting the Ca2+ chelator ethylene glycol tetraacetic acid (EGTA) into the larval haemolymph just before PF (Extended Data Fig. 1g). These findings suggest that the underlying cause of the elongated pupal phenotype is neuromuscular Ca2+ deficiency resulting from decreased haemolymph Ca2+ concentration.

a, Schematic of the methods of rearing larvae on SHM (+) or CFHM (−) from various larval stages. b–f, Larvae reared on CFHM showed reduced haemolymph Ca2+ levels and locomotor activity, along with elongated pupal phenotypes that correlated inversely with haemolymph Ca2+ concentration. b–d, The haemolymph Ca2+ concentration (b) and locomotor activity (c) of wandering L3 larvae and the pupal axis ratio (length/width; d) of pupae reared on SHM (+) or CFHM (−) from various larval stages. e, Representative images of wandering L3 larvae and pupae reared on SHM or CFHM from 0 h after L3 ecdysis (0 h AL3E). Scale bar, 1 mm. f, Representative images of skeletal muscles in white prepupae reared on SHM or CFHM from 0 h AL3E. Mhc-GFP was used to visualize skeletal muscles. Enlarged images correspond to the boxed areas. Scale bar, 1 mm. g–i, Larvae reared on HM containing low Ca2+ (0.2 mM) maintain normal circulating Ca2+ levels and form puparia with normal pupal axis ratios. g, Schematic of rearing larvae on HM containing varying amounts of Ca2+ from 0 h AL3E. h,i, The haemolymph Ca2+ concentration of wandering L3 larvae (h) and the pupal axis ratio of pupae (i) reared on HM containing varying amounts of Ca2+ from 0 h AL3E. w1118 flies were used in all of the experiments except for in f. Data are mean ± s.d. Sample sizes (n) and P values (P < 0.05) are shown in the graphs. Statistical analysis was performed using two-tailed Mann-Whitney U-tests in comparison to the SHM (+) group (b–d) and one-way analysis of variance (ANOVA) with Tukey’s multiple-comparisons test (h and i); *P < 0.0001; NS, not significant (P > 0.05).
To assess the effects of more severe Ca2+ deficiency on larval development and physiology, we reared larvae on CFHM from earlier instar stages (that is, the first or second instars, L1 or L2) (Fig. 1a). Under these conditions, the larvae exhibited increased mortality proportional to CFHM rearing duration (Extended Data Fig. 1h,i). Moreover, surviving escapers reared on CFHM from early instars showed marked reductions in haemolymph Ca2+ and locomotor activity (Fig. 1b,c and Supplementary Video 1), along with elongated pupal phenotypes that correlated inversely with haemolymph Ca2+ levels (Fig. 1d and Extended Data Fig. 1j). We were surprised to see any larvae achieve successful PF when reared on CFHM from L1 (Extended Data Fig. 1h–j). By contrast, when we reared larvae in HM lacking Mg2+, another divalent alkaline earth metal cation, most died at an early stage and the few that did escape did not produce elongated pupae (Extended Data Fig. 1h,i,k). Considering the necessity of Ca2+, these results suggest the existence of both a Ca2+ reservoir and a mobilization system in D. melanogaster that can help to compensate for dietary Ca2+ deficiency.
To further explore such a mechanism that helps larvae cope with reduced Ca2+ conditions, we measured haemolymph Ca2+ concentrations in larvae reared on HM containing varying amounts of Ca2+ from the onset of the L3 (Fig. 1g,h). As described above, larvae reared on CFHM (0 mM) exhibited hypocalcaemia. By contrast, those raised on diets containing 0.2 mM Ca2+, a concentration lower than one-tenth of standard fly food (Extended Data Fig. 1a), maintained haemolymph Ca2+ levels comparable to those reared on 1.0 or 5.0 mM Ca2+ diets (Fig. 1h). They also formed pupae with normal axial ratios and survival rates (Fig. 1i and Extended Data Fig. 1l,m). These observations strongly support the existence of an internal Ca2+ mobilization system in D. melanogaster that buffers against fluctuations in dietary Ca2+ availability.
Capa from VaNs regulates haemolymph Ca2+
On the basis of the observations above, we hypothesized that knockdown of a hypothetical PTH-like Ca2+ mobilization factor—if it exists in D. melanogaster—should reduce haemolymph Ca2+ and motor activity while also triggering the induction of elongated pupae. We therefore conducted an in vivo RNA interference (RNAi) screen for genes encoding neuropeptides and peptide hormones of which knockdown leads to elongated pupal phenotypes. Using a ubiquitous driver (TubP-Gal4), we knocked down 51 genes encoding neuropeptides or peptide hormones specifically in the L3 stage and then analysed their resulting pupal axis ratios (Fig. 2a,b and Supplementary Table 2). Among all of the genes tested, knockdown of Capability (Capa) substantially increased the pupal axis ratio (Fig. 2b and Extended Data Fig. 2a,b), leading to elongated pupal phenotypes like those of CFHM-reared animals (Fig. 1d,e and Extended Data Fig. 1j). We confirmed recent reports that knockdown of insulin-like peptide 8 (ilp8) or Hugin (Hug) increased the pupal axis ratio11,12, but neither increase was as large as the increase induced by knockdown of Capa (Fig. 2b). Moreover, Capa RNAi larvae showed significantly less locomotor activity compared with the controls (Extended Data Fig. 2c and Supplementary Video 2). We were able to confirm the elongated pupal phenotype across multiple independent Capa RNAi lines (Extended Data Fig. 2a,b), and we observed reduced haemolymph Ca2+ in Capa RNAi larvae (Extended Data Fig. 2d). On the basis of these results, we focused on Capa as a candidate endocrine factor regulating haemolymph Ca2+ in D. melanogaster.

a,b, In vivo RNAi screen to identify endocrine regulators of haemolymph Ca2+. a, Schematic of the in vivo RNAi screen. b, The pupal axis ratio of pupae with knockdown of genes encoding neuropeptides and peptide hormones. TubP-Gal4TS>UAS-dicer2 was used as a ubiquitous driver. c, The relative expression levels of Capa in various tissues, as assessed using quantitative PCR with reverse transcription (RT–qPCR). Car, carcass; FB, fat body; ID, imaginal discs; SG, salivary glands; Tra, tracheae; WB, whole body. d,e, Expression of Capa-2A-Gal4 (d) and a VaN-specific driver (R18D09∩R54A09-Gal4TS) (e) in the CNS of wandering L3 larvae. Gal4 drivers were crossed to UAS-myr::GFP flies. The white arrows and arrowheads indicate the SEGNs and VaNs, respectively. The yellow arrow indicates the CC, and the yellow arrowhead indicates the TVNs. The two rightmost images show a magnified view of the region within the dotted square. Nuclei were stained with DAPI (magenta). Capa-producing neurons were labelled with the Capa precursor (P) antibody (yellow; e). For d and e, scale bars, 100 µm. f, VaN-specific Capa knockdown (Capa-i; UAS-Capa RNAi, UAS-dicer2) reduced the locomotor activity of wandering L3 larvae. g–i, Genetic manipulations of the VaN affected pupal morphology and Ca2+ concentration. Representative images (g) and the pupal axis ratio (h) of pupae, and the haemolymph Ca2+ concentration of wandering L3 larvae (i) after VaN-specific Capa knockdown (Capa-i), genetic ablation (grim), inactivation (Kir2.1) and activation (TrpA1). For g, scale bar, 1 mm. R18D09∩R54A09-Gal4TS was used as a VaN-specific driver. Data are mean ± s.d. Sample sizes (n) and P values (P < 0.05) are shown in the graphs. Statistical analysis was performed using one-way ANOVA with Dunnett’s multiple-comparisons test in comparison to the control group (b, h and i) and two-tailed Mann–Whitney U-tests in comparison to the control group (f); *P < 0.0001; NS, not significant (P > 0.05).
Analysis of tissue-specific expression of Capa revealed its specific expression in the central nervous system (CNS) (Fig. 2c). Using Capa-2A-Gal4, which mimics endogenous Capa expression, we were able to observe Capa expression only in a restricted set of neurons in the CNS (Fig. 2d and Extended Data Fig. 2e). Consistent with previous reports13,14, two neural clusters within the CNS—a pair of suboesophageal ganglion neurons (SEGNs) and three pairs of ventroabdominal neurons (VaNs)—express high levels of Capa (Fig. 2d and Extended Data Fig. 2e,f). The SEGNs extend axons to the corpora cardiaca (CC), while the VaNs extend axons to the transverse nerves (TVNs), which are known neurohemal release sites located outside the CNS in Drosophila larvae14,15,16 (Fig. 2d). This suggests that Capa peptides are secreted into the haemolymph from these axons. To assess the importance of each Capa-expressing neural cluster in determining the elongated pupal phenotype, we selected several different Gal4 drivers for the SEGNs and VaNs from the FlyLight database (https://www.janelia.org/project-team/flylight) (Extended Data Fig. 3a–d). We observed elongated pupae with Capa knockdown using drivers expressed in the VaNs, but not using drivers expressed in the SEGNs (Extended Data Fig. 3e). To confirm this, we next used the Gal80 system to analyse tissue- and neuron-specific requirements for Capa17. We found that pan-neuronal Gal80 (nSyb-Gal80) suppressed the elongated pupal phenotype observed after ubiquitous knockdown of Capa (TubP-Gal4) (Extended Data Fig. 2g). Furthermore, VaN Gal80 (R18D09-LexA>LexAop-Gal80) suppressed the elongated pupal phenotype induced by Capa knockdown through either pan-neuronal (nSyb-Gal4) or VaN (R54A09-Gal4) drivers (Extended Data Fig. 2g–i). These results indicate that Capa expressed in the VaNs is responsible for the elongated pupal phenotype.
To further demonstrate the importance of Capa secreted from the VaNs, we generated a VaN-specific driver, R18D09∩R54A09-Gal4 (that is, αTub84B-FRT.Gal80.FRT, R18D09-LexA>LexAop-FLP, R54A09-Gal4) through the genetic intersection of two VaN drivers (R18D09-LexA and R54A09-Gal4) using the flip-out recombinase technique18 (Fig. 2e). We were able to confirm a VaN-specific loss of Capa production when using this driver to knock down Capa (Extended Data Fig. 2j). Importantly, we found that a loss of Capa production in only three pairs of VaNs reduced larval locomotion and induced the elongated pupal phenotype (Fig. 2f–h and Supplementary Video 3). We next ablated the VaNs by overexpressing the proapoptotic gene grim and inactivated them by overexpressing the strong inward rectifier potassium channel Kir2.1. We found that both VaN ablation and inactivation led to the formation of elongated pupae like those observed after induction of Capa RNAi (Fig. 2g,h). However, VaN activation through expression of the heat-sensitive channel TrpA1 did not alter the pupal axis ratio (Fig. 2g,h). Notably, we found that knockdown of Capa, VaN ablation and VaN inactivation reduced haemolymph Ca2+ levels, while VaN activation slightly increased haemolymph Ca2+ levels (Fig. 2i). These results indicate that Capa secreted from VaN is responsible for regulating haemolymph Ca2+.
CapaR in IS regulates haemolymph Ca2+
Capa encodes the Capa precursor, which is cleaved into three PRX-amide peptides—Capa-1, Capa-2 and Pyrokinin-1 (PK-1)13,14 (Extended Data Fig. 4a). PRX-amide peptides are evolutionarily conserved, and neuromedin U (NmU) is a well-known example of a vertebrate PRX-amide that exists even in mammals19. Capa-1 and Capa-2 act specifically on the Capa receptor (CapaR), while PK-1 acts on the Pyrokinin 1 receptor (PK1-R)20,21,22 (Extended Data Fig. 4a). Furthermore, a direct peptide profiling analysis suggested that, although Capa-1, Capa-2 and PK-1 are all secreted from the VaN into the haemolymph, only PK-1 is secreted from the SEGNs15. Considering the role of the VaNs in regulating haemolymph Ca2+ levels (Fig. 2i), we expect that Capa-1 and Capa-2 act through CapaR to regulate Ca2+ levels. As expected, we found that ubiquitous knockdown of CapaR reduced larval locomotion and induced the formation of elongated pupae (Extended Data Fig. 4b–d and Supplementary Video 4). We also found that CapaR RNAi larvae exhibited significantly reduced haemolymph Ca2+ levels (Extended Data Fig. 4e). By contrast, we did not observe any of these phenotypes after knockdown of PK1-R (Extended Data Fig. 4c–e). These results strongly suggest that Capa-1 and Capa-2 secreted from the VaNs regulate haemolymph Ca2+ concentration through CapaR.
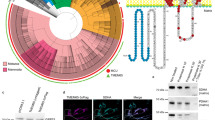
To determine the tissues targeted by Capa-1 and Capa-2, we examined the expression pattern of CapaR and found predominant expression in the Malpighian tubules (MT) (Fig. 3a). The MT is a pair of tubular tissues extending anteriorly and posteriorly from the gut that act as functional equivalents of mammalian kidneys23. We found strong CapaR-Gal4 expression in the distal region of the anterior MT (A-MT) called the initial segments (IS) (Fig. 3b). When we further examined the localization of endogenous CapaR tagged with eGFP (CapaR-eGFP), we found specific localization to the basal membranes of the principal cells in the IS but not in the main segments (Fig. 3c). To analyse the role of CapaR in the IS, we knocked down CapaR using multiple drivers expressed in the MT24,25,26 (Fig. 3d and Extended Data Fig. 4f–i). Knockdown of CapaR using drivers expressed in the principal cells of the IS (CapaR-Gal4, NP1093-Gal4 and Debcl-Gal4) resulted in the elongated pupal phenotype, whereas neither the knockdown of CapaR using drivers expressed in the principal cells of the main segment (Uro-Gal4) nor in the stellate cells (c724-Gal4) produced any obvious phenotype (Fig. 3d,e and Extended Data Fig. 4f–i). Furthermore, we did not observe the elongated pupal phenotype after silencing CapaR expression in other tissues (Extended Data Fig. 4j), including the CC and muscles, which reportedly require CapaR in adult flies27,28. Moreover, we confirmed that knockdown of CapaR, specifically in the principal cells of the IS, reduced larval locomotion and the haemolymph Ca2+ concentration (Fig. 3f,g and Supplementary Video 5). These results clearly indicate that CapaR is required in the IS of the A-MT for haemolymph Ca2+ regulation. Notably, although the TVN terminals extending from the Capa-producing VaNs did not project directly into the IS of the A-MT, they terminated nearby (Extended Data Fig. 5a–c), suggesting the secretion of Capa peptides in the vicinity of the IS of the A-MT.

a–c, CapaR was predominantly expressed in the IS of the A-MT. a, The relative expression levels of CapaR in various tissues, as assessed using RT–qPCR. b, The CapaR-Gal4 expression pattern in wandering L3 larvae. The left image shows all internal tissues, and the right image shows an isolated A-MT. The white arrows indicate the A-MT, and the yellow arrow indicates the posterior MT. The Gal4 driver was crossed to UAS-myr::GFP flies. Nuclei were stained with DAPI (magenta). Scale bars, 500 µm. c, The localization of endogenous CapaR tagged with eGFP (CapaR-eGFP). CapaR–eGFP was localized to the basal membrane of principal cells (PC) in the IS but not in the main segment. No GFP signal was observed in stellate cells (SC). Cell membranes were stained with phalloidin (F-actin, yellow), and nuclei were stained with DAPI (magenta). Scale bars, 25 µm. d,e, CapaR knockdown in the principal cells of the IS led to the induction of elongated pupae. d, Schematic of the expression patterns of various Gal4 drivers expressed in the MT. e, The pupal axis ratio of pupae with CapaR RNAi under the control of various Gal4 drivers. f,g, Specific knockdown of CapaR in the IS reduced locomotor activity (f) and haemolymph Ca2+ concentration (g) in wandering L3 larvae. CapaR-Gal4TS>UAS-dicer2 was used as an IS driver. Data are mean ± s.d. Sample sizes (n) are shown in the graphs. Statistical analysis was performed using one-way ANOVA with Dunnett’s multiple-comparison test in comparison to the control group (e and g) and two-tailed Mann–Whitney U-tests in comparison to the control group (f); *P < 0.0001; NS, not significant (P > 0.05).
IS of the A-MT as a major Ca2+ reservoir
We next examined the role of the IS of the A-MT. The lumen of the MT reportedly contains mineralized granules in many insect species29,30, including Diptera31,32,33,34,35,36,37. Indeed, we observed abundant white granules in the lumen of the IS in wandering L3 larvae in D. melanogaster (Fig. 4a,b). We also found that the IS of the A-MT contained approximately half of the Ca2+ in the whole body (Fig. 4c). Scanning electron microscopy (SEM) analysis revealed a pearl-like appearance for these white granules in the lumen of the IS (Fig. 4d). Further analysis using an electron probe microanalyser (EPMA) revealed that the granules were primarily composed of Ca2+, but they also included phosphorus, oxygen and carbon (Fig. 4e). This is consistent with previous reports suggesting that many dipterans, including Drosophilidae species, store Ca2+ in the MT in the form of calcium phosphate and calcium carbonate34,35,36,37. These results indicate that the IS of the A-MT in D. melanogaster is a specialized tissue for storing Ca2+ as pearl-like calcium granules (PCGs).

a,b, The lumen of the IS was filled with white granules. a, Representative image of the A-MT of a wandering L3 larva. Scale bar, 250 µm. b, Cross-sectional image of the IS. Cell membranes were stained with phalloidin (F-Actin, green), and nuclei were stained with DAPI (magenta). Differential interference contrast (DIC) image showing the lumen packed with granules. Scale bars, 25 µm. c, The Ca2+ content per larva in the whole body or in various regions of the MT. P-MT, posterior MT. d, SEM images of PCGs within the lumen of the IS. Scale bars, 10 µm (top) and 1 µm (bottom). e, PCG ionic composition as analysed by EPMA. w1118 flies were used in a–e. f,g, The effects of Capa peptides on ex vivo cultured A-MT. f, Schematic of ex vivo A-MT culture. The two tubules from the same larva (24 h AL3E) were separated so that one could be cultured as a control (−) and the other with Capa peptides (Capa-1, Capa-2 and PK-1) for 30 min. g, Ca2+ content in the MT from control and CapaR RNAi flies after ex vivo culture. h, Specific knockdown of CG17759 or Pkc98E in the IS reduced the haemolymph Ca2+ concentration in wandering L3 larvae. i, The Ca2+ content in the MT from control, CG17759 RNAi and Pkc98E RNAi after ex vivo culture. CapaR-Gal4TS>UAS-dicer2 was used as an IS driver. Data are mean ± s.d. Sample sizes (n) and P values (P < 0.05) are shown in the graphs. Statistical analysis was performed using two-tailed Mann-Whitney U-tests in comparison to the negative control group (g and i) and one-way ANOVA with Dunnett’s multiple-comparison test in comparison to the control group (h); *P < 0.0001; NS, not significant (P > 0.05).
Next, we examined whether Capa peptides directly reduced the Ca2+ stored in the MT. To test this, we dissected a pair of A-MTs, culturing one as a negative control and the other with Capa peptides (Capa-1, Capa-2 and PK-1) (Fig. 4f). After 30 min of ex vivo culture, Capa-1 and Capa-2, but not PK-1, significantly reduced the amount of Ca2+ stored in the tubules (Fig. 4g). Furthermore, we found that CapaR knockdown in the IS counteracted this effect (Fig. 4g), indicating that Capa-1 and Capa-2 act directly through CapaR expressed in the IS to mobilize stored Ca2+.
To elucidate the downstream signalling components of CapaR that mediate Ca2+ mobilization in the IS, we conducted an in vivo RNAi screen targeting Gα proteins and protein kinases in D. melanogaster (Supplementary Tables 3 and 4). Tissue-specific knockdown of CG17759 (encoding Gαq) and Pkc98E (encoding protein kinase C epsilon, PKCε) in the IS of the MT resulted in elongated pupae and reduced Ca2+ levels in the haemolymph (Fig. 4h and Extended Data Fig. 6). Notably, the Ca2+-mobilizing effect of Capa peptides was abolished in A-MTs after IS-specific knockdown of CG17759 or Pkc98E (Fig. 4i). These findings strongly suggest that Capa peptides activate the CapaR–Gαq–PKC signalling cascade in the IS, leading to mobilization of luminal Ca2+ into the haemolymph.
Capa–CapaR mobilizes Ca2+ under deficiency
Our findings suggest that Capa peptides secreted from the VaNs act directly on the IS of the A-MTs to mobilize stored Ca2+. Notably, despite the Ca2+ independence of Capa expression (Extended Data Fig. 7a,b), mature Capa peptide immunoreactivity in the VaNs was reduced in flies reared on CFHM compared with those reared on SHM (Extended Data Fig. 7c–e). As the immunoreactivity of peptide hormones, including Capa, is inversely correlated with their secretion in D. melanogaster28,38, this observation suggests that Ca2+ deficiency promotes Capa secretion from the VaNs.
To determine whether the Ca2+ stored in the MT is indeed mobilized when larvae experience Ca2+ deficiency, we transferred larvae to CFHM at various larval stages and analysed the resulting levels of Ca2+ stored in the A-MT. As a result, the stored Ca2+ levels in the A-MT substantially decreased depending on the CFHM rearing duration (Extended Data Fig. 8a–c). This decrease was suppressed by knocking down CapaR specifically in the IS (Fig. 5a,b), indicating that the mobilization of stored Ca2+ from the MT is Capa–CapaR dependent. When we reared CapaR RNAi larvae on CFHM, both dietary and stored Ca2+ were unavailable, reducing haemolymph Ca2+ levels even further (Extended Data Fig. 8d,e). Not only did this cause a considerable increase in larval mortality (Fig. 5c), but the few escaping survivors produced pupae that were even more elongated (Extended Data Fig. 8f,g). These results imply that the neuroendocrine mobilization of stored Ca2+ through the action of Capa is crucial to surviving Ca2+ deficiency.

a–c, Ca2+ stored in the MT was mobilized in a CapaR-dependent manner under Ca2+-deficient conditions. a, Schematic of rearing larvae on SHM or CFHM from 0 h AL3E. b,c, The A-MT Ca2+ content (b) and survival rate (PF rate) (c) for control or CapaR RNAi (CapaR-i) larvae reared on SHM or CFHM. d–f, Specific knockdown of Capa in the VaNs disrupted haemolymph Ca2+ homeostasis in wandering L3 larvae. d, Schematic of rearing larvae on HM containing varying amounts of Ca2+ from 0 h AL3E. e,f, The haemolymph Ca2+ concentration (e) and survival rate (PF rate) (f) for control or Capa RNAi larvae reared on HM containing varying amounts of Ca2+. g–j, Feeding larvae a high-Ca2+ diet rescued the elongated pupal phenotype induced by Capa RNAi. g, Schematic of rearing larvae on standard fly food (SFF), high-Ca2+ food (HCF) and high-Mg2+ food (HMF) from 0 h AL3E. h, Representative images of control or Capa RNAi pupae reared on SFF or food supplemented with 50 mM CaCl2 (HCF) or 50 mM MgCl2 (HMF). Scale bar, 1 mm. i, The pupal axis ratio of control or Capa RNAi pupae reared on SFF (0 mM) or food supplemented with 10, 25 or 50 mM CaCl2. j, The haemolymph Ca2+ concentrations for control or Capa RNAi wandering L3 larvae reared on SFF or food supplemented with 50 mM CaCl2 (HCF). CapaR-Gal4TS>UAS-dicer2 was used as an IS driver, and R18D09∩R54A09-Gal4TS was used as a VaN-specific driver. Data are mean ± s.d. Sample sizes (n) and P values (P < 0.05) are shown in the graphs. Statistical analysis was performed using two-way (b, c, e, f and i) and one-way (j) ANOVA with Tukey’s multiple-comparison test; *P < 0.0001; NS, not significant (P > 0.05).
If Capa–CapaR is involved in maintaining Ca2+ homeostasis, its absence should cause aberrant fluctuations in haemolymph Ca2+ levels when the dietary Ca2+ concentration varies. Consistent with this hypothesis, control larvae maintained stable haemolymph Ca2+ levels regardless of the dietary Ca2+ concentration (0.2, 1.0 or 5.0 mM) (Fig. 5d,e and Extended Data Fig. 9a,b), as shown in wild-type (w1118) larvae (Fig. 1h). By contrast, Capa and CapaR RNAi larvae exhibited disruption of haemolymph Ca2+ homeostasis, leading to a direct correlation between dietary and haemolymph Ca2+ levels (Fig. 5e and Extended Data Fig. 9b). These RNAi larvae also showed a marked reduction in survival rates in larvae reared on low-Ca2+ HM (Fig. 5f and Extended Data Fig. 9c). Thus, Capa–CapaR-dependent Ca2+ mobilization is essential for the maintenance of haemolymph Ca2+ homeostasis.
Importantly, we found that both haemolymph Ca2+ levels and survival rates of Capa and CapaR RNAi larvae were restored by rearing them on high-Ca2+ HM (25 mM) (Fig. 5e,f and Extended Data Fig. 9b,c). Thus we finally examined whether high dietary Ca2+ intake can indeed reduce the severity of Capa/CapaR loss-of-function phenotypes. The Ca2+ concentration of standard fly food (approximately 2.5 mM) is lower than that of the haemolymph of wandering L3 larvae (approximately 5 mM) (Extended Data Fig. 1a). Thus, in this analysis, we examined the response to diets with Ca2+ concentrations (10 mM or higher) higher than those of the haemolymph. We found that a dietary Ca2+-concentration-dependent rescue of the elongated pupal phenotype was associated with both Capa and CapaR RNAi using food containing increasing levels of CaCl2 (Fig. 5g–i and Extended Data Fig. 9d–f), concomitant with the restoration of haemolymph Ca2+ levels (Fig. 5j and Extended Data Fig. 9g). By contrast, diets enriched with MgCl2 did not rescue the elongated pupal phenotype (Fig. 5h and Extended Data Fig. 9e,h,i). These results demonstrate that the elongated pupal phenotype associated with Capa/CapaR deficiency is due to Ca2+ deficiency.
Notably, larvae reared on diets of higher Ca2+ concentration maintained haemolymph Ca2+ levels of around 5.0 mM regardless of dietary Ca2+ content (Fig. 5e,j and Extended Data Fig. 9b,g). We also observed this Ca2+ buffering in Capa and CapaR RNAi larvae. These results suggest the existence of an unidentified, Capa–CapaR-independent mechanism that acts to prevent excess Ca2+ accumulation in the haemolymph.
Discussion
Terrestrial animals, in contrast to those dwelling in mineral-rich water, rely on dietary intake and internal storage to replenish Ca2+. Consequently, terrestrial vertebrates have evolved endocrine systems that control the use of Ca2+ stored in the bones during periods of Ca2+ deficiency4. While invertebrates lacking bones, and therefore bone Ca2+ stores, are widely distributed on land, their endocrine systems that regulate Ca2+ mobilization remain largely unknown. Here, we identified a neuroendocrine mechanism that regulates extracellular fluid Ca2+ in the fruit fly D. melanogaster (Extended Data Fig. 10). Specifically, in D. melanogaster larvae, the IS of the A-MT functions as a specialized Ca2+ storage organ. The neuroendocrine hormones Capa-1 and Capa-2 act directly on CapaR in this Ca2+ reservoir, activating the PKC pathway to trigger Ca2+ release into the haemolymph. Capa peptides were first identified as cardioacceleratory peptides in the tobacco hornworm Manduca sexta39, and the gene Capa is currently designated Capability in D. melanogaster13. We propose that Capa can also act as an abbreviation for Ca2+-regulatory PRX-amide. Although we used D. melanogaster larvae as a model in this study, a similar Ca2+ regulatory endocrine system likely exists in adult D. melanogaster and in other insect species. Indeed, high CapaR expression was also observed in the IS of adult D. melanogaster25. Considering the diversity of terrestrial habitats, it will be interesting to investigate whether adult D. melanogaster and other insect species use similar mechanisms for Ca2+ regulation.
Mineralized granules were first observed in the MT of various insects, including Diptera, over a century ago31. Our analysis of the ion composition of these granules in D. melanogaster larvae was consistent with studies of other dipteran larvae34,35,36,37 and indicated that, like vertebrate bone, they are composed primarily of calcium phosphate and calcium carbonate. In the dipteran face fly Musca autumnalis, Ca2+ stored in the MT is transported to the cuticle through haemolymph to harden the puparium40. While Drosophilidae species do not use Ca2+ for puparium hardening36, we found that Ca2+ mobilization from PCGs to the haemolymph is important in maintaining Ca2+ homeostasis. Although it remains unclear how Capa peptides induce Ca2+ flux from PCGs to the haemolymph, previous research suggests direct Ca2+ efflux from the distal IS of the MT into the haemolymph in D. melanogaster larvae41, suggesting that Capa–CapaR signalling may trigger dissolution of PCGs. Given that PCGs dissolve under acidic conditions, we hypothesized that Capa–CapaR signalling through PKC activation may regulate the activity or localization of channels that acidify the luminal environment, as well as transporters involved in Ca2+ mobilization. Notably, Pkc98E—the protein kinase that we identified acting downstream of Capa–CapaR—belongs to the PKC family, specifically PKCε, which is activated in a Ca2+-independent manner42. Considering the specialized role of IS cells in Ca2+ transport, they probably use robust signalling mechanisms that can be enhanced independent of Ca2+. By contrast, in the main segment of the adult MT, Capa signalling promotes diuresis through a different mechanism, activating the nitric oxide–cGMP–PKG pathway through intracellular Ca2+ elevation27,43. This functional divergence within a single organ suggests spatially distinct Capa–CapaR signalling pathways: Ca2+ mobilization in the IS versus fluid secretion in the main segment.
The Capa–CapaR endocrine system regulating Ca2+ in invertebrates is similar to the PTH–PTHR system in vertebrates, which mobilizes Ca2+ during hypocalcaemia5. Our results suggest Ca2+ depletion triggers the release of Capa peptides from the VaNs (Extended Data Fig. 7), although the mechanism sensing haemolymph Ca2+ remains unclear. The fruit fly lacks any orthologues of Ca2+-sensing receptor (CaSR)44, which regulates PTH secretion from the parathyroid glands in vertebrates3, suggesting the existence of an alternative Ca2+-sensing mechanism. As the Ca2+ concentration of the CNS differs from that of the haemolymph due to the presence of the blood–brain barrier45, peripheral tissues may sense haemolymph Ca2+ and transmit signals to the VaNs, which lie within the CNS. Alternatively, Ca2+ sensing may occur at TVN axon terminals, which are neurohemal release sites that extend from the VaNs. Future research will be necessary to identify the Ca2+-sensing systems of D. melanogaster and other invertebrates.
The phenotypes induced by Capa/CapaR loss of function are reminiscent of the symptoms of human hypoparathyroidism, which is associated with neuronal and muscular abnormalities7. In hypoparathyroidism, Ca2+ supplements are used clinically to increase and maintain serum Ca2+ concentrations7. We similarly found that the Capa and CapaR knockdown phenotypes are rescued by dietary Ca2+ supplementation (Fig. 5g–j and Extended Data Fig. 9d–g). This strongly supports the hypothesis that the Capa–CapaR system is crucial for maintaining physiological Ca2+ homeostasis. In contrast to vertebrate PTH, which acts on multiple organs including bone, kidney and intestine5, Capa appears to act primarily on the IS of the MT to regulate Ca2+ mobilization. The absence of a phenotype after CapaR knockdown in the gut and other tissues (Extended Data Fig. 4j), along with the consistent phenotypic similarity between VaN-specific Capa RNAi and IS-specific CapaR RNAi, support the conclusion that Capa-mediated Ca2+ regulation is IS specific.
Despite the functional similarity between Capa peptides and vertebrate PTH, they belong to structurally and evolutionarily distinct peptide families. Capa peptides belong to the PRX-amide family, which includes the vertebrate neuropeptide NmU19. NmU regulates various physiological functions, including feeding behaviour, muscle contraction and bone remodelling46. Although it remains unclear whether NmU’s role in bone remodelling is direct or indirect47,48,49, a recent study associated nucleotide polymorphisms in NmU with bone density and serum Ca2+ levels in children50. It is therefore possible that the evolutionary origin of the function of PRX-amide peptides, including NmU, may relate to extracellular fluid Ca2+ regulation. Collectively, we conclude that, although it seems to have evolved independent of the PTH–PTHR system in vertebrates, the Capa–CapaR system functions as an endocrine mechanism through which terrestrial invertebrates regulate Ca2+ levels even in the absence of bone Ca2+ stores.
Methods
Fly stocks and husbandry
w1118 (BDSC, 5905; the control strain), Akh-Gal4 (BDSC, 25684), btl-Gal4 (BDSC, 78328), c57-Gal4 (BDSC, 32556), c179-Gal4 (BDSC, 6450), CaLexA (BDSC, 66542), Capa-2A-Gal4 (BDSC, 84597), Capa-2A-LexA (BDSC, 84358), Cg-Gal4 (BDSC, 7011), Debcl-Gal4 (BDSC, 81163), Eip71CD-Gal4 (BDSC, 6871), LexAop-FLP (BDSC, 55819), LexAop-Gal80 (BDSC, 32214), LexAop-myr::GFP (BDSC, 32209), Mef2-Gal4 (BDSC, 27390), Mhc-GFP (BDSC, 38462), nSyb(R57C10)-Gal4 (BDSC, 90854), OK6-Gal4 (BDSC, 64199), R9F03-Gal4 (BDSC, 40734), R16E04-Gal4 (BDSC, 47326), R18D09-Gal4 (BDSC, 48814), R18D09-LexA (BDSC, 54752), R20C04-Gal4 (BDSC, 48882), R22H11-Gal4 (BDSC, 48043), R53E05-Gal4 (BDSC, 48196), R54A09-Gal4 (BDSC, 50460), R82E06-Gal4 (BDSC, 40149), αTub84B-FRT.Gal80.FRT (BDSC, 38879), TubP-Gal4 (BDSC, 5138), TubP-Gal80TS (on the third chromosome) (BDSC, 7017), TubP-Gal80TS (on the second chromosome) (BDSC, 7019), UAS-Ca-α1D RNAi (BDSC, 33413), UAS-Ca-α1T RNAi (BDSC, 39029), UAS-Ca-β RNAi (BDSC, 43292), UAS-cac RNAi (BDSC, 77174), UAS-Capa RNAi (3) (BDSC, 28345), UAS-GCaMP6s (BDSC, 42746), UAS-Kir2.1 (BDSC, 6596), UAS-mCD8::RFP (BDSC, 32219), UAS-myr::GFP (on the second chromosome) (BDSC, 32198), UAS-myr::GFP (on the third chromosome) (BDSC, 32197), UAS-PK1-R RNAi (BDSC, 27539), UAS-TrpA1 (BDSC, 26263) and Vglut (OK371)-Gal4 (BDSC, 26160) flies were obtained from the Bloomington Drosophila Stock Center (BDSC). UAS-Capa RNAi (1) (VDRC, 41124; unless otherwise noted, this line was used in all Capa RNAi experiments), UAS-Capa RNAi (2) (VDRC, 101705), UAS-CapaR RNAi (1) (VDRC, 330441; unless otherwise noted, this line was used in all CapaR RNAi experiments), UAS-CapaR RNAi (2) (VDRC, 105556), UAS-dicer2 (on the second chromosome) (VDRC, 60008) and UAS-dicer2 (on the third chromosome) (VDRC, 60009) flies were obtained from the Vienna Drosophila Resource Center (VDRC). Myo1A-Gal4 (DGRC, 112001) and NP1093-Gal4 (DGRC, 103880) flies were obtained from the Kyoto Stock Center (Department of Drosophila Genomics and Genetic Resources, Kyoto Institute of Technology). CapaR-Gal427, tsh(c724)-Gal451 and Uro-Gal452 flies were obtained from T. Koyama and K. Halberg. elav-Gal4(3A3), phm22-Gal4 and UAS-grim flies were obtained from Na. Yamanaka. nSyb(R57C10)-Gal80 was obtained from James W. Truman (University of Washington). All of the RNAi lines used for the in vivo RNAi screens are listed in Supplementary Table 2–4. CapaR-eGFP flies were generated through CRISPR-mediated mutagenesis as described below.
All fly stocks were maintained on standard fly food containing 5.5 g agar (Daishin, P700), 100 g glucose (Showa Sangyo), 40 g dry yeast (Asahi Beer, HB-P02), 90 g cornmeal (Sunny Maize, Yellow-No.4 M), 3 ml propionic acid (Nacalai Tesque, 29018-55), 3.5 ml 10% butylparaben (in 70% ethanol) (Nacalai Tesque, 06327-15) and 1 l of water. All of the experiments were conducted under non-crowded conditions.
For all of the knockdown experiments except for those in Extended Data Fig. 1f, larvae carried the TubP-Gal80TS transgene, which encodes a temperature-sensitive transcriptional repressor17. Unless otherwise specified, these larvae were raised at 18 °C during the early instar stages (L1 or L2). The larvae were then shifted to 29 °C from 0 h AL3E to induce RNAi from L3. For the genetic manipulation of the VaNs, the larvae were reared at 18 °C during the early larval stages and shifted to 29 °C from 12 h after L2 ecdysis to induce grim expression and from 24 h AL3E to induce expression of Kir2.1 or TrpA1.
Unless otherwise specified, progeny from crosses between w1118 and either Gal4 lines or UAS RNAi lines were used as controls.
Preparation of a Ca2+-free HM
A Ca2+-free diet was prepared by modifying the components of the chemically defined diet for D. melanogaster known as HM8,53,54. Agar, commonly used in the preparation of standard fly food and HM, is rich in minerals, including Ca2+. When we prepared HM using standard agar (Daishin, P700), we detected high levels of Ca2+ even after excluding CaCl2·2H2O from the ingredients (Extended Data Fig. 1a). We therefore measured the Ca2+ concentrations of several purified agar/agarose products (product-A: Nacalai Tesque, 01028-85; product-B: Nacalai Tesque, 01162-15; product-C: Nacalai Tesque, 01163-05; and product-D: Sigma-Aldrich, A9539). We then prepared HM using product-D (Sigma-Aldrich, A9539), which did not contain detectable Ca2+ (Extended Data Fig. 1a). Moreover, we used sodium pantothenate as a substitute for calcium pantothenate. As Ca2+ was undetectable in the resulting HM when we excluded CaCl2·2H2O, we defined it as CFHM. Mg2+-free HM was prepared by excluding MgSO4·7H2O. A complete list of the components used in the preparation of HM is provided in Supplementary Table 1.
Measuring Ca2+ concentrations
For haemolymph samples, wandering L3 larvae were rinsed in Milli-Q water and wiped on Kimwipes. The larval cuticle was carefully torn to release the haemolymph onto a parafilm membrane. Haemolymph (2 µl) was collected from 5 larvae, mixed with 8 µl of ice-cold UltraPure distilled water (Thermo Fisher Scientific, 10977015) and centrifuged at 4 °C for 10 min. The resulting supernatant was stored at −80 °C for later use in the Ca2+ assay.
For whole-body samples, wandering L3 larvae were rinsed in Milli-Q water, wiped onto Kimwipes, transferred individually into tubes, immediately flash-frozen in liquid nitrogen and stored at −80 °C. Dissected tissues or ex vivo cultured tissues were rinsed in UltraPure distilled water and immediately collected in tubes along with UltraPure distilled water. MT samples were collected individually in tubes along with 9 μl of UltraPure distilled water. Other tissues were collected in 45 µl of UltraPure distilled water, flash-frozen in liquid nitrogen and stored at −80 °C until use. Before the calcium assay, whole-body samples were homogenized for 3 min with 50 µl of 0.1 M HCl (diluted with UltraPure distilled water). MT samples were vortexed for 3 min after adding 1 µl of 1.0 M HCl (diluted with UltraPure distilled water to a total of 10 µl). Other tissues were homogenized for 3 min after adding 5 µl of 1.0 M HCl (diluted with UltraPure distilled water to a total of 50 µl). The samples were centrifuged at 4 °C for 10 min, and the resulting supernatants were used for the Ca2+ assay.
Fly food, agar and agarose samples were weighed, placed into sample tubes, melted at 50 °C and used for the Ca2+ assay.
Ca2+ concentrations were measured with the QuantiChrom Calcium Assay kit (Funakoshi, DICA-500) according to the manufacturer’s instructions with the included Ca2+ solution as a standard. Then, 2 μl of each sample and the standard samples were added to a 96-well plate (Thermo Fisher Scientific, 269620) with 100 µl of working solution. After incubation for 3 min at room temperature, the optical density at 612 nm was measured using the Multiskan GO (Thermo Fisher Scientific). For fly food, agarose and agar samples, the assay was performed using prewarmed working solution to prevent solidification. Multiple samples were collected in duplicate and included in the analysis for each experiment.
Analysis of pupal axis ratio
Pupae were affixed to glass slides using double-sided tape, dorsal side up. Images were captured alongside a ruler under a dissecting microscope equipped with a Flexacam C3 12MP microscope camera (Leica). The pupal length and width were analysed using Fiji (ImageJ2, v.2.16.0, National Institute of Health; https://imagej.net/fiji)55. The pupal length was measured from the anterior-most point to the junction of the posterior spiracles. The pupal width was measured along the plane perpendicular to the midpoint of the pupal length axis. Although female pupae were slightly larger than males, we did not consider animal sex because both males and females are known to exhibit equal pupal axis ratios11.
Analysis of muscle contraction activity
Larvae (Mef2-Gal4>UAS-GCaMP6s) were reared on either SHM or CFHM containing red dye (Kyoritsu Foods, 4901325001245) from 0 h AL3E. Prepupal wandering L3 larvae, from which red dye food had been completely purged from the gut, were then used for analysis. Larvae were rinsed with Milli-Q water and dried using Kimwipes. One larva reared on SHM and one larva reared on CFHM were placed side by side on a 35-mm glass-bottom dish (IWAKI, 3910-035, 27 mm glass), and GCaMP imaging was performed simultaneously for 5 min using a Nikon SMZ25 stereomicroscope equipped with a Digital Sight 10 camera (Nikon). At least three independent experiments were performed, with different larvae used for each recording. Only larvae that underwent PF within 1 h after imaging were included in the analysis. In the Ca2+ imaging time-series data, the onset times of body wall peristaltic waves, propagating from posterior to anterior or vice versa, were labelled through visual observation using ImageJ. Propagations across more than three segments were classified as peristaltic waves. The wave frequency was calculated by taking the inverse of the time interval between the onsets of two successive waves. The maximal GCaMP fluorescence signal intensity within the larval body for each wave was measured using Fiji (v.2.16.0) and R (v.4.4.2).
EGTA injections
Larvae (w1118) were reared on standard fly food containing red dye (Kyoritsu Foods, 4901325001245) from 0 h AL3E. Then, prepupal wandering L3 larvae, from which red dye food had been completely purged from the gut, were used for injection. To confirm the injection of the solution into the larval body, 1x phosphate-buffered saline (PBS) (Nacalai Tesque, 27575-31) was coloured blue by adding Erioglaucine disodium salt (final conc. 0.1 g per 10 ml) (Sigma-Aldrich, 861146). Under a dissecting microscope, wandering L3 larvae affixed to glass slides with double-sided tape were injected with EGTA (BMS, BR-401201271) diluted in the blue-coloured PBS. Glass needles were made from glass capillary filament (Narishige, GD-1) using a puller (Narishige, PC-10), and the tips were sharpened using a microgrinder (Narishige, EG-401). Although the exact volume could not be controlled due to injector limitations, each larva was injected with a volume of approximately 100–200 nl. After the injected larvae were rinsed with water to remove any residual glue, they were transferred to vials containing standard fly food and those that successfully formed pupae were analysed to determine their pupal axis ratios.
Larval locomotion assay
Wandering L3 larvae were rinsed in Milli-Q water and wiped on Kimwipes to dry them. Subsequently, 10 larvae were placed together in the centre of a 1% agar plate with a 9-cm diameter (Sansei Medical, 01-013). Water (10 μl) was then added to the larval aggregate in the centre of the plate. The entire plate was recorded for 3 min from above using a fixed-position video camera (SONY, FDR-AX45) with an in-frame ruler. At least three independent experiments were conducted for each genotype, with ten larvae recorded simultaneously in each experiment. Different larvae were used for each recording, resulting in a minimum of 30 larvae recorded for each genotype. The resulting video files were edited using iMovie (v.10.3.5; Apple), Adobe Photoshop (v.24.7.4) and Adobe Premiere Rush (v.2.10.0). The coordinates of the centres of individual larvae in the binarized videos were recorded. Except for the larvae reared on the Ca2+-free diet from the first instar, individual larvae were tracked using the FIMTrack software56. For the larvae reared on the Ca2+-free diet from the first instar, the centres of the larvae were labelled manually using Fiji (ImageJ2, v.2.16.0). This was required because the larvae had such low locomotor activity that they did not separate well enough for FIMTrack to detect individual larvae. Larval locomotor speed was obtained by dividing the distance each larva moved in 10 s by 10 s. The speed of individual larvae was calculated using Python 3.9 by averaging 2–4 speed measurements along a consecutive trajectory.
Analysis of the larval survival rate
The larval survival rate was calculated as the proportion of larvae developed into the pupal stage in each vial. Larvae were transferred into small food vials containing HM at a density of 25 larvae per vial for newly moulted L2 and L3, or 50 larvae per vial for newly hatched L1. The vials were maintained in a humidified incubator, and the number of individuals that successfully formed pupae in each vial was recorded.
Total RNA extraction and RT–qPCR analysis
Animals or dissected tissues from white prepupae (w1118) were collected in 1.5 ml tubes and immediately flash-frozen in liquid nitrogen. Total RNA from animals or tissues was extracted using TRIzol (Thermo Fisher Scientific, 15596-026) according to the manufacturer’s instructions. cDNA was generated from purified total RNA using ReverTra Ace qPCR RT Master Mix with gDNA Remover (TOYOBO, FSQ-301) according to the manufacturer’s instructions. RT–qPCR was performed on the CFX Duet Real-Time PCR machine (Bio-Rad) using THUNDERBIRD Next SYBR qPCR Mix (TOYOBO, QPX-201). For absolute mRNA quantification, serial dilutions of pGEM-T (Promega, A362A) plasmids containing the coding sequences of a target gene or of rp49 were used for standards. After the molar amounts were calculated, the transcript levels of each target mRNA were normalized to the rp49 levels in the same samples. Three separate samples were collected for each experiment and duplicate measurements were conducted. The following primers were used: Capa forward, 5’-AGAAGAACCGTGACCGTTCCGAGG-3’; Capa reverse, 5’-TCGTGGTCCGTCTCAGCTGTACTG-3’; CapaR forward, 5’-TTCGTGTGCTGGTTCCCGTTCCAC-3’; CapaR reverse, 5’-TCCCGCAATCGAGAAGAGTGCCTC-3’. The primers to detect rp49 levels were previously reported57.
Immunostaining
Tissues were dissected in 1× PBS (Nacalai Tesque, 27575-31), fixed with 4% paraformaldehyde (PFA) (Nacalai Tesque, 02890-45) in PBS (4% PFA/PBS) containing 0.1% Triton X-100 for 20 min at room temperature and then washed multiple times with PBS containing 0.1% Triton X-100 (PBST). Tissues were blocked with 5% normal goat serum (NGS) (Sigma-Aldrich, G9023) in PBST for at least 1 h at room temperature, incubated overnight at 4 °C with primary antibodies mixed in PBST containing 5% NGS, washed multiple times with PBST, incubated for 2 h at room temperature with secondary antibodies mixed in PBST containing 5% NGS and again washed multiple times with PBST. When necessary, DNA was stained with DAPI (Sigma-Aldrich, D9542) at 1:2,000 and F-actin was stained with Alexa Fluor 488 or 568 Phalloidin (Thermo Fisher Scientific, A12379 or A12380) at 1:200 for 30 min at room temperature. After washing, tissues were mounted in Vectashield H-1000 (Vector Laboratories) and observed using the Zeiss Axio Imager M2 equipped with ApoTome.2 or the Zeiss LSM 700 confocal microscope. The specificity of the signals was established by comparison with appropriate controls.
Chicken anti-GFP (1:2,000 dilution, Abcam, ab13970) and rabbit anti-RFP (1:2,000, Medical and Biological Laboratories, PM005) were used as primary antibodies. Rabbit anti-Capa precursor antibodies (1:2,000) and guinea pig anti-Capa mature peptide antibodies (1:500) were generated by BioGate. The rabbit anti-Capa precursor antibodies were generated against the peptide NH2-LDGIYGDASQEDYNEADFQ-COOH. The guinea pig anti-Capa mature peptide antibodies were generated against the peptide NH2-GLYAFPRV-CONH2, which is identical to Capa-1 and matches all of Capa-2 except for one amino acid (Y), suggesting that the resulting antibodies can recognize both Capa-1 and Capa-2. Alexa Fluor 488 goat anti-chicken (1:1,000, Thermo Fisher Scientific, A32931), Alexa Fluor 555 goat anti-rabbit (1:1,000, Thermo Fisher Scientific, A32732) and Alexa Fluor 555 goat anti-guinea pig (1:1,000, Thermo Fisher Scientific, A21435) were used as secondary antibodies.
Generation of a C-terminal eGFP-tagged CapaR CRISPR knock-in line
An eGFP-tagged CapaR line (CapaR-eGFP) was generated by WellGenetics using a slightly modified published method58. In brief, gRNA sequences (AAGGTTGTTCTCGACGTCCT[TGG]) were cloned into U6 promoter plasmids. Two homology arms and an eGFP-3xP3-RFP cassette containing eGFP and a floxed 3xP3-RFP were cloned into pUC57-Kan as a repair donor template. CapaR-targeting gRNAs and hs-Cas9 were supplied in DNA plasmids, together with the donor plasmid, for microinjection into embryos of a w1118 control strain (LWG228). F1 flies carrying the selection marker indicating the presence of 3xP3-RFP were further validated by genomic PCR and sequencing. CRISPR generated a break in CapaR that was then repaired by replacement with the eGFP-3xP3-RFP cassette. The resulting eGFP-tagged CapaR line was homozygous viable and did not exhibit an elongated pupal phenotype, suggesting that the tagged CapaR remained functional.
SEM analysis of MT granules and analysing their ion composition using EPMA
A-MTs were dissected in 1× PBS, rinsed with UltraPure water and immediately collected on a carbon-coated film along with UltraPure water. The initial segment of the A-MT was carefully torn to release the granules in its lumen. Then, after discarding the A-MT tissue, the granules were immediately dried on a carbon-coated film. Before analysis, the granules on carbon-coated film attached to a copper grid were vacuum processed and coated with carbon. The granules were analysed using SEM and ion-composition analysis using a field emission electron probe microanalyzer (FE-EPMA) (JEOL, JXA-8530F) operated at a voltage of 10.0 kV. This analysis revealed the ion composition of the granules, excluding their oxygen and carbon content. Each measurement was performed at least three times on different granules. For the oxygen and carbon composition analysis, samples were collected on copper-coated films without adding a carbon coating. The measured Ca2+ values were used as standard values, and the measured oxygen and carbon values were included in a recalculation of the composition of all ions.
Peptide synthesis and ex vivo culture of MTs
Mature amidated Capa peptides in D. melanogaster—Capa-1 (NH2-GANMGLYAFPRV-CONH2), Capa-2 (NH2-ASGLVAFPRV-CONH2) and PK-1 (NH2-TGPSASSGLWFGPRL-CONH2)—were synthesized by Eurofins Genomics.
Control and CapaR RNAi larvae were reared at 18 °C and shifted to 29 °C from 0 h AL3E to induce RNAi from L3. After 24 h of RNAi induction (24 h AL3E), the larvae were washed with 1× PBS. Then, the two A-MTs and the gut were carefully dissected in Schneider’s Drosophila medium (SDM) (Thermo Fisher Scientific, 21720024) supplemented with 10% FBS (Thermo Fisher Scientific, 10270106). Fat bodies attached to the MTs were carefully removed, and the dissected MT–gut complexes were pre-incubated in SDM containing 10% FBS in a Petri dish (AS ONE, 1-8549-01) for 10 min. Only intact MTs were selected for analysis to prevent leakage of Ca2+ granules from MTs ruptured during the dissection process. After pre-incubation, the MTs were severed at the base where they connect to the gut. Both MTs obtained from the same larva were transferred to separate wells of a 24-well clear flat-bottom tissue culture plate (TPP, 92424). To avoid damaging the MTs during transfer, each MT was transferred along with medium. Finally, six MTs per well were placed in a total of 450 µl of medium. Synthetic Capa peptides (Capa-1, Capa-2 and PK-1) in SDM with 10% FBS were added to a final concentration of 10 μM (50 μl per peptide; total volume, 500 μl per well) and incubated at 25 °C for 30 min. After the incubation, the MTs were washed twice with UltraPure distilled water (Thermo Fisher Scientific, 10977015) and collected individually in tubes along with 9 μl of UltraPure distilled water before being frozen at −80 °C.
Statistics and reproducibility
All statistical analyses were carried out using GraphPad Prism (v.9.5.1). Pairwise comparisons were made using the two-tailed Mann–Whitney U-tests, and multiple samples were compared using the one-way ANOVA with Dunnett’s or Tukey’s multiple-comparison test. In some cases, the two-way ANOVA with Tukey’s multiple-comparison test was performed to examine interactions between variables. All of the experiments were performed independently at least twice using independently reared populations. In each experiment, we analysed three or more specimens. Animals and samples were randomly assigned to experimental groups. Representative images were chosen from at least three options. Data collection and analysis were not performed blinded to the experimental conditions. Although no statistical method was used to predetermine sample size, sample sizes were determined based on significance obtained from previous studies with similar experimental setups. Comparable sample sizes were used in each experiment. No datapoints or animals were excluded from analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated or analysed during this study are available within the Article and its and Supplementary Information. Further information can be requested from the corresponding authors. Source data are provided with this paper.
References
Berridge, M. J., Bootman, M. D. & Roderick, H. L. Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529 (2003).
Clapham, D. E. Calcium signaling. Cell 131, 1047–1058 (2007).
Hofer, A. M. & Brown, E. M. Extracellular calcium sensing and signalling. Nat. Rev. Mol. Cell Biol. 4, 530–538 (2003).
Bouillon, R. & Suda, T. Vitamin D: calcium and bone homeostasis during evolution. Bonekey Rep. 3, 480 (2014).
Murray, T. M., Rao, L. G., Divieti, P. & Bringhurst, F. R. Parathyroid hormone secretion and action: evidence for discrete receptors for the carboxyl-terminal region and related biological actions of carboxyl- terminal ligands. Endocr. Rev. 26, 78–113 (2005).
Wein, M. N. & Kronenberg, H. M. Regulation of bone remodeling by parathyroid hormone. Cold Spring Harb. Perspect. Med. 8, a031237 (2018).
Gafni, R. I. & Collins, M. T. Hypoparathyroidism. N. Engl. J. Med. 380, 1738–1747 (2019).
Piper, M. D. W. et al. A holidic medium for Drosophila melanogaster. Nat. Methods 11, 100–105 (2014).
Ball, E., Ball, S. & Sparrow, J. A mutation affecting larval muscle development in Drosophila melanogaster. Dev. Genet. 6, 77–92 (1985).
Bunch, T. A. et al. The PS2 integrin ligand tiggrin is required for proper muscle function in Drosophila. Development 125, 1679–1689 (1998).
Heredia, F. et al. The steroid-hormone ecdysone coordinates parallel pupariation neuromotor and morphogenetic subprograms via epidermis-to-neuron Dilp8-Lgr3 signal induction. Nat. Commun. 12, 3328 (2021).
Ohhara, Y. et al. A neuropeptide signaling network that regulates developmental timing and systemic growth in Drosophila. J. Comp. Neurol. 532, e25677 (2024).
Kean, L. et al. Two nitridergic peptides are encoded by the gene capability in Drosophila melanogaster. Am. J. Physiol. Regul. Integr. Comp. Physiol. 282, R1297–R1307 (2002).
Predel, R. & Wegener, C. Biology of the CAPA peptides in insects. Cell. Mol. Life Sci. 63, 2477–2490 (2006).
Wegener, C., Reinl, T., Jänsch, L. & Predel, R. Direct mass spectrometric peptide profiling and fragmentation of larval peptide hormone release sites in Drosophila melanogaster reveals tagma-specific peptide expression and differential processing. J. Neurochem. 96, 1362–1374 (2006).
Santos, J. G., Pollák, E., Rexer, K. H., Molnár, L. & Wegener, C. Morphology and metamorphosis of the peptidergic Va neurons and the median nerve system of the fruit fly, Drosophila melanogaster. Cell Tissue Res. 326, 187–199 (2006).
Suster, M. L., Seugnet, L., Bate, M. & Sokolowski, M. B. Refining GAL4-driven transgene expression in Drosophila with a GAL80 enhancer-trap. Genesis 39, 240–245 (2004).
Gordon, M. D. & Scott, K. Motor control in a Drosophila taste circuit. Neuron 61, 373–384 (2009).
Jurenka, R. The PRXamide neuropeptide signalling system: conserved in animals. Adv. Insect Physiol. 49, 123–170 (2015).
Iversen, A., Cazzamali, G., Williamson, M., Hauser, F. & Grimmelikhuijzen, C. J. Molecular cloning and functional expression of a Drosophila receptor for the neuropeptides capa-1 and -2. Biochem. Biophys. Res. Commun. 299, 628–633 (2002).
Park, Y., Kim, Y.-J. & Adams, M. E. Identification of G protein-coupled receptors for Drosophila PRXamide peptides, CCAP, corazonin, and AKH supports a theory of ligand-receptor coevolution. Proc. Natl Acad. Sci. USA 99, 11423–11428 (2002).
Cazzamali, G., Torp, M., Hauser, F., Williamson, M. & Grimmelikhuijzen, C. J. P. The Drosophila gene CG9918 codes for a pyrokinin-1 receptor. Biochem. Biophys. Res. Commun. 335, 14–19 (2005).
Dow, J. A. T., Simons, M. & Romero, M. F. Drosophila melanogaster: a simple genetic model of kidney structure, function and disease. Nat. Rev. Nephrol. 18, 417–434 (2022).
Wang, C. & Spradling, A. C. An abundant quiescent stem cell population in Drosophila Malpighian tubules protects principal cells from kidney stones. eLife 9, e54096 (2020).
Xu, J. et al. Transcriptional and functional motifs defining renal function revealed by single-nucleus RNA sequencing. Proc. Natl Acad. Sci. USA 119, e2203179119 (2022).
Yin, S., Qin, Q. & Zhou, B. Functional studies of Drosophila zinc transporters reveal the mechanism for zinc excretion in Malpighian tubules. BMC Biol. 15, 12 (2017).
Terhzaz, S. et al. Mechanism and function of Drosophila capa GPCR: a desiccation stress-responsive receptor with functional homology to human neuromedinU receptor. PLoS ONE 7, e29897 (2012).
Koyama, T. et al. A nutrient-responsive hormonal circuit mediates an inter-tissue program regulating metabolic homeostasis in adult Drosophila. Nat. Commun. 12, 5178 (2021).
Wigglesworth, V. B. The Principles of Insect Physiology (Chapman and Hall, 1972).
Brown, B. E. The form and function of metal-containing ‘granules’ in invertebrate tissues. Biol. Rev. Camb. Philos. Soc. 57, 621–667 (1982).
Keilin, D. On the calcium carbonate and the calcospherites in the Malpighian tubes and the fat body of Dipterous larvae and the ecdysial elimination of these products of excretion. Q. J. Microsc. Sci. 65, 611–625 (1921).
Eastam, L. Peristalsis in the Malpighian tubules of Diptera, preliminary account: with a note on the elimination of calcium carbonate from the Malpighian tubules of Drosophila funebris. Q. J. Microsc. Sci. 69, 5–98 (1925).
Waterhouse, D. F. Studies of the physiology and toxicology of blowflies. XTV. The composition, formation, and fate of the granules in the Malpighian tubules of Lucilia cuprina larvae. Aust. J. Sci. Res. Ser. B 3, 76–113 (1950).
Grodowitz, M. J., Broce, A. B. & Kramer, K. J. Morphological and biochemical composition of mineralized granules from the Malpighian tubules of Musca autumnalis de Geer larvae (Diptera: Muscidae). Insect Biochem. 17, 335–345 (1987).
Krueger, R. A., Broce, A. B. & Hopkins, T. L. Dissolution of granules in the Malpighian tubules of Musca autumnalis DeGeer, during mineralization of the puparium. J. Insect Physiol. 33, 255–263 (1987).
Wessing, A., Zierold, K. & Hevert, F. Two types of concretions in Drosophila Malpighian tubules as revealed by X-ray microanalysis: a study of urine formation. J. Insect Physiol. 38, 543–554 (1992).
Wessing, A. & Zierold, K. The formation of type-I concretions in Drosophila Malpighian tubules studied by electron microscopy and X-ray microanalysis. J. Insect Physiol. 45, 39–44 (1999).
Géminard, C., Rulifson, E. J. & Léopold, P. Remote control of insulin secretion by fat cells in Drosophila. Cell Metab. 10, 199–207 (2009).
Tublitz, N. J. & Truman, J. W. Insect cardioactive peptides. I. Distribution and molecular characteristics of two cardioacceleratory peptides in the tobacco hawkmoth, Manduca sexta. J. Exp. Biol. 114, 365–379 (1985).
Krueger, R. A., Broce, A. B., Hopkins, T. L. & Kramer, K. J. Calcium transport from Malpighian tubules to puparial cuticle of Musca autumnalis. J. Comp. Physiol. B 158, 413–419 (1988).
Browne, A. & O’Donnell, M. J. Segment-specific Ca2+ transport by isolated Malpighian tubules of Drosophila melanogaster: a comparison of larval and adult stages. J. Insect Physiol. 87, 1–11 (2016).
Steinberg, S. F. Structural basis of protein kinase C isoform function. Physiol. Rev. 88, 1341–1378 (2008).
Davies, S. A. et al. Signaling by Drosophila capa neuropeptides. Gen. Comp. Endocrinol. 188, 60–66 (2013).
Bjarnadóttir, T. K., Fredriksson, R. & Schiöth, H. B. The gene repertoire and the common evolutionary history of glutamate, pheromone (V2R), taste (1) and other related G protein-coupled receptors. Gene 362, 70–84 (2005).
Abbott, N. J., Patabendige, A. A., Dolman, D. E., Yusof, S. R. & Begley, D. J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 37, 13–25 (2010).
Martinez, V. G. & O’Driscoll, L. Neuromedin U: a multifunctional neuropeptide with pleiotropic roles. Clin. Chem. 61, 471–482 (2015).
Sato, S. et al. Central control of bone remodeling by neuromedin U. Nat. Med. 13, 1234–1240 (2007).
Rucinski, M., Ziolkowska, A., Tyczewska, M., Szyszka, M. & Malendowicz, L. K. Neuromedin U directly stimulates growth of cultured rat calvarial osteoblast-like cells acting via the NMU receptor 2 isoform. Int. J. Mol. Med. 22, 363–368 (2008).
Hsiao, Y. T. et al. Neuromedin U (NMU) regulates osteoblast differentiation and activity. Biochem. Biophys. Res. Commun. 524, 890–894 (2020).
Gianfagna, F. et al. Understanding the links among neuromedin U gene, beta2-adrenoceptor gene and bone health: an observational study in European children. PLoS ONE 8, e70632 (2013).
Sözen, M. A. et al. Functional domains are specified to single-cell resolution in a Drosophila epithelium. Proc. Natl Acad. Sci. USA 94, 5207–5212 (1997).
Terhzaz, S. et al. Mislocalization of mitochondria and compromised renal function and oxidative stress resistance in Drosophila SesB mutants. Physiol. Genom. 41, 33–41 (2010).
Piper, M. D. W. et al. Matching dietary amino acid balance to the in silico-translated exome optimizes growth and reproduction without cost to lifespan. Cell Metab. 25, 1206 (2017).
Kosakamoto, H. et al. Early-adult methionine restriction reduces methionine sulfoxide and extends lifespan in Drosophila. Nat. Commun. 14, 7832 (2023).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Risse, B., Berh, D., Otto, N., Klämbt, C. & Jiang, X. FIMTrack: an open source tracking and locomotion analysis software for small animals. PLoS Comput. Biol. 13, e1005530 (2017).
Okamoto, N. et al. A membrane transporter is required for steroid hormone uptake in Drosophila. Dev. Cell 47, 294–305 (2018).
Kondo, S. & Ueda, R. Highly improved gene targeting by germline-specific Cas9 expression in Drosophila. Genetics 195, 715–721 (2013).
Jetti, S. K. et al. Molecular logic of synaptic diversity between Drosophila tonic and phasic motoneurons. Neuron 111, 3554–3569 (2023).
Acknowledgements
We thank the members of the Vienna Drosophila Resource Center, the Bloomington Drosophila Stock Center, the National Institute of Genetics Fly Stock Center, the Kyoto Stock Center, the Transgenic RNAi Project at Harvard Medical School, and M. Enomoto, T. Koyama, K. Halberg, J. W. Truman and N. Yamanaka for fly stocks; the staff at FlyBase for providing comprehensive and invaluable resources on Drosophila; the members of the Chemical Analysis Division in the University of Tsukuba for help with the chemical analysis of the Ca2+ granules; F. Obata for providing us with a basic protocol to prepare holidic media; R. Tajiri, T. Kondo, N. Yamanaka, Y.-J. Kim and K. Halberg for discussions; and the staff at Write Labs (https://write-labs.com) for help with editing the manuscript. This study was supported by the Japan Science and Technology Agency (JST) FOREST Program (JPMJFR224M), the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant-in-Aid for Research Activity Start-up (20K22645), the JSPS KAKENHI Grant-in-Aid for Scientific Research (C) (22K06290), the JSPS KAKENHI Grant-in-Aid for Scientific Research (B) (25K02307), the 10th Tomizawa Jun-ichi & Keiko Fund of MBSJ for Young Scientists, the Takeda Science Foundation to N.O., the JSPS KAKENHI Grant-in-Aid for JSPS Fellow to Y.M. (23KJ0252) and A.W. (24KJ0466), the JST SPRING Program (JPMJSP2124) to Y.M. and A.W., the JSPS KAKENHI Grant-in-Aid for Scientific Research (C) (23K05959), the JSPS KAKENHI Grant-in-Aid for Transformative Research Areas (A) (21H05301) to H.K. and the AMED AMED-CREST grant (20gm1110001s0304) to R.N.; Y.M. and A.W. are recipients of a fellowship from the JSPS.
Author information
Authors and Affiliations
Contributions
N.O. conceived the study. N.O. and R.N. led the project, designed the experiments, and discussed and interpreted the data. N.O. performed most of the experiments, analysed the data, prepared the figures and wrote the manuscript with contributions from Y.M., A.W., H.K. and R.N.; Y.M. contributed to the preparation of HM. A.W. and H.K. performed the GCaMP analysis to quantify muscle contraction activity. H.K. performed the larval tracking analysis to quantify locomotor activity. N.O. and R.N. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Makoto Kuro-o and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Physiological response to calcium (Ca2+) deficiency during larval development in D. melanogaster.
a, Table showing the Ca2+ concentrations of standard fly food, standard holidic medium (SHM) or Ca2+-free holidic medium (CFHM) prepared using standard agar, standard agar alone (Agar), purified agar/agarose products (product-A: Nacalai tesque #01028-85, product-B: Nacalai tesque #01162-15, product-C: Nacalai tesque #01163-05, and product-D: Sigma-Aldrich #A9539), and standard or calcium-free HM prepared with product-D. HM used in this study was prepared with product-D agarose, in which Ca2+ was undetectable. Standard fly food and the chemically defined diet shown in grey were used. Data shown as means ± SD (n = 3). ND: not detected. b–d, Larvae reared on food lacking dietary Ca2+ showed reduced activity and frequency of skeletal muscle contractions. b, Schematic for the imaging of GCaMP6s expressed specifically in muscles using Mef2-Gal4. c,d, Box and whisker plot showing maximal GCaMP activity during peristalsis (b) and the time interval between peristaltic waves (c) of wandering third (final) instar (L3) larvae just before puparium formation (PF). These larvae were reared on SHM (+) or CFHM (−) from 0 h after L3 ecdysis (0 hAL3E). At least three independent experiments were performed, each with different larvae used for the recordings. Only larvae that underwent PF within 1 h after imaging were included in the analysis. Box plots show the median, the bounds of the box (25th and 75th percentiles), and whiskers representing minimum and maximum values. A. U., arbitrary unit. e,f, Motor neuron-specific knockdown of genes encoding voltage-gated Ca2+ channels (VGCCs) resulted in the elongated pupal phenotype. e, Schematic showing the VGCCs of D. melanogaster, which are classified into high-voltage activated (HVA) and low-voltage activated (LVA) types. f, Pupal axis ratios of pupae with knockdown of the VGCCs via a motor neuron driver (OK6-Gal4). Motor neuron-specific knockdown of the HVA-type VGCC components Ca-α1D, cac, and Ca-β resulted in the elongated pupal phenotype. Motor neurons express higher levels of HVA-type VGCCs than LVA-type VGCCs59. Among these, cac is the most highly expressed, followed by Ca-α1D59. This explains why knockdown of HVA-type channels, rather than LVA-type channels, induces the pupal elongation phenotype, and why the phenotype of cac RNAi is stronger than that of Ca-α1D RNAi. g, Injection of the Ca2+ chelator EGTA into the haemolymph of wandering L3 larvae just before PF caused a concentration-dependent induction of the elongated pupal phenotype. Injected larvae that successfully formed pupae were analysed to determine their pupal axis ratios. h, Schematic for the rearing of larvae on SHM, CFHM, or Mg2+-free holidic medium (MFHM) from various larval stages. L1, first instar; L2, second instar. Larvae were transferred into small food vials containing HM at a density of 25 larvae per vial for newly moulted L2 and L3, or 50 larvae per vial for newly hatched L1. i, Survival rate (PF rate) for larvae reared on SHM, CFHM, or MFHM from various larval stages. j, Representative images of pupae reared on SHM or CFHM from various larval stages. Scale bar, 1 mm. k, Pupal axis ratio of pupae reared on SHM (+) or MFHM (−) from 0 hAL3E. l, Schematic for the rearing of larvae on HM containing varying amounts of Ca2+ from 0 hAL3E. m, Survival rate (PF rate) for larvae reared on HM containing varying amounts of Ca2+ from 0 hAL3E. w1118 was used in all experiments except b–f. Data shown as means ± SD. Sample sizes (n) and p values (p < 0.05) are shown in the graphs, with * indicating p < 0.0001 and ns denoting no significant difference (p > 0.05). Statistics: two-tailed Mann-Whitney U-test in comparison to SHM (+) (c,d,k); one-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons test in comparison to control (f) or 0 mM (g) or SHM (i); one-way ANOVA with Tukey’s multiple comparisons test (m).
Extended Data Fig. 2 Capa expressed in the VaN is responsible for the elongated pupal phenotype.
a–d, Ubiquitous knockdown of Capa induced the formation of elongated pupae (a,b), reduced locomotor activity (c), and reduced haemolymph Ca2+ (d). a,b, Representative images (a) and pupal axis ratio (b) of control and Capa RNAi pupae. Scale bar, 1 mm. c, Locomotor activity of control and Capa RNAi wandering L3 larvae. d, Haemolymph Ca2+ concentration of control and Capa RNAi wandering L3. TubP-Gal4TS>UAS-dicer2 was used as a ubiquitous driver. Three independent Capa RNAi lines were used to confirm the phenotype (a,b,d). e,f, Capa is specifically expressed in two clusters of neurosecretory cells, SEGN (white arrows) and VaN (white arrowheads). e, Capa-2A-Gal4 expression pattern in wandering L3 larva viewed ventrally. The Gal4 driver was crossed to UAS-myr::GFP. The rightmost image shows a magnified view of the region within the yellow-dotted square. f, Capa precursor (P) immunoreactivity (yellow) in the CNS of wandering L3 larvae (w1118). The right image shows a magnified view of the region within the white-dotted square. Nuclei were stained with DAPI (magenta). Scale bars, 500 µm (e), 100 µm (f). g, Pupal axis ratio of pupae inducing Capa RNAi under the control of various Gal4 drivers with the Gal80 system. Capa expressed in the VaN is responsible for the elongated pupal phenotype. TubP-Gal4TS>UAS-dicer2 was used as a ubiquitous driver, nSyb-Gal4TS>UAS-dicer2 as a pan-neuronal driver, and R54A09-Gal4TS>UAS-dicer2 as a VaN driver. Pan-neuronal and VaN-specific inhibition of Gal4 activity was achieved using nSyb-Gal80 and R18D09-LexA>LexAop-Gal80, respectively. h–j, Capa (P) immunoreactivity (yellow) in the CNS of control and Capa RNAi wandering L3 larvae. h, Knockdown of Capa using a VaN driver (R54A09-Gal4TS>UAS-dicer2) abolished Capa (P) immunoreactivity in the VaN and increased the pupal axis ratio (g). i, Combining a pan-neuronal driver (nSyb-Gal4TS>UAS-dicer2) with VaN Gal80 (R18D09-LexA>LexAop-Gal80) abolished Capa (P) immunoreactivity in all neurons except for the VaN. This did not alter the pupal axis ratio (g). j, VaN-specific Capa knockdown using a VaN-specific driver (R18D09∩R54A09-Gal4TS) abolished Capa (P) immunoreactivity only in the VaN. White arrows and arrowheads in (h–j) indicate the SEGN and VaN, respectively. Enlarged images correspond to the white dotted areas. Nuclei were stained with DAPI (magenta). Scale bars, 100 µm (h–j). Data shown as means ± SD. Sample sizes (n) are shown in the graphs. * indicates p < 0.0001, and ns denotes no significant difference (p > 0.05). Statistics: one-way ANOVA with Dunnett’s multiple comparisons test in comparison to control (b,d), two-tailed Mann-Whitney U-test in comparison to control (c), and one-way ANOVA with Tukey’s multiple comparisons test (g).
Extended Data Fig. 3 Expression of VaN and SEGN drivers and Capa RNAi phenotypes.
a–c, Expression of VaN Gal4 (a), SEGN Gal4 (b), and VaN LexA (c) (green), as well as Capa precursor (P) immunoreactivity (yellow) in the CNS. Gal4 and LexA drivers were crossed to UAS-myr::GFP and LexAop-myr::GFP lines, respectively. Nuclei were stained with DAPI (magenta). The two rightmost images show magnified views of the region within the white dotted square in the leftmost images. The white dotted lines in the second image from the left indicate the CNS. For R53E05-Gal4, section images of the yellow outlined square region are included due to the difficulty of distinguishing GFP-positive cell bodies in the Z-stack images. Scale bars, 100 µm. d, Numbers of GFP-expressing cells in the VaN and the SEGN for each Gal4 and LexA driver line. e, Pupal axis ratios of pupae with Capa knockdown via a pan-neuronal driver (nSyb-Gal4), VaN drivers (R16E04-Gal4, R18D09-Gal4, R53E05-Gal4, or R54A09-Gal4), or SEGN drivers (R9F03-Gal4, R20C04-Gal4, R22H11-Gal4, or R82E06-Gal4). Knockdown of Capa via various VaN drivers increased the pupal axis ratio to a level comparable to knockdown of Capa via pan-neuronal driver. Knockdown of Capa via various SEGN drivers did not affect the pupal axis ratio. Each Gal4 line was crossed to UAS-Capa RNAi; UAS-dicer2, TubP-Gal80TS. Data shown as means ± SD. Sample sizes (n) are shown in the graphs. * indicates p < 0.0001, and ns denotes no significant difference (p > 0.05). Statistics: one-way ANOVA with Dunnett’s multiple comparisons test in comparison to control (e).
Extended Data Fig. 4 The effects of CapaR RNAi and the expression of multiple MT drivers.
a, Schematic showing the receptor specificity of the three mature Capa peptides (Capa-1, -2, and PK-1) derived from the Capa precursor. b, Ubiquitous knockdown of CapaR reduced locomotor activity in wandering L3 larvae. c–e, Ubiquitous knockdown of CapaR, but not PK1-R, led to the induction of elongated pupae accompanied by reduced haemolymph Ca2+. c,d, Representative images (c) and pupal axis ratio (d) of control, CapaR RNAi, and PK1-R RNAi pupae. Scale bar, 1 mm. e, Haemolymph Ca2+ concentration of control, CapaR RNAi, and PK1-R RNAi wandering L3 larvae. TubP-Gal4TS>UAS-dicer2 was used as a ubiquitous driver (b–e), and the phenotypes were confirmed using two independent CapaR RNAi lines (c–e). f–i, Expression of NP1093-Gal4 (f), Debcl-Gal4 (g), Uro-Gal4 (h), and tsh(c724)-Gal4 (i) in the MT. Each Gal4 was crossed to the UAS-myr::GFP line. Nuclei were stained with DAPI (magenta). NP1093-Gal4 is expressed in the principal cells of the whole MT (f). Debcl-Gal4 is expressed in the principal cells of the initial segments of the anterior MT, but also in other tissues, including the hindgut (white arrow) (g). Uro-Gal4 is expressed in the principal cells of the main segments of the MT (h). tsh(c724)-Gal4 is expressed in the stellate cells of the MT (i). Immunostaining was performed at least twice, and representative images are shown. Scale bars, 500 µm. j, Pupal axis ratios of pupae with knockdown of CapaR via the following tissue-specific Gal4 driver lines: TubP (ubiquitous), elav and nSyb (pan-neuronal), Vglut (OK371) (motor neurons), phm22 (prothoracic gland), Akh (corpora cardiaca), Mef2, c179, and c57 (muscle), Myo1A (gut enterocytes), Cg (fat body), btl (tracheal cells), and Eip71CD (epidermis). Knockdown of CapaR using tissue-specific drivers other than those driving expression in the initial segments of the anterior MT did not alter pupal axis ratio. Each Gal4 line was crossed to UAS-CapaR RNAi, TubP-Gal80TS. Data shown as means ± SD. Sample sizes (n) are shown in the graphs. * indicates p < 0.0001, and ns denotes no significant difference (p > 0.05). Statistics: two-tailed Mann-Whitney U-test in comparison to control (b), one-way ANOVA with Dunnett’s multiple comparisons test in comparison to control (d,e,j).
Extended Data Fig. 5 The terminals of the transverse nerves (TVN) extending from the VaN reach the vicinity of the initial segments (IS) of the anterior MT (A-MT).
a,b, Ventral (a) and lateral (b) views of the TVN extending from the VaN and approaching the IS of the A-MT. TVN from the VaN (green) and the IS of the A-MT (red) were observed simultaneously using Capa-2A-LexA>LexAop-myr::GFP and CapaR-Gal4>UAS-mCD8::RFP, respectively. This analysis was performed using L2, which have a more transparent cuticle than L3 that facilitates external observations. Immunostaining was performed at least twice, and representative images are shown. Scale bars, 100 µm. c, Schematic of the lateral view of the TVN extending from the VaN and approaching the IS of the A-MT.
Extended Data Fig. 6 In vivo RNAi screen to identify signalling components acting downstream of CapaR to mediate Ca2+ mobilization.
a,b, In vivo RNAi screen for genes encoding Gα proteins. a, Pupal axis ratios of pupae with knockdown of genes encoding each Gα in the D. melanogaster genome. Two independent RNAi lines were used for each gene. Knockdown of the Gαq-encoding gene CG17759 induced the elongated pupal phenotype. According to FlyAtlas2 (https://flyatlas.gla.ac.uk/FlyAtlas2/index.html) and Fly kidney single-cell RNA-seq (https://www.flyrnai.org/scRNA/kidney/), among the three Gαq-encoding genes (CG17759, CG17760, and CG30054), only CG17759 is highly expressed in the Malpighian tubules. b, Representative images of control and CG17759 RNAi pupae. Scale bar, 1 mm. c,d, In vivo RNAi screen for genes encoding protein kinases. c, Pupal axis ratios of pupae with knockdown of all five PKC-encoding genes, all five PKA-encoding genes, and all three PKG-encoding genes in the D. melanogaster genome. Two independent RNAi lines were used for each gene. Knockdown of the PKCε-encoding gene Pkc98E induced the elongated pupal phenotype. Pkc98E is the only PKC in the D. melanogaster genome that encodes PKCε. d, Representative images of control and Pkc98E RNAi pupae. Scale bar, 1 mm. Data shown as means ± SD. Sample sizes (n) are shown in the graphs. * indicates p < 0.0001, and ns denotes no significant difference (p > 0.05). Statistics: one-way ANOVA with Dunnett’s multiple comparisons test in comparison to control (a,c).
Extended Data Fig. 7 Ca2+ deficiency promotes Capa secretion from the VaN.
a, Schematic for rearing larvae on SHM or CFHM for Capa expression analysis. b, Relative Capa expression levels in larvae reared on SHM or CFHM for long durations (24 h). Capa expression levels do not change after 24 h on SHM or CFHM compared to 0 hAL3E. c, Schematic for rearing larvae on SHM or CFHM for Capa secretion analysis. d,e, Mature Capa peptide immunoreactivity in the VaN of larvae reared on SHM or CFHM for short durations (6 h). Representative images for each condition (d) and quantification of mature Capa peptide immunoreactivity (e) are shown. Mature Capa peptide immunoreactivity in the VaN decreased on CFHM but was restored upon SHM re-feeding, suggesting Ca2+ deficiency promotes Capa secretion from the VaN. w1118 was used in all experiments. Scale bars, 25 µm. Data shown as means ± SD. Sample sizes (n) and p values (p < 0.05) are shown in the graphs, with * indicating p < 0.0001 and ns denoting no significant difference (p > 0.05). Statistics: one-way ANOVA with Tukey’s multiple comparisons test (b,e).
Extended Data Fig. 8 Ca2+ accumulated in the A-MT is mobilized in a Capa/CapaR-dependent manner.
a–c, Accumulated Ca2+ in the A-MT is dramatically reduced when larvae are reared on CFHM. a, Representative images of the A-MT from wandering L3 larvae reared on SHM or CFHM from 0 hAL3E. Enlarged images correspond to the white dotted areas. Scale bars, 500 µm. b, Schematic for rearing larvae on SHM (+) or CFHM (−) from various larval stages. c, Ca2+ content per A-MT in wandering L3 larvae reared from various larval stages on SHM (+) or CFHM (−). d–g, When CapaR knockdown larvae were reared on CFHM, depriving them of Ca2+ from both dietary and stored sources, haemolymph Ca2+ levels fell, and the surviving animals formed even more elongated pupae. d, Schematic for rearing control or CapaR RNAi larvae on SHM (+) or CFHM (−). e, Haemolymph Ca2+ concentrations for control or CapaR RNAi (CapaR-i) larvae reared on SHM or CFHM. f,g, Representative images (f) and the pupal axis ratio (g) of control or CapaR RNAi (CapaR-i) pupae reared on SHM or CFHM. Data shown as means ± SD. Sample sizes (n) are shown in the graphs. * indicates p < 0.0001, and ns denotes no significant difference (p > 0.05). Statistics: two-tailed Mann-Whitney U-test in comparison to SHM (+) (c), one-way ANOVA with Tukey’s multiple comparisons test (e,g).
Extended Data Fig. 9 Mobilization of stored Ca2+ via Capa/CapaR is crucial to surviving Ca2+ deficiency.
a–c, Specific knockdown of CapaR in the IS disrupted haemolymph Ca2+ homeostasis in wandering L3 larvae. a, Schematic showing the rearing of larvae on HM containing varying amounts of Ca2+ from 0 hAL3E. b,c, Haemolymph Ca2+ concentration (b) and survival rate (PF rate) (c) for control or CapaR RNAi larvae reared on HM containing varying amounts of Ca2+. d–g, Feeding larvae a high-Ca2+ diet rescued the elongated pupal phenotype induced by CapaR RNAi. d, Schematic showing the rearing of larvae on standard fly food (SFF), high-Ca2+ food (HCF), and high-Mg2+ food (HMF) from 0 hAL3E. e, Representative images of control or CapaR RNAi pupae reared on SFF or food supplemented with 50 mM CaCl2 (HCF) or 50 mM MgCl2 (HMF). Scale bar, 1 mm. f, Pupal axis ratio of control or CapaR RNAi pupae reared on SFF (0 mM) or food supplemented with 10, 25, 50 mM CaCl2. The elongated pupal phenotype induced by CapaR RNAi is rescued in a dose-dependent manner by CaCl2 (mM) supplementation. g, Haemolymph Ca2+ concentrations for control or CapaR RNAi wandering L3 larvae reared on SFF or food supplemented with 50 mM CaCl2 (HCF). h,i, Pupal axis ratio of control or Capa RNAi (h) or CapaR RNAi (i) pupae reared on SFF (0 mM) or food supplemented with 10, 25, 50 mM MgCl2. Food supplemented with MgCl2 did not rescue the elongated pupal phenotype induced by Capa RNAi and CapaR RNAi. CapaR-Gal4TS>UAS-dicer2 was used as an IS driver, and R18D09∩R54A09-Gal4TS was used as a VaN-specific driver. Sample sizes (n) and p values (p < 0.05) are shown in the graphs, with * indicating p < 0.0001 and ns denoting no significant difference (p > 0.05). Statistics: two-way (b,c,f,h,i) or one-way (g) ANOVA with Tukey’s multiple comparisons test.
Extended Data Fig. 10 Schematic showing the neuroendocrine mechanism governing haemolymph Ca2+ regulation in Drosophila larvae.
Capa peptides (Capa-1 and -2) secreted from ventroabdominal neurons (VaN) acts on the initial segments (IS) of the anterior Malpighian tubules (A-MT), activating Gαq and PKCε to trigger Ca2+ mobilization into the haemolymph. The lumen of the IS is a specialized for storing Ca2+ as pearl-like calcium granules (PCGs). PC, principal cells.
Supplementary information
Supplementary Table 1 (download XLSX )
Compositions of the holidic medium used in this study.
Supplementary Table 2 (download XLSX )
RNAi lines targeting neuropeptide/peptide hormone-encoding genes for in vivo RNAi screening.
Supplementary Table 3 (download XLSX )
RNAi lines targeting G-protein alpha subunit-encoding genes for in vivo RNAi screening.
Supplementary Table 4 (download XLSX )
RNAi lines targeting protein-kinase-encoding genes for in vivo RNAi screening.
Supplementary Video 1 (download MP4 )
Larvae reared on a Ca2+-free diet exhibited reduced locomotor activity. Representative video showing the locomotion of wandering third instar (L3) larvae (w1118) reared from various larval stages on standard holidic medium (SHM) or Ca2+-free holidic medium (CFHM). L1, first instar; L2, second instar. The locomotion of 10 wandering L3 larvae placed together on the centre of an agar plate (9 cm diameter) was recorded for 3 minutes. The video plays at 20x speed.
Supplementary Video 2 (download MP4 )
Ubiquitous knockdown of Capa led to reduced larval locomotor activity. Representative video showing the locomotion of Control and Capa RNAi wandering L3 larvae. TubP-Gal4TS>UAS-dicer2 was used as a ubiquitous driver. Larvae were reared at 18°C during early larval stages (L1 and L2) and shifted to 29°C from 0 hr after L3 ecdysis (0 hAL3E) to induce RNAi from L3. The locomotion of 10 wandering L3 larvae placed together on the centre of an agar plate (9 cm diameter) was recorded for 3 minutes. The video plays at 20x speed.
Supplementary Video 3 (download MP4 )
Specific knockdown of Capa in the ventroabdominal neurons (VaN) led to reduced larval locomotor activity Representative video showing the locomotion of Control and Capa RNAi wandering L3 larvae. R18D09∩R54A09-Gal4TS>UAS-dicer2 was used as a VaN-specific driver. Larvae were reared at 18°C during early larval stages (L1 and L2) and shifted to 29°C from 0 hAL3E to induce RNAi from L3. The locomotion of 10 wandering L3 larvae placed together on the centre of an agar plate (9 cm diameter) was recorded for 3 minutes. The video plays at 20x speed.
Supplementary Video 4 (download MP4 )
Ubiquitous knockdown of CapaR led to reduced larval locomotor activity. Representative video showing the locomotion of Control and CapaR RNAi wandering L3 larvae. TubP-Gal4TS>UAS-dicer2 was used as a ubiquitous driver. Larvae were reared at 18°C during early larval stages (L1 and L2) and shifted to 29°C from 0 hAL3E to induce RNAi from L3. The locomotion of 10 wandering L3 larvae placed together on the centre of an agar plate (9 cm diameter) was recorded for 3 minutes. The video plays at 20x speed.
Supplementary Video 5 (download MP4 )
Specific knockdown of CapaR in the initial segment (IS) led to reduced larval locomotor activity. Representative video showing the locomotion of Control and CapaR RNAi wandering L3 larvae. CapaR-Gal4TS>UAS-dicer2 was used as an IS driver. Larvae were reared at 18°C during early larval stages (L1 and L2) and shifted to 29°C from 0 hAL3E to induce RNAi from L3. The locomotion of 10 wandering L3 larvae placed together on the centre of an agar plate (9 cm diameter) was recorded for 3 minutes. The video plays at 20x speed.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Okamoto, N., Mizuno, Y., Watanabe, A. et al. Neuroendocrine control of calcium mobilization in the fruit fly. Nature 649, 122–130 (2026). https://doi.org/10.1038/s41586-025-09670-z
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09670-z
