
Abstract
Heterochromatin, marked by histone H3 lysine 9 methylation, can be epigenetically inherited through cell division1,2,3, maintaining gene repression that preserves cell identity and enables adaptation to environmental challenges2,3,4,5,6. Studies on Schizosaccharomyces pombe have shown that heterochromatin propagation depends on the read–write mechanism, wherein a sufficient density of H3K9me3-modified nucleosomes, stabilized by histone deacetylases, concentrates Clr4SUV39H on chromatin to promote further deposition of H3K9 methylation7,8,9. Whether other mechanisms control heterochromatin propagation by means of Clr4SUV39H, a subunit of the E3 ubiquitin ligase complex ClrC10,11,12, was unknown. Here we uncover a ubiquitin-dependent heterochromatin heritability regulatory hub (HRH) that broadly governs heterochromatin propagation, even without histone deacetylase activity. The HRH is tuned by the limiting factor Raf1DDB2, a substrate receptor for the ClrC ubiquitin ligase. In addition to linking Clr4SUV39H to other ClrC components on chromatin, Raf1DDB2 acts in a dosage-dependent manner to promote ubiquitination of histone H3 at lysine 14 (H3K14ub), which is critical for heterochromatin self-propagation. HRH is intricately linked to environmentally responsive pathways, including nonsense-mediated decay (NMD) and target of rapamycin (TOR) signalling, enabling cells to adapt to changing conditions. By modulating heterochromatin propagation, cells leverage the HRH to gain resistance to antifungal agents and adapt to high temperature. Thus, heterochromatin self-propagation is actively regulated by means of H3K14ub in response to external stimuli, with broad implications for understanding mechanisms governing rapid changes in the epigenetic landscape in physiology and disease.
Similar content being viewed by others


Main
Heterochromatin, a repressive chromatin state, is crucial for various chromosomal processes in eukaryotes1. Heterochromatin suppresses transcription and recombination at repetitive DNA elements and prevents lineage-inappropriate gene expression during development1,3. Heterochromatin also enables adaptive, heritable gene repression in response to environmental change without genetic mutation2,4,5,6,13,14. Epigenetic inheritance of heterochromatin occurs through a self-templating mechanism, enabling persistent gene repression1. Despite its central role in cell identity and adaptation, the mechanisms underlying stable heterochromatin propagation remain to be fully elucidated.
Schizosaccharomyces pombe offers an ideal genetic system for studying heterochromatin propagation. Clr4, an orthologue of Drosophila Su(var)3–9 and mammalian SUV39H, methylates histone H3 at lysine 9 (H3K9)15,16. H3K9 methylation (H3K9me) enables chromatin association of HP1 proteins and their associated effectors to assemble both discrete facultative heterochromatin islands and major heterochromatin domains found at the silent mating-type (mat) region, centromeres and telomeres15,17. At the silent mat region, heterochromatin is nucleated at the centromere-homologous (cenH) element through RNA interference (RNAi)-mediated targeting of Clr4SUV39H, followed by spreading of H3K9me and HP1 to adjacent sequences18.
Clr4SUV39H binds to pre-existing trimethylated H3K9 (H3K9me3)-marked nucleosomes through its chromodomain (‘read’) and catalyses further H3K9me deposition (‘write’)8. This read–write mechanism is crucial for the spreading and epigenetic inheritance of heterochromatin8. Efficient self-propagation of heterochromatin requires a critical H3K9me3 density to maintain sufficient Clr4SUV39H bound to chromatin7,9. Histone deacetylases (HDACs), such as the class II HDAC Clr3, help maintain H3K9me3 density9,19 by blocking access to nucleosome-disrupting chromatin remodellers20. Whether other mechanisms control heterochromatin propagation, including those acting at the level of Clr4-containing E3 ubiquitin ligase complex ClrC, has not been explored.
Here we identify an H3K14ub-dependent heterochromatin HRH operating directly at the level of ClrC. This regulatory hub integrates signals from growth-condition-responsive pathways to govern the spread and epigenetic inheritance of heterochromatin.
NMD affects heterochromatin propagation
The S. pombe ClrC shares structural resemblance to CUL4-DDB1-DDB2 E3 ubiquitin ligase complexes. Beyond the Clr4SUV39H methyltransferase, ClrC includes the WD-40 protein Raf1, the Zn finger protein Raf2, the β-propeller protein Rik1 and the cullin family protein Cul4 (refs. 10,11,12,21,22). Rik1 closely resembles human DDB1, which together with CUL4 participates in histone methylation23. Raf1 is similar to human DDB2 and probably a DCAF (DDB1 and CUL4 associated factor), serving as the substrate receptor for E3 ubiquitin ligases23,24,25. Raf1 features two conserved WDxR motifs23,24,25 (residues 515–518 and 573–576).
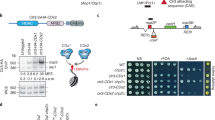
To probe WDxR motif function in Raf1, we generated raf1R518H and raf1R576H alleles and assessed heterochromatic silencing at the mat region using the sensitive REIIΔ mat2P::ura4+ reporter (Extended Data Fig. 1a). In mat1-M cells lacking the local REII silencer, defects in heterochromatin assembly result in derepression of mat2P and haploid meiosis, indicated by dark brown staining of cells when exposed to iodine vapour7,9,26. By contrast, cells with functional heterochromatic silencing stain yellow. The nearby mat2P::ura4+ reporter provides an extra readout for heterochromatic silencing when tested on medium lacking uracil (−URA) or counter-selective 5-fluoroorotic acid (FOA) medium. Both raf1R518H and raf1R576H showed loss of mat2P::ura4+ silencing and dark iodine staining (Extended Data Fig. 1a). However, their effects on silencing of ura4+ inserted at the cenH nucleation site (Kint2::ura4+) differed. Whereas Kint2::ura4+ in raf1R518H showed severe silencing defects with loss of H3K9me3 across the mat region, Kint2::ura4+ in raf1R576H cells was only weakly affected (Extended Data Fig. 1a,b). Notably, raf1R576H maintained H3K9me3 around the cenH nucleation centre but failed to spread it to surrounding sequences (Extended Data Fig. 1b).
We then investigated how raf1R576H affects heterochromatin spreading. We performed an unbiased genetic screen for factors that restore silencing of REIIΔ mat2P::ura4+ in raf1R576H cells (Fig. 1a). Three suppressors reestablished silencing, indicated by increased growth on FOA medium and light iodine staining. Two mutations mapped to esl1 (also called ebs1), and one to upf1 (Fig. 1b). Upf1 and Esl1 are components of the NMD pathway27,28,29. Deleting upf1 or esl1 reproduced suppression of the raf1R576H silencing defects (Fig. 1c), confirmed by PCR with reverse transcription (RT–PCR) analysis of the ura4+ reporter (Fig. 1d). Chromatin immunoprecipitation with sequencing (ChIP–seq) showed increased H3K9me3 and Swi6HP1 across the silent mat region in raf1R576H upf1Δ and raf1R576H esl1Δ (Fig. 1e,f), with a similar increase in heterochromatin spreading at subtelomeric regions (Extended Data Fig. 1c). By contrast, centromeric H3K9me3 levels were comparable to wild-type (WT) cells in the single and double mutants, consistent with persistent RNAi-directed ClrC targeting to pericentromeric repeats (Extended Data Fig. 1d). These results show that loss of NMD components can rescue the heterochromatin propagation defect of raf1R576H.

a, Genetic screen to identify suppressor mutants that restore silencing at the mat region in the raf1R576H mutant. Staining of colonies with iodine vapours as well as growth on uracil-deficient and FOA-containing media were used to assess rescue of silencing defects. Percentage of cells undergoing haploid meiosis is indicated. b, Domain organization of Upf1 and Esl1 showing the mutations (in red) identified in the suppressor screen. c, Restoration of heterochromatic silencing on loss of NMD components in raf1R576H mutant cells. Serial dilutions were plated on non-selective, uracil-deficient and FOA-containing media to assess mat2P::ura4+ expression. Haploid meiosis was assessed by iodine staining. d, RT–qPCR analysis of ura4+ expression (data are presented as mean ± s.d.; n = 3 independent experiments, adjusted P value by one-way analysis of variance followed by Holm–Sidak multiple comparisons test). e,f, ChIP–seq analysis of H3K9me3 (e) and Swi6HP1 (f) distribution at the silent mat region in the indicated strains. Data are representative of two independent experiments. +FOA, FOA-containing; N/S, non-selective; −Ura, uracil-deficient.
NMD controls ClrC subunit Raf1DDB2 levels
To explain how NMD loss suppresses heterochromatin defects in raf1R576H, we considered that NMD competes with RNAi for centromeric repeat transcripts, so NMD defects would enhance RNAi-mediated small RNA production and heterochromatin assembly. However, Upf1 loss did not increase small RNA production (Extended Data Fig. 2a). Moreover, the RNAi factor Ago1 was dispensable for restored silencing and H3K9me3 enrichment across the silent mat region in raf1R576H upf1Δ cells (Extended Data Fig. 2b,c). In addition, H3K9me3 levels and heterochromatic silencing were increased in upf1Δ cells relative to WT cells, even when the RNAi-dependent heterochromatin nucleation centre cenH was replaced with ura4+ (K∆::ura4+) (Extended Data Fig. 2d). Thus, the suppression of heterochromatin defects in raf1R576H cells on loss of NMD components is not due to increased RNAi-mediated heterochromatin assembly.
We asked whether NMD regulates the expression of a critical factor necessary for heterochromatin propagation. RNA sequencing (RNA-seq) showed reduced raf1 mRNA levels in raf1R576H compared with WT (Fig. 2a and Extended Data Fig. 2e). raf1 mRNA levels increased in raf1R576H upf1Δ cells, exceeding WT levels (Fig. 2a and Extended Data Fig. 2f). This effect was specific to raf1, as transcripts encoding other ClrC components were unaffected (Supplementary Table 1). RNA immunoprecipitation (RIP) showed that Upf1 associates with raf1 mRNA, suggesting NMD directly controls raf1 expression (Fig. 2b). Indeed, NMD pathway defects increased raf1 mRNA and Raf1 protein levels (Fig. 2c and Extended Data Fig. 2g). Loss of Upf1 resulted in higher raf1 expression, not only in raf1R576H but also in raf1WT cells (Extended Data Fig. 2g). Therefore, although our genetic screen using the low-expression raf1R576H mutant allele was pivotal in identifying raf1 as an NMD target, these analyses show that NMD also suppresses raf1WT expression. Thus, raf1 is a bona fide NMD target involved in regulating heterochromatin propagation.

a, RNA-seq expression profile of raf1 and a neighbouring gene in the indicated strains. b, RIP to detect Upf1 association with raf1 transcript. Data normalized to act1 (data are presented as mean ± s.d.; n = 3 independent experiments, P value by two-tailed unpaired t-test). c, Western blot analysis of Raf1 in the indicated strains. Cdc2 and Ponceau S serve as loading controls. d, Analysis of heterochromatic silencing in raf1R576H mutant cells on overexpression of WT (raf1-oe) or mutant (raf1R576H-oe) raf1 under the control of nmt1 promoter. Serial dilution monitored mat2P::ura4+ expression, and iodine staining assessed haploid meiosis. e,f, ChIP–seq analysis of H3K9me3 distribution across the silent mat region (e) and subtelomeric region of Chromosome 2R (f) in the indicated strains. Data are representative of two independent experiments. Raw western blots are presented in Supplementary Fig. 1. RPM, reads per million mapped reads.
We noticed raf1 mRNA contains cryptic, inefficiently spliced introns linked to RNA decay30 (Extended Data Fig. 3a). These introns are detected in upf1Δ cells, consistent with the appearance of cryptic introns when target RNA decay pathways are disabled30 (Extended Data Fig. 3a). Our analyses also revealed that, like NMD, splicing machinery affects raf1 mRNA. From the screen that initially implicated NMD in raf1 regulation (Fig. 1a), we characterized two more mutants: one carried a mutation in upf2, which also functions in NMD, and the other carried a mutation in sap49, which encodes a U2 snRNP–associated RNA-binding splicing factor. Introducing the sap49A175V mutation in raf1R576H cells restored heterochromatic silencing and H3K9me3 spreading at the silent mat region (Extended Data Fig. 3b,c) and partially restored H3K9me3 spreading at subtelomeres (Extended Data Fig. 3d). No major changes in H3K9me3 were observed at centromeres in which RNAi persistently targets heterochromatin (Extended Data Fig. 3e). The sap49A175V markedly upregulated raf1 mRNA and Raf1 protein, mirroring upf1Δ (Extended Data Fig. 3f,g). Together with previous links between splicing and NMD31,32, these results indicate that Sap49 may promote raf1 mRNA decay through NMD.
Raf1 drives heterochromatin propagation
To determine whether NMD loss restores heterochromatin propagation simply by increasing Raf1 levels, we ectopically overexpressed raf1R576H under an inducible promoter (raf1R576H-oe) and, for comparison, overexpressed the raf1WT (raf1-oe). Raf1 expression at levels comparable to those in upf1Δ cells restored silencing in raf1R576H cells (Extended Data Fig. 3h,i). raf1R576H-oe rescued silencing to the same extent as raf1WT-oe (Fig. 2d). Restoration of silencing was accompanied by H3K9me3 spreading across the silent mat region (Fig. 2e), and H3K9me3 levels were comparable between raf1R576H-oe and raf1WT-oe cells. These findings indicate that the R576H mutation does not impair Raf1 function; rather, heterochromatin defects in raf1R576H cells reflect reduced Raf1 abundance. raf1R576H-oe also rescued subtelomeric heterochromatin spreading defects in raf1R576H cells (Fig. 2f). Thus, NMD-mediated control of Raf1 abundance is a key control point in the regulation of heterochromatin propagation.
Raf1DDB2 abundance governs ClrC binding
We then investigated how Raf1 abundance affects H3K9me3 and heterochromatin propagation. Our analyses suggested that Raf1 is limiting for ClrC assembly, particularly in recruiting Clr4SUV39H, among other functions (below). Glycerol-gradient fractionation revealed a substantial fraction of monomeric Clr4SUV39H, indicating a weak association with other ClrC components (Fig. 3a). On Raf1 overexpression, Clr4SUV39H shifted to higher molecular weight fractions (Fig. 3a). This shift correlated with increased incorporation of Clr4SUV39H into ClrC, evidenced by enhanced Clr4SUV39H association with the ClrC subunit Raf2 in both raf1-oe and upf1Δ cells (Fig. 3b and Extended Data Fig. 4a). Therefore, Raf1 levels probably govern the association of Clr4SUV39H with other ClrC components.

a, Glycerol-gradient fractionation showing Clr4SUV39H distribution from low to high molecular weight fractions on overexpression of raf1 (raf1-oe) under the control of nmt1 promoter. b, Co-immunoprecipitation of Raf2 with Clr4SUV39H in the indicated strains. OE, overexpression. c, Co-immunoprecipitation showing Raf2 and Clr4SUV39H association in cells expressing different levels of Raf1. The asterisk * denotes the non-specific band. d, ChIP–seq analysis of H3K14ub distribution across the silent mat region, centromere 2 and subtelomeric region 1R in WT and raf1Δ cells. e, ChIP–seq analysis of H3K14ub and H3K9me3 distribution across subtelomeric region 1R in the indicated strains. f, ChIP–seq analysis of H3K14ub distribution across the silent mat region in the indicated strains. g, Heterochromatic silencing in ubc4-1 cells on overexpression of raf1 (raf1-oe) under the control of adh1 promoter. Serial dilutions on indicated media were used to assess mat2P::ura4+ expression. Haploid meiosis was evaluated by iodine staining. h, ChIP–seq analysis of H3K9me3 distribution across the silent mat region in the indicated strains. All data are representative of two independent experiments. IP, immunoprecipitation.
The Raf1 WD-repeat domain adopts a β-propeller fold that mediates protein interactions33. We wondered whether Raf1 directly engages Clr4SUV39H. The Raf1 WD-repeat region bound Clr4SUV39H in vitro (Extended Data Fig. 4b). In vivo, Clr4SUV39H association with Raf2 correlated with Raf1 abundance. Clr4 failed to associate with Raf2 in cells lacking Raf1, and only weak association was detected in raf1R576H cells with low Raf1 levels (Fig. 3c). By contrast, Clr4–Raf2 association increased with higher Raf1 levels in cells carrying the raf1WT allele, and Raf1 overexpression produced a marked enhancement of Clr4 association with Raf2. These findings further suggest that Raf1 is a limiting factor for incorporation of Clr4SUV39H into ClrC.
To determine whether Raf1 levels influence the chromatin-bound state of ClrC, we tracked Raf2 dynamics in live cells. Raf2 tagged with HaloTag (Raf2-Halo) formed two to three foci, corresponding to clustered heterochromatic loci at the nuclear periphery (Extended Data Fig. 4c). Single-molecule tracking (SMT) revealed slow and fast diffusing Raf2-Halo particles and their diffusion coefficients (D) (Extended Data Fig. 4d and Supplementary Videos 1 and 2). As a chromatin-bound control, histone H2B tagged with Halo (H2B-Halo) showed two similar diffusive states. On Raf1 overexpression, the slow-diffusing Raf2 population increased, indicating a redistribution of ClrC to a less mobile, chromatin-bound state. Moreover, the survival distribution indicated reduced dissociation of chromatin-bound ClrC on overexpression of Raf1 (Extended Data Fig. 4e).
To further analyse ClrC mobility, we classified Raf2 trajectories into different diffusive states using a systems-level classification algorithm perturbation-expectation maximization (pEM) (Extended Data Fig. 4f). Three subdiffusive Raf2 mobility states were identified (Extended Data Fig. 4f). State 3 showed a radius of confinement (Rc) corresponding to the average nuclear radius, consistent with the free state that was not observed for H2B. States 1 and 2 had smaller Rc values and were considered confined. Notably, the D and Rc of Raf2-Halo confined states 1 and 2 were similar to the two H2B states, indicating that they are the chromatin-bound states (Extended Data Fig. 4g,h). Raf1 overexpression led to an increased proportion of the Raf2 confined states 1 and 2 and a reduction in the free state (Extended Data Fig. 4i), indicating a shift of Raf2 molecules towards the chromatin-associated population. Together, these results indicate that higher levels of Raf1 not only promote ClrC integrity but also increase the proportion of ClrC bound to chromatin.
The enhanced chromatin association of ClrC observed with increased Raf1 levels was also evident from direct visualization of Raf2 localization. The number and intensity of Raf2-Halo foci increased considerably in raf1-oe and upf1Δ cells, both of which contain higher Raf1 levels (Extended Data Fig. 5a,b). In raf1-oe cells, higher Raf2 foci intensity correlated with enhanced Raf2 spreading, especially at subtelomeric regions lacking defined heterochromatin boundaries (Extended Data Fig. 5c), whereas Raf2 distribution at heterochromatic regions flanked by boundary elements, such as the silent mat region, remained unchanged (Extended Data Fig. 5d). Raf1 overexpression also increased ClrC binding at heterochromatin islands, including genes responsive to developmental or environmental signals, such as pho1 (Extended Data Fig. 5e).
Collectively, these analyses suggest that elevated Raf1 stabilizes and prolongs ClrC chromatin association, concentrating this histone methyltransferase and ubiquitin ligase complex, which ultimately promotes the read–write activity that drives heterochromatin propagation.
Raf1DDB2 DCAF directs H3K14ub
ClrC monoubiquitylates histone H3 at lysine 14 (H3K14ub), which enhances Clr4SUV39H methyltransferase activity in vitro34,35,36, however, its in vivo relevance remained unclear. We asked whether histone H3 is the physiological substrate of the ClrC E3 ligase and whether Raf1 promotes H3K14ub beyond its dose-dependent effects on ClrC chromatin association.
We profiled H3K14ub genome-wide by ChIP–seq with an anti-H3K14ub antibody37. H3K14ub was enriched across major heterochromatin domains, including the silent mat region, pericentromeres and subtelomeres, but was completely abolished in cells lacking Raf1 (Fig. 3d). This result indicates that H3K14ub is a prominent heterochromatin mark whose deposition requires the DCAF Raf1. Raf1 overexpression markedly increased H3K14ub, particularly at subtelomeres that lack boundary elements, and this increase correlated with enhanced H3K9me3 spreading (Fig. 3e). Thus, Raf1 dosage is critical for H3K14ub and H3K9me3, and merely increasing Raf1 enhances heterochromatin propagation.
To test the requirement for H3K14ub in heterochromatin propagation, we examined the E2 ubiquitin-conjugating enzyme Ubc4 mutant ubc4-1 (ref. 38). H3K14ub was markedly reduced across heterochromatin domains, including the mat region (Fig. 3f). Because Ubc4 is not a core component of the dually functioning ClrC, which mediates both ubiquitylation and H3K9 methylation, this mutation isolates the contribution of H3K14ub to heterochromatin assembly. In ubc4-1, loss of H3K14ub correlated with impaired heterochromatin spreading and silencing at the silent mat region (Fig. 3g,h). Although H3K9me3 was established at the cenH nucleation site, where the RNAi machinery recruits ClrC, it failed to spread in the H3K14ub-deficient ubc4-1 mutant (Fig. 3h). Raf1 overexpression did not restore H3K14ub, H3K9me3 spreading or heterochromatic silencing in ubc4-1 cells (Fig. 3f,g,h).
H3K14ub stimulates Clr4SUV39H methyltransferase activity in vitro34,35,36 and is therefore expected to hyperactivate Clr4SUV39H, leading to H3K9me3 accumulation that stimulates the read–write mechanism of heterochromatin propagation. Accordingly, ubc4-1 probably disrupts the H3K14ub and Clr4SUV39H crosstalk, reducing H3K9me3 below the threshold required for efficient heterochromatin propagation1,7.
H3K14ub defines a new regulatory pathway
Clr3, the HDAC that deacetylates H3K14, is essential for heterochromatin propagation9,19. By suppressing histone turnover, Clr3 maintains sufficient H3K9me3 density required for Clr4 read–write activity1. Loss of Clr3 reduces H3K9me3 density and impairs heterochromatin propagation and silencing9,19 (Fig. 4a). We also found reduced H3K14ub in clr3Δ cells, particularly across regions surrounding the cenH nucleation site (Fig. 4b), prompting us to test whether elevating Raf1 could restore heterochromatin assembly.

a, Rescue of silencing defects in clr3Δ cells by raf1 overexpression (raf1-oe) from the adh1 promoter or by deletion of upf1 (upf1Δ). Serial dilution assessed mat2P::ura4+ expression, and iodine staining assessed haploid meiosis. b,c, ChIP–seq profiles of H3K14ub (b) and H3K9me3 (c) across the silent mat region in the indicated strains. d, Schematic illustrating restoration of heterochromatin propagation in clr3Δ cells on increased Raf1 expression through raf1-oe or upf1Δ. ClrC, recruited to the nucleation site, deposits H3K9me3 and H3K14ub. In WT cells, the Clr3 HDAC suppresses histone turnover, preserving H3K14ub, H3K9me3 and associated Clr4SUV39H, to support heterochromatin spreading through the read–write mechanism (top). Loss of Clr3 lowers H3K9me3 and H3K14ub density, compromising Clr4SUV39H read–write activity and heterochromatin propagation (middle). Elevated Raf1 restores Clr4/ClrC, H3K14ub and H3K9me3 density, enabling heterochromatin propagation even in the absence of Clr3 (bottom). e, Schematic of ectopic heterochromatin establishment and maintenance in the indicated strains (top). A TetR-Clr4 fusion, expressed from a thiamine-repressible promoter (tetR-clr4ON), nucleates heterochromatin at a tetO-ade6+ reporter. Heterochromatin inheritance by read–write activity of endogenous Clr4SUV39H was tested after TetR-Clr4 release in the presence of thiamine (tetR-clr4OFF). Red colonies on low-adenine medium indicate ade6+ repression, whereas white colonies indicate expression (middle). ChIP analysis of H3K9me3 at the ade6+ reporter in the indicated strains (bottom). Data are mean ± s.d. (n = 3 independent experiments); P values by two-tailed unpaired t-test. All data are representative of at least two independent experiments.
Increasing Raf1, by means of raf1-oe or upf1Δ, reinstated heterochromatic silencing of the REIIΔ mat2P::ura4+ reporter in clr3Δ cells (Fig. 4a) and correlated with increased H3K14ub levels across the silent mat region (Fig. 4b). Whereas clr3Δ cells showed H3K9me3 enrichment mainly near cenH, H3K9me3 spread across the entire domain in clr3Δ raf1-oe and clr3Δ upf1Δ cells (Fig. 4c). This suggests a parallel mechanism such that boosting Raf1 levels, which enhances ClrC chromatin association and H3K14ub, bypasses the requirement for the Clr3 HDAC activity in heterochromatin propagation (Fig. 4d).
Raf1 upregulation in clr3Δ cells phenocopies loss of the anti-silencing factor Epe1 (ref. 39). Because Epe1 undergoes ubiquitin-dependent proteolysis40,41, we asked whether Raf1 alters Epe1 abundance. Epe1-GFP (green fluorescent protein) foci number and intensity were similar in WT and raf1-oe cells (Extended Data Fig. 6a), indicating Raf1 does not act by reducing Epe1 levels. Genetic analyses showed that Epe1 and Raf1 act independently to suppress the silencing defect of Clr3 catalytic mutant (clr3D232N) cells. Deleting epe1 or upf1 each partially restored REIIΔ mat2P::GFP reporter silencing, and the double deletion produced an additive effect and near-complete rescue (Extended Data Fig. 6b). Together with the unchanged Epe1 levels on Raf1 overexpression, these results support a distinct Raf1-mediated pathway controlling heterochromatin propagation.
To further validate a Raf1-based mechanism for controlling heterochromatin propagation, we tested whether increasing Raf1 mitigates defects in other contexts. In cells carrying a mutation in one of the three histone H3 copies (hht2G13D), the reduction in H3K9me3 density impairs heterochromatin propagation7. Notably, introducing raf1-oe or upf1Δ into hht2G13D cells restored REIIΔ mat2P::ura4+ silencing (Extended Data Fig. 7a). hht2G13D cells also showed defective H3K14ub across the silent mat region (Extended Data Fig. 7b), a defect rescued by increased Raf1 levels in upf1Δ cells and accompanied by restored H3K9me3 spreading across the domain (Extended Data Fig. 7c,d).
We further explored whether elevating Raf1 bypasses other requirements for heterochromatin propagation. Indeed, raf1-oe or upf1Δ restored silencing in cells lacking the FACT (facilitates chromatin transcription) subunit Pob3, the SMARCAD1 SNF2 remodeller Fft3 or the nuclear rim protein Amo1NUPL2 (Extended Data Fig. 7e), factors that preserve H3K9me3 density26,42,43. Collectively, these results identify Raf1-mediated control of ClrC chromatin association and H3K14ub as a previously unrecognized mechanism supporting heterochromatin maintenance and propagation.
Raf1 levels control self-propagation
We tested whether Raf1 abundance influences sequence-independent heterochromatin inheritance. Heterochromatin was nucleated at an ectopic site by reversibly tethering TetR-Clr4 (Clr4SUV39H fused to the TetR DNA-binding domain) to six tetracycline operators located upstream of the ade6+ reporter (Fig. 4e). Without tetracycline (tetR-clr4ON), TetR-Clr4 induced H3K9me3 and silenced ade6+ (red colonies). On release of TetR-Clr4 (tetR-clr4OFF), H3K9me3 and ade6+ silencing were rapidly lost, probably because the local chromatin concentration of endogenous Clr4SUV39H was insufficient to sustain read–write activity and maintain ectopic heterochromatin9,26,44,45. By contrast, releasing TetR-Clr4 in upf1Δ cells with elevated Raf1 levels allowed ade6+ silencing and H3K9me3 at the ectopic site to persist for several generations (Fig. 4e), indicating that increased Raf1 abundance, which enhances ClrC chromatin association, is sufficient to support heterochromatin self-propagation.
Raf1 abundance is regulated in adaptation
NMD-regulated Raf1 levels substantially affect heterochromatin propagation, suggesting this mechanism operates under physiological conditions. Self-propagation of heterochromatin enables heritable gene reprogramming and environmental adaptation2,4,6,13,14. A recent study demonstrated that cells acquire resistance to caffeine through heterochromatin propagation14. Because caffeine attenuates NMD activity in other systems46,47, we investigated whether caffeine treatment affects NMD in S. pombe. NMD-regulated transcripts29 were upregulated in the presence of caffeine (Extended Data Fig. 8a). Moreover, the raf1R576H mutant transcript was derepressed, as in upf1Δ (Fig. 2a), with a corresponding increase in protein levels (Extended Data Fig. 8b,c). Caffeine treatment also markedly increased WT Raf1 levels (Fig. 5a).

a, Western blot of Raf1 in cells treated with 16 mM caffeine for 2 weeks. Cdc2 and Ponceau S serve as loading controls. b, Cells with or without raf1 overexpressed from the adh1 promoter (raf1-oe) were plated on caffeine and caffeine-resistant colonies were quantified (mean ± s.d.; n = 3 independent experiments; P values by two-tailed unpaired t-test). c, Caffeine, fluconazole (FLZ) and clotrimazole (CLZ) resistance in raf1-oe and upf1Δ cells assessed by serial dilutions. d,e, Heterochromatin assembly and silencing at 30 °C and 37 °C with or without raf1-oe assessed by serial dilution analysis of mat2P::ura4+ expression (d) and by H3K9me3 ChIP–seq across the silent mat region (e). f, Western blot analysis of Gad8 S546 phosphorylation at 30 °C and 37 °C. Cdc2 and Ponceau S serve as loading controls. g,h, Serial dilution analysis of mat2P::ura4+ expression in upf1Δ and tor1Δ single and double mutant cells (g) and H3K9me3 ChIP–seq across the silent mat region in the indicated strains (h). i, Western blot of Raf1 at 30 °C and 37 °C in the indicated strains. Cdc2 and Ponceau S serve as loading controls. j, Model showing the environmental cues converge on the heterochromatin HRH, where Raf1DDB2 abundance, regulated by TOR and the NMD pathway, modulates heterochromatin propagation to reprogram gene expression and enable adaptation. Raf1DDB2, a limiting factor for ClrC chromatin association, also mediates H3K14ub, which together maintain high local H3K9me3 density and associated Clr4SUV39H, critical for heterochromatin spreading and inheritance. e,f,i, Cells were grown at 37 °C for 16 h. Data are representative of two independent experiments.
We asked if Raf1 abundance alone favours the development of caffeine resistance. Raf1 overexpression correlated with increased caffeine-resistant colonies (Fig. 5b) and resistance to the antifungal agents, fluconazole and clotrimazole (Fig. 5c). Similar resistance was observed in upf1Δ cells (Fig. 5c). By contrast, Raf1 overexpression failed to confer caffeine resistance in cells lacking Clr4SUV39H, suggesting a heterochromatin-dependent mechanism (Extended Data Fig. 8d,e). ChIP–seq of H3K9me3 in raf1-oe caffeine-resistant cells revealed increased H3K9me3 and spreading at known heterochromatin islands and at new genomic locations (Extended Data Fig. 8f), including loci whose silencing confers caffeine resistance, such as cup1 implicated in mitochondrial dysfunction14 (Extended Data Fig. 8g). Thus, Raf1 upregulation is part of a natural mechanism to enhance heterochromatin stabilization, enabling cells to adapt to unfavourable conditions.
TOR-Raf1 node modulates heterochromatin
We explored whether other conditions affect Raf1 levels. Culturing cells at an elevated temperature (37 °C) reduced Raf1 levels (Extended Data Fig. 9a), which is intriguing given the temperature-dependent alterations in heterochromatin observed in various systems48,49,50,51. This Raf1 reduction at 37 °C resulted in impaired heterochromatin spreading and disruption in gene silencing (Fig. 5d,e). Raf1 overexpression restored both silencing and H3K9me3 distribution across the silent mat interval (Fig. 5d,e), suggesting that cells downregulate Raf1 in response to elevated temperatures, thereby affecting heterochromatin propagation.
We next examined signalling pathways that modulate Raf1. Loss of Tor1, a TORC2 complex subunit52, severely reduced Raf1 protein and transcript levels (Extended Data Fig. 9b,c). By contrast, Raf1 levels were not affected by mutations in other kinases, including Tor2, the TORC1 subunit that targets RNA processing machinery for H3K9me at meiotic genes but is dispensable for heterochromatin assembly elsewhere53 (Extended Data Fig. 9b). To test temperature effects on Tor1 activity, we examined Gad8 (human AKT orthologue)54,55 phosphorylation at 30 °C and 37 °C. Gad8 phosphorylation was high at 30 °C, but undetectable at 37 °C (Fig. 5f). Cells lacking Gad8 also showed reduced Raf1, similar to tor1Δ (Extended Data Fig. 9b). These findings suggest that high temperature inactivates the Tor1 signalling cascade, which is required for proper Raf1 expression. The reduction in Raf1 levels in both tor1Δ and gad8Δ cells also resulted in a decrease in caffeine-resistant colonies (Extended Data Fig. 9d,e), a phenotype reversed by Raf1 overexpression (Extended Data Fig. 9f).
On the basis of our results, we hypothesized that reduced Raf1 abundance drives the heterochromatin defects previously reported in tor1Δ cells56. Indeed, increasing Raf1 in upf1Δ suppressed heterochromatin propagation and silencing defects in a tor1Δ background (Fig. 5g,h). However, Raf1 levels were lower in the upf1Δ tor1Δ double mutant than in upf1Δ alone (Fig. 5i), indicating that Tor1 affects Raf1 independently of NMD. Supporting this, inactivating Tor1 by culturing upf1Δ cells at 37 °C reduced Raf1 expression compared with 30 °C (Fig. 5i). As expected, shifting upf1Δ tor1Δ cells from 30 °C to 37 °C caused no further decrease (Fig. 5i). Thus, Upf1 and Tor1 operate through separate pathways to control Raf1 abundance. These findings support a model in which many inputs converge on the control of Raf1 levels, positioning Raf1 as a key component of a regulatory hub controlling heterochromatin robustness and heritability (Fig. 5j).
Preserving heterochromatin under stress
Given the pivotal role of Raf1-based HRH in heterochromatin propagation, we wondered whether factors influencing Raf1 expression affect epigenetic inheritance of heterochromatin. Because high temperature considerably reduces Raf1 levels, we proposed that this would disrupt the self-propagation of an ectopic heterochromatin domain. We tested this using a robust reporter containing one or two copies of Clr3-attracting sequences (CAS) inserted adjacent to the tetO-ade6+ gene. CAS recruits the Clr3 HDAC, facilitating efficient heterochromatin propagation9.
We established heterochromatin by tethering TetR-Clr4 (tetR-clr4ON) to the CAS-tetO-ade6+ reporter in cells cultured at 30 °C, then assessed propagation of the silenced heterochromatic state after TetR-Clr4 release (tetR-clr4OFF) at 30 °C or 37 °C. At 30 °C, the heterochromatic state was epigenetically inherited, with propagation efficiency increasing with the number of CAS elements, such that two copies of CAS3 offered the most efficient heterochromatin maintenance (Extended Data Fig. 10a). Conversely, cells cultured at 37 °C failed to maintain the silenced state (Extended Data Fig. 10a).
We then asked whether increasing Raf1 rescues the high-temperature defect in heterochromatin propagation. Using the 2xCAS3-tetO-ade6+ reporter, we compared the propagation of the ectopic heterochromatin domain in WT and upf1Δ cells. After releasing TetR-Clr4, upf1Δ cells propagated silenced ade6+ even at 37 °C, whereas WT maintained inheritance of the silenced state only at 30 °C (Extended Data Fig. 10b). ChIP revealed greater preservation of H3K9me3 in upf1Δ than WT (Extended Data Fig. 10b).
These findings establish that the control of Raf1 levels under different growth conditions represents a bona fide heterochromatin HRH. This hub modulates heterochromatin stability by governing ClrC chromatin association and H3K14ub levels, and is exploited during adaptation to environmental challenges.
Discussion
The remarkable ability of heterochromatin to be epigenetically inherited across many cell divisions stabilizes gene repression, maintaining committed cell states and enabling adaptation without genetic changes2,3,4,5,6,14,51. We have previously shown that histone deacetylation by HDACs suppresses histone turnover, thereby maintaining H3K9me3 density required for heterochromatin propagation through the Clr4SUV39H read–write mechanism1,7,8,9. Here, we identify a previously unrecognized regulatory hub for heterochromatin heritability, the HRH, that responds to environmental signals to control heterochromatin self-propagation.
Given the central role of heterochromatin in epigenetic gene regulation, our discovery of the HRH, with Raf1 DCAF as a key control point, marks a major advance. NMD and TOR signalling, which respond to environmental and developmental cues27,28,52,57, converge on Raf1, whose abundance ultimately dictates the robustness of heterochromatin propagation. How does elevated Raf1 promote heterochromatin propagation? Raf1 is a limiting factor that links Clr4SUV39H to other ClrC components and facilitates their chromatin association, which alone may create the high local concentration of the H3K9me3 needed to sustain heterochromatin propagation. However, Raf1 DCAF also acts in a dose-dependent manner to promote H3K14ub, a modification required for propagation of heterochromatic structures. This is important because in vitro studies showed that H3K14ub notably increases Clr4SUV39H activity by inducing a conformational shift, enhancing affinity for the histone substrate34,35,36, and also provides a binding site for chromodomain-containing amino-terminal region of Clr4/SUV39H34,37. Therefore, Raf1 promotes heterochromatin propagation by directly influencing ClrC chromatin association and mediating H3K14ub, which are both essential for achieving the critical H3K9me3 threshold and supporting the read–write activity of Clr4SUV39H. As a DCAF, Raf1 may also target further substrates for ClrC-mediated ubiquitination, including factors that destabilize heterochromatin. Consistently, increasing Raf1 abundance bypasses the requirement for the Clr3 HDAC, mirroring effects observed when histone acetyltransferases or SWI/SNF remodelling enzymes that disrupt nucleosomes are absent9,20.
Two distinct pathways, HDACs and HRH, maintain the high H3K9me and Clr4SUV39H density needed for read–write propagation of heterochromatin. Under normal growth conditions, cells with basal Raf1 levels probably rely more on HDACs, which safeguard H3K9me3 nucleosomes by blocking access to remodellers and enhance Clr4SUV39H activity9,20, thereby promoting heterochromatin propagation. This pathway is most effective at loci with high HDAC concentrations9. By contrast, the HRH becomes critical under stress or other specific conditions that demand broader reinforcement of heterochromatin heritability. To this end, elevating Raf1 alone is sufficient to promote heterochromatin self-propagation at an ectopic site, even under stress.
The HRH pathway enhances heterochromatin propagation, enabling resistance to caffeine and to the antifungal agents, fluconazole and clotrimazole. Caffeine increases Raf1 levels (this study) and has also been reported to reduce levels of the negative heterochromatin regulator, Epe1 (ref. 41). We show that these factors operate independently. Raf1 overexpression alone increases caffeine resistance without affecting Epe1 levels. HRH also responds to other stresses. High temperature deactivates TORC2, lowers Raf1 and impairs heterochromatin propagation. Further stress-responsive pathways may also signal to HRH to protect against environmental stressors.
Given the emerging role of epigenetic mechanisms in phenotypic plasticity across diverse systems58, our discovery that HRH governs stimulus-responsive heterochromatin self-propagation has important implications. Raf1 and ClrC are present in pathogenic fungi and may drive antifungal drug resistance by altering heterochromatin heritability. H3K14ub, a key HRH mark, is conserved in mammalian heterochromatin37, which undergoes pronounced H3K9me3 changes during development and in response to stress or oncogenic transformation3,59. Similar pathways probably operate in other species, including plants, in which ubiquitin ligases enable adaptation to temperature extremes, drought and pathogens60.
Methods
Strains and media
Standard methods were used for the culture, sporulation, genetic crossing and manipulation of S. pombe. Strains were generated through transformations or genetic crosses followed by tetrad dissection. Strains used in this study are listed in Supplementary Table 2. Gene deletion strains were obtained from Bioneer haploid deletion library version 4.0, except for tor1 and gad8, which were deleted using pFA6a-natMX6 construct. Most epitope-tagged strains were generated through PCR module-based methods. GFP or GFP-raf1 was cloned in the pDUAL-Pnmt1 or pUC119-Padh1 vector using In-Fusion cloning. Pombe Minimal Glutamate medium supplemented with adenine, uracil, leucine, histidine and lysine (PMG-5S) was used to overexpress protein under the nmt1 promoter. All other experiments were performed in yeast extract-rich medium supplemented with adenine (YEA) at 30 °C unless otherwise specified. For western blotting analysis, the tor2-ts6 mutant was cultured at 26 °C overnight and then transferred to 30 °C for 16 h. For experiments with antifungal agents, cells were treated with 16 mM caffeine, 0.4 mM fluconazole or 0.3 μM clotrimazole in YEA medium.
Genetic screen
Genetic screens were conducted using a heterothallic strain (mat1M-smt0) harbouring the raf1R576H mutation and a deletion of the local silencer (REIIΔ) adjacent to the mat2P cassette (Fig. 1a). In addition, the strain contained a ura4+ reporter downstream of mat2P (mat2P::ura4+). Exponentially growing cells were treated with the chemical mutagen methylnitronitrosoguanidine at a concentration of 0.5 mg ml−1 for 60 min at room temperature or irradiated with ultraviolet light (UV) at 100 J m−2 using UV crosslinker. Colonies formed on YEA medium were replica plated onto PMG-5S medium and assayed for haploid meiosis by means of iodine staining. Light colonies were restreaked to single colonies and assessed for growth on −Ura and FOA media. Suppressor mutants were subjected to whole-genome sequencing using the Illumina NextSeq500 platform. Genomic sequencing reads were quality trimmed using fastp61 and aligned to the S. pombe ASM294v2.30 reference sequence62 with the BWA aligner63 using default parameters. Duplicate reads in the resulting BAM files were marked using Picard tools (http://broadinstitute.github.io/picard/) ‘MarkDuplicates’. Mutations were called from the duplicate-marked BAM files using samtools ‘mpileup’ and subsequently processed with bcftools64 to generate a single VCF file65 containing mutations identified in the WT and mutant genomes. Mutation impacts were predicted using SnpEff66. Variations that were present in the mutants but absent in the WT controls and were predicted to have ‘HIGH’ or ‘MODERATE’ impact were flagged for further investigation.
Serial dilution assay for heterochromatic silencing
To assess the silencing of the ura4+ reporter in mat1M-smt0 REIIΔ mat2P::ura4+ cells, tenfold serial dilutions of cultures were spotted on uracil-deficient medium and PMG-5S medium supplemented with FOA. The derepression of mat2P was evaluated by exposing the cells to iodine vapour. Cells undergoing haploid meiosis on PMG-5S medium, which results from mat2P derepression, accumulate a starch-like compound and stain dark brown when exposed to iodine vapour. Images of serial dilution plates were presented with Adobe Photoshop v.22.4.2 and Adobe Illustrator v.2024.
RT–qPCR
Total RNA was extracted from 2 × 108 cells using the MasterPure Yeast RNA Purification Kit (Biosearch Technologies) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized from 1 μg of DNase-treated RNA using SuperScript III reverse transcriptase with gene-specific primers. The resulting cDNA was subjected to quantitative PCR (qPCR) using iTaq Universal SYBR Green Supermix (Bio-Rad), following the manufacturer’s instructions. The act1+ gene served as the internal control for normalization. Oligonucleotides used for qPCR are listed in Supplementary Table 3.
ChIP–qPCR
For the H3K9me3 ChIP assay, 5 × 108 cells were crosslinked with 3% paraformaldehyde (PFA) for 30 min at room temperature, followed by quenching with 125 mM glycine. For Swi6 or Raf2 or H3K14ub ChIPs, 1 × 109 of cells were incubated for 2 h at 18 °C, fixed in 3% PFA and washed in ice-cold PBS. Cells were treated with 10 mM dimethyl adipimidate at room temperature for 45 min.
Fixed cells were resuspended in ChIP lysis buffer (50 mM HEPES/KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate) supplemented with 1 mM PMSF and protease inhibitors (cOmplete Mini Protease Inhibitor Cocktail, Roche) and lysed by bead-beating (Biospec Mini-Beadbeater-16). For H3K14ub, ChIP lysis buffer was also supplemented with DUB inhibitor 100 μM PR-619 and 100 mM N-ethylmaleimide. The lysates were sonicated with a Diagenode Bioruptor for 14 cycles on medium power setting (30 s ON, 30 s OFF) to shear DNA into 0.4–0.6 kilobase (kb) fragments. Cellular debris was removed by centrifugation and 2% of the supernatant was kept for whole cell extract input control. The remaining lysates were precleared with protein A and protein G agarose beads for 1 h at 4 °C. After bead removal, lysates were incubated with the appropriate antibody (2–10 μg) overnight at 4 °C with slow rotation. Antibodies used include anti-H3K9me3 (Abcam), anti-c-Myc (Santa Cruz), anti-H3K14ub, as described in ref. 37, and custom affinity-purified anti-Swi6. Prewashed protein A and protein G agarose beads were added to the lysates and incubated for 4 h at 4 °C. The beads were sequentially washed, twice with wash buffer I (50 mM HEPES/KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate), twice with wash buffer II (50 mM HEPES/KOH pH 7.5, 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate), twice with wash buffer III (10 mM Tris/HCl pH 8.0, 250 mM LiCl, 0.5% IGEPAL, 0.5% sodium deoxycholate, 1 mM EDTA) and once with TE buffer (50 mM Tris/HCl pH 8.0, 10 mM EDTA). The chromatin was eluted from the beads twice with 50 μl of elution buffer (50 mM Tris/HCl pH 8.0, 10 mM EDTA, 1% SDS) at 65 °C for 30 min each, with shaking (roughly 1,200 rpm). Eluted chromatin and input control samples were decrosslinked overnight at 65 °C. Samples were treated sequentially with RNase A (10 μg, 2 h, 37 °C) and Proteinase K (20 μg, 2 h, 37 °C), and subsequently purified using the Qiagen PCR purification kit. qPCR was conducted with iTaq Universal SYBR Green Supermix (Bio-Rad) according to the manufacturer’s instructions. Fold enrichment was calculated using the ΔΔCt method, with leu1+ serving as the reference. Oligonucleotide sequences for qPCR are provided in Supplementary Table 3.
ChIP–seq and data processing
Genome-wide analysis of ChIP DNA was conducted as previously described in ref. 9. One ng of DNA was used for library preparation using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs) following the manufacturer’s instructions. Libraries were purified with AMPure XP magnetic beads (Beckman Coulter), and their quality was analysed on the TapeStation System 4150 (Agilent) before sequencing using the Illumina MiSeq sequencing platform. Sequenced reads were quality trimmed with fastp61 and aligned with the BWA aligner63 to the S. pombe ASM294v2.30 reference sequence62 to which we added a separate contig containing the sequence of the full 40-kb mat region. Both the Pombase provided mat region on chromosome 2 (chr. II:2109748–2138781) and the mat1M locus on the 40-kb mat contig (MAT 4,489–5,615) were masked from alignment. ChIP–seq alignments for strains lacking the REII element in the mat region were made to the standard genome as described but with the sequence of the REII element removed. Bedgraphs of ChIP enrichment over input were produced using the MACS2 (ref. 67) ‘callpeaks’ function to make broad calls with options ‘-nomodel–extsize 147’, followed by the MACS2 ‘bdgcmp’ function to compute fold enrichment over the input background. As the sequence of the constitutively euchromatic mat1 locus is identical to that of its counterpart within silent mat3, the euchromatic copy of mat1 was masked when aligning reads to prevent read misalignment. The Integrative Genomics Viewer68 was used to plot ChIP–seq data.
RNA-seq analysis
Transcriptome analysis was performed as previously described in ref. 30. Briefly, total RNA was isolated using a MasterPure Yeast RNA Purification Kit (Lucigen) and then ribosomal RNA (rRNA) was removed using a Ribo-Zero magnetic gold rRNA removal kit (yeast; Illumina). The library was constructed using a ScriptSeq v.2 RNA-seq library preparation kit (Illumina) or NEBNext Ultra II directional RNA library prep kit for Illumina (NEB). The final library was analysed using an Agilent 2100 BioAnalyzer and sequenced on the Illumina NextSeq500 platform. Single ended short reads from RNA-seq experiments were quality trimmed using fastp61 and aligned using the STAR aligner69. Variable interval bedgraphs, normalized to counts per million mapped reads and including both uniquely and multi-mapping reads, were generated by STAR and were further processed to produce 10-base pair fixed interval versions for purposes of figure construction. Read counts for transcripts were computed from BAMs using Rsubread70 and differential expression with respect to WT controls was computed using DEGseq71. The Volcano plot was constructed from log2 fold-changes and P values produced by DEGseq using the ‘EnhancedVolcano’ R library (https://github.com/kevinblighe/EnhancedVolcano).
Northern blotting
Northern blot analysis of centromeric small interfering RNAs (siRNAs) was performed as described previously in ref. 72. Briefly, small RNAs (less than 200 nt) were purified from mid-log phase cells with mirVana microRNA isolation kit (Thermo Fisher Scientific). Then 20 μg of small RNAs were resolved on a 15% denaturing acrylamide gel and transferred to Hybond-N+ (Thermo Fisher Scientific) membrane in 0.5× TBE for 1 h at 100 V. After UV crosslinking, the membrane was hybridized with α-P32-UTP (PerkinElmer) labelled RNA probes (roughly 50 nucleotides) corresponding to the dg sequence in ULTRAhyb-Oligo hybridization buffer (Thermo Fisher Scientific). The membrane was exposed and scanned using Typhoon FLA 9500 phosphor imager (GE Healthcare).
For northern blot analysis of raf1 transcript, exponentially growing cells were resuspended in LETS buffer (100 mM LiCl, 10 mM EDTA, 10 mM Tris/HCl pH 7.5, 0.2% SDS), combined with equal volume of LETS-saturated phenol–chloroform and lysed by bead-beating. Total RNA was purified through repeated extraction with LETS-saturated phenol–chloroform and collected by precipitation after the addition of LiCl to a final concentration of 0.5 M and 2.5 volumes of 100% ethanol. The RNA was resuspended in NorthernMax formaldehyde loading dye (Invitrogen), resolved on a 1% agarose gel and transferred onto a BrightStar-Plus (Invitrogen) membrane by following the NorthernMax kit (Invitrogen) protocol. Blotted RNA was detected using α-P32-UTP (PerkinElmer) labelled RNA probe. RNA probe was prepared by in vitro transcription with the Maxiscript T7 kit (Invitrogen). Probe was hybridized to blotted RNAs in ULTRAhyb hybridization buffer (Invitrogen) and the blots were visualized using the Typhoon FLA 9500 phosphor imager (GE Healthcare).
RIP–qPCR
For the Myc-tagged Upf1 RIP assay, 1 × 109 cells were incubated for 2 h at 18 °C, fixed in 3% PFA and washed in ice-cold PBS. Cells were treated with 10 mM Dimethyl adipimidate at room temperature for 45 min. Fixed cells were resuspended in ChIP lysis buffer (50 mM HEPES/KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate) supplemented with 1 mM PMSF, complete EDTA-free proteinase inhibitor cocktail (Roche) and 40 units of RNase inhibitor (Thermo Fisher Scientific, AM2694) and lysed by bead-beating (Biospec Mini-Beadbeater-16). The lysates were sonicated with a Diagenode Bioruptor for 14 cycles on medium power setting (30 s ON, 30 s OFF). Cellular debris was removed by centrifugation, and 5% of the supernatant was reserved for whole cell extract input control. The remaining lysates were precleared with 0.9 mg of prewashed protein G Dynabeads (Invitrogen, 10004D) at 4 °C for 1 h. After bead removal, lysates were incubated with 5 μg of anti-c-Myc antibody (Santa Cruz, 9E10) overnight at 4 °C with slow rotation. Antibody–protein complexes were captured using 1.2 mg of protein G Dynabeads for 2 h at 4 °C. The beads were sequentially washed in 900 μl of each buffer, once with wash buffer I (50 mM HEPES/KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate), once with wash buffer II (50 mM HEPES/KOH pH 7.5, 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate), once with wash buffer III (10 mM Tris/HCl pH 8.0, 250 mM LiCl, 0.5% IGEPAL, 0.5% sodium deoxycholate, 1 mM EDTA) and once with TE buffer (50 mM Tris/HCl pH 7.0, 10 mM EDTA). Each wash was done for 7 min at slow rotation. Beads were eluted twice in 75 μl of RIP elution buffer (50 mM Tris/HCl pH 8, 10 mM EDTA, 300 mM NaCl, 1% SDS) at 37 °C for 10 min. To 50-μl input samples, 100 μl of RIP elution buffer was added to make a final volume of 150 μl. Then 20 μg of proteinase K (Thermo Fisher Scientific, AM2548) was added to both immunoprecipitation and input samples, and the mixtures were incubated at 37 °C for 1 h followed by de-crosslinking at 65 °C for 1 h. The samples were then extracted with phenol–chloroform, precipitated with ethanol and the pellet was resuspended in 80 μl of DEPC-treated water. The samples were further treated with 20 units of RNase-free DNase I (Thermo Fisher Scientific, AM2222) at 37 °C for 1 h, extracted with phenol–chloroform and ethanol precipitated as above. Immunoprecipitation and input samples were resuspended in 20 µl and 50 µl water, respectively. RNA was used for performing RT–qPCR as mentioned above. Fold enrichment was calculated using the ΔΔCt method, with act1+ serving as the reference. Oligonucleotide sequences for qPCR are provided in Supplementary Table 3.
Western blotting
The 4 × 107 mid-log phase S. pombe cells were lysed in 20% trichloroacetic acid by bead-beating (Biospec Mini-Beadbeater-16). The precipitated proteins were washed with ethanol. For Flag-Clr4, cells were resuspended in 150 μl TBS (50 mM Tris/HCl pH 7.5, 150 mM NaCl), heated for 5 min at 95 °C and lysed by bead-beating. Lysate was resuspended in SDS–PAGE sample buffer, and resolved using polyacrylamide gel electrophoresis. Anti-c-Myc 9E10 (Santa Cruz, sc-40), anti-Flag M2 (Sigma Aldrich, F3165) and mouse IgG HRP-linked (GE Healthcare, NA931) antibodies were used for probing the epitope-tagged proteins. For endogenous proteins, affinity-purified anti-Swi6 (in-house), antibody specific for Gad8 phosphorylated at Ser546 (gift from R. Weisman) and Rabbit IgG HRP-linked (GE Healthcare, NA934) antibodies were used. Primary antibody dilution was 1:1,000 and secondary antibody dilution was 1:2,500.
Glycerol-gradient fractionation
The 1 × 109 S. pombe cells grown to mid-log phase were pelleted and flash frozen in liquid nitrogen. The frozen cells were lysed using a CryoMill (Retsch) with the parameters: three cycles at 30 Hz for 1 min followed by 5 Hz for 1 min, and the resulting cell powder was resuspended in lysis buffer (20 mM Tris/HCl, pH 8.0; 2 mM EDTA; 1% IGEPAL; 2 mM β-mercaptoethanol; 137 mM NaCl and 2× complete proteinase inhibitor). The lysate was centrifuged at 27,000g at 4 °C for 30 min, and protein concentrations of the supernatant were measured using the Bradford assay. A discontinuous glycerol gradient (1.8 ml) was prepared by layering glycerol solutions with decreasing concentrations (200 μl each of 50%, 45%, 40%, 35%, 30%, 25%, 20%, 15% and 10% glycerol) in lysis buffer within a polyallomer ultracentrifuge tube (Beckman Coulter). The gradient was allowed to form at room temperature for 2 h, followed by cooling at 4 °C for an extra 2 h. Subsequently, 120 μl of whole cell extract was loaded atop the gradient. The tubes were inserted into precooled buckets and centrifuged at 35,000 rpm in a Beckman TLS-55 rotor at 4 °C for 19 h. Fractions (150 μl) were collected starting from the top of the gradient, and 10 μl from each fraction was resolved on a 4–12% SDS–PAGE Bis-Tris gel, followed by western blotting to detect specific proteins.
Immunoprecipitation analysis
A 2-l culture of S. pombe was harvested at an optical density at 600 nm (OD600) of 1. Cells were washed with ice-cold distilled water and resuspended in 2 ml of lysis buffer (300 mM HEPES/KOH pH 7.6; 100 mM KCl; 2 mM EDTA; 2 mM PMSF; 1 mM DTT (dithiothreitol); 0.2% IGEPAL; Roche complete mini protease inhibitor cocktail) and flash frozen in liquid nitrogen as nuggets. The frozen cell nuggets were then lysed using a CryoMill (Retsch) with the following parameters: nine cycles at 30 Hz for 1 min followed by 5 Hz for 1 min. The resulting cell powder was resuspended in 10 ml of lysis buffer. The lysate was centrifuged at 4,000 rpm for 10 min, and the supernatant was further clarified by ultracentrifugation at 27,000g for 1 h at 4 °C. Two hundred microlitres of M2-Flag beads (Sigma Aldrich, A2220) were washed three times with wash buffer (150 mM HEPES/KOH pH 7.6, 250 mM KCl, 1 mM EDTA, 1 mM PMSF, 0.5 mM DTT, 0.1% IGEPAL) and subsequently added to the clarified lysate. After 2 h of incubation at 4 °C, beads were washed twice with wash buffer, followed by two washes with AC buffer (20 mM HEPES/KOH, pH 7.6, 200 mM KCl, 1 mM EDTA, 2 mM MgCl2, 0.5 mM DTT, 0.1% IGEPAL). Beads were resuspended in 200 μl of AC buffer containing 500 μg ml−1 Flag peptide for elution of bound protein. After overnight incubation at 4 °C, proteins in the eluate were precipitated by adding 10% trichloroacetic acid on ice for 10 min. The protein pellet was washed with acetone, air-dried, resuspended in 200 μl of SDS–PAGE sample buffer, boiled at 95 °C for 5 min and analysed by western blotting.
Ectopic heterochromatin silencing assay
The reporter strains contain 6xtetO-ade6+ inserted at the ura4+ locus and Pnmt81-tetR-2xflag-clr4+ integrated at the leu1+ locus. Strains were grown in Edinburgh Minimal Medium (EMM) liquid media for 48 h to induce TetR-Clr4 expression. Expression and heterochromatization of the ade6+ reporter were assayed by serial dilution and ChIP–qPCR analyses, respectively. Tenfold serial dilutions were spotted on low-adenine (7.5 mg l−1) EMM medium lacking thiamine (tetR-clr4ON) or supplemented with 5 mg l−1 thiamine (tetR-clr4OFF), and ade6+ expression status was determined by observing colony coloration. White colonies indicate ade6+ expression, whereas ade6+-repressed cells form red colonies. H3K9me was analysed in cells grown for 48 h in EMM and collected at OD600 of 0.5 (establishment, tetR-clr4ON cells). For heritability of H3K9me, cells were further cultivated in liquid EMM supplemented with tetracycline (2.5 mg l−1) and thiamine (5 mg l−1) for 10 generations and fixed with 3% PFA at OD600 of 0.5. Fixed cells were processed for ChIP as described earlier. Oligonucleotides used for qPCR are listed in Supplementary Table 3.
Live-cell imaging
Cells were grown overnight at 26 °C in PMG-5S medium until they reached logarithmic phase. For Halo tagged proteins, cells were stained with 25 nM Janelia Fluor 646 HaloTag ligand (Promega) for 1 h. For mat2P::GFP imaging, YEA medium was used. Cells were mounted on a 2% agarose pad formed on a glass slide. Images were acquired on a DeltaVision Elite microscope (Leica) with a ×100, 1.35 numerical aperture oil lens (Olympus). Optical z sections were acquired (0.2-μm step size, 20 sections) for each field. Images were deconvolved and all z-stacks were projected into a single-plane as maximum-intensity projections. Fiji (ImageJ2 v.1.53, National Institutes of Health (NIH)) was used for processing the images.
Image acquisition for SMT
Cells were grown in PMG-5S medium to mid-log phase and stained with 100 pM of Janelia Fluor JFX650 HaloTag ligand (JFX650 Promega) for 1 h at 30 °C. Cells were then briefly washed twice, resuspended in fresh media, mounted on a chambered coverglass (Lab Tek II) and covered by a 1% agarose patch. Imaging was performed on custom-built HiLO microscope73. This custom-built microscope from the CCR, LRBGE Optical Microscopy Core facility is controlled by μManager v.2.0 software (Open Imaging Inc.), equipped with an Okolab state top incubator for temperature control (30 °C) with an objective heater, a ×100, 1.49 numerical aperture objective (Olympus Scientific Solutions), 647-nm and 488-nm lasers (Coherent OBIS), and EM-CCD cameras (Evolve 512 Delta, Photometrics). Images for GFP and JFX650 (for HaloTag) were acquired simultaneously with an exposure time of 100 ms, and a time-lapse interval of 200 ms for a total of 2 min, with laser powers of 3 mW for the 647-nm laser and 100 mW for the 488-nm laser.
SMT analysis
The custom-made software TrackRecord74 in MATLAB (v.R2024a, The MathWorks Inc.) was used for analysis (https://sourceforge.net/projects/single-molecule-tracking/). Briefly, to analyse each time series, data were filtered using top-hat, Wiener and Gaussian filters. A region of interest was defined to encompass the nuclei based on the nuclear GFP staining, then nuclear particles were detected, fitted to two-dimensional Gaussian function for subpixel localization and finally tracked using a nearest neighbour algorithm75. The tracking parameters were as follows: window size for particle detection 3 pixels, maximum frame-to frame displacement of 6 pixels, shortest track 2 frames and gaps to close 2. Diffusion coefficients of individual tracks were estimated by calculating the mean-squared displacement over time and fitting the first four points in the curve to a line, the slope of which gives the average diffusion coefficient. Only those fits whose confidence intervals included only positive numbers were retained for the analysis.
Track segments were classified into distinct diffusive states using pEM (v.2)76. Assumptions about the type of diffusion shown by the tracked particles are not required with pEM and the number of diffusive states can be deduced from the analysis. pEM analysis requires all analysed tracks to be of the same length, and it is preferable to use shorter tracks that do not include transitions between states. Therefore, tracks were split into seven frame segments and the pEM classification analysis was performed on the set of all these track segments. The minimum number of states for the system to converge to was set at two and the maximum at seven. If the optimal number of states that the analysis converged to was seven, the algorithm was rerun with a higher number of maximum states. The number of reinitializations was set to 50 with 200 perturbation trials. The maximum number of iterations was 1,000 with a convergence criterion for the change of log likelihood of 10−7. The number of features for the covariance matrix was set to three for tracks of length seven. A motion blur coefficient was calculated as (1/6)(Δe/Δt), where Δe corresponds to the exposure time and Δt the acquisition interval.
Survival distributions were calculated from the immobile portions of particle tracks as in ref. 77. Immobile segments of tracks were identified on the basis of their displacements compared with HaloTag-H2B data, which was acquired with identical imaging conditions to the Raf2. Because the motion of H2B includes the thermal motion of chromatin, it can be used to determine the maximum distance a DNA-bound molecule will be allowed to move. Therefore, we used the jump distribution of H2B tracks to define three parameters: Rmin, Rmax and Nmin. Rmin is the maximum distance for which histone molecules can move between two consecutive frames, calculated as the 99th percentile for H2B frame-to-frame displacements. Rmax is the maximum distance for which bound histone molecule may move in Nmin frames, calculated as the 99th percentile of displacements of histone molecules after Nmin frames. Nmin is the minimum frame number of a shortest bound track, and is calculated to obtain a low probability, P = 0.01, that a diffusing molecule is classified as bound using the following equation:
The photobleaching rate was estimated as the slowest exponential decay obtained from fitting the H2B survival distribution to a triple exponential decay. The survival distribution of the Raf2 was corrected for photobleaching as follows (\(S(t)={\rm{SE}}(t)/\gamma (t)\), where S(t) corresponds to the survival distribution after photobleaching correction, SE(t) the empirical survival distribution and γ(t) is the normalized photobleaching decay).
In vitro affinity pulldown
Recombinant His6-Clr4, glutathione S-transferase (GST) and GST-Raf1 (amino acids (aa) 211–638) were expressed and purified from Escherichia coli (BL21). His6-Clr4 was purified by Ni-bead affinity pulldown followed by high-performance liquid chromatography size fractionation (GenScript). GST and GST-Raf1 (aa 211–638) were purified by affinity pulldown using Glutathione Sepharose 4B resin (Cytiva, 17075601). Purified His6-Clr4 was added to GST or GST-Raf1 (aa 211–638) bound Glutathione Sepharose beads in affinity binding buffer (20 mM Tris/HCl pH 7.5, 100 mM NaCl, 1 mM EDTA, 0.05% NP-40, 2 mM DTT and complete protease inhibitors) at a protein ratio of 4:1. After 60 min of incubation with gentle rotation at 4 °C, beads were pulled down by a short spin at 500g. Beads were washed twice with 500 µl of affinity binding buffer and resuspended in SDS sample buffer. Bound proteins were analysed by SDS–PAGE and western blotting for Clr4 with an affinity-purified rabbit α-Clr4 antibody (dilution 1:1,000) and Alexa-Fluor 647 labelled secondary antibody (Invitrogen, A21246) (dilution 1:2,500). Fluorescence signals were captured by scanning using Typhoon FLA 9500 (GE Healthcare).
Quantification and statistical analysis
Quantification and statistical tests used are described in the figure legends. The n represents the number of independent biological replicates. GraphPad Prism v.10 software was used to plot all the graphs and calculate statistical significance.
Materials availability
Strains and plasmids generated in this study are available from the corresponding author upon reasonable request.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
ChIP–seq and RNA-seq datasets are deposited in the Gene Expression Omnibus with accession numbers GSE280646 and GSE280607. Source data are provided with this paper.
Code availability
The software used to analyse the data is listed in the Methods and is publicly available. Picard tools is available through GitHub at http://broadinstitute.github.io/picard/; the EnhancedVolcano R library is available through GitHub at https://github.com/kevinblighe/EnhancedVolcano and the software TrackRecord74 in MATLAB is available through sourceforge at https://sourceforge.net/projects/single-molecule-tracking/.
References
Grewal, S. I. S. The molecular basis of heterochromatin assembly and epigenetic inheritance. Mol. Cell 83, 1767–1785 (2023).
Fitz-James, M. H. & Cavalli, G. Molecular mechanisms of transgenerational epigenetic inheritance. Nat. Rev. Genet. 23, 325–341 (2022).
Nicetto, D. & Zaret, K. S. Role of H3K9me3 heterochromatin in cell identity establishment and maintenance. Curr. Opin. Genet. Dev. 55, 1–10 (2019).
Chang, Z., Yadav, V., Lee, S. C. & Heitman, J. Epigenetic mechanisms of drug resistance in fungi. Fungal Genet. Biol. 132, 103253 (2019).
Voss, T. S., Bozdech, Z. & Bartfai, R. Epigenetic memory takes center stage in the survival strategy of malaria parasites. Curr. Opin. Microbiol. 20, 88–95 (2014).
Wang, J., Jia, S. T. & Jia, S. New insights into the regulation of heterochromatin. Trends Genet. 32, 284–294 (2016).
Cutter DiPiazza, A. R. et al. Spreading and epigenetic inheritance of heterochromatin require a critical density of histone H3 lysine 9 tri-methylation. Proc. Natl Acad. Sci. USA 118, e2100699118 (2021).
Zhang, K., Mosch, K., Fischle, W. & Grewal, S. I. S. Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat. Struct. Mol. Biol. 15, 381–388 (2008).
Zofall, M., Sandhu, R., Holla, S., Wheeler, D. & Grewal, S. I. S. Histone deacetylation primes self-propagation of heterochromatin domains to promote epigenetic inheritance. Nat. Struct. Mol. Biol. 29, 898–909 (2022).
Horn, P. J., Bastie, J. N. & Peterson, C. L. A Rik1-associated, cullin-dependent E3 ubiquitin ligase is essential for heterochromatin formation. Genes Dev. 19, 1705–1714 (2005).
Jia, S., Kobayashi, R. & Grewal, S. I. S. Ubiquitin ligase component Cul4 associates with Clr4 histone methyltransferase to assemble heterochromatin. Nat. Cell Biol. 7, 1007–1013 (2005).
Hong, E. J., Villen, J., Gerace, E. L., Gygi, S. P. & Moazed, D. A cullin E3 ubiquitin ligase complex associates with Rik1 and the Clr4 histone H3-K9 methyltransferase and is required for RNAi-mediated heterochromatin formation. RNA Biol. 2, 106–111 (2005).
Gallagher, P. S. et al. Iron homeostasis regulates facultative heterochromatin assembly in adaptive genome control. Nat. Struct. Mol. Biol. 25, 372–383 (2018).
Torres-Garcia, S. et al. Epigenetic gene silencing by heterochromatin primes fungal resistance. Nature 585, 453–458 (2020).
Nakayama, J., Rice, J. C., Strahl, B. D., Allis, C. D. & Grewal, S. I. S. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292, 110–113 (2001).
Rea, S. et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599 (2000).
Cam, H. P. et al. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat. Genet. 37, 809–819 (2005).
Hall, I. M. et al. Establishment and maintenance of a heterochromatin domain. Science 297, 2232–2237 (2002).
Aygun, O., Mehta, S. & Grewal, S. I. S. HDAC-mediated suppression of histone turnover promotes epigenetic stability of heterochromatin. Nat. Struct. Mol. Biol. 20, 547–554 (2013).
Sahu, R. K. et al. Nucleosome remodeler exclusion by histone deacetylation enforces heterochromatic silencing and epigenetic inheritance. Mol. Cell 84, 3175–3191 (2024).
Li, F. et al. Two novel proteins, Dos1 and Dos2, interact with Rik1 to regulate heterochromatic RNA interference and histone modification. Curr. Biol. 15, 1448–1457 (2005).
Thon, G. et al. The Clr7 and Clr8 directionality factors and the Pcu4 cullin mediate heterochromatin formation in the fission yeast Schizosaccharomyces pombe. Genetics 171, 1583–1595 (2005).
Higa, L. A. et al. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol. 8, 1277–1283 (2006).
Angers, S. et al. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443, 590–593 (2006).
Buscaino, A. et al. Raf1 Is a DCAF for the Rik1 DDB1-like protein and has separable roles in siRNA generation and chromatin modification. PLoS Genet. 8, e1002499 (2012).
Holla, S. et al. Positioning heterochromatin at the nuclear periphery suppresses histone turnover to promote epigenetic inheritance. Cell 180, 150–164 (2020).
Kurosaki, T., Popp, M. W. & Maquat, L. E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 20, 406–420 (2019).
Karam, R., Wengrod, J., Gardner, L. B. & Wilkinson, M. F. Regulation of nonsense-mediated mRNA decay: implications for physiology and disease. Biochim. Biophys. Acta 1829, 624–633 (2013).
Matia-Gonzalez, A. M., Hasan, A., Moe, G. H., Mata, J. & Rodriguez-Gabriel, M. A. Functional characterization of Upf1 targets in Schizosaccharomyces pombe. RNA Biol. 10, 1057–1065 (2013).
Lee, N. N. et al. Mtr4-like protein coordinates nuclear RNA processing for heterochromatin assembly and for telomere maintenance. Cell 155, 1061–1074 (2013).
Aznarez, I. et al. Mechanism of nonsense-mediated mRNA decay stimulation by splicing factor SRSF1. Cell Rep. 23, 2186–2198 (2018).
Wen, J. & Brogna, S. Splicing-dependent NMD does not require the EJC in Schizosaccharomyces pombe. EMBO J. 29, 1537–1551 (2010).
Kuscu, C. et al. CRL4-like Clr4 complex in Schizosaccharomyces pombe depends on an exposed surface of Dos1 for heterochromatin silencing. Proc. Natl Acad. Sci. USA 111, 1795–1800 (2014).
Oya, E. et al. H3K14 ubiquitylation promotes H3K9 methylation for heterochromatin assembly. EMBO Rep. 20, e48111 (2019).
Stirpe, A. et al. SUV39 SET domains mediate crosstalk of heterochromatic histone marks. eLife 10, 62682 (2021).
Du, Y. et al. Mechanistic insights into the stimulation of the histone H3K9 methyltransferase Clr4 by proximal H3K14 ubiquitination. Sci. Adv. 11, eadu1864 (2025).
Huang, Y. et al. A conserved H3K14ub-driven H3K9me3 for chromatin compartmentalization. Nature 647, 786–797 (2025).
Kim, H. S. et al. Clr4(SUV39H1) ubiquitination and non-coding RNA mediate transcriptional silencing of heterochromatin via Swi6 phase separation. Nat. Commun. 15, 9384 (2024).
Zofall, M. & Grewal, S. I. S. Swi6/HP1 recruits a JmjC domain protein to facilitate transcription of heterochromatic repeats. Mol. Cell 22, 681–692 (2006).
Braun, S. et al. The Cul4-Ddb1(Cdt)(2) ubiquitin ligase inhibits invasion of a boundary-associated antisilencing factor into heterochromatin. Cell 144, 41–54 (2011).
Yaseen, I. et al. Proteasome-dependent truncation of the negative heterochromatin regulator Epe1 mediates antifungal resistance. Nat. Struct. Mol. Biol. 29, 745–758 (2022).
Murawska, M. et al. The histone chaperone FACT facilitates heterochromatin spreading by regulating histone turnover and H3K9 methylation states. Cell Rep. 37, 109944 (2021).
Taneja, N. et al. SNF2 family protein Fft3 suppresses nucleosome turnover to promote epigenetic inheritance and proper replication. Mol. Cell 66, 50–62 (2017).
Audergon, P. N. et al. Epigenetics. Restricted epigenetic inheritance of H3K9 methylation. Science 348, 132–135 (2015).
Ragunathan, K., Jih, G. & Moazed, D. Epigenetics. Epigenetic inheritance uncoupled from sequence-specific recruitment. Science 348, 1258699 (2015).
Lentini, L., Melfi, R., Cancemi, P., Pibiri, I. & Di Leonardo, A. Caffeine boosts Ataluren’s readthrough activity. Heliyon 5, e01963 (2019).
Yamashita, A., Ohnishi, T., Kashima, I., Taya, Y. & Ohno, S. Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev. 15, 2215–2228 (2001).
Berry, S. & Dean, C. Environmental perception and epigenetic memory: mechanistic insight through FLC. Plant J. 83, 133–148 (2015).
Gowen, J. W. & Gay, E. H. Effect of temperature on eversporting eye color in Drosophila melanogaster. Science 77, 312 (1933).
Rizzi, N. et al. Transcriptional activation of a constitutive heterochromatic domain of the human genome in response to heat shock. Mol. Biol. Cell 15, 543–551 (2004).
Torres, J. R. & Sanchez, D. H. Emerging roles of plant transcriptional gene silencing under heat. Plant J. 119, 1197–1209 (2024).
Otsubo, Y. & Yamamato, M. TOR signaling in fission yeast. Crit. Rev. Biochem. Mol. Biol. 43, 277–283 (2008).
Wei, Y. et al. TOR targets an RNA processing network to regulate facultative heterochromatin, developmental gene expression and cell proliferation. Nat. Cell Biol. 23, 243–256 (2021).
Cohen, A., Kupiec, M. & Weisman, R. Glucose activates TORC2-Gad8 protein via positive regulation of the cAMP/cAMP-dependent protein kinase A (PKA) pathway and negative regulation of the Pmk1 protein-mitogen-activated protein kinase pathway. J. Biol. Chem. 289, 21727–21737 (2014).
Matsuo, T., Kubo, Y., Watanabe, Y. & Yamamoto, M. Schizosaccharomyces pombe AGC family kinase Gad8p forms a conserved signaling module with TOR and PDK1-like kinases. EMBO J. 22, 3073–3083 (2003).
Cohen, A. et al. TOR complex 2 in fission yeast is required for chromatin-mediated gene silencing and assembly of heterochromatic domains at subtelomeres. J. Biol. Chem. 293, 8138–8150 (2018).
Roux, P. P. & Topisirovic, I. Signaling pathways involved in the regulation of mRNA translation. Mol. Cell. Biol. 38, e00070-18 (2018).
Feinberg, A. P. Phenotypic plasticity and the epigenetics of human disease. Nature 447, 433–440 (2007).
Hunter, R. G. et al. Acute stress and hippocampal histone H3 lysine 9 trimethylation, a retrotransposon silencing response. Proc. Natl Acad. Sci. USA 109, 17657–17662 (2012).
Miricescu, A., Goslin, K. & Graciet, E. Ubiquitylation in plants: signaling hub for the integration of environmental signals. J. Exp. Bot. 69, 4511–4527 (2018).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Wood, V. et al. The genome sequence of Schizosaccharomyces pombe. Nature 415, 871–880 (2002).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Thorvaldsdottir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 14, 178–192 (2013).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47 (2019).
Wang, L., Feng, Z., Wang, X., Wang, X. & Zhang, X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 (2010).
Sugiyama, T. et al. SHREC, an effector complex for heterochromatic transcriptional silencing. Cell 128, 491–504 (2007).
Mehta, G. D. et al. Single-molecule analysis reveals linked cycles of RSC chromatin remodeling and Ace1p transcription factor binding in yeast. Mol. Cell 72, 875–887 (2018).
Mazza, D., Ganguly, S. & McNally, J. G. Monitoring dynamic binding of chromatin proteins in vivo by single-molecule tracking. Methods Mol. Biol. 1042, 117–137 (2013).
Presman, D. M. et al. Quantifying transcription factor binding dynamics at the single-molecule level in live cells. Methods 123, 76–88 (2017).
Koo, P. K. & Mochrie, S. G. Systems-level approach to uncovering diffusive states and their transitions from single-particle trajectories. Phys. Rev. E 94, 052412 (2016).
Garcia, D. A. et al. An intrinsically disordered region-mediated confinement state contributes to the dynamics and function of transcription factors. Mol. Cell 81, 1484–1498 (2021).
Acknowledgements
We are grateful to R. Weisman for the generous gift of antibody against Gad8 phosphorylated at Ser546; K. Zhang, R. Pandian, A. Anil, R. Sahu and L. Sun for helpful contributions; members of the Grewal laboratory for valuable discussions and J. Barrowman for editing the paper. This study used the computational resources of the NIH-HPC. This research was supported by the Intramural Research Program of the NIH. The contributions of the NIH authors are considered Works of the United States Government. The findings and conclusions presented in this paper are those of the authors and do not necessarily reflect the views of the NIH or the US Department of Health and Human Services.
Author information
Authors and Affiliations
Contributions
S.I.S.G., Y.W. and B.B. conceived the project and designed experiments. B.B., Y.W., A.K.P., M.Z., J.D., S.J., H.X., D.V. and H.D.F. performed experiments. D.A.B. and T.S.K. helped with SMT data acquisition and data analyses, D.W. performed bioinformatic analyses. H.Q. and J.W. generated anti-H3K14ub antibody. All authors contributed to data interpretation. B.B. and S.I.S.G. wrote the paper with input from all authors. All authors read and approved the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Ivan Topisirovic and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Mutations in components of the NMD pathway affect heterochromatin propagation.
a, Heterochromatic silencing in raf1R518H or raf1R576H mutant cells. Serial dilutions on indicated media were used to assess mat2P::ura4+ (top) or Kint2::ura4+ expression (bottom). Haploid meiosis was assessed by iodine staining. b, c, d, ChIP-seq analysis of H3K9me3 distribution across the silent mat region (b), the subtelomeric region 2 R (c) and centromere 2 (d) in the indicated strains. Data are representative of two independent experiments. Source data are provided.
Extended Data Fig. 2 NMD affects heterochromatin independently of the RNAi pathway.
a, Northern blot analysis of dg siRNA in WT and upf1Δ cells. tRNA was used as a loading control. b, Heterochromatic silencing in raf1R576H mutant cells with simultaneous loss of NMD and RNAi components. Serial dilution evaluated mat2P::ura4+ expression, and iodine staining assessed haploid meiosis. c, ChIP-seq analysis of H3K9me3 distribution across the silent mat region in the indicated strains. d, Effect of upf1Δ on KΔ::ura4+ expression. Serial dilutions were plated on indicated media to assess KΔ::ura4+ expression (top). ChIP-qPCR analysis of H3K9me3 across the mat region (bottom) (data are presented as mean ± SD; n = 3 independent experiments, P value by two-stage step-up Benjamini, Krieger, and Yekutieli unpaired t test). e, f, Volcano plot from RNA-seq analyses showing the fold change in the expression of heterochromatin-related genes in the raf1R576H over WT cells (e), and raf1R576H upf1Δ double mutant over WT cells (f) (Log2FC and two-sided P values were computed using the MARS model as implemented in DEGseq. The P value significance threshold is set to 1e-5 (-log10 (P value) = 5) to correct for multiple comparisons). g, Northern blot analysis of raf1 transcript in the indicated strains. Ribosomal RNA served as the loading control. Data are representative of two independent experiments. Source data are provided.
Extended Data Fig. 3 Splicing factor mutation mimics NMD loss in causing raf1 upregulation.
a, RNA-seq splice junctions reveal cryptic introns in raf1. Numbers indicate the percentage of spliced reads. b, Mutation in the Sap49 splicing factor or the NMD component Upf2 suppresses the heterochromatic silencing defect in raf1R576H cells. Serial dilutions assessed mat2P::ura4+ expression, and iodine staining assessed haploid meiosis (top). Domain organizations of Sap49 and Upf2 are shown (bottom). c, d, e, ChIP-seq profiles of H3K9me3 across the silent mat region (c), the subtelomeric region 1R (d), and centromere 2 (e) in the indicated strains. f, Northern blot of raf1 transcripts in the indicated strains; ribosomal RNA served as the loading control. g, h, Western blot analysis of Raf1 in the indicated strains. Cdc2 and Ponceau S served as loading controls. i, Expressing Raf1 in raf1-oe (Pnmt1-raf1) cells at levels comparable to those in upf1Δ cells is sufficient to suppress the heterochromatic silencing defect in raf1R576H cells. Serial dilutions on the indicated media were used to assess mat2P::ura4+ expression. Data are representative of two independent experiments. Source data are provided.
Extended Data Fig. 4 Raf1 overexpression enhances the association of ClrC with chromatin.
a, Co-IP of Raf2 with Clr4 upon deletion of upf1. * denotes the non-specific band. b, In-vitro (GST) pulldown of purified Clr4 with purified Raf1 (211-628aa). c, Live-cell fluorescence imaging of Raf2-Halo with Halo ligand JFX650. Scale bars, 2 µm. d, Distribution of diffusion coefficients (D, log10) from individual tracks (gray bars) of H2B in WT (top), Raf2 in control (Pnmt1-GFP) (middle) and Raf2 in raf1-oe (Pnmt1-raf1) (bottom), all labeled with HaloTag and JFX650, and fit with a mixture of two Gaussians population (red line). Dashed black lines show the two individual sub-populations. e, Survival probability curves (1-CDF [cumulative distribution function]) from apparent dwell times of single-molecule chromatin-binding events of Raf2-Halo in control and raf1-oe cells. f, g, Comparison of diffusion coefficients and confinement radii for the three diffusive states of Raf2 (f) or two diffusive states of H2B (g) as obtained by pEM analysis of SMT data (P values from two-sided Z-test comparing). h, Proportion of H2B molecular states in WT (mean ± 95% CI). i, Proportion of Raf2 molecular states in control and raf1-oe cells (mean ± 95% CI, *p = 4.7 × 10−3, **p = 5.5 × 10−4, and ***p = 2.0 × 10−12, P values from two-sided Z-test). a, b, c, Data are representative of two independent experiments. d, f, i, Data are representative of 5 independent experiments which included 310 tracks for control and 1,019 tracks raf1-oe cells. d, g, h, Data are representative of 3 independent experiments which included 547 tracks of H2B. Source data are provided.
Extended Data Fig. 5 Increase in ClrC localization to heterochromatin regions upon Raf1 overexpression.
a, b, Fluorescence live-cell imaging of Raf2-HaloTag (JFX650) in the indicated cells and quantification of the number of Raf2 foci and their intensity. Number of foci are represented with mean ± SD, n = 3 independent experiments, P value by two-stage step-up Benjamini, Krieger, and Yekutieli unpaired t test (a,b, top right). Intensity of individual foci represented with median; >100 foci are represented; n = 3 independent experiments, P value by two-tailed unpaired t test (a,b, bottom right). c, d, e, ChIP-seq analysis of myc-tagged Raf2 distribution across the subtelomeric region 1 R (c), the silent mat region (d) and the heterochromatin islands (e) upon raf1-oe (Pnmt1-raf1). Data are representative of two independent experiments. Source data are provided.
Extended Data Fig. 6 Raf1 regulates heterochromatin independent of Epe1.
a, Live-cell fluorescence imaging of Epe1-GFP in WT, raf1-oe (Pnmt1-raf1) and swi6-115 mutant cells. Note that the number and intensity of Epe1-GFP foci are similar in WT and raf1-oe cells. Epe1-GFP foci are abolished in swi6-115 mutant cells included as a control. Scale bars, 5 µm. Dashed boxes indicate zoomed-in areas in the bottom panel. b, Fluorescence imaging analysis of mat2P::GFP expression in the indicated strains. The percentage of cells expressing GFP (GFP-ON) is indicated. Scale bar, 5 µm. Haploid meiosis was assessed by iodine staining. Data are representative of two independent experiments.
Extended Data Fig. 7 Increased Raf1 abundance alleviates heterochromatin propagation defects in various mutants.
a, Heterochromatic silencing upon Raf1 overexpression (raf1-oe; Pnmt1-raf1) or loss of upf1 in hht2G13D (H3.2G13D) mutant cells. Serial dilution assessed mat2P::ura4+ expression, and iodine staining assessed haploid meiosis. b, c, ChIP-seq analysis of H3K14ub (b) and H3K9me3 (c) distribution across the silent mat region in the indicated strains. Note that the WT H3K14ub data is the same as in Fig. 4b as the experiments were performed at the same time. d, Schematic illustrating restoration of heterochromatin propagation upon Raf1 overexpression or loss of Upf1 in hht2G13D cells. Heterochromatin initiated at the nucleation site through recruitment of ClrC spreads across an extended domain through the Clr4SUV39H read-write activity, requiring a critical density of H3K9me3 as well as H3K14ub (top). Incorporation of hht2G13D causes reduction not only in H3K9me3 density, but also H3K14ub, thus disrupting heterochromatin propagation (middle). Elevated Raf1 levels enhance chromatin association of ClrC, H3K14ub and H3K9me3 density, reestablishing heterochromatin spreading (bottom). e, Elevating Raf1 expression in raf1-oe or upf1Δ cells bypass the requirement of other factors for heterochromatin propagation. Serial dilution measured mat2P::ura4+ expression, and iodine staining assessed haploid meiosis. Data are representative of two independent experiments. Source data are provided.
Extended Data Fig. 8 Increase in Raf1 abundance promotes caffeine resistance through heterochromatin formation.
a, b, Real-time qPCR analysis of indicated genes in cells treated with 16 mM caffeine for 2 weeks in WT or raf1R576H cells. Data are presented as mean ± SD; n = 3 independent experiments, P value by two-stage step-up Benjamini, Krieger, and Yekutieli unpaired t test for (a) and one-way ANOVA followed by Tukey’s multiple comparisons test for (b). c, Western blot analysis of Raf1 in raf1R576H cells treated with 16 mM caffeine for 2 weeks. Cdc2 and Ponceau S staining included as loading controls. d, Serial dilutions of the indicated strains were plated on nonselective (N/S) or 16 mM caffeine-containing medium. e, Quantification of caffeine-resistant colonies in raf1-oe cells (Padh1-raf1) with or without clr4 (data are presented as mean ± SD; n = 3 independent experiments, P value by two-tailed unpaired t test). f, ChIP-seq analyses of H3K9me3 enrichment in cells treated with 16 mM caffeine for 2 weeks, with or without raf1-oe. Facultative heterochromatin islands with increased H3K9me3 levels in raf1-oe cells are indicated. g, Representative plots of H3K9me3 distribution at facultative islands responsive to caffeine. Open reading frame (ORF) positions are indicated. Data are representative of two independent experiments. Source data are provided.
Extended Data Fig. 9 Tor1-Gad8 signaling axis modulates Raf1 levels and caffeine resistance.
a, b, Western blot analysis of Raf1 under stress conditions for 16 h (a) or in the indicated strains (b). c, Real-time qPCR analysis of raf1 expression in WT or tor1Δ cells (data are presented as mean ± SD; n = 3 independent experiments, P value by two-tailed unpaired t test). d, WT, tor1Δ, and gad8Δ cells plated on 16 mM caffeine-containing medium. e, Quantification of caffeine resistant colonies (data are presented as mean ± SD; n = 3 independent experiments, P value by one-way ANOVA followed by Dunnett’s multiple comparisons test). f, Caffeine resistance in tor1Δ cells upon raf1-oe (Padh1-raf1). Serial dilutions were plated on nonselective (N/S) and 16 mM caffeine containing media. Data are representative of two independent experiments. Source data are provided.
Extended Data Fig. 10 Elevated Raf1 expression enables epigenetic inheritance of heterochromatin under heat stress.
a, Effect of high temperature (37 °C) on heterochromatin propagation at an ectopic site in the indicated strains. Establishment of silenced heterochromatin was assessed by serial dilution in low-adenine-containing medium. TetR-Clr4 was expressed (tetR-clr4ON) from a thiamine-repressible promoter in cells containing the tetO-ade6+ reporter, with or without CAS sites as indicated. Maintenance of ade6+ gene silencing was assessed by serial dilution in thiamine-supplemented low-adenine medium (tetR-clr4OFF). b, Maintenance of ade6+ gene silencing in cells containing 2xCAS3-tetO-ade6+ with or without upf1 at 30 °C and 37 °C was assessed by serial dilution in thiamine-supplemented low-adenine medium (tetR-clr4OFF). H3K9me3 enrichment at the ade6+ was determined by ChIP-qPCR (data are presented as mean ± SD; n = 3 independent experiment, P value by one-way ANOVA followed by Tukey’s multiple comparisons test). Data are representative of three independent experiments. Source data are provided.
Supplementary information
Supplementary Information (download PDF )
This file contains Supplementary Fig. 1 and Tables 1–3.
Supplementary Video 1 (download MP4 )
Representative video of Raf2-Halo SMT.
Supplementary Video 2 (download MP4 )
Representative video of H2B-Halo SMT.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bhatt, B., Wei, Y., Pradhan, A.K. et al. Stress controls heterochromatin inheritance via histone H3 ubiquitylation. Nature 650, 768–778 (2026). https://doi.org/10.1038/s41586-025-09899-8
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09899-8
