
Abstract
Tissue inflammation or nerve injury at the periphery can cause chronic pain. Although the spinal-cord-projecting neurons in the rostral ventromedial medulla (RVMSC neurons) can promote pain chronification1,2,3,4, the pathway by which peripheral injury signals drive these neurons is poorly understood1,2,3,5. Here we report a circuit loop that extends from the spinal cord to the ventral posterolateral thalamus and posterior complex of the thalamus, proceeds to the primary somatosensory cortex and returns to the spinal cord via the lateral superior colliculus, which in turn connects to μ-opioid-receptor-expressing RVMSC neurons. Silencing any node along this multisynaptic circuit has minimal effects on nociception in healthy mice, but can eliminate mechanical hypersensitization and restore normal nociceptive response thresholds in mouse models of inflammatory and neuropathic pain. In healthy mice, repetitive—but not acute—activation of each node in this circuit is sufficient to cause robust chronic mechanical hypersensitization. Our findings reveal a spino-brain–spinal cord circuit loop that links ascending and descending pathways and specifically drives chronic mechanical pain. This could enable the identification of cellular targets for treating chronic pain.
Similar content being viewed by others

Main
Tissue inflammation or nerve injury typically results in temporary allodynia and hyperalgesia, which attracts attention to the affected area to prevent further damage and promote healing. Sometimes, however, this sensitization persists and leads to chronic pain, imposing large psychological and socioeconomic burdens. Although tissue damage often occurs at the periphery, accumulating evidence supports the crucial role of an endogenous pain-modulation network in the central brain in the initiation and maintenance of a state of chronic pain1,2,3,4,5. The diffused brain regions in this network converge onto the periaqueductal grey (PAG)–rostral ventromedial medulla (RVM, including the raphe magnus and gigantocellular reticular nuclei) system, then interact with local pain circuitry in the spinal cord through RVMSC neurons1,2,3,4,5. However, little is known with regard to how information about peripheral injury is transmitted to these descending RVMSC neurons.
Classic in vivo electrophysiological studies have identified a group of RVMSC neurons, known as on-cells, that are activated by nociceptive stimuli and inhibited by morphine6,7. The μ-opioid receptor (encoded by the Oprm1 gene), the primary target of morphine, is a Gi-coupled G-protein-coupled receptor whose activation suppresses neural activity. Therefore, these morphine-inhibited RVMSC neurons should express OPRM1. Together, these response properties have led to a widely accepted working model hypothesizing that the activity of OPRM1+ RVMSC neurons facilitates pain, and potentially promotes chronic pain1,2,3,4. However, selective activation of OPRM1+ RVMSC neurons in behaving animals has not been done, so this model has not been directly tested4. Moreover, if the model is correct, how PAG inputs modulate the activity of these neurons becomes a puzzle, because the net effect of PAG activation is to reduce rather than to promote pain8,9.
Here we developed genetic and viral tools to access OPRM1+ RVMSC neurons, and found that they have a specific role in driving pain sensitization after injury, but not in acute pain. Using OPRM1+ RVMSC neurons as a starting point, and combining monosynaptic retrograde tracing with pathway manipulation, we then mapped out a multisynaptic circuit that passes from the spinal cord to the primary somatosensory cortex through the spinothalamic tract, then returns to the spinal cord from the cortex through the OPRM1+ RVMSC neurons via the lateral superior colliculus (lSuC), but not the PAG. This circuit loop is specifically involved in injury-caused mechanical hypersensitization, but has a limited role in acute nociceptive pain in healthy conditions. Our findings show that targeting this circuit could hold potential for treating chronic pain while sparing protective nociceptive pain.
Accessing OPRM1+ RVMSC neurons
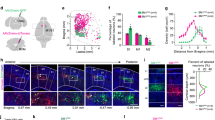
To access OPRM1+ RVMSC neurons, we generated a knock-in mouse line to express Cre recombinase from the endogenous Oprm1 locus (Oprm1cre mice; Fig. 1a, Extended Data Fig. 1a–c and Supplementary Note). However, besides RVMSC neurons, local interneurons or ascending neurons in the RVM could also express the μ-opioid receptor (Extended Data Fig. 1d). To gain projection-specific access to the RVMSC neurons, we optimized a neonatal spinal virus injection procedure and achieved broad spreading of adeno-associated virus (AAV) in the dorsal horn between the cervical and lumbar spinal cord10 (Extended Data Fig. 2a–c). We also developed a new retrograde AAV by inserting the decapeptide LADQDYTKTA between N590 and T591 of the AAV8 capsid (AAV8retro)11 (Extended Data Fig. 2d). Compared with AAV2retro, this modification led to about 2.3-fold and 3.5-fold increases in retrogradely labelled RVMSC neurons and cortical–thalamic projecting neurons, respectively (Extended Data Fig. 2e–g). AAV8retro also labelled twice as many neurons as the co-injected chemical tracer CTB did, and did not transport anterogradely into postsynaptic neurons (Extended Data Fig. 2h,i). We injected AAV8retro-H2B-Clover3-FLEX(LoxP)-H2B-Ruby3 into the spinal cord of Oprm1cre mice at postnatal day (P) 1.5 to express nuclear-localized Ruby3 and Clover3 in OPRM1+ neurons and in all spinal projecting neurons, respectively (Fig. 1b). We labelled about 5,600 RVMSC neurons per mouse, ranging from Bregma −4.7 to −6.5 mm; 65% of these expressed the μ-opioid receptor, and these OPRM1+ RVMSC neurons were located mainly in the raphe magnus (Fig. 1c,d and Extended Data Fig. 2j). Using the transparent embedding solvent system (TESOS) method and single-axon tracing12, we found that many of these neurons travel together in the lateral funiculus before they enter the spinal cord from the deep laminar, then branch into thin axon terminals to innervate the superficial laminar of the dorsal horn, suggesting that it has a role in somatosensation (Extended Data Fig. 3a–c). Most OPRM1+ RVMSC neurons are GABAergic, consistent with previous observations13 (Fig. 1e and Extended Data Fig. 3d). Although the OPRM1+ RVMSC neurons are not serotoninergic, intraspinal injection of AAV8retro could robustly label descending serotoninergic neurons concentrated in the median raphe and in the lateral paragigantocellular nucleus (Fig. 1e and Extended Data Fig. 3d,e). DAMGO, a highly selective μ-opioid-receptor agonist, inhibited the firing of action potentials in OPRM1+ RVMSC neurons, confirming that they express functional μ-opioid receptor (Fig. 1f). Proenkephalin (Penk) is expressed in a group of GABAergic RVMSC neurons that can form direct axo-axonic interactions with peripheral sensory inputs from dorsal root ganglia14. Notably, most OPRM1+ RVMSC neurons do not express Penk, suggesting that at least two distinct descending GABAergic pathways from the RVM modulate sensory processing in the spinal cord (Fig. 1e and Extended Data Fig. 3d).

a, Design of Oprm1cre mice. b–d, Labelling (b), reconstruction (c) and distribution (d) of RVMSC neurons (n = 4). Scale bar, 1 mm. e, RNAscope detection of Oprm1, vGat (also known as Slc32a1), tryptophan hydroxylase 2 (Tph2) and Penk in OPRM1+ RVMSC neurons, visualized by HA immunostaining (green). n = 5–8 slides from 4 mice. Scale bars, 50 μm. Insets: magnified images of representative neuron(s). Scale bars, 10 μm. f, DAMGO inhibition of OPRM1+ RVMSC neurons. Left, biocytin labelling of a recorded neuron. Scale bar, 50 μm. Example traces (middle) and quantification (right) of action potential (AP) firing before and after treatment with DAMGO (1 μM, n = 6, P = 0.0313) or DAMGO + CTAP (1 μM + 1 μM, n = 5, P = 0.4375). Scale bars, 50 mV and 1 s. g, Experimental timeline for h–j, and representative image of fibre track and jGCaMP7s expression. Scale bar, 500 μm. h, Mechanical (von Frey) and thermal (Hargreaves, plantar cold) response traces. Single trials (black), mean (red), s.e.m. (shading); n = 7–8. Arrows indicate stimulus-evoked (black) or spontaneous (red) paw withdrawal. Scale bars, 5 s and 1% ΔF/F (single trial); 2 s and 1% ΔF/F (average). i, Mechanical response traces (left) and area under curve (AUC; right) before and after SNI (n = 8). (Before and day 7, P = 0.0005; before and day 14, P = 0.0005, before and day 28, P = 0.0038; day 2 and day 7, P = 0.0493). Scale bars, 2 s and 2% ΔF/F. j, Calcium activity (top left), speed (bottom left) and their correlation (right) in a normalized movement bout in mice with SNI. Dashed line, movement start; red line (right), linear fitting of dots (average speed calcium signal). Pearson’s r = −0.8212 (locomotion) or 0.9188 (escape). Scale bars, 50% of movement bout, 2% ΔF/F and 5 cm s−1. See Supplementary Table 1 for detailed statistics. Two-sided Wilcoxon signed-rank test (f); non-parametric analysis of variance (ANOVA) (i). *P < 0.05, **P < 0.01, ***P < 0.001. Mean ± s.e.m.
Noxious stimuli activate OPRM1+ RVMSC neurons
Besides the RVM, many neurons in the locus coeruleus (LC) were also retrogradely labelled (Extended Data Fig. 3f), consistent with robust expression of Oprm1 in the LC15. We therefore devised an intersectional strategy to specifically target OPRM1+ RVMSC neurons while sparing OPRM1+ LCSC neurons16 (Methods). We first transduced OPRM1+ RVMSC neurons with genetically encoded neuronal activity sensor jGCaMP7s (ref. 17), then used fibre photometry to record their in vivo activity during von Frey, Hargreaves and plantar cold tests18,19,20 (Fig. 1g and Supplementary Video 1). In healthy mice, these neurons exhibited calcium transients time-locked to paw withdrawal in response to all three types of noxious stimuli (Fig. 1h and Extended Data Fig. 4a,b). After spared nerve injury (SNI) to induce a severe and persistent state of neuropathic pain21, the neurons showed heightened responses to mechanical and cold stimuli, but not to heat (Fig. 1h and Extended Data Fig. 4c). Calcium responses in the OPRM1+ RVMSC neurons increased from day 2 to day 7 after SNI and remained high (Fig. 1i). By contrast, in a model of inflammatory pain induced by complete Freund’s adjuvant (CFA), calcium responses peaked at day 2 before gradually returning to baseline over 21 days (Extended Data Fig. 4d,e). Notably, these neurons showed minimal activity during voluntary locomotion or in the absence of paw withdrawal, but robust activity during pain-evoked behaviours (Fig. 1h–j and Extended Data Fig. 4a,f,g). The amplitude of this activity positively correlated with the speed of the escape response (Fig. 1j and Extended Data Fig. 4f,g) and was also observed during spontaneous pain episodes (for example, paw flinching) at levels comparable with evoked responses (Fig. 1h and Extended Data Fig. 4h). Collectively, these observations indicate that OPRM1+ RVMSC neurons are specifically activated by nociception and sensitized by peripheral injury, but not active during locomotion.
Mechanical and cold hypersensitization
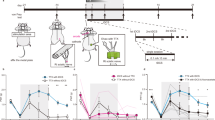
The ability to effectively access the OPRM1+ RVMSC neurons offers the opportunity to determine their roles in normal nociception and pain sensitization caused by nerve injury and inflammation. Using chemogenetic tools, we found that acute inhibition or activation of OPRM1+ RVMSC neurons in these mice had little effect on their behavioural responses to mechanical or temperature stimuli, or on their locomotion, indicating that the descending OPRM1+ pathway contributes minimally to protective acute nociception and locomotor function4,22 (Fig. 2a,b and Extended Data Figs. 5a–c and 6a,b). This is different from the activation of descending serotoninergic neurons in the RVM, in which single activation can cause prolonged hypersensitivity23.

a, Timeline for b–h (Oprm1cre mice). b, Effects of saline (black) or clozapine (CLZ) on mechanical threshold (von Frey test; left) and plantar cold latency (right) in hM4D-expressing (blue; n = 8 (left, P = 0.0078) or n = 9 (right, P = 0.0039)) or hM3D-expressing (red; n = 9 (right, P = 0.4258) or n = 10 (left, P = 0.9004)) healthy mice. c, Representative images for caspase ablation. OPRM1+ RVM neurons express Ruby3. OPRM1+ RVMSC neurons (yellow, left) are ablated by caspase (right). Dashed line indicates the nucleus raphe magnus (NRM). MARN, magnocellular reticular nucleus; Py, pyramid; RPa, nucleus raphe pallidus. Scale bars, 100 μm. d,e, Mechanical thresholds of control (black, n = 7) and caspase 3-expressing (orange, n = 8) mice with SNI (d) or CFA injection (e). Images show OPRM1+ RVMSC terminals and FOS in the dorsal horn (dashed line) of control but not caspase 3-expressing mice. Scale bars, 100 μm (P = 0.2146, 0.5175, 0.0003, 0.0003, 0.0003 and 0.0003 (left to right; d); P = 0.0252, 0.11, 0.0003, 0.0003 and 0.0003 (left to right; e)). f,g, Mechanical thresholds (f) and plantar cold latency (g) after CLZ treatment in hM4D mice (n = 8 in f; n = 9 in g) and mCherry mice (n = 8) after SNI (P = 0.0103, 0.0002, 0.0002, 0.0002 and 0.0002 (left to right in f); P < 0.0001 in g). h, CPA in control (n = 5) but not hM4D (n = 5) mice after SNI (P = 0.0159). i, Mechanical thresholds after daily injection of CLZ 23 h before each von Frey test for 7 consecutive days (shaded) in hM3D (n = 5) and mCherry (n = 9) healthy mice. See Supplementary Table 1 for detailed statistics. Two-sided Wilcoxon signed-rank test (b); two-sided Mann–Whitney test (d–i). *P < 0.05, **P < 0.01, ***P < 0.001. Non-parametric ANOVA (i), #P < 0.05. Mean ± s.e.m.
We next investigated the roles of OPRM1+ RVMSC neurons in neuropathic and inflammatory pain. We reasoned that if OPRM1+ RVMSC neurons are required for the development of persistent pain, ablation of these neurons before injury should prevent its initiation. We expressed activated caspase 3 in OPRM1+ RVMSC neurons to induce cell-autonomous apoptosis24, which eliminated their cell bodies in the RVM and their terminals in the spinal cord (Fig. 2c,d and Extended Data Fig. 7a,b). These mice have normal mechanical thresholds and nociceptive-stimulus-induced FOS in the dorsal horn (Fig. 2c,d and Extended Data Fig. 7c), but neither SNI nor CFA injection decreased mechanical withdrawal thresholds in the von Frey test (Fig. 2d,e). Mechanical stimulation after injury no longer caused increased FOS expression in the dorsal horn of the spinal cord (Fig. 2d and Extended Data Fig. 7d). Notably, ablation of OPRM1+ RVMSC neurons had only a partial effect on CFA-induced thermal hypersensitivity (Extended Data Fig. 7e). Thus, OPRM1+ RVMSC neurons are required for the initiation of SNI- and CFA-induced mechanical hypersensitization.
Similarly, if activity of OPRM1+ RVMSC neurons is required for maintaining the persistent pain state, silencing these neurons after injury should alleviate established pain. SNI produces a severe chronic neuropathic pain state. This model is characterized not only by somatosensory hypersensitivity, which drives heightened affective–motivational responses to evoked stimuli21,25, but also by spontaneous pain, which is triggered by the synchronized cluster firing of injured sensory neurons26. We again used chemogenetic silencing of OPRM1+ RVMSC neurons and evaluated its effects on sensory hypersensitivity, affective behaviours and spontaneous pain. We found that clozapine infusion completely reversed sensitized mechanical and cold pain thresholds, returning them to normal levels, in SNI mice expressing hM4D but not mCherry in OPRM1+ RVMSC neurons (Fig. 2f,g). The same treatment also normalized stimulus-evoked affective behaviours, including paw attending and escape response, and blocked mechanical-stimulus-induced conditioned place aversion (CPA)4 (Fig. 2h and Extended Data Figs. 6c and 8a). We further quantified spontaneous pain by monitoring stimulus-independent episodes of licking and flinching of the injured paw, and by using an automated facial grimacing scale27. SNI mice exhibited significantly increased spontaneous pain behaviours and grimace scores, and silencing OPRM1+ RVMSC neurons reduced both of these indicators of spontaneous pain down to the levels seen in non-injured control mice (Extended Data Fig. 8b,c). Together, these observations indicate that both sensory and affective components of pain are reduced after silencing of OPRM1+ RVMSC neurons. Notably, at 28 days after SNI, the time point when SNI-induced mechanical hypersensitivity becomes morphine resistant28, silencing these neurons still robustly alleviated neuropathic pain (Fig. 2f). Moreover, examining FOS expression evoked by mechanical stimuli in several pain-related brain regions, we found that chemogenetic silencing of OPRM1+ RVMSC neurons in SNI mice restored FOS expression to the same level as seen in the mice that underwent sham surgery, revealing a brain-wide reduction of pain responses after silencing of these neurons (Extended Data Fig. 6d,e).
In contrast to the minimal effect on mechanical threshold after acute activation (Fig. 2b), repetitive chemogenetic activation of OPRM1+ RVMSC neurons in healthy mice induced profound and long-lasting mechanical hypersensitivity and drove heightened stimulus-evoked affective behaviours (Fig. 2i and Extended Data Fig. 8a). In addition, these mice also became sensitized to the plantar cold stimulus but did not change their response to heat (Extended Data Fig. 6f,g). Together, these results reveal that the descending OPRM1+ pathway is necessary and sufficient for both the initiation and the maintenance of mechanical hypersensitivity and cold allodynia4,22. Of note, these mice did not show more spontaneous licking and flinching or facial grimacing than control mice did, and also did not show generalized sensitization to air puffs to the cornea (Extended Data Fig. 8b–d).
Beyond sensory hypersensitivity, chronic pain is frequently comorbid with anxiety and depression, which affect approximately 40% of adults with chronic pain29. Consistent with this, we observed heightened anxiety- and depression-like states in SNI mice six weeks after surgery, evidenced by reduced centre time in the open field test, less time in the open arm of the elevated plus maze (EPM) and increased immobility in the tail suspension test30 (Extended Data Fig. 8e–g). Given that manipulating OPRM1+ RVMSC neurons modulates both sensory and affective components of pain, we examined whether they also contribute to these comorbid states. We found that repetitive chemogenetic activation of OPRM1+ RVMSC neurons in healthy mice was sufficient to induce anxiety-like, but not depression-like, behaviours. By contrast, chemogenetic silencing of these neurons in SNI mice had no effect on the established anxiety- and depression-like states, suggesting that although their activity can drive anxiety, they are not required for maintaining these comorbid mental-health conditions after nerve injury (Extended Data Fig. 8e–g).
Input of the lSuC onto OPRM1+ RVMSC neurons
We next used the OPRM1+ RVMSC neurons as a starting point to map the neuronal circuits that send injury information from the periphery to this crucial descending output for driving chronic mechanical pain. Because peripheral stimuli strongly activate OPRM1+ RVMSC neurons (Fig. 1h), we focused on identifying the excitatory monosynaptic inputs onto these neurons that drive chronic pain. Current models emphasize the role of the PAG to RVM pathway in descending pain modulation1,2,3. We therefore first examined the effect of silencing the excitatory PAG terminals in the RVM, validated using ex-vivo slice recordings, on nociception and mechanical hypersensitivity (Fig. 3a, Extended Data Fig. 5d–f and Supplementary Fig. 1a,b). Notably, chemogenetic silencing of this pathway had no effect on either the baseline mechanical threshold or SNI-induced mechanical hypersensitivity (Fig. 3b), consistent with the predominantly analgesic effect of PAG stimulation. This indicates that other excitatory inputs are responsible for driving the engagement of OPRM1+ RVMSC neurons in mechanical hypersensitivity9,31,32.

a, Schematic of chemogenetic silencing of the PAG→RVM pathway (top) and representative VGLUT2+ terminals (red) from PAG in the RVM (bottom; vGlut2cre mouse) (vGlut2 is also known as Slc17a6). Scale bar, 500 μm. b, Mechanical thresholds of saline (black) and CLZ (blue) infusion in hM4D-expressing mice (n = 8). c–e, Experimental timeline (Oprm1cre mouse; c), representative images from rabies tracing (d) and quantification (e) of monosynaptic inputs onto OPRM1+ RVMSC neurons (n = 6). Scale bars, 250 μm. Inset (e): mCherry+ neurons in intermediate (iSuC) and deep (dSuC) superior colliculus (Bregma −3.64 mm). MRN, midbrain reticular nucleus; ZI, zona incerta; HY, hypothalamus; APN, anterior pretectal nucleus; CTX, cortex; BNST, bed nucleus of the stria terminalis. f, Representative images (left) and quantification (right; percentage) of RNAscope detection of vGlut2 and vGat in mCherry+ input neurons from the lSuC, PAG and ZI (6 slides from 3 mice). Scale bar, 50 μm. g, Quantification of mechanical thresholds of saline (black, n = 6) and CLZ (blue, n = 6) infusion in the RVM of vGlut2cre mice with hM4D expression in the lSuC for chemogenetic silencing (P = 0.5625, 0.0313 and 0.0313, left to right). h, Quantification of mechanical thresholds after daily injection of CLZ into the RVM 23 h before each von Frey test for 7 consecutive days (shaded), in non-injured vGlut2cre mice with hM3D expression in the lSuC for chemogenetic activation (n = 6). i, Time for two phases of paw licking and biting after formalin injection into the hind paw. CLZ was infused into the RVM of vGlut2cre mice with eGFP (black, n = 5) and hM4D (blue, n = 5) expression in the lSuC for chemogenetic silencing. See Supplementary Table 1 for detailed statistics. Two-sided Wilcoxon signed-rank test (g); non-parametric ANOVA (h). *P < 0.05, **P < 0.01. Mean ± s.e.m.
We then performed cTRIO (cell-type-specific tracing of the relationship between input and output) experiments to reveal all monosynaptic inputs onto OPRM1+ RVMSC neurons13,16. We found that intermediate and deep grey layers of the lSuC (ranging from Bregma −3.45 to −3.65 mm) contained a similar number of retrogradely labelled neurons to the PAG (Fig. 3c–e). Our cTRIO tracing labelled very few neurons in the lateral parabrachial nucleus (lPBN), which has been shown to provide direct inputs onto on-cells in the RVM5,33. By contrast, cTRIO tracing from the entire population of GABAergic RVMSC neurons revealed input from the parabrachial nucleus13. This discrepancy could reflect the tropism of the rabies virus, or could be because lPBN neurons prefer to innervate local on-cells or OPRM1− RVMSC neurons, rather than OPRM1+ RVMSC neurons33,34. We further examined these brain regions using RNAscope probing for inhibitory (VGAT) and excitatory (VGLUT2), neurons and found that about 30% of retrogradely labelled neurons in the PAG were VGLUT2+, whereas this number was more than 90% in the lSuC (Fig. 3f). Therefore, the lSuC provides more excitatory input onto OPRM1+ RVMSC neurons than the PAG does.
Consistent with their robust anatomical connection, the functional connection between the lSuC and the RVM is essential for mechanical hypersensitization. The excitatory RVM-projecting neurons in the lSuC showed a significantly larger response to von Frey stimuli after SNI (Extended Data Fig. 4i–k). We chemogenetically silenced excitatory lSuC terminals in the RVM after SNI, and found that this restored normal mechanical thresholds (Fig. 3g and Supplementary Fig. 1a,b) and eliminated pain-induced CPA in SNI mice (Extended Data Fig. 9a), but had no effect on locomotion (Extended Data Fig. 5g). Similarly, silencing of this pathway eliminated CFA-induced mechanical but not thermal sensitization, indicating that it has a key role in inflammatory mechanical pain (Extended Data Fig. 6h,i). A previous study reported that the lSuC is part of orienting circuits for defensive response, and that lSuC lesion prevented licking and biting directed towards a paw injected with formalin35. It is therefore possible that the lSuC→RVM pathway is part of this defensive circuit, and that silencing this pathway would make mice less attentive to their pain. To examine this possibility, we chemogenetically silenced the excitatory lSuC→RVM pathway in formalin-injected mice and found that this had no effect on either licking and biting directed towards the formalin-injected paw (Fig. 3i). Furthermore, optogenetic activation of this pathway had no effect on locomotion, turning, heart rate or breathing rate (Extended Data Fig. 5h–k).
Finally, repetitive chemogenetic activation of excitatory lSuC inputs in the RVM caused long-lasting mechanical hypersensitization and pain-induced CPA in non-injured healthy mice (Fig. 3h, Extended Data Fig. 9b and Supplementary Fig. 1a,b). Together, these results show that the excitatory lSuC→RVM pathway has a causal role in driving mechanical hypersensitivity.
SS drives chronic mechanical pain through the lSuC
We next investigated the upstream inputs onto the lSuC that drive chronic mechanical pain. We again used the cTRIO approach to map brain-wide monosynaptic inputs onto the RVM-projecting excitatory lSuC neurons. This revealed two prominent clusters of retrogradely labelled neurons in the cortex, located in layer 5 of the somatosensory cortex (SS) and the anterior motor cortex (MOa) (Fig. 4a,b). Retrogradely labelled lSuC-projecting SS neurons were concentrated between Bregma −1.2 and Bregma −2.2 mm, whereas retrogradely labelled MOa neurons were located within the anterior cortex region, overlapping with the region identified by retrograde labelling from the lateral rostral medulla, which collaterally innervates the same lSuC regions that project to the RVM36 (Fig. 4a, inset). Chemogenetic silencing of the SS→lSuC pathway, but not the MOa→lSuC pathway, eliminated SNI-induced mechanical hypersensitivity and CPA (Fig. 4c–e and Supplementary Fig. 1c,d), whereas repetitive chemogenetic activation of the SS→lSuC pathway induced chronic mechanical pain and pain-induced aversion in healthy mice (Fig. 4f,g and Supplementary Fig. 1c,d). Notably, this same activation protocol did not drive mechanical hypersensitization in mice in which OPRM1+ RVMSC neurons had been ablated, highlighting that an intact descending pathway from the SS to the spinal cord is required for pain sensitization (Fig. 4f). Together, these findings extend the descending pathway for chronic mechanical pain one synapse upstream, from lSuC→RVM→spinal cord to SS→ lSuC→RVM→spinal cord. The fact that the SS drives the lSuC→RVM pathway for chronic pain further supports the anatomical and functional complexity of the superior colliculus.

a,b, Representative images (a) and quantification (b) of monosynaptic inputs onto lSuCRVM neurons (n = 4). Scale bar, 500 μm. Inset: three-dimensional (3D) reconstruction of cortical input neurons. Dashed circles indicate MOa and SS. MO, motor cortex; SNr, substantia nigra; LHA, lateral hypothalamus; GRN, gigantocellular reticular nucleus; VNC, vestibular nuclei; VM, ventral medial thalamus; PRNc, caudal-pontine reticular nucleus; PB, parabrachial nucleus; RSP, retrosplenial area. c,d, Mechanical thresholds of saline (black, n = 6 in c, n = 7 in d) and CLZ (blue, n = 6 in c, n = 7 in d) infusion into the lSuC of mice expressing hM4D in MOa (c) or SS (d, P = 0.8438, 0.0156 and 0.0156, left to right) for chemogenetic silencing. e, Locomotor traces and CPA scores in mice expressing mCherry (white, n = 6) and hM4D (blue, n = 7) in the SS after SNI (P = 0.0012). f, Mechanical thresholds in non-injured mice (control mice (red, n = 7) or mice with OPRM1+ RVMSC neurons ablated (black, n = 8)) expressing hM3D in SS after daily injection of CLZ into the lSuC 23 h before each von Frey test for 7 consecutive days (shaded). g, Locomotor traces and CPA scores in hM3D (red, n = 7) and mCherry (white, n = 6) mice after repetitive infusion of CLZ into the lSuC (P = 0.0012). See Supplementary Table 1 for detailed statistics. Two-sided Wilcoxon signed-rank test (c,d); two-sided Mann–Whitney test (e,g and control versus ablation in f); Dunn’s multiple comparisons test (before versus after CLZ in f). *P < 0.05, **P < 0.01, ***P < 0.001; #P < 0.05, ##P < 0.01, ###P < 0.001. Mean ± s.e.m.
Spinothalamic pathways for chronic mechanical pain
The spinothalamic tract (STT) has conventionally been seen as the major ascending pathway for transmitting innocuous sensory discriminative information and pain from the peripheral to the SS through the thalamus37,38. The prominent target of the STT in the thalamus is the ventral posterolateral nucleus (VPL), with some minor innervation of other thalamic nuclei, including the posterior complex of the thalamus (Po)37,38,39. The Po→SS pathway is involved in injury-induced pain sensitization, but whether this function requires input from the STT is not known40. Moreover, besides inputs from the spinal cord, both the VPL and the Po relay other somatosensory inputs to the SS41,42. We thus sought to selectively manipulate the VPL→SS and Po→SS pathways that convey information from the STT (STT→VPL→SS and STT→Po→SS). To achieve that, we used high-titre AAV1-Cre, which has the property of monosynaptic, anterograde travel across synapes43. We injected AAV1-Cre into the lumbar region of the spinal cord dorsal horn and AAV8-FLEX-hM4D into the VPL, the Po or both, so that hM4D would be expressed only in VPL or Po neurons that receive STT inputs (Fig. 5a). Electrophysiological recording of retrogradely labelled lSuC-projecting neurons in layer 5 of the SS (SSL5) revealed robust functional connectivity in both VPL→SSL5 and Po→SSL5 pathways (Fig. 5b). We next injected clozapine to selectively silence the STT→VPL→SS pathway, the STT→Po→SS pathway or both. We found that chemogenetic silencing of either pathway alone had no effect on mechanical hypersensitization after SNI and pain-induced CPA (Fig. 5c,e and Extended Data Fig. 9c). However, chemogenetic silencing of both pathways together abolished SNI-induced chronic mechanical pain and CPA (Fig. 5d,e and Extended Data Fig. 9c). These observations suggest that either the STT→VPL→SS or the STT→Po→SS pathway is sufficient to drive mechanical hypersensitization after injury. Indeed, repetitive chemogenetic activation of either pathway drove chronic mechanical pain in healthy mice, and this pain sensitization effect also required the OPRM1+ RVMSC neurons (Fig. 5f,g and Extended Data Figs. 9d and 10a,b). Finally, six weeks after repetitive chemogenetic activation of the STT pathway, the mice showed a heightened anxiety state, but not depression-like behaviour, similar to what was seen with the activation of OPRM1+ RVMSC neurons (Extended Data Figs. 8e–g and 10c,d). In summary, we found two parallel thalamic pathways that connect the STT with the SS to produce chronic mechanical pain, supporting the hypothesis that SNI sensitizes both STT→SS pathways, which operate in a redundant or parallel manner to initiate and maintain injury-induced pain sensitization.

a, Representative image of transsynaptically labelled thalamic neurons and their terminals in the SS. AAV1-Cre was injected into the spinal cord and AAV8-FLEX(LoxP)-mScarlet into the thalamus. Scale bar, 500 μm. SSp, primary somatosensory cortex; SSs, secondary somatosensory cortex. b, Functional connectivity between the Po or the VPL and retrogradely labelled lSuC-projecting neurons in the SS. Top, optical excitatory postsynaptic current (oEPSC) traces. Bottom, quantification of connectivity. Numbers indicate cells with oEPSC/total recorded. Scale bars, 50 ms and 50 pA. c,d, Mechanical thresholds after injection of saline (black, n = 7 (c, left), n = 5 (c, right), n = 7 (d)) or CLZ (blue, n = 7 (c, left), n = 5 (c, right), n = 7 (d)) into mice expressing hM4D in the SC→VPL pathway (c, left), the SC→Po pathway (c, right) or both pathways (d: P = 0.8125, 0.0156 and 0.0156, left to right). e, CPA scores after silencing the SC→VPL pathway (n = 6), the SC→Po pathway (n = 5) or both pathways (n = 5). f, Mechanical thresholds after daily injection of CLZ into the RVM 23 h before each von Frey test for 7 consecutive days (shaded), in non-injured mice expressing hM3D in the SC→Po pathway (left, n = 5) or in the SC→VPL pathway (right, n = 5). g, CPA scores after repetitive activation of the SC→VPL (n = 5) or SC→Po (n = 5) pathway. Control (black, n = 5). h, Diagram (left) and quantification of texture discrimination (right) in mice in which both the SC→VPL and the SC→Po pathway (yellow, n = 5) were silenced, mice in which the DCN→VPL pathway (white, n = 6) was silenced or wild-type (WT) mice without manipulation (grey, n = 5) (P = 0.0625, 0.0313 and 0.1250, left to right). i, Summary circuitry diagram for pain sensitization and touch or tactile sensation. Dashed line indicates hypothetical interneurons connecting the OPRM1+ inhibitory inputs to the excitatory STT output pathway. See Supplementary Table 1 for detailed statistics. Two-sided Wilcoxon signed-rank test (c,d,h); one-way ANOVA (e); non-parametric ANOVA (f,g). *P < 0.05, **P < 0.01, ***P < 0.001. Mean ± s.e.m.
Chemogenetic silencing of the STT→VPL→SS and the STT→Po→SS pathway, either separately or simultaneously, had no effect on nociceptive thresholds before SNI, indicating that neither pathway is involved in nociception in healthy mice (Fig. 5c,d, before SNI and before SNI + CLZ). Because the conventional view also suggests that the STT has a role in somatosensory discrimination, we examined the contribution of these pathways to behaviours that require the discrimination of innocuous tactile stimuli. Using a behavioural chamber with two compartments that have distinct floor textures (smooth Plexiglass and fine-grit sandpaper), we found that freely moving mice spent significantly more time in the compartment with the sandpaper floor (Fig. 5h). We also performed a texture-based novel object recognition test and found that mice spent more time examining objects with novel textures44 (Extended Data Fig. 10e). Silencing both the STT→VPL→SS and the STT→Po→SS pathway together had no effect on the preference of mice for the sandpaper floor or for objects with novel textures (Fig. 5h and Extended Data Fig. 10e). These results reveal the limited contribution of the STT→thalamus→SS pathway in tactile discrimination, further supporting its selective role in mediating chronic mechanical pain after injury.
The VPL is known to be important for somatosensory discrimination42. Because tactile discrimination is not affected by the manipulation of STT inputs onto the VPL (Fig. 5h and Extended Data Fig. 10e), it could be regulated by other inputs. One candidate input could be from the dorsal column nucleus (DCN), which integrates tactile information directly from low-threshold mechanoreceptors and indirectly from postsynaptic dorsal column neurons42,45,46. Indeed, we found that silencing the excitatory DCN→VPL pathway abolished the preference of mice for the sandpaper floor and for objects with novel textures (Fig. 5h, Extended Data Fig. 10e and Supplementary Fig. 1e). Notably, silencing this pathway had no effect on nociceptive response and SNI-induced pain sensitization (Extended Data Fig. 10f). The DCN and the STT inputs onto the thalamus thus have distinct roles, in tactile discrimination and in injury-induced pain sensitization, respectively.
Discussion
In conclusion, this study establishes the causal role of a multisynaptic pathway from spinal cord→thalamus (VPL + Po)→SS→lSuC→RVM→spinal cord that selectively mediates chronic mechanical pain after nerve injury or inflammation but is not involved in setting nociceptive thresholds in healthy mice (Fig. 5i). Silencing any node in this long-range circuit loop completely eliminates injury-induced mechanical hypersensitivity and restores normal mechanical responses in SNI mice. Conversely, repetitive activation of each node is sufficient to drive chronic mechanical hypersensitization in non-injured healthy mice. Thus, this long-range circuit loop is necessary and sufficient for mechanical hypersensitization after injury or inflammation, rendering it a promising cellular target for treating chronic mechanical pain. Our elucidation of this core circuit loop for chronic pain lays the foundation for future studies investigating its interaction with the broader ‘pain matrix’ network.
Current models emphasize the important role of the PAG–RVM system in descending pain modulation. Our cTRIO tracing and in situ characterization revealed that more inhibitory than excitatory PAG neurons innervate OPRM1+ RVMSC neurons. Silencing excitatory PAG terminals in the RVM had no effect on either nociceptive pain or injury-induced pain sensitization. Of note, a previous study using non-selective ablation of both excitatory and inhibitory PAG→RVM pathways also reported no effect on SNI-induced mechanical hypersensitivity47. Together, these two observations suggest that the inhibitory PAG→RVM pathway also has a limited role in pain sensitization. These results align with the well-documented analgesic effect of PAG stimulation, which might alleviate injury-induced pain sensitization through the activation of inhibitory PAG inputs that suppress OPRM1+ RVMSC neurons. PAG stimulation also suppresses acute pain, an effect that could be mediated by OPRM1− neurons in the RVM8,9,32.
Our results reveal the specific role of the STT in injury-induced pain sensitization, but not in nociceptive pain and discrimination of innocuous tactile stimuli. By contrast, the DCN→VPL pathway is specifically required for non-painful tactile discrimination, but is dispensable for both acute and chronic pain. Together, our findings support a model in which distinct aspects of somatosensation are transmitted through separate ascending pathways (Fig. 5i), and highlight the need to identify ascending pathway(s) that are responsible for acute nociceptive pain. The spinoparabrachial pathway is a compelling candidate, given the important role of the parabrachial nucleus in mediating nocifensive responses to noxious stimuli34,38,48.
Finally, although we have delineated, in the context of chronic pain, the anatomical and functional connections that run from the spinal cord to the brain, then back to the spinal cord, the local spinal circuits that connect these two ends remain unclear (Fig. 5i). Given that OPRM1+ RVMSC neurons are GABAergic and inhibitory, but chronic mechanical pain requires persistent STT activation, we propose a plausible mechanism: these neurons might innervate a specific group of inhibitory interneurons that act as a ‘gate’, normally preventing non-painful somatosensory input from reaching the thalamus through the STT. Repetitive activation of OPRM1+ RVMSC neurons could then induce long-term inhibition of this gating mechanism, permitting non-painful inputs to aberrantly activate the STT and drive pain. A crucial next step is to identify these inhibitory interneurons, which could constitute the long-sought gate circuit proposed in the gate control theory of pain more than 60 years ago49,50, and elucidate their connection to the STT. In addition, we note that cTRIO tracing identified the zona incerta, a region containing mostly inhibitory neurons that directly innervates both OPRM1+ RVMSC neurons and their presynaptic lSuC neurons (Figs. 3e and 4b). Activation of these inhibitory inputs should be able to suppress activity in both nodes of the descending pathway, thereby offering a powerful therapeutic target for alleviating chronic mechanical pain.
Methods
Mice
All procedures were in accordance with the US National Institutes of Health (NIH) guidelines for the care and use of laboratory animals and were approved by Stanford University’s Administrative Panel on Laboratory Animal Care. Mice (1.5 days–10 weeks) of both sexes were used in experiments. Genetically engineered mouse lines used in this study included Oprm1cre/+ and vGlut2-IRES-cre (JAX 016963). The Oprm1cre/+ knock-in mouse line was generated in the Stanford University Transgenic, Knockout and Tumor Model Center (TKTC) using conventional embryonic-stem-cell-targeting strategies. The Cre recombinase cDNA, followed by the rabbit β-globin poly-A signal, was introduced by homologous recombination immediately after the start codon in exon 1 of the mouse Oprm1 gene51 (Extended Data Fig. 1a). Heterozygous mice were generated by mating chimeric mice with C57BL/6 mice.
Viruses
The following viruses were produced and packaged in the laboratory and used in this study: AAV8retro-hSyn-H2B-Clover3-FLEX(LoxP)-H2B-mRuby3 (2.0 × 1013 genome copies (gc) per ml), AAV8retro-CAG-mCherry (5.0 × 1012 gc ml−1), AAV2retro-CAG-eGFP (5.0 × 1012 gc ml−1), AAV8-hSyn-FLEX(FRT)-mCherry (1.0 × 1013 gc ml−1), AAV8-hSyn-FLEX(FRT)-Clover3 (1.0 × 1013 gc ml−1), AAV8-hSyn-FLEX(FRT)-hM4D-IRES-EGFP (2.0 × 1013 gc ml−1), AAV8-hSyn-FLEX(FRT)-hM3D-EYFP (3.0 × 1013 gc ml−1), AAV8-hSyn-FLEX(FRT)-taCaspase3-TEVp (1.0 × 1013 gc ml−1), AAV8-hSyn-FLEX(LoxP)-Ruby3-FLEX(FRT)-Clover3 (1.0 × 1013 gc ml−1), AAV8retro-hSyn-FLEX-mTagBFP2-P2A-FlpO (5.0 × 1013 gc ml−1), AAV8retro-hSyn-mTagBFP2-P2A-Cre (5.0 × 1013 gc ml−1), AAV8-hSyn-FLEX(LoxP)-hM4D-IRES-GFP (5.0 × 1013 gc ml−1), AAV8-hSyn-FLEX(LoxP)-hM3D-IRES-mCherry (1.0 × 1013 gc ml−1), AAV8-hSyn-FLEX(LoxP)-hM3D-IRES- EGFP (1.0 × 1013 gc ml−1), AAV8-hSyn-FLEX(FRT)-jGCaMP7s (5.0 × 1013 gc ml−1), AAV1-hSyn-Cre (1.0 × 1013 gc ml−1), AAV8-hSyn-hM4D-mCherry (5.0 × 1012 gc ml−1), AAV8-hSyn-hM3D-mCherry (5.0 × 1012 gc ml−1), AAV8-EF1α-FLEX(LoxP)-mScarlet (1.0 × 1013 gc ml−1), AAV8-Ef1a-DIO-hChR2(H134R)-EYFP (5.0 × 1012 gc ml−1), AAV8-hSyn-FLEX(FRT)-EGFP-P2A-TVA-T2A-oG (5.0 × 1013 gc ml−1). SADΔG-mCherry(EnvA) (2.0 × 108 infectious unit (IU) ml−1) was purchased from Salk viral core, CVS-N2cΔG-mCherry(EnvA) (5.0 × 108 IU ml−1) was purchased from Jefferson Center for Vaccines. AAV5-Ef1a-DIO-hChR2(H134R)-mCherry (1.2 × 1013 gc ml−1, Addgene 20297) and AAV2-hSyn-DIO-hM4D(Gi)-mCherry (1.2 × 1013 gc ml−1, Addgene 44362) were purchased from Addgene.
Surgery
Stereotaxic injection and implantation of optical fibres and cannulas
Stereotaxic surgeries were performed on five- to seven-week old mice under ketamine and xylazine (100 mg kg−1 and 5 mg kg−1, intraperitoneally; i.p.) anaesthesia using a stereotaxic instrument (BenchMARK Digital, Leica). Virus was injected into the RVM (200 nl AAV, Bregma −5.60 mm, lateral ±0.1 mm, ventral 5.75 mm), lSuC (200 nl AAV, Bregma −3.45 mm, lateral ±1.65 mm, ventral 2.40 mm), PAG (250 nl AAV, Bregma −4.65 mm, lateral ±0.5 mm, ventral 3 mm), SSp and SSs (200 nl AAV at site 1: Bregma −1.45 mm, lateral ±3.75 mm, ventral −1.45 mm; 200 nl AAV at site 2: Bregma −1.65 mm, lateral ±3.85 mm, ventral −1.55 mm), MOa (200 nl AAV, Bregma +1.8 mm, lateral ±1.5 mm, ventral −1.35 mm), VPL (200 nl, Bregma −1.8 mm, lateral ±2.0 mm, ventral 3.5 mm), Po (200 nl, Bregma −1.8 mm, lateral ±1.6 mm, ventral 2.85 mm), DCN (70 nl per injection, at site 1: obex 0.0 mm, lateral ±0.6 mm, ventral −0.25 mm; site 2: obex 0.25 mm, lateral ±0.8 mm, ventral −0.3 mm; site 3: obex 0.5 mm, lateral ±1.1 mm, ventral −0.35 mm) with a pulled glass capillary at a slow rate (100 nl min−1) using a pressure microinjector (Micro4 system, World Precision Instruments). The injection capillary was removed 5 min after the end of the injection. For mice used for terminal chemogenetic manipulation or fibre photometry, an infusion cannula (PlasticsOne) or optical fibre (Inper) was placed at least 200 μm above the target brain region and cemented to the skull using dental cement (Lang Dental Manufacturing). After surgery, a dummy cannula was inserted and a cap was screwed on to keep the guide cannula from becoming occluded. Mice were allowed at least two weeks to recover and to express the virus before behavioural training commenced.
SNI
SNI surgery were performed as previously described21 on five- to seven-week old mice under ketamine and xylazine (100 mg kg−1 and 5 mg kg−1, i.p.) anaesthesia. In brief, after skin incision and blunt dissection to expose the sciatic nerve, the tibial and common peroneal branches of the sciatic nerve were ligated with a 5.0 silk suture and transected distally. The sural nerve was left intact. For sham surgery, only skin incision and blunt dissection were performed. After injury, skin was sutured, and mice were left to recover on a heated pad before being returned to their home cages. Mechanical and thermal thresholds were measured two days after the surgery.
CFA injection
As previously described52, mice were anaesthetized with isoflurane (2%). Five microlitres of CFA was injected into the plantar surface of the left hind paw. Mechanical and thermal thresholds were measured one to three days after CFA treatment.
Neonatal spinal cord injection
The neonatal intraspinal cord injection method was modified from a previously published method10. Neonatal pups were injected within 1.5–2.5 days after birth. The pups were covered in aluminium foil, then surrounded in ice for 3–4 min until all movement stopped and the skin colour changed from pink to purple. One to three microlitres of rAAV vector containing 0.04% Trypan blue (for visualization of the injection site) was slowly injected into the spinal cord of cryoanaesthetized neonates using 5-μl syringes (1 inch needle, 30° bevel; Hamilton Company). Only pups in which a blue line in the back of the body could be clearly seen were used for experiments. After injection, pups were recovered on a warming blanket until their skin colour changed to pink; their body movements were observed and they were then returned to the home cage. The neonatal injection did not lead to retrograde labelling of extra neurons in the RVM (Extended Data Fig. 2b,c).
Adult spinal cord injection
The experiments in Fig. 5 were performed using adult spinal cord injection to avoid potential toxicity from long-term AAV1-Cre expression. A dorsal laminectomy was performed at vertebra level T12 to expose the spinal cord, followed by removal of the dura mater. Then, 250 nl of AAV1-hSyn-Cre was injected into the spinal cord 0.3–0.4 mm lateral to the central blood vessel and 0.3 mm below the surface of the spinal cord. Four injections were performed on each side of the spinal cord at a space of 0.5 mm in the anterior-to-posterior direction.
Fibre photometry
Fibre photometry experiments were performed as previously described53 at least four weeks after AAV8-hSyn-FLEX(FRT)-jGCaMP7s was injected into the RVM of Oprm1cre/+ mice receiving neonatal intraspinal cord injection of AAV8-retro-hSyn-FLEX(LoxP)-mTagBFP-P2A-FlpO, or after AAV8-hSyn-FLEX(FRT)-jGCaMP7s was injected into the lSuC of vGlut2cre/+ mice receiving RVM injection of AAV8-retro-hSyn-FLEX(LoxP)-mTagBFP-P2A-FlpO. The implanted fibre was connected to a fibre optic metre (FOM-02M, C-Light) through an optical fibre patch cord (400 μm, 0.50 NA; Inper). Mice were habituated for three days (30 min each) to fibre tethering before fibre photometry recording. To record fluorescence signals, a beam from a 480-nm LED was reflected with a dichroic mirror, and a lens connected to a CMOS detector (DCC3240M, Thorlabs) was used to focus the beam to record fluorescence data. At the tip of the patch cord, the LED power was less than 50 μW. A LabVIEW program (v.2018) was used to control the CMOS camera to record the fluorescent signal. Using an RZ5D processor (Tucker-Davis Technologies), the analogue voltage signal was digitalized, filtered (200 Hz low-pass) and sampled at 3 kHz. OpenEx software (v.2.36, Tucker-Davis Technologies) was used to record the fibre photometry data, and a custom MATLAB script (MathWorks) was used for data analysis. To record mechanical-stimulation-evoked responses in uninjured and SNI mice, 4 g and 0.4 g von Frey fibre, respectively, was applied to the lateral part of the plantar surface of the hind paw four to six times, with a 30-s interval between each stimulation. For thermal stimulation, an infrared laser (70% of maximum power, Ugo Basile) or dry ice was applied to the plantar surface through a glass plane underneath the hind paw four to six times with a 30-s interval between each stimulation. For the CFA-injected group, 4 g (before, and day 21 after CFA) and 0.4 g (day 2 and day 7 after CFA) von Frey fibre was applied to the plantar surface of the hind paw four to six times with a 30-s interval between each stimulation. For thermal stimulation, an infrared laser (70% of maximum power, Ugo Basile) was applied to the plantar surface through a glass plane underneath the hind paw four to six times with a 30-s interval between each stimulation. The video recording was synchronized with the fibre photometry recording through a TTL signal. The fluorescence change (ΔF/F) was calculated as (F − F0)/F0, where F0 is the averaged fluorescence signal during a 3-s baseline period before the onset of withdrawal reflex in each trial. The area under the curve (AUC) was calculated as the integration of ΔF/F during each trial.
To assess the correlation between fibre photometry signals and locomotor speed, each individual mouse was placed in a 25 × 45-cm arena. Locomotion bouts (four to six per mouse) were identified, and the time course of each bout was normalized to a 0–100% scale. Data from a peri-event window (−50% to 150%) were extracted for analysis. The corresponding photometry signals and locomotor speeds were then obtained. Pearson’s correlation analysis was performed between the average photometry signal and the average locomotor speed during the first half of the locomotion bout (−50% to 50%) to quantify the relationship between neuronal activity and movement (spontaneous locomotion or pain-evoked escape).
Chemogenetic manipulation
For chemogenetic activation or silencing experiments, 0.1 mg kg−1 clozapine was injected (i.p.) 30 min before behaviour tests. For terminal silencing experiments, 300 nl of 5 μM clozapine was infused into each target through a cannula 30 min before behaviour tests.
Behavioural tasks
von Frey test
Each mouse was habituated in a red plastic cylinder on an elevated wire grid for at least one hour before testing. Mechanical sensitivity was determined with a set of calibrated von Frey filaments (0.02–4 g, Ugo Basile). For the SNI model, filaments were applied to the lateral part of the left hind paw. Between individual measurements, von Frey filaments were applied at least 3 s after the mice had returned to their initial resting states. A positive withdrawal response was defined as a brisk paw withdrawal, shaking or licking directed at the stimulated paw. Movements related to general locomotion, weight shifting, grooming or exploratory behaviour were not considered withdrawal responses and were excluded from the analysis. The 50% paw withdrawal threshold was determined using Dixon’s up-and-down method18.
Hargreaves test
Each mouse was habituated in a red plastic cylinder on a glass floor for at least one hour before to testing. A radiant heat beam (Hargreaves apparatus, Ugo Basile) was focused onto the left hind paw. The latency to hind paw withdrawal was recorded with at least two trials per mouse repeated at least 10 min apart. A cut-off latency of 20 s was set to avoid tissue damage.
Plantar cold test
Each mouse was habituated in a red plastic cylinder on a glass floor for at least one hour before testing. The test was performed by applying a dry-ice pellet to the plantar surface of the hind paw through a glass floor. The pellet was prepared by packing powdered dry ice into a modified syringe. The latency to paw withdrawal was recorded.
Formalin test
eGFP- or hM4D-expressing mice were infused with clozapine 30 min before the test. Twenty microlitres of 2.5% formalin was injected into the dorsal surface of the hind paw. Paw licking or biting duration were quantified during phase I (0–10 min after formalin injection) and phase II (11–40 min after formalin injection).
Affective–motivational pain behaviours
Affective–motivational responses were evaluated as previously described25. Each mouse was habituated in a red plastic cylinder on a glass floor for at least one hour before testing. Mice received a series of mechanical stimuli using von Frey filaments (0.07 g, light touch; 0.4 g, mild touch; 2.0 g, moderate touch). Each filament was applied 10 times per session at 20–30-s intervals, with 60-s pauses between stimulus blocks. The time spent attending to the injured paw (intentional lifting or licking) and performing escape behaviours (accelerating away or rearing toward chamber openings) was quantified.
Spontaneous pain behaviours
Spontaneous pain behaviours were evaluated as previously described26. Mice were habituated to the observation chamber for three days (30 min per day) before video recording. Mice were placed in a transparent plastic box (10 × 10 × 10 cm) on a glass floor. Spontaneous pain was quantified from 30-min video recordings (bottom view). Flinching and licking behaviours were scored as 1 and 2, respectively; licking during grooming was excluded.
Mouse facial-grimace scale
Facial grimacing was assessed as described27. Mice were habituated to the observation chamber for three days (30 min per day) before video recording. In brief, each mouse was placed in a white wooden chamber (9 × 9 × 9 cm with a 2-cm floor extension) for an 8-min video recording. Videos were scored online (https://painface.net) using a 0–2 scale for orbital tightening, ear position, whisker change and nose bulge.
Air-puff-evoked blink response
The air-puff test was based on a previously described method54. Head-fixed mice were habituated on a running wheel for three days. The periocular area was recorded under infrared illumination. Compressed air (20 psi) was delivered in 60-ms pulses every 3 s via polyethylene tubing positioned 2.5 mm from the right eye. Each trial consisted of eight puffs, and blink responses were quantified from three or four consecutive trials, with at least one minute between trials. Bonsai-rx software (v.2.8) was used to control simultaneous video recording and air-puff timing, and to crop the recording area.
Conditioned place aversion test
The conditioned place aversion (CPA) assay was based on a previously described method55. In brief, mice were habituated for three days (30 min each) to a custom-designed two-compartment CPA apparatus (30 cm × 25 cm × 20 cm) placed on an elevated mesh rack. Each chamber contained unique visual cues (black and grey stripes or plain grey walls). On the final day of habituation, baseline preferences were video-recorded for 10 min and movement was tracked using the custom tracking software running on MATLAB (MathWorks). After baseline measurements, mice were confined to their preferred side of the chamber and paired with repeated stimulation of the left hind paw once every 10 s for 10 min using a 0.16-g filament. After pairing, mice were returned to their home cage for 20 min before being re-exposed to the CPA chamber with free access to both side of the CPA chamber for 10 min. CPA scores were calculated by subtracting the time spent in the filament-stimulation-paired side of the chamber during baseline from the time spent in the same side of the chamber during the re-exposure.
Open field test
Each mouse was placed in a square arena (50 × 50 cm) with a defined centre zone (30 × 30 cm). Locomotor activity and time spent in the centre were recorded and analysed as measures of exploratory drive and anxiety-like behaviour.
Elevated plus maze
The elevated plus maze (EPM) consisted of two open and two enclosed arms (each 30 cm long), elevated above the floor. Mice were placed at the central junction facing an open arm and allowed to explore for 5 min. The time spent in the open arms was analysed as an index of anxiety-like behaviour.
Tail suspension test
Mice were suspended by the tail for 6 min in an enclosed chamber (55 cm (H) × 15 cm (W) × 11.5 cm (D). Total immobility time was analysed.
Locomotion test
Mice were habituated for three days (30 min each) to a custom-designed apparatus (50 cm × 50 cm × 30 cm). On consecutive days, mice were injected (i.p.) with saline, clozapine (0.1 mg kg−1, Cayman Chemical) or morphine (15 mg kg−1, Hikma), and locomotor activity was measured for 10 min (for chemogenetic manipulation) or 30 min (for morphine-induced locomotion).
Textured novel object recognition test
The textured novel object recognition test (NORT) was based on a previously described method44. Bilateral whisker plucking was performed three days before the habituation session. Mice were habituated to the behaviour chamber for 10 min 24 h before the familiarization session. In the familiarization session, the mouse was placed in the chamber containing two copies of identical objects for 10 min, and was then removed from the chamber and placed in a transport cage for 5 min. Next, the mouse was reintroduced to the chamber and presented with the familiar object and a novel object that differ only in texture. For both sessions, the experiments end when there has been a 20-s physical interaction of both objects or when a 10-min period has been reached. The discrimination index was calculated as the change in the percentage of time spent on the selected object in each session.
Textured conditioned place preference
In brief, mice were habituated for three days (30 min each) to a custom-designed two-compartment CPA apparatus with unique visual cues in each compartment (30 cm × 25 cm × 20 cm). Baseline preferences were video-recorded for 10 min and movement was tracked using the custom tracking software running on MATLAB (MathWorks). Next, the floor of the less-preferred side of the chamber was covered with fine-grit sandpaper, whereas the other side was left as smooth plexiglass. For texture pairing, the mouse was then reintroduced to the chamber, and movement was video-recorded and tracked. Thirty minutes after clozapine infusion, the mouse was placed back into the chamber, and the difference in the time spent in the sandpaper side between the pairing and the test was calculated.
Measurement of heart rate and breathing rate
Measurements of heart rate were performed as previously described56. In brief, after anaesthetization with isoflurane, two electrodes were connected to the forelimb and hindlimb of the mice. The electrocardiogram signal was filtered (10 Hz–500 Hz), amplified (×100) with an amplifier, digitized (10 kHz) and stored with Spike 2 software (v.7.03). For breathing rate, the skin on top of the most posterior rib was tied and connected to a customized force transducer, which vibrates during each breathing cycle. The changes in voltage caused by the vibration of the force transducer were filtered (10 Hz–500 Hz), amplified (×100) with an amplifier, digitized (10 kHz) and stored with Spike 2 software (v.7.03).
Electrophysiology
Four weeks after AAV injection, mice (eight to nine weeks old) were deeply anaesthetized and transcardially perfused with ice-cold cutting solution containing (in mM) 92 N-methyl-d-glucamine (NMDG), 2.5 KCl, 1.25 NaH2PO4, 25 d-glucose, 20 HEPES, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 30 NaHCO3, 12 N-acetyl-l-cysteine, 10 MgSO4, 0.5 CaCl2 and 0.005 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline (NBQX) (pH 7.2). Acute coronal slices (250 μm) containing RVM were made using a vibratome (VT1000S, Leica), and recovered in cutting solution without NBQX, and then for at least one hour in aCSF containing (in mM) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 10 d-glucose, 1 MgCl2, 26 NaHCO3 and 2 CaCl2 (pH 7.3) at room temperature. All slice recordings were done in room-temperature artificial cerebrospinal fluid (aCSF) continuously perfused with O2 balanced with 5% CO2.
Whole-cell patch-clamp recordings were obtained in RVM cells located adjacent to midline (less than 150 μm lateral from midline). For current clamp recording, glass microelectrodes (3–7 MΩ) were filled with internal solution containing (in mM) 130 K-gluconate, 1 KCl, 10 HEPES, 10 EGTA, 1 MgCl2, 1 CaCl2, 2 Mg-ATP and 0.2 Na-GTP (pH 7.3). Neurobiotin (0.3% w/v, Vector Laboratories) was also included to label recorded neurons. Descending RVM neurons were identified by the expression of Clover3 under a 40× water-immersion objective lens using a fluorescence microscope (BX51WI, Olympus). The baseline firing rate was monitored for 3 min before and after application of drugs. DAMGO (1 μM, Tocris) was bath-applied with or without CTAP (1 μM, Cayman Chemical) for 10 min. Cells exhibiting more than 20% variance in baseline firing rate were excluded from further analyses. Action potentials were detected and counted using Clampfit v.10.2 (Molecular Devices).
For voltage-clamp recordings, internal solution containing (in mM) 130 CsMeSO3, 1 MgCl2, 1 CaCl2, 11 EGTA, 10 HEPES, 2 Mg-ATP, 0.2 Na-GTP and 5 QX314 (Tocris) was used. Picrotoxin (100 μM) was bath-applied throughout the recordings. RVM cells were held at −70 mV, and a 2-ms pulse of 470-nm blue light was emitted with an LED driver (M470L2, Thorlabs) every 15 s to activate ChR2-expressing axon terminals. After monitoring baseline oEPSCs for 5 min, clozapine-N-oxide (5 μM, Cayman Chemical) was bath-applied for 15 min to activate hM4D. Amplitudes of oEPSCs from baseline and the last 5 min of drug application were analysed. All chemicals were obtained from Sigma unless specified otherwise.
After whole-cell patch-clamp recording, slices were fixed with 4% paraformaldehyde (PFA) in PBS at 4 °C overnight. Fixed slices were further sectioned to 50 μm using a vibratome and washed with PBS three times. Tissues were then incubated with streptavidin conjugated to Alexa Fluor 647 (1:1,000, Thermo Fisher Scientific) in PBS containing 0.5% Triton X-100 overnight at room temperature. Sections were then washed with PBS three times. Z-stacks of confocal fluorescent images were taken with Airyscan2 (LSM980, ZEISS).
For experiments in Fig. 5b, AAV8-retro-mCherry was injected into the lSuC and AAV8-ChR2 was injected into the Po or VPL. Slices were prepared four or five weeks after virus injection. A 2-ms pulse of 470-nm blue light was emitted with an LED driver (M470L2, Thorlabs) every 15 s to activate ChR2-expressing axon terminals.
cTRIO tracing and analysis
Experiments in Figs. 3c–e and 4a,b were performed in Oprm1cre+/− mice with neonatal spinal cord injection of AAV8-retro-hSyn-FLEX(LoxP)-mTagBGFP-P2A-FlpO into the spinal cord, and in vGlut2cre+/− mice with injection of AAV8-retro-hSyn-FLEX(LoxP)-mTagBGFP-P2A-FlpO into the RVM, respectively. Two to six weeks later, 200 nl of AAV8-hSyn-FLEX(FRT)-EGFP-P2A-TVA-T2A-oG was injected into the RVM or lSuC using a stereotaxic instrument (BenchMARK Digital, Leica). Four weeks later, 200 nl SADΔG-mCherry(EnvA) or CVS-N2cΔG-mCherry(EnvA) was injected into the same area of the RVM or lSuC using the procedure described above. Mice were housed in a biosafety level 2 (BSL2) facility for seven days before being euthanized. For quantifications of long-range-input brain regions, brain regions 1 mm anterior or posterior to the injection site were excluded from analysis. Images were taken from consecutive 50-μm coronal sections using a ZEISS Axioplan2 with a 2.5× or 5× objective. Cell counting was performed manually using Fiji. For quantifications of subregions, boundaries were based on the Allen Institute’s reference atlas (https://mouse.brain-map.org/experiment/thumbnails/100048576?image_type=atlas). No adjustment was made for potential double counting of cells from consecutive sections. Fractional input was calculated by dividing the number of labelled neurons in each brain region by the total number of labelled neurons throughout the entire brain.
Ablation of OPRM1+ RVMSC neurons
AAV8-hSyn-FLEX(FRT)-taCaspase3-TEVp and AAV8-hSyn-FLEX(LoxP)-Ruby3-FLEX(FRT)-Clover3 were co-injected into the RVM of Oprm1cre/+ mice with neonatal injection of AAV8-retro-hSyn-FLEX-mTagBFP-P2A-FlpO (1 μl) into the spinal cord. For control mice, AAV8-hSyn-FLEX(LoxP)-Ruby3-FLEX(FRT)-Clover3 was injected into the RVM. The infected OPRM1+ RVM neurons express mRuby3 (red), and OPRM1+ RVMSC neurons express both mRuby3 and Clover3 (yellow). Four weeks later, after behavioural tests, the brains and spinal cords were collected from both groups for histological analysis.
Immunostaining and RNAscope in situ hybridization
Mice were deeply anaesthetized with pentobarbital sodium solution and transcardially perfused with PBS, followed by 4% PFA in 1× PBS at room temperature. Brains and spinal cords were dissected from perfused mice and post-fixed in 4% PFA in 1× PBS at 4 °C overnight, cryoprotected in 30% sucrose in 1× PBS at 4 °C overnight, embedded in OCT compound, frozen using dry ice and kept at –80 °C. Brains and spinal cords were cryosectioned (14 μm for RNAscope in situ hybridization or 50 μm for immunostaining) using a cryostat (Leica). For immunostaining, 50-μm sections were washed three times for 5 min each with 1× PBS. They were then incubated in a blocking solutions (0.3% PBST containing 10% normal donkey serum (LAMPIRE Biological Products 7332100)) for two hours at room temperature. Subsequently, the sections were incubated with primary antibodies diluted in 0.3% PBST containing 3% normal donkey serum overnight at 4 °C. After three 10-min washes with PBS, the sections were incubated with secondary antibodies diluted in 0.3% PBST containing 5% normal donkey serum for two hours at room temperature. After this, the sections were washed three times for 10 min each with PBS; Hoechst 33342 solution (Thermo Fisher Scientific) was included in the second wash at a 1:10,000 dilution. Finally, the sections were mounted with Fluoromount-G (Southern Biotech). The primary antibodies used in this study included rabbit anti-mCherry (1:1,000, 600401397, Rockland), rabbit anti-FOS (1:1,000, 226003, Synaptic Systems), goat anti-TPH2 (1:500, ab121013, Abcam) and mouse anti-HA (1:1,000, 901514, Biolegend). Secondary antibodies included Alexa-594- or 647-conjugated donkey anti-rabbit antibodies, Alexa-647-conjugated donkey anti-mouse antibodies and Alexa-647-conjugated anti-goat antibodies. All secondary antibodies were purchased from Life Technologies and used at a 1:1,000 dilution. For RNAscope in situ hybridization, 14-μm sections were collected on glass slide and mRNA transcripts were detected using the RNAscope Fluorescent Multiplex Assay (Advanced Cell Diagnostics) and RNAscope Fluorescent Multiplex Reagent Kit v2 (323100). The RNAscope catalogue probes were used to detect Oprm1 (493251), vGat (319191), vGlut2 (319171), Tph2 (318691) and Penk (318761) RNA molecules. Images were obtained using a ZEISS 710 confocal microscope using either 10× (Plan-Apochromat 10×, NA 0.45) or 20× (Plan-Apochromat 20×, NA 0.8) objectives. For imaging large sections of spinal cord and brain, the tile-scan function was used and the tile images were stitched using ZEISS Zen microscope software.
For FOS immunostaining, each SNI mouse was habituated in a red plastic cylinder on a glass floor for at least one hour before to testing. The left hindpaws were repeatedly stimulated once every 10 s for 10 min using a 0.16-g filament (SNI mice), or stimulated with a 1.4-g filament 90 times for 10 min (non-injured mice). Mice were perfused 1.5 h after the delivery of each stimulation, then processed for FOS immunohistochemical analysis.
Two-dimensional registration of RVM descending neurons and 3D visualization
Thirty to forty brain slices (50 μm) containing the RVM were scanned using Olympus VS120 for two-dimensional (2D) registration. Custom MATLAB software was used to remove all image features outside the brain slices. Background subtraction and contrast enhancement of the Neurotrace 640 channel were then applied. The processed Neurotrace 640 images for each section were then serially analysed using a combination of automated and manual methods. For a more detailed description of this procedure, see a previous report57. Instructions and custom codes written in MATLAB for 2D image registration can be found at https://sites.google.com/view/brain-mapping/home/code.
TESOS imaging and single-cell tracing of OPRM1+ RVMSC neurons
AAV8-EF1α-FLEX(LoxP)-mScarlet was injected into the brainstem of Oprm1cre/+ mice (200 nl at Bregma −5.6 mm, lateral ±0.1 mm, ventral 5.75 mm). Six weeks after AAV injection, Mice were deeply anaesthetized with pentobarbital sodium solution and transcardially perfused with PBS, followed by 4% PFA in 1× PBS at room temperature. Brains and spinal cords were dissected and post-fixed in 4% PFA in 1× PBS at 4 °C overnight. The brain and spinal tissues were then subjected to the TESOS clearing procedure through passive immersion, and the transparent embedding was performed 48 h after sample transparency, as previously described12. Confocal images of transparently embedded samples were obtained using an upright Nikon confocal microscope combined with a rotary microtome using the 40× objective, and reconstructed using custom software provided by the laboratory of H. Zhao. Manual tracing of axons was performed using Lychnis (https://github.com/SMART-pipeline/Lychnis-tracing)58.
Statistical analysis
No statistical methods were used to predetermine sample size. However, the sample sizes were comparable to those in previous publications. All analyses were performed using GraphPad Prism software and statistical methods are indicated when used. Normality was assessed for each dataset before analysis. In cases in which data did not meet the assumption of normality, appropriate non-parametric tests were used. When a t-test was used, it was performed as a two-tailed test. No method of randomization was used in any of the experiments. Experimenters were not blinded to group allocation in behavioural experiments, but CPA scores were measured automatically by custom tracking software running in MATLAB (MathWorks). All mice that completed the entire behavioural training and testing were included in the analysis. Unless otherwise indicated in the figure legend, data points are derived from individual mice and are presented as mean ± s.e.m.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Images showing the expression of OPRM1 in pain-related regions in the brain, spinal cord and sensory neurons from the Oprm1cre × Ai14 mice are available at Zenodo: https://doi.org/10.5281/zenodo.17110833 (ref. 59). Source data are provided with this paper.
Code availability
The code for tracking locomotion is available on GitHub using the link: https://github.com/thepenglab/posTracker. Instructions and custom codes written in MATLAB for 2D image registration can be found at https://sites.google.com/view/brain-mapping/home/code.
References
Fields, H. State-dependent opioid control of pain. Nat. Rev. Neurosci. 5, 565–575 (2004).
Heinricher, M. M. Pain modulation and the transition from acute to chronic pain. Adv. Exp. Med. Biol. 904, 105–115 (2016).
Ossipov, M. H., Dussor, G. O. & Porreca, F. Central modulation of pain. J. Clin. Invest. 120, 3779–3787 (2010).
Dogrul, B. N. et al. Descending facilitation from rostral ventromedial medulla mu opioid receptor-expressing neurons is necessary for maintenance of sensory and affective dimensions of chronic neuropathic pain. Pain 166, 153–159 (2025).
Chen, Q. & Heinricher, M. M. Shifting the balance: how top-down and bottom-up input modulate pain. Front. Pain Res. 3, 932476 (2022).
Fields, H. L., Bry, J., Hentall, I. & Zorman, G. The activity of neurons in the rostral medulla of the rat during withdrawal from noxious heat. J. Neurosci. 3, 2545–2552 (1983).
Heinricher, M. M., Morgan, M. M. & Fields, H. L. Direct and indirect actions of morphine on medullary neurons that modulate nociception. Neuroscience 48, 533–543 (1992).
Hosobuchi, Y., Adams, J. E. & Linchitz, R. Pain relief by electrical stimulation of the central gray matter in humans and its reversal by naloxone. Science 197, 183–186 (1977).
Reynolds, D. V. Surgery in the rat during electrical analgesia induced by focal brain stimulation. Science 164, 444–445 (1969).
Ayers, J. I. et al. Widespread and efficient transduction of spinal cord and brain following neonatal AAV injection and potential disease modifying effect in ALS mice. Mol. Ther. 23, 53–62 (2015).
Tervo, D. G. et al. A designer AAV variant permits efficient retrograde access to projection neurons. Neuron 92, 372–382 (2016).
Yi, Y. et al. Mapping of individual sensory nerve axons from digits to spinal cord with the transparent embedding solvent system. Cell Res. 34, 124–139 (2024).
François, A. et al. A brainstem–spinal cord inhibitory circuit for mechanical pain modulation by GABA and enkephalins. Neuron 93, 822–839 (2017).
Zhang, Y. et al. Identifying local and descending inputs for primary sensory neurons. J. Clin. Invest. 125, 3782–3794 (2015).
Arvidsson, U. et al. Distribution and targeting of a mu-opioid receptor (MOR1) in brain and spinal cord. J. Neurosci. 15, 3328–3341 (1995).
Schwarz, L. A. et al. Viral-genetic tracing of the input-output organization of a central noradrenaline circuit. Nature 524, 88–92 (2015).
Dana, H. et al. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods 16, 649–657 (2019).
Chaplan, S. R., Bach, F. W., Pogrel, J. W., Chung, J. M. & Yaksh, T. L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63 (1994).
Cheah, M., Fawcett, J. W. & Andrews, M. R. Assessment of thermal pain sensation in rats and mice using the Hargreaves test. Bio Protoc. 7, e2506 (2017).
Brenner, D. S., Golden, J. P. & Gereau, R. W. A novel behavioral assay for measuring cold sensation in mice. PLoS ONE 7, e39765 (2012).
Decosterd, I. & Woolf, C. J. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158 (2000).
Porreca, F. et al. Inhibition of neuropathic pain by selective ablation of brainstem medullary cells expressing the mu-opioid receptor. J. Neurosci. 21, 5281–5288 (2001).
Cai, Y. Q., Wang, W., Hou, Y. Y. & Pan, Z. Z. Optogenetic activation of brainstem serotonergic neurons induces persistent pain sensitization. Mol. Pain 10, 70 (2014).
Yang, C. F. et al. Sexually dimorphic neurons in the ventromedial hypothalamus govern mating in both sexes and aggression in males. Cell 153, 896–909 (2013).
Corder, G. et al. An amygdalar neural ensemble that encodes the unpleasantness of pain. Science 363, 276–281 (2019).
Zheng, Q. et al. Synchronized cluster firing, a distinct form of sensory neuron activation, drives spontaneous pain. Neuron 110, 209–220 (2022).
McCoy, E. S. et al. Development of PainFace software to simplify, standardize, and scale up mouse grimace analyses. Pain 165, 1793–1805 (2024).
Cheng, L. et al. Identification of spinal circuits involved in touch-evoked dynamic mechanical pain. Nat. Neurosci. 20, 804–814 (2017).
Aaron, R. V. et al. Prevalence of depression and anxiety among adults with chronic pain: a systematic review and meta-analysis. JAMA Netw. Open 8, e250268 (2025).
Zhou, W. et al. A neural circuit for comorbid depressive symptoms in chronic pain. Nat. Neurosci. 22, 1649–1658 (2019).
Samineni, V. K. et al. Divergent modulation of nociception by glutamatergic and GABAergic neuronal subpopulations in the periaqueductal gray. eNeuro 17, ENEURO.0129-16.2017 (2017).
Basbaum, A. I., Clanton, C. H. & Fields, H. L. Opiate and stimulus-produced analgesia: functional anatomy of a medullospinal pathway. Proc. Natl Acad. Sci. USA 73, 4685–4688 (1976).
Chen, Q. et al. Optogenetic evidence for a direct circuit linking nociceptive transmission through the parabrachial complex with pain-modulating neurons of the rostral ventromedial medulla (RVM). eNeuro 19, ENEURO.0202-17.2017 (2017).
Palmiter, R. D. Parabrachial neurons promote nociplastic pain. Trends Neurosci. 47, 722–735 (2024).
Wang, S. & Redgrave, P. Microinjections of muscimol into lateral superior colliculus disrupt orienting and oral movements in the formalin model of pain. Neuroscience 81, 967–988 (1997).
Yang, W., Kanodia, H. & Arber, S. Structural and functional map for forelimb movement phases between cortex and medulla. Cell 186, 162–177 (2023).
Wang, L. H., Ding, W. Q. & Sun, Y. G. Spinal ascending pathways for somatosensory information processing. Trends Neurosci. 45, 594–607 (2022).
Chen, H. et al. The functional and anatomical characterization of three spinal output pathways of the anterolateral tract. Cell Rep. 43, 113829 (2024).
Hodge, C. J. & Apkarian, A. V. The spinothalamic tract. Crit. Rev. Neurobiol. 5, 363–397 (1990).
Zhu, X. et al. Distinct thalamocortical circuits underlie allodynia induced by tissue injury and by depression-like states. Nat. Neurosci. 24, 542–553 (2021).
Ding, W. Q. et al. Single-neuron projectome reveals organization of somatosensory ascending pathways in the mouse brain. Neuron 113, 2083–2101 (2025).
Lee, K. S. et al. Transformation of neural coding for vibrotactile stimuli along the ascending somatosensory pathway. Neuron 112, 3343–3353 (2024).
Zingg, B. et al. AAV-mediated anterograde transsynaptic tagging: mapping corticocollicular input-defined neural pathways for defense behaviors. Neuron 93, 33–47 (2017).
Orefice, L. L. et al. Peripheral mechanosensory neuron dysfunction underlies tactile and behavioral deficits in mouse models of ASDs. Cell 166, 299–313 (2016).
Li, L. et al. The functional organization of cutaneous low-threshold mechanosensory neurons. Cell 147, 1615–1627 (2011).
Abraira, V. E. & Ginty, D. D. The sensory neurons of touch. Neuron 79, 618–639 (2013).
Huang, J. et al. A neuronal circuit for activating descending modulation of neuropathic pain. Nat. Neurosci. 22, 1659–1668 (2019).
Huang, T. et al. Identifying the pathways required for coping behaviours associated with sustained pain. Nature 565, 86–90 (2019).
Melzack, R. & Wall, P. D. Pain mechanisms: a new theory. Science 150, 971–979 (1965).
Braz, J., Solorzano, C., Wang, X. & Basbaum, A. I. Transmitting pain and itch messages: a contemporary view of the spinal cord circuits that generate gate control. Neuron 82, 522–536 (2014).
Liu, S. et al. Neural basis of opioid-induced respiratory depression and its rescue. Proc. Natl Acad. Sci. USA 118, e2022134118 (2021).
Fehrenbacher, J. C., Vasko, M. R. & Duarte, D. B. Models of inflammation: carrageenan- or complete Freund's adjuvant (CFA)-induced edema and hypersensitivity in the rat. Curr. Protoc. Immunol. 56, 5.4.1–5.4.7 (2012).
Gunaydin, L. A. et al. Natural neural projection dynamics underlying social behavior. Cell 157, 1535–1551 (2014).
Kauvar, I. et al. Conserved brain-wide emergence of emotional response from sensory experience in humans and mice. Science 388, eadt3971 (2025).
Donnelly, C. R. et al. STING controls nociception via type I interferon signalling in sensory neurons. Nature 591, 275–280 (2021).
Shang, C. et al. A parvalbumin-positive excitatory visual pathway to trigger fear responses in mice. Science 348, 1472–1477 (2015).
Xiong, J., Ren, J., Luo, L. & Horowitz, M. Mapping histological slice sequences to the Allen Mouse Brain Atlas without 3D reconstruction. Front. Neuroinform. 12, 93 (2018).
Xu, F. et al. High-throughput mapping of a whole rhesus monkey brain at micrometer resolution. Nat. Biotechnol. 39, 1521–1528 (2021).
Chen, X. Oprm1-Cre x Ai14. Zenodo https://doi.org/10.5281/zenodo.17110833 (2025).
Acknowledgements
We thank H. L. Fields, L. Luo and members of the X.C. laboratory for comments on the manuscript; H. Zhao for single-axon tracing, the Microscope Core at Stanford’s Wu Tsai Neurosciences Institute for microscope services; and Y. Q. Peng for sharing locomotor tracking code. This work was supported by grants from the NIH (R01DA045664, R01MH116904, R01HL150566, R01NS129834, R01DA059602, RF1NS126076 and R01MH135934), a Terman Fellowship, a Firmenich Scholarship, the Wu Tsai Neurosciences Institute’s NeuroChoice Initiative and the ChEM-H Innovative Medicines Accelerator from Stanford University. X.C. also acknowledges support from the Tianqiao and Chrissy Chen Institute.
Author information
Authors and Affiliations
Contributions
X.C. conceived the study. Q.W., G.N. and X.C. designed the experiments. Q.W., J.H.L., G.N., Y.Y., M.A.M., L.Y. and W.Q. performed experiments. J.X. and M.A.H. developed image registration algorithm. Q.W., J.H.L., G.N. and X.C. analysed data. Q.W., G.N. and X.C. wrote the paper, with inputs from all authors.
Corresponding author
Ethics declarations
Competing interests
X.C., Q.W. and G.N. have filed a patent covering some components used in this study (US application: PCT/US2022/07356; Stanford file: S21-124).
Peer review
Peer review information
Nature thanks Cheng-Chang Lien, Gerald Zamponi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Generation and characterization of the Oprm1Cre knock-in mouse line.
a, Gene targeting strategy used to generate the Oprm1Cre knock-in mouse line. b, Morphine produced similar analgesic effect in WT (black) and Oprm1Cre/+ (blue), but not in Oprm1Cre/Cre (red) mice (Non-parametric ANOVA (Friedman test), post-hoc pairwise comparisons were conducted using Dunn’s multiple comparison test. WT Saline vs. 2.5 mg/kg morphine: P = 0.1733; WT Saline vs. 5 mg/kg morphine: P = 0.0133; WT 2.5 mg/kg morphine vs. 5 mg/kg morphine: P > 0.9999; Oprm1Cre/+ Saline vs. 2.5 mg/kg morphine: P = 0.6177; Oprm1Cre/+ Saline vs. 5 mg/kg morphine: P = 0.0080; Oprm1Cre/+ 2.5 mg/kg morphine vs. 5 mg/kg morphine: P = 0.2460; Oprm1Cre/Cre Saline vs. 2.5 mg/kg morphine: P > 0.9999; Oprm1Cre/Cre Saline vs. 5 mg/kg morphine: P > 0.9999; Oprm1Cre/Cre 2.5 mg/kg morphine vs. 5 mg/kg morphine: P > 0.9999. n = 5 mice for each genotype). c, Morphine increased locomotion in WT (black) and Oprm1Cre/+(blue), but not in Oprm1Cre/Cre (red) mice. Two-sided Wilcoxon signed-rank test (WT saline vs. WT 15 mg Kg−1 morphine, P = 0.0313; Oprm1Cre/+ saline vs. Oprm1Cre/+ 15 mg Kg−1 morphine, P = 0.0313; Oprm1Cre/Cre saline vs. Oprm1Cre/Cre 15 mg Kg−1 morphine, P = 0.0625). n = 6 mice for WT and Oprm1Cre/+, n = 5 mice for Oprm1Cre/Cre. d, Representative images from three independently performed experiments show the Ruby3-expressing terminal in the thalamus from OPRM1+ ascending RVM neurons after injection of AAV9-FLEX(LoxP)-Ruby3 into the RVM of the Oprm1Cre mice. Scale bar: 1 mm. * P < 0.05, ** P < 0.01. Data are presented as mean ± s.e.m.
Extended Data Fig. 2 Developing and characterization of the neonatal intraspinal injection strategy and AAV8retro.
a, Images of dissected spinal cord from mice injected with 3 μl (left) and 1 μl (right) of AAV8retro-mCherry. Neonatal injection of 1 μl or 3 μl of AAV8retro-mCherry led the virus infection of the entire lumbar region or both lumber and thoracic regions of the spinal cord, respectively. Inset shows the expression of mCherry was restricted largely in the dorsal horn. Scale bar: 1 mm. b, Representative images of RVM section from three independently performed experiments following spinal cord injection of AAV8retro-H2B-Ruby3 at Postnatal day 1(neonatal, P1) and injection of AAV8retro-H2B-Clover3 at P28 (adult), and Venn diagram illustrated the number of Ruby3-labelled, Clover3-labelled, and colocalized neurons in the RVM. Scale bar: 100 μm. c, Representative medulla section from three independently performed experiments following neonatal and adult spinal cord injection of AAV8retro-H2B-Ruby3 and AAV8retro-H2B-Clover3, respectively. Retrogradely labelled neurons in RVM and facial motor nucleus (FMN). Scale bar: 1 mm. d, Plasmid map shows the site for inserting the decapeptide LADQDYTKTA between N590 and T591 of the AAV8 capsid (AAV8retro). e, Representative images of retrogradely labelled RVM neurons by AAV8retro-mCherry (red), AAV2retro-eGFP (green), and merge (yellow) after co-injection of AAV8retro-mCherry and AAV2retro-eGFP into the spinal cord of P1.5 wild-type mice. Scale bar: 200 μm. GRN: gigantocellular reticular nucleus; MARN: magnocellular reticular nucleus. f, Quantification of e (n = 3). g, Representative images (left) from three independently performed experiments and quantification (right) of retrogradely labelled neurons in the prefrontal cortex (PFC) when co-inject AAV8retro-mCherry (red) and AAV2retro-eGFP (green) into the paraventricular nucleus of the thalamus (PVT) (inset). Scale bar: PVT: 100 μm; PFC: 200 μm. h, Quantification of retrogradely labelled neurons in the RVM when co-inject AAV8retro-mCherry (red) and CTB488 (green) into the adult spinal cord. i, Representative image from three independently performed experiments of retrogradely labelled neurons in the LC and axon terminals in parabrachial nucleus (PBN) when inject AAV8retro-Cre in the spinal cord of Ai14 mice. Scale bar: 500 μm. j, Representative images three independently performed experiments of retrogradely labelled neurons in the RVM when inject AAV8retro-H2B-Clover3-FLEX(LoxP)-H2B-Ruby3 in the spinal cord of Oprm1Cre/+ mice. Majority of OPRM1+ RVMSC neurons were located outside the lateral paragigantocellular nucleus (LPGi). Scale bar: 200 μm.
Extended Data Fig. 3 Characterization of OPRM1+ RVMSC neurons.
a, Schematic showing spinal injection of AAV8-retro-FLEX(LoxP)-Flp at P1.5 in Oprm1Cre mice, then four weeks later, RVM injection of AAV8-FLEX(FRT)-Clover3. b, Representative images from three independently performed experiments show the terminals of descending OPRM1+ RVMSC neurons in the spinal cord. Notably, axons of OPRM1+ RVMSC neurons are clustered in lateral funiculus (dash line) before gets into the spinal cord, and its terminals are enriched in the dorsal horn but not in the motor area in the spinal cord. Scale bar: 200 μm. c, Representative TESOS images and single-cell tracing of OPRM1+ RVMSC neurons from three independently performed experiments. Axons of OPRM1+ RVMSC neurons of both red and pink neurons are travelled along the left lateral funiculus, then enter the spinal cord to innervate both sides of the dorsal horn. Scale bar: 500 μm. d, Quantification of molecular markers expressed in the OPRM1+ RVMSC neurons. Related to Fig. 1e. e, Representative images from three independently performed experiments of retrogradely labelled spinal cord projecting Tph2+ neurons in the RVM (left) and median raphe (MR, right) after spinal injection of AAV8-retro-mCherry. More Tph2+ neurons were labelled in MR than in RVM. mCherry labelled neurons are in red, Tph2+ neurons are visualized by immunostaining in green. Scale bar: 500 μm. f, Representative image from three independently performed experiments shows OPRM1+ LCSC neurons labelled by intraspinal injection of AAV8-retro-FLEX(LoxP)-Rpl22-3XHA. RNAscope probes were used to visualize Oprm1 (red), HA tag (green) was visualized by immunostaining. Scale bar: 100 μm.
Extended Data Fig. 4 Fibre photometry recording.
a, Representative average traces of non-withdrawal and withdrawal trails Pre-SNI (left) and Post-SNI (right) in OPRM1+ RVMSC neurons. Scale bar: 1% ΔF/F, 3 sec. b, Quantification of AUC of a. n = 6, Two-sided Paired t-test (Normal non-withdrawal vs. withdrawal: P = 0.006; SNI non-withdrawal vs. withdrawal: P = 0.0017). c, Quantification of OPRM1+ RVMSC neurons respond to mechanical (von Frey) and thermal (Hargreaves and Plantar cold) stimuli in normal and SNI mice (n = 7–8). Two-sided Mann–Whitney test (Von Frey normal vs. SNI: P = 0.0003; Hargreaves normal vs. SNI: P = 0.62; Plantar cold normal vs. SNI: P = 0.0041). Related to Fig. 1h. d,e, Representative traces (d) and quantification (e) of mechanical (von Frey) and thermal (Hargreaves) stimulation evoked calcium response in OPRM1+ RVMSC neurons of before, 2, 7, and 21 days after CFA injection. n = 6, Non-parametric ANOVA (Friedman test), post-hoc pairwise comparisons were conducted using Dunn’s multiple comparison test (von Frey Pre vs. 2D post CFA: P = 0.0048; von Frey Pre vs. 7D post CFA: P > 0.9999; von Frey Pre vs. 21D post CFA: P > 0.9999; von Frey 2D post CFA vs. 7D post CFA: P = 0.1521; von Frey 2D post CFA vs. 21D post CFA: P = 0.0834; von Frey 7D post CFA vs. 21D post CFA: P > 0.9999; Hargreaves Pre vs. 2D post CFA: P = 0.4418; Hargreaves Pre vs. 7D post CFA: P > 0.9999; Hargreaves Pre vs. 21D post CFA: P > 0.9999; Hargreaves 2D post CFA vs. 7D post CFA: P = 0.7051; Hargreaves 2D post CFA vs. 21D post CFA: P = 0.2650; Hargreaves 7D post CFA vs. 21D post CFA: P > 0.9999). Scale bar: 1% ΔF/F, 2 sec. f, Representative traces of calcium response (left, upper panel) and movement speed (left, lower panel), and correlation of calcium response vs. movement speed (right) in normal mice. Dash line indicates the time of movement start. Dot indicates the average speed calcium signal during each movement event. Red line: linear fitting of dots. Pearson’s r = −0.1453 or 0.9176 for locomotion and escape, respectively. Scale bar: 50 % of normalized movement bout, 1% ΔF/F, 5 cm/s. Related to Fig. 1j. Locomotion: spontaneous movement without stimulus; Escape: escape response after von Frey stimulus. g, Quantification of f and Fig. 1j. n = 6, one-way ANOVA, post-hoc pairwise comparisons were conducted using Tukey’s multiple comparisons test (Locomotion BL vs. escape BL: P = 0.0169; Locomotion BL vs. Locomotion SNI: P = 0.9686; Locomotion BL vs. escape SNI: P = 0.0083; Escape BL vs. Locomotion SNI: P = 0.0026; Escape BL vs. Escape SNI: P = 0.0351; Locomotion SNI vs. Escape SNI: P = 0.0039). h, Representative traces (left) and quantification (right, n = 7) of calcium response in OPRM1+ RVMSC neurons evoked by von Frey stimulation or occurring spontaneously following SNI. Scale bar: 1% ΔF/F, 3 sec. Two-sided Paired t-test, P = 0.1553. i, Experimental paradigm for j and k. j, Representative image from six independently performed experiments shows fibre track and jGCaMP7s expression in RVM-projecting vGlut2+ lSuC neurons. Scale bar: 500 μm (lSuC), 200 μm (RVM). k, Representative traces (left) and quantification (right) of calcium response in RVM-projecting vGlut2+ lSuC neurons evoked by von Frey stimulation before and after SNI. Scale bar: 1% ΔF/F, 3 sec. n = 6, two-sided paired t-test, P = 0.0069. * P < 0.05, ** P < 0.01, *** P < 0.001. Data are presented as mean ± s.e.m.
Extended Data Fig. 5 The activity of OPRM1+ RVMSC neurons and the lSuC→RVM pathway have no effect on locomotion.
a,b, Example traces (a) and quantification (b) of total distance travelled in open field for 10 min after saline or clozapine (CLZ, 0.1 mg/Kg) injection for chemogenetic manipulation of OPRM1+ RVMSC neurons (n = 5). c, Locomotion dynamics of WT mice after i.p. injection of clozapine (CLZ, 0.1 mg/Kg) or saline (n = 6). d, Electrophysiological validation of terminal silencing of the vlPAG→RVM pathway. AAV5-DIO-ChR2 and AAV2-DIO-hM4D were co-injected into the vlPAG of vGlut2Cre mice. e,f, Representative optogenetic stimulation evoked EPSC (oEPSC) traces (e) and quantification (f) recorded on the RVM slices containing ChR2-expressing terminals from the vlPAG before (baseline, black) and after infusion of CNO (5 μM). Scale bar: 50 pA, 25 ms. n = 6 cells from 3 mice. Two-sided Wilcoxon signed-rank test (Baseline vs. after CNO: P = 0.0313). g, Example traces (left) and quantification (right) of total distance travelled in open field for 10 min after saline or clozapine (0.1 mg/Kg) injection for chemogenetic inhibition of the lSC→RVM pathway (n = 6). h,i, Unilateral optogenetic activation of lSuC→RVM pathway (h) did not alter the locomotion trajectory and average speed of the ChR2-expressed mice (i). Control n = 6, ChR2 n = 6. j,k, Unilateral Optogenetic activation of lSuC→RVM pathway did not alter the heart rate (j, control n = 6, ChR2 n = 5) and breath rate of the ChR2-expressed mice (k, control n = 6, ChR2 n = 6). * P < 0.05. Data are presented as mean ± s.e.m.
Extended Data Fig. 6 Chemogenetic manipulation of OPRM1+ RVMSC neurons or VGLUT2+ RVM-projecting lSuC neurons.
a, Quantification of Hargreaves withdrawal latency in healthy mice expressing hM4D (blue, n = 8) or hM3D (red, n = 10) in OPRM1+ RVMSC neurons, following saline (black) or clozapine (0.1 mg/Kg) treatment. b, Temporal dynamics of mechanical thresholds in mice expressing hM3D in OPRM1+ RVMSC neurons from 1 to 12 h after a single clozapine injection (0.1 mg/kg, i.p.) (n = 10). c, Example CPA trace after SNI in mice expressing mCherry- or hM4D- in OPRM1+ RVMSC neurons. Related to Fig. 2h. d,e, Representative images (d) and quantification (e) of von Frey fibre (0.16 g) induced FOS-expressing neurons in the spinal cord of SNI (black, n = 4), SNI+hM4D (blue, n = 4) expressed in OPRM1+ RVMSC neurons, and sham control mice (orange, n = 4). Scale bar: 100 μm. (SC: spinal cord; mPFC: medial prefrontal cortex; ACC: anterior cingulate cortex; VPL: ventral posterolateral nucleus of thalamus; Po: posterior complex of the thalamus; MD: medial dorsal nucleus of thalamus; SSp: primary somatosensory cortex; SSs: secondary somatosensory cortex; Amy: amygdala; LPBN: lateral parabrachial nucleus). f, Quantification of plantar cold withdrawal latency in mice expressing mCherry- (black, n = 6) or hM3D- (red, n = 10) in OPRM1+ RVMSC neurons following repetitive clozapine administration (7 daily injections, 0.1 mg/kg). Two-sided Mann–Whitney test (CLZ-mCherry vs. CLZ-hM3D: P < 0.0119). g, Quantification of thermal withdrawal latency during and after repetitive daily injection of clozapine in mice expressing hM3D in OPRM1+ RVMSC neurons (n = 5). Clozapine was injected 23 h before each von Frey test for 7 consecutive days (shade area), related to Fig. 2i. h,i, Quantification of mechanical thresholds (h) and Hargreaves withdrawal latency (i) after clozapine infusion in terminals of vGlut2+ RVM-projecting lSuC neurons expressing hM4D (blue, n = 8) or mCherry (black, n = 8) pre and post CFA injection. Two-sided Mann–Whitney test (von Frey CLZ-mCherry vs. CLZ-hM4D pre CFA: P = 0.7389; CLZ-mCherry vs. CLZ-hM4D 1 d post CFA: P = 0.0002; CLZ-mCherry vs. CLZ-hM4D 2 d post CFA: P = 0.0002; CLZ-mCherry vs. CLZ-hM4D 3 d post CFA: P = 0.0002). * P < 0.05, *** P < 0.001. Data are presented as mean ± s.e.m.
Extended Data Fig. 7 Quantification of FOS expression and withdrawal latency after ablation of OPRM1+ RVMSC neurons.
a, Experiment timeline for b–e. b, Quantification of the percentage of OPRM1+ descending neurons in total OPRM1+ RVM neurons (left) of control- (white, n = 5) and Caspase- (orange, n = 5) expressing mice with SNI. Two-sided Mann–Whitney test (Control vs. caspase: P = 0.0079). Related to Fig. 2c,d. c, Representative images (left) and quantification (right) of von Frey fibre (1.4 g) induced FOS-expressing neurons in the spinal cord of control- (white, n = 3) and Caspase- (orange, n = 3) expressing mice without injury. Dash line indicates the dorsal horn. d, Quantification of the von Frey fibre (0.16 g) induced FOS-expressing neurons in the spinal cord of control- (white, n = 4) and Caspase- (orange, n = 4) expressing mice with SNI. Two-sided Mann–Whitney test (Control vs. caspase: P = 0.0286). Related to Fig. 2c,d. e, Quantification of thermal withdrawal latency of control- (black, n = 7) and Caspase-3- (orange, n = 8) expressing mice after CFA injection. Two-sided Mann–Whitney test (1 d post CFA control vs. caspase: P = 0.0025; 2 d post-SNI control vs. caspase: P = 0.0101; 3 d post-SNI control vs. caspase: P = 0.0005). Related to Fig. 2e. * P < 0.05, ** P < 0.01, *** P < 0.001. Data are presented as mean ± s.e.m.
Extended Data Fig. 8 Affective pain, spontaneous pain and anxiety-like behaviours after manipulation of OPRM1+ RVMSC neurons.
a, Attending and escape behaviours assessed with three different von Frey fibres stimuli. Groups: eGFP + 7 daily CLZ (white, n = 5), hM3D + 7 daily CLZ (red, n = 6), SNI + eGFP + CLZ (grey, n = 6), SNI + hM4D + CLZ (blue, n = 5). One-way ANOVA test (0.007 g eGFP+7 daily CLZ vs. hM3D + 7 daily CLZ: P < 0.0001; 0.007 g eGFP+7 daily CLZ vs. SNI_eGFP+CLZ: P < 0.0001; 0.007 g eGFP+7 daily CLZ vs. SNI_hM4D+CLZ: P = 0.0265; 0.007 g hM3D + 7 daily CLZ vs. SNI_eGFP+CLZ: P = 0.9424; 0.007 g hM3D + 7 daily CLZ vs. SNI_hM4D+CLZ: P = 0.0001; 0.007 g SNI_eGFP+CLZ vs. SNI_hM4D+CLZ: P < 0.0001; 0.4 g eGFP+7 daily CLZ vs. hM3D + 7 daily CLZ: P < 0.0001; 0.4 g eGFP+7 daily CLZ vs. SNI_eGFP+CLZ: P < 0.0001; 0.4 g eGFP+7 daily CLZ vs. SNI_hM4D+CLZ: P = 0.3070; 0.4 g hM3D + 7 daily CLZ vs. SNI_eGFP+CLZ: P = 0.9579; 0.4 g hM3D + 7 daily CLZ vs. SNI_hM4D+CLZ: P = 0.0004; 0.4 g SNI_eGFP+CLZ vs. SNI_hM4D+CLZ: P = 0.0004; 2 g eGFP+7 daily CLZ vs. hM3D + 7 daily CLZ: P = 0.0136; 2 g eGFP+7 daily CLZ vs. SNI_eGFP+CLZ: P = 0.0111; 2 g eGFP+7 daily CLZ vs. SNI_hM4D+CLZ: P = 0.9975; 2 g hM3D + 7 daily CLZ vs. SNI_eGFP+CLZ: P = 0.9996; 2 g hM3D + 7 daily CLZ vs. SNI_hM4D+CLZ: P = 0.0088; SNI_eGFP+CLZ vs. SNI_hM4D+CLZ: P = 0.0072). b, Histogram shows facial-grimace score in different groups: eGFP + CLZ (white, n = 9), hM3D + CLZ (red, n = 10), SNI + eGFP + CLZ (grey, n = 8), SNI + hM4D + CLZ (blue, n = 10). One-way ANOVA test, Turkey’s multiple comparisonOne-way ANOVA, followed by Tukey’s multiple comparisons test (eGFP+7 daily CLZ vs. hM3D + 7 daily CLZ: P = 0.7413; eGFP+7 daily CLZ vs. SNI_eGFP+CLZ: P = 0.0024; eGFP+7 daily CLZ vs. SNI_hM4D+CLZ: P > 0.9999; hM3D + 7 daily CLZ vs. SNI_eGFP+CLZ: P = 0.0001; hM3D + 7 daily CLZ vs. SNI_hM4D+CLZ: P = 0.7338; SNI_eGFP+CLZ vs. SNI_hM4D+CLZ: P = 0.0018). c, Histogram shows spontaneous pain in different groups: eGFP + CLZ (white, n = 8), hM3D + CLZ (red, n = 8), SNI + eGFP + CLZ (grey, n = 10), SNI + hM4D + CLZ (blue, n = 8). Non-parametric ANOVA (Kruskal–Wallis test), followed by post-hoc pairwise comparisons using Dunn’s multiple comparison test (eGFP+7 daily CLZ vs. hM3D + 7 daily CLZ: P > 0.9999; eGFP+7 daily CLZ vs. SNI_eGFP+CLZ: P = 0.0147; eGFP+7 daily CLZ vs. SNI_hM4D+CLZ: P > 0.9999; hM3D + 7 daily CLZ vs. SNI_eGFP+CLZ: P = 0.0023; hM3D + 7 daily CLZ vs. SNI_hM4D+CLZ: P > 0.9999; SNI_eGFP+CLZ vs. SNI_hM4D+CLZ: P = 0.0019). d, Eye blink responses to 20 PSI air puff directed at the cornea show no difference between each group: eGFP + CLZ (white, n = 6), hM3D + CLZ (red, n = 5), SNI + eGFP + CLZ (grey, n = 5), SNI + hM4D + CLZ (blue, n = 5). e, Quantification of total distance travelled (left) and time in centre (right) in open field test. Groups: eGFP + 7 daily CLZ (white, n = 9), hM3D + 7 daily CLZ (red, n = 10), SNI + eGFP + CLZ (grey, n = 9), SNI + hM4D + CLZ (blue, n = 7). Non-parametric ANOVA (Kruskal–Wallis test), followed by followed by post-hoc pairwise comparisons using Dunn’s multiple comparison test (Total distance: eGFP+7 daily CLZ vs. hM3D + 7 daily CLZ: P = 0.2431; eGFP+7 daily CLZ vs. SNI_eGFP+CLZ: P = 0.3374; eGFP+7 daily CLZ vs. SNI_hM4D+CLZ: P = 0.1490; hM3D + 7 daily CLZ vs. SNI_eGFP+CLZ: P > 0.9999; hM3D + 7 daily CLZ vs. SNI_hM4D+CLZ: P > 0.9999; SNI_eGFP+CLZ vs. SNI_hM4D+CLZ: P > 0.9999; % time in centre: eGFP+7 daily CLZ vs. hM3D + 7 daily CLZ: P = 0.0179; eGFP+7 daily CLZ vs. SNI_eGFP+CLZ: P = 0.009; eGFP+7 daily CLZ vs. SNI_hM4D+CLZ: P = 0.0023; hM3D + 7 daily CLZ vs. SNI_eGFP+CLZ: P = 0.9832; hM3D + 7 daily CLZ vs. SNI_hM4D+CLZ: P = 0.6992; SNI_eGFP+CLZ vs. SNI_hM4D+CLZ: P = 0.8834). f, Quantification of time in open arm in elevated plus maze test. Groups: eGFP + 7 daily CLZ (white, n = 7), hM3D + 7 daily CLZ (red, n = 10), SNI + eGFP + CLZ (grey, n = 9), SNI + hM4D + CLZ (blue, n = 9). One-way ANOVA, post-hoc pairwise comparisons were conducted using Tukey’s multiple comparisons test (eGFP+7 daily CLZ vs. hM3D + 7 daily CLZ: P = 0.023; eGFP+7 daily CLZ vs. SNI_eGFP+CLZ: P = 0.0042; eGFP+7 daily CLZ vs. SNI_hM4D+CLZ: P = 0.0002; hM3D + 7 daily CLZ vs. SNI_eGFP+CLZ: P = 0.9987; hM3D + 7 daily CLZ vs. SNI_hM4D+CLZ: P = 0.7408; SNI_eGFP+CLZ vs. SNI_hM4D+CLZ: P = 0.6675). g, Quantification of immobility time in tail suspension test. Groups: eGFP + 7 daily CLZ (white, n = 6), hM3D + 7 daily CLZ (red, n = 8), SNI + eGFP + CLZ (grey, n = 8), SNI + hM4D + CLZ (blue, n = 9). Non-parametric ANOVA (Kruskal–Wallis test), followed by post-hoc pairwise comparisons using Dunn’s multiple comparison test (eGFP+7 daily CLZ vs. hM3D + 7 daily CLZ: P > 0.9999; eGFP+7 daily CLZ vs. SNI_eGFP+CLZ: P = 0.4322; eGFP+7 daily CLZ vs. SNI_hM4D+CLZ: P = 0.0086; hM3D + 7 daily CLZ vs. SNI_eGFP+CLZ: P = 0.3926; hM3D + 7 daily CLZ vs. SNI_hM4D+CLZ: P = 0.0048; SNI_eGFP+CLZ vs. SNI_hM4D+CLZ: P > 0.9999). * P < 0.05, ** P < 0.01; *** P < 0.001; **** P < 0.0001. Data are presented as mean ± s.e.m.
Extended Data Fig. 9 Effects of manipulating the activity of the excitatory lSuC→RVM pathway or the SS→lSuC pathway on pain-induced CPA.
a, Example traces (left) and quantification (right) of mechanical stimuli (0.16 g von Frey filament) evoked CPA in control mice (black, n = 6) and mice with the lSuC→RVM pathway silenced (blue, n = 6). Related to Fig. 3g. Two-sided Mann–Whitney test (CLZ-mCherry vs. CLZ-hM4D: P = 0.0022). b, Example traces (left) and quantification (right) of mechanical stimuli (0.16 g von Frey filament) evoked CPA in control mice (black, n = 6) and mice after repetitive activation of the excitatory lSuC→RVM pathway (red, n = 5). Related to Fig. 3h. Two-sided Mann–Whitney test (CLZ-mCherry vs. CLZ-hM3D: P = 0.0022). c, Example traces of mechanical stimuli (0.16 g von Frey filament) evoked CPA for Fig. 5e. d, Example traces of mechanical stimuli (0.16 g von Frey filament) evoked CPA for Fig. 5g. Data are presented as mean ± s.e.m.
Extended Data Fig. 10 Anxiety-like behaviours after repetitive activation of the STT, and chemogenetic silencing of the VPL-projecting DCN neurons.
a, Experimental timeline for b. b, Quantification of mechanical thresholds after repetitive chemogenetic activation of the spinal cord to VPL pathway in mCherry expressing control mice (Control, red, n = 6) but not in OPRM1+ RVMSC neurons ablation mice (Ablation, black, n = 6). CLZ was infused 23 h before each von Frey test for 7 consecutive days (shade area). Pre vs. post clozapine infusion: nonparametric ANOVA (Friedman test), followed by Dunn’s multiple comparison test (control pre vs. D1 on CLZ: P > 0.9999; control Pre vs. D2 on CLZ: P = 0.4633; control Pre vs. D3 on CLZ: P = 0.0354; control Pre vs. D5 on CLZ: P = 0.0027; control Pre vs. D7 on CLZ: P = 0.0008; control Pre vs. D7 post CLZ: P = 0.0008; control Pre vs. D14 post CLZ: P = 0.0104; control Pre vs. D21 post CLZ: P = 0.0491). # P < 0.05, ## P < 0.01, ### P < 0.001. Control vs. ablation: two-sided Mann–Whitney test (Pre CLZ-control vs. Pre CLZ-ablation: P = 0.05779; 1D on CLZ-control vs. CLZ-ablation: P = 0.0065; 2D on CLZ-control vs. CLZ-ablation: P = 0.0022; 3D on CLZ-mCherry vs. CLZ-hM3D: P = 0.0022; 5D on CLZ-mCherry vs. CLZ-hM3D: P = 0.0022; 7D on CLZ-mCherry vs. CLZ-hM3D: P = 0.0022; 7D post CLZ-mCherry vs. CLZ-hM3D: P = 0.0022; 14D post CLZ-mCherry vs. CLZ-hM3D: P = 0.0022; 21D post CLZ-mCherry vs. CLZ-hM3D: P = 0.0022). ** P < 0.01. c, Quantification of total distance travelled and time in centre in open field test. eYFP + 7 daily CLZ (white, n = 7), hM3D + 7 daily CLZ (red, n = 7). Two-sided Mann–Whitney test (Total distance: eYFP + 7 daily CLZ vs. hM3D + 7 daily CLZ: P = 0.7104; % time in centre: eYFP + 7 daily CLZ vs. hM3D + 7 daily CLZ: P = 0.0017). ** P < 0.05. d, Quantification of time in open arm in elevated plus maze test (left) and immobility time in tail suspension test (TST; right). eYFP + 7 daily CLZ (white, n = 8), hM3D + 7 daily CLZ (red, n = 8). Two-sided Mann–Whitney test (EPM eYFP + 7 daily CLZ vs. hM3D + 7 daily CLZ: P = 0.003; TST eYFP+7 daily CLZ vs. hM3D + 7 daily CLZ: P = 0.1949). ** P < 0.05. e, Diagram (left) and quantification (right) of silencing the SC→VPL + SC→Po pathways together (yellow, n = 6) or the DCN→VPL pathway (white, n = 6) or wild-type mice without manipulation (WT, grey, n = 5) on object with novel texture. One-way ANOVA, *** P < 0.001. f, Chemogenetic silencing of the vGLut2+ VPL-projecting DCN neurons did not affect the mechanical thresholds of mice under normal or SNI conditions (n = 6). Data are presented as mean ± s.e.m.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figure 1, Supplementary Note and Supplementary Table 1
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Q., Lee, J.H., Nachtrab, G. et al. Deconstruction of a spino-brain–spinal cord circuit that drives chronic pain. Nature (2026). https://doi.org/10.1038/s41586-026-10296-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41586-026-10296-y
