
Abstract
The initial development of adenine base editors (ABEs), which facilitate A•T to G•C base pair changes in the genome, used directed evolution to install 14 mutations into the wild-type deaminase TadA, producing the first-of-its-kind editor ABE7.10. Here we study the installed mutations’ impacts on TadA fitness using comprehensive reversion analysis and apply our results to engineer more efficient, precise editors. By measuring activity in both human and Escherichia coli host systems, we categorize mutations as critical, dispensable or host dependent. We show that up to five mutations can be reverted back to wild type, generating minimally evolved ABEs (ME-ABEs). ME-ABEs show narrow editing windows (similar to that of ABE7.10) and enhanced on-target editing (matching activities of the high-activity editor variants ABE8e and ABE8.20 in most sequence contexts) and exhibit low levels of guide-RNA-dependent and guide-RNA-independent off-target activity. ME-ABEs efficiently target six sites of clinical interest that had previously proved challenging to edit with ABE7.10, ABE8e or ABE8.20.
Similar content being viewed by others


Main
Base editors (BEs) comprise a set of genome editing tools that fuse a catalytically impaired clustered regularly interspaced short palindromic repeat (CRISPR) enzyme to a single-stranded DNA (ssDNA)-modifying enzyme. BEs combine the programmability of their CRISPR component with the efficiency and precision of an ssDNA-editing enzyme, allowing for site-specific introduction of single-nucleotide variants (SNVs) into the genome of live cells without introducing deleterious double-strand DNA breaks (DSBs). To date, two main classes of BEs are widely used: cytosine base editors (CBEs) and adenine base editors (ABEs), which convert C•G to T•A and A•T to G•C, respectively1,2. BEs are uniquely suited to model and correct SNVs—traditional CRISPR technologies struggle to introduce SNVs efficiently and precisely1, and while prime editors can accomplish similar edits, their editing approach requires considerable optimization3,4. The utility of BEs as a class of tools is further supported by their use in several clinical applications, despite their relatively recent development5,6,7. Given that SNVs account for the majority of human genetic variation, and current CBEs and ABEs are theoretically capable of correcting 62% of known pathogenic SNVs2,8,9,10, the improvement of existing BEs and the development of new BEs capable of facilitating additional base changes remain areas of active research.
The first BEs were developed following the hypothesis that if a mutagenic intermediate could be site-specifically introduced, native cellular repair pathways would convert the mutagenic mismatched base pair into a canonical DNA base pair1,2. For CBEs, this mutagenic intermediate is a U•G mismatch, which is resolved into a T•A base pair. Similarly, ABEs use a deoxyinosine (I) intermediate to facilitate A•T to I•T to G•C transition mutations. To site-specifically introduce these intermediates, deaminases were fused to Streptococcus pyogenes nickase CRISPR associated protein 9 (Sp-nCas9, a nicking endonuclease that installs a break in only one DNA strand rather than a DSB). The Sp-nCas9 scans the genome to locate a protospacer adjacent motif (PAM, for the Sp-nCas9 variant, this is NGG) and neighboring protospacer with a matching sequence to the user-supplied guide RNA (gRNA). Once found, Sp-nCas9 facilitates the formation of an R-loop, unwinding the local DNA helix to expose a stretch of ssDNA on the non-target strand (Fig. 1). A small window of this ssDNA becomes accessible for deamination, and any C’s or A’s within the window are converted to U’s by CBEs or I’s by ABEs. Nicking of the opposing gRNA-bound strand by Sp-nCas9 biases repair of the U•G or I•T intermediates in favor of T•A or G•C outcomes, respectively. While initial CBEs used the cytidine deaminase enzyme Rattus norvegicus APOBEC1, which shows substrate promiscuity for both ssDNA and RNA, no naturally occurring enzyme existed that could deaminate deoxyadenosine in an ssDNA context. As such, the Escherichia coli TadA enzyme, a deaminase that natively functions as a homodimer to target the UAC anticodon loop of tRNAArg (ref. 11), was used as a starting point. When fused to Sp-nCas9 (to produce the editor ABE0.1), wtTadA showed extremely low levels of DNA editing12. However, following 7 rounds of directed evolution in an E. coli host system, 14 mutations within the 167-amino-acid protein were introduced to produce ABE7.10, with vastly improved A•T to G•C editing activity (Fig. 1 and Supplementary Fig. 1).

The ABE7.10 base editor is a fusion protein between nCas9 and the engineered deaminase TadA*7.10 (where TadA* indicates a mutated TadA variant). nCas9 and a user-supplied gRNA direct the editor to a region of interest, unwinding dsDNA to reveal a small window of accessible ssDNA on the non-target strand. The engineered TadA*7.10 acts on this substrate, converting adenosines to inosines, which are further processed to guanines, resulting in an overall A•T to G•C edit. Monomeric ABE7.10 was modeled in silico using ChimeraX, starting with the crystal structure of ABE8e (PDB ID: 6VPC) and replacing the catalytic TadA*8e domain with Alphafold2-predicted TadA*7.10. For simplicity, only the TadA domain and a short section of the ssDNA from the R-loop are shown (right). The residues that were mutated during the development of ABE7.10 are highlighted and color coded by the round of evolution they first emerged in. Relative stringencies for a given round of evolution were determined by kinetic pressure and number of selection targets used to evolve residues (left). Note that evolutionary rounds 4 and 6 did not produce any mutations that remained in the final iteration of ABE7.10.
Additional engineering efforts building on ABE7.10 have resulted in the more widely used ABE8e (ref. 13) and ABE8.20 (ref. 14) editors (Supplementary Fig. 1). While both ABE8e and ABE8.20 enhance absolute editing, they concurrently evolved expanded editing windows. Counting from the PAM-distal end of the protospacer, ABE7.10 edits positions 4–8, ABE8.20 expands to include positions 3–9 and ABE8e further encompasses positions 3–11. These wider editing windows have been attributed to increases in catalytic efficiency and enzyme processivity by TadA, acquired during the evolutionary process13,15. Wide windows make efficient single-nucleotide editing challenging owing to concurrent editing of adjacent, or ‘bystander’, A’s. Furthermore, these highly evolved ABEs are reported to increase rates of gRNA-dependent13 and gRNA-independent off-target DNA editing14, and show high levels of RNA off-target editing16. An editor with the appropriate balance of high on-target activity, low off-target activity and minimal window size would be of great value to the field. We hypothesized that a systematic analysis of the impact each accumulated mutation conferred on editing profiles would facilitate the design of an editor that meets these needs.
Due to its development by directed evolution, our ability to rationalize the benefit of each of the 14 acquired mutations in ABE7.10 is limited. Directed evolution campaigns are notorious for applying unexpected selection pressures, while selection stringencies and library biases often allow dispensable mutations to pass selections17,18. Such mutations may neither help nor hurt the desired activity but can impact subsequent evolutionary trajectories by altering the enzyme’s fitness landscape. To assess the importance of enriched mutations, they can be reverted to wild type (in a method called reversion analysis) or mutated to alanine, and the resulting variants evaluated for activity, stability and/or expression. Here we perform a systematic reversion analysis for all 14 mutations acquired during the evolution of ABE7.10. We evaluated each reversion editor for base editing activity in two different host systems to account for differences between their E. coli evolutionary host and human cells where BEs are frequently applied and characterized. These comparisons allowed us to identify dispensable mutations, as well as mutations that appear to have host-system-dependent beneficial activity. Combining these findings, we produced and characterized several multi-residue reversion editors. A subset of these ME-ABEs shows increased editing activity relative to ABE7.10 (with average editing efficiencies within error of ABE8e and ABE8.20 at the central editing position), while maintaining narrow editing windows (to avoid bystander editing), and gRNA-independent off-target activity levels >10-fold lower than that of ABE8e.
Results
Activity of ABE7.10 single-residue reversion variants at episomal reporter and endogenous sites identifies both essential and dispensable mutations
We previously reported that reversion of the single D108N mutation in ABE7.10 (to produce the editor that we call ABE7.10-N108D) resulted in an average 146-fold decrease in editing activity, as measured across 6 endogenous genomic sites in mammalian cells19. This suggests that the D108N mutation introduced in the first round of directed evolution of ABE7.10 was critical for both enhancing DNA editing activity by TadA and maintaining this activity even in the context of the 13 other acquired mutations. To investigate how the additional mutations in ABE7.10 impact base editing activity, we generated all 14 single-residue ABE7.10 reversion editors and evaluated their activities using a previously reported set of 8 episomal, highly sensitive, fluorescence turn-on reporters (Fig. 2a and Supplementary Discussion 1)12.

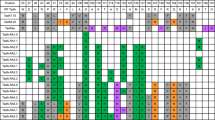
a, BE activity was assessed using a series of fluorescent turn-on plasmid reporters. Each reporter plasmid (Rep) coded for a transfection marker mCherry fluorescent protein linked to EGFP reporter protein via a P2A ribosomal skipping sequence. The deactivated EGFP reporter protein (dEGFP) required an A•T to G•C correction at the DNA level to restore EGFP fluorescence. The target A was placed such that the base either directly 5′ (−1) or 3′ (+1) fell at a wobble codon position, allowing for substitution of any base N at this position without impacting the resulting protein sequence and fluorescence. For the −1 family of reporters, the target A fell at position 5 while in the +1 family of reporters the target A was at position 6. HEK293Ts were transfected with Rep, base editor plasmid and appropriate gRNA and incubated for 72 h, after which EGFP fluorescence turn-on was evaluated using flow cytometry. b, ABE7.10 single-residue reversion editors were evaluated for editing activity by calculating their fold change in activity relative to that of ABE7.10. Relative activities of reversion editors were determined using BE activity scores (top set of heat maps) and the MFI of the double-color (mCherry and EGFP)+ cell population (bottom set of heat maps). Data are separated by reporter family, with A111V (−1 base reporters) on the left and H182Y (+1 base reporters) on the right. Heat map scales have been set so white indicates equivalent activity to ABE7.10, gray scale indicates activity less than ABE7.10 and color indicates activity greater than ABE7.10. Editor variants are ordered by round of evolution their mutation was introduced in, with color coding adherent to Fig. 1. Values represent the average of fold changes calculated for n = 3 independent biological replicates.
We performed our reversion analyses using monomeric forms of ABE (TadA*-nCas9, where TadA* indicates a mutated TadA variant). While wild-type TadA is known to act as a dimer on its native tRNAArg substrate in E. coli11,20, and the crystal structure of ABE8e captures TadA* in a homodimeric form15, many researchers have found no significant difference in editing activity when delivering monomeric versus dimeric ABE7.10 and ABE8 variants12,14,19,21. HEK293T cells were transfected with a mixture containing one of the eight reporter plasmids, its respective gRNA plasmid and a monomeric base editor plasmid encoding one of the 14 ABE7.10 single-residue reversion editors or ABE7.10. Cells were incubated for 72 h to allow for editing and fluorescent protein expression before analysis by flow cytometry (Supplementary Fig. 2). Cells were analyzed for both mCherry and EGFP fluorescence, and a ‘BE activity’ score was calculated by dividing the percentage of edited cells by the total percentage of transfected cells (Fig. 2a). While this BE activity score is not equivalent to editing efficiency, it allows for comparisons of relative editing activities among multiple BE variants and helps account for variation resulting from differences in transfection efficiency12 (Supplementary Figs. 3a,b, 4 and 5). The median EGFP fluorescence intensity (MFI) of double-color-positive cells was used as a secondary marker of BE activity (Supplementary Figs. 3c,d, 6 and 7).
All BE activity scores and MFIs were normalized to that of ABE7.10 within a given biological replicate to enable fold-change comparisons of how each reversion variant impacted activity relative to ABE7.10 (Fig. 2b and Supplementary Fig. 8). In general, we observed more drastic changes in relative activities of the reversion variants with the A111V (−1) family of reporters compared with the H182Y (+1) family of reporters. This is consistent with previous studies that show the identity of the −1 base to have a much stronger impact on relative ABE editing efficiency12,22. While the H182Y reporters showed a smaller dynamic range, they still captured a dramatic reduction in activity for the ABE7.10-N108D variant, and significant differences in activity between several reversion editors and ABE7.10 (Fig. 2b and Supplementary Figs. 5 and 7). Overall, this suggests that these two families of reporters can detect changes in editing activity across diverse sequence contexts.
The data from all 14 ABE7.10 single-residue reversion editors tested against all eight fluorescent turn-on reporter plasmids are shown in Fig. 2b and Supplementary Figs. 3–9. Notably, the ABE7.10-N108D reversion editor had the greatest impact on editing activity for all reporters, showing average normalized activity scores relative to the parental ABE7.10 editor (1.00 ± 0.10) of (2.07 ± 0.95) × 10−4 for the −1 family of reporters and (1.04 ± 0.31) × 10−3 for the +1 family of reporters. Similar extreme decreases in editing activity for the ABE7.10-N108D variant were reflected with MFI values (Fig. 2b and Supplementary Figs. 3c,d and 8b). While no other single-residue reversion variant had as deleterious an impact on editing activity as N108D, the reversions V155E, F84L and F156I also resulted in significantly decreased editing activity across all reporters. We observed average normalized activity scores relative to ABE7.10 of 0.66 ± 0.05, 0.74 ± 0.06 and 0.85 ± 0.07 across all eight reporters for the V155E, F84L and F156I reversion variants, respectively. The V106A, Y147D and R23W reversion editors also reduced editing activity but to a lesser degree, with average activity scores relative to ABE7.10 of 0.88 ± 0.08, 0.89 ± 0.08 and 0.93 ± 0.09, respectively, across all eight reporters.
From these data, we also identified five reversions (C146S, L36H, A48P, P152R and L51R) that had minimal impact on editing efficiency (for example, an overall normalized average base editing activity score between 0.95 and 0.99 relative to ABE7.10), regardless of sequence motif (Supplementary Fig. 8). Finally, we found two reversion variants (Y123H and N157K) that increased activity scores relative to ABE7.10, an effect that was more apparent with the −1 reporters (Fig. 2b and Supplementary Fig. 8). ABE7.10-Y123H had an increased BE activity score relative to ABE7.10 of 1.10 ± 0.20 with the −1 reporters and 1.04 ± 0.10 over all reporters, while ABE7.10-N157K had an increased BE activity score relative to ABE7.10 of 1.21 ± 0.23 with the −1 reporters and 1.09 ± 0.12 overall. Trends in MFI changes mirrored those of the activity scores (Fig. 2b and Supplementary Figs. 3, 8 and 9).
These data allowed for some assessment of editor sequence motif preference, with a greater focus placed on the identity of the base 5′ of the target A. Generally, we noted that the sequence motif preferences of most single-residue reversion editors matched that of ABE7.10: TAC > CAC > AAC ~ GAC (Supplementary Fig. 10 and Supplementary Discussion 2). These data suggested that no reversion strongly favored any one sequence motif over others. Taken together, these data identified that certain mutations accumulated during the directed evolution of ABE7.10 remained necessary for base editing activity (most crucially D108N and to a lesser degree E155V, L84F and I156F), while others may be dispensable (S146C, H36L, P48A, R152P and R51L) as their reversions had minimal impact on base editing activity across these eight reporters. We also identified two mutations that appear detrimental to base editing activity (H123Y and K157N), as their reversions on average increased editing activity across all eight reporters.
We next evaluated each ABE7.10 single-residue reversion editor for activity at three genomic sites (selected for their varied target motifs) to confirm that the results from the episomal editing assay extended to endogenous editing (Extended Data Fig. 1a,b and Supplementary Fig. 11). The trends in these data were consistent with what we observed for episomal editing (Supplementary Discussion 3). For a subset of reversion variants that altered editing activity relative to ABE7.10, we developed alanine substitution variants and similarly profiled editing activity at these three endogenous sites (Extended Data Fig. 1c–f). While alanine substitution allowed for investigation into biochemical interactions that may drive changes in deaminase activity (Supplementary Figs. 12 and 13, and Supplementary Discussion 4), no alanine substitution consistently improved editing activity at all three sites. Together, these results at endogenous genomic sites support observations from the episomal fluorescent turn-on reporter assay, suggesting that some reversions have minimal impact on or even improve ABE editing activity.
Stability of expression assays shows no drastic decreases in protein expression levels for ABE7.10 single-residue reversion variants
It has been hypothesized that during the course of directed evolution, mutations that increase activity can often recompense for this change with a concurrent loss in protein stability17,23. This hypothesis has not been investigated in cellular contexts for mutations accumulated by TadA during the development of ABEs. In addition, it is hypothesized that some mutations acquired during directed evolution, particularly those distal from an enzyme’s active site, compensate for destabilizing mutations by providing allosteric support or additional stability without directly impacting catalytic activity24,25. While some mutations acquired during the evolution from ABE7.10 into ABE8e show this behavior26, none of the initial mutations in ABE7.10 have previously been identified as compensatory. Reversion of such a compensatory mutation could lead to additional destabilizing effects, impacting the reversion variant’s activity. Given our findings that several ABE7.10 single-residue reversions modified base editing activity, we sought to evaluate whether altered intracellular ABE protein levels could account for these changes in editing activity.
To this end, we adapted a fluorescent reporter assay in which the ratio of EGFP/mCherry fluorescence measured relative intracellular expression levels of the ABE reversion variants in HEK293T cells (Extended Data Fig. 2a and Supplementary Discussion 5)27. Preliminary studies with this assay found diminishing ABE expression for more evolved editors (ABE0.1 > ABE7.10 > ABE8e; Supplementary Fig. 14 and Extended Data Fig. 2b), results that were verified by independent western blot analysis (Extended Data Fig. 2c and Supplementary Discussion 5). We extended the fluorescent reporter assay to examine stability of expression for all 14 ABE7.10 single-residue reversion variants (Extended Data Fig. 2d and Supplementary Fig. 15). Generally, we noted a pattern in which reversion of mutations acquired in earlier rounds of evolution tended to increase expression and stability while reversion of mutations acquired in later rounds of evolution tended to decrease expression and stability (Supplementary Discussion 5). This lends support to the hypothesis that some later mutations may act in a compensatory manner with regard to protein expression and stability. Of note, none of the ABE7.10 single-residue reversion variants that increased editing activity or were deemed neutral in the fluorescent reporter EGFP turn-on assay (indicated with asterisks in Extended Data Fig. 2d) showed significantly increased EGFP/mCherry fluorescence intensity ratios. Some of these single-residue reversion editors were further evaluated for ABE protein expression levels via western blot (using an ABE-P2A-mCherry construct) and were confirmed to show similar expression levels to ABE7.10 (Extended Data Fig. 2c). Taken together, these data indicate that the changes in ABE activity caused by the reversion mutations were not driven by changes in expression and stability, but rather by changes in enzyme activity.
ABE7.10 single-residue reversion variants show disparate behavior in a bacterial host
Although the majority of ABE applications are in mammalian systems, the directed evolution scheme used to develop ABE7.10 occurred in an E. coli bacterial host2. This is notable owing to the possibility of endogenous TadA dimerizing in trans with the evolving TadA of the ABE, an interaction not possible in mammalian hosts that lack a TadA homolog11,15,26,28. Given these complex interactions, as well as additional differences between mammalian and bacterial systems (including presence or absence of chaperone proteins, differences in posttranslational modifications, availability of critical cofactors or differences in DNA repair pathways), we hypothesized that alternate editor activity may be observed between these two hosts29,30,31. We therefore developed an assay to quantify editing activities of the single-residue reversion variants in a bacterial host system that used bacterial survival rate as a measure of ABE activity (Fig. 3a and Supplementary Discussion 6).

a, Base editing in bacteria was measured using an antibiotic survival assay. Sequence encoding a dead chloramphenicol acetyltransferase (CamR) requiring an A•T to G•C correction to restore antibiotic resistance was placed on a carrier plasmid, pCamR. Two versions of this were developed: one with the deactivating mutation H193Y, where the target A had the sequence motif TA6G, and the other a Q122* mutation where the target A had the sequence motif TA6C. S1030 E. coli harboring pCamR were transformed with various ABE7.10 single-residue reversion plasmids optimized for bacterial editing. ABE plasmids also contained appropriate gRNAs for targeting the mutation in CamR to reactivate chloramphenicol resistance, as well as a theophylline-inducible riboswitch to temporally control expression of the base editor. Bacteria were allowed to grow overnight in liquid culture with the base editor expressed, before splitting the culture and dilution plating each sample on +/− chloramphenicol plates (+/− chlor). Colony counts on +/− chlor plates were used to determine the survival rate of a given single-residue reversion editor in bacteria. b,c, Heat maps showing average fold-change values in survival rate for single-residue reversion editors against the H193Y TAG reporter (b) and Q122* TAC reporter (c). d,e, Fold change in survival rates relative to ABE7.10 for bacterial base editing activity assay with the H193Y TAG (d) and Q122* TAC reporters (e). Fold-change survival rate measurements were calculated for n = 9 plating pairs per sample with ROUT 1% outliers removed, and all remaining fold-change values plotted individually. The box plot center illustrates median fold change, hinges show quartiles and whiskers show minimum and maximum fold-change values.
For most reversion variants, we observed similar trends in relative editing activity in the bacterial system as in the mammalian episomal and endogenous editing assays (Fig. 3b–e and Supplementary Fig. 16). Specifically, ABE7.10-N108D was found to be the most deleterious reversion variant, showing an activity that was on average four orders of magnitude lower than that of ABE7.10 in both tested reporters, with survival rates on par with that of ABE0.1 (Fig. 3d,e and Supplementary Fig. 16). The three reversion editors that moderately decreased editing in mammalian systems, V155E, F84L and F156I, generally followed these same trends in bacteria. In the Q122* TAC reporter system, all three showed average survival rates two orders of magnitude below that of ABE7.10. For the H193Y TAG reporter, the F84L and F156I reversion variants both lowered average survival rates by an order of magnitude relative to ABE7.10, while the V155E variant’s activity showed no significant difference. Overall, agreement in activity trends across the three base editing activity reporters analyzed suggests critical roles for the N108D, E155V, L84F and I156F mutations independent of the host system or DNA context (episomal versus chromosomal).
The reversion variant with the greatest relative increase in editing activity in bacteria was ABE7.10-Y147D, which decreased editing slightly in both mammalian-based assays (Fig. 2b and Extended Data Fig. 1b), but resulted in an average 200-fold increase in editing activity compared with ABE7.10 in the H193Y TAG bacterial system, and a less drastic 13-fold increase measured by the Q122* TAC reporter (Fig. 3b–e). Notably, this residue was further evolved during the development of both ABE8e and ABE8.20. These variants were both evolved in bacteria (starting from ABE7.10), with the Y147D reversion mutation selected for in ABE8e and the Y147R mutation selected for in ABE8.20 (Supplementary Fig. 1). In light of these data, these mutations may be causing host-cell-specific increases in editing efficiency. Of further interest, neither the ABE7.10-N157K nor ABE7.10-Y123H reversion variants, which caused the largest increases in editing activity in HEK293Ts, had statistically significant impacts in bacteria, resulting in survival rates within error of ABE7.10 (Fig. 3b–e). These host-system-dependent differences in editing activity may explain why some ABE7.10 mutations that were identified during bacterial-directed evolution (such as mutations acquired during evolutionary rounds four and six) did not result in high activity during validation in mammalian systems2. Furthermore, these results highlight a potential pitfall in using a different host system for evolution or engineering efforts than the intended end-use system.
Multi-residue reversion editors increased editing activities at endogenous sites
Given that several reversion variants had a minimal or sometimes beneficial impact on base editing activity, we postulated that combinations of these reversions may lead to a similar or synergistic effect on editing activity. Following preliminary studies on a series of combination reversion variants using an episomal reporter and at a subset of endogenous sites (Extended Data Fig. 3 and Supplementary Fig. 17), we identified a core set of reversions for further investigation (Supplementary Discussion 7). We renamed the set of ABE7.10 multi-residue reversion editors listed in Fig. 4a, collectively calling these ME-ABEs. The ME-ABEs, ABE7.10-N157K and benchmark editors ABE7.10, ABE8e and ABE8.20 were further characterized at a panel of 27 additional endogenous genomic sites in HEK293T cells (Supplementary Figs. 18 and 19). For further discussion on site selection and normalization of ABE activity data, see Supplementary Discussion 8. For this set of editors, the average normalized editing values across all sites as a function of position in the protospacer are presented in Fig. 4b, Extended Data Fig. 4 and Supplementary Fig. 20. Using our metric for editing window (Supplementary Discussion 8), we found ABE7.10 and all five ABE7.10 reversion variants to robustly edit between positions 4 and 7, ABE8.20 between positions 3 and 8, and ABE8e between positions 3 and 11.

a, Renaming legend for a set of ABE7.10 reversion editors. HEK293T cells were transfected with a single ABE plasmid containing either one of the editors listed, ABE7.10, ABE8e or ABE8.20, along with 1 of 27 gRNA plasmids targeting an endogenous site or an NT control (Supplementary Fig. 18). After a 72-h incubation, genomic DNA was collected, target sites were amplified and sequences were analyzed by NGS. Absolute editing values (percentage of reads with a given A•T pair converted to G•C) were normalized to the absolute editing of ABE7.10 at the H2.0 A5 site (the maximally edited A in all experiments) within a given replicate. Editing activity was examined in n = 3 biological replicates for all ABE7.10 reversion editors, and n = 6 for ABE7.10, ABE8e and ABE8.20. b, Normalized editing scores for all 205 A’s were sorted by position and further averaged across all gRNAs to identify editing windows. The number of A’s considered for each position is given below the x-axis. The horizontal dotted line shows the 15% cutoff used to determine editing windows. c, Sequences of all on-target gRNAs and their predicted gRNA-dependent DNA off-target sites. The most editable A base is shown in bold. Mismatches between on-target and off-target site are shown in red. HEK293T cells were transfected with plasmids encoding each of the ABE variants indicated and an on-target gRNA. Both the on-target and off-target loci were amplified and evaluated for A•T to G•C editing with NGS 72 h after transfection. Bars show average editing measured for n = 3 biological replicates, with error bars showing standard deviation. Dashed horizontal lines indicate 0.1% editing activity, demarcating the limit of reliable detection. d, The orthogonal R-loop assay compares ME-ABE gRNA-independent off-target activity with canonical editors. Briefly, this assay uses two orthogonal Cas constructs; Sp-Cas9 ABE and Sp-gRNA edit an on-target site, while Sa-dCas9 and Sa-gRNA expose an off-target ssDNA R-loop for errant deamination (cartoon, above). HEK293T cells were transfected with plasmids encoding an ABE, an on-target Sp-gRNA, Sa-dCas9, and an off-target Sa-gRNA. After 72 h, genomic DNA was collected and on-target and off-target loci were sequenced. The x–y plot indicates the average off-target activity for the most edited A across three independent off-target sites, and average on-target activity at the most edited A across two independent on-target sites, accounting for n = 3 biological replicates with error bars showing propagated standard deviation.
When comparing average normalized A•T to G•C editing efficiencies at each position within the editing window, all ME-ABEs were nearly equivalent to one another. They showed notable increases in activity compared with ABE7.10 at positions 4 and 5 while performing similar to ABE7.10 at positions 6 and 7 (Fig. 4b and Supplementary Fig. 20). Of particular note at position 5 (the most highly edited position), the single-residue reversion variant ABE7.10-K had a 1.14-fold increase in editing activity over ABE7.10, while the dual-residue reversion variant ABE7.10-HK and the maximally reverted ABE7.10-HRHSK variant showed 1.41-fold and 1.36-fold increases in editing activity compared with ABE7.10, respectively. Furthermore, all four ME-ABEs on average performed similarly to ABE8e and ABE8.20 at position 5 (ABE8e was only 1.01-fold more active than ABE7.10-HK). Overall, the ME-ABEs show identically narrow editing windows to ABE7.10 and provide an editing efficiency advantage over ABE7.10 at positions 4 and 5. It was previously hypothesized that increases in editing activity by highly evolved ABE variants were always accompanied by an increase in editing window size16. While this is the case when comparing ABE7.10 against ABE8e and ABE8.20, the ME-ABEs decouple this behavior, increasing editing activity while maintaining a narrow editing window.
This series of endogenous target sites also allowed for analysis of editor sequence motif preference (Extended Data Fig. 5, Supplementary Fig. 21 and Supplementary Discussion9). When considering the subset of editable A’s (A’s present in the editing window for a given ABE variant), we found that at GA, AA, TA, AA, AT and AC motifs, the ME-ABEs had average editing activities similar to or higher than that of the ABE8 variants. ME-ABE editing efficiencies were improved with respect to ABE7.10 at CA and AG motifs, but were lower than the ABE8 variants in these sequence contexts. We also use this set of endogenous target sites to look for undesired editing outcomes. Given previous findings that highly active TadA variants can deaminate bystander cytosines within the editing window32, we analyzed these data for cytosine editing. While we noted slightly higher levels of C•G to T•A editing for ABE8e and ABE8.20 compared with ME-ABEs and ABE7.10, global cytosine editing for all editors was within error of non-target controls (Supplementary Figs. 22 and 23 and Supplementary Discussion10). A similar analysis of indels across all 27 sites found median indel rates for all editors to be below 0.3%, with ME-ABEs facilitating indel rates comparable to that of ABE7.10 and slightly lower than those of ABE8e and ABE8.20 (Supplementary Figs. 24 and 25).
Altogether, the four ME-ABEs behaved quite similarly to each other. They maintained the same narrow editing window as ABE7.10 (positions 4–7), but improved editing efficiencies compared with ABE7.10 at most sequence motifs. Measuring the activity of the single-residue reversion editor ABE7.10-K allowed us to see that alone, the N157K mutation resulted in activity between that of ABE7.10 and the multi-residue reversion variants. This supported our hypothesis that the Y123H and N157K reversions combined may be responsible for the improved editing activities of the ME-ABEs and the modification in sequence context specific activity, while the additional L36H, L51R and C146S reversions appeared to have a neutral impact on editing activity.
ME-ABEs are evaluated for off-target activity
Previous efforts to evolve editors from ABE7.10 for enhanced on-target activity have co-evolved increased gRNA-dependent and gRNA-independent off-target editing activity13. Furthermore, TadA-derived editors maintain the ability to deaminate RNA transcriptome-wide16,33. While certain mutations have been found to reduce this off-target activity, it is often accompanied by a reduction in on-target editing from the parent-derived editor13,21,34. We therefore sought to assess whether our ME-ABEs, which showed improved on-target editing activities relative to ABE7.10, also increased their off-target propensities. To assess gRNA-dependent off-target activity, we selected five previously validated off-target sites associated with three different on-target sites. These potential off-targets were evaluated with all four ME-ABEs and the benchmark editors ABE7.10, ABE8e and ABE8.20 (Fig. 4c and Supplementary Fig. 26)2,14,35,36,37,38,39,40. HEK293T cells were transfected with plasmids encoding an ABE variant and one of the three gRNAs (or a non-targeting control). Cells were incubated for 72 h before genomic DNA was collected and the on-target and relevant off-target site(s) were amplified and sequenced. ME-ABE on-target editing was within error of ABE8e and ABE8.20 at two of the three sites (Hi.0 and H2.0) and ranged from 50.2% to 63.7% of the efficiency of ABE8e at the third site (H3.0). Despite similarities in on-target editing, significant off-target editing was detected at all five off-target loci for ABE8e (ranging from 0.08 ± 0.02% to 4.64 ± 0.58%), while for all four ME-ABEs, off-target editing was only statistically significantly above background levels at the Hi.0 off-target 1 site (on-target editing was within error for all editors at this site). This activity ranged from 1.21 ± 0.28% for ABE7.10-HK to 1.81 ± 0.84% for ABE7.10-HSK, which was higher than that of ABE7.10 (0.19 ± 0.04%) but lower than that of ABE8.20 (3.18 ± 0.94%) and ABE8e (4.64 ± 0.58%). Reduced off-target activity at the Hi.0 site and negligible off-target activity at all other tested sites suggest that the improved on-target activity of the ME-ABEs is not coupled with an equivalent increase in their gRNA-dependent off-target editing profiles as is the case with ABE8e and ABE8.20.
To further evaluate gRNA-independent off-target activity, we used a previously described orthogonal R-loop assay (Supplementary Fig. 27a and Supplementary Discussion 11)13,41. Average on-target activity at two independent sites is plotted against average off-target activity at three independent, previously validated sites in Fig. 4d. A breakdown of these data by an on-target–off-target site combination can be found in Supplementary Fig. 27b–g. Across all three off-targets, the ME-ABEs showed similar levels of off-target editing activity to one another and to that of the parent ABE7.10, with substantially less off-target activity than ABE8e or ABE8.20 (Supplementary Fig. 28a). Averaged across all off-targets, editing efficiencies were 1.17 ± 0.14% for ABE7.10, 1.00 ± 0.09% for ABE7.10-HK, 0.91 ± 0.07% for ABE7.10-HRHSK, 10.15 ± 2.11% for ABE8e and 5.03 ± 0.22% for ABE8.20. The maximally reverted ABE7.10-HRHSK editor showed 11.2-fold and 5.6-fold reductions in average off-target editing activity compared with ABE8e and ABE8.20, respectively. For both on-target sites, the ME-ABEs facilitated higher editing efficiencies than ABE7.10 (Supplementary Figs. 28b,c and 29a). As expected, off-target editing efficiencies were independent of the on-target gRNA used (Supplementary Fig. 29b–d). Overall, the orthogonal R-loop data suggest that the ME-ABEs increased on-target editing activity independently of gRNA-independent off-target editing.
Finally, we assessed RNA off-target activity by performing whole-transcriptome RNA sequencing (RNA-seq) on the benchmark editors ABE7.10, ABE8e, ABE8.20 and the maximally modified ME-ABE ABE7.10-HRHSK. We found that all editors produced similar levels of A to I RNA off-target editing at levels significantly above that of the nCas9 control (Supplementary Fig. 30 and Supplementary Discussion 12). Taken together, these off-target analyses suggest that while the ME-ABEs reduce gRNA-dependent and gRNA-independent off-target DNA activity, they maintain RNA off-target editing levels at rates comparable to currently widely used editors.
ME-ABE reversions are ineffectual in ABE8e and ABE8.20 backgrounds, but offer advantages over other reported precision editors
Given the beneficial activity of ME-ABEs, we investigated combining the minimal and maximal sets of residue reversions by installing them into ABE8e and ABE8.20 (Extended Data Fig. 6). We found that applying these reversion combinations to the ABE8e and ABE8.20 backgrounds neither improved activity nor altered the editing window width, suggesting that the identified reversions’ activity was specific to the context of ABE7.10. Subsequent evolutionary changes in ABE8e and ABE8.20 may epistatically interfere with or override the beneficial interactions provided by ME-ABE reversions (Extended Data Fig. 6, Supplementary Figs. 20 and 31–35, and Supplementary Discussion 12). With this finding, we sought to further compare ME-ABEs with several additional editors engineered for precision (Fig. 5a). The ABE8e-V106W variant is reported to greatly reduce DNA and RNA off-target editing while maintaining high on-target activity, making it a favored editor for clinical applications7,13. To our knowledge, extensive editing window studies have not been published for this variant, although analogous TadCBE-V106W variants have been shown to have narrowed editing windows relative to their parent TadCBEs34. While the V106W variant was co-evolved alongside ABE8e, the ABE9 editor was rationally engineered from ABE8e for precision editing37. ABE9 contains two additional mutations (N108Q and L145T) and is reported to have an extremely narrow editing window centered at position 5 in the protospacer, alongside reduced off-target activity. To facilitate a direct comparison, these editors and maximally reverted editor ABE7.10-HRHSK were transfected into HEK293T cells with one of 12 gRNAs targeting a diverse set of endogenous genomic sites (Supplementary Fig. 18), and data were treated as previously described (Supplementary Discussion 8) to generate a representative editing window for all three editors (Fig. 5b–g and Supplementary Fig. 36). Using our defined metric for editing window, we found ABE8e-V106W to have an editing window spanning positions 3–11, matching that of its parent editor ABE8e. By contrast, ABE9 showed a narrower window encompassing only positions 5–6. When comparing average editing efficiencies at position 5, ABE7.10-HRHSK showed a 1.6-fold increase in editing over ABE9, while the ABE8e-V106W variant showed an additional 1.1-fold increase over ABE7.10-HRHSK.

a, The subset of editors analyzed for precision editing are shown, highlighting derivation ancestry. Residues that differentiate each precision editor from their parent editor are listed—those that correspond to the wtTadA identity (reversions) are underlined, those that have been modified multiple times over the course of engineering (‘evolutionary hot spots’) are shown in bold, and all others are in plain text. b, HEK293T cells were transfected with plasmids encoding one of the listed ABEs and one of 12 gRNAs targeting an endogenous site or an NT control (Supplementary Fig. 18; indicated by asterisks). After a 72-h incubation, genomic DNA was collected, target sites were amplified and sequences were analyzed by NGS. Editing was performed in n = 3 biological replicates and normalized as described in Fig. 4. Normalized editing scores for all 96 A’s were then sorted by A position, and scores were further averaged across gRNAs to identify the editing window. The number of A’s considered for each position is given below the x-axis. The horizontal dotted line shows the 15% cutoff used to determine the editing window for each editor. c–g, Data from b are shown for editors ABE7.10 (c), ABE7.10-HRHSK (d), ABE8e (e), ABE8e + V106W (f) and ABE9 (g). Points show normalized editing efficiency for a given editor at a given site and are shown for all replicates. Box hinges illustrate quartiles, lines show medians and bars show minimum and maximum editing. h, Orthogonal R-loop assay was used to analyze gRNA-independent off-target activity of precision editors as described in Fig. 4d. The x–y plot indicates the average off-target activity for the most edited A position across three independent off-target sites, and average on-target activity at the most edited A position across two independent on-target sites, accounting for n = 3 biological replicates with error bars showing propagated standard deviation.
We additionally evaluated the ABE7.10-HRHSK variant against ABE8e-V106W and ABE9 using the orthogonal R-loop assay to assess gRNA-independent off-target editing activity (Fig. 5g and Supplementary Figs. 37 and 38). Using the same on- and off-target editing sites as before, we found that ABE9 had nearly undetectable levels of gRNA-independent off-target editing activity, with an average off-target efficiency of 0.05 ± 0.02%. ABE8e-V106W showed an average off-target efficiency of 4.77 ± 0.48%, representing a 2.3-fold decrease from ABE8e. However, the average off-target efficiency of ABE7.10-HRHSK was reduced 4.2-fold compared with ABE8e-V106W. The average on-target editing activities at the two selected sites suggest that ABE7.10-HRHSK and ABE8e-V106W have similar on-target editing efficiencies (36.6 ± 2.4% and 36.8 ± 1.4%, respectively), while that of ABE9 (21.1 ± 2.0%) was 1.7-fold less than that of ABE7.10-HRHSK. These comparisons with other precision editors suggest the ME-ABEs strike a unique balance between precision and activity. While the ME-ABE editing window is larger than that of ABE9, on-target editing is greatly improved in comparison, and on-target activity is comparable to that of ABE8e-V106W but with reduced bystander and gRNA-independent off-target editing.
ME-ABEs facilitate modeling of challenging SNVs of clinical interest
Given our findings that the ME-ABEs generally increased editing activity relative to ABE7.10, improved editing at multiple sequence contexts, and on average matched the efficiency of ABE8e and ABE8.20 at the central A5 position without widening the editing window, we reasoned that these alternative tools could expand our ability to robustly and precisely edit target A’s beyond well-established sites in HEK293T cells. We found that the ABE7.10-HRHSK ME-ABE maintained similar editing profiles in different cell types (Fig. 6a,b, Extended Data Fig. 7 and Supplementary Discussion 14). In addition, ME-ABEs successfully edited six SNVs of clinical interest that had previously evaded modeling with base editors ABE7.10, ABE8e and ABE8.20 owing to the presence of missense-mutation-inducing bystanders, target A sequence context, overall low editing efficiencies or a combination of these issues (Fig. 6c–g and Supplementary Figs. 39–44). For a further discussion on site selection and data generation and normalization, see Supplementary Discussion 15.

a, Activity of ABE7.10-HRHSK was measured at six genomic sites in both HEK293T and HeLa cells. HEK293T cells were treated as described in Fig. 4. HeLa cells were transfected with plasmids encoding ABE-P2A-mCherry and one of six gRNAs. The mCherry fluorescence marker enabled sorting for transfected cells before genomic DNA collection and NGS analysis. Activity is presented as an average fold change in activity relative to ABE7.10 for the indicated A; n = 3 biological replicates, and error bars show standard deviation. b,Editing activity of various ABEs at the Hi.4 site in HeLa cells. Bars show average editing of n = 3 biological replicates, and error bars represent the standard deviation. c, Six SNVs of clinical interest with challenging-to-edit target A’s were selected for modeling with ABEs. The target A is shown in bold, while bystander A’s whose edit would result in missense mutations are shown in red. d, Summary of editing activity at six SNVs of clinical interest. Data for each editor are presented as fold changes in the percentage of DNA sequencing reads with the desired allele sequence (that is, target A edited and no bystander editing) with respect to ABE7.10. Bars show mean fold change for n = 3 independent sets of transfections, error bars indicate standard deviation and the horizontal dashed line shows average ABE7.10 activity. e, Summary of editing precision at six SNVs of clinical interest. Precision scores for each editor were calculated by determining the percentage of all edited sequencing reads in which only target A was appropriately modified. Fold-change values were determined by taking the average precision score for each editor and comparing it with that of ABE7.10 for n = 3 biological replicates; error bars show standard deviation and fold-change values from individual replicates are shown as points. f, Absolute editing efficiencies of ABEs used to introduce the ERCC2E95G SNV. Absolute editing was measured in n = 3 biological replicates, shown as individual points; bars show means, and error is reported as standard deviation. g, The absolute editing efficiency data from f was normalized to total editing (all sequencing reads with any type of edit) to report precision of desired editing among all edited sequences. Bars show average precision scores for n = 3 biological replicates, and error bars represent standard deviation
At five of the six tested sites, both ME-ABEs showed higher absolute editing efficiencies than ABE7.10 (Fig. 6d), ranging from a 1.17 ± 0.15-fold higher editing rate for ABE7.10-HRHSK at the SCN5AI1835T site to a 4.26 ± 1.21-fold higher editing rate for the ABE7.10-HRHSK editor at the ERCC2T76A site. The ERCC2Y542C edit was the only site tested where activity of the ME-ABEs matched that of ABE7.10 without further improvement. However, we will note that the target A for this site lies at position 7 rather than 5. These results therefore are consistent with our characterization studies (Fig. 4b), which showed that the ME-ABEs have enhanced editing relative to ABE7.10 at positions 4–6, while maintaining similar editing rates at the remaining editable positions.
At all sites, both ME-ABEs showed vastly improved precision over ABE8e (Fig. 6e). This ranged from a 6.6- and 6.3-fold increase in precision over ABE8e for ABE7.10-HK and ABE7.10-HRHSK, respectively, for the ERCC2T76A mutation, to a 28.9- and 30.2-fold increase over ABE8e for ABE7.10-HK and ABE7.10-HRHSK, respectively, for the SCN5AH558R mutation. Similarly, although to a lesser degree, the ME-ABEs improved precision over that of ABE8.20 at all sites, ranging from 1.5-fold increases for both ABE7.10-HK and ABE7.10-HRHSK at the ERCC2D673G mutation to a 5.0- and 5.1-fold increase for ABE7.10-HK and ABE7.10-HRHSK, respectively, at the ERCC2Y542C mutation. While the ABE8 variants were often more ‘active’ than the ME-ABEs, resulting in a greater number of edited sequencing reads, their tendency to edit multiple A’s present in their wider editing windows led to less precise editing outcomes. This was particularly well illustrated at the ERCC2E95G site (Fig. 6f,g and Supplementary Fig. 42). While the target A5 was readily edited by ABE8e and ABE8.20 (with overall bulk editing efficiencies of the target A of 34.0 ± 4.8% and 32.1.0 ± 7.2%, respectively), adjacent bystanders including a readily edited A7 were also modified in many sequences, resulting in high levels of undesired editing efficiency and low precision scores (9.5 ± 1.28% and 15.1 ± 4.4% of edited reads had only the target A•T, and no bystanders, converted to G•C for ABE8e and ABE8.20, respectively). While bulk editing efficiency of the target A by ABE7.10-HRHSK at this site was 23.8 ± 7.5%, which is lower than that of ABE8e and ABE8.20, 63.8 ± 4.8% of edited reads had only the target A, and no bystanders, edited, a 6.7- and 4.2-fold increase in precision editing compared with ABE8e and ABE8.20, respectively. At all six sites tested, the ME-ABEs had precision scores on par or greater than ABE7.10, and always greater than ABE8e and ABE8.20. Furthermore, at all but one site, ME-ABEs showed higher precise allele editing efficiencies than ABE8 editors. Overall, we present use of the ME-ABEs in different Cas architecture contexts and show the utility and advantages of editors with improved editing efficiency but narrow editing windows for modeling SNVs of clinical interest.
Discussion
We have completed a systematic and comprehensive reversion analysis of all 14 mutations in the widely used ABE7.10 editor. Using three independent assays in two different host systems, we were able to categorize the 14 single-residue reversion editors into 3 groups with respect to ABE activity: inhibiting reversions, neutral reversions and enhancing reversions (Extended Data Fig. 8). Interestingly, by measuring the activity of these reversion variants in different host systems, we noted host-system-dependent activity. In particular, the ABE7.10-Y147D reversion editor enhanced ABE activity in our bacterial reporters, but inhibited mammalian cell editing in both genomic and episomal contexts. Host-dependent characteristics such as unique posttranslational modifications, system-specific chaperones and cofactors, and altered aggregation properties have been an acknowledged challenge when using directed evolution to evolve proteins in E. coli for eventual use in mammalian systems30. In the case of base editing, the differences in host DNA repair mechanisms and the context of DNA substrate (chromatin) add additional layers of complexity. While it is unclear which, if any, of these factors are responsible for the ABE7.10-Y147D variant’s differential activity, our findings show that the TadA enzyme and ABE activity fitness landscape are constrained by their host system. Future development of new genome editing tools via directed evolution may consider designing more precision-focused selection strategies and coupling these with rigorous evaluation of accumulated mutations following each round of selection in the end-use system. Alternatively, the field may benefit from the development of strategies to perform directed evolution outside of bacteria. While evolution in yeast may bridge some of the differences between prokaryotic and eukaryotic systems with regard to DNA structure and repair, the future standard may be to evolve genome editors in mammalian cells. While this currently faces challenges brought on by limitations in delivering libraries with sufficient coverage, lengthy culturing and selection periods, and high levels of researcher intervention, development of an editor in the native mammalian context may avoid unfavorable evolutionary trajectories.
Systematic analysis of the 14 acquired mutations’ contributions to ABE activity allowed us to engineer several ME-ABEs. Specifically, simultaneous reversion of the residues categorized as neutral and enhancing in our single-residue reversion studies in mammalian cells resulted in a set of ME-ABEs with favorable properties. These ME-ABEs maintained the narrow editing window and minimal gRNA-dependent and gRNA-independent off-target editing profile of ABE7.10, but with increased on-target editing efficiencies at central positions (most notably position 5) within the editing window similar to those of ABE8e and ABE8.20. In particular, these ME-ABEs edit target A’s within DA and AH (D = A, G or T; H = A, C or T) motifs on par or higher than the ABE8 variants. Most notably, this included the ABE7.10-HRHSK variant in which over a third of the residues initially introduced to ABE7.10 are reverted. This reversion strategy may have found success in decoupling increases in editing efficiency from increases in TadA enzyme processivity as it did not rely on selection pressure to alter the enzyme’s fitness landscape. Instead, we investigated the local fitness landscape for mutations that may have provided advantage only under unique selection conditions and identified a set of core mutations critical for ABE activity. Notably, four of the five ABE7.10-HRHSK reversions came from the fifth round of directed evolution, in which the selection required editing at two sites with target A’s in different positions (positions 6 and 9) and embedded in unique target motifs (GA and TA). It is possible that this level of stringency led to mutants passing the selection via indirect, bacterial-specific mechanisms. Subsequent removal of these mutations may have refocused enzyme activity to Cas9-driven deamination and an improved editing profile that enabled decoupling of on-target editing and processivity. Future base editor evolution attempts may consider confining selection system targets to a single protospacer position to increase on-target editing and maintain a narrow editing window, or incorporate negative selections to avoid mutants with wider editing windows. The identification of these beneficial and neutral reversions was limited to the background of the ABE7.10 editor—introducing these same reversions into ABE8e and ABE8.20 did not improve editing activity nor narrow the editing window. This highlights the importance of studying variants in the appropriate context, taking into account the effects of epistatic compensatory mutations in TadA’s fitness landscape. Given that many widely used editors such as the ABE8 variants and TadCBEs are further evolved from ABE7.10, future work performing reversion analyses on these editors has the potential to identify similar dispensable mutations from earlier rounds of evolution against these backgrounds.
The ME-ABEs introduce an alternative tool to the genome editing toolbox. They are compatible with additional Sp-Cas9 variants (such as the PAM-expanded variants Cas9-NG and SpRY-Cas9, which allow for easier positioning of target A’s at the optimal position 5 within the protospacer), and they function well in multiple cell types. Given this, combined with their increased editing activity and narrow editing window, we anticipate that they will be useful for applications in which efficient editing without bystanders is required. We illustrate this with a series of six clinically relevant SNVs; ME-ABEs showed up to 15.7-fold increases in precision over both ABE8 variants, and an average 2.7-fold increase in editing efficiency over ABE7.10 when the target A is placed at position 5 within the editing window. While all clinically relevant SNVs analyzed in this study were targeted with editors delivered via plasmid and without selection for transfection, we hypothesize that optimization of delivery methods could enhance overall editing activity while maintaining the favorable editing profile of the ME-ABEs. We note that this benefit is strongly linked to positioning and, to a lesser degree, to the sequence motif. We therefore encourage use of ME-ABEs when the target A is in a DA motif and can be placed at position 5 within the protospacer, and particularly when bystanders nearby produce non-silent mutations. We anticipate that these tools may allow for efficient and precise editing of SNVs previously inaccessible by base editors due to bystander editing, while our single-residue reversion strategy used to identify key residues may prove useful for characterization of mutational effect in other evolved editors.
Methods
Cloning and plasmid materials
A variety of cloning methods were used to produce the plasmids in this study including blunt-end cloning, USER cloning and golden gate assembly. For all base editor plasmids, a reservoir-to-vector backbone strategy was used in which modifications to the TadA deaminase portion of the editor could be made in smaller reservoir plasmids to facilitate optimized insertion of the entire TadA cassette to a variety of base editor vector backbones for preparation of mammalian editors (both S. pyogenes nCas9-NG and SpRY variants), stability of expression constructs, or bacterial editors. All cloning and Sanger sequencing primers were ordered through Integrated DNA Technologies. Phusion High-Fidelity DNA Polymerase (ThermoFisher, F534L) or Phusion U Hot Start DNA Polymerase (ThermoFisher, F556L) was used for fragment amplification, as appropriate. PCR products were purified using the QIAquick PCR Purification Kit (Qiagen, number 28104), and all plasmids were prepared using the ZymoPURE II Midiprep kit, endotoxin-free protocol (Zymo Research, D4201). Vector backbone plasmids were sequenced in full by Plasmidsaurus using Oxford Nanopore Technology. Reservoir plasmids’ TadA cassette modifications and golden gate assembly plasmid junctions were sequence verified with Sanger sequencing provided by Azenta Life Sciences.
ABE variants were constructed in pUC reservoir plasmids containing only TadA and an ampicillin resistance gene. Four starting reservoir plasmids were constructed with TadA gene fragments ordered from Integrated DNA Technologies (ABEmax/GenScript codon optimization42). TadA fragments were inserted into the reservoir plasmid via USER assembly (New England Biolabs protocol). The four reservoirs included a wtTadA reservoir, a TadA*7.10 reservoir, a TadA*8e reservoir and a TadA*8.20 reservoir. From these starting plasmids, single-residue modifications were introduced using one-piece blunt-end cloning in a site-directed mutagenesis strategy1. Blunt-end primers containing the single-residue modification were phosphorylated with T4 polynucleotide kinase (New England Biolabs, M0201) before reservoir amplification with Phusion polymerase. The PCR product was treated with DpnI (New England Biolabs, R0176) to remove leftover template, QiaQuick column purified, followed by re-ligation with Quick ligase (New England Biolabs, M2200). Reversions were matched to the codon optimization used in the wtTadA gene fragment. Modifications were introduced sequentially for multi-residue reversion variants. TadA cassette fragments were then amplified with BsaI cut site flanking primers to facilitate golden gate assembly into an appropriate base editor vector backbone.
Briefly, vector backbones were designed with BsaI recognition cut sites placed such that golden gate assembly (New England Biolabs protocol) with amplified TadA led to re-constitution of a functional base editor. Mammalian expression ABE and stability of expression ABE plasmids were adapted from nCas9-NG_P2A-mCherry (Addgene, 219934) or nCas9-SpRY_P2A-mCherry (Addgene, 219935) plasmids. Bacterial expression ABE plasmids were adapted from GFP-itis pBE-t (Addgene, 195342). Coding sequences and further cloning information for all new base editor plasmids created can be found in Supplementary Data 1. The remaining plasmids used in the study were constructed as previously described—gRNA plasmids43 (protospacer sequences listed in Supplementary Data 1), fluorescence turn-on reporter plasmids12 and bacterial antibiotic resistance survival plasmids2. Key ME-ABE plasmids are available from Addgene (https://www.addgene.org/Alexis_Komor/).
Cell culture
HEK239T (ATCC, CRL-3216) and HeLa cells (ATCC, CCL-2) were cultured at 37 °C with 5% CO2 in Dulbecco’s modified Eagle medium (DMEM) high-glucose GlutaMAX Supplement (ThermoFisher, 10566016) with 10% fetal bovine serum (FBS, ThermoFisher, A5256701) and 1% penicillin–streptomycin (ThermoFisher, 15140122). Cells were passaged as needed with TrypLE (ThermoFisher, 12605010) to maintain confluency below 90%. All cell lines were obtained directly from and authenticated by the supplier (ATCC). The passage number was limited so as not to exceed 29 sub-passages from the commercial stock. All cell lines tested negative for mycoplasma with Mycostrip (InvivoGen, rep-mys-10) every 2 months.
Transfections, genomic DNA collection and preparation for RNA-seq
HEK293T cells were plated in 48-well plates in penicillin–streptomycin-free media at a density of 35,000 cells per well 24 h before transfection. All transfections used Lipofectamine 2000 (ThermoFisher, 11668030) in reduced serum Opti-MEM media (ThermoFisher, 31985062), prepared according to the manufacturer’s protocol. Transfection mixes for fluorescence turn-on reporters contained 750 ng ABE plasmid, 500 ng reporter plasmid and 250 ng gRNA plasmid. For testing editing at endogenous mammalian sites, transfection mixes contained 750 ng ABE plasmid and 250 ng gRNA plasmid. For stability of expression studies, 750 ng ABE stability reporter (Extended Data Fig. 1) was delivered. HEK293T cells were transfected at approximately 50–60% confluency. The media was changed for DMEM with 10% FBS and 1% penicillin–streptomycin 18 h post-transfection. Cells assayed for stability of ABE expression were collected 48 h post-transfection, while all other HEK293T transfections added an additional 250 µl DMEM with 10% FBS and 1% penicillin–streptomycin at 48 h, and were collected 72 h post-transfection.
Cells collected for genomic DNA had media removed and were gently washed with phosphate-buffered saline (PBS, ThermoFisher, 10010023). Following removal of PBS, 100 µl of lysis solution was applied for approximately 2–5 min. Lysis solution was prepared in molecular-biology-grade water with 100 mM Tris–HCl (pH 7.0), 0.05% sodium dodecyl sulfate and 25 μg ml−1 Proteinase K (New England Biolabs, P8107S). Cell lysis solution was then transferred to PCR strips and incubated at 37 °C for 1 h followed by 80 °C denaturation for 30 min, with long-term storage at 4 °C.
HeLa cells were plated in 6-well plates in penicillin–streptomycin-free media at a density of 350,000 cells per well 24 h before transfection. TransIT-HeLaMONSTER (Fisher, MIR2904) transfection reagent was used according to the manufacturer’s protocol to deliver 2,500 ng ABE plasmid and 500 ng gRNA plasmid in Opti-MEM. Cells were transfected at approximately 60–70% confluency and collected 72 h post-transfection, with no media change following transfection.
For RNA-seq experiments, HEK293T cells were plated in a 6-well plate format (penicillin–streptomycin-free media) at a density of 350,000 cells per well. Subsequently, 24 h after plating, when cells were ~50–60% confluent, they were transfected with a mixture of 2.5 µg ABE plasmid (or control mCherry expression vector) and 0.5 µg H2.0 gRNA plasmid in reduced serum Opti-MEM with 9 µl Lipofectamine 2000, prepared according to the manufacturer’s instructions. Then, 18 h post-transfection, the media was changed for DMEM with 10% FBS and 1% penicillin–streptomycin. Cells were collected and RNA was obtained with the Zymo Quick RNA/DNA Miniprep kit 72 h post-transfection (Zymo Research, D7001), with the suggested DNAse I treatment. RNA samples were stored at −80 °C until library preparation.
ABE modeling
Molecular graphics and analyses were performed with UCSF ChimeraX (v1.9), developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases). Monomeric ABE7.10 was modeled in silico using ChimeraX44, starting with the crystal structure of ABE8e (Protein Data Bank (PDB) ID: 6VPC)15, and replacing the catalytic TadA*8e domain with Alphafold2-predicted TadA*7.10 (as done in Ranzau et al.)12. The ‘docking’ domain was modeled using the swapaa function in ChimeraX to replace TadA*8e residues with TadA*7.10. A comparison of ABE0.1 and ABE7.10 structure was done by substituting the modeled TadaA*7.10 with wtTadA (PDB ID: 1Z3A with AlphaFold predictions for unmodeled regions)20. All residue reversions were modeled using the ChimeraX swapaa function.
Flow cytometry and fluorescence-activated cell sorting
Flow cytometry and fluorescence-activated cell sorting were performed on a BioRad S3e cell sorter equipped with 488-nm, 561-nm and 640-nm lasers, using the Biorad ProSort software v1.6. Cells were collected and prepared for sorting 48 h (stability studies) or 72 h (fluorescence turn-on and HeLa editing activity) post-transfection. The media was removed and cells were gently washed with PBS, which was then removed to allow for disassociation with Accumax (ThermoFisher, 00-4666-56)—50 µl in a 48-well plate setting and 500 µl in 6-well plates. Cells were resuspended to approximately 1 × 106 cells per ml in resuspension buffer (PBS, 2% FBS, 1 mM EDTA), before passing through a 0.35-µM filter cap round-bottomed test tube (Fisher, 352235) and stored on ice until analyzed.
Scatter gating was first performed to isolate live, singlet cells (Supplementary Figs. 2 and 11b,c). For dual-color fluorescence experiments, single-color controls helped establish color gates. mCherry was detected with the FL3 channel (602–627 nm), while EGFP used FL1 (510–540 nm). Single-color and no-color controls were established with cell samples transfected with plasmids matching the experimental samples with inactivating mutations in either or both fluorophores: for mCherry, the Y72H deactivating mutation was introduced, and for EGFP, the A111V or H182Y mutation was used as appropriate for the reporter being tested. A minimum of 50,000 cells passing scatter gating were analyzed for all flow cytometry experiments. For episomal fluorescent reporter assays, approximately 10–50% of cells were considered double color (EGFP+ and mCherry+), while approximately 50–70% were considered transfected (mCherry+). Data were analyzed with FlowJo v9 software and visualized with PRISM GraphPad 9.
For cell sorting, similar scatter gating protocols were used to establish a population of live, singlet HeLa cells. An ABE0.1-P2A-dmCherry (Y72H)-transfected sample was used to establish the single-color mCherry+ gate. For each sample, approximately 10,000 mCherry+ cells (representing approximately 3–6% of total cells) were sorted into 500 µl cold PBS using the ‘Enrich’ sort mode on the Biorad ProSort software v1.6. Samples were then pelleted at 300 relative centrifugal force (rcf) for 10 min, and approximately 500 µl of supernatant was removed, followed by the transfer of the remaining cell solution to a PCR strip. Cells were further pelleted with a benchtop PCR-strip centrifuge, and the remaining supernatant was carefully removed, following which the cells were resuspended in 5 µl lysis buffer and incubated at 37 °C for 1 h followed by 80 °C denaturation for 30 min, with long-term storage at 4 °C.
Western blot analysis
HEK29T cells were seeded in penicillin–streptomycin-free media in a 6-well plate at a density of 350,000 cells per well 24 h before transfection. Lipofectamine2000 (ThermoFisher, 11668030) and reduced serum Opti-MEM media (ThermoFisher, 31985062) were used according to the manufacturer’s instructions to deliver 2.5 µg of ABE plasmid or pUC19 control to HEK293Ts at ~40–50% confluency. The media was changed 18 h post-transfection for DMEM with 10% FBS and 1% penicillin–streptomycin. The cells were collected 48 h post-transfection with 100 µl RIPA lysis buffer (ThermoFisher 89900) prepared with Halt Protease Inhibitor (ThermoFisher, 78429), according to the manufacturer’s instructions. Cells were gently agitated for 30 min via a tube rotator at 4 °C and pelleted at 4 °C for 10 min at 16,000 rcf. Protein was then collected from the supernatant. Protein was quantified using the Pierce BCA Protein Assay Kit with Dilution-Free BSA Protein Standards (ThermoFisher, A55864). In duplicate, 40 µg total protein was prepared in reducing conditions with Laemmli Buffer (BioRad, 1610747) and 100 mM DTT and denatured by heating at 70 °C for 10 min. Samples were loaded on a 12% Mini-PROTEAN TGX gel 12-well gel (BioRad, 4561045) alongside a BioRad Precision Plus Protein Dual Color Standard ladder (BioRad, 1610374) and run in Tris–Glycine–SDS buffer (BioRad, 1610772) for 60–70 min at 140 V. Protein was transferred to 0.45-µm nitrocellulose membranes (BioRad, 1620145) at 4 °C using Tris–glycine transfer buffer (BioRad, 1610771) for 3 h at 60 V. Membranes were blocked with SuperBlock (ThermoFisher, 37515) for 1 h following which proteins were sequentially stained. All antibody dilutions were made in SuperBlock. The first membrane was stained for Cas9 using a 1:1,000 dilution of mouse IgG1, κ anti-Cas9 (BioLegend, 698301), the second membrane for mCherry using a 1:2,000 dilution of rabbit IgG anti-mCherry (AbCam, ab213511). Primary staining took place overnight at 4 °C. Membranes were washed with PBST (0.02% Tween20) before secondary staining. The first membrane used a 1:10,000 dilution of goat-anti-mouse Licor IRDye 800CW (Licor, 926-32210), and the second membrane used a 1:10,000 dilution of goat-anti-rabbit Licor IRDye 800CW (Licor, 926-32211), applied for 1 h at room temperature with rocking. Membranes were imaged using a Licor FC Odyssey with Image Studio (v 6.1) with a 2-min exposure time. Following staining for proteins of interest, membranes were both stained for the housekeeping protein β-actin. A 1:5,000 dilution of rat IgG2a, κ anti-β-actin (BioLegend, 664801) was applied for 3 h at room temperature, followed by PBST washing and secondary staining with a 1:10,000 dilution of goat-anti-rat Licor IRDye 680RD (Licor, 926-68076), applied for 1 h at room temperature, followed by imaging with a 2-min exposure. Uncropped western images are shown in Supplementary Fig. 45 and are available as source data.
Bacterial survival assay
Measurement of ABE activity in bacteria was adapted from a previously reported protocol12. Briefly, two sets of bacterial base editor plasmids were prepared—one set containing gRNA against pCamR H193Y and the other against pCamR Q122*. The two pCamR plasmids were independently heat shock transformed into S1030 E. coli (Addgene, 10506) following which two lines of chemically competent S1030-pCamR bacterial stocks were prepared45. gRNA-appropriate bacterial base editor plasmids were then heat shock transformed into the S1030-pCamR E. coli, allowed to recover in EZ Rich media (Teknova, M2105) for 1 h at 37 °C, before cultures were diluted 1:1,000 in 5 ml EZ Rich media containing 50 ng µl−1 kanamycin, 50 ng µl−1 carbenicillin (plasmid maintenance antibiotics) and 1 mM theophylline (for base editor induction). Cultures were grown for 16–18 h with shaking at 37 °C until visually saturated. Cultures were then diluted 1- to 1 × 107-fold in PBS, and 5 µl of each dilution was plated on 2× YT agar plates with 50 ng µl−1 kanamycin, 50 ng µl−1 carbenicillin and ±25 ng µl−1 chloramphenicol. Plates were grown at 37 °C for 16–18 h, following which colonies were counted at the highest possible dilution with single colonies visible. Colony counts were compared on chloramphenicol ± plates to determine survival rates. Three independent transformations were performed, with a further three sets of independent dilutions for each sample making for a total of 9 sets of dilutions for each bacterial base editor. Samples with visually disrupted dilution patterns were not included; the ROUT 1% outlier test was performed on calculated survival rates within a given bacterial editor. All data were then visualized with PRISM GraphPad 9.
Orthogonal R-loop
gRNA-independent off-target activity was determined using an orthogonal R-loop assay. HEK293T cells were plated in antibiotic-free media at 35,000 cells per well in a 48-well plate format 24 h before transfection. Cells were concurrently transfected with 300 ng Sa-dCas9 plasmid, 300 ng Sp-ABE plasmid, 200 ng Sp-gRNA and 200 ng Sa-gRNA with Lipofectamine 2000 reduced serum Opti-MEM media, prepared according to the manufacturer’s protocol. Sa-gRNA sites were selected for previous validation of ABE7.10 gRNA-independent off-target activity46 (off-target 1, site 25; off-target 2, site 26; off-target 3, site 29). Cells were cultured for 72 h, following which they were collected and lysed for genomic DNA, and targeted amplicons were amplified and sequenced.
Next-generation sequencing and data analysis
Genomic DNA was prepared for target amplicon sequencing following adapted protocol 6.1.2 from Osgood et al.43. Briefly, round 1 PCRs were carried out using 1–3 µl of 10-fold-diluted gDNA for all bulk lysis samples, and 1–3 µl of gDNA lysis (approximately 2,000–6,000 cells) for all sorted HeLA samples. Primer concentrations were adjusted for a final concentration of 0.125 µM, and cell cycle amplification was limited to 28–30 cycles (the minimal amount needed to see round 1 PCR product on a 2% agarose TAE gel without amplification of equivalently treated lysis buffer only ‘template’ negative control). All unique round 1 amplification primers used can be found in Supplementary Data 1. Round 2 PCR was carried out in adjusted 20-µl-volume reactions with 1–3 µl of round 1 PCR product, and 0.125 µM barcoding round 2 PCR primers. Round 2 PCR products were pooled, purified with gel extraction, quantified using Qubit dsDNA HS assay (ThermoFisher, Q32854) and run on an Illumina MiniSeq. Libraries were prepared per manufacturer’s protocol for 2 × 150 paired-end reads.
Next-generation sequencing data were first processed using Illumina Local Run Manager Generate FASTQ analysis module (v2.0) on MiniSeq control software (v2.2.1) to demultiplex and trim Fastq files. Data processing on individual amplicons was further performed using CRISPResso2 (version 2.2.14), batch mode analysis47. Base editing efficiencies were determined using CRISPResso2 nucleotide percent summary output. For all amplicons, a non-target gRNA (NT) control was used to identify any modifications present in the cell line independent of base editing. When the NT editing efficiency was greater than 1%, the average NT editing efficiency at a given position was subtracted out of the reported efficiency for a given editor at the same site. Allele editing efficiencies were determined from the CRISPResso2 allele frequency table: sequences were filtered for those with percentages of reads greater than 0.1% (those higher than the error rate) then further processed to consider only those with A•T to G•C changes within the protospacer with appropriate reference sequence alignment. Percentage reads for this subset of sequences were then averaged over three independent biological replicates, and resulting allele changes were determined. Note that for the SCN5A H558R allele, laboratory strain HEK293Ts are heterozygous (approximately 5% of bulk sequenced reads already contain the desired A•T to G•C change), so a non-targeting control was used to subtract the fraction of the bulk cell population already containing the edit from samples treated with the editor. All data were then visualized with PRISM GraphPad 9.
RNA-seq sample library preparation and sequencing were performed by Azenta Life Sciences. Briefly, a 2 × 150-bp strand-specific library was generated using poly-A selection and run on an Illumina NovaSeq. RNA-seq data were first quality checked with FastQC (v0.12.1) before adapters and homopolymeric tails were trimmed with FastP (v1.0.1)48. Reads were aligned using splice-aware STAR (2.7.11b+galaxy0)49 with alignment to hg38 and the settings suggested by REDITools in Lo Giudice et al.50,51,52. Alignment quality was monitored with MultiQC (v1.27)53, then deduplicated with the samtools RmDup function (v2.0.1)54. Variants were called with REDITools3 (v3.4). All samples were analyzed using reverse-strand orientation with strand correction, with a minimum coverage requirement of 10 reads. Control mCherry samples were analyzed with a minimum edit frequency of 0 (-me 0) and all variants were collected (-v all), while all experimental samples were analyzed for A to I (-v AG) variation only. Significantly edited sites were then called using a custom Python script (Supplementary Note 1) that added the following additional requirements: (1) control sample sites needed to match the reference at 99% or more of the collected reads, (2) editing frequency in the experimental samples needed to be greater than 1% of the collected reads and (3) sites of common genetic variants (SNVs) were removed using dbSNP151 as a reference. Significant sites were called using Fisher’s exact t-test comparing experimental samples with the transfection-relevant mCherry control, P < 0.05. Data were visualized using Python (v3.9) and plotted with Seaborn (0.13.2), or Logomaker (0.8.7)55.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Next-generation sequencing data are available in the NCBI Sequencing Read Archive database under project number PRJNA1247975 (ref. 56). ME-ABE plasmids from this study are available in Addgene (numbers 238103 and 238068—SpRY variants; numbers 238104 and 238067—NG variants; https://www.addgene.org/Alexis_Komor/). Sequences of all constructs, gRNAs and primers are included in Supplementary Data 1. Statistical source data for all supplementary figures can be found in Supplementary Data 2. Source data are provided with this paper.
Code availability
The custom Python script for determining significantly edited RNA transcripts collected from whole-transcriptome RNA-seq is available in Supplementary Note 1.
References
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Gaudelli, N. M. et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017).
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019).
Chen, P. J. & Liu, D. R. Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. 24, 161–177 (2023).
Porto, E. M. & Komor, A. C. In the business of base editors: evolution from bench to bedside. PLoS Biol. 21, e3002071 (2023).
Georgiadis, C. et al. Base-edited CAR T cells for combinational therapy against T cell malignancies. Leukemia 35, 3466–3481 (2021).
Musunuru, K. et al. Patient-specific in vivo gene editing to treat a rare genetic disease. N. Engl. J. Med. 392, 2235–2243 (2025).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Landrum, M. J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868 (2016).
Sherry, S. T., Ward, M. & Sirotkin, K. dbSNP—database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 9, 677–679 (1999).
Wolf, J., Gerber, A. P. & Keller, W. tadA, an essential tRNA-specific adenosine deaminase from Escherichia coli. EMBO J. 21, 3841–3851 (2002).
Ranzau, B. L., Rallapalli, K. L., Evanoff, M., Paesani, F. & Komor, A. C. The wild-type tRNA adenosine deaminase enzyme TadA is capable of sequence-specific DNA base editing. ChemBioChem 24, e202200788 (2023).
Richter, M. F. et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 38, 883–891 (2020).
Gaudelli, N. M. et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 38, 892–900 (2020).
Lapinaite, A. et al. DNA capture by a CRISPR–Cas9-guided adenine base editor. Science 369, 566–571 (2020).
Xiao, Y.-L., Wu, Y. & Tang, W. An adenine base editor variant expands context compatibility. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01994-3 (2024).
Stimple, S. D., Smith, M. D. & Tessier, P. M. Directed evolution methods for overcoming trade-offs between protein activity and stability. AIChE J. 66, e16814 (2020).
Bloom, J. D. & Arnold, F. H. In the light of directed evolution: Pathways of adaptive protein evolution. Proc. Natl Acad. Sci. USA 106 (supplement_1), 9995–10000 (2009).
Rallapalli, K. L., Komor, A. C. & Paesani, F. Computer simulations explain mutation-induced effects on the DNA editing by adenine base editors. Sci. Adv. 6, eaaz2309 (2020).
Kim, J. et al. Structural and kinetic characterization of Escherichia coli TadA, the wobble-specific tRNA deaminase. Biochemistry 45, 6407–6416 (2006).
Grünewald, J. et al. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol. 37, 1041–1048 (2019).
Arbab, M. et al. Determinants of base editing outcomes from target library analysis and machine learning. Cell 182, 463–480.e30 (2020).
Fasan, R., Meharenna, Y. T., Snow, C. D., Poulos, T. L. & Arnold, F. H. Evolutionary history of a specialized P450 propane monooxygenase. J. Mol. Biol. 383, 1069–1080 (2008).
Wang, X., Minasov, G. & Shoichet, B. K. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J. Mol. Biol. 320, 85–95 (2002).
DePristo, M. A., Weinreich, D. M. & Hartl, D. L. Missense meanderings in sequence space: a biophysical view of protein evolution. Nat. Rev. Genet. 6, 678–687 (2005).
Arantes, P. R. et al. Dimerization of the deaminase domain and locking interactions with Cas9 boost base editing efficiency in ABE8e. Nucleic Acids Res. 52, 13931–13944 (2024).
Ngo, A., Wang, R. & Puschnik, A. Dual fluorescence cytometry assay to assess cellular protein levels. Bio Protoc. 10, e3597 (2020).
Losey, H. C., Ruthenburg, A. J. & Verdine, G. L. Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat. Struct. Mol. Biol. 13, 153–159 (2006).
Sahdev, S., Khattar, S. K. & Saini, K. S. Production of active eukaryotic proteins through bacterial expression systems: a review of the existing biotechnology strategies. Mol. Cell. Biochem. 307, 249–264 (2007).
Hendel, S. J. & Shoulders, M. D. Directed evolution in mammalian cells. Nat. Methods 18, 346–357 (2021).
Pourmir, A. & Johannes, T. W. Directed evolution: selection of the host organism. Comput. Struct. Biotechnol. J. 2, e201209012 (2012).
Kim, H. S., Jeong, Y. K., Hur, J. K., Kim, J.-S. & Bae, S. Adenine base editors catalyze cytosine conversions in human cells. Nat. Biotechnol. 37, 1145–1148 (2019).
Grünewald, J. et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569, 433–437 (2019).
Neugebauer, M. E. et al. Evolution of an adenine base editor into a small, efficient cytosine base editor with low off-target activity. Nat. Biotechnol. 41, 673–685 (2023).
Tsai, S. Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR–Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015).
Lam, D. K. et al. Improved cytosine base editors generated from TadA variants. Nat. Biotechnol. 41, 686–697 (2023).
Chen, L. et al. Engineering a precise adenine base editor with minimal bystander editing. Nat. Chem. Biol. 19, 101–110 (2023).
Cowan, Q. T. et al. Development of multiplexed orthogonal base editor (MOBE) systems. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02240-0 (2024).
Kuscu, C., Arslan, S., Singh, R., Thorpe, J. & Adli, M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 32, 677–683 (2014).
Hsu, P. D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013).
Doman, J. L., Raguram, A., Newby, G. A. & Liu, D. R. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat. Biotechnol. 38, 620–628 (2020).
Koblan, L. W. et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 36, 843–846 (2018).
Osgood, N. R. B. et al. in Methods in Enzymology Vol. 712 (eds Ke, A. & Tang, W.) Ch. 14 (Elsevier, 2025).
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Chung, C. T., Niemela, S. L. & Miller, R. H. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc. Natl Acad. Sci. USA 86, 2172–2175 (1989).
Yu, Y. et al. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat. Commun. 11, 2052 (2020).
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Lo Giudice, C., Tangaro, M. A., Pesole, G. & Picardi, E. Investigating RNA editing in deep transcriptome datasets with REDItools and REDIportal. Nat. Protoc. 15, 1098–1131 (2020).
Fonzino, A. et al. REDInet: a temporal convolutional network-based classifier for A-to-I RNA editing detection harnessing million known events. Brief. Bioinform. 26, bbaf107 (2025).
Picardi, E. & Pesole, G. REDItools: high-throughput RNA editing detection made easy. Bioinformatics 29, 1813–1814 (2013).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Tareen, A. & Kinney, J. B. Logomaker: beautiful sequence logos in python. Preprint at bioRxiv https://doi.org/10.1101/635029 (2019).
Evanoff, M., Korpal, S., Krill, Z. D., Cowan, Q. T. & Komor, A. C. Precise, minimally evolved adenine base editors through mutation reversion analysis. NCBI Bioproject http://www.ncbi.nlm.nih.gov/bioproject/PRJNA1247975 (2025).
Acknowledgements
This research was supported by the University of California, San Diego and the National Institutes of Health (NIH) through grant number 2R35GM138317 (to A.C.K.). M.E. was supported by the Cell and Molecular Genetics Training Program, NIH Grant T32 GM007240-41. Q.T.C. was supported by the Molecular Biophysics Training Grant, NIH Grant T32 GM008326. Z.D.K. was supported by the Genetics Training Program, NIH Grant T32 GM145427. We gratefully acknowledge all current and former members of the Komor Lab for their advice, support and input. Thanks to B. L. Ranzau for support with fluorescent reporter applications, K. L. Rallapalli for TadA modeling support and N. Osgood for gRNA plasmid materials. We would additionally like to thank B. Yee and S. Aigner for support with RNA-seq analysis, and N. Zawalick and M. Holay for editing contributions.
Author information
Authors and Affiliations
Contributions
M.E. contributed to the conceptualization of the research project, experimental design, data curation, data analysis and writing of the paper. S.K. contributed to data curation and experimental design. Z.D.K. contributed to experimental design and data analysis. Q.T.C. contributed to data curation, experimental design and data analysis. A.C.K. contributed to the conceptualization of the research project, experimental design, data analysis, supervision of the work, writing of the paper and acquisition of funding. All authors contributed to the editing of the paper.
Corresponding author
Ethics declarations
Competing interests
A.C.K. is a member of the SAB of Pairwise Plants and an equity holder for Pairwise Plants and Beam Therapeutics, and receives royalties from Pairwise Plants, Beam Therapeutics and Editas Medicine via patents licensed from Harvard University. The interests of A.C.K. have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. All other authors declare no competing interests.
Peer review
Peer review information
Nature Biotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 ABE7.10 single-residue reversion and alanine mutants evaluated for editing efficiencies at endogenous genomic loci.
a. Table of gRNAs and target motifs used for evaluating editing efficiency at endogenous sites in mammalian cells. The most-edited A (considered the target A) for each of these sites was position 5, highlighted in blue. b. HEK293T cells were transfected with plasmids encoding one of the ABEs indicated and gRNA targeting one of three protospacers (listed in a). After 72 h, genomic DNA was harvested, target sites were amplified, and A•T to G•C editing efficiencies were analyzed via NGS. Heat maps show the average (n = 3) fold-change in sequencing reads with target A•T converted to G•C for each single-residue reversion base editor compared to that of ABE7.10. c. Heat maps show the average (n = 3) fold-change in sequencing reads with target A•T converted to G•C for select single-residue reversion base editors and their corresponding alanine substitution mutants, compared to that of ABE7.10. Heat maps in c/d are colored so that greyscale indicates activity less than ABE7.10, white activity on par with ABE7.10, and green (c) or maroon (d) indicates activity greater than ABE7.10. d-f. Average fold-changes in A•T to G•C editing activities compared to ABE7.10 for several single-residue reversion editors and corresponding alanine substitution mutants at the s15.0 (d), Ps.2 (e) and Ps.0 (f) endogenous genomic sites. Bars and error bars represent the average and standard deviation for n = 3 biological replicates. Data were analyzed with unpaired one-tailed, parametric t-tests (comparing each reversion editor to its corresponding alanine mutant), with significant p-values reported (p ≥ 0.05 not significant, not indicated).
Extended Data Fig. 2 ABE7.10 single-residue reversion variants assessed for expression and stability.
a. Construct map of plasmids used for expression and stability evaluation. Cartoon illustrates how ribosomal skipping P2A sequence allows for normalization of ABE-EGFP protein expression relative to amount of delivered reporter. mCherry shown in red, ABE-EGFP fusion shown in blue-green. Changes in ABE-EGFP expression modify the mCherry/EGFP ratio. b. 48 h following transfection of HEK293T with stability reporter plasmids, cells were analyzed via flow cytometry for EGFP and mCherry fluorescence intensities. The EGFP/mCherry fluorescence intensity ratio was calculated for all mCherry & EGFP positive cells. An EGFP/mCherry ratio frequency of distribution plot (bin width 0.08) is shown for the benchmark editors ABE0.1 (black), ABE7.10 (red), and ABE8e (fuchsia). c. Representative western blot (n = 3) monitoring ABE expression. HEK293T cells were transfected with plasmids expressing negative control pUC19 or the indicated editor in an ABE-P2A-mCherry architecture. Cells grew for 48 h before harvesting and quantifying total protein. Samples were evenly split and ran in parallel to assess expression of ABE (protein of interest, stained for with a Cas9-specific antibody) relative to β-Actin (housekeeping protein) or mCherry (transfection indicator) relative to β-Actin. HRHSK is the ABE7.10-L36H/L51R/Y123H/C146S/N157K multi-residue reversion editor. d. The average (n = 3) median EGFP/mCherry fluorescence intensity values for the subset of double color positive cells for each single-residue reversion editor are reported for cells treated as described in a-b. Reversion variants are ordered by round of evolution in which they first arose, left to right. Bars and error bars show the average median EGFP/mCherry ratio with associated standard deviation reported for n = 3 biological replicates. * symbols indicate reversion variants found to have neutral or improved activity with respect to ABE7.10. Data were analyzed with an ordinary one-way ANOVA with Holm-Šídák multiple comparison test to ABE7.10, with significant p-values reported (p ≥ 0.05 not significant, not indicated).
Extended Data Fig. 3 ABE7.10 multi-residue reversion variants evaluated in mammalian cells.
a. Table identifying ABE7.10 multi-residue combination reversions tested. b-c. Activity of combination reversion editors as measured by episomal reporter. Plasmids harboring ABE7.10 or ABE variants listed in a, were transfected into HEK293T cells alongside the EGFP A111V TAC fluorescent turn-on reporter and appropriate gRNA. Cells incubated for 72 h prior to analysis via flow cytometry where BE activity was calculated as described in Fig. 2a. Activity is reported as fold-change in BE activity (b) or fold-change in EGFP MFI, with respect to ABE7.10 (c). Dotted horizontal line shows normalized activity of ABE7.10. Bars and error bars represent the average and associated standard deviation for n = 5 biological replicates for ABE7.10 and C1, n = 3 biological replicates for all others. Data were analyzed using one-way ANOVA with Holm-Šídák multiple comparison test to ABE7.10 with significant p-values reported (p ≥ 0.05 not significant, not indicated) d. Heat map showing activity of a subset of combination reversion editors (those indicated by highlighted cells in a) against three endogenous genomic sites. ABE plasmids were transfected into HEK293T cells alongside one of three gRNA plasmids (protospacers shown in Extended Data Fig. 1a) and allowed to incubate for 72 h. Genomic DNA was then harvested, target amplicons amplified, and BE activity (percent of reads with target A•T converted to G•C) analyzed via NGS. Activity is reported as a fold-change in BE activity for a given editor with respect to ABE7.10 in a given replicate. Heat map colors represent the average fold-change in activity for n = 3 biological replicates, with greyscale indicative of activity less than ABE7.10, white indicative of activity on par with ABE7.10, and cyan indicative of activity greater than ABE7.10.
Extended Data Fig. 4 Normalized editing activity for ABEs across 27 endogenous sites, broken down by target A position.
HEK293T cells were transfected with an ABE plasmid encoding ABE7.10 or variant editor indicated along with one of the 27 gRNA plasmids from Supplementary Fig. 18, or a non-target (NT) gRNA. Cells were harvested after 72 h and genomic loci of interest analyzed for editing activity with NGS. Within a given transfection replicate, absolute editing efficiency for all A’s was normalized to the editing activity of ABE7.10 at the H2.0 A5 position (the maximally edited A, across all sites and in every replicate). The normalized editing efficiencies for all 205 A’s are shown as a function of A position within the protospacer. Data are represented by box and whiskers plot where center shows median, hinges show quartiles, and whiskers show minimum and maximum. ABE7.10-derived and ABE8-derived reversion editors were tested in n = 3 biological replicates, ABE7.10, ABE8e, and ABE8.20 in n = 6. All H2.0 normalized editing values from all replicates are indicated with overlaying points. The horizontal dotted line indicates the 15% relative editing efficiency cut-off used to demarcate the editing window for each editor.
Extended Data Fig. 5 Sequence motif preferences of multi-residue reversion editors at endogenous sites.
The normalized editing activities of ABE editors were sorted by the 5’ or 3’ motif of the edited A’s. Target A’s were limited to ‘editable A’s’ - those A’s in positions where the average normalized editing across all sites for a given editor was greater than 15%. Legend highlights inclusive positions for each editor, resulting in 19 sites interrogated between positions 4–7, 34 sites between positions 3–11, and 26 sites between positions 3–8. The normalized average editing activity for individual sites is shown by points, with each site measured in n = 3 biological replicates for all ABE7.10 reversion editors, and n = 6 for ABE7.10, ABE8e, and ABE8.20. Whiskers show the minimum and maximum average editing for a given editor/motif, boxes show quartiles, and lines show median editing activity.
Extended Data Fig. 6 Characterization of ABE8 multi-residue reversion variants in mammalian cells.
a. Naming legend for ABE8-derived reversion editors. b. HEK293T cells were transfected with a single ABE plasmid containing either one of the editors listed in part a, ABE7.10, ABE8e, or ABE8.20, along with one of 27 gRNAs plasmids targeting an endogenous site or a non-targeting (NT) control (See Supplementary Fig. 18). After a 72-h incubation, genomic DNA was harvested, target sites were amplified, and sequences analyzed by NGS. Absolute editing values (percent of reads with a given A•T pair converted to G•C) were normalized to the absolute editing of ABE7.10 at the H2.0 A5 site (the maximally edited A in all experiments) within a given replicate. Editing activity was examined in n = 3 biological replicates for all ABE8 reversion editors, and n = 6 for ABE7.10, ABE8e and ABE8.20. Horizontal dotted line shows the 15% cut-off used to determine the editing window for each editor. c-d. Normalized editing activities of a subset of ABE8-derived editors sorted by 5’ (c) or 3’ (d) motif are shown. Target A’s were limited to ‘editable A’s’ - those A’s in positions where the average normalized editing across all sites for a given editor was greater than 15%. Legend highlights inclusive positions for each editor, resulting in 19 sites interrogated between positions 4–7, 34 sites between positions 3–11, and 26 sites between positions 3–8. The normalized average editing activity for individual sites is shown by points, with each site measured in n = 3 biological replicates for all ABE8-derived reversion editors, and n = 6 for ABE7.10, ABE8e, and ABE8.20. Whiskers show the minimum and maximum average editing for a given editor/motif, boxes show quartiles, and lines show median editing activity. e. Editing activity of a subset of ABE8e-derived combination reversion editors measured by orthogonal R-loop off-target assay, as described in Fig. 4d. x-y plot indicates the average off-target activity for the most edited A position across three independent off-target sites, and average on-target activity at the most edited A position across two independent on-target sites, accounting for n = 3 biological replicates with error bars showing propagated standard deviation.
Extended Data Fig. 7 Editing activity at 6 endogenous genomic sites in HeLa cells.
HeLa cells were transfected with plasmids encoding an ABE (ABE-P2A-mCherry architecture) and a gRNA with the indicated protospacer sequence targeting an endogenous site, or a non-targeting (NT) control. Cells were allowed to incubate for 72 h. To normalize for variations in transfection efficiencies and low overall transfection efficiencies, cells were then sorted for mCherry fluorescence marker prior to harvest of genomic DNA and amplification and analysis of loci of interest. Only A’s where editing for one of the editors tested was greater than 1% are shown. Bars show the average of n = 3 independent transfection biological replicates, error bars show standard deviation, points show individual replicates.
Extended Data Fig. 8 Summary of ABE7.10 mutated residues reversion impact on editing activity.
Modeled structure of TadA*7.10 (prepared as described in Fig. 1), highlighting how individual reversion mutations impacted ABE editing activity. Scale shows most inhibitory reversions to ABE editing activity (red) through residues where reversions had minimal impact on editing activity (grey), and residues where reversions enhanced editing activity (green). The Y147D reversion is highlighted in magenta, as its activity was strongly host-dependent.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–45, Supplementary Discussions 1–15 and Supplementary Note 1.
Supplementary Data 1 (download XLSX )
Sequences, plasmids and primers.
Supplementary Data 2 (download XLSX )
Source data for Supplementary Figs. 1–45.
Source data
Source Data Figs. 1–6 and Extended Data Figs. 1–7 (download XLSX )
Statistical source data for Figs. 1–6 and Extended Data Figs. 1–7.
Source Data Extended Data Fig. 2 (download TIF )
Uncropped western blot for Extended Data Fig. 2c.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Evanoff, M., Korpal, S., Krill, Z.D. et al. Precise, minimally evolved adenine base editors generated through mutation reversion analysis. Nat Biotechnol (2026). https://doi.org/10.1038/s41587-026-03045-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41587-026-03045-z
