
Abstract
Targeting cancer stem cells (CSCs) is crucial for effective cancer treatment, yet resistance mechanisms to LGR5+ CSC depletion in WNT-driven colorectal cancer (CRC) remain elusive. In the present study, we revealed that mutant intestinal stem cells (SCs) depart from their canonical identity, traversing a dynamic phenotypic spectrum. This enhanced plasticity is initiated by oncofetal (OnF) reprogramming, driven by YAP and AP-1, with subsequent AP-1 hyperactivation promoting lineage infidelity. The retinoid X receptor serves as a gatekeeper of OnF reprogramming and its deregulation after adenomatous polyposis coli (APC) loss of function establishes an OnF ‘memory’ sustained by YAP and AP-1. Notably, the clinical significance of OnF and LGR5+ states in isolation is constrained by their functional redundancy. Although the canonical LGR5+ state is sensitive to the FOLFIRI regimen, an active OnF program correlates with resistance, supporting its role in driving drug-tolerant states. Targeting this program in combination with the current standard of care is pivotal for achieving effective and durable CRC treatment.
Similar content being viewed by others


Main
Colorectal cancer (CRC) is the second leading cause of cancer-related mortality worldwide1. Treatment failure in WNT-driven CRC has traditionally been attributed to the malignant features of LGR5+ (leucine-rich repeat-containing G-protein-coupled receptor 5-positive) cancer stem cells (CSCs)2,3,4,5. However, recent evidence contests this notion, demonstrating that selective ablation of this population is insufficient for achieving durable tumor regression6,7. Notably, phenotypic plasticity is increasingly recognized as a mechanism of tumor escape from targeted therapies in various cancers8,9,10,11. Yet, it remains unclear whether tumor evolution and adaptability in CRC are driven by cellular plasticity, distinct cell populations, or a combination of both. In the present study, we elucidate the evolution of neoplastic cell states and their underlying epigenetic mechanisms throughout CRC progression and provide a mechanistic foundation to enhance the effectiveness and durability of current therapies.
Results
A dynamic phenotypic continuum during CRC evolution
The presence of an LGR5− fetal-like population has been well documented in BRAFmut/MMRd (mismatch repair-deficient) CRC12,13. Although considered a feature of WNT-independent tumors14,15, recent work suggests that the fetal-like state can be adopted by LGR5+ stem cells in Apc mutant premalignant lesions16. As a similar aberrant state emerges only transiently in the regenerating epithelium17,18, its persistence beyond the precancerous stage in WNT-driven CRC remains unclear. Without a comprehensive understanding of its temporal dynamics during tumor evolution and its biological importance, assessing the clinical relevance of this aberrant state is challenging.
To elucidate cell state dynamics along the entire malignancy continuum, we generated isogenic mouse organoid models that mimic the clinical progression of WNT-driven CRC (Fig. 1a and Extended Data Fig. 1a). Specifically, we induced common driver mutations in intestinal SCs using Lgr5-driven CreERT2 (Extended Data Fig. 1a). Transcriptome profiling of single cells from normal wild-type (WT) intestinal organoids, precancerous Apcnull (A) and advanced malignant Apcnull::KrasG12D::Smad4null::Trp53null (AKSP) tumoroids revealed the emergence of tumor-specific cell clusters (Extended Data Fig. 1b,c and Supplementary Table 1a,b). Notably, cells within this neoplastic metacluster re-expressed markers of fetal intestinal progenitors19 (Extended Data Fig. 1d and Supplementary Table 1c). We henceforth refer to this population as oncofetal (OnF) and define a 51-gene signature (Extended Data Fig. 1e and Supplementary Table 1d) to chart and track its dynamics during tumorigenesis. Changes in cellular composition along the CRC malignancy continuum were characterized by a significant reduction in enterocytes, indicative of a differentiation block in tumoroids (Fig. 1b, Extended Data Fig. 1f and Supplementary Table 1e). Moreover, although a burst of LGR5+ SCs was triggered by adenomatous polyposis coli (APC) loss of function (LoF), a subset of these mutant cells adopted an OnF state (Fig. 1b, Extended Data Fig. 1g,h and Supplementary Table 1e). This observation unveils a heterogeneous response to APC depletion, reconciling divergent claims regarding both the expansion of LGR5+ cells14 and fetal-like reprogramming16 in WNT-driven precancerous tumors. Intriguingly, the progression to a malignant stage (AKSP) was marked by a decline in canonical LGR5+ SCs and a noticeable expansion of OnF cells (Fig. 1b, Extended Data Fig. 1g,h and Supplementary Table 1e). These data indicate that, contrary to the transient emergence of fetal-like cells in response to injury17, the OnF state is perpetuated in CRC. This raises the question of whether OnF reprogramming generates a discrete, de novo cellular entity. Analysis of the evolution of SC phenotypes along the CRC malignancy continuum confirmed that healthy intestinal SCs are exclusively in an LGR5+ state (Fig. 1c,d). However, their neoplastic counterparts did not distinctly adopt either this canonical state or the OnF state but rather appeared to traverse a phenotypic spectrum delimited by these cellular states (Fig. 1c,d and Extended Data Fig. 1g–i). Although most cells along this spectrum retain Lgr5 expression, extreme OnF states are Lgr5− (Extended Data Fig. 1j). Collectively, these findings indicate that, although cell plasticity is tightly regulated in the healthy epithelia, APC LoF is sufficient to lift these restrictive barriers. The subsequent expansion of plastic potential during CRC progression suggests that accumulation of additional mutations may instruct cell state dynamics. To investigate the effects of various genetic events subsequent to APC LoF, we analyzed the transcriptome of dual mutant tumoroids (that is, Apcnull::Smad4null = AS, Apcnull::KrasG12D = AK and Apcnull::Trp53null = AP). This analysis indicated that SMAD4 LoF has a limited effect on fate decision, whereas KRASG12D mutation and TP53 LoF favor the OnF and canonical LGR5+ states, respectively (Extended Data Fig. 1k).

a, Schematics of the organoid models used to recapitulate the malignancy continuum of CRC. b, The percentage of various cell types and states in the indicated models (n = 3 independent organoid cultures per group). Cell numbers per group are given in Supplementary Table 1a. The bar plots are mean ± s.e.m. Linear regression tested for significant differences in log(transformed cell-type proportions) between genotypes, with P values adjusted using the Benjamini–Hochberg method. c, Percentage of cells in stem and OnF states along diffusion component 1 (DC1), grouped into 500 bins by increasing DC1 values. The bottom color bars show the percentage of cells from each genotype per bin. d, Scatter plot showing cell state distribution along the SC–OnF phenotypic spectrum across the CRC malignancy continuum. Horizontal and vertical box plots indicate OnF and LGR5+ SC module score enrichment, respectively (n = 3 independent organoid cultures per group). e–g, Scatter plot depicting cell state distribution along the SC–OnF phenotypic spectrum in the Broad (n = 36 patients) (e), SMC/KUL3 (n = 10 patients) (f) and Guangzhou (n = 5 patients) (g) cohorts. The horizontal and vertical box plots indicate OnF and SC module score enrichment, respectively. PT, primary tumor; Met, metastasis. In d–g, box plots show the center line as the median, box limits as the interquartile range (IQR: 25th to 75th percentiles), the whiskers the ±1.5× the IQR and individual points the outliers. The P values were calculated using two-sided, paired Student’s t-test comparing sample means. h,i, Proportions of cells expressing lineage-specific signatures of the indicated gastrointestinal tract tissues, in individual patients from the Broad (h) and SMC/KUL3 (i) cohorts: J, junction; M, mucosa; S, small; Sig, sigmoid; Trans, transverse. j–l, Correlation between the OnF and lineage plasticity scores (Methods) in the Broad (j; n = 36 patients), SMC/KUL3 (k; n = 10 patients) and Guangzhou (l; n = 5 patients) normal CRC-matched cohorts, respectively. Two-sided Pearson’s correlation for significance is used. m, Schematic model depicting the evolution of the phenotypic spectrum of neoplastic SCs throughout CRC progression. In b–d, WT = 4,058 cells, A = 5,890 cells and AKSP = 10,946 cells. *P < 0.05, **P < 0.01; ***P < 0.001; ****P < 0.0001; exact P values are given in Supplementary Table 7. Illustrations in a created using BioRender.com.
These genetically engineered models offer an ideal platform for accurately investigating cell state dynamics, free from confounding factors typically associated with human datasets, such as treatment exposure and inconsistent temporal sampling. Nevertheless, given the inherent genetic heterogeneity and microenvironmental diversity among patients, we defined a human OnF signature to investigate OnF reprogramming in patients with CRC (Methods and Supplementary Table 1f). Our analysis of matched normal colons and adenocarcinomas from the Cancer Genome Atlas Colon Adenocarcinoma (TCGA/COAD) dataset confirmed activation of the OnF program in CRC (Extended Data Fig. 2a). Moreover, single-cell analysis of the Broad and SMC/KUL3 CRC cohorts20,21 unequivocally demonstrated that the OnF state is significantly enriched in tumors (Fig. 1e,f). Further examination of 27 samples of matched primary tumors and liver metastases from five patients with CRC22 confirmed its persistence in metastatic tumors (Fig. 1g). Notably, patient stratification based on both mismatch repair (MMR) and APC mutation status proved that OnF reprogramming is a common feature in APCmut/MSS (microsatellite-stable) CRC (Extended Data Fig. 2b–d). These data address a long-standing ambiguity surrounding OnF reprogramming in WNT-driven CRC12,13,14,15,16,18,23,24 and indicate that lack of comparison to adjacent healthy tissue in previous studies led to inaccurate conclusions.
Despite the widespread occurrence of OnF reprogramming in CRC, its biological significance remains largely elusive. We reasoned that this developmental rewind may endow neoplastic cells with enhanced plasticity, enabling them to transcend their typical lineage restraints. Comparative transcriptomic analysis of CRC tumors and various developmental stages of the human gastrointestinal tract25 revealed remarkable similarities across tumors, fetal intestines, and adult stomach tissues (Extended Data Fig. 2e). Overall, we noticed a discernible shift in neoplastic tissue identity from its posterior colonic origins toward more anterior regions of the gut tube (Extended Data Fig. 2f). Further examination of single-cell transcriptomic data using lineage-specific signatures26 (Supplementary Table 1g) corroborated that CRC tumors have acquired a metaplastic multiregional identity (Fig. 1h,i). It is interesting that we noted a significant correlation between the OnF score and lineage plasticity, characterized by a loss of the original colonic identity and acquisition of noncanonical cell fates (Fig. 1j–l and Extended Data Fig. 2g–j).
Collectively, these findings reveal the prevalence of the OnF state in WNT-driven CRC and shed light on its functional significance. During tumor initiation, OnF reprogramming extends the plastic potential of neoplastic SCs beyond their normally restricted capability. The resulting phenotypic spectrum continues to broaden throughout disease progression, leading to lineage infidelity (Fig. 1m). These observations offer insights into why more advanced tumors often exhibit increased resistance to therapies.
Distinct roles of YAP and AP-1 in driving phenotypic plasticity
Despite the potential clinical relevance of OnF reprogramming, its molecular drivers remain poorly characterized. Activation of the Hippo pathway effector YAP (yes-associated pathway) in response to injury17,23 or tumor initiation23,24,27 induces fetal-like reprogramming. However, our data show a dynamic evolution of OnF states during CRC progression (Fig. 1c,d), indicating a more intricate regulation at play.
To investigate the molecular mechanisms underlying cell state dynamics in CRC, we analyzed the chromatin accessibility landscapes across its malignancy continuum (Extended Data Fig. 3a). Assay for transposase-accessible chromatin using sequencing (ATAC–seq) data revealed a striking evolution of chromatin landscapes across the adenoma–adenocarcinoma sequence. Although most changes induced by APC LoF at the premalignant stage were maintained, additional events emerged during the progression to cancer (Extended Data Fig. 3b). Unsupervised hierarchical clustering of ATAC–seq peaks identified two primary patterns of chromatin accessibility (Fig. 2a). Genomic regions in clusters 1 (c1) and 2 (c2) exhibited reduced and increased accessibility, respectively, in the mutant tumoroids (Fig. 2a and Supplementary Table 2a,b). The progressive nature of these changes reflects the dynamic evolution of neoplastic cell states in CRC (Fig. 1d). To examine their functional significance, we performed transcription factor (TF) footprinting analysis, using TOBIAS28, and calculated a ‘combined binding score’ to assess TF activity across the malignancy continuum (Methods). C1 was enriched for footprints of the caudal-related homeobox (CDX) and hepatocyte nuclear factor family (HNF) members (Fig. 2b,c and Extended Data Fig. 3c) involved in establishing the caudal identity of intestinal cells and their maturation, respectively29,30,31. The progressive reduction in activity of these TFs (Extended Data Fig. 3d,e) aligns with the gradual regression to a more primitive state and loss of intestinal identity during CRC evolution (Fig. 1). Intriguingly, we noted a significant decrease in the activity of several ligand-regulated nuclear receptors, including PPAR, RXR, LXR, VDR and FXR, all of which require retinoid X receptor (RXR) dimerization to become functional (Fig. 2b,c and Extended Data Fig. 3f–i).

a, Heatmap of ATAC–seq signal in WT, A and AKSP organoids at genomic DARs (top). Average ATAC–seq signal profile ±2 kb is around the peak center in clusters c1 and c2 (bottom). RPKM, reads per kilobase million. b–e, Elbow plots showing TF activity in A versus WT (b and d) and AKSP versus WT (c and e) using DARs from clusters c1 (b and c) and c2 (d and e). For a–e, n = 2 independent organoid cultures per group. CEBP, CCAAT-enhancer-binding protein; NRs, nuclear receptors; BATF, basic leucine zipper transcription factor, ATF-like; JUND, JunD proto-oncogene, AP-1 transcription factor subunit. f,g, Relative OnF signature enrichment during tumor initiation following knockdown (kd) of YAP or FOS (f) or the ectopic expression of a dominant-negative (DN) FOS (g). Each data point is a gene in the OnF signature (n = 4 independent experiments; ***P < 0.001, two-sided Wilcoxon’s rank-sum test). h, Line plot of differential TF activity (combined score), from d and e, during CRC evolution. Two-sided, paired Student’s t-tests were used for significance. JUN, Jun proto-oncogene, AP-1 transcription factor subunit; MAFK, v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog K. i–k, Motif activity of ASCL2 (i), FOS/AP-1 (j) and TEAD1 (k) in AKSP single cells across the SC–OnF phenotypic spectrum determined by chromVAR z-scores for the indicated TFs. The correlation coefficients between TF activity and the LGR5+ SC or OnF score are indicated in orange and green, respectively. Multiome data are from two independent organoid cultures (n = 11,030 cells). l–n, Relative enrichment of the OnF (l), esophagus (m) and small intestine (n) signatures in WT organoids following ectopic expression of YAP or YAP then FOS, sequentially. Each data point is a gene (n = 4 independent experiments; NS, not significant, ***P < 0.001, two-sided Wilcoxon’s rank-sum test). EV, empty vector. o–q, GSEA of OnF (o), esophagus (p) and small intestine (q) gene signatures in YAP overexpression (OE) versus YAP + FOS sequential OE organoids, relative to l–n. Two-sided, permutation-based test was used for significance; P values were adjusted via the Benjamini–Hochberg method. r, Model of phenotypic spectrum evolution in neoplastic cells during CRC progression highlighting key TFs at play. Various gradients of ASCL2, YAP and FOS activity are pivotal to establishing a phenotypic continuum in neoplastic cells. In the box plots in f, g and l–n, the center line is the median, the box limits the IQR (25th to 75th percentiles) and the whiskers the highest and lowest values within ±1.5× the IQR. *P < 0.05; ***P < 0.001; exact P values are given in Supplementary Table 8. NES, normalized enrichment score; TPM, transcripts per million.
In contrast, tumoroid-specific events in C2 were predominantly driven by three TF families (T cell factor/lymphoid enhancer factor (TCF/LEF), transcriptional enhanced associate domains (TEADs) and activator protein 1 (AP-1)) (Fig. 2d,e). TCF/LEF operates under canonical WNT signaling and, together with ASCL2, sustains the canonical LGR5+ state32. TEADs, on the other hand, are the cognate DNA-binding partners of YAP, recently implicated in transient activation of a fetal-like program after injury and in precancerous lesions16,18,23. Meanwhile, although AP-1 has been associated with promoting aggressiveness in various cancer types, including CRC33,34, its role in regulating neoplastic cell states remains largely unexplored. To investigate its potential implication in OnF reprogramming, we initially conducted HOMER motif analysis on promoter regions of OnF markers compared with a set of random genes. This revealed a significant enrichment of both TEAD- and AP-1-binding motifs in the former (Extended Data Fig. 3j). Next, we examined the effects of YAP and FOS (Fos proto-oncogene, AP-1 transcription factor subunit) depletion on OnF reprogramming (Extended Data Fig. 4a) and found that both perturbations were sufficient to impede OnF reprogramming after APC LoF (Fig. 2f, Extended Data Fig. 4b–d and Supplementary Table 2c). Stronger inhibition of AP-1 using a dominant-negative form of FOS led to a complete repression of the OnF program (Fig. 2g, Extended Data Fig. 4e and Supplementary Table 2d). Together, these findings support a cooperative model whereby AP-1 acts in concert with YAP to drive OnF reprogramming during tumor initiation.
Notably, although binding sites of both TFs became more accessible in precancerous tumoroids (A), accessibility of AP-1 target regions further increased in the advanced AKSP model (Fig. 2h), suggesting a more intricate interplay during tumor evolution. The dynamic shift in the activity of these TFs supports a role of AP-1 in expanding the OnF state during CRC progression (Fig. 1d). To achieve a deeper understanding of the molecular mechanisms governing the establishment of a phenotypic spectrum in CRC, we performed single-cell multiome (single-cell (sc)ATAC–seq + scRNA-seq) analysis on AKSP tumoroids. This simultaneous examination of the transcriptome and chromatin accessibility landscape at single-cell resolution allowed us to chart TF activity across neoplastic cell states. We first ranked cells along the SC–OnF phenotypic spectrum using scRNA-seq (Extended Data Fig. 4f,g). Then, we projected scATAC–seq cells on to this continuum and used chromVAR35 to map TF motif activity. This analysis revealed that the SC master regulator, ASCL2, and AP-1 (FOS) motif activities were most enriched at either extreme of the phenotypic spectrum, correlating with SC and OnF scores, respectively (Fig. 2i,j). It is interesting that TEADs exhibited a broader activity profile characteristic of transitional states (Fig. 2k). Moreover, we found a substantially higher correlation between AP-1 activity and lineage infidelity (Extended Data Fig. 4h,i). These data indicate that, although the transition to a continuum of hybrid states is predominantly facilitated by YAP, increased AP-1 activity reinforces the OnF state, enabling lineage plasticity.
To functionally validate the distinct roles of YAP and AP-1 in OnF reprogramming, we sequentially expressed a constitutively active YAP (S6A) and FOS in WT organoids (Extended Data Fig. 4j). It is interesting that ectopic YAP expression induced an upregulation of several AP-1 subunits (Fos, Fosl1, Fosl2, Atf3 and Batf2) (Extended Data Fig. 4k and Supplementary Table 2e) and initiated OnF reprogramming (Fig. 2l and Extended Data Fig. 4l–n). The subsequent addition of FOS further enhanced the OnF program (Fig. 2l,o), triggering lineage infidelity (Fig. 2m,n,p,q).
Collectively, these findings demonstrate that the phenotypic spectrum in CRC is governed by varying gradients of OnF and SC state drivers. They support an adaptive bet-hedging model in which the cooperative action of YAP and AP-1 serves as a molecular switch facilitating OnF reprogramming during tumor initiation. Further hyperactivation of AP-1 during CRC progression promotes phenotypic heterogeneity and unleashes lineage plasticity (Fig. 2r).
RXR is a gatekeeper of early OnF reprogramming
Next, we set out to determine the molecular mechanisms underpinning activation of the OnF drivers YAP and AP-1 in CRC. In the context of injury, inhibition of the nuclear receptor RXR sustains the typically transient fetal-like state by maintaining YAP in an active form26. However, the role of RXR during intestinal tumorigenesis remains unexplored.
To address this, we first investigated RXR deregulation in CRC. TCGA/COAD dataset analysis indicated significantly lower RXRa levels in CRC tumors compared with matched healthy intestinal tissues (Fig. 3a). Our murine CRC models revealed an early downregulation of Rxra after APC LoF (Fig. 3b). Together with the reduced accessibility of RXR target sites and those of its partners in precancerous tumoroids (Fig. 2b), these data indicate that RXR operates downstream of APC in CRC.

a, RXRa expression in matched human normal colon and tumors from TCGA/COAD (n = 41 patients). P values were calculated using two-sided, paired Wilcoxon’s test. b, Rxra expression during tumorigenesis (WT and A, n = 6 independent organoid cultures; AKSP, n = 3). The P values were calculated using two-sided, unpaired Wilcoxon’s test. In the box plots, the center line is the median, the box limits the IQR (25th to 75th percentiles) and the whiskers the highest and lowest values within ±1.5× the IQR. c, Experimental approach for the comparative analyses of Anull and RXRi (+HX531) versus WT organoids. d,e, Spearman’s correlation of the top 2,000 (d) or 1,000 (e) highly variable genes. READ, rectum adenocarcinoma. f,g, Elbow plots depicting TF activity in RXRi (f) versus WT organoids (g). TF combined binding score = −log10(P value) × log2(fold-change). Significantly more accessible or less accessible regions were used in f and g, respectively (WT, n = 2; RXR, n = 3 independent ATAC–seq experiments). h, Experimental design summary for Fig. 2i. i, GSEA of OnF genes following the APC–RXR axis perturbation during tumor initiation (n = 4 independent experiments). j,k, GSEA of OnF genes in CRC tumoroids (AKSP), following RXR-OE (j) or inhibition (RXRi) (k) (n = 4 and 3 independent experiments, respectively). l, Experimental design summary for Fig. 2m–o. m, GSEA of OnF genes >5 weeks post-RXRi washout (n = 3 independent cultures). In i–m, a two-sided permutation-based test for significance was used; P values were adjusted via the Benjamini–Hochberg method. n, ATAC–seq signal intensity in RXRi, woRXRi (>5 weeks post-HX531 withdrawal), A and AKSP versus WT organoids. o, Elbow plot of more accessible regions in RXR versus WT organoids ranked by their log2(fold-change) in woRXRi versus WT. Resolved peaks were subset to obtain the 1,342 regions (mean of number of suppressed and persistent regions) with log2(fold-change) closest to 0. The HOMER motif enrichment analysis indicates the top enriched TFBSs of the persistent ‘memory’ regions (>5 weeks post-RXRi washout (right)). n,o, RXR and woRXRi (n = 3 independent cultures; WT, A, AKSP, n = 2). FC, fold change. Illustrations in c, h and l created using BioRender.com.
Next, we conducted comparative analyses of RXR inhibition and APC LoF to determine whether RXR deregulation is responsible for activation of the OnF program during tumor initiation (Fig. 3c). Similar to APC depletion, treatment with the RXR antagonist HX531, hereafter referred to as RXRi, altered the budding structure of WT organoids, imposing a spheroid morphology (Extended Data Fig. 5a,b) reminiscent of fetal organoids19 and mutant tumoroids (Extended Data Fig. 5c). Transcriptomic analyses uncovered a striking resemblance between RXRi organoids and both human CRC and mouse tumoroids (Fig. 3d,e and Supplementary Table 3a). Furthermore, hallmarks of OnF reprogramming were significantly enriched in RXRi organoids (Extended Data Fig. 5d). Intriguingly, despite causing a similar block in differentiation, RXR inhibition induced a more prominent increase in OnF genes compared with APC depletion and did not result in the expansion of LGR5+ SCs (Extended Data Fig. 5e and Fig. 1b).
To better understand how RXR inhibition favors a complete reversion to an OnF-like state, we investigated its effects on global TF dynamics. Our ATAC–seq data revealed remarkable similarities in chromatin accessibility landscapes between Apc mutant tumoroids and RXRi organoids (Extended Data Fig. 5f–h). TF footprinting analysis (TOBIAS28) demonstrated that RXRi recapitulated most TF activity changes induced by APC LoF in tumorigenesis (Fig. 3f,g and Supplementary Table 3b), including reduced activity of retinoid X receptor alpha (RXRa) itself, CDX2 and HNF4 (Extended Data Fig. 5i–k).
Notably, RXR blockade induced a significant hyperactivation of the OnF TFs AP-1 and TEADs (Extended Data Fig. 5l,m) without activating WNT-related TFs (Fig. 3f,g and Extended Data Fig. 5n). HOMER analysis confirmed enrichment of the TCF/LEF DNA-binding motif in less accessible regions in RXRi organoids (Extended Data Fig. 5o and Supplementary Table 3c). These findings support a divergent regulation of the WNT signaling pathway (Extended Data Fig. 6a–c) after RXR inhibition and APC depletion. While hyperactivation of WNT signaling is known to reinforce the canonical SC state32,36, these data demonstrate that diminished RXR activity induces OnF reprogramming through activation of YAP and AP-1. Deregulation of both circuitries downstream of the gatekeeper APC (Fig. 2b,d and Extended Data Fig. 5o) elucidates the emergence of a continuum of neoplastic cell states during CRC tumorigenesis.
Consistent with these findings, blocking the APC–RXR regulatory axis through ectopic expression of RXRa impaired OnF reprogramming during tumor initiation (Fig. 3h,i and Supplementary Table 3d). Intriguingly, RXR perturbation in advanced AKSP tumoroids exhibited no significant effects on the OnF program (Fig. 3j,k and Supplementary Tables 3e,f), indicating its diminished relevance once the OnF state has been established. This suggests that early RXR deregulation, after APC LoF (Fig. 3b), is sufficient to epigenetically reprogram cells into an OnF state. Indeed, intestinal organoids temporarily exposed to RXRi largely maintained features of OnF reprogramming for >5 weeks post-inhibitor withdrawal (Fig. 3l,m). Intriguingly, although transcriptional changes were largely maintained, only a small fraction of chromatin accessibility changes persisted (Extended Data Fig. 6d,e and Supplementary Table 3g,h). Notably, the persistently more accessible regions were also more accessible in A and AKSP tumoroids (Fig. 3n). HOMER analysis of these ‘memory sites’ revealed a significant enrichment of YAP- and AP-1-binding motifs (Fig. 3o).
Together these findings indicate that RXR serves as a gatekeeper of OnF reprogramming during tumor initiation. Following its deregulation, YAP and AP-1 activation is sufficient to sustain the OnF state during tumor progression. Indeed, unlike RXR perturbation (Fig. 3j,k), knockdown of either YAP or AP-1 in VAKSP (Villin-Cre AKSP) tumoroids induced significant depletion of the OnF program (Extended Data Fig. 6f–h and Supplementary Table 3i).
Therapeutic relevance of the OnF state in CRC
In contrast to the well-characterized LGR5+ SC state in CRC, the functional relevance and potential therapeutic implications of the OnF state remain, to the best of our knowledge, unknown. Thus far, inference of the OnF state using transcriptional signatures has provided only a static snapshot of their presence. To comprehensively characterize the temporal dynamics of this cellular state, a well-defined and measurable tool is needed.
We have recently developed a new strategy to genetically trace cell fate transitions within a heterogeneous tissue37,38. To this end, we leveraged our in-depth understanding of the molecular underpinnings of the OnF program (Figs. 2 and 3) to construct a synthetic locus control region (sLCR) that contains specific cis-regulatory elements (CREs) reflecting the transcriptional output and activity of OnF state-associated TFs (Extended Data Fig. 7a,b and Supplementary Table 4a). Then, we fused this genetic tracing cassette to an enhanced green fluorescent protein (eGFP; Fig. 4a) to allow visualization and tracking of this cellular state. Flow cytometry analysis of WT organoids expressing this phenotypic reporter confirmed that only a small fraction of cells exhibited low-to-moderate levels of GFP expression (Extended Data Fig. 7c). In contrast, WT organoids treated with RXRi and tumor-derived VAKSP organoids displayed a notable shift in reporter activity, evidenced by a higher proportion of GFP+ cells and increased mean fluorescence intensity (Extended Data Fig. 7c,d). Moreover, transcriptomic analysis of sorted GFPhigh and GFP− cells from VAKSP tumoroids confirmed the enrichment of the OnF signature and markers of lineage plasticity in the former (Fig. 4b, Extended Data Fig. 7e and Supplementary Table 4b). These data support the validity and specificity of our phenotypic reporter as an effective tool for tracing OnF cells.

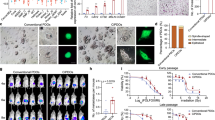
a, Schematic of the OnF phenotypic reporter structure. PGK, phosphoglycerate kinase promoter; NEO, neomycin resistance gene. b, Experimental flow diagram (top) and gene-set enrichment in GFPhigh versus GFP− cells sorted from VAKSPOnF (Villin-Cre AKSP) tumoroids expressing the OnF reporter (bottom) (n = 3 independent sorts). c, Representative pseudocolor plots from flow cytometry of VAKSP tumoroids co-expressing OnF–GFP (y axis) and stem/STAR–mCherry (x axis) phenotypic reporters. VAKSPOnF/STAR tumoroids were treated with dimethyl sulfoxide or FOLFIRI for 3 days before analysis. d, Quantitative representation of flow cytometry data from c. In c and d, n = 3 independent experiments; error bars = s.d.; P values are by paired, two-sided Student’s t-tests. e, Heatmap reporting log2(fold-change) in WNT (left) and YAP or AP-1 (right) target gene expression (n = 3 independent experiments). f, Summary of the VAKSP tumoroid models used for functional studies. g,i, Schematic of the experimental strategy to genetically target the OnF state, stem state or both, either transiently (g) or persistently (i), relevant to h–j. h–j, Growth rate of subcutaneous VAKSP tumors with indicated reporter combinations, in response to DT treatment. Mean tumor volume ± s.e.m. The dotted lines indicate saline treatment (all models, n = 6 tumors). h, Dashed lines indicate transient DT treatment (5 d; 3 doses on alternate days): OnFDTR (n = 11), STARDTR (n = 9), OnFDTR/STARDTR (n = 10). j, The solid lines indicate persistent DT treatment, on alternate days, throughout the experiment: OnFDTR (n = 12), STARDTR (n = 11), OnFDTR/STARDTR (n = 9). Bottom, DT dosing schedule; the dashed red line indicates treatment duration. k, Schematic of the experimental strategy used in l. l, Dotted line, vehicle (n = 8 tumors); solid lines, DT (n = 6), FOLFIRI (n = 7) and DT + FOLFIRI (n = 8). Mice received three doses per week on alternate days. Values are mean tumor volume ± s.e.m. In h, j and l, the P values were calculated using a mixed-effects linear model with Tukey’s adjustment for multiple comparisons (two sided). m–p, Heatmaps showing percentage viability of VAKSP tumoroids with the indicated drug combinations. A single dose of MRTX1133 and IAG933 was used in n and p, respectively. The effects of these single doses of each drug are highlighted in m (MRTX1133) and o (IAG933), respectively. IC, inhibitory concentration. q, Dot plot indicating the combination index from drug combinations in m–p. The P values were calculated using two-sided, paired Student’s t-tests. In m–q, n = 3 independent experiments. Illustrations in b, g, i and k created using BioRender.com.
Next, to investigate the intricacies of their potential interplay with LGR5+ SCs, we sought to develop a tracing system that enables simultaneous tracking of both populations. To this end, we replaced the OnF sLCR cassette with the STAR minigene39 driving mCherry expression (Extended Data Fig. 7f) and generated a tumoroid line that expressed both reporters (VAKSPOnF/STAR). This strategy allowed us to visualize the continuum of neoplastic phenotypes (Extended Data Fig. 7g).
To trace cell state dynamics under therapeutic pressure, we treated VAKSPOnF/STAR tumoroids with FOLFIRI, a chemotherapy regimen commonly employed in clinical settings. Flow cytometry analysis of reporter activity revealed a significant enrichment of hybrid states at the expense of canonical SCs (Fig. 4c,d). This observation is consistent with the upregulation of key OnF markers and YAP or AP-1 target genes, alongside reduced expression of WNT-related genes (Fig. 4e, Extended Data Fig. 7h and Supplementary Table 4c). In contrast, the GFPhigh OnF population was largely preserved, suggesting intrinsic resistance.
To further investigate the response of these cell states to current therapies, we sorted individual clones from each population (GFPhigh, hybrid, and mCherryhigh) (Extended Data Fig. 7i) and treated their respective cultures with FOLFIRI. Although cells with an active OnF program (GFP+–OnF state and GFP+/mCherry+–hybrid state) remained largely unaffected under therapeutic pressure (Extended Data Fig. 7j,k,m–p), we noted a significant depletion of LGR5+ cells, which coincided with the enrichment of hybrid cells (Extended Data Fig. 7l,q,r). These findings confirm the key role of the OnF program in driving drug-tolerant states (that is, pure OnF or hybrid states) in CRC. Intriguingly, in the absence of treatment, we observed marginal phenotypic fluctuations in the sorted clones, indicating plasticity among neoplastic states (Extended Data Fig. 7i,m,o,q). Therefore, it is possible that activation of the OnF program in canonical SCs may contribute to adaptive resistance.
To investigate resistance mechanisms in human CRC, we used three independent patient-derived organoids (PDOs) (colo13, colo31 and colo41) exhibiting varying sensitivity levels to treatment (Extended Data Fig. 8a). Consistent with our findings in mouse tumoroids, exposure to FOLFIRI led to increased expression of OnF genes across all PDO lines, albeit to varying degrees (Extended Data Fig. 8b). Notably, colo13 displayed a more consistent upregulation of OnF markers and a conspicuous depletion of canonical SC markers and WNT target genes (Extended Data Fig. 8c), indicating a greater potential for adaptability. It is interesting that, although YAP activation levels were comparable across all PDOs, we observed a stronger activation of AP-1 targets in colo13 (Extended Data Fig. 8d). These data support a pivotal role of AP-1 in driving phenotypic plasticity and therapy resistance in CRC.
Although these findings underscore its potential therapeutic significance, the consequences of targeting the OnF state have yet to be uncovered. Previous studies provided unexpected evidence that ablation of the LGR5+ cells altogether is insufficient to achieve a durable regression of CRC6,7, which suggests that targeting their phenotypic plasticity alone is not enough to overcome resistance. Our data from organoid models (Fig. 4c,d and Extended Data Fig. 7j–l) imply that, in such scenarios, the pre-existing OnF cells may play a crucial role in sustaining tumor growth. To test this hypothesis, we developed a diphtheria toxin (DT) receptor (DTR)-expressing version of the OnF and canonical (STAR+) SC-tracing cassettes. We then generated VAKSP tumoroids that express various combinations of these reporters to allow selective attrition of either or both cellular states (Fig. 4f). In the absence of DTR expression, DT treatment did not affect cellular composition or viability of VAKSPOnF/STAR tumoroids (Extended Data Fig. 8e,f). However, the respective target populations, including hybrid cells, were efficiently ablated in the DTR+ models (Extended Data Fig. 8e,f).
To investigate the therapeutic potential of targeting the OnF state in CRC, we transplanted the different VAKSP reporter lines (Fig. 4f) into the flanks of immunocompromised mice. We first allowed tumors to reach a relatively large volume (350-400 mm3) before administering DT every other day for 5 days (Fig. 4g). Consistent with previous work6,7, selective ablation of cells with an active LGR5+ program led to tumor stasis in the VAKSPSTAR-DTR model, followed by prompt regrowth on DT cessation (Fig. 4h). More importantly, attrition of OnF+ cells halted tumor growth transiently but was not sufficient to induce tumor regression (Fig. 4h). In contrast, co-targeting both cell states resulted in tumor shrinkage and delayed resurgence significantly (Fig. 4h). It is interesting that sustained depletion of either OnF+ or STAR+ cells alone using continuous DT treatment (Fig. 4i and Extended Data Fig. 8g) failed to maintain tumor stasis indefinitely (Fig. 4j), suggesting that either population can fuel tumor growth in the absence of the other. However, we noted a prominent tumor regression in the VAKSPOnF-DTR/STAR-DTR model that was maintained for as long as DT treatment continued (Fig. 4j).
In the light of these findings, we reasoned that targeting the OnF program could sensitize tumors to chemotherapies, offering substantial clinical benefits. To test this hypothesis, we investigated the combined effects of FOLFIRI and DT on VAKSPOnF-DTR tumoroids in vivo (Fig. 4k). Although each treatment alone reduced tumor growth, their combination resulted in a durable tumor regression throughout the treatment period (Fig. 4l).
It is interesting that pharmacological inhibition of either YAP–TEAD with IAG933—a potent first-in-class and selective disruptor of the YAP–TEAD interaction40—or AP-1’s upstream regulator KRAS with the selective KRASG12D inhibitor MRTX1133 (ref. 41), marginally improved VAKSP tumoroid response to FOLFIRI (combination index (CI) = 0.96 and 0.94, respectively) (Fig. 4m,o,q). However, the trio combinations were highly synergistic (CI = ~0.40) (Fig. 4n,p,q), indicating that concurrent inhibition of YAP and AP-1 is key to enhancing the effectiveness of FOLFIRI. These data further support the cooperative, yet distinct, functions of both TFs or pathways in regulating the OnF program (Fig. 2).
Collectively, our findings shed light on the functional interplay between the canonical (LGR5+) SC and noncanonical OnF states. Unexpectedly, we found that the LGR5+ state is sensitive to FOLFIRI and tumors require an active OnF program to survive treatment. Importantly, the clinical relevance of each population in isolation is limited by their functional redundancy. However, we provide evidence that targeting the OnF program is key to enhancing effectiveness and durability of the current standard of care (SOC) in mouse models and patient-derived CRC tumoroids.
Discussion
Despite notable advancements in CRC molecular classification42,43,44, their translational significance remains unclear. We and others have recently described the existence of two CSC populations across CRC subtypes15,45. In the present study, we revealed that the canonical LGR5+ SC and noncanonical OnF states are not distinct entities but rather extremes of a phenotypic continuum. This SC–OnF spectrum is prevalent among patients with CRC regardless of molecular characteristics, namely MMR and WNT mutation status.
We challenge the prevailing belief that canonical LGR5+ CSCs are the sole drivers of malignancy in WNT-driven CRC46,47,48, which assumes a rigid cellular hierarchy within tumors akin to that of the homeostatic intestinal epithelium. We demonstrate that, unlike their healthy counterparts, CSCs do not maintain a discrete state but traverse an evolving SC–OnF phenotypic spectrum. Extreme OnF states, prominent in advanced disease, are induced by AP-1 hyperactivation and exhibit features of lineage plasticity, contributing to therapy resistance. It is interesting that mixed lineage states have been observed in pancreatic49,50, lung51,52,53 and prostate cancers54. Therefore, we propose that early OnF reprogramming may represent a potential universal mechanism of phenotypic plasticity in cancer. This hypothesis is further supported by the role of YAP and AP-1 as orchestrators of an aberrant pan-cancer enhancerome55,56,57,58,59,60. Although the molecular circuits at play may be context dependent, the activation of regenerative and inflammatory pathways emerges as a recurring mechanism promoting epigenetic plasticity50,54,61,62. Our studies identify RXR as a master regulator of these pathways and a gatekeeper of OnF reprogramming in CRC, potentially linking statins’ protective effects in patients with hyperlipidemia63,64 to an early blockade of this program through activation of the RXR::PPAR complex.
Targeting the OnF program in fully developed CRC has marginal effects as a result of functional redundancy with the canonical LGR5+ state. However, in tumors exposed to FOLFIRI, this leads to sustained CRC regression. Notably, unlike the OnF state, the recently described high relapse cell state is not triggered by FOLFIRI65. Although these states differ in their dependence on YAP activity, both are induced by KRAS mutations, warranting further investigation of potential functional similarities, particularly in metastatic dissemination.
Collectively, our findings propose OnF reprogramming as a crucial driver of drug-tolerant states in CRC and indicate that success of the current SOC treatment hinges on effectively targeting this program. Notably, although our organoid data and recent in vivo data66 suggest that OnF reprogramming in the tumor precedes that of its ecosystem67,68, the mechanisms underlying such crosstalk remain to be determined.
Methods
Ethics statement
Mouse experiments to derive the VAKPS tumoroids followed UK Home Office regulations (project license no. 70/8646) and ARRIVE guidelines with ethical review at the University of Glasgow. Experiments to derive all other organoid models were approved by the Institutional Animal Care and Use Committee (A*STAR, Singapore). Experiments using the subcutaneous tumor models were approved by the Icahn School of Medicine at Mount Sinai (ISMMS) Institutional Animal Care and Use Committee (protocol no. 2018-0013).
Application to access patient tissue and organoids was authorized by the National Health Service (NHS) Greater Glasgow and Clyde Biorepository under their NHS Research Ethics Committee approval with ethical approval granted in biorepository application no. 602, following strict measures to ensure privacy and anonymity.
Mouse strains
To derive Lgr5-2a-CreERT2 organoids, mice were generated through homologous recombination with a 2a-CreERT2 cassette at the Lgr5-ORF stop codon69. These mice were crossbred with Rosa26-tdTomato (Ai14)70, LSL-KrasG12D71, Apc-loxP flanked72, Trp53-loxP flanked73 and Smad4-loxP flanked74 mice from the Jackson Laboratory. All were bred on to the C57B6/J background. Mice were maintained under a 12-h light:dark cycle, at 21 ± 1 °C, 55–70% humidity and placed in ventilated cages for pathogen-free conditions.
To derive the villin-CreER tumoroids, a 12-week-old male villin-CreERApcfl/+KrasG12D/+Trp53fl/flSmad4fl/fl mouse on a C57BL/6 background underwent intracolonic induction under general anesthesia. A single 70-µl dose of 100 µM 4-hydroxytamoxifen (4-OH; Sigma-Aldrich, cat. no. H7904-5MG) was injected into the colonic submucosa. At clinical endpoint (weight loss), colonic tumor tissue was collected and organoid cell lines were generated as described previously75.
Organoid derivation and culture
Organoids were generated as previously described76. Briefly, small intestines were opened, washed with phosphate-buffered saline (PBS) and villi removed. The tissue was cut into ~2-mm fragments and incubated in 2.5 mM EDTA shaking at 4 °C for 30 min, then resuspended in advanced Dulbecco’s modified Eagle’s medium (DMEM)/F12 with 0.1% bovine serum albumin (BSA) and vigorously shaken. The suspension was then passed through a 70-μm strainer. This process was repeated to collect four fractions, which were centrifuged (300g for 5 min at 4 °C) and pellets resuspended in 7 mg ml−1 of Matrigel (Corning, cat. no. 35234). Matrigel was plated into 24-well plates and polymerized at 37 °C before adding mouse IntestiCult Medium (STEMCELL Technologies, cat. no. 06005) with Y-27632 (10 μM). Organoids were passaged every 3–4 days by trituration in Gentle Cell Dissociation Reagent (GCDR, STEMCELL Technologies, cat. no. 100-0485) before washing with basal medium (advanced (a)DMEM/F12, N2/B27, N-acetylcysteine, 1 mM Hepes and 100 μg ml−1 of penicillin–streptomycin). Cell pellets were then resuspended in Matrigel.
Isogenic tumoroid line generation and treatment
ApcKO tumoroids were generated by treating Lgr5-2a-CreERT2::Apcflox/flox organoids with 4-OHT (500 nM; Sigma-Aldrich, cat. no. SML1666-1ML) for 24 h, activating tdTomato in recombined intestinal SCs. Tumoroids were then cultured in selection medium (basal medium, epidermal growth factor (EGF; 50 ng ml−1) and Noggin (100 ng ml−1) (PeproTech)) for 2–3 passages. AKSP tumoroids were generated by treating Lgr5-2a-CreERT2::Apcflox/flox::Lox-Stop-Lox-KrasG12D::Smad4flox/flox organoids with 4-OHT. After selection in basal medium, organoids were edited by clustered regularly interspaced short palindromic repeats (CRISPR)–Cas9 to knock out the Trp53 gene. AKSP tumoroids were selected in 10 μM Nutilin-3. All established lines were maintained in complete mouse IntestiCult medium.
RXR blockade: WT organoids were plated in HX531 (1.25 μM) and maintained in half-dose after the first passage. FOLFIRI treatment: tumoroids were treated with 5 μM 5-fluorouracil (5FU) and irinotecan (1:1) for 3 days before collection and analysis. DT treatment: VAKPS tumoroids were treated with 100 ng ml−1 of DT or demi-water for 3 days before collection and analysis.
Patient-derived organoids
Models and genetic data were obtained from the Human Cancer Models Initiative (HCMI): https://ocg.cancer.gov/programs/HCMI, dbGaP accession no. phs001486. PDO lines were deposited at the American Type Culture Collection.
Phenotypic reporter design and generation
The OnF sLCR was designed using the logical design of synthetic cis-regulatory DNA (LSD) method (https://gitlab.com/gargiulo_lab/sLCR_selection_framework)38, with customized deviations. First, we focused on three top OnF markers only—S100a6, Anxa1 and Ly6a—retrieving their genomic boundaries (S100a6: chr3:90,612,893-90,614,414; Anxa1: chr19:20,373,433- 203,906,71; Ly6a: chr15:749,948,76-749,980,31) from mm10 genome. Second, transcriptional regulators were curated based on differential gene expression across OnF and SC populations, ingenuity pathway analysis and gene ontology. Finally, we queried their respective known TF-binding sites (TFBSs) and associated position weight matrices (PWMs). This allowed LSD to generate potential CREs with a fixed length (default is a 150-bp window sliding with a 50-bp step), determined by CTCF sites surrounding each marker. Next, LSD assigned TFBSs to CREs using FIMO (default --output-pthresh 1e-4 --no-qvalue), creating a matrix of putative CREs × TFBSs. Finally, LSD selected the minimal number of CREs representing the complete set of TFBSs by sorting and selecting the best CRE by the sum of the affinity score (−log10(P)) and TFBS diversity (number of different TFBSs). The first CRE was prioritized based on 5ʹ-CAGE data (ENCODE) to increase the chances of successful transcriptional firing. The top seven CREs were synthesized in the same order, using the same strand for ‘plus’ and the reverse complement for ‘minus’.
The OnF sLCR was synthesized and cloned into a third-generation pLV lentiviral backbone at VectorBuilder. To generate the STAR–mCherry reporter, eGFP was substituted by mCherry (VectorBuilder) and the OnF sLCR with the STAR minigene (Addgene, cat. no. 136255) using BamHI-based Gibson cloning.
Virus production and transductions
Lentiviral packaging plasmids (pCMV-D8.9 and pCMV-VSVG) and reporter plasmids were transfected into HEK293T cells using Lipofectamine 3000. Virus-containing supernatant was concentrated with Lenti-X Concentrator and titered using GoStix-Plus (Takara Bio, cat. no. 631280). For infection, organoids or tumoroids were dissociated, mixed with lentivirus and TransDux MAX (SBI-LV860A-1), centrifuged at 600g —32 °C for 15 min (organoids) and 60 min (tumoroids)—and incubated at 37 °C for 6 h. Cells were then washed and replated in Matrigel with IntestiCult medium and Y-27632 (10 μM). Infected lines were selected with antibiotics. Plasmids and small hairpin RNA details are given in Supplementary Table 5.
Subcloning of dominant-negative FOS
A dominant-negative Fos was amplified by high-fidelity PCR from Addgene (cat. no. 33353) and subcloned into a lentiviral backbone (Addgene, cat. no. 25890), using BmtI and SpeI restriction enzymes.
Quantitative PCR
RNA was converted to complementary DNA using Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, cat. no. K1642). Then quantitative reverse transcription PCR (RT–qPCR) was performed on a QuantStudio5 (Applied Biosystems) in 10-μl reactions containing 5 μl of 2× SYBR Green Fast qPCR mix (Abclonal, cat. no. RK21203), 1 μl of primer mix (4 μM) and 8 ng of cDNA. Primer sequences are listed in Supplementary Table 6.
Flow cytometry
Matrigel-embedded organoids were collected in cold basal medium and centrifuged (500g for 5 min at 4 °C). Cells were dissociated in TrypLE at 37 °C, washed with cold basal medium (500g for 5 min at 4 °C), and stained with live or dead dye (Invitrogen, cat. no. L34957) at 1:2,000 in 1 ml of FACS buffer (PBS with 2% fetal bovine serum, 1 mM EDTA) for 10 min at room temperature (RT). In some experiments, Zombie NIR (BioLegend, cat. no. 423106) was used. Cells were washed with FACS buffer, passed through a 40-μm strainer and resuspended in 200 μl of FACS buffer, transferred to FACS tubes and acquired on a BD LSRFortessa (BD Biosciences). For sorting, a 35-μm filter was added and cells sorted on a CytoFLEX SRT (Beckman Coulter). Unstained cells and fluorescence − 1 controls were used to determine fluorescence intensity. Data were analyzed using FlowJo software (v.10.7.1).
Subcutaneous transplantation
Small clusters of VAKSP tumoroids were resuspended in a 1:1 Matrigel:basal medium mix; 5 × 105 cells were injected subcutaneously into both flanks of 7- to 8-week-old female NSG mice (Jackson Laboratory, cat. no. 005557). The tumor volume was calculated as 0.5 × (length × width2). DT was administered Intraperitoneally (i.p.) at 50 μg kg−1 every other day. FOLFIRI (5FU 30 mg kg−1, irinotecan 75 mg kg−1 and leucovorin 90 mg kg−1) was prepared as previously described77 and given i.p. 3× a week alone or with DT. Controls received vehicle. Mice were monitored and euthanized following ISMM Institutional Animal Care and Use Committee regulations, including clinical signs of distress or pain, ≥20% body weight, tumor size ≥1 cm3 or tumor ulceration.
Immunohistochemistry
Tumors were harvested from euthanized mice at a humane endpoint or experiment conclusion. A tumor piece was fixed in 10% formalin for 24 h (RT), then transferred to 70% ethanol. Samples were sent to HistoWiz, Inc., where they were in embedded in paraffin, sliced and stained with anti-mCherry (1:500; Rockland, cat. no. 600-401-379) and anti-GFP (1:100; abcam, cat. no. ab183734) antibodies.
RNA-seq library preparation
Organoids or tumoroids were grown in complete mouse IntestiCult medium for 3–4 d, before RNA extraction using TRIzol and PureLink RNA Mini Kit (Thermo Fisher Scientific, cat. no. 12183025). Treatments were applied as indicated in the figure legends. RNA quality was assessed with a bioanalyzer. Libraries were prepared using the PerkinElmer NextFlex Rapid Directional RNA-Seq Kit 2.0 (PerkinElmer, cat. no. NOVA-5198-02) and sequenced on a NovaSeq 6000 (SE75).
ATAC–seq library preparation
ATAC–seq was performed as described previously78. In brief, organoids were extracted from Matrigel using GCDR, pretreated with 200 U ml−1 of DNase (Worthington) for 30 min at 37 °C, then washed 3x with cold PBS. Viable cells, 50,000, were resuspended in cold lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40, 0.1% Tween-20 and 0.01% digitonin) and incubated on ice for 3 min. Nuclei were resuspended in 50 µl of transposition mix (nuclease-free water (22.5 µl), TD buffer (25 µl), Tn5 transposase (2.5 µl)) and incubated for 30 min at 37 °C. Transposed DNA fragments were then purified using MinElute PCR Purification Kit (QIAGEN), barcoded with Nextera dual indices (Illumina) and PCR amplified up to 11 cycles. Amplified libraries were purified using QIAGEN MinElute PCR Purification Kit (cat. no. 28004), checked for fragment sizes on a Bioanalyzer using D1000 DNA High-Sensitivity Chips (Agilent, cat. no. 5067-5585) and underwent size selection before a second quality check and quantification (Qubit). Normalized libraries were then pooled and sequenced on a Novaseq 6000 (Illumina).
Sample preparation for scRNA-seq and multiome
Organoids were washed in cold basal medium, resuspended in Cell Recovery Solution (Corning, cat. no. 76332-050) and incubated on a shaker (15 min at 4 °C). Pellets were dissociated into single cells using TrypLE and washed twice in basal medium and resuspended in PBS with 0.04% BSA. Single-cell preparations were counted and processed per the manufacturer’s instructions (10x Genomics, single-cell 3ʹ v.3.1 protocol). In brief, single cells were resuspended in a master mix and loaded with gel beads and partitioning oil into the chip to generate gel bead-in-emulsions (GEMs). Poly(A) RNA from each GEM was retrotranscribed to cDNA, containing an Illumina R1 primer sequence, a unique molecular identifier (UMI) and the 10× barcode. Pooled barcoded cDNA was purified with Silane DynaBeads, amplified by PCR and appropriately sized fragments selected for library construction, which included adding Illumina R2 primer sequence, paired-end constructs (P5 and P7 sequences) and a sample index.
For the multiome experiments, after nuclei dissociation, scATAC–seq was performed using Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v.1.1 (10x Genomics, cat. no. 1000175) and single-nucleus (sn)RNA-seq was performed using Chromium Next GEM Single Cell 3′ Reagent Kits v.3.1 (10x Genomics, cat. no. 1000121). Samples were sequenced on a NovaSeq 6000 (Illumina).
Bulk RNA-seq data processing and analysis
Quality control was conducted using FastQC (v.0.11.8,). Adapter sequences were trimmed using Trim Galore! (v.0.6.6) with a quality threshold of 20 (ref. 79). The mouse genome reference, GRCm38.p6 and GENCODE release M25 were used for alignment with STAR aligner (v.2.7.5b)80. Gene-level read counts were obtained using Salmon (v.1.2.1)81. Sample normalization was performed using the median ratio normalization method from DESeq2 R (v.1.28.1) and differential expression analysis was conducted using DESeq2 (ref. 82). Genes with fewer than five reads across all samples were filtered out. A gene was considered differentially expressed if the Benjamini–Hochberg-adjusted P value (Padj) < 0.01 and the absolute log2(fold-change) > 1. The ggplot2 (v.3.3.5) and VennDiagram (v.1.6.20) were used to plot and visualize differentially expressed gene data83,84. Gene-set enrichment analysis (GSEA) was performed on gene lists sorted by −log10(P) × log2(fold-change) using clusterProfiler (v.4.2.2) and enrichment or bubble plots visualized using gseaplot2 from enrichplot (v.1.14.2)85,86. All bioinformatics analyses were conducted on R (v.4.1.0).
Bulk ATAC–seq data processing and analysis
Quality control was performed using FastQC (v.0.11.8). Adapter sequences were trimmed using Trim Galore! (v.0.6.6) with a quality threshold of 20. Paired-end 75-bp reads were aligned to the mouse genome (GRCm38.p6 and GENCODE release M25) using Bowtie2 (v.2.2.8). Reads were sorted with SAMtools (v.1.11), removing mitochondrial alignments and retaining reads with MAPQ 30 (refs. 87,88). Duplicates were eliminated with Picard (v.2.2.4)89. Post-filtering BAM files were merged using SAMtool and peaks were called using MACS (v.2.1.0)90. Read quantification in significant peaks was conducted using BedTools multicov (v.2.29.2)91 and differential peak analysis was performed using DESeq2 (Padj < 0.01; absolute log2(fold-change) ≥ 1.5). Coverage tracks (Bigwig files) were generated using deepTools (v.3.2.1) bamCoverage92. Hierarchical clustering of average accessibility in differentially accessible regions (DARs) was performed with Rʹ ‘hclust’ function and visualized using deepTools plotHeatmap.
Motif enrichment and TF footprinting analysis
Motif analysis was performed using findMotifsGenome.pl from HOMER (v.4.10) on the peak summit (-size 200)93. Footprinting analysis was performed with Tobias (v.0.13.2) on aggregated BAM files per condition28. The ATACorrect function was used to correct Tn5 insertion bias and ScoreBigwig to calculate TF-binding scores in peak regions. Differential binding fold-change and P value for each TF were determined using BINDetect function and selected TF results were visualized using PlotAggregate. Elbow plots were generated using the ggplot2 (ref. 84).
TCGA bulk RNA-seq data processing
STAR raw gene counts were downloaded for TCGA-COAD and TCGA-READ. Data were normalized using median ratios normalization and differential expression analysis was performed using DESeq2. GSEA was conducted on gene lists sorted by −log10(P) × log2(fold-change) using clusterProfiler (v.4.2.2) and data visualized using gseaplot2 from enrichplot (v.1.14.2). To assess correlation, log(c.p.m.) counts were generated using the counts per min function from edgeR (v.3.36.0)94,95,96. Average expression was calculated for each gene in TCGA samples. The log(c.p.m.) values of TCGA samples and mouse organoids were merged and the top 2,000 most varied genes were selected for Spearman’s correlation analyses.
Public human bulk RNA-seq data processing
FPKM (fragments per kilobase of transcript per million) data from the human gastrointestinal tract (https://github.com/hilldr/Finkbeiner_StemCellReports2015/tree/master/DATA)25 were merged with TCGA FPKM data. Batch correction was applied using removeBatchEffect from limma R package (v.3.50.1), and principal components analysis (PCA) was performed using prcomp97.
Sc- and snRNA-seq data processing
Sequenced FASTQ files were processed using Cell Ranger (v.7.0.1, scRNA-seq) and Cell Ranger (v.2.0.0, snRNA-seq) from 10x Genomics with mm10 as the mouse genome reference (RRID: SCR_017344). Cells with ≥1,000 UMIs, ≥400 genes expressed and <10% mitochondrial reads were retained. UMI counts were then normalized to a total of 10,000 UMIs per cell across all genes and log(transformed) using the ‘LogNormalize’ function in Seurat. The top 2,000 most highly variable genes were identified with ‘FindVariableFeatures’ and counts were scaled with ‘ScaleData’. Datasets were processed using Seurat (v.4.0.3)98.
PCA was performed on the top 2.000 variable features using ‘RunPCA’ and the top 30 PCs were used for downstream analysis. Datasets were integrated using the ‘RunHarmony’ function (v.0.1.0)99 to correct for multiplexed samples and biological replicates. The k-nearest neighbor graphs were created with ‘FindNeighbors’ and Uniform Manifold Approximation and Projection (UMAP) results were generated using ‘RunUMAP’. The Louvain algorithm clustered cells based on expression similarity with a resolution set at 1.6.
Differential markers for each cluster in the scRNA-seq were identified using Wilcoxon’s test (‘FindAllMarkers’; Padj < 0.01, absolute log2(fold-change) > 0.25, 10% ≥ cells expressing the gene in comparison groups) using 1,000 random cells per cluster. Gene expression signature scores were calculated using ‘AddModuleScore’ and cells were annotated based on respective maximum module scores (Fig. 1b).
The mouse OnF signature was defined by overlapping genes upregulated in the neoplastic metacluster (clusters 6, 7, 10, 16, 19 and 20 combined) with mouse fetal genes from Mustata et al.19. Human tissue-type-specific signatures from Lukonin et al.26 were used to calculate signature scores with Seurat AddModuleScore. Lineage plasticity score was calculated as the esophagus mucosa module score − colon module score.
Data processing of scRNA-seq human datasets
Raw count matrices for the SMC/KUL3 (ref. 20) and Broad21 cohorts were obtained from the Gene Expression Omnibus (GEO) (accession nos. GSE132465 and GSE178341). Seurat objects were created using these matrices, with the SMC/KUL3 dataset filtered to retain cells from patients with matching normal and tumor tissues. Both datasets were subset to include only epithelial cells based on available annotations and data were analyzed following the same workflow as the mouse scRNA-seq above. Tissue-specific signatures from Lukonin et al.26 were used for cell- or tissue-type assignment.
To identify human OnF (huOnF) gene signature, a Seurat object was created from Gao et al.’s100 transcript per kilobase million counts and filtered to retain fetal (6- to 11-week-old embryos) and adult epithelial cells. Mouse OnF gene orthologues with normalized counts <0.5 in >98% of cells were filtered out. Human fetal and adult cells were then pseudobulked and orthologues highly expressed in fetal but low or not expressed in adult cells were selected as huOnF.
ScATAC–seq data processing and motif analysis
FASTQ files were processed using Cell Ranger ARC (v.2.0.0) with mm10 genome reference. Peaks were called using ‘CallPeaks’ (MACS2) function in Signac (v.1.3.0)101. Cells were filtered based on the following criteria: fragments in peaks >1,000 and <50,000, >20% reads in peaks, blacklist ratio <0.002, nucleosome signal <1.5 and transcription start site (TSS) enrichment score >2. The blacklist ratio = reads aligned to blacklist region or total reads. The nucleosome signal was calculated using ‘NucleosomeSignal’ in Signac and TSS enrichment score by ‘TSSEnrichment’. Latent semantic indexing (LSI) was applied to the top 95% variable peaks, combining the term frequency-inverse document frequency used for normalization and singular-value decomposition used for dimensional reduction. The first LSI component was excluded due to correlation with sequencing depth. De novo clustering (resolution 1.6) was done with ‘FindNeighbors’ and ‘FindClusters’ (Signac). Motif position frequency matrices from JASPAR 2020 were added to the data and refs. 102,103. ChromVAR motif analysis was performed using ‘RunChromVAR’. The results of selected TFs were visualized on UMAP using FeaturePlot35.
Statistics and reproducibility
Experiments were reproduced as indicated in the figure legends, with data from representative panels reproduced at least two or three times. No blinding or predetermined sample size calculations were used, but our sample sizes align with those in previous studies6,7. Animals or injection numbers were estimated based on the variability in tumor take rate and growth and are indicated in the respective figure legends. Animals were randomized among treatment groups once tumors reached the mean volume specified in Fig. 4h,j,l. Exclusion criteria included death or humane endpoints (for example, ≥20% weight loss, respiratory distress, persistent hypothermia or tumor size >1 cm3). Data distributions were assumed to be normal, but this was not formally tested. Statistical analyses were conducted using R (v.4.1.0, RRID, accession no. SCR_001905)104 and PRISM (v.9.5.1). Computational data figures were generated using ggplot2 (v.3.3.5, RRID, accession no. SCR_014601), ggpubr (v.0.4.0, RRID, accession no.SCR_021139) and patchwork (v.3.0.1)84,105,106. All statistical tests are detailed in the figure legends. Significance levels are defined as follows: NS (not significant): P > 0.05, ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001 and ∗∗∗∗P ≤ 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data needed to evaluate the conclusions in the present study are present in the paper and/or its Supplementary Information. RNA-seq, ATAC–seq, scRNA-seq and scATAC–seq data supporting the findings of the present study have been deposited into the National Center for Biotechnology Information, GEO under series accession no. GSE237954. The human reference genome GRCh38 GENCODE, release 36, and mouse reference genome GRCm38 GENCODE, release M25, were used to align raw data from human and mouse samples. They were accessed through https://www.gencodegenes.org/human/release_36.html and https://www.gencodegenes.org/mouse/release_M25.html, respectively. Other datasets referenced in the study are available from the GEO database under the accession nos. GSE132465 (ref. 20) and GSE178341 (ref. 21). Data used from Finkbeiner et al.25 are accessed through https://github.com/hilldr/Finkbeiner_StemCellReports2015/tree/master/DATA. Data from TCGA were accessed through the database of Genotypes and Phenotypes under accession no. phs000178.v11.p8 (TCGA-READ and TCGA-COAD). Source data are provided with this paper.
Code availability
This paper did not generate or use any custom code or software. All code and software packages utilized in the study are referenced in the Methods and Reporting Summary.
References
Sinicrope, F. A. Increasing incidence of early-onset colorectal cancer. N. Engl. J. Med. 386, 1547–1558 (2022).
Merlos-Suarez, A. et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 8, 511–524 (2011).
Vermeulen, L. et al. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc. Natl Acad. Sci. USA 105, 13427–13432 (2008).
Zhou, Y. et al. Cancer stem cells in progression of colorectal cancer. Oncotarget 9, 33403–33415 (2018).
Ohta, Y. et al. Cell–matrix interface regulates dormancy in human colon cancer stem cells. Nature 608, 784–794 (2022).
de Sousa e Melo, F. et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 543, 676–680 (2017).
Shimokawa, M. et al. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature 545, 187–192 (2017).
Kemper, K. et al. Monoclonal antibodies against Lgr5 identify human colorectal cancer stem cells. Stem Cells 30, 2378–2386 (2012).
Junttila, M. R. et al. Targeting LGR5+ cells with an antibody-drug conjugate for the treatment of colon cancer. Sci. Transl. Med. 7, 314ra186 (2015).
Davies, A., Zoubeidi, A., Beltran, H. & Selth, L. A. The transcriptional and epigenetic landscape of cancer cell lineage plasticity. Cancer Discov. 13, 1771–1788 (2023).
Perez-Gonzalez, A., Bevant, K. & Blanpain, C. Cancer cell plasticity during tumor progression, metastasis and response to therapy. Nat. Cancer 4, 1063–1082 (2023).
Reischmann, N. et al. BRAF(V600E) drives dedifferentiation in small intestinal and colonic organoids and cooperates with mutant p53 and Apc loss in transformation. Oncogene 39, 6053–6070 (2020).
Leach, J. D. G. et al. Oncogenic BRAF, unrestrained by TGFbeta-receptor signalling, drives right-sided colonic tumorigenesis. Nat. Commun. 12, 3464 (2021).
Chen, B. et al. Differential pre-malignant programs and microenvironment chart distinct paths to malignancy in human colorectal polyps. Cell 184, 6262–6280.e26 (2021).
Vasquez, E. G. et al. Dynamic and adaptive cancer stem cell population admixture in colorectal neoplasia. Cell Stem Cell 29, 1213–1228.e8 (2022).
Bala, P. et al. Aberrant cell state plasticity mediated by developmental reprogramming precedes colorectal cancer initiation. Sci. Adv. 9, eadf0927 (2023).
Nusse, Y. M. et al. Parasitic helminths induce fetal-like reversion in the intestinal stem cell niche. Nature 559, 109–113 (2018).
Yui, S. et al. YAP/TAZ-dependent reprogramming of colonic epithelium links ECM remodeling to tissue regeneration. Cell Stem Cell 22, 35–49.e7 (2018).
Mustata, R. C. et al. Identification of Lgr5-independent spheroid-generating progenitors of the mouse fetal intestinal epithelium. Cell Rep. 5, 421–432 (2013).
Lee, H. O. et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet. 52, 594–603 (2020).
Pelka, K. et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell 184, 4734–4752.e20 (2021).
Wang, F. et al. Single-cell and spatial transcriptome analysis reveals the cellular heterogeneity of liver metastatic colorectal cancer. Sci. Adv. 9, eadf5464 (2023).
Gregorieff, A., Liu, Y., Inanlou, M. R., Khomchuk, Y. & Wrana, J. L. Yap-dependent reprogramming of Lgr5+ stem cells drives intestinal regeneration and cancer. Nature 526, 715–718 (2015).
Roulis, M. et al. Paracrine orchestration of intestinal tumorigenesis by a mesenchymal niche. Nature 580, 524–529 (2020).
Finkbeiner, S. R. et al. Transcriptome-wide analysis reveals hallmarks of human intestine development and maturation in vitro and in vivo. Stem Cell Rep. 4, 1140–1155 (2015).
Lukonin, I. et al. Phenotypic landscape of intestinal organoid regeneration. Nature 586, 275–280 (2020).
Cheung, P. et al. Regenerative reprogramming of the intestinal stem cell state via Hippo signaling suppresses metastatic colorectal cancer. Cell Stem Cell 27, 590–604.e9 (2020).
Bentsen, M. et al. ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat. Commun. 11, 4267 (2020).
Chen, L. et al. HNF4 factors control chromatin accessibility and are redundantly required for maturation of the fetal intestine. Development https://doi.org/10.1242/dev.179432 (2019).
Verzi, M. P., Shin, H., San Roman, A. K., Liu, X. S. & Shivdasani, R. A. Intestinal master transcription factor CDX2 controls chromatin access for partner transcription factor binding. Mol. Cell. Biol. 33, 281–292 (2013).
Stringer, E. J. et al. Cdx2 determines the fate of postnatal intestinal endoderm. Development 139, 465–474 (2012).
Schuijers, J. et al. Ascl2 acts as an R-spondin/Wnt-responsive switch to control stemness in intestinal crypts. Cell Stem Cell 16, 158–170 (2015).
Eferl, R. & Wagner, E. F. AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859–868 (2003).
Rampioni Vinciguerra, G. L. et al. Role of Fra-2 in cancer. Cell Death Differ. 31, 136–149 (2024).
Schep, A. N., Wu, B., Buenrostro, J. D. & Greenleaf, W. J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017).
Barker, N. et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007 (2007).
Schmitt, M. J. et al. Phenotypic mapping of pathologic cross-talk between glioblastoma and innate immune cells by synthetic genetic tracing. Cancer Discov. 11, 754–777 (2021).
Company, C. et al. Logical design of synthetic cis-regulatory DNA for genetic tracing of cell identities and state changes. Nat. Commun. 15, 897 (2024).
Oost, K. C. et al. Specific labeling of stem cell activity in human colorectal organoids using an ASCL2-responsive minigene. Cell Rep. 22, 1600–1614 (2018).
Chapeau, E. A. et al. Author correction: direct and selective pharmacological disruption of the YAP–TEAD interface by IAG933 inhibits Hippo-dependent and RAS–MAPK-altered cancers. Nat. Cancer https://doi.org/10.1038/s43018-024-00797-y (2024).
Wang, X. et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J. Med. Chem. 65, 3123–3133 (2022).
Guinney, J. et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 21, 1350–1356 (2015).
Joanito, I. et al. Single-cell and bulk transcriptome sequencing identifies two epithelial tumor cell states and refines the consensus molecular classification of colorectal cancer. Nat. Genet. 54, 963–975 (2022).
Malla, S. B. et al. Pathway level subtyping identifies a slow-cycling biological phenotype associated with poor clinical outcomes in colorectal cancer. Nat. Genet. 56, 458–472 (2024).
Qin, X. et al. An oncogenic phenoscape of colonic stem cell polarization. Cell 186, 5554–5568.e18 (2023).
Barker, N. et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, 608–611 (2009).
Schwitalla, S. et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152, 25–38 (2013).
Schepers, A. G. et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 337, 730–735 (2012).
Cui Zhou, D. et al. Spatially restricted drivers and transitional cell populations cooperate with the microenvironment in untreated and chemo-resistant pancreatic cancer. Nat. Genet. 54, 1390–1405 (2022).
Burdziak, C. et al. Epigenetic plasticity cooperates with cell-cell interactions to direct pancreatic tumorigenesis. Science 380, eadd5327 (2023).
LaFave, L. M. et al. Epigenomic state transitions characterize tumor progression in mouse lung adenocarcinoma. Cancer Cell 38, 212–228.e13 (2020).
Laughney, A. M. et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med. 26, 259–269 (2020).
Marjanovic, N. D. et al. Emergence of a high-plasticity cell state during lung cancer evolution. Cancer Cell 38, 229–246.e13 (2020).
Chan, J. M. et al. Lineage plasticity in prostate cancer depends on JAK/STAT inflammatory signaling. Science 377, 1180–1191 (2022).
Zanconato, F. et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 17, 1218–1227 (2015).
He, L. et al. YAP and TAZ are transcriptional co-activators of AP-1 proteins and STAT3 during breast cellular transformation. eLife https://doi.org/10.7554/eLife.67312 (2021).
Park, J. et al. YAP and AP-1 cooperate to initiate pancreatic cancer development from ductal cells in mice. Cancer Res. 80, 4768–4779 (2020).
Koo, J. H. et al. Induction of AP-1 by YAP/TAZ contributes to cell proliferation and organ growth. Genes Dev. 34, 72–86 (2020).
Della Chiara, G. et al. Epigenomic landscape of human colorectal cancer unveils an aberrant core of pan-cancer enhancers orchestrated by YAP/TAZ. Nat. Commun. 12, 2340 (2021).
Shao, D. D. et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158, 171–184 (2014).
Alonso-Curbelo, D. et al. A gene–environment-induced epigenetic program initiates tumorigenesis. Nature 590, 642–648 (2021).
Ge, Y. et al. Stem cell lineage infidelity drives wound repair and cancer. Cell 169, 636–650.e14 (2017).
Poynter, J. N. et al. Statins and the risk of colorectal cancer. N. Engl. J. Med. 352, 2184–2192 (2005).
Chang, W. T., Lin, H. W., Lin, S. H. & Li, Y. H. Association of statin use with cancer- and noncancer-associated survival among patients with breast cancer in Asia. JAMA Netw. Open 6, e239515 (2023).
Canellas-Socias, A. et al. Metastatic recurrence in colorectal cancer arises from residual EMP1+ cells. Nature 611, 603–613 (2022).
Goto, N. et al. SOX17 enables immune evasion of early colorectal adenomas and cancers. Nature 627, 636–645 (2024).
Sharma, A. et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell 183, 377–394.e21 (2020).
Li, Z. et al. Presence of onco-fetal neighborhoods in hepatocellular carcinoma is associated with relapse and response to immunotherapy. Nat. Cancer 5, 167–186 (2024).
Leushacke, M. et al. Lgr5-expressing chief cells drive epithelial regeneration and cancer in the oxyntic stomach. Nat. Cell Biol. 19, 774–786 (2017).
Madisen, L. et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010).
Johnson, L. et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410, 1111–1116 (2001).
Shibata, H. et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science 278, 120–123 (1997).
Jonkers, J. et al. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 29, 418–425 (2001).
Yang, X., Li, C., Herrera, P. L. & Deng, C. X. Generation of Smad4/Dpc4 conditional knockout mice. Genesis 32, 80–81 (2002).
Jackstadt, R. et al. Epithelial NOTCH signaling rewires the tumor microenvironment of colorectal cancer to drive poor-prognosis subtypes and metastasis. Cancer Cell 36, 319–336.e7 (2019).
Sato, T. et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009).
Alvarez-Varela, A. et al. Mex3a marks drug-tolerant persister colorectal cancer cells that mediate relapse after chemotherapy. Nat. Cancer 3, 1052–1070 (2022).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Trim Galore! version 0.6.6 (Babraham Institute, 2012).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Chen, H. & Boutros, P. C. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 12, 35 (2011).
ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, 2016).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Yu, G., Wang, L. G. & He, Q. Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience https://doi.org/10.1093/gigascience/giab008 (2021).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Picard Tools version 2.2.4 (Broad Institute, 2019).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Chen, Y., Lun, A. T. & Smyth, G. K. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res. 5, 1438 (2016).
McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Gao, S. et al. Publisher correction: tracing the temporal-spatial transcriptome landscapes of the human fetal digestive tract using single-cell RNA-sequencing. Nat. Cell Biol. 20, 1227 (2018).
Stuart, T., Srivastava, A., Madad, S., Lareau, C. A. & Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021).
Fornes, O. et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 48, D87–D92 (2020).
Baranasic, D. JASPAR2020: data package for JASPAR database (version 2020). R package version 0.99.8 https://bioconductor.org/packages/release/data/annotation/html/JASPAR2020.html (2022).
R Core Team R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2023).
Kassambara, A. ggpubr: ‘ggplot2’ based publication ready plots. R package version 0.6.0 https://rpkgs.datanovia.com/ggpubr/ (2023).
Pedersen, T. L. patchwork: the composer of plots. R package version 1.1.1 https://patchwork.data-imaginist.com/ (2023).
Acknowledgements
Research reported in this publication was supported by ISMMS seed fund to E.G. and S.M. and the National Cancer Institute (NCI) of the National Institutes of Health (NIH; grant no. R01CA292376-01 to S.M. and E.G.). We gratefully acknowledge the use of the services and facilities of the Tisch Cancer Institute supported by the NCI Cancer Center Support grant (no. P30 CA196521). This work was supported in part by the Bioinformatics for Next Generation Sequencing (BiNGS) shared resource facility within the Tisch Cancer Institute at the Icahn School of Medicine at Mount Sinai, which is partially supported by the NIH (grant no. P30 CA196521). This work was also supported in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai and supported by the Clinical and Translational Science Awards (CTSA; grant no. UL1TR004419 from the National Center for Advancing Translational Sciences). Research reported in this paper was in part supported by the Office of Research Infrastructure of the NIH under award no. S10OD026880. Part of this research was supported by an International Accelerator Award, ACRCelerate, jointly funded by Cancer Research UK (grant nos. A26825 and A28223), FC AECC (grant no. GEACC18004TAB) and AIRC (grant no. 22795). A.M.T.’s lab acknowledges funding from the American Cancer Society (Research Scholar Grant RSG-23-1039063-01-MM). G.G.’s lab acknowledges funding from MDC, Helmholtz (grant no. VH-NG-1153), ERC StG (grant no. 714922) and ERC PoC (grant no. 101069235). We acknowledge the use of models and data derived by the HCMI, dbGaP accession no. phs001486. We are grateful to the members of the Human Immune Monitoring Center and the Genomics Core Facility at the Icahn School of Medicine at Mount Sinai for their expertise and assistance with scRNA-seq and multiome-seq, as well as the Dean’s Flow Cytometry CoRE at Mount Sinai. We thank the Garnett Lab at the Wellcome Sanger Institute for deriving the human organoids as part of the HCMI (https://www.sanger.ac.uk/collaboration/human-cancer-models-initiative-hcmi). We also thank A. Rialdi, F. Cantatore, F. Fonseca, K. Beaumont, S. Campaner, D. Pasini, R. DasGupta and M. Dawson for fruitful discussions.
Author information
Authors and Affiliations
Contributions
S.M. and E.G. conceptualized the project, designed experiments and supervised research. They also wrote the manuscript, with critical input from all authors. S.M., M.S., H.Z., K.M. and F.D.T. performed the experiments and acquired the data. S.M. and K.M. prepared ATAC–seq and RNA-seq libraries, respectively. D.D., X.W., G.U., D.T., D.H., A.M.T. and B.G. performed computational and -omics analyses with input from S.M. and E.G. G.G. designed the OnF reporter with input from S.M., C.C. and Y.D. T.R.L. and O.J.S. generated the VAKSP tumoroids. M.L. and N.B. generated the remaining genetic organoid models. R.S. and J.C.M. provided support for multi-omic data acquisition. D.L.O. provided guidance for flow cytometry data collection and acquisition. All authors provided scientific input and contributed to writing the materials and methods.
Corresponding authors
Ethics declarations
Competing interests
E.G.’s laboratory received research funds from AZ and Prelude Therapeutics (for unrelated projects). E.G. is a cofounder and shareholder of Immunoa Pte. Ltd and cofounder shareholder, consultant and advisory board member of Prometeo Therapeutics. G.G. is inventor on the patent application no. EP18192715 by the MDC for the design and use of sLCRs. R.S. is cofounder and CEO of Panacent Bio, Inc. and equity holder in GeneDx, neither of which has contributed to or been involved in the research in this publication. The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Itay Tirosh and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Oncofetal reprogramming during intestinal tumorigenesis.
a, Schematics of the genetic approach used to generate isogenic mouse organoid models mimicking the adenoma-to-adenocarcinoma sequence. b, UMAP of scRNA-seq of the indicated models. c, Cells from the neoplastic metacluster are indicated in green on the UMAP. d, Volcano plot of differentially expressed genes (DEGs) in the neoplastic metacluster vs. all other clusters. p-values calculated using DESeq2, Wald test (two-sided, unpaired); Benjamini-Hochberg adjustments. e, Venn diagram depicting the strategy used to define the mouse oncofetal (OnF) signature. f-h, UMAPs showing enrichment scores of enterocytes (f), stem cell (g) and OnF (h) signatures throughout CRC progression. i, Heatmap of SC and OnF module scores across cell clusters and genotypes. In panels b-c and f-h WT = 4058 cells; A = 5890 cells; AKSP 10946 cells. j, Relative Lgr5 expression along the SC-OnF continuum. k, Enrichment of the OnF and SC module scores across genotypes (n = 3 independent organoid cultures per group). boxplots: center line, median; box limits, interquartile range (IQR: 25th to 75th percentile); whiskers, ±1.5x IQR; individual points, outliers. p-values calculated using two-sided unpaired t-test. Illustrations in a created using BioRender.com.
Extended Data Fig. 2 Enhanced oncofetal reprogramming triggers lineage plasticity in advanced human CRC.
a, Human OnF score enrichment in matched normal and tumors tissues (TCGA/COAD; n = 41 patients). Two-sided paired t-tests for significance. b-d, Scatter plots depicting cell state distribution along the SC-OnF spectrum in matched healthy colons and tumors stratified by APC mutation and MMR status, respectively. b; SMC/KUL3 cohort, n = 7 patients; c-d; Broad cohort, n = 15 MMRp patients (c) and n = 21 MMRd patients (d). Two-sided paired t-tests for significance. e, Principal Component Analysis (PCA) of integrated bulk RNA-seq data from human gut developmental stages, definitive endoderm (Def. end), embryonic stem cells (ESC) (Finkbeiner et al., 2015) normal colon and CRC (TCGA/COAD). f, Gene Set Enrichment Analysis (GSEA) of lineage-specific signatures in tumor (n = 455) vs. normal (n = 41) specimens (TCGA/COAD). NES: normalized enrichment score. g-j, Scatter plots (left) and boxplots (right) of lineage specific signature enrichment across the SC-OnF continuum in matched tumors and normal colons from the SMC/KUL3 (g and h, n = 10) and Broad (i and j, n = 36) cohorts. Two-sided paired t-tests for significance. In panels a, b-d and g-j, boxplots: center line, median; box limits, interquartile range (IQR: 25th to 75th percentile); whiskers, ±1.5x IQR; individual points, outliers.
Extended Data Fig. 3 A multi-omics approach elucidates the molecular determinants of cell state dynamics during CRC evolution.
a, Schematic of the experimental strategy, integrating multiomics with functional validation, and the genetic models used. b, Differentially accessible regions (DARs) in A vs. WT and AKSP vs. WT; shared DARs shown in faded colors (data from 2 independent organoid cultures per group). c, HOMER motif analysis for CDX2 and HNF4a, using DARs from cluster c1 in Fig. 2a. d-e, TF occupancy strength for CDX2 (d) and HNF4a (e) shown as aggregated footprinting plot matrix centered around binding motifs. f, HOMER motif analysis using DARs from cluster c1 in Fig. 2a. g-i, TF occupancy strength for RXR (g), PPAR (h) and VDR (i) shown as aggregated footprinting plot matrix centered around binding motifs. j, HOMER motif enrichment analysis in promoter regions of OnF genes or random promoters (TSS + /- 1 kb). Illustrations in panel a created using BioRender.com.
Extended Data Fig. 4 Functional validation of YAP and AP-1 as drivers of OnF reprogramming.
a, Schematic summary of the experimental design related to Fig. 2f, g. b-e, GSEA of OnF genes following YAP (b-c) depletion and FOS depletion (d) or inhibition (e) during tumor initiation, related to Fig. 2f, g. Two-sided permutation-based test for significance; p-values adjusted via the Benjamini-Hochberg method. f-g, Scatter plots showing enrichment of the SC (left) and small intestine (right) (f) and the OnF (left) and esophagus mucosa (right) module scores (g) along the SC-OnF continuum in scRNA-seq cells from the AKSP Multiome. h-i, Scatter plots showing the correlation between FOS (h) or TEAD (i) activity and lineage infidelity score. Two-sided Pearson correlation for significance. j, Schematic summary of the experimental design related to Fig. 2l-n and o-q. k, Log2 median-of-ratios (DESeq2) normalized counts of AP-1 genes following YAP (S6A) overexpression in WT organoids (n = 4 independent experiments). Boxplots: center line, median; box limits, interquartile range (IQR: 25th to 75th percentile); whiskers, highest and lowest values within ±1.5x IQR. P-values calculated via two-sided Student’s t-test with multiple comparison adjustments. l-n, GSEA of OnF (l), esophagus (m) and small intestines (n) genes following YAP (S6A) overexpression in WT organoids, related to Fig. 2l-n. Two-sided permutation-based test for significance; p-values adjusted via the Benjamini-Hochberg method. Illustrations in a and j created using BioRender.com.
Extended Data Fig. 5 Comparative analyses of RXR inhibition versus APC depletion.
a-c, Representative images of WT organoids treated with DMSO (a) or RXR antagonist HX531 (RXRi) (b), and A tumoroids (c). Scale bars, 1 mm. Lower panels show close-ups of dashed line areas, highlighting morphological changes. d, GSEA of OnF genes in RXRi vs. WT organoids (n = 3 independent cultures). e, Heatmap of differentially expressed marker genes of the indicated markers in RXRi vs. WT and A vs. WT (n = 3 independent cultures per group). f, g, Venn diagrams showing overlaps of the more (f) and less (g) accessible regions in RXRi and A organoids. Punctuated areas represent changes in the same direction but not meeting the log2 fold change cutoff ≥1.5. h, ATAC-seq signal heatmap in WT, A and RXRi organoids at differentially accessible regions (upper panel). Average ATAC-seq signal within a ±2Kb region around the peak center (lower panel). i-n, TF occupancy strength (aggregated footprinting plot matrix) for the RXRa (i), CDX2 (j), HNF4a (k), FOS (l), TEAD3 (m) and TCF7 (n) in WT, A and RXRi organoids. Plots are centered around binding motifs. o, HOMER motif enrichment analysis of the indicated TFs in A and RXRi models vs. WT. In f-o, RXRi, n = 3; WT, n = 2 and A, n = 2 independent cultures.
Extended Data Fig. 6 RXR inhibition phenocopies APC depletion excluding WNT activation.
a-b, Venn diagrams showing overlap of up-regulated (a) and down-regulated (b) genes following RXR inhibition and APC depletion in WT organoids. Punctuated areas represent changes not meeting the log2 fold change ≥1 cutoff. KEGG pathway analysis reveals common regulation in both models, except for WNT. c, GSEA of KEGG pathway enrichment in RNA-seq data from RXRi vs. WT and A vs. WT. NES, normalized enrichment score. In a-c n = 3 independent cultures per group. d-e, Differentially expressed genes (DEGs) (d) and regions (DARs) (e) in RXRi vs. WT and woRXRi vs. WT organoids. Faded colors indicate persistent DEGs and DARs. Log2 fold change (f.c) and p-values thresholds for DEGs and DARs are indicated in the figure. p-values calculated using DESeq2, Wald test (two-sided, unpaired); Benjamini-Hochberg adjustments. f-h, GSEA of OnF genes following YAP (f-g) or FOS (h) depletion in CRC tumoroids (AKSP) (n = 3-4 independent experiments). Two-sided permutation-based test for significance; p-values adjusted via the Benjamini-Hochberg method.
Extended Data Fig. 7 Design and functional validation of the oncofetal phenotypic reporter.
a, Design method the oncofetal (OnF) synthetic locus control regions (sLCR). b, Genome Browser view of cis-regulatory elements used in the OnF sLCR. c, Normalized distribution of OnF-GFP+ cells and eGFP fluorescence intensity measured by flow cytometry. d, eGFP geometric mean fluorescence insensitivity (MFI) quantification. Error bars, s.d; n = 3 independent cultures, p-values calculated using Fisher/s LSD post-hoc test after one-sided ANOVA (p < 0.05). e, Scaled OnF gene expression in GFPhigh and GFPNeg cells sorted from VAKSPOnF tumoroids, n = 3 independent sorts. f, Schematic of the OnF sLCR substitution with the STAR minigene. g, Representative fluorescence microscopy images of VAKSP tumoroids co-expressing both reporters (VAKSPOnF/STAR). Scale bars, 200μm. h, Volcano plot of differentially expressed genes (DEGs) in VAKSP tumoroids post-3-day FOLFIRI treatment (n = 3 independent experiments). Significant DEGs are on both sides of dashed lines (log2FC > 1; p-value < 0.01); p-values, DESeq2, Wald test (two-sided, unpaired); Benjamini-Hochberg adjustments. i, The GFP/mCherry gating strategy to sort clones used in (j-l). j-l, Quantification of predominantly cell states in response to FOLFIRI in 3 independent clones sorted from GFPhigh (j), mCherryhigh (k) or GFP/mCherryhigh (hybrid) (l) populations. p-values calculated using paired two-sided t-tests. m-r, Representative pseudocolor plots of flow-cytometry analysis of GFPhigh (m-n) mCherryhigh (o-p) and hybrid clones (q-r) before (m, o and q) and after FOLFIRI tretament for 3 days (n, p and r), related to (j-l).
Extended Data Fig. 8 A dual phenotypic reporter system to trace and target the OnF and Stem Cell states.
a, Heatmap of day 5 FOLFIRI dose-response in patient-derived organoids (PDOs); n = 3 independent experiments. b-d, DEGs in response to FOLFIRI. b, OnF genes; c, stem cell and WNT-related genes; d, YAP and AP-1 target genes. PDOs treated with their respective IC70 for 5 days. Data are log2 fold change in FOLFIRI vs. DMSO; n = 3 independent experiments. e, Fluorescence microscopy images of the indicated models treated with demi-water or DT for 3 days. Scale bars, 400 µm. f, Quantification of the predominantly stem, OnF and hybrid states post-DT treatment (day3); n = 4-7 independent experiments per group. p-values calculated using paired two-sided t-tests, adjusted for multiple comparisons using Benjamini-Hochberg method. Center, median; boxes, lower and upper quartiles; whiskers min and max values. g, Representative immunohistochemistry staining of GFP and mCherry in tumors from mice treated with saline or DT (n = 2 tumors for the saline; n = 3 tumors for DT), relative to Fig. 4l. Scale bars, 2 mm.
Supplementary information
Source data
Source Data Fig. 2 (download XLSX )
Data underlying Fig. 2h.
Source Data Fig. 3 (download XLSX )
Data underlying Fig. 3o.
Source Data Fig. 4 (download XLSX )
Data underlying Fig. 4b,d,h,j,l,m–q.
Source Data Extended Data Fig. 1 (download XLSX )
Data underlying Extended Data Fig. 1k.
Source Data Extended Data Fig. 2 (download XLSX )
Data underlying Extended Data Fig. 2a,f.
Source Data Extended Data Fig. 3 (download XLSX )
Data underlying Extended Data Fig. 3c,f,j.
Source Data Extended Data Fig. 7 (download XLSX )
Data underlying Extended Data Fig. 7d,j–l.
Source Data Extended Data Fig. 8 (download XLSX )
Data underlying Extended Data Fig. 8a–d,f.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mzoughi, S., Schwarz, M., Wang, X. et al. Oncofetal reprogramming drives phenotypic plasticity in WNT-dependent colorectal cancer. Nat Genet 57, 402–412 (2025). https://doi.org/10.1038/s41588-024-02058-1
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41588-024-02058-1
This article is cited by
-
Single-cell multi-omics reveals DUSP9 as a key regulator of cancer stemness and a potential therapeutic target in hepatocellular carcinoma
Journal of Translational Medicine (2026)
-
Cancer stem cell-driven drug resistance in colorectal carcinoma: molecular aspects and therapeutic potentials
Molecular Cancer (2026)
-
Fetal reversion from diverse lineages sustains the intestinal stem cell pool and confers stress resilience
Communications Biology (2026)
-
Dynamic remodelling of epithelial plasticity in colorectal cancer from single-cell and spatially resolved perspectives
Journal of Translational Medicine (2025)
-
Defining the mucosal ecosystem: epithelial–mesenchymal interdependence in gastrointestinal health and disease
Nature Reviews Gastroenterology & Hepatology (2025)
