
Abstract
Small nuclear RNAs (snRNAs) combine with specific proteins to generate small nuclear ribonucleoproteins (snRNPs), the building blocks of the spliceosome. U4 snRNA forms a duplex with U6 and, together with U5, contributes to the tri-snRNP spliceosomal complex. Variants in RNU4-2, which encodes U4, have recently been implicated in neurodevelopmental disorders. Here we show that heterozygous inherited and de novo variants in RNU4-2 and in four RNU6 paralogs (RNU6-1, RNU6-2, RNU6-8 and RNU6-9), which encode U6, recur in individuals with nonsyndromic retinitis pigmentosa (RP), a genetic disorder causing progressive blindness. These variants cluster within the three-way junction of the U4/U6 duplex, a site that interacts with tri-snRNP splicing factors also known to cause RP (PRPF3, PRPF8, PRPF31), and seem to affect snRNP biogenesis. Based on our cohort, deleterious variants in RNU4-2 and RNU6 paralogs may explain up to ~1.4% of otherwise undiagnosed RP cases. This study highlights the contribution of noncoding RNA genes to Mendelian disease and reveals pleiotropy in RNU4-2, where distinct variants underlie neurodevelopmental disorder and retinal degeneration.
Similar content being viewed by others


Main
While approximately2 million individuals worldwide are affected by retinitis pigmentosa (RP), it is estimated that 30% to 50% remain without a conclusive genetic diagnosis, even after exome or genome sequencing is performed1,2,3,4. This reflects high genetic heterogeneity, limited testing access and as-yet-unidentified disease genes, which in general carry pathogenic variants that are exceedingly rare in the control population5,6,7.
Noncoding RNAs are essential to many cellular processes, including pre-messenger RNA (pre-mRNA) splicing, which is ensured by the spliceosome, a macromolecular complex that in its major form is composed of five small nuclear RNAs (snRNAs), U1, U2, U4, U5 and U6, and ~300 proteins8. Each snRNA associates with a specific set of proteins to form a small nuclear ribonucleoprotein (snRNP), the functional unit of the spliceosome. Variants in RNU4-2, one of the two paralogs encoding U4, have been linked to a common neurodevelopmental disorder (NDD) known as ReNU syndrome (OMIM: 620851). These variants account for up to 0.4% of all NDD cases and lead to systematic misrecognition of donor splice sites by the spliceosome9,10,11. Likewise, RNU2-2 and RNU5B-1 have been recently associated with NDDs11,12,13.
Several spliceosomal proteins are also known to be involved in a wide range of hereditary diseases, including RP, as first noted by McKie and colleagues14. Specifically, of the ~100 genes that are currently associated with nonsyndromic RP5, the tri-snRNP splicing factor genes PRPF3, PRPF4, PRPF8, PRPF31 and SNRNP200 underlie the autosomal dominant form of the condition (adRP), with variants in PRPF31 accounting for 10–20% of all adRP cases3,15.
Here, we identify both inherited and de novo variants in RNU4-2 and four paralogs of RNU6, encoding the U6 snRNA, as the molecular cause of adRP in 153 individuals across 67 families. We demonstrate that all identified variants cluster within the U4/U6 duplex, in a region that binds directly to PRPF31 and PRPF3 and indirectly to PRPF6 and PRPF816,17. Furthermore, we show that such variants increase the association of U4 and U6 snRNAs with the splicing factors SART3 and PRPF31, suggesting impaired snRNP biogenesis.
Results
RNU4-2 variants underlie adRP
We initially examined a nonconsanguineous family with adRP (Family M1-A; Supplementary Fig. 1), in which seven of eight siblings (II:1–II:7) and their father (I:1) displayed classical RP features (Supplementary Fig. 2 and Supplementary Data 1). Genome sequencing was negative for pathogenic variants in known retinal disease-associated genes, but selective DNA variant filtering and shared haplotype analysis revealed a total of 55 variants that were absent from gnomAD v.4.17,18 and co-segregated with RP. Of these, none was predicted to impact splicing (SpliceAI > 0.2)19 and only one was evolutionarily conserved (GERP = 4.03 and phyloP-vertebrate = 3.18)20,21, a single-nucleotide insertion in the gene RNU4-2 (NR_003137.2:n.18_19insA; Fig. 1a, Supplementary Fig. 1 and Supplementary Tables 1 and 2). This DNA change was present in one individual from the All of Us database22.

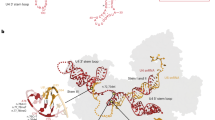
a, Two-dimensional structure of the U4/U6 duplex, with recurrent variants identified in RP cases (in red for U4 and in green for U6), all clustering within the three-way junction. Nucleotides affected by variants previously observed in NDD cases are underlined. b, Rare variants affecting RNU4-1, defined as AF < 0.1% in gnomAD v.4.1, identified in RP cases and in controls. c, Same as in b for RNU4-2, with recurrent pathogenic variants displayed in red. d, Same as b for all five RNU6 paralogs combined, with recurrent causative variants displayed in green. Significant P values for variants enriched in RP cases versus controls from gnomAD are indicated (two-sided Fisher’s test with Bonferroni correction).
To find additional families, we first screened by Sanger sequencing a cohort of 1,891 individuals from the European Retinal Disease Consortium (www.erdc.info) with RP or Leber congenital amaurosis who remained undiagnosed after a large high-throughput screening using single molecule Molecular Inversion Probes23. This analysis led to the identification of three additional families comprising 15 affected individuals segregating the same pathogenic variant (Supplementary Fig. 1 and Supplementary Tables 1 and 2). The n.18_19insA allele was significantly enriched in the RP cohort compared with both the gnomAD and the All of Us databases (analyzed control genomes: 76,215 and 414,000, respectively; Bonferroni-corrected P values = 2.6 × 10−3 and 6.9 × 10−5, respectively, by two-sided Fisher’s test; Supplementary Table 3). Additional screening of the RNU4-2 sequence in the same cohort led to the identification of 28 other variants, one of which (n.56T>C) recurred in eight individuals from four families (Fig. 1a, Supplementary Fig. 1 and Supplementary Tables 1 and 2), was absent in controls and was significantly enriched in patients versus controls (Bonferroni-corrected P values = 6.4 × 10−5 (gnomAD) and 7.9 × 10−8 (All of Us); Supplementary Table 3).
Additional screening of 2,830 RP cases without previous genetic diagnosis from our respective institutions’ cohorts, the UK National Genomic Research Library (hosting data from the Genomics England 100,000 Genomes Project24 and from the NHS Genomic Medicine Service) uncovered an additional patient harboring n.18_19insA (for whom the variant was de novo) and six families (nine affected individuals) carrying the n.56T>C variant (Supplementary Fig. 1 and Supplementary Tables 1 and 2). Altogether, recurrent variants in RNU4-2 were identified in 41 affected individuals from 15 families (Supplementary Fig. 3 and Supplementary Tables 1 and 2). Of note, incomplete penetrance was observed for nine obligate carriers, without visual symptoms (Supplementary Fig. 1). One carrier of n.56T>C was asymptomatic, with subnormal electroretinogram, diffuse atrophic changes in the periphery and attenuated vessels. Another individual with the same variant showed no clinical signs of disease upon examination, and seven (among whom four were deceased) were not clinically evaluated to determine their disease status. Our combined screening of RNU4-2 also revealed 24 other unique rare DNA changes in 27 families, which were classified as variants of uncertain significance (VUS), as well as three benign changes (Supplementary Table 3).
Because U4 snRNA can also be transcribed from its paralog RNU4-1, which differs from RNU4-2 at only four positions (n.37, n.88, n.99 and n.113; Supplementary Table 4), we next examined its sequence in our initial cohort and identified 63 variants, none of which were significantly enriched in cases compared with controls; also, these changes did not include variants at sites corresponding to n.18_19 and n.56 of RNU4-2 (Fig. 1b and Supplementary Table 3). Notably, RNU4-1 appears to be more tolerant to variation compared with RNU4-2, as evidenced by the numerous and frequent variants that are present in genomes from the general population (cumulative allele frequency of 20.4% in RNU4-1 versus 1.2% in RNU4-2; gnomAD v.4.1) (Fig. 1b,c and Supplementary Fig. 4), as already noted previously9.
Variants in U6 paralogues also cause RP
In the di-snRNP and the tri-snRNP complexes of the major spliceosome, U4 binds to U6 to form the U4/U6 RNA duplex. We therefore hypothesized that variants in U6 could also underlie adRP and extended our analysis to all five identical paralogous genes producing the U6 snRNA, scattered across the genome (RNU6-1, RNU6-2, RNU6-7, RNU6-8 and RNU6-9; Supplementary Table 4). A screening of these genes by Sanger sequencing in our initial cohort of 1,891 RP families revealed 94 DNA changes in total. The n.55_56insG insertion recurred at the exact relative position in RNU6-2, RNU6-8 and RNU6-9 (four families per gene, 34 cases in total; Supplementary Fig. 1 and Supplementary Tables 1 and 2) and was significantly enriched in cases versus controls, who were all negative for this change (Bonferroni-corrected P value = 2.6 × 10−18 (gnomAD) and 5.1 × 10−27 (All of Us); Supplementary Table 3). Since this variant was identical in three U6 genes, we reasoned that the specific DNA change, rather than any particular paralog, was relevant to the etiology of the disease. We therefore repeated our analysis by collapsing the five RNU6 genes and detected 66 unique variants. Another insertion, n.56_57insG, was identified in two unrelated families (once in RNU6-2 and once in RNU6-9, four cases in total; Supplementary Fig. 1 and Supplementary Table 2) and found to be significantly enriched in cases versus controls (Bonferroni-corrected P value = 1.8 × 10−3 (gnomAD, a single RNU6-2 positive individual of unknown status) and 2.1 × 10−5 (All of Us, no positive individuals); Supplementary Table 3). We then extended our analysis to the same international cohorts of patients that were previously analyzed (n = 2,830) and identified 74 additional cases from 38 families who were positive for either n.55_56insG or n.56_57insG (Supplementary Table 2).
In total, these two variants were detected in 112 affected individuals from 52 families, involving all RNU6 paralogs except RNU6-7. The n.55_56insG insertion was present in most cases (102 individuals from 47 families), occurring in four of the five RNU6 paralogs: RNU6-1, RNU6-2, RNU6-8 and RNU6-9, while n.56_57insG was present in ten individuals from five families, in RNU6-1, RNU6-2 and RNU6-9 (Supplementary Tables 1 and 2 and Supplementary Figs. 1 and 3). Notably, n.55_56insG was confirmed to be a de novo event in eight individuals, clinically identified as sporadic cases. In 14 additional pedigrees, it was also observed in individuals born to unaffected parents, for which de novo inheritance was suspected but could not be confirmed, due to the lack of parental DNA. In contrast, no de novo events could be detected for n.56_57insG, which was identified exclusively in families with adRP (Supplementary Fig. 1). Similar to the screening of the RNU4 paralogs, our analysis of RNU6 paralogs revealed 66 VUSs and 23 benign variants, validated by Sanger sequencing (Supplementary Table 3).
In summary, we identified variants in RNU4-2 or RNU6 paralogs that underlie de novo or inherited dominant RP in 67 families. The overall phenotype across all cases was consistent with classical RP, based on clinical examination and electrophysiological testing, with symptomatic onset predominantly in adolescence (Supplementary Table 5). In addition, other concurrent ocular disease features were noted across individuals in the cohort: cystoid macular edema (55.9%), non-age-related lens opacities (23.6%) and various vitreomacular complications (30.6%) (Supplementary Table 5). Based on our data from these 4,722 RP cases, mostly of European descent and lacking a genetic diagnosis, we estimate that RNU4- and RNU6-associated RP could be responsible for ~1.4% of all molecularly undiagnosed individuals with this disease. Furthermore, considering that approximately 30% of RP diagnoses correspond to adRP25,26 and that our positive families include 24 isolated individuals, we can further infer that these variants may account for approximately 3.0% of undiagnosed adRP families.
Predicted effects of variants on the U4/U6 duplex
All RP variants are predicted to map in spatial proximity with each other, within the three-way junction delimited by stem-I and stem-II of the U4/U6 duplex and the 5′ stem-loop of U4 (Figs. 1a and 2a). In particular, they are located in a different region compared with those underlying NDD (Fig. 1a). In silico two-dimensional modeling of RNA secondary structure predicted as well that the RNU4-2 variant n.18_19insA inserts a nucleotide between stem-II and the U4 5′ stem-loop (Supplementary Fig. 5a,b), while n.56T>C disrupts the first base-pairing of the U4/U6 duplex within stem-I (Supplementary Fig. 5a,c). Both changes lead to the extension of the internal loop, an event that is predicted to impact the overall stability of the duplex. In addition, n.18_19insA slightly modifies the orientation of the 5′ stem-loop relative to stem-I and stem-II (Supplementary Fig. 5a,b).
In contrast, both n.55_56insG and n.56_57insG in RNU6 paralogs are predicted to extend the length of stem-I by three additional base pairs, reduce the size of the internal loop and drastically change the orientation of the 5′ stem-loop (Supplementary Fig. 5a,d,e). Interestingly, we observed that a benign insertion at the same position, n.55_56insT, was present in gnomAD v.4.1 in all five RNU6 paralogs with a cumulative frequency of 0.12% (n = 181) (Supplementary Fig. 5f). While these models provide a coherent structural rationale for the observed clustering, the precise effects of the variants on U4/U6 architecture remain to be experimentally verified.
Analysis of cryo-electron microscopy data (PDB 6QW6)27 confirmed that all RP variants identified reside in a region critical for binding of the U4/U6 duplex to the splicing factors PRPF31, PRPF3 and PRPF8, all previously associated with adRP16,17 (Fig. 2b). Specifically, this region first engages PRPF31 or the PRPF3/PRPF4 complex, initiating the assembly interface, and is subsequently stabilized in its native orientation upon the coordinated binding of additional tri-snRNP components, including PRPF6 and PRPF828. The mutated and neighboring U4 and U6 nucleotides detected in RP cases directly participate in the binding of PRPF31 and PRPF3 (Fig. 2c,d), via hydrogen bonds with eight and three residues of these proteins, respectively. Notably, by querying the ClinVar database29, we detected a missense variant affecting one of these residues, p.(Arg449Gly) of PRPF3, identified in a three-generation family with seven affected individuals having clinical features similar to those observed in most cases from our study30.

a, Naked U4/U6 pairing, showing the proximity of the causative variants identified (red and green). b, Same as in a with interacting PRPF proteins. c, Direct interactions of nucleotides of the U4/U6 duplex with PRPF31, via hydrogen bonds. d, Same as c but for PRPF3.
Expression of RNU4 and RNU6 genes
Since the human genome contains several RNU4 and RNU6 pseudogenes31, we investigated whether any of these might be incorrectly annotated and could instead produce functional RNA, potentially contributing to the disease. In addition, we sought to understand why the various U4 and U6 paralogs appear to be differentially mutated, with RNU4-1 and RNU6-7 displaying none of the recurrent pathogenic variants. We used RNA sequencing (RNA-seq) data from human neurosensory retina (NSR), retinal pigment epithelium (RPE) and choroid that were enriched for small RNAs, applying stringent and paralog-aware bioinformatics analyses designed to mitigate the complexities associated with reads aligning against multiple paralogs and/or pseudogenes (Methods). RNU4-2 was more highly expressed than RNU4-1 in all tissues (average ratio: 1.63; Fig. 3a). Conversely, individual expression of RNU6 genes and pseudogenes in the retina could not be reliably quantified by RNA-seq, since their sequences are identical, except for the last nucleotide. Therefore, we compared the total expression of RNU4 and RNU6, regardless of their respective paralogs and pseudogenes. RNU6 expression was on average 3.39× higher across the three tissues, compared with RNU4 (Fig. 3b). Of note, NSR and RPE had higher expression of RNU4 (2.51×) and RNU6 (6.09×) with respect to the choroid, an ocular tissue not directly involved in vision, used as a control (Fig. 3b). This observation is in agreement with previous data showing that snRNA expression in the retina is approximately sixfold higher compared with muscle, testis, heart and brain32, indicating a high demand for snRNAs in these two retinal layers.

a, Expression of RNU4-1 and RNU4-2 from RNA-seq of human donor choroid (n = 13), NSR (n = 4) and RPE (n = 16). For these boxplots, the tick line within boxes indicates the median (also expressed numerically), boxes represent the first and the third quartiles and whiskers indicate the largest observation smaller than or equal to the first quartile − 1.5 × IQR and the smallest observation greater than or equal to the third quartile + 1.5 × IQR. b, Same as in a for RNU4 genes (RNU4-1, RNU4-2 and pseudogenes) and for all RNU6 genes (RNU6-1, RNU6-2, RNU6-7, RNU6-8, RNU6-9 and pseudogenes). c, ATAC-seq and H3K27ac signals for RNU4-1, RNU4-2, RNU4ATAC (red) and 105 RNU4 pseudogenes (black). d, Same as in c for five RNU6 genes and RNU6ATAC (red), as well as for 1,312 RNU6 pseudogenes (black). IQR, interquartile range.
In addition, we analyzed the Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) and H3K27ac chromatin immunoprecipitation followed by sequencing (ChIP–seq) data from retinal tissues33 in genomic regions spanning all RNU4 and RNU6 sequences. ATAC-seq assesses chromatin accessibility across the genome, while H3K27ac ChIP–seq reveals the presence of active enhancers. These data, combined, indicate potential active transcription at promoter regions. Our analysis showed clear transcription marks in all paralogous RNU4 and RNU6 genes in the retina (Fig. 3c,d). Conversely, these signatures were absent from the 105 U4 pseudogenes and the 1,312 U6 pseudogenes, except for RNU4-8P, which displayed strong signals, but probably by virtue of its close proximity to the ACTR1B promoter. Of note, RNU6-92P and RNU6-656P had high ATAC-seq signals but very low H3K27ac signals at their respective promoters (Fig. 3d).
We performed the same analysis for other snRNA genes present in the human genome, which revealed a similar trend: all RNU genes, with the exception of RNU5F-1, had marks of active transcription and only a few among the thousands of RNU pseudogenes displayed signals compatible with potential expression, therefore representing plausible candidate genes for retinal disease (Supplementary Fig. 6). In addition, RNU2-2 was recently implicated in NDD, yet without evidence of ocular involvement13. Interestingly, the same type of analysis, based on conservation and expression data from GTEx, was recently performed by others, showing similar results34.
For RNU6-7, both ATAC-seq and H3K27ac signals were within the same range as those observed for other RNU6 genes (Fig. 3d), and, therefore, the absence of pathogenic variants could not be explained by a potential differential expression. We thus analyzed the genetic landscape of variations in healthy individuals in all five U6 paralogs and observed that RNU6-7 displayed a lower number of variants, compared with the others (Supplementary Fig. 7). We also identified the recurrent variant n.55_56insG in RNU6-7 in six control individuals of African or African American ancestry in gnomAD v.4.1 (allele frequency (AF) = 0.014%) and in 14 individuals of African origin in the All of Us database (AF = 0.013%). These seemingly contradictory observations merit further investigations in future studies.
Transcriptome analysis in patients
We performed transcriptome analysis following the collection of RNA from circulating leukocytes in nine affected individuals carrying variants in RNU4-2, RNU6-1 and RNU6-9 (three individuals per gene), as well as from 14 healthy controls (Supplementary Table 6). To avoid systematic errors linked to the use of different collection kits35 across our cohort (Methods), we performed independent case–control tests for samples collected with PAXgene kits (three RNU4-2 cases, six controls) or Tempus kits (six RNU6 cases, eight controls). We identified 27 and eight differentially expressed genes in the two datasets, respectively, with no gene overlap (fold-change > 2 and false discovery rate (FDR) P value < 0.05; Supplementary Table 7), indicating no major differences in global gene expression in leukocytes in cases versus controls. We then further explored the data by investigating potential bias in pre-mRNA splicing, as performed in ref. 11. This analysis led to the identification of 107 upregulated and 67 downregulated 5′ splice sites in the PAXgene set, and 37 upregulated and 13 downregulated 5′ splice sites in the Tempus set, with only two sites in common between the datasets (in the genes CLEC2D and SNHG29; Supplementary Table 8). At these two sites the expression differences, although statistically significant, were below 10%.
We then examined the DNA sequences of the 224 differentially expressed splice sites, focusing on the occurrence of the ‘AG’ dinucleotide at positions −2/−1, which was previously reported to be enriched in sites with increased splicing in RNU4-2 in patients with NDD11. No differences in ‘AG’ frequency were observed between sites with increased versus decreased usage in either the PAXgene or Tempus samples (two-tailed Fisher’s test, P = 0.63 and P = 0.52, respectively). We also compared nucleotide frequencies at each position between upregulated and downregulated splice sites, separately for the two groups, and found no significant differences (two-tailed Fisher’s test with FDR correction, P < 0.05). A similar analysis of overlapping dinucleotides across the region flanking the 5′ splice site (for example, positions −4/−3, −3/−2, up to +7/+8) revealed no significant differences.
Functional effects of RP variants
Since PRPF variants associated with RP affect primarily spliceosomal assembly32, we investigated whether the same phenomenon could be driven by the variants detected in this work. Specifically, we immunopurified ectopically expressed U4 and U6 snRNAs containing the RP variants and analyzed their association with specific markers for the U6 snRNP (SART3), the U4/U6 di-snRNP (SART3 and PRPF31), the U4/U6.U5 tri-snRNP (PRPF31 and SNRNP200) and the U5 snRNP (SNRNP200). The combined results showed an increased association of snRNA constructs with RP variants with SART3 and partially with PRPF31, while the interaction with SNRNP200 was unchanged or reduced (Fig. 4). For comparison, we included in our assays the U4 n.64_65insT variant, which causes NDD, and observed no significant alteration in the association with any of the proteins tested, compared with wild type (Fig. 4a). Additionally, no significant differences were detected between NDD and RP variants, pointing to the need for targeted functional studies to delineate their respective impacts on spliceosome dynamics. Similarly, U6 RNA bearing the n.55_56insT and n.57T>G variants, observed in healthy control individuals, presumably did not affect spliceosome formation, since the low amount of protein associated with them implies that they entered the spliceosome assembly process only minimally (Fig. 4b). Taken together, the results indicate that RP pathogenic variants have potentially a specific dominant effect on snRNP biogenesis and delay the assembly process at the di-snRNP stage.

a,b, Immunoprecipitation of U4-MS2 (WT and variants) (a) and U6-MS2 (WT and variants) (b). snRNPs were immunoprecipitated via MS2-YFP by anti-GFP antibodies and co-precipitated proteins were detected by western blotting. The position of the MS2 loop (green) in snRNAs is indicated. Four independent experiments were quantified. Immunoprecipitated proteins are normalized to input and U4 or U6 WT controls. Middle bars indicate average values and error bars the s.e.m. Statistical significance was analyzed by the two-tailed unpaired t-test and the P values were adjusted using the Benjamini–Hochberg FDR method to control for false discoveries. P values ≤ 0.05 are indicated. Full-length blots and antibody validation are provided as Source Data. Ctrl, control; IP, immunoprecipitation; WT, wild type.
Discussion
The numerous genes associated with RP and allied diseases belong to a wide range of functional classes, from retina-specific biochemical pathways to ubiquitous cellular processes5. Yet, how these defects ultimately lead to retinal degeneration often remains unclear. The link between pathogenic variants in splicing factors of the tri-snRNP complex (RP-PRPFs), essential for survival in all eukaryotes, and RP, a phenotype limited to the eye, represents perhaps the most intriguing of these biological enigmas.
In this study, we identified recurrent heterozygous variants in RNU4-2, encoding U4 RNA, and in multiple paralogs of the U6 RNA as a cause of RP. Interestingly, these snRNAs are also an integral part of the di- and tri-snRNP and directly interact with some RP-PRPF proteins. In addition, similar to RP-PRPFs, they are also associated with the same specific phenotype: de novo or inherited adRP, with reduced penetrance for RNU4-2 variants. Importantly, the clinical presentation of patients with RNU4-2 and RNU6 variants overlaps with that of other spliceosome-related forms of adRP, particularly showing an earlier onset—contrasting with the generally milder prognosis observed in most other adRP types36,37—and a relatively high co-occurrence of features such as cataracts and cystoid macular edema, found in cases with PRPF3138,39, PRPF840 and SNRNP20041 variants. Prevalence estimations indicate that these snRNA pathogenic changes may account for an elevated number of undiagnosed cases, and it is therefore surprising that the RNU4 and RNU6 genes have escaped disease association until now. A partial explanation for this phenomenon is that mainstream sequencing approaches are biased towards DNA-capturing procedures that do not include snRNA genes. Furthermore, although genome sequencing is increasingly being adopted in routine diagnostics, variants in snRNA genes may have remained undetected because they affect noncoding transcripts, which are more challenging to interpret and are often overlooked or deprioritized by standard analytical pipelines.
An intriguing feature of pathogenic changes in RNU4-2 is their pleiotropy with respect to NDD (ReNU syndrome) and RP. Chen et al.9 described that more than half of the patients with ReNU also display some visual abnormalities, although only three were documented as having retinal phenotypes (one had an abnormal electroretinogram response, one had Leber congenital amaurosis and one presented with macular dysfunction). However, most cases were too young to display the typical symptoms of RP, which usually manifest during adolescence or early adulthood42. Although the exact mechanism for this phenotypic selectivity is unknown, RNU4-2 variants represent a clear and new allelic series involving noncoding RNA genes. A recent preprint highlighted a strong effect of ReNU variants in an RNU4-2 saturation genome editing experiment, while the RP variant n.56T>C in the same gene did not show any statistically significant effect43. In addition, the region of RNU4-2 containing RP variants showed function scores within the neutral range of the saturation genome editing assay43, suggesting a potentially milder pathogenic effect compared with ReNU changes. It is therefore plausible that the RP variants identified in RNU4-2 and RNU6 paralogs could lead to photoreceptor death and subsequent visual loss, while having no influence on the development of the brain. Additionally, ReNU variants are located in the stem-III and the T-loop of the U4/U6 duplex and interfere with the proper recognition of intronic 5′ splice signals, likely because these regions are involved in pairing pre-mRNA with U6 (ref. 9). In contrast, RP variants cluster in spatial proximity to the three-way junction, in regions not directly engaged in interactions with pre-mRNA but that are involved in the binding of various proteins, including RP-associated splicing factors.
Consistent with this evidence, we did not observe any major splicing anomalies in transcripts from patients with RP, with the only two significant events showing differences below 10% in expression. Conversely, our biochemical assays support a role for RP-associated variants in altering spliceosomal assembly. As the magnitude of the observed changes was in all instances rather moderate (less than 1.5-fold with respect to controls), we interpret these data as indicating that snRNA variants associated with RP are unlikely to prevent the assembly of spliceosomal complexes. Rather, they may cause a subtle alteration in snRNP dynamics, possibly affecting the efficiency of their biogenesis or recycling steps. In particular, the increased association with SART3 and, to a lesser extent, with PRPF31, together with unchanged or slightly reduced interaction with SNRNP200, may indicate a modest delay in the transition from the di-snRNP to the tri-snRNP form. Moreover, the pathogenic variants identified in this study lie within regions of the U4/U6 duplex that directly contact the PRPF3 and PRPF31 proteins, two splicing factors linked to adRP whose mutations also delay spliceosomal complex assembly32.
In terms of specific molecular effect, our functional data show that snRNAs bearing RP variants display enhanced interaction with di-snRNP protein markers, suggesting that pathogenesis could result from a gain-of-function or dominant-negative mechanism, rather than from haploinsufficiency. This hypothesis is strengthened by the evidence that molecularly similar but benign variants, commonly observed in the general population, seem not to bind efficiently to di-snRNP markers and potentially not to be incorporated into the spliceosome, supporting the idea that spliceosomal functions could be haplosufficient with respect to heterozygous and snRNA-depleting variants.
Although DNA changes associating RNU4-2 to ReNU syndrome have been primarily reported as de novo events9,10, in our study most families with RP (61%) bore RNU4-2 and RNU6 changes as inherited variants. In part, this difference can be explained by the reduced reproductive fitness associated with NDD versus RP. Unlike ReNU syndrome, symptomatic onset (night-blindness and peripheral vision loss) in nonsyndromic adRP begins later in life, with severe central vision loss usually occurring after the onset of reproductive age. Another difference involves the inheritance of dominant variants, which in ReNU seems to be almost exclusively of maternal origin9. We did not observe the same trend for RP, with variants being inherited from either of the parents, possibly indicating the absence of any sex-specific negative selection during gametogenesis or at the embryonic stage.
The human genome contains two RNU4 paralogs and five RNU6 paralogs. This indicates that, assuming equal expression within paralogs, the presence of only ~25% of mutant U4 (heterozygous genotype, over two copies) or ~10% of mutant U6 (heterozygous genotype, over five copies) is sufficient to lead to a disease phenotype, again in support of a gain-of-function or dominant-negative molecular mechanism. This could be a crucial consideration for the development of potential gene-based therapies, as gene-augmentation strategies may be suboptimal compared with gene correction or antisense oligonucleotide approaches. Our data also highlight the existence of mutational hotspots outside the coding regions of the human genome, emphasizing the need for further research into these parts of our genetic material, and show that the clustering of de novo pathogenic variants is not restricted to severe diseases with childhood onset44, but may extend to milder pathologies, such as RP.
In conclusion, we identified four recurrent pathogenic variants in RNU4-2 and in four of the five paralogs of the U6 snRNA as a frequent cause of de novo or inherited adRP. The immediate impact of these findings involves improved diagnosis and genetic counseling for patients with hereditary visual loss, especially for isolated cases who could potentially bear heterozygous de novo events. More fundamentally, this work substantially broadens our understanding of the genetic landscape of human disease, paving the way for the development of new molecular therapeutic approaches.
Methods
Patients and DNA samples
This study adhered to the tenets of the Declaration of Helsinki, and signed, informed consent was obtained from all participants. All procedures were conducted in accordance with Institutional Review Board-approved human research protocols and were approved by the ethics committees of the Radboud University Medical Center (Nijmegen, the Netherlands) and the Rotterdam Eye Hospital (Rotterdam, the Netherlands) (MEC-2010-359; OZR protocol no. 2009-32), and the local ethics committees of all other participating institutions.
Clinical characterization and analysis
Complete ophthalmic examinations were performed by a retinal specialist, which included measurement of best-corrected visual acuity and intraocular pressures, and examination of anterior segment and fundus (dilated). Color fundus photographs and montages were captured using the FF450plus Fundus Camera (Carl Zeiss Meditec) and Optos 200 Tx (Optos). Fundus autofluorescence images (488-nm excitation) and high-resolution spectral-domain optical coherence tomography (SD-OCT) scans were acquired using the Spectralis HRA+OCT module (Heidelberg Engineering). Hyper-autofluorescent ring contours were analyzed using a custom program in FIJI software (National Institute of Mental Health)45. Progression rates were calculated using linear mixed-effects regression in R (v.4.0.4) with time (years) since baseline as the primary independent variable, baseline ring size as a covariate and inter-ocular differences as a random effect. Photoreceptor+ thickness was assessed on horizontal SD-OCT scans through the fovea using a semi-automated procedure46. Photoreceptor+ was defined as the distance between the Bruch’s membrane/choroid interface and the inner nuclear layer/outer plexiform layer boundary. Layer segmentation was performed in a semi-automated manner using a custom software in MatLab (MathWorks). Full-field electroretinogram recordings were conducted using the Espion Visual Electrophysiology System (Diagnosys) according to International Society for Clinical Electrophysiology of Vision (ISCEV) standards47.
Genome sequencing and annotation
Genomic DNA from probands was isolated from peripheral blood lymphocytes according to standard procedures. Sequencing was performed by BGI Tech Solutions using the DNBseq Sequencing Technology, with a minimal median coverage per genome of 30×. The processing of the sequencing data was performed by using BWA mem (v.0.7.17)48, Picard (v.2.14.0-SNAPSHOT) (http://broadinstitute.github.io/picard) and GATK (v.4.1.4.1)49 for mapping to the human genome reference sequence (build hg19/GRCh37) and variant calling2. For variant annotation, we used ANNOVAR50 with the addition of splicing predictions by MaxEntScan51 and SpliceAI19.
Assessment of variants
Human Genome Variation Society (HGVS) notations of the variants were retrieved using VariantValidator52 and American College of Medical Genetics and Genomics (ACMG) classification53 was applied according to the ACGS Best Practice Guidelines for Variant Classification in Rare Disease 202354. In particular, we used the PS4_strong criterion for variants significantly enriched in cases versus controls (gnomAD v.4.1 and All of Us), as assessed for each variant by two-tailed Fisher’s exact test in R (fisher.test function), in agreement with the ACMG recommendations53 (odds ratio > 5.0, lower bound of the confidence interval > 1.0, corrected P value < 0.05), but only for variants present in at least three probands to avoid any bias from imbalanced case–control sets. This assessment was made using probands only (n = 1,891) for all variants, except for those in Supplementary Table 1, which were assessed in 4,722 individuals. PM6 and PP1 were applied according to ClinGen Sequence Variant Interpretation (SVI) recommendations. Specifically, PM6_sup was applied when two unrelated families had de novo variants without parental confirmation, given that RP is a ‘phenotype consistent with gene but not highly specific and high genetic heterogeneity’. PP1_sup, PP1_mod and PP1_strong were assigned when the variant segregated with disease in ≥1, ≥2 and ≥5 informative meioses, respectively55. We defined thresholds for PM2_sup and BS2 based on the frequency of RHO p.(Pro23His), the most prevalent variant causing adRP, which was detected once in gnomAD v.4.1 and 13 times in All of Us. Specifically, PM2_sup was assigned to variants that were present fewer than two times in gnomAD v.4.1 and fewer than 14 times in All of Us. BS2 was applied to variants that were observed more than four times in gnomAD v.4.1 or more than 28 times in All of Us, that is, twice the values of p.(Pro23His). PM2_sup was not applied to variants for which the PS4 criterion had already been used, to avoid double-counting evidence related to their low frequency in gnomAD. BA1 was considered for variants with allele frequency >5% in gnomAD v.4.1 or the All of Us databases, whereas BS1 was assigned to variants with allele frequencies greater than expected for disease (1/2,000 = 0.05%).
Screening by Sanger sequencing
Genomic DNA was collected, and RNU4-1, RNU4-2, RNU6-1, RNU6-2, RNU6-7, RNU6-8 and RNU6-9 genes were amplified using standard PCR procedures. RNU4-1, RNU4-2, RNU6-1, RNU6-2, RNU6-7, RNU6-8 and RNU6-9 PCR fragments were sequenced using Sanger sequencing and screened for novel variants in these genes.
Two-dimensional modeling of the effect of variants and three-dimensional representation
We utilized RNAfold WebServer to model the effect of variants with default parameters56 and RNAcanvas was used for drawing the structure57. We used ChimeraX with PDB file, using PDB file 6QW6 to draw three-dimensional representation of the U4/U6 duplex with and without surrounding PRPF proteins.
RNA-seq experiments and analysis
RNA was isolated from human donor eye tissue, which was collected and dissected according a reported procedure58 from an ethically approved Research Tissue Bank (UK NHS Health Research Authority reference no. 15/NW/0932). Total RNA was isolated from four NSR samples, 16 pelleted RPE samples and 13 choroid samples that had been stored in RNAlater (Thermo Fisher Scientific), using an Animal Tissue RNA Purification kit (Norgen Biotek), as per manufacturer’s instructions. Sequencing libraries were prepared using the NEBnext multiplex small RNA library preparation kit, as per manufacturer’s protocols, with size selection performed using Ampure beads. Paired-end sequencing (2 × 75 base pairs (bp)) was performed on an Illumina HiSeq 4000.
NEBnext adapters were removed from sequencing reads using trimmomatic (v.0.39) before alignment against the GRCh38 reference genome with bowtie59 (v.1.3). No mismatches between sequencing reads and the reference genome were allowed, and no restriction was set on multi-mapping reads. Sequence read counts were restricted to primary alignments using samtools (v.1.21)60, and therefore only counted once if they aligned to multiple RNU4 (n = 90) or RNU6 (n = 1,277) genes or pseudogenes. Calculations were drawn from read 1 datasets and normalized for the total read count achieved for the sample. Total RNU4 and RNU6 expression was based on all annotated genes and pseudogenes in GENCODE v.38.
ATAC-seq and H3K27ac ChIP–seq data
ATAC-seq data from ref. 61 (eight different experiments) and H3K27ac ChIP–seq data from ref. 62 (five different experiments) were downloaded as bigwig files from the RegRet database (http://genome.ucsc.edu/s/stvdsomp/RegRet)63. For both data types, the signal (the genes and 500 bp on each side) was extracted using bedtools (v.2.27.1) after conversion using bigWigToWig (v.469). We quantified the signal for all RNU genes and pseudogenes first by normalizing the signal of each experiment to the maximum and then summing them. For RNU4, we quantified two genes and 105 pseudogenes, while for RNU6 we assessed five genes and 1,312 pseudogenes, in addition to RNU4ATAC and RNU6ATAC.
RNA-seq from blood RNA
Peripheral blood samples were collected from affected individuals and controls using either Tempus Blood RNA tubes (Applied Biosystems) or PAXgene Blood RNA tubes (Qiagen). Total leukocyte RNA was extracted with the Tempus Spin RNA Isolation Kit (Applied Biosystems) or the Preserved Blood RNA Purification Kit II (Norgen Biotek), respectively, following the manufacturers’ protocols. Following the quality assessment of RNA integrity and concentration, 100 ng of input RNA per sample was subsequently processed for library preparation using the KAPA RNA HyperPrep Kit with RiboErase (HMR) and KAPA Globin Depletion Hybridization Oligos (Roche). Sequencing was performed on an Illumina NovaSeq 6000 platform with 2 × 101-bp paired-end reads. To improve quality score calculations for the final base, one additional base was sequenced in both read 1 and read 2. The Q30 value for all RNA-seq data was ≥91.1%. Adapters were trimmed with Skewer (v.0.2.2)64.
Reads were aligned to reference transcripts from Ensembl (v.110, GRCh38) using STAR (v.2.7.11a) with the option --twopassMode Basic. DESeq2 (v.1.46.0) with default options was used for differential expression analysis between different groups according to sample origin (Tempus or PAXgene tubes) and presence/absence of the pathogenic RNU genotypes, with fold-change > 2 and FDR P value < 0.05. We used rMATS65 to assess differential alternative splicing, separately for the Tempus and PAXgene sets and with specific options (--allow-clipping --variable-read-length --anchorLength 1 --novelSS --task both --libType fr-unstranded -t paired --readLength 101). We further used the Python scripts from ref. 11 to process the rMATS output and filter the data according to a mean coverage > 7, an FDR P value < 0.1 and a deltaPSI value > 0.05. The R function fisher.test with default parameters was used to assess differences in base compositions at splicing sites, at each position, as well as differences for 2-mers (for example, positions −4/−3 to +7/+8).
U4 and U6 snRNP analysis
U4 n.18_19insA, n.56T>C and n.64_65insT variants were introduced by site-directed mutagenesis into the plasmid expressing U4-MS266. The full-length U6 sequence, including 256 bp upstream and 93 bp downstream of the RNU6-1 gene, was inserted into the pcDNA3 plasmid lacking the CMV promoter. The MS2 loop was inserted between nucleotides 10 and 11. U6 n.55_56insG, n.55_56insT, n.56_57insG and n.57G>T variants were introduced by site-directed mutagenesis. U4- and U6-expressing plasmids were transfected into HeLa cells stably expressing MS2-YFP protein. At 24 h after transfection, snRNAs were immunoprecipitated using anti-GFP antibodies and co-precipitated proteins were analyzed by western blotting66.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Research on the de-identified patient data used in this publication from the Genomics England 100,000 Genomes Project and the NHS GMS dataset can be carried out in the Genomics England Research Environment subject to a collaborative agreement that adheres to patient-led governance. All interested readers will be able to access the data in the same manner that the authors accessed the data. For more information about accessing the data, interested readers may contact research-network@genomicsengland.co.uk or access the relevant information on the Genomics England website: https://www.genomicsengland.co.uk/research. Sharing of additional sequencing or blood RNA-seq data is subject to the European General Data Protection Regulation (GDPR) applicable in the countries of residence of the tested individuals and may become available upon a data transfer agreement approved by local ethical committees. Patient sample identifiers from this study can be released upon reasonable request from ‘M1-A to M9-B’ to the corresponding local ‘DNA-number’. Specific variant requests or other data are available from the corresponding author (S.R.) upon reasonable request. The data generated during this study (causative variants from Supplementary Table 1) are submitted to the Leiden Open (source) Variation Database (LOVD) (http://www.lovd.nl) and ClinVar (accession codes SCV006562526 to SCV006562534). Sequences of primers used in this study are listed in Supplementary Table 9. Additional details regarding PCR conditions or primer design are available upon request. Detailed information on antibodies used are provided in Supplementary Table 10. Small RNA-seq datasets analyzed in this study are available at the NCBI Sequence Read Archive through accession PRJNA1256119 (https://www.ncbi.nlm.nih.gov/sra/PRJNA1256119). The genes and pseudogenes analyzed are present in Supplementary Table 11 and the read counts are available in Supplementary Table 12. Source data are provided with this paper.
References
Verbakel, S. K. et al. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 66, 157–186 (2018).
Peter, V. G. et al. The first genetic landscape of inherited retinal dystrophies in Portuguese patients identifies recurrent homozygous mutations as a frequent cause of pathogenesis. PNAS Nexus 2, pgad043 (2023).
Perea-Romero, I. et al. Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci. Rep. 11, 1526 (2021).
Conti, G. M. et al. Genetics of retinitis pigmentosa and other hereditary retinal disorders in western Switzerland. Ophthalmic Res. 67, 172–182 (2024).
Rivolta, C. et al. RetiGene, a comprehensive gene atlas for inherited retinal diseases. Am. J. Hum. Genet. 112, 2253–2265 (2025).
Hanany, M., Rivolta, C. & Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl Acad. Sci. USA 117, 2710–2716 (2020).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Rogalska, M. E. et al. Transcriptome-wide splicing network reveals specialized regulatory functions of the core spliceosome. Science 386, 551–560 (2024).
Chen, Y. et al. De novo variants in the RNU4-2 snRNA cause a frequent neurodevelopmental syndrome. Nature 632, 832–840 (2024).
Greene, D. et al. Mutations in the U4 snRNA gene RNU4-2 cause one of the most prevalent monogenic neurodevelopmental disorders. Nat. Med. 30, 2165–2169 (2024).
Nava, C. et al. Dominant variants in major spliceosome U4 and U5 small nuclear RNA genes cause neurodevelopmental disorders through splicing disruption. Nat. Genet. 57, 1374–1388 (2025).
Jackson, A. et al. Analysis of R-loop forming regions identifies RNU2-2 and RNU5B-1 as neurodevelopmental disorder genes. Nat. Genet. 57, 1362–1366 (2025).
Greene, D. et al. Mutations in the small nuclear RNA gene RNU2-2 cause a severe neurodevelopmental disorder with prominent epilepsy. Nat. Genet. 57, 1367–1373 (2025).
McKie, A. B. et al. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Hum. Mol. Genet. 10, 1555–1562 (2001).
Weisschuh, N. et al. Genetic architecture of inherited retinal degeneration in Germany: a large cohort study from a single diagnostic center over a 9-year period. Hum. Mutat. 41, 1514–1527 (2020).
Mozaffari-Jovin, S. et al. The Prp8 RNase H-like domain inhibits Brr2-mediated U4/U6 snRNA unwinding by blocking Brr2 loading onto the U4 snRNA. Genes Dev. 26, 2422–2434 (2012).
Liu, S. et al. Binding of the human Prp31 Nop domain to a composite RNA-protein platform in U4 snRNP. Science 316, 115–120 (2007).
Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625, 92–100 (2024).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535–548 (2019).
Pollard, K. S., Hubisz, M. J., Rosenbloom, K. R. & Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20, 110–121 (2010).
Davydov, E. V. et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6, e1001025 (2010).
Denny, J. C. et al. The “All of Us” Research Program. N. Engl. J. Med. 381, 668–676 (2019).
Panneman, D. M. et al. Cost-effective sequence analysis of 113 genes in 1,192 probands with retinitis pigmentosa and Leber congenital amaurosis. Front. Cell Dev. Biol. 11, 1112270 (2023).
Caulfield, M. et al. National Genomic Research Library. Figshare https://doi.org/10.6084/m9.figshare.4530893.v7 (2024).
Daiger, S. P., Bowne, S. J. & Sullivan, L. S. Genes and mutations causing autosomal dominant retinitis pigmentosa. Cold Spring Harb. Perspect. Med. 5, a017129 (2014).
Sullivan, L. S. et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest. Ophthalmol. Vis. Sci. 47, 3052–3064 (2006).
Charenton, C., Wilkinson, M. E. & Nagai, K. Mechanism of 5′ splice site transfer for human spliceosome activation. Science 364, 362–367 (2019).
Hardin, J. W., Warnasooriya, C., Kondo, Y., Nagai, K. & Rueda, D. Assembly and dynamics of the U4/U6 di-snRNP by single-molecule FRET. Nucleic Acids Res. 43, 10963–10974 (2015).
Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985 (2014).
Zhong, Z. et al. Two novel mutations in PRPF3 causing autosomal dominant retinitis pigmentosa. Sci. Rep. 6, 37840 (2016).
Denison, R. A., Van Arsdell, S. W., Bernstein, L. B. & Weiner, A. M. Abundant pseudogenes for small nuclear RNAs are dispersed in the human genome. Proc. Natl Acad. Sci. USA 78, 810–814 (1981).
Tanackovic, G. et al. PRPF mutations are associated with generalized defects in spliceosome formation and pre-mRNA splicing in patients with retinitis pigmentosa. Hum. Mol. Genet. 20, 2116–2130 (2011).
D’Haene, E. et al. Comparative 3D genome analysis between neural retina and retinal pigment epithelium reveals differential cis-regulatory interactions at retinal disease loci. Genome Biol. 25, 123 (2024).
Prasetyo, N. K. & Gardner, P. P. Assessing the robustness of human ncRNA notation at HGNC. Preprint at bioRxiv https://doi.org/10.1101/2024.12.08.627405 (2024).
Skogholt, A. H. et al. Gene expression differences between PAXgene and Tempus blood RNA tubes are highly reproducible between independent samples and biobanks. BMC Res. Notes 10, 136 (2017).
Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 1, 40 (2006).
Grover, S. et al. Visual acuity impairment in patients with retinitis pigmentosa at age 45 years or older. Ophthalmology 106, 1780–1785 (1999).
Bodenbender, J. P. et al. Clinical and genetic findings in a cohort of patients with PRPF31-associated retinal dystrophy. Am. J. Ophthalmol. 267, 213–229 (2024).
Waseem, N. H. et al. Mutations in the gene coding for the pre-mRNA splicing factor, PRPF31, in patients with autosomal dominant retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 48, 1330–1334 (2007).
Maubaret, C. G. et al. Autosomal dominant retinitis pigmentosa with intrafamilial variability and incomplete penetrance in two families carrying mutations in PRPF8. Invest. Ophthalmol. Vis. Sci. 52, 9304–9309 (2011).
Yusuf, I. H. et al. Clinical characterization of retinitis pigmentosa associated with variants in SNRNP200. JAMA Ophthalmol. 137, 1295–1300 (2019).
Berson, E. L. Retinitis pigmentosa. The Friedenwald Lecture. Invest. Ophthalmol. Vis. Sci. 34, 1659–1676 (1993).
De Jonghe, J. et al. Saturation genome editing of RNU4-2 reveals distinct dominant and recessive neurodevelopmental disorders. Preprint at medRxiv https://doi.org/10.1101/2025.04.08.25325442 (2025).
Veltman, J. A. & Brunner, H. G. De novo mutations in human genetic disease. Nat. Rev. Genet. 13, 565–575 (2012).
Petersen-Jones, S. M. et al. Patients and animal models of CNGβ1-deficient retinitis pigmentosa support gene augmentation approach. J. Clin. Invest. 128, 190–206 (2018).
Lee, W. et al. Cis-acting modifiers in the ABCA4 locus contribute to the penetrance of the major disease-causing variant in Stargardt disease. Hum. Mol. Genet. 30, 1293–1304 (2021).
Robson, A. G. et al. ISCEV Standard for full-field clinical electroretinography (2022 update). Doc. Ophthalmol. 144, 165–177 (2022).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Yeo, G. & Burge, C. B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11, 377–394 (2004).
Freeman, P. J., Hart, R. K., Gretton, L. J., Brookes, A. J. & Dalgleish, R. VariantValidator: accurate validation, mapping, and formatting of sequence variation descriptions. Hum. Mutat. 39, 61–68 (2018).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Ellard, S. et al. ACGS Best Practice Guidelines for Variant Classification 2019. ACGS Best Practice Guidelines for Variant Classification 2019 (ACGS, 2019); https://www.acgs.uk.com/media/11285/uk-practice-guidelines-for-variant-classification-2019-v1-0-3.pdf
Biesecker, L. G. et al. ClinGen guidance for use of the PP1/BS4 co-segregation and PP4 phenotype specificity criteria for sequence variant pathogenicity classification. Am. J. Hum. Genet. 111, 24–38 (2024).
Gruber, A. R., Lorenz, R., Bernhart, S. H., Neuböck, R. & Hofacker, I. L. The Vienna RNA websuite. Nucleic Acids Res. 36, W70–W74 (2008).
Johnson, P. Z. & Simon, A. E. RNAcanvas: interactive drawing and exploration of nucleic acid structures. Nucleic Acids Res. 51, W501–W508 (2023).
McHarg, S. et al. Mast cell infiltration of the choroid and protease release are early events in age-related macular degeneration associated with genetic risk at both chromosomes 1q32 and 10q26. Proc. Natl Acad. Sci. USA 119, e2118510119 (2022).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Wang, J. et al. ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration. Nat. Commun. 9, 1364 (2018).
Cherry, T. J. et al. Mapping the cis-regulatory architecture of the human retina reveals noncoding genetic variation in disease. Proc. Natl Acad. Sci. USA 117, 9001–9012 (2020).
Van de Sompele, S. et al. Multi-omics approach dissects cis-regulatory mechanisms underlying North Carolina macular dystrophy, a retinal enhanceropathy. Am. J. Hum. Genet. 109, 2029–2048 (2022).
Jiang, H., Lei, R., Ding, S. W. & Zhu, S. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15, 182 (2014).
Shen, S. et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-seq data. Proc. Natl Acad. Sci. USA 111, E5593–E5601 (2014).
Roithová, A. et al. The Sm-core mediates the retention of partially-assembled spliceosomal snRNPs in Cajal bodies until their full maturation. Nucleic Acids Res. 46, 3774–3790 (2018).
Acknowledgements
We thank all patients and their family members for their help and participation in this study. We thank S. Föhr for technical and administrative support; S. van der Velde-Visser, E. Blokland and M. Jacobs-Camps for sample registration and administration; and T. Rosseel (computational) and S. Van de Sompele (clinical reporting) for their support. The title page was formatted using AuthorArranger, a tool developed at the National Cancer Institute (National Institutes of Health, NIH). C.R. was supported by the Swiss National Science Foundation (SNSF) Grant No. 310030_204285 entitled ‘Genomics of inherited retinal diseases’. S.R. was supported by the Foundation Fighting Blindness Career Development Award (grant no. CD-GE-0621-0809-RAD), Radboudumc Starter grant (no. OZI-23.009) and NWO Aspasia (grant no. 015.021.028). S.R., C.R., E.D.B., M.B., S.B. and D. Stanek were supported by HORIZON-MSCA-2022-DN (grant no. 101120562, ProgRET). E.D.B., S.R. and C.R. were supported by the EJPRD19-234 Solve-RET. This work has been funded by a Foundation Fighting Blindness Program Project Award (grant no. PPA-0622–0841-UCL) (to A.J.H., S.R. and S.E.d.B.). S.R. and F.P.M.C. were supported by the Gelderse Blindenstichting, the Algemene Nederlandse Vereniging ter voorkoming van Blindheid, Oogfonds, Landelijke Stichting voor Blinden en Slechtzienden, Rotterdamse Stichting Blindenbelangen, Stichting Blindenhulp, Stichting tot Verbetering van het Lot der Blinden and Stichting Blinden-Penning. M.Q. was supported by the RetinAward 2021. S.H.T. – Jonas Children’s Vision Care (JCVC) is supported by the National Institute of Health grant nos. U01EY030580, U01EY034590 R24EY028758, R24EY027285, 5P30EY019007, R01EY033770 and R01EY018213, R01EY024698; the Foundation Fighting Blindness grant no. TA-GT-0321-0802-COLU-TRAP; Richard Jaffe; the NYEE Foundation; the Rosenbaum Family Foundation; the Gebroe Family Foundation; the Piyada Phanaphat Fund; the Research to Prevent Blindness (RPB) Physician-Scientist Award; and unrestricted funds from RPB, New York, NY, USA. C.A. was supported by Instituto de Salud Carlos III (ISCIII) of the Ministerio de Ciencia e Innovación and Unión Europea – European Regional Development Fund (FEDER) (grant nos. PI22/00321 and IMP/00009), Centro de Investigación Biomédica en Red Enfermedades Raras (CIBERER, grant no. 06/07/0036), IIS-FJD BioBank (grant no. PT23/00114), the Organización Nacional de Ciegos Españoles (ONCE), the European Regional Development Fund (FEDER) and the University Chair UAM-IIS-FJD of Genomic Medicine. This work was performed by using the data contained in the ‘Programa Infraestructura de Medicina de Precisión asociada a la Ciencia y la Tecnología en Medicina Genómica (IMPaCT-GENóMICA)’, coordinated by the CIBERER and founded by ISCIII. L.F.-C. was supported by Centro de Investigación Biomédica en Red (CIBER). R.A. was supported by the National Eye Institute (NEI) (grant nos. RO1 EY030591, RO1 EY031663, T32 EY026590 and P30 EY22589). C.C.W.K. was supported by the Combined Ophthalmic Research Rotterdam grant no. 8.2.0. S.B. was supported by the Italian Telethon Foundation and by the European Union HORIZON-MSCA-2021-DN-01 (grant no. 101073316, RETORNA). T.S.B. was supported by ZonMw Vidi (grant no. 09150172110002), and acknowledges support from Stichting 12q. G.J.F. and N.C. were supported by Fighting Blindness Ireland (grant nos. FB22FAR, FB16FAR), Fighting Blindness Ireland – Health Research Charities Ireland (grant no. MRCG-2016-14) and the Science Foundation Ireland (grant nos. 16/IA/4452 and 22/FFP-A/10544). J.M.E. was supported by the Macular Society (United Kingdom), the National Institute for Health and Care Research (NIHR) Manchester Biomedical Research Centre (BRC) (grant no. NIHR203308) and the University of Manchester Core Genomics Technology Facility. T.B.-Y. was supported by the Israel Science Foundation (grant no. 331/24). A.C.B.-J. is supported by the University of Melbourne Research Fellowship. K.M.B. was supported by the National Eye Institute (NEI) (grant nos. RO1 EY035717 and P30 EY014104 (MEE core support)), the Iraty Award 2023, the Lions Foundation and RPB (Unrestricted Grant). L.S.S., E.L.C. and S.P.D. were supported by grants from the Foundation Fighting Blindness (grant no. EGI-GE1218-0753-UCSD) and the Brett & Jane Eberle Foundation. E.D.B. and B.P.L. were supported by Ghent University Special Research Fund (grant no. BOF20/GOA/023) and E.D.B. (grant no. 1802220N) and B.P.L. (grant no. 1803816N) are Senior Clinical Investigators of the Research Foundation-Flanders (FWO). N.M. and S.S. are Ph.D. fellows of HORIZON-MSCA-2022-DN ProgRET (grant no. 101120562). R.A. was supported by the Foundation Fighting Blindness. J.L.D. was supported by the UCSF Vision Core shared resource of the NIH/NEI grant no. P30 EY002162, the Foundation Fighting Blindness, an unrestricted grant from RPB and the All May See Foundation. T.I. was supported by research grants from the Japan Agency for Medical Research and Development (AMED) (grant nos. 20ek0109493h0001, 21ek0109493h0002, 22ek0109493h0003, 23ek0109617h0002, 24ek0109617h0003). R.K.K. was supported by The Montreal Children’s Hospital Foundation, The Vision Sciences Research Network (VSRN), The NIH (grant no. R01 EY030499-01, Dr. Lentz), The Canadian Institutes for Health Research (CIHR), Fighting Blindness Canada (FBC) and Fonds de Recherche du Québec - Santé (FRQS). R.K.K. participates in the NAC Attack clinical trial, which is funded by the NIH via grant nos. UG1EY033286, UG1EY033293, UG1EY033286 and UG1EY033292. T.M.L., T.L.M. and J.N.D.R. were supported by Retina Australia (awarded to the Australian Inherited Retinal Disease Registry and DNA Bank). O.A.M. was supported by the Wellcome Trust (grant no. 206619/Z/17/Z). M.P. was supported by the BrightFocus Foundation (grant no. M2024009N). E.A.P. was supported by the National Eye Institute (NEI) (grant no. R01 EY012910). R.R. was supported by Retina South Africa and the South African Medical Research Council (MRC). S.G.S. and E.M.V. were supported by the Italian Ministry of Health (grant no. PNRR-MR1-2023-12377314). D. Stanek was supported by the Project P JAC grant no. CZ.02.01.01/00/22_008/0004575 RNA for therapy, Co-Funded by the European Union. T.B.H. was supported by the European Commission (Recon4IMD – grant no. GAP-101080997) and the Deutsche Forschungsgemeinschaft (German Research Foundation, DFG, grant nos. 418081722 and 433158657 to T.B.H.). P.L. and L.D. were supported by a research grant (no. NW24-06-00083) from the Ministry of Health of the Czech Republic and grant no. UNCE/24/MED/022. V.R.d.J.L.-R. and J.C.Z. were supported by the Velux Stiftung Grant no. 1860. M.A., M.M., C.F.I., J.C.G., A.J.H. and C.T. are supported by Retina UK and Fight for Sight UK (RP Genome Project Grant no. GR586). A.J.H., J.C.G., M. Michaelides, O.A.M., A.R.W., G. Arno and S.L. were supported by the National Institute for Health Research Biomedical Research Centre at Moorfields Eye Hospital and UCL Institute of Ophthalmology. S.L. was funded by a Medical Research Council (MRC) Clinician Scientist Fellowship (grant no. UKRI440). J.M.M. was supported by Instituto de Salud Carlos III (ISCIII) of the Spanish Ministry of Health (grant no. PI22/00213), CIBERER (grant no. 06/07/1030), grant no. CIPROM/2023/26 from the Generalitat Valenciana and IMPaCT-GENOMICA (grant no. IMP/00009) co-funded by ISCIII and FEDER. S.R., C.C.W.K., C.J.F.B. and A.G. were supported by the Dutch Ministry of Education, Culture and Sciences, Gravitation grant no. 024.006.034 Lifelong VISION. W.L. was supported by the National Institute of Health/National Eye Institute (grant no. 1K99EY036930-01). This work is supported by partners of the European Reference Network for Rare Eye Diseases ERN-EYE (Grant Agreement No. 101085439, C.J.F.B., E.D.B., C.B.H., S.K., B.P.L., P.L., L.H.-W., K. Stingl, L.I.v.d.B.). Novartis contributed funding for the preceding RP-LCA smMIPs panel design and subsequent sequencing (to F.P.M.C., S.R. and D.M.P.). Novartis was not involved in the study design; collection, analysis or interpretation of data; the writing of this article; or the decision to submit it for publication. This research was made possible through access to data generated by the 2025 French Genomic Medicine initiative and present in the National Genomic Research Library, which is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The National Genomic Research Library holds data provided by patients and collected by the NHS as part of their care, and data collected as part of their participation in research. The National Genomic Research Library is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also funded research infrastructure.
Author information
Authors and Affiliations
Contributions
M.Q., F.P.M.C., S.R. and C.R. conceived and designed the study. M.Q., K.R., K.K. and M.U. analyzed pedigrees and genotypes. J.B., S.H.T., C.L., A.I.J., K.B.F. and W.L. contributed to the collection of material and clinical data for family M1-A. F.P.M.C., S.E.d.B., D.M.P., R.J.H.-M. and S.R. coordinated the collection of DNA samples. K.R., S.E.d.B., E.G.M.B., N.Z., L.K.H., Z. Corradi, S.S., D.M.P. and R.J.H.-M. performed the majority of the Sanger sequencing screening. K.K., A.B.I.-R. and M.F. carried out the molecular biology experiments. M.Q. and C.R. performed the statistical analyses. M.Q. and J.M.E. retrieved and analyzed large-scale data. K.R. and S.R. coordinated the genotyping of the cohorts. W.L. was responsible for clinical data analysis. Z. Cvackova and D. Stanek conducted all in vitro experiments. F.P.M.C., C.R. and S.R. supervised the project. M.Q., K.R., F.P.M.C., J.M.E., D. Stanek, S.R. and C.R. prepared the original draft of the manuscript. M.Q., K.R., S.R. and C.R. reviewed and edited the manuscript. M.A., A.A., S.A., G. Ansari, G. Arno, G.D.N.A., C.A., R.A., S.B., E.B., T.S.B., M.T.S.B., M.B., T.B.-Y., V.B., D.G.B., P.B., F.B.-K., B.B., C.J.F.B., K.B., D.B.-G., A.C.B.-J., K.M.B., C.B.d.R., E.L.C., G.C., F.C., L.C., N.C., P.C.I., L.C.-S., S.P.D., E.D.B., M.D.B., B.d.l.C., J.N.D.R., J.D.Z., R.D., C.-M.D., L.D., J.L.D., G.J.F., N.F., B.J.F., L.F.-C., J.M.F.S., S.G., A.G., J.C.G., C.G., R.G.-D., K.G., S.G.-J., T.B.H., L.H.-W., A.J.H., T.H., E.H., L.H.H., A.H., J.P.H., C.B.H., M.B.B.I., C.F.I., T.I., B.O.J., K.J., V.K., S. Kamakari, M.K., U.K., C.C.W.K., K.K., R.K.K., S. Kohl, T.K., L.K., T.M.L., R.L., B.P.L., S.L., P.L., I.L., V.R.d.J.L.-R., Q.M., O.A.M., G.M., L. Mansard, M.P.M.-G., N.M., L. Mauring, M. McKibbin, T.L.M., I.M., M. Michaelides, J.M.M., K.M., R.M., Z.Z.N., K.N., M. Ołdak, M. Oorsprong, Y.P., A. Papachristou, A. Percesepe, M.P., E.A.P., E.P., R.R., F.R., F.A.R., G.I.R., L.R., M.R.-H., J.R.-E., A.H.S., A.F.S., A.I.S.-B., A.S.S., R.S., C.M.S., M. Scarpato, H.P.N.S., D. Sharon, S.G.S., F.S., A.B.S., M. Stefaniotou, K. Stefansson, K. Stingl, A.S., P.S., L.S.S., V.S., J.P.S., G.T., A.A.H.J.T., C.T., V.H.T., M.K.T., P.T., V.V., M.V., S. Valeina, E.M.V., C.V., R.V., S. Valleix, J.v.A., L.I.v.d.B., M.V.H., V.J.M.V., A.L.V., A.R.W., L.W., B.W., G.G.Y., K.Y., J.C.Z., R.Z. and T.Z. were instrumental in acquiring funding, clinical data, validation of genetic or molecular data, and reviewing and editing the final manuscript. All authors approved the final content of this work.
Corresponding author
Ethics declarations
Competing interests
The authors affiliated with deCODE genetics/Amgen Inc. (B.O.J., K. Stefansson, P.S.) are employed by the company. The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–7 and Data 1.
Supplementary Table 1 (download XLSX )
Supplementary Tables 1–12.
Source data
Source Data Fig. 4 (download PDF )
Unprocessed blots and gels and PRPF31 antibody validation.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Quinodoz, M., Rodenburg, K., Cvackova, Z. et al. De novo and inherited dominant variants in U4 and U6 snRNA genes cause retinitis pigmentosa. Nat Genet 58, 169–179 (2026). https://doi.org/10.1038/s41588-025-02451-4
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41588-025-02451-4
This article is cited by
-
Cause of vision loss discovered in overlooked genes
Nature (2026)
