
Abstract
Cells fine-tune gene expression in response to cellular stress, a process critical for tumorigenesis. However, mechanisms governing stress-responsive transcription remain incompletely understood. This study shows that the MED1 subunit of the Mediator coactivator complex is acetylated in its intrinsically disordered region (IDR). Under stress, SIRT1 associates with the super elongation complex to deacetylate MED1 in promoter-proximal regions. The deacetylated (or acetylation-defective mutant) MED1 amplified stress-activated cytoprotective genes and rescued stress-suppressed growth-supportive genes in estrogen-receptor-positive breast cancer (ER+ BC) cells. Mechanistically, deacetylated MED1 promotes chromatin incorporation of RNA polymerase II (Pol II) through IDR-mediated interactions. Functionally, ER+ BC cells with deacetylated MED1 exhibit faster growth and enhanced stress resistance in culture and in an orthotopic mouse model. These findings advance our understanding of Pol II regulation under cellular stress and potentially suggest therapeutic strategies targeting oncogenic transcription driven by MED1 and Mediator.

Similar content being viewed by others

Main
Eukaryotic cells continuously face environmental stresses that disrupt homeostasis. To survive, they adjust gene expression, downregulating growth-supportive genes and activating cytoprotective genes1,2. While RNA polymerase II (Pol II) pause–release is known to modulate this response2,3, the full mechanisms remain unclear. Given the role of stress response in cancer progression4, targeting these pathways presents a promising therapeutic strategy.
The 30-subunit Mediator complex, recruited by transcription factors (TFs), assembles preinitiation complexes (PICs) at core promoters to initiate Pol II transcription5,6. The MED1 subunit of Mediator is essential for Pol II transcription in multiple cell types including estrogen-receptor-positive (ER+) breast cancer (BC) cells7,8,9,10,11, where ER interaction with MED1-containing Mediator drives target gene activation9,12,13. In addition, recent findings have suggested broader effects of MED1 in regulating Pol II pause–release and transcription elongation14, as well as Pol II recycling and reinitiation15. SIRT1, a key NAD+-dependent deacetylase for histone and nonhistone proteins, modulates stress responses in aging and disease16. The super elongation complex (SEC) recruits positive transcription elongation factor B (P-TEFb) to release paused Pol II and transcribe stress-induced genes17. Whether SIRT1, SEC, ER and Mediator cooperate in stress responses in ER+ BC remains unknown.
Here, we show that MED1 is acetylated in ER+ BC cells, with six acetylation sites located within its intrinsically disordered region (IDR), the low-complexity fragments of proteins that support condensate formation18. In response to multiple types of stress, SIRT1 is guided by the SEC to deacetylate MED1 IDR. Consequently, deacetylated MED1 (represented by the acetylation-defective 6KR mutant) enhances the expression of MED1-dependent genes under normal growth conditions and amplifies cytoprotective gene induction, while attenuating the silencing of growth-promoting genes during stress. Mechanistically, MED1 IDR deacetylation alters its liquid–liquid phase separation (LLPS) properties and incorporates more RNA Pol II through IDR-mediated interaction with the Pol II CTD (C-terminal domain). The enhanced growth and a higher tolerance to stress conditions of ER+ BC cells with acetylation-defective mutant MED1 in culture, as well as an accelerated tumor growth and remote metastasis with these cells in an animal model, suggest a MED1 deacetylation-driven mechanism in cellular fitness and tumorigenesis. Targeting the SIRT1–SEC–MED1 axis, a proposed integration hub for cellular stress response, might offer an innovative line of therapies for cancers involving stress-induced gene expression reprogramming.
Results
MED1 is essential for oncogene expression in ER+ BC cells
Corroborating the essential role of MED1 in ER target gene transcription9,12,13, CRISPR–Cas9-mediated MED1 depletion in MCF7 and T47D cells (Extended Data Fig. 1a,b) attenuated cell growth (Extended Data Fig. 1c) and colony formation (Extended Data Fig. 1d) without altering ERα levels (Extended Data Fig. 1b). Sequencing of poly(A) RNA (RNA-seq) in MED1-knockout (KO) MCF7 cells (Extended Data Fig. 2a,b) identified differentially expressed genes (DEGs) and revealed downregulation of genes in cell cycle and DNA replication pathways (Extended Data Fig. 2c), underscoring the importance of MED1 for optimal cell growth. As expected, the estrogen signaling pathway is also enriched in downregulated DEGs (Extended Data Fig. 2c). Although MED1 was identified as a critical factor supporting Pol II transcription in ER+ BC cells9,13, many genes were unaffected by MED1 KO, which is potentially a result of residual MED1 expression or compensatory post-transcriptional mechanisms.
Both chromatin immunoprecipitation followed by sequencing (ChIP-seq) and cleavage under targets and release using nuclease followed by sequencing (CUT&RUN-seq) were used to map the genomic occupancy of MED1 in MCF7 cells. The most robust MED1 binding sites (peaks) used for downstream analyses were identified by integrating ChIP-seq and CUT&RUN-seq replicates (Methods). ChIP-seq was also conducted in MCF7 cells to map MED17, a core Mediator scaffolding protein19,20, and transcriptionally engaged RNA Pol II, the Pol II complex with phosphorylated S5 in the CTD of RPB1 (referred to as S5P). There were 1,077 peak regions co-occupied by MED1, MED17 and Pol II (Extended Data Fig. 2d) and these peaks were overlapped with cis-regulatory elements of genes, as defined by ChIP-seq of histone marks21,22 (Methods). Peaks co-occupied by MED1, MED17 and Pol II (MED1+MED17+Pol II+) localized primarily to active promoters, while MED1 peaks without both MED17 and Pol II were enriched more at enhancers (Extended Data Fig. 2e,f), suggesting distinct roles of the partially assembled Mediator PIC in transcription initiation. By examining the association with DEGs of MED1-KO cells, relative to Pol II-free MED1 peaks, peaks co-occupied by MED1 and Pol II (regardless of MED17 presence) were associated more with downregulated genes compared to upregulated genes (Extended Data Fig. 2g), suggesting that the gene-regulatory potential of MED1 is enhanced by Pol II association. All expressed genes were stratified into five tiers according to differential expression in MED1-KO cells and MED1 occupancy at active promoters of genes was the highest in the tier of the strongly downregulated genes (Extended Data Fig. 2h), supporting the notion that MED1-containing Mediator is more involved in the activation rather than suppression of target genes.
MED1 is acetylated at six lysines in the IDR
Many transcriptional activators and coactivators were identified as substrates of acetyltransferases23, which prompts the question of whether Mediator subunits are also acetylated. Among Mediator subunits, we detected acetylation of FLAG-tagged MED1, MED17 and MED23, which showed enhancement by both trichostatin A (TSA; HDAC inhibitor) and nicotinamide (NAM; Sirtuin inhibitor) in HEK293T cells (Extended Data Fig. 3a,b). Given that the MED1 subunit has been reported to have an essential role in transcription in certain cell types including ER+ BC cells9,11,24,25 and, in light of the above results, the present study focused on MED1 acetylation. Two additional biological replicates for FLAG–MED1 acetylation induced by TSA and NAM are shown in Supplementary Fig. 1. The acetylation of FLAG–MED1 was also induced by overexpression of acetyltransferases p300 and CBP but not by a catalytically inactive mutant p300 (ref. 26) (Fig. 1a). The p300-induced acetylation of FLAG–MED1 was attenuated by A485, an HAT (histone acetyltransferase) inhibitor of p300, in contrast to other inhibitors (Fig. 1b). Other acetyltransferases did not induce acetylation of FLAG–MED1 (Fig. 1c), demonstrating the specificity. The endogenous MED1 also showed increased acetylation by p300/CBP (Fig. 1d,e). MED1 acetylation was also examined in an in vitro assay with purified Mediator (Extended Data Fig. 3c). Incubation of Mediator with p300 and acetyl-CoA resulted in acetylation of MED1, while the previously described autoacetylation of p300 (ref. 27) was also evident (Fig. 1f), demonstrating the direct action of p300 on Mediator and MED1.

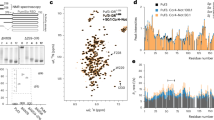
a, Immunoblotting showing enhanced lysine acetylation (referred to as AceK) of FLAG–MED1 in HEK293T cells following overexpression of WT but not D1399Y mutant p300 (human and mouse, with 98% sequence identity to human CBP). IB, immunonlot. b, Immunoblotting showing enhanced acetylation of FLAG–MED1 in HEK293T cells following overexpression of p300 and its suppression by the p300 inhibitor A485 but not C646 or SGC-CBP30. Cells were treated with these inhibitors at the concentrations indicated for 24 h. c, Immunoblotting showing enhanced acetylation of FLAG–MED1 following overexpression of p300 or CBP but not GCN5, PCAF, TIP60 and MOF in HEK293T cells. d, Immunoblotting showing enhanced acetylation of endogenous MED1 in HEK293T cells following overexpression of p300 or CBP. e, Immunoblotting showing enhanced acetylation of endogenous MED1 from HEK293T cells with overexpression of p300 at variable concentrations. f, Immunoblotting showing enhanced acetylation of MED1 (within the purified Mediator complex) by p300 and acetyl-CoA. Effects of variable p300 concentrations (top) and variable incubation times (bottom) were tested. Autoacetylation of p300 was also observed. g, Signal of digested peptides with acetylation plotted as a function of amino acid sequence. Signals from control samples and from p300 overexpression samples with or without A485 were compared and the two acetylation clusters are indicated. h, Analysis of MED1 protein sequence by MobiDB showing a disordered region (amino acids 947–1573). i, Immunoblotting showing enhanced acetylation of WT but not 6KR FLAG–MED1 in HEK293T cells following overexpression of p300. Effects of variable p300 concentrations were tested. For d,e, IgG was used as an IP control. For a–e,i, densitometry analysis was performed on immunoblotting images and the normalized protein levels (relative to the IP target protein, either FLAG-tagged or endogenous MED1) are shown. For a–f,i, immunoblots are representative of three independent experiments.
To identify the acetylated lysine residues in MED1, we used a liquid chromatography–tandem mass spectrometry (LC–MS/MS) procedure to examine immunoprecipitated endogenous MED1 from HEK293T cells (Extended Data Fig. 3d,e). Acetylation levels at K1076, K1113 and K1152 (referred to as ‘island 1’) and at nearby K1300, K1309 and K1311 (referred to as ‘island 2’) were appreciably higher in p300-overexpressing cells than in untreated or A485-treated control cells (Fig. 1g). It is noteworthy that these six lysines are located in a MED1 fragment that is predicted to be an IDR by the VL-XT algorithm28 (Extended Data Fig. 3f) and the mobiDB database29 (Fig. 1h). Given the importance of IDR-mediated LLPS of MED1 in transcriptional regulation30,31, these six lysines became the focus of our study. These six residues are conserved in MED1 from various vertebrate species, indicative of their evolutionary importance (Extended Data Fig. 3g). To specifically determine how these six lysines contribute to MED1 acetylation and function, the acetylation-defective 6KR mutant in which the six lysines were replaced by arginines was tested. Like WT MED1, the vast majority of FLAG-tagged 6KR MED1 was also localized in cell nuclei in MCF7 cells (Extended Data Fig. 3h), suggesting that these six lysines do not control MED1 function by nuclear entry. Ectopic p300 alone induced robust acetylation of WT FLAG–MED1 but showed only a limited effect on 6KR FLAG–MED1 (Fig. 1i; biological replicate in Supplementary Fig. 2). In summary, our results identify six major acetylation sites in the MED1 IDR and further show that these acetylation events are mediated by p300/CBP.
SIRT1–SEC controls MED1 IDR deacetylation upon stress
Sirtuin-class deacetylases regulate histones and numerous nonhistone proteins involved in transcription regulation16. SIRT1 controls the deacetylation of MED1, as FLAG–MED1 showed increased acetylation with Ex527 (SIRT1 inhibitor) treatment in HEK293T cells (Extended Data Fig. 4a). A similar Ex527-induced acetylation of endogenous MED1 occurred in MCF7 cells, prevented by p300 inhibitor A485 (Fig. 2a; biological replicate in Supplementary Fig. 3). MED1 acetylation was also induced by Sirtinol, another SIRT1 inhibitor (Extended Data Fig. 4b). Demonstrating acetylation site specificity, Ex527 induced acetylation of WT but not 6KR FLAG–MED1 in both MCF7 (Fig. 2b) and T47D (Fig. 2c) cells. Endogenous MED1 was replaced with ectopic WT or 6KR MED1 in the tested T47D cells, as described below.

a, Immunoblotting showing enhanced acetylation of endogenous MED1 in MCF7 cells treated with Ex527, where the effect was attenuated by cotreatment with A485 (6 μM, 24 h). b, Immunoblotting showing enhanced acetylation of WT but not 6KR FLAG–MED1 in MCF7 cells treated with Ex527 (100 μM, 24 h). A control sample (ctrl IP) was included, in which the lysate of normal MCF7 cells was immunoprecipitated with anti-FLAG. c, Immunoblotting showing enhanced acetylation of WT but not 6KR MED1 in T47D cells where endogenous MED1 was depleted by CRISPR–Cas9 (referred to as WT/KO and 6KR/KO) treated with Ex527 (25 and 100 μM for 4 h). Unmodified T47D cells with IgG used as the IP control. d, Immunoblotting showing reduced acetylation of WT FLAG–MED1 in MCF7 cells treated with various concentrations of DFOM (24 h). HIF1α was detected to demonstrate the induction of hypoxic response. e, Immunoblotting showing reduced acetylation of WT FLAG–MED1 in MCF7 cells treated with DFOM (100 μM, 24 h), while cotreatment of Ex527 (50 μM) prevented this effect. f, Immunoblotting showing reduced acetylation of WT FLAG–MED1 in MCF7 cells under 42 °C heat shock for various durations. g, Immunoblotting showing reduced acetylation of WT FLAG–MED1 in MCF7 cells treated with various concentrations of H2O2 (4 h). h, Immunoblotting showing reduced acetylation of endogenous MED1 in MCF7 cells treated with H2O2 or DFOM (either with 200 μM, 24 h), while cotreatment of Ex527 (50 μM, 24 h) prevented this effect. i, Immunoblotting showing elevated SIRT1 detected for association with endogenous AFF4 in MCF7 cells treated with DFOM (16 h), while equivalent association of cyclin T1 with AFF4 with or without DFOM treatment was detected. j, AlphaFold 3 prediction of structure of the SEC associated with SIRT1 (green). The overall structure (top) is shown in cartoon form and the close-up views of the SIRT1–SEC interface (bottom left and bottom right) are shown in cartoon form and surface representation. The extended fraction of SIRT1 inserting into the pocket formed by AFF4 (dark blue) and CDK9 (light blue) is indicated with a dashed circle. For a,h, IgG was used as an IP control. For a–i, densitometry analysis was performed on immunoblotting images and the normalized protein levels (relative to the IP target protein, FLAG tag of MED1 or endogenous MED1 or AFF4) are shown; these immunoblots are representative of three independent experiments.
Cells reprogram gene expression to increase stress fitness1,3. The acetylation of FLAG–MED1 was reduced in MCF7 cells exposed to deferoxamine mesylate (DFOM)32 and cobalt33, two typical hypoxia-mimicking agents that stabilize HIF1α (Fig. 2d and Extended Data Fig. 4c). This deacetylation of FLAG–MED1 was SIRT1 dependent, as Ex527 cotreatment reversed it (Fig. 2e). Similar FLAG–MED1 deacetylation occurred during thermal (heat shock) or oxidative (H2O2 treatment) stress in MCF7 cells (Fig. 2f,g), which was also prevented by Ex527 (Extended Data Fig. 4d). Endogenous MED1 showed parallel deacetylation with DFOM or H2O2 treatment, reversible by Ex527 (Fig. 2h; biological replicate in Supplementary Fig. 4). Therefore, diverse stress conditions may collectively regulate gene expression through MED1 deacetylation.
The SEC, through CDK9 (P-TEFb) recruitment17, enables stress-responsive gene regulation34, prompting us to investigate its role in stress-induced MED1 deacetylation by SIRT1. The coimmunoprecipitation (co-IP) assay demonstrated dramatically enhanced association of SIRT1 with AFF4, a core component of the SEC, in DFOM-treated MCF7 (Fig. 2i; biological replicate in Supplementary Fig. 5) and T47D (Extended Data Fig. 4e) cells relative to untreated control cells. Like AFF4, CDK9 also pulled down much more SIRT1 in stressed MCF7 cells compared to untreated cells (Extended Data Fig. 4f). Under thermal stress, SIRT1–AFF4 association was also dramatically induced in MCF7 cells (Extended Data Fig. 4g). Notably, cyclin T1–CDK9–AFF4 interactions remained unchanged under stress (Fig. 2i and Extended Data Fig. 4f,g), indicating that stress specifically enhances SIRT1–SEC binding without altering SEC assembly. To complement the co-IP assays, we performed MS to identify AFF4-associated proteins in DFOM-treated and control MCF7 cells. This test confirmed constitutive interaction of the other SEC components with AFF4, while SIRT1 showed stress-dependent interaction (Extended Data Fig. 4h). The other chromatin factors that also displayed stress-induced binding (Extended Data Fig. 4h) suggests the existence of a dynamic protein interaction network connecting SIRT1 and the SEC under stress conditions. Although the contribution of additional factors to SIRT1–SEC association cannot be excluded, AlphaFold 3 (ref. 35) predicts direct SIRT1–SEC binding wherein a segment near the SIRT1 N terminus inserts into a pocket formed by AFF4 and CDK9 (Fig. 2j). CUT&RUN-seq revealed increased occupancy of SIRT1 and AFF4 in promoter-proximal regions of representative genes in DFOM-treated relative to control MCF7 cells (Extended Data Fig. 4i), demonstrating the chromatin context of SIRT1–SEC association. Together, these findings demonstrate that the stress-induced SIRT1–SEC chromatin association enables MED1 IDR deacetylation.
Acetylation-defective mutant MED1 modifies gene expression
As the six conserved lysines in the MED1 IDR were identified as acetylation substrates, we next sought to determine the functional importance of MED1 acetylation and deacetylation in ER+ BC cells. Using CRISPR–Cas9, endogenous MED1 was depleted in pools of MCF7 or T47D cells that ectopically expressed CRISPR-resistant wild-type (WT) or acetylation site mutant MED1 (Extended Data Fig. 5a). In addition to acetylation-defective 6KR mutant, a 6KQ mutant in which the lysines were replaced by polar and neutral glutamines to mimic acetylation was also studied. In these MED1 addback cells (referred to as WT/KO, 6KR/KO and 6KQ/KO, respectively), the ectopic HA-tagged WT and mutant MED1 proteins replaced the endogenous MED1 with similar levels of expression and ERα expression showed no notable alteration (Extended Data Fig. 5b,c).
Poly(A) RNA-seq was then conducted for WT/KO and 6KR/KO MCF7 cells in steady-state culture (Extended Data Fig. 5d). Gene set enrichment analysis (GSEA) comparing the 6KR/KO versus WT/KO gene rank list with KEGG (Kyoto Encyclopedia of Genes and Genomes) and GOBP (Gene Ontology biological processes) gene lists showed an enrichment of mitochondrial respiration-related genes among the DEGs in 6KR/KO cells, indicating a mechanism by which 6KR MED1 alters the growth of MCF7 cells (Extended Data Fig. 5e). GSEA also showed that the upregulated genes in 6KR/KO versus WT/KO cells were enriched in genes downregulated in MED1-KO cells versus MED1 WT cells, while the downregulated genes in 6KR/KO versus WT/KO cells were enriched in genes upregulated in MED1-KO versus MED1 WT cells (Fig. 3a). Furthermore, GSEA using DEGs in MED1-KO cells showed consistent results when compared to the rank list of 6KR/KO versus WT/KO cells (Extended Data Fig. 5f). These results lead us to envision that the acetylation-defective 6KR mutant MED1 (and, by extrapolation, natural deacetylated MED1) acts as a more active form of MED1 for gene activation in the steady state.

a, GSEA showing that the upregulated (left) and downregulated (right) genes in 6KR/KO versus WT/KO cells were enriched in the set of downregulated and upregulated genes of MED1-KO (versus control) cells, respectively. The cutoff for defining DEGs was fold change (FC) > 2 and adjusted P < 0.05. NES (normalized enrichment score) values are shown. b,c, Detection by RT–qPCR showing stronger induction of representative stress-induced genes in 6KR/KO versus WT/KO MCF7 cells. b, Genes induced by DFOM. c, Genes induced 42 °C heat shock for 60 or 120 min. For each of WT/KO or 6KR/KO cells, expression of the gene at each condition was normalized by its expression at control condition in this cell type. For b,c, n = 4 (biological replicates) for each group. Bars report the mean ± s.e.m. If <0.05, the P values of a two-sided unpaired t-test comparing cell types are shown. d–i, Poly(A) RNA-seq for MCF7 cells under H2O2 stress, comparing 6KR/KO versus WT/KO or 6KQ/KO versus WT/KO. Three biological replicates were detected for each cell type in each condition. d, Volcano plots showing the log2 FC versus Wald statistics (two-sided) after H2O2 treatment for each detected gene in tests of WT/KO, 6KR/KO and 6KQ/KO cells, with the cutoff for defining DEGs (FC > 2, P < 0.05) indicated. e, A clustered heat map of z scores of genes showing differential expression by H2O2 treatment (adjusted P < 0.05) in any of the WT/KO, 6KR/KO or 6KQ/KO cells. A division of six groups for these genes is shown. Colors in the scale represent relative expression levels: blue (z ≤ −3), white (z = 0), and red (Z ≥ 3), with a linear gradient for intermediate values. f, A box plot showing the FC by H2O2 treatment for group B genes (n = 1,601) in WT/KO, 6KR/KO and 6KQ/KO cells. g, FC values (H2O2 treatment versus control condition in each cell type) of representative stress-protective genes in group B. If <0.05, the P values from a two-sided unpaired t-test are shown. h, A box plot showing FC by H2O2 treatment for group F genes (n = 8481) in WT/KO, 6KR/KO and 6KQ/KO cells. i, GO analyses with adjusted P values for GOBP (left) and KEGG (right) gene lists for group F genes. For f,h, boxes span the 25th–75th percentiles, the center line indicates the median, whiskers extend to 1.5× the interquartile range (IQR) and points beyond represent outliers. P values of a two-sided unpaired t-test comparing cell types are shown.
In addition to the steady state, we investigated the effect of MED1 acetylation site mutation on stress-responsive gene expression. Most representative stress-activated genes are cytoprotective; the antiapoptotic gene NDRG1 and proangiogenic genes ANGPTL4 and ADM are induced by hypoxia, while the protein chaperone genes HSPA1A, HSPA1B and BAG3 induced by heat shock. These stress-activated genes collectively exhibited augmented induction in 6KR/KO versus WT/KO cells (Fig. 3b,c and Extended Data Fig. 5g).
To systematically investigate the effects of MED1 mutations on stress-responsive genes, poly(A) RNA-seq was conducted in WT/KO, 6KR/KO and 6KQ/KO MCF7 cells with oxidative stress induced by H2O2. The 6KR/KO and 6KQ/KO cells were tested separately, with WT/KO cells examined as control in each test. Spike-in of Drosophila RNA was included to ensure accurate normalization of reads across conditions (Fig. 3d). The DEGs showing H2O2-altered expression in any of WT/KO, 6KR/KO or 6KQ/KO cells were clustered into six groups (Fig. 3e, assigned as groups A to F). Among these groups, group B genes were predominantly upregulated by H2O2 in WT/KO cells, with this induction further amplified in 6KR/KO cells but diminished in 6KQ/KO cells (Fig. 3f). Although the limited number of genes precluded identification of a discrete pathway, we identified several key cytoprotective genes, such as EGF (a critical growth factor), TNFRSF1A and TNFRSF11B (TNF pathway), showing enhanced induction in 6KR/KO and attenuated induction in 6KQ/KO (Fig. 3g).
The largest group, group F, comprised genes that were generally downregulated by H2O2 in WT/KO cells and this downregulation was attenuated in 6KR/KO but exacerbated in 6KQ/KO cells (Fig. 3h), underscoring the role of MED1 acetylation in gene suppression. Pathway analysis revealed that this group of genes were enriched in multiple cell-growth-supportive pathways, such as RNA-related processes, DNA replication and repair, cell cycle and metabolism (Fig. 3i). Taken together, the stress-associated deacetylation of MED1 (represented by the 6KR mutant) both mitigates the suppression of essential growth-supportive genes and augments the induction of cytoprotective genes in stressed cells, which could protect the cells from growth retardation and death.
Acetylation-defective MED1 promotes Pol II association
To investigate the mechanism by which acetylation-defective 6KR MED1 regulates gene expression under steady-state and stress conditions, we examined the genomic occupancies of MED1, MED17, RNA Pol II (RPB1) and ER in WT/KO and 6KR/KO MCF7 cells. These two types of cells showed comparable levels of ectopic (HA-tagged) WT and 6KR MED1, MED17, RPB1 and ER, as well as efficient depletion of endogenous MED1 (Supplementary Fig. 6). We performed ChIP-seq to test MED1, MED17 and transcriptionally engaged Pol II (RPB1 S5P) and CUT&RUN-seq to test the HA tag of MED1 and elongating Pol II (RPB1 with CTD S2 phosphorylated, referred to as S2P). Most of these tests were conducted with biological replicates, which showed high correlation for each factor in each cell type (Supplementary Fig. 7). Sites occupied by MED1, MED17 and Pol II were predefined according to the previously described genomic mapping data from untreated MCF7 cells (Methods) and used in the subsequent analyses. The occupancy of 6KR MED1 (detected by either MED1 or HA) showed little or no change at MED1+ peaks compared to WT MED1 (Fig. 4a). MED17 and ER occupancies showed little or no change at these peaks as well (Fig. 4a). In contrast, an appreciable enhanced Pol II occupancy at the MED1+MED17+Pol II+ peaks was observed in 6KR/KO cells relative to WT/KO cells (Fig. 4a). By restricting the analyses to ER-occupied and Mediator-occupied active enhancers (predefined as ER+MED1+MED17+H3K27ac+H3K4me+H3K4me3− peaks) (Fig. 4b) or all active promoters (monitored by H3K4me3+ within ±3 kb of the transcription start site (TSS)) (Fig. 4c,d), we found analogous results for occupancies of these factors.

a–c, Density metaplots for predefined MED1 peaks (further classified according to their co-occupancy with MED17 and Pol II) (a), ER+ active enhancers (b) and active promoters (c), showing the occupancy of MED1 (by detecting MED1 or its HA tag), MED17, Pol II (RPB1 S5P) and ERα (only in b), with a comparison between 6KR/KO and WT/KO MCF7 cells. d, Heat maps of predefined active promoters showing occupancy of (ectopic HA-tagged) MED1, MED17 and Pol II (RPB1 S5P) with comparisons between 6KR/KO and WT/KO MCF7 cells. e–g, Metagene density plot (e) and heat map (f) for genes associated with predefined active promoters showing elevated occupancy of transcriptionally engaged Pol II (RPB1 S5P) across genic regions in 6KR/KO versus WT/KO MCF7 cells. The pausing index for each gene associated with these promoters was calculated accordingly and is shown with a box plot (g). The boxes span the 25th–75th percentiles, the center line indicates the median, whiskers extend to 1.5× the IQR and points beyond represent outliers. The P value of a two-sided unpaired t-test comparing cell types is shown (n = 11,395 for each box). h,i, Metagene density plot (h) and heat map (i) for genes associated with predefined active promoters showing comparable occupancy of elongating Pol II (by detecting phosphorylation of RPB1 CTD S2 across genic regions in 6KR/KO versus WT/KO MCF7 cells. j, IGV snapshots of regulatory regions of two representative genes, TFF1 and GREB1, with occupancy of HA (MED1), MED17 and Pol II (RPB1 S5P) shown for WT/KO and 6KR/KO cells. Epigenetic marks to identify enhancers and promoters are also shown.
The metagene analysis for genes associated with the active promoters (Fig. 4e,f) showed elevated Pol II (RPB1 S5P) density across gene bodies. The pausing index was calculated for each active promoter-associated gene according to RPB1 S5P density. 6KR/KO cells exhibited a globally increased pausing index compared to WT/KO cells (Fig. 4g). In contrast, the density of elongating Pol II (RPB1 S2P) did not show observable difference comparing WT/KO and 6KR/KO (Fig. 4h,i). These results suggest that, in the absence of additional stress signals, the excess Pol II recruited by hypoacetylated MED1 accumulates in promoter-proximal regions instead of proceeding into productive elongation.
To illustrate the findings above, occupancies of the tested factors in regulatory regions of representative ER target genes (Fig. 4j) and stress-activated genes (Extended Data Fig. 6a) are shown in IGV (Integrative Genomic Viewer) genomic snapshots. To determine whether the differential Pol II association resulted from altered Mediator assembly, we performed a co-IP assay for Mediator subunits in WT/KO, 6KR/KO and 6KQ/KO cells. Similar levels of major subunits, including MED1, were detected in the MED17 immunoprecipitates (Extended Data Fig. 6b; biological replicate in Supplementary Fig. 8), indicating that the mutation of the acetylation sites does not meaningfully impact Mediator complex formation. The observed (mild) decrease in MED17 occupancy relative to MED1 might result from IDR-mediated weak interaction.
Motivated by the differential genomic occupancies of 6KR and WT MED1, we next explored the genomic distribution of acetylated MED1 (relative to total MED1) in MCF7 cells. Limited by the availability of acetylation site-specific antibodies, we used a sequential ChIP assay as an alternative. Thus, anti-FLAG IP from MCF7 cells expressing FLAG–MED1 were eluted with FLAG peptide and followed with a second IP with pan-anti-acetylated lysine antibodies (Extended Data Fig. 6c). The acetylation level of MED1 (and/or its binding proteins) at each of the preidentified ChIP-seq MED1 peaks (from untreated MCF7 cells) can be evaluated by comparing the read densities from sequencing of the first and second ChIP samples. Accordingly, we stratified the MED1 peaks by ranking the read density ratios of the second ChIP to first ChIP and taking the upper 75th, middle 50th and lower 25th percentiles (referred to as high Ac, mid Ac and low Ac, respectively) (Extended Data Fig. 6d,e). The high-Ac group was accompanied by higher MED17 and lower Pol II (RPB1 S5P) occupancy (Extended Data Fig. 6d–f). The p300 occupancy was lower for the low-Ac group but the difference was marginal (Extended Data Fig. 6e,f), demonstrating that, in addition to p300, SIRT1 (or another deacetylase) may also have a critical role for levels of acetylation of MED1 and associated factors. Low-Ac MED1 peaks exhibited less MED17 co-occupancy (total MED17+Pol II+ and MED17+Pol II− peaks) (Extended Data Fig. 6g) and these peaks were associated with less active enhancers (Extended Data Fig. 6h).
These results suggest that MED1 deacetylation enhances Pol II occupancy at promoters, thereby enabling the upregulation of steady-state MED1-dependent genes, amplifying stress-activated genes and restricting stress-downregulated genes in ER+ BC cells.
Acetylation controls physicochemical properties of MED1 IDR
IDRs are low-complexity fragments of proteins that support dynamic protein interactions (either homotypic or heterotypic)18 and are overrepresented in TFs and cofactors6. MED1 IDR promotes condensation of MED1 at superenhancers of genes and corresponding MED1 condensates incorporate Pol II (ref. 30) and elongation factors CTR9 and SPT6 (ref. 31) to facilitate transcription. As the six major MED1 acetylation sites are located in the MED1 IDR, we anticipated that acetylation would change the physicochemical properties of the MED1 IDR and, thus, impact MED1 condensation. To address this issue, MED1 IDR segments (residues 963–1320 as predicted by VL-XT; Extended Data Fig. 3f) in either 6KR or 6KQ mutants fused with EGFP were purified from Escherichia coli (Extended Data Fig. 7a). Both IDRs formed droplets in a crowding agent-free buffer but 6KQ exhibited higher condensate partitioning than 6KR (Fig. 5a), supporting that 6KQ IDR is more prone to condensate formation than 6KR IDR. This was further supported by the increased absorbance of 6KQ relative to 6KR IDR across varying crowding agents and protein concentrations in a turbidity assay (Fig. 5b) and the measurement of saturation concentration36, which was lower for 6KQ than 6KR IDR (Extended Data Fig. 7b). Fluorescence recovery after photobleaching (FRAP) showed faster recovery of the 6KQ IDR relative to the 6KR IDR in the MED1 IDR droplets (Fig. 5c,d), further supporting the notion that both the acetylation-mimicking 6KQ IDR and the acetylation-defective 6KR IDR undergo condensation but with the 6KQ IDR exhibiting more dynamic and fluid features.

a, Droplet formation of EGFP-fused MED1 IDR showing enhanced partitioning of 6KQ versus 6KR MED1 IDR. Left, representative images are shown. Scale bar, 10 μm. Right, the partition coefficient of MED1 IDR (inside versus outside droplets) is plotted. Bars report the mean ± s.e.m. P values (two-sided unpaired t-test) and the numbers of droplets analyzed (across three biological replicates) are indicated. b, Differed turbidity (absorbance at 350 nm) for 6KQ versus 6KR MED1 IDRs. PEG and protein concentrations are indicated (n = 7 for each condition; across three biological replicates). Bars indicate the mean ± s.e.m. If <0.05, the P values of a two-sided unpaired t-test are shown. c,d, Faster recovery of 6KQ relative to 6KR MED1 IDR droplets observed by FRAP. Images for representative droplets (c) are shown. Scale bar, 10 μm. In recovery curves (d), data points report the mean ± s.e.m. 6KR and 6KQ IDRs were tested for 10 and 12 droplets (across three biological replicates), respectively. P values (two-sided unpaired t-test) for each time point are shown. e,f, Increased incorporation of a mCherry-fused Pol II CTD heptad fragment into droplets formed by EGFP-fused 6KR (e) or nonacetylated WT (f) MED1 IDR, compared to the droplets formed by 6KQ (e) or acetylated WT (f) MED1 IDR. Left, representative images are shown. Scale bar, 10 μm. Right, the partition coefficient of the mCherry–Pol II fragment (inside versus outside MED1 IDR droplets) is plotted. P values of a two-sided unpaired t-test are shown. The parentheses show the numbers of MED1 IDR droplets analyzed across three biological replicates. g, Increased RNA Pol II enrichment was observed in condensates formed by the 6KR MED1 IDR compared to 6KQ in cells. Left, schematic of LacO array in cells where LacI fused with MED1 IDR forms condensates. Middle, immunofluorescence staining of Pol II in LacO array reporter cells with ectopic expression of GFP–LacI (control or fused with 6KR/Q MED1 IDR). Representative images are shown. Scale bar, 10 μm. Right, the partition coefficient of Pol II (intensity inside versus outside LacI puncta) is plotted for control and 6KR/Q MED1 IDR-fused GFP–LacI. Numbers of cells analyzed in each group (across three biological replicates) are shown in parentheses. P values of a two-sided unpaired t-test are shown. h,i, Metaplots (h) and heat maps (i) showing Pol II occupancy at high-Ac or low-Ac MED1 peaks (taking top and bottom 10%, respectively, from sequential ChIP-seq experiment) with or without 1,6-hexanediol (Hexn) treatment in MCF7 cells. j, ChIP–qPCR detection of Pol II occupancy at TSS of TFF1 gene showing increasing in 6KR/KO versus WT/KO MCF7 cells and reduction in both cells with 1,6-hexanediol treatment (n = 6 biological replicates for Pol II and n = 2 biological replicates for nonspecific IgG control in each group). Bars report the mean ± s.e.m. P values of a two-sided unpaired t-test are shown.
To confirm that the difference between 6KR and 6KQ MED1 IDRs reflected the effect of natural WT MED1 IDR acetylation, WT MED1 IDR was purified from E. coli and was subjected to acetylation by mouse CBP HAT domain (Extended Data Fig. 7c). The CBP-acetylated WT MED1 IDR formed droplets with increased condensate partitioning (Extended Data Fig. 7d) and was saturated at a lower concentration (Extended Data Fig. 7e) compared to untreated WT MED1 IDR. In the FRAP assay, CBP-acetylated WT MED1 IDR exhibited a slower recovery rate relative to untreated WT MED1 IDR in the droplets (Extended Data Fig. 7f,g). These results paralleled the differential phase behavior between 6KR and 6KQ MED1 IDRs and again pointed to the effect of acetylation on physicochemical properties that support condensate formation.
Pol II and MED1 cocondensates at active transcription sites have been reported37. To gain further insights into possible connections to MED1 acetylation status, we purified a human Pol II CTD heptad fragment38 fused to mCherry that was expressed in E. coli (Extended Data Fig. 7h) and imaged its cocondensation with EGFP–MED1 IDR. More Pol II CTD heptads were incorporated into 6KR relative to 6KQ MED1 IDR droplets, indicated by a higher partition coefficient (Fig. 5e). Similarly, an increased incorporation of Pol II CTD heptads within droplets formed by untreated WT MED1 IDR relative to CBP-acetylated WT MED1 IDR was also detected (Fig. 5f). As the supraphysiological concentration of MED1 IDR used in our in vitro assays (>90 μM) must be taken into account, we also monitored the incorporation of Pol II into MED1 condensates in vivo. We used a U2OS reporter cell with a genomically integrated tandem LacO array39, where MED1 IDR—fused to LacI and GFP—forms condensates on the LacO array31 (Fig. 5g, left). As detected by immunofluorescence, RNA Pol II (RPB1 S5P) was incorporated into the puncta of GFP–MED1 IDR–LacI but not the puncta of control GFP–LacI. Furthermore, the puncta of 6KR MED1 IDR incorporated more Pol II than the puncta of 6KQ MED1 IDR, as indicated by the increased partition coefficient (Fig. 5g, middle and right).
Motivated by the above results, we next investigated the IDR-mediated interactions involved in Pol II–Mediator assembly. Mediator complex in association with Pol II was purified from HeLa-S cells (Methods) and the bead-bound complex was washed with buffers containing increasing concentrations of salt (Extended Data Fig. 7i) or 1,6-hexanediol (taking 2,5-hexanediol as a control treatment) (Extended Data Fig. 7j). High salt disrupts electrostatic interactions, whereas 1,6-hexanediol weakens hydrophobic interactions. Immunoblot monitoring of Pol II and Mediator subunits remaining on beads revealed that Pol II (RPB1), along with MED1 and MED25, is more sensitive than other select Mediator subunits (for example, MED12) to dissociation by high salt (Extended Data Fig. 7i), whereas Pol II (RPB1), along with MED23 and MED25 but not MED1, is more sensitive to 1,6-hexanediol (Extended Data Fig. 7j). Therefore, Pol II association with Mediator involves contributions from both electrostatic and hydrophobic interactions.
These in vitro studies prompted us to examine the dependency of Pol II recruitment on IDR-mediated interactions in cells. To this end, ChIP-seq for Pol II (RPB1 S5P) was conducted in MCF7 cells treated briefly with 1,6-hexanediol. Comparing MED1 peaks with high versus low acetylation, defined as the top 10% and bottom 10% of peaks according to the results of sequential ChIP-seq, respectively (Extended Data Fig. 6d,e), revealed that Pol II occupancy was reduced by 1,6-hexanediol at low-Ac but not high-Ac peaks (Fig. 5h,i). A ChIP–qPCR result for the TSS of a representative gene, TFF1, confirmed increased Pol II occupancy in 6KR/KO relative to WT/KO MCF7 cells and further showed that 1,6-hexanediol reduced Pol II occupancy to equally lower levels in these two cells (Fig. 5j). These results support the view that MED1 with lower acetylation promotes Pol II recruitment through IDR-mediated interaction, an effect similarly observed for Pol II occupancy elevation in 6KR MED1 cells.
Acetylation-defective MED1 promotes ER+ BC
To determine the tumorigenic role of MED1 deacetylation in ER+ BC, we examined the effects of acetylation-defective 6KR MED1 both in cell culture and in a mouse model. In culture, 6KR/KO MCF7 cells grew faster (Fig. 6a) and formed more colonies (Fig. 6b) than WT/KO MCF7 cells. Increased colony formation was also detected in 6KR/KO relative to WT/KO T47D cells (Extended Data Fig. 8a). The 6KR/KO MCF7 cells exhibited a greater tolerance to DFOM-mimicked hypoxia (Fig. 6c) and H2O2-induced or paraquat-induced oxidative stress (Fig. 6d,e) compared to WT/KO MCF7 cells. The increased tolerance to these stress conditions for 6KR/KO versus WT/KO T47D cells was also evident (Extended Data Fig. 8b,c). Notably, 6KR/KO cells survived high-dose H2O2 but showed no added growth at moderate doses (Fig. 6d), implying that 6KR MED1 supports cell survival specifically under stress conditions rather than generally promoting cell growth. Cell migration in culture was also examined, which detected appreciably enhanced migration of 6KR/KO compared to WT/KO MCF7 cells (Extended Data Fig. 8d,e).

a, Elevated growth rate of 6KR/KO versus WT/KO MCF7 cells monitored by CellTiter Blue assay (n = 4 biological replicates). Data points report the mean ± s.e.m. The P value from a one-way analysis of variance (ANOVA) comparing two curves is shown. b, More colonies formed by 6KR/KO versus WT/KO MCF7 cells stained with crystal violet following 10 days of growth after initial seeding. c–e, Declined viability of 6KR/KO and WT/KO MCF7 cells treated with various concentrations of DFOM (24 h) (c), H2O2 (24 h) (d) or paraquat (48 h) (e) monitored by MTT assays showing tolerance of 6KR/KO (n = 4 biological replicates in each group). Bars report the mean ± s.e.m. If <0.05, the P values of a two-sided unpaired t-test comparing cell types are shown. f,g, Enhanced growth of tumors formed from 6KR/KO versus WT/KO MCF7 cells implanted into inguinal mammary fat pad in NSG mice. Images of the mice with orthotopic tumors (18 and 16 tumors for WT/KO and 6KR/KO cells, respectively) (f) and curves of tumor volume (g) are shown. Data points report the mean ± s.e.m. The P value from a one-way ANOVA comparing two groups before 95 days after implantation is indicated. h, Poly(A) RNA-seq for 6KR/KO and WT/KO MCF7 tumors developed in NSG mice. Three tumor samples (biological replicates) were examined for each type of tumor. A volcano plot is presented of the adjusted P value (two-sided) and FC for each detected gene, with the cutoff for defining DEGs (FC > 2, adjusted P < 0.05) indicated. i–k, GSEA showing that the upregulated genes in hypoxia-exposed (data from GSE153557) (i), paraquat-treated (data from GSE241672) (j) or H2O2-treated (data from study described above) (k) versus control MCF7 cells were enriched in the set of upregulated genes of 6KR/KO versus WT/KO MCF7 tumors. Adjusted P values are noted in addition to the NES. l,m, Increased lung metastasis of 6KR/KO versus WT/KO MCF7 cells in NSG mice with implantation of these cells originally into inguinal mammary fat pad. l, Representative hematoxylin and eosin staining images of lung sections within which metastatic foci are indicated. Scale bar, 100 μm. m, Quantification of metastatic foci. Data are the mean ± s.e.m. and the P value of a Wilcoxon rank-sum test (two-sided) is shown.
To assess the tumorigenic effect of 6KR MED1 in vivo, WT/KO and 6KR/KO MCF7 cells were injected into the inguinal mammary fat pads in NSG mice. The development of primary xenograft tumors was much faster for 6KR/KO cells relative to WT/KO cells (Fig. 6f,g), although WT/KO cells eventually formed tumors with similar size (Fig. 6g and Extended Data Fig. 9a). Immunoblotting confirmed equivalent MED1 expression in size-matched tumors (Extended Data Fig. 9b), ruling out clonal selection as the cause of enhanced 6KR/KO tumor growth. The 6KR/KO tumors also exhibited elevated expression of NRF2 and NDRG1, demonstrating a stronger response to oxidative stress and hypoxia (Extended Data Fig. 9c).
Poly(A) RNA-seq for size-matched 6KR/KO versus WT/KO MCF7 tumors (Fig. 6h) implied the contribution of pathways related to cell cycle, ribosome, RNA splicing and ribosomal RNA in 6KR MED1-promoted tumorigenesis in mice (Extended Data Fig. 9d). To determine the contribution of stress-induced gene reprograming, we took advantage of public RNA-seq results from the Gene Expression Omnibus for MCF7 cells in stress response. Upregulated genes associated with either hypoxia (Fig. 6i) or paraquat-induced oxidative stress (Fig. 6j) were both enriched in upregulated genes of 6KR/KO versus WT/KO tumors as revealed by GSEA. Similarly, H2O2-upregulated genes (Fig. 3d) were also enriched in upregulated genes of 6KR/KO versus WT/KO tumors (Fig. 6k). RNA-seq confirmed elevated expression for representative stress-activated genes in 6KR/KO tumors (Extended Data Fig. 9e). These results corroborate an important role for MED1 deacetylation in stress-responsive gene expression in tumorigenesis.
ER+ BC cells exhibit metastasis to lymph nodes or other remote organs40,41. Thus, metastatic nodules in lungs from our NSG model mice could be identified as aggregations of cells with a diminished cytosol and enlarged nucleus, which frequently showed signs of division and angular shapes (Fig. 6l). A higher number of such nodules were observed in lungs from mice bearing 6KR/KO tumors compared to mice bearing size-matched WT/KO tumors (Fig. 6l,m).
These findings on 6KR MED1 provide support for a role for MED1 deacetylation, which is regulated by SEC-associated SIRT1 upon stress, not only in facilitating MED1/Mediator-dependent transcription in steady-state and stress conditions but also as a determinant for growth, metastasis and stress responses in ER+ BC.
Discussion
Eukaryotic cells including cancer cells rely on rapid transcriptional reprogramming to survive cellular stress1, yet how stress signals are transmitted to the transcription machinery remains incompletely understood. While Pol II pause–release is known to modulate stress-responsive gene expression42, a critical gap persists in identifying the mechanisms that govern Pol II recruitment to promoters under stress conditions. Our work resolves this by uncovering MED1 deacetylation as a molecular switch that bridges stress sensing with Pol II recruitment.
Upon identifying MED1 acetylation sites, we examined the effects of the acetylation-defective mutant (6KR) and the acetylation-mimicking mutant (6KQ) on stress-responsive gene expression. While the difference in steady-state gene expression between 6KR/KO and WT/KO cells was modest, the impact of MED1 acetylation mutants became more pronounced under stress. Specifically, 6KR/KO cells showed enhanced induction of cytoprotective genes and reduced suppression of growth-supportive genes compared to WT/KO cells, whereas 6KQ/KO cells exhibited the opposite effect. These findings suggest that, like the 6KR mutant, naturally deacetylated MED1 may fine-tune stress-responsive gene expression. Notably, we also observed elevated expression of stress-activated genes in 6KR/KO versus WT/KO tumors in mice, implying a critical role of MED1 IDR deacetylation in stress response both in vitro and in vivo.
We also report that stress induces SIRT1 recruitment by the SEC to promoters, where SIRT1 deacetylates the IDR of MED1 to enhance Pol II recruitment. This process represents a cascade of transient promoter-proximal events, as the SEC rapidly moves away from the promoter upon Pol II elongation43 while MED1 and the Mediator complex remain stably anchored in the promoter-proximal region5,6. Consequently, the above-described events can occur only during the brief window when the SEC is present at the promoter. Although our genomic mapping detected elevated transcriptionally engaged Pol II (RPB1 S5P) across entire gene bodies, pausing index calculations and RPB1 S2P mapping indicated that elongation kinetics in 6KR/KO cells are comparable to or even lower than that in WT/KO cells. Thus, while reduced MED1 acetylation strengthens Pol II association, it alone is insufficient to drive productive elongation and transcriptional activation. Instead, an additional signal, such as cellular stress, is required to release the recruited Pol II into elongation. This explains why 6KR MED1 has only a modest impact on gene expression under steady-state, low-stress conditions. Regarding the prevailing view that the SEC acts as a positive cofactor for stress-induced genes, 6KR MED1-mediated effects on stress-inhibited genes suggest either context-dependent SEC repression or SEC-independent regulation through altered Pol II recruitment—a dichotomy requiring further investigation. While ER occupancy was unaffected by MED1 deacetylation, its known cooperation with stress-responsive TFs (such as p53 (ref. 44) and NRF2 (ref. 45)) and ability to activate preloaded Pol II (refs. 46,47) suggest potential coordination with the SEC–SIRT1–MED1 axis that merits further exploration.
Involvement of the MED1 IDR acetylation in Pol II regulation might be manifested through IDR-mediated interaction. The acetylation-defective 6KR MED1 IDR exhibits LLPS properties distinct from those of the acetylation-mimicking 6KQ MED1 IDR. Notably, the untreated WT MED1 IDR and the 6KR MED1 IDR behaved similarly in the in vitro tests (Fig. 5a,d–f and Extended Data Fig. 7d,g). Thus, increasing the net charge by substituting the six lysines to arginines per se does not alter the phase behavior of MED1 IDR. It was reported that condensates per se do not activate gene transcription but that incorporation of other transcription components determines the functional outcome48. This notion was supported by the enrichment of multiple transcription machinery components, particularly Pol II, into MED1 condensates, which facilitates gene activation37. Consistent with these results, we showed the incorporation of the Pol II CTD into MED1 IDR droplets, which is dramatically enhanced by 6KR and deacetylated WT MED1 compared to 6KQ and CBP-acetylated WT MED1.
Collectively, this study demonstrates (1) dynamic regulation and function of the Mediator MED1 subunit, a model transcription coactivator, beyond PIC assembly; (2) an SEC–SIRT1–MED1 regulatory axis for Pol II occupancy that acts concomitantly with SEC-facilitated Pol II pause–release to support stress-responsive gene expression; and (3) IDR acetylation-mediated regulation of components of the transcription machinery through dynamic phase behavior transitions (Extended Data Fig. 10). The overall functional importance of MED acetylation control is evidenced by the findings that 6KR MED1 ER+ BC cells display accelerated cell growth both in culture and in an orthotopic mouse model, as well as increased tolerance to various stress conditions and an increased capability for metastasis. Therefore, our discovery may offer a distinctive opportunity for therapeutic development for diseases with MED1/Mediator-related transcription control upon cellular stress, such as ER+ BC.
Methods
Orthotopic tumor growth and lung metastasis in mice
Animal care and experimentation were conducted according to National Institutes of Health (NIH) guidelines and were approved by the Institutional Animal Care and Use Committee at The Rockefeller University (protocol 23007-H). MCF7 cells (with WT/KO or 6KR/KO transgenes) were resuspended in a 1:1 mix of PBS and Matrigel (Corning) at a concentration of 4 × 107 cells per ml before being injected into the fourth mammary fat pad on the both sides of 7-week-old female NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; The Jackson Laboratory). In total, 4 × 106 WT/KO or 6KR/KO cells (0.1 ml) were injected per side. Animals were palpated weekly for tumor formation, after which sizeable tumors were measured with a caliper once a week. Tumor volume was calculated as (short diameter)2 × (long diameter)/2.
At the time when WT/KO and 6KR/KO MCF7 tumors reached comparable size (near the humane endpoint), mice were killed. Tumors and lungs were isolated (with intracardial PBS perfusion), fixed with 4% paraformaldehyde overnight at 4 °C and then stored in 70% ethanol. Lungs were then paraffin-embedded, sectioned and stained with hematoxylin and eosin. Images of the sections from high-resolution scanning were used to observe metastatic nodules, which were identified as aggregation of cells with a larger, angular and dividing nucleus and diminished cytosol.
Cell culture
Human MCF7 and T47D cells (American Type Culture Collection) were cultured in DMEM (Gibco) supplemented with 10% FBS (Gibco) in incubator at 37 °C, with 5% CO2 and 95% humidity, and tested regularly for Mycoplasma. For heat-shock experiments, the incubator was set at 42 °C, with all other conditions the same.
Stable transfection, pool selection and KO pool preparation
The two-plasmid lentivirus packaging system was used to generate HEK293T or MCF7 cells with stable transgenic expression of FLAG–MED1 (WT or mutant), HA–MED1 (WT or mutant) or FLAG–Cas9. The pLenti (CMV promoter) or pLX313 (EF1α promoter) constructs with inserted genes were cotransfected with vectors expressing lentiviral gag/pol (psPAX2) and vsvg (pMD2.G) genes in HEK293T cells to produce lentiviruses by TransIT-LT1 transfection reagent (Mirus). Supernatants containing virus particles were collected at 48 h and 72 h after transfection. After filtration through a 0.45-μm filter, collected virus-containing culture medium was used to infect HEK293T or MCF7 cells, facilitated by polybrene, followed by a selection in puromycin (Sigma-Aldrich) for 3 days or hygromycin (Sigma-Aldrich) for 7 days.
MED1 depletion in cells
To generate cell pool with MED1 gene depletion, cells with stable transgenic expression of FLAG–Cas9, as well as HA–MED1 (with mutated gRNA targeting sites) in MED1 addback experiments, were infected by collected culture medium carrying lentivirus expressing gRNA targeting MED1 (or with mouse Rosa26 sequence as a nontargeting infection control) (sequence in Supplementary Table 2) where GFP was coexpressed. Cells were checked for near 100% GFP positivity, indicating the infection efficiency, 3 days after infection and total protein from cells was extracted 4 or 5 days after infection for immunoblotting to confirm the depletion efficiency (by detecting MED1 expression in depletion-only versus infection control cells) and expression level of added back ectopic MED1 (by detecting HA). Cell growth and colony formation assays were started with cells 5 days after infection and RNA-seq and ChIP-seq samples were all collected at 5 days after infection. To avoid selection and expansion of advantageous clones from the KO addback pool, which would change the expression level of MED1, all experiments were conducted with cells collected immediately after KO of MED1 by lentiviral infection.
MTT and CellTiter blue assays
To analyze cell growth, cells were digested and plated in 96-well plates at a density of 500 or 1,000 cells per well in 100 μl of culture medium. Cell growth was measured on day 0 and at various time points after seeding. Using the CellTiter blue assay kit (Promega, G8081), 20 μl of CellTiter blue reagent was added to each well. After 4 h of incubation at 37 °C, fluorescence was measured by a SpectraMax M5 (Molecular Device) at 560/590 nm. Cell viability was calculated by subtracting background fluorescence of the culture medium and normalized against that on day 0. Cell growth was also monitored by traditional MTT assay. Briefly, 20 μl of thiazolyl blue tetrazolium bromide (Sigma-Aldrich) (5 mg ml−1 in PBS) was added to each well. After 4 h of incubation at 37 °C, culture medium was removed carefully and 200 μl of DMSO was added to each dried well. After 45 min of mixing at room temperature, the absorbance at 560 nm was measured by a SpectraMax M5.
To monitor cell viability with stress-inducing chemical treatment, cells were digested and plated in 96-well plates at a density of 1,000 cells per well in 50 μl of culture medium. The following day, 50 μl of culture medium with the chemicals dissolved was added to each well. After a given duration of treatment, cell viability was monitored by traditional MTT assay as described above.
Colony formation assay
To analyze colony formation, cells were digested and plated in six-well plates at a density of 1,000 cells per well in 2 ml of culture medium. After incubation for 1.5–2.5 weeks, culture medium was removed and colonies were stained with 0.1% crystal violet (Sigma-Aldrich) in 10% ethanol for 30 min.
Wound-healing assay
Full confluent monolayer cells were wounded with a sterile plastic pipette tip. Cells were then cultured in medium without serum. Cell migration was observed for multiple time points later by microscopy. Images were taken on a Nikon Eclipse Ti microscope and processed with NIS-elements BR software (Nikon).
Immunofluorescence staining
U2OS (2-6-3) reporter cells were transiently transfected with GFP–LacI (Addgene, 183920; as control or further fused with 6KR/Q MED1 IDR) and incubated for 2 days for staining. MCF7 cells with stable integration of FLAG-tagged MED1 were stained directly. Cells were cultured in six-well plates to the appropriate density (<50% confluency), fixed with 4% paraformaldehyde (Sigma-Aldrich) for 15 min at room temperature and then permeabilized with 0.02% Triton X-100 for 5 min at 4 °C. After blocking with 0.5% BSA for 30 min, cells were incubated with mouse anti-FLAG (Sigma-Aldrich, F1804, 1:2,000) or anti-Pol II (CTD4H8, Santa Cruz 47701, 1:100) antibodies diluted in 0.5% BSA solution at 4 °C overnight. After washing with TBST (0.05% Tween-20, 50 mM Tris-HCl and 150 mM NaCl, pH 7.6) three times (5 min each), cells were incubated with Alexa Fluor 488-conjugated goat anti-mouse antibodies (Invitrogen, A11001) diluted (1:2,000) in PBS for 1 h. Then, cells were incubated with DAPI (Sigma-Aldrich) at 0.1 μg ml−1 for 5 min and then washed with PBS three times (5 min each). Images were taken on a Nikon Eclipse Ti microscope and image acquisition was performed using NIS-elements BR software (Nikon).
Immunoblotting
Whole-tissue lysate of MCF7 tumors collected from NSG mice was prepared with a bead mill homogenizer (Bead Ruptor Elite, OMNI International). Whole-cell lysates of postconfluent MCF7, T47D and HEK293T cells were lysed directly in Laemmli buffer. Whole-tissue lysate, whole-cell lysate or immunoprecipitated samples were resolved by SDS–PAGE (6–10%) and transferred onto methanol-treated immune-blot PVDF membrane (Bio-Rad). The membranes were incubated overnight at 4 °C with corresponding primary antibodies (antibody information in Supplementary Table 1). Membranes were then washed three times with TBST (0.05% Tween-20, 50 mM Tris-HCl and 150 mM NaCl, pH 7.6), incubated with enhanced chemiluminescence (ECL) horseradish-peroxidase-labeled anti-rabbit (Amersham NA934) or anti-mouse (Amersham NA931) IgG at a dilution of 1:10,000 for 1 h at room temperature, washed three times with TBST, exposed with mixed ECL prime peroxide solution and ECL prime luminol enhancer solution (Amersham) and exposed to X-ray film for visualization.
For quantification, ImageJ was used to monitor the signal intensity of bands in images. Background signal was also monitored to correct the band signal. The signal intensity of detected proteins (or acetylated proteins) was normalized by the IP target proteins (for IP and co-IP assays) or loading control (for testing whole-cell and whole-tissue lysates).
IP, co-IP and acetylation examination
Cells were lysed in RIPA lysis buffer (50 mM Tris, 300 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 0.1% SDS and 0.25% deoxycholate sodium, pH 8.0) with protease inhibitors (Roche). The insoluble fraction in lysate was spun down at 13,523g for 15 min at 4 °C to discard. For FLAG IP, the transferred supernatant was incubated with anti-FLAG M2 affinity resin (Sigma-Aldrich, A2220) (20 μL per 1 ml of lysate) at 4 °C overnight. For IP of endogenous MED1, MED17, AFF4 or CDK9, transferred supernatant was incubated with the antibodies (antibody information in Supplementary Table 1) at 4 °C overnight and then mixed with prewashed protein A Sepharose CL-4B beads (Amersham, 17-0963-03) or protein G Sepharose 4B beads (Invitrogen, 101241) (20 μL per 1 ml of lysate) for 4 h of additional incubation. After three washes with ice-cold RIPA buffer, beads were resuspended with Laemmli buffer (2% SDS, 50 mM Tris-HCl, 10% glycerol, 5% β-mercaptoethanol and 0.0005% bromophenol blue, pH 6.8) and boiled at 100 °C for 10 min for immunoblotting.
To detect protein acetylation, FLAG-tagged (ectopically expressed) or endogenous protein was immunoprecipitated from the whole-cell lysate with this protocol and detected by immunoblotting for the immunoprecipitates with a pan-anti-acetylated lysine antibody (Supplementary Table 1). The FLAG tag or the antibody was detected by immunoblotting, which was used to normalize the acetylation signal in densitometry quantification. For endogenous protein detection, nonspecific IgG (from same species as the antibody for target protein) was used as the IP control.
IP–MS
To identify residues of MED1 with acetylation, the endogenous MED1 was immunoprecipitated from whole-cell lysate of HEK293T by anti-MED1 antibody (Bethyl/Fortis, A300-793, 1:800) followed by conjugation of protein A Sepharose CL-4B beads (Amersham, 17-0963-03). The affinity-captured sample was subjected to SDS–PAGE and stained by GelCode Blue stain reagent (Thermo Fisher Scientific). The bands corresponding to MED1 were subjected to in-gel trypsin and chymotrypsin digestions.
To identify AFF4-binding proteins, the endogenous AFF4 was immunoprecipitated from nuclear lysate of MCF7 cells (lysis buffer: 50 mM Tris, 300 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 0.1% SDS and 0.25% deoxycholate sodium, pH 8.0) by anti-AFF4 antibody (Bethyl/Fortis, A302-538, 1:300) followed by conjugation of protein G Sepharose 4B beads (Invitrogen, 101241). The affinity-captured proteins on beads underwent partial on-bead digestion with trypsin for 3 h. The supernatant was extracted and underwent reduction and alkylation followed by digestion using Lys-C and trypsin overnight. Digestion reactions were stopped with neat TFA. Samples then underwent solid-phase extraction before being analyzed by LC–MS/MS.
The LC–MS/MS analysis was performed on an Easy-nLC 1200 LC system (Thermo Fisher Scientific) coupled to an Orbitrap Fusion Lumos MS instrument (Thermo Electron) equipped with a nanoelectrospray ion source (Thermo Fisher Scientific). Tryptic peptides were dissolved in loading buffer (1% acetonitrile and 0.1% TFA), injected and separated using a C18 column (15 cm × 100 μm × 100 μm, 1.9 μm, 120 Å; for acetylation site identification) or an a EasySprayer column (C18 reversed phase, 25 cm, 75 μm; for AFF4-binding protein identification) using a linear gradient created by mixing buffer B (80% acetonitrile in 0.1% formic acid) and buffer A (0.1% formic acid). Eluted peptides were analyzed by data-dependent MS/MS acquisition. The MS instrument (ASCEND) was operated in high-resolution high-mass-accuracy mode. The scan range was set from m/z 375 to m/z 1,500. Spectral data were searched against human protein UniProt database in Proteome Discoverer 1.4 suites with Mascot software (version 2.4, Matrix Science) and filtered for a false discovery rate of <5%. The mass tolerance was set to 7 ppm for the precursor and 0.0.02 Da for the tolerance of product ions. For MED1 acetylation analysis, lysine acetylation (N-terminal), acetylation and phosphorylation were chosen as variable modifications. For AFF4-binding protein analysis, the area values were calculated as the average area of the three most intense peptides for the protein, a proxy for the protein amount.
Protein expression and purification from E. coli
The consensus fragment between VL-XT and MobiDB predictions (residues 963–1320 from reference sequence NM_004774.4) was used as the MED1 IDR. The pET22b constructs of His-tagged EGFP–MED1 IDR (WT or mutant) or EGFP alone, the pET28c constructs of His-tagged mCherry–Pol II CTD (subcloned from Addgene 98678) or mCherry alone and the pGEX2T constructs of GST-tagged CBP HAT domain (Addgene, 21093) were introduced into BL21(DE3) E. coli cells. The transformants were grown at 37 °C in LB medium (10 g L−1 tryptone, 5 g L−1 yeast extract and 10 g L−1 NaCl) until the optical density of the medium at 600 nm reached 0.6. Protein expression in E. coli was stimulated by 1 mM IPTG (Sigma-Aldrich) at 16 °C overnight. E. coli cells were spun down and resuspended in cold imidazole extraction buffer (50 mM Tris-HCl, 500 mM NaCl and 10 mM imidazole, pH 7.4) for His-tagged proteins or BC100 buffer (20 mM Tris-HCl, 100 mM KCl, 0.2 mM EDTA and 20% glycerol, pH 7.9) for GST-tagged proteins with the addition of 1% Triton X-100, 1 mM DTT and protease inhibitor cocktail (Roche), then lysed in a high-pressure homogenizer (EmulsiFlex-C5, Avestin) and centrifuged at 13,523g for 15 min at 4 °C to remove insoluble debris. The fusion protein in the lysate was affinity-purified by Ni-NTA agarose beads (Qiagen, 30210) capturing the His tag or by glutathione Sepharose 4B beads (Amersham, 17-0756-01) capturing the GST tag after 4 h of incubation at 4 °C. After washing with cold imidazole extraction buffer or BC100 buffer three times, respectively, the His-tagged proteins were eluted twice by imidazole elution buffer (50 mM Tris-HCl, 500 mM NaCl and 250 mM imidazole, pH 7.4) for 10 min at 4 °C and the GST-tagged proteins were eluted twice by glutathione elution buffer (50 mM Tris-HCl, 10 mM reduced glutathione and 1 mM DTT, pH 8.0) for 10 min at room temperature. The purity of recombinant proteins was verified by SDS–PAGE with staining by GelCode Blue stain reagent (Thermo Fisher Scientific).
To prepare the acetylated WT MED1 IDR, the Ni-NTA agarose beads bound with HIS-tagged MED1 IDR (about 500 μl) were resuspended with 500 μl of BC100 buffer after washing with cold imidazole extraction buffer and GST–CBP was added to a final concentration of 0, 0.708, 7.08 or 70.8 μg ml−1. After incubation at 30 °C for 1 h, the His-tagged MED1 IDR was eluted by imidazole elution buffer for 10 min at 4 °C for twice. The purity of eluted proteins was verified by SDS–PAGE with staining by GelCode Blue stain reagent (Thermo Fisher Scientific) and acetylation was verified by immunoblotting.
The eluted His-tagged MED1 IDR proteins and their controls (2–3 ml each) were transferred into a 15.5-mm cellulose dialysis tube (BioDesign) and dialyzed twice (overnight the first time and for 6 h the second time) with 1 L of dialysis buffer (for MED1 IDR: 50 mM Tris-HCl, 150 mM NaCl, 5% glycerol and 1 mM DTT, pH 7.4; for Pol II fragment: 50 mM Tris-HCl, 200 mM NaCl, 10% glycerol and 1 mM DTT, pH 7.4) to remove imidazole and reduce salt concentration. Dialyzed samples were concentrated in Ultracel centrifugal filters with a cellulose membrane (10-kDa molecular weight cutoff; Amicon, UFC501096) at 845g at 4 °C for 20–30 min. The concentration of purified proteins (with or without dialysis) was determined using a Nanodrop A280 (for MED1 IDR) or the Bradford method (for CBP or Pol II fragment). The ratio of absorbance at 260/280 nm was also monitored using Nanodrop for potential DNA or RNA contamination (ratio should be lower than 0.8).
p300 purification
For recombinant p300, the protocol from a previous publication was followed49. In brief, His-fused p300 was expressed from pFastBac1 vector in High-Five cells and purified on Ni-NTA agarose beads (Qiagen, 30210) in BC buffer (20 mM HEPES pH 7.9, 1 mM EDTA, 10% glycerol and 1 mM DTT) with 100 mM NaCl and 300 mM imidazole. HIS-p300 was further purified on a Q sepharose column (HiTrap, GE Healthcare) in BC buffer and eluted between 100 and 600 mM NaCl. The purity of the protein preparation was assessed on gels and the pure fractions (>90%) were collected and flash-frozen until use.
In vitro droplet formation, saturation concentration and turbidity measurement
After dialysis, droplet formation from EGFP–MED1 IDR (WT, acetylated WT, 6KR or 6KQ) was visible under fluorescence microscopy. No crowding agent (such as PEG) was added. MED1 IDR concentration was determined using a Nanodrop A280 after dilution and the concentration of WT, acetylated WT, 6KR or 6KQ MED1 IDR was adjusted to 6 mg ml−1 (about 94 μM) with dialysis buffer and imaged for droplets at this concentration.
For MED1 IDR and Pol II heptad codroplet tests, two types of dialyzed and concentrated fragments were mixed with equal volume for 3 min at room temperature (reaching the solution with 47 μM EGFP–MED1 IDR, 0.19 μM mCherry–Pol II heptad, 50 mM Tris-HCl, 175 mM NaCl, 7.5% glycerol and 1 mM DTT, pH 7.4).
For imaging these droplets or codroplets, samples were loaded onto a glass slide and covered by a no. 1.5 coverslip. The slides were imaged using a Nikon Eclipse Ti microscope with a ×4 lens and image acquisition was performed using NIS-elements BR software (Nikon). Saturation of images was avoided by adjusting exposure time. ImageJ was used to measure the fluorescence intensity. The partition coefficient of MED1 IDR condensates was calculated by dividing the EGFP intensity inside droplets by the EGFP intensity outside of those droplets. The partition coefficient of the cocondensates of MED1 IDR and Pol II heptads was calculated by dividing the mCherry intensity inside MED1 IDR droplets by the mCherry intensity outside of those droplets.
To monitor the saturation concentration, MED1 IDR solution (25 μl in dialysis buffer) was centrifuged at 10,000g for 10 min at room temperature; then, the supernatant was carefully isolated to measure the protein concentration using a Nanodrop A280. For each trial, the concentration was measured three times and the mean value was recorded.
To monitor turbidity, MED1 IDR solution (in dialysis buffer) was diluted to various concentrations and mixed with various concentrations of PEG in 96-well plates; the absorbance at 350 nm was immediately measured using a SpectraMax M5 (Molecular Device).
FRAP for in vitro droplets
The WT, acetylated WT, 6KR or 6KQ MED1 IDR, all adjusted to 6 mg ml−1 (about 94 μM) in dialysis buffer, was tested for FRAP. Droplets formed by these MED1 IDRs were loaded onto a glass slide and covered by a no. 1.5 coverslip. The slide was inverted and imaged. The prebleach image was acquired, followed by pulses of a 488-nm laser line for photobleaching target droplets. Postbleach images were collected at 0.05-s intervals lasting 60 s. Samples were imaged using a DeltaVision Image Restoration Microscope (Leica) equipped with an inverted IX-71 microscope stand (Olympus), SSI light source (Insight), CoolSNAP HQ monochrome charge-coupled device camera (Photometrics), ×60/1.42 oil objective and bandpass filter sets for detection of EGFP (525/50) fluorescence. Image acquisition was performed using Softworx software (Softworx). Fluorescence intensity at the photobleaching target center was measured by ImageJ.
Purification of Mediator complex from HeLa-S and following assay
Nuclear extracts were prepared as described previously50 from a HeLa-derived cell line that stably expresses a FLAG-tagged human MED10, a core Mediator subunit. Mediator complex in the nuclear extract was affinity-purified by anti-FLAG M2 affinity gel (Sigma-Aldrich, A2220), washed extensively using BC100 buffer (20 mM Tris-HCl, 100 mM KCl, 0.2 mM EDTA and 20% glycerol, pH 7.9) with 0.1% NP-40 and then eluted by FLAG peptide (0.3 mg ml−1) in BC100 three times.
When the bead-bound proteins were tested, the Mediator complex remaining conjugated on beads (after BC100/NP-40 washing) was washed five times using (1) BC100, BC150, BC200 or BC300 buffers (20 mM Tris-HCl, 100, 150, 200 or 300 mM KCl, 0.2 mM EDTA and 20% glycerol, pH 7.9) with 0.1% NP-40 (for high-salt test) or (2) BC100 buffer with 0.1% NP-40 and 2% or 10% 1,6-hexanediol or 2,5-hexanediol (for hexanediol test) and then the components remaining on beads were eluted with Laemmli buffer (2% SDS, 50 mM Tris-HCl, 10% glycerol, 5% β-mercaptoethanol and 0.0005% bromophenol blue, pH 6.8) and boiled at 100 °C for 10 min for immunoblotting.
In vitro acetylation assay
Purified Mediator complex (0.2 μg) was incubated with or without p300 (0–0.2 μg, amount indicated in relevant text or figure) and 50 μM acetyl-CoA in 12.5 μL of BC100 buffer (20 mM Tris-HCl, 100 mM KCl, 0.2 mM EDTA and 20% glycerol, pH 7.9) with the addition of 0.1% NP-40 and 1 mM DTT. After 0–1 h (indicated in relevant text or figure) at 30 °C, samples were subjected to SDS–PAGE and immunoblotting.
RNA extraction, reverse transcription (RT)–qPCR and RNA-seq
Total RNA was extracted from postconfluent cells using TRIzol reagent (Ambion). The RNA template was converted into complementary DNA using iScript RT SupemMix (Bio-Rad) with a blend of oligo(dT) and random primers for complete RNA coverage. Real-time qPCR reactions were performed using a 7300 real-time PCR system (Applied Biosystems) with QuantiTech SYBR green PCR kit (Qiagen, 204141). The PCR protocol involved warming up at 50 °C for 2 min, preheating at 95 °C for 10 min, denaturation at 95 °C for 10 s and combined annealing and extension at 60 °C for 1 min over 40 cycles. The melting curve was generated after these cycles (95 °C for 10 s, 60 °C for 1 min and 95 °C for 15 s) to ensure that the amplification in each reaction was specific. GAPDH was taken as the internal reference. The fold changes (FCs) of RNA levels were calculated using the \(\varDelta \varDelta \,{C}_{t}\) method (primer sequences for RT–qPCR in Supplementary Table 2).
For RNA-seq, total RNA was extracted using RNeasy Mini Kit (Qiagen, 74104) according to the manufacturer’s instructions. Cultured cells were lysed with lysis buffer in the culture dish or plate. Total RNA from mouse MCF7 tumors was prepared with a bead mill homogenizer (Bead Ruptor Elite, OMNI International). In RNA-seq experiments for WT/KO, 6KR/KO and 6KQ/KO MCF7 cells with H2O2 treatment, spike-in RNA (Drosophila melanogaster total RNA for 6KR experiments, and Drosophila biarmipes total RNA for 6KQ tests) was added with the appropriate amount proportional to the cell numbers in each group counted before RNA extraction. A lLibrary of poly(A)-containing mRNA (size: 350 bp) was prepared using the TruSeq stranded mRNA library prep kit (Illumina) according to the manufacturer’s instructions. The library quality was evaluated using the Bioanalyzer and Illumina NovaSeq SP paired-end (100 bp × 2) (for MCF7 MED1 KO versus control and steady-state 6KR/KO versus WT/KO), Illumina NextSeq 2000 P3 paired-end (100 bp × 2) (for mouse tumors and MCF7 6KR/KO versus WT/KO with H2O2 treatment) or Element AVITI B1 paired-end (75 bp × 2) sequencing was performed.
ChIP, ChIP–qPCR and ChIP-seq
Postconfluent cells (control normal cells or after treatment) were placed at room temperature for 10 min (for hexanediol experiments, cells were also treated with 5% 1,6-hexanediol in culture medium at room temperature for 10 min) and then fixed with 1% formaldehyde for 10 min at room temperature, followed by quenching with 125 mM glycine and washing with cold TBS (20 mM Tris-HCl and 150 mM NaCl, pH 7.4) twice. Cells were lysed with cell lysis buffer (10 mM Tris-HCl, 10 mM NaCl and 0.5% NP-40, pH 7.5) while sitting on ice for 10 min. Cell lysates were spun down and resuspended in 500 μl of MNase digestion buffer (20 mM Tris-HCl, 15 mM NaCl, 60 mM KCl and 1 mM CaCl2, pH 7.5) with MNase (New England Biolabs, M0247S; 0.25 U per 1,000 cells). After 20 min of incubation at 37 °C with continuous mixing, digestion was stopped by the addition of 500 μl of 2× STOP/ChIP buffer (100 mM Tris-HCl, 20 mM EDTA, 200 mM NaCl, 2% Triton X-100 and 0.2% sodium deoxycholate, pH 8.1). Samples were sonicated for seven cycles (30 s on, 30 s off) with 40% amplitude (Branson Digital Sonifier) and then centrifuged at 13,523g for 15 min at 4 °C to remove insoluble debris. Apart from input samples (1% volume for each ChIP reaction) kept for reverse crosslinking, lysates were incubated with antibodies (antibody information in Supplementary Table 1) at 4 °C overnight.
For sequential ChIP, the lysates were incubated with anti-FLAG M2 affinity gel (Sigma-Aldrich, A2220) (20 μl per 1 ml of lysate, equivalent to 4 × 106 cells) at 4 °C overnight, then washed and resuspended with STOP/ChIP buffer (50 mM Tris-HCl, 10 mM EDTA, 100 mM NaCl, 1% Triton X-100 and 0.1% sodium deoxycholate, pH 8.1) with the addition of FLAG peptide (1 mg ml−1) for 5 h of incubation at 4 °C. After spinning down to discard the beads, the eluate was further incubated with anti-acetylated lysine antibody (Cell Signaling, 9681) with 1:500 dilution at 4 °C overnight.
Antibody-bound complexes were then captured by incubation with protein A Sepharose CL-4B beads (Amersham, 17-0963-03) for rabbit antibodies or protein G Sepharose 4B beads (Invitrogen, 101241) for mouse antibodies (both 20 μl per 1 ml of lysate) for 4 h at 4 °C. The beads were washed twice sequentially with STOP/ChIP buffer (50 mM Tris-HCl, 10 mM EDTA, 100 mM NaCl, 1% Triton X-100 and 0.1% sodium deoxycholate, pH 8.1), high-salt buffer (50 mM Tris-HCl, 10 mM EDTA, 500 mM NaCl, 1% Triton X-100 and 0.1% sodium deoxycholate, pH 8.1), LiCl2 buffer (10 mM Tris-HCl, 0.25 M LiCl2, 0.5% NP-40, 0.5% sodium deoxycholate and 1 mM EDTA, pH 8.0) and TE buffer (50 mM Tris-HCl and 10 mM EDTA, pH 8.0). Bound DNA was eluted and reverse-crosslinked at 65 °C overnight in ChIP elution buffer (10 mM Tris-HCl, 150 mM NaCl, 10 mM EDTA, 5 mM DTT and 1% SDS, pH 8.0). After treatment with RNase A (0.2 mg ml−1, 37 °C, 1 h) and proteinase K (2 mg ml−1, 37 °C, 2 h), DNA used for ChIP-seq was purified using a MiniElute PCR purification kit (Qiagen, 28004) and DNA used for ChIP–qPCR was purified using a QIAquick PCR purification kit (Qiagen, 28106). DNA quality was assessed using an Agilent Bioanalyzer and quantified using a Qubit fluorometer (Invitrogen).
For ChIP–qPCR, the PCR reactions were performed using a 7300 real-time PCR system (Applied Biosystems) with QuantiTech SYBR green PCR kit (Qiagen, 204141). The PCR protocol involved warming up at 50 °C for 2 min, preheating at 95 °C for 10 min, denaturation at 95 °C for 10 s and combined annealing and extension at 60 °C for 1 min over 40 cycles. The melting curve was generated after these cycles (95 °C for 10 s, 60 °C for 1 min and 95 °C for 15 s) to ensure that the amplification in each reaction was specific.
For ChIP-seq, the libraries were generated using DNA SMART ChIP-seq kits (Takara Bio, 634865) according to the manufacturer’s instructions. After size selection (for 250–600 bp) by AMPure XP beads (Beckman Coulter, A63880), the library quality was evaluated using the Bioanalyzer and sequenced on Illumina NovaSeq SP (100 bp, two ends) for sequential ChIP-seq and Illumina NextSeq High (75 bp, single end) for all other ChIP samples by the Genomics Resource Center in Rockefeller University.
CUT&RUN-seq
Postconfluent cells (500,000 cells per reaction) were harvested and fixed with freshly prepared 0.5% PFA for 2 min. After stopping the crosslinking using 500 μl of 2.5 M glycine, cells were washed three times with digitonin wash buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 0.5 mM spermidine, protein inhibitor cocktail, 0.005% digitonin, 0.05% SDS and 1% Triton X-100) and bound to concanavalin-A-coated beads (Polysciences, 86057-10) prewashed with binding buffer (20 mM HEPES pH 7.4, 10 mM KCl, 1 mM CaCl2 and 1 mM MnCl2). Cells on beads were then incubated with antibodies on a nutator overnight at 4 °C. After overnight incubation, cells were washed three times with digitonin wash buffer, resuspended in 100 μl of digitonin buffer containing protein A/G–MNase fusion protein (pAG-MNase; EpiCypher, 15-1016) and nutated in 4 °C for 90 min. After pAG-MNase binding, cells were washed three times with digitonin wash buffer for 5 min. Finally, cells were resuspended in 100 μl of ice-cold digitonin wash buffer in a heating block precooled in an ice-water bath. Digestion of the chromatin was initiated by addition of 2 mM CaCl2 in the tube and lasted for 4 h at 0 °C. The digestion was stopped by addition of 100 μl of 2× STOP buffer (340 mM NaCl, 20 mM EDTA, 4 mM EGTA, 0.005% digitonin, 100 μg ml−1 RNase A and 50 μg ml−1 glycogen). Digested DNA was released by incubation at 37 °C for 30 min and the supernatant was collected. Then, 5 μl of proteinase K (20 mg ml−1) and 200 μl of 2× elution buffer (20 mM Tris-HCl pH 8.0, 300 mM NaCl, 20 mM EDTA, 2% SDS and 10 mM DTT) was added to each sample for reverse crosslinking at 65 °C overnight. DNA was extracted using phenol–chloroform and dissolved in nuclease-free water.
For sequencing, the libraries were generated using a ThruPLEX DNA-seq kit (Takara Bio, 400674) and unique dual index kit (Takara Bio, 634756) according to the manufacturer’s instructions. After size selection (for 250–600 bp) by AMPure XP beads (Beckman Coulter, A63880), the library quality was evaluated using the Bioanalyzer and sequenced on Illumina NextSeq High (75 bp, single end) by Genomics Resource Center in Rockefeller University.
AlphaFold prediction for protein structure
Initial models of the ELL2–EAF1–AF9–AFF4–CDK9–CCNT1–SIRT1 complex were generated using the AlphaFold 3 server. These models were subsequently aligned with the published crystal structures of the P-TEFb complex in complex with AFF4 and Tat (PDB 4OGR) and the ELL2 occludin domain with an AFF4 peptide (PDB 5JW9). The best model was selected on the basis of optimal alignment results. All structural data were analyzed and figures were generated using PyMOL (version 3.0).
Bioinformatic data analysis
RNA-seq analysis
For the cultured cell RNA-seq experiments, transcript abundance was determined from FASTQ files using Salmon (version 0.8.1) and the GENCODE reference transcript sequences; then, transcript counts were imported into R with the tximport R Bioconductor package (version 1.8.0). For the H2O2 RNA-seq experiments in MCF7 cells after reexpression of 6KR MED1 or 6KQ MED1, spike-in controls were used. In both cases, combined Drosophila and human FASTA and GTF files were created. The human/hg38 FASTA and GTF files were derived from the BSgenome.Hsapiens.UCSC.hg38 (version 1.4.1) and TxDb.Hsapiens.UCSC.hg38.knownGene (version 3.4.0) R Bioconductor packages, respectively. When testing reexpression of the 6KR MED1, D. melanogaster (dm6) FASTA and GTF files were derived from the BSgenome.Dmelanogaster.UCSC.dm6 (version 1.4.1) and TxDb.Dmelanogaster.UCSC.dm6.ensGene (version 3.2.0) R Bioconductor packages, respectively. When testing reexpression of 6KQ MED1, D. biarmipes FASTA and GTF files were retrieved from the RU_DBia_V1.1 genome assembly (RefSeq assembly GCF_025231255.1). For the xenograft tumor RNA-seq experiments, combined mm10 and hg38 FASTA and GTF files were retrieved from the 10X Genomics public reference repository (version 2024-A). For all combined genomes (spike-in and xenograft), FASTQ files were aligned to the combined reference with STAR (version 2.5.2b)51 using default settings, except that the ‘outSAMmultNmax’ argument was set to 1. The reads that preferentially aligned to the hg38 chromosomes were extracted from the BAM file using the ‘view’ function from SAMtools (version 1.10)52 and gene-level counts were obtained using the ‘summarizeOverlaps’ function from the GenomicAlignments R Bioconductor package (version 1.28.0)53.
For all RNA-seq experiments, differential gene expression was performed with the DESeq2 R Bioconductor package (version 1.20.0)54. For the spike-in experiments, Drosophila genes were used to estimate size factors in the DESeq2 differential expression workflow. Normalized counts were retrieved from the DESeq2 results and z scores for the indicated gene sets were visualized with heat maps generated using the pheatmap R package (version 2.8.0). For GSEA, the Wald statistic from the DESeq2 analysis was used to generate the ranked list of genes. The clusterProfiler R Bioconductor package (version 4.0.5) was then used to perform GSEA to determine the enrichment of the indicated gene lists within this ranked list55,56. The enrichKEGG function from clusterProfiler was used for over representation analysis (Fisher test) of KEGG gene lists in DEG sets. The enrichplot R Bioconductor package (version 1.12.3) was used to visualize the GSEA result. Volcano plots showing the log2 FC versus log10 of the adjusted P value and the plot showing the correlation between RNA-seq datasets were generated using the ggplot2 R package (version 3.3.5). When genes were divided into categories on the basis of the degree of their gene expression changes, the groups were defined as follows: strong up, FC is greater than 2 and adjusted P value is less than 0.05; mild up, FC is between 0 and 2 and adjusted P value is less than 0.05; strong down, FC is less than 2 and adjusted P value is less than 0.05; mild down, FC is between 0 and 2 and adjusted P value is less than 0.05; no change, adjusted P value is greater than 0.05.
For the heat map showing RNA-seq expression values from 6KR/KO or WT/KO MED1 and MED1 KO or WT MED1, hierarchical clustering was performed using any DEG (P < 0.05) in the 6KR/KO MED1 versus WT/KO MED1 or MED1 KO versus WT MED1 (overlap of all three clones) comparisons. The values used in the heat map are z scores of normalized expression values (using DESeq2 vst function). Six clusters were identified and shown in a heat map using the clusterProfiler R Bioconductor package (version 4.0.5). Motif analysis was then performed using the promoter region (−300 bp/+50 bp) for genes within each cluster using the ‘findMotifs.pl’ function from the Homer suite of tools (version 4.11).
For the heat map showing RNA-seq expression values from the 6KR/KO or WT/KO MED1 and 6KQ/KO or WT/KO comparisons, hierarchical clustering was performed using any DEG (P < 0.05) across any of the samples. This was achieved using the likelihood ratio test and DESeq2. The values used in the heat map are z scores of normalized expression values (using DESeq2 vst function).
ChIP-seq and CUT&RUN-seq analysis
ChIP-seq and CUT&RUN-seq reads were aligned using the Rsubread R Bioconductor package (version 1.30.6) and predicted fragment lengths were calculated using the ChIPQC R Bioconductor package (version 1.16.2)57,58. Normalized (reads at each loci per million mapped reads), fragment-extended signal bigWig files were created using the rtracklayer R Bioconductor package (version 1.40.6). For ChIP-seq experiments, peaks were called using MACS2 (version 2.1.1)59,60. For CUT&RUN-seq experiments, peaks were called with SEACR (version 1.3)61 using the ‘stringent’ setting and the top 1% of peaks as a function of the total signal within peaks were kept for further analysis. The high-confidence MED1 peak set was generated by taking the intersecting peaks from MED1 ChIP-seq, FLAG–MED1 ChIP-seq and the union of peaks from the three MED1 CUT&RUN replicates. This was used throughout the manuscript except for analysis involving the sequential FLAG + AceK ChIP-seq experiment, where the MED1 ChIP-seq peaks were used. Further normalization of bigWig files (normalized to input) was performed using the deepTools (version 3.5.1) bigwigCompare function62 with the ‘operation’ argument set to log2. Correlation of the signal from ChIP-seq replicates was performed using the deepTools multiBigwigSummary function with the ‘binSize’ argument set to 5,000, followed by the ‘plotCorrelation’ function using a Pearson correlation. Any regions included in the ENCODE blacklisted regions of the genome were excluded from all region-specific analyses63.
Range-based heat maps, line plots and box plots showing the normalized signal over genomic regions were generated using the profileplyr R Bioconductor package (version 1.8.1) and ggplot2. Annotation of peaks to genes was performed with the rGREAT R Bioconductor package (version 1.24.0).
The classes of cis-regulatory elements are defined as follows: active enhancers, H3K4me+H3K4me3−H3K27ac+; primed enhancers, H3K4me+H3K4me3−H3K27ac−; active promoters, H3K4me3+ within TSS ± 3 kb; silent promoters, H3K4me3− within TSS ± 3 kb.
The pausing index was calculated by taking the ratio of counts of Pol II (RPB1 S5P) ChIP-seq reads over promoters (±150 kb around TSS) versus the gene body (500 kb from TSS to the end of the gene). The counts for both promoter and gene-body regions were normalized to the length of the region being counted over. Genes less than 800 kb were excluded to guarantee that gene-body regions were at least 300 kb—the same as the promoter region.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All RNA-seq, CUT&RUN-seq and ChIP-seq data were deposited to the Gene Expression Omnibus (GSE245869) and are open to public access. Source data are provided with this paper.
Change history
24 March 2026
This paper was originally published under a standard Springer Nature license (© The Author(s), under exclusive licence to Springer Nature America, Inc.). It is now available as an open-access paper under an Attribution-NonCommercial-NoDerivatives 4.0 International licence, © The Author(s).
References
Pessa, J. C., Joutsen, J. & Sistonen, L. Transcriptional reprogramming at the intersection of the heat shock response and proteostasis. Mol. Cell 84, 80–93 (2024).
Himanen, S. V. & Sistonen, L. New insights into transcriptional reprogramming during cellular stress. J. Cell Sci. 132, jcs238402 (2019).
Vihervaara, A., Duarte, F. M. & Lis, J. T. Molecular mechanisms driving transcriptional stress responses. Nat. Rev. Genet. 19, 385–397 (2018).
Senft, D. & Ronai, Z. E. Adaptive stress responses during tumor metastasis and dormancy. Trends Cancer 2, 429–442 (2016).
Malik, S. & Roeder, R. G. Regulation of the RNA polymerase II pre-initiation complex by its associated coactivators. Nat. Rev. Genet. 24, 767–782 (2023).
Richter, W. F., Nayak, S., Iwasa, J. & Taatjes, D. J. The Mediator complex as a master regulator of transcription by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 23, 732–749 (2022).
Ge, K. et al. Transcription coactivator TRAP220 is required for PPARγ2-stimulated adipogenesis. Nature 417, 563–567 (2002).
Malik, S. et al. Structural and functional organization of TRAP220, the TRAP/Mediator subunit that is targeted by nuclear receptors. Mol. Cell. Biol. 24, 8244–8254 (2004).
Zhang, X. et al. MED1/TRAP220 exists predominantly in a TRAP/Mediator subpopulation enriched in RNA polymerase II and is required for ER-mediated transcription. Mol. Cell 19, 89–100 (2005).
Chen, W. & Roeder, R. G. The Mediator subunit MED1/TRAP220 is required for optimal glucocorticoid receptor-mediated transcription activation. Nucleic Acids Res. 35, 6161–6169 (2007).
Chu, C. S. et al. Unique immune cell coactivators specify locus control region function and cell stage. Mol. Cell 80, 845–861 (2020).
Boija, A. et al. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 175, 1842–1855 (2018).
Murakami, S., Nagari, A. & Kraus, W. L. Dynamic assembly and activation of estrogen receptor α enhancers through coregulator switching. Genes Dev. 31, 1535–1548 (2017).
Lu, X. et al. Multiple P-TEFbs cooperatively regulate the release of promoter-proximally paused RNA polymerase II. Nucleic Acids Res. 44, 6853–6867 (2016).
Chen, Z. et al. Phosphorylated MED1 links transcription recycling and cancer growth. Nucleic Acids Res. 50, 4450–4463 (2022).
Jing, H. & Lin, H. Sirtuins in epigenetic regulation. Chem. Rev. 115, 2350–2375 (2015).
Luo, Z., Lin, C. & Shilatifard, A. The super elongation complex (SEC) family in transcriptional control. Nat. Rev. Mol. Cell Biol. 13, 543–547 (2012).
Banani, S. F., Lee, H. O., Hyman, A. A. & Rosen, M. K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 (2017).
Chen, X., et al. Structures of the human Mediator and Mediator-bound preinitiation complex. Science 372, eabg0635 (2021).
Abdella, R. et al. Structure of the human Mediator-bound transcription preinitiation complex. Science 372, 52–56 (2021).
Creyghton, M. P. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA 107, 21931–21936 (2010).
Rada-Iglesias, A. et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283 (2011).
Narita, T., Weinert, B. T. & Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 20, 156–174 (2019).
Ito, K. et al. Critical roles of transcriptional coactivator MED1 in the formation and function of mouse adipose tissues. Genes Dev. 35, 729–748 (2021).
Cui, J. et al. Cross-talk between HER2 and MED1 regulates tamoxifen resistance of human breast cancer cells. Cancer Res. 72, 5625–5634 (2012).
Delvecchio, M., Gaucher, J., Aguilar-Gurrieri, C., Ortega, E. & Panne, D. Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat. Struct. Mol. Biol. 20, 1040–1046 (2013).
Ortega, E. et al. Transcription factor dimerization activates the p300 acetyltransferase. Nature 562, 538–544 (2018).
Xue, B., Dunbrack, R. L., Williams, R. W., Dunker, A. K. & Uversky, V. N. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 1804, 996–1010 (2010).
Piovesan, D. et al. MobiDB: intrinsically disordered proteins in 2021. Nucleic Acids Res. 49, D361–D367 (2021).
Sabari, B. R., et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018).
Lyons, H. et al. Functional partitioning of transcriptional regulators by patterned charge blocks. Cell 186, 327–345 (2023).
Lee, K. H., Choi, E., Chun, Y. S., Kim, M. S. & Park, J. W. Differential responses of two degradation domains of HIF-1α to hypoxia and iron deficiency. Biochimie 88, 163–169 (2006).
Yuan, Y., Hilliard, G., Ferguson, T. & Millhorn, D. E. Cobalt inhibits the interaction between hypoxia-inducible factor-α and von Hippel–Lindau protein by direct binding to hypoxia-inducible factor-α. J. Biol. Chem. 278, 15911–15916 (2003).
Core, L. & Adelman, K. Promoter-proximal pausing of RNA polymerase II: a nexus of gene regulation. Genes Dev. 33, 960–982 (2019).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Keenen, M. M., Larson, A. G. & Narlikar, G. J. Visualization and quantitation of phase-separated droplet formation by human HP1α. Methods Enzymol. 611, 51–66 (2018).
Guo, Y. E. et al. Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572, 543–548 (2019).
Janke, A. M. et al. Lysines in the RNA polymerase II C-terminal domain contribute to TAF15 fibril recruitment. Biochemistry 57, 2549–2563 (2018).
Janicki, S. M. et al. From silencing to gene expression: real-time analysis in single cells. Cell 116, 683–698 (2004).
Harrell, J. C. et al. Estrogen receptor positive breast cancer metastasis: altered hormonal sensitivity and tumor aggressiveness in lymphatic vessels and lymph nodes. Cancer Res. 66, 9308–9315 (2006).
Zhang, X. H., Giuliano, M., Trivedi, M. V., Schiff, R. & Osborne, C. K. Metastasis dormancy in estrogen receptor-positive breast cancer. Clin. Cancer Res. 19, 6389–6397 (2013).
Adelman, K. & Lis, J. T. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat. Rev. Genet. 13, 720–731 (2012).
Lin, C. et al. Dynamic transcriptional events in embryonic stem cells mediated by the super elongation complex (SEC). Genes Dev. 25, 1486–1498 (2011).
Das, G. M., Oturkar, C. C. & Menon, V. Interaction between estrogen receptors and p53: a broader role for tamoxifen? Endocrinology 166, bqaf020 (2025).
Czogalla, B., et al. Interaction of ERα and NRF2 impacts survival in ovarian cancer patients. Int. J. Mol. Sci. 20, 112 (2018).
Kininis, M., Isaacs, G. D., Core, L. J., Hah, N. & Kraus, W. L. Postrecruitment regulation of RNA polymerase II directs rapid signaling responses at the promoters of estrogen target genes. Mol. Cell. Biol. 29, 1123–1133 (2009).
Hah, N. et al. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 145, 622–634 (2011).
Trojanowski, J. et al. Transcription activation is enhanced by multivalent interactions independent of phase separation. Mol. Cell 82, 1878–1893 (2022).
Wang, J. et al. EZH2 noncanonically binds cMyc and p300 through a cryptic transactivation domain to mediate gene activation and promote oncogenesis. Nat. Cell Biol. 24, 384–399 (2022).
Dignam, J. D., Martin, P. L., Shastry, B. S. & Roeder, R. G. Eukaryotic gene transcription with purified components. Methods Enzymol. 101, 582–598 (1983).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Lawrence, M. et al. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9, e1003118 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Carroll, T. S., Liang, Z., Salama, R., Stark, R. & de Santiago, I. Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front. Genet. 5, 75 (2014).
Liao, Y., Smyth, G. K. & Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47 (2019).
Lawrence, M., Gentleman, R. & Carey, V. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics 25, 1841–1842 (2009).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Meers, M. P., Tenenbaum, D. & Henikoff, S. Peak calling by sparse enrichment analysis for CUT&RUN chromatin profiling. Epigenetics Chromatin 12, 42 (2019).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Amemiya, H. M., Kundaje, A. & Boyle, A. P. The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep. 9, 9354 (2019).
Acknowledgements
We thank members of the R.G.R. lab for technical assistance, helpful discussions and comments regarding the manuscript. We thank D. Spector at the Cold Spring Harbor Laboratory for sharing U2OS LacO reporter cells and related reagents. We thank B. Sabari at the University of Texas Southwestern Medical Center for helpful discussions and comments. We thank H. Molina (director) and colleagues in The Rockefeller University Proteomics Resource Center for assistance with protein MS analyses, P. Banerjee, C. Pyrgaki and A. North (director) in The Rockefeller University Bioimaging Resource Center for assistance with droplet imaging and FRAP assays, T. Carroll (director) and colleagues in The Rockefeller University Bioinformatics Resource Center for assistance with sequencing data analysis, the laboratory of L. Zhao at The Rockefeller University for providing Drosophila RNA for spike-in control of RNA-seq experiments, R. Gong in the laboratory of G. Alushin at The Rockefeller University for assistance with bacterial cell lysis by high-pressure homogenization and J. Rodriguez in the laboratory of H. Steller Lab at The Rockefeller University for assistance with use of microplate readers. We also thank F. Yang at Albert Einstein College of Medicine for sharing MED15 plasmids. This work was supported by NIH grants (CA234575, CA273709) and Leukemia and Lymphoma Society (7037-25) grants to R.G.R.
Author information
Authors and Affiliations
Contributions
R.L. conceptualized and designed the project, conducted the in vitro biochemical, cell-based and animal experiments, analyzed the data and wrote the paper (supervised by R.G.R.). Y.M. conducted the molecular biology experiments (supervised by Z.Z. and E.A.). D.B. conducted the bioinformatic analyses related to the sequencing experiments. W.M. conducted the animal experiments (supervised by S.F.T.). T.O. conducted the in vitro biochemical experiments (supervised by R.G.R.). J.S. conducted the protein structure prediction (supervised by R.G.R.). Z.Z., E.A. and S.F.T. were responsible for overall supervision. R.G.R. conceptualized and designed the project, wrote the paper, funded the project and was responsible for overall supervision.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemical Biology thanks Julie Soutourina and the other, anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 MED1 is essential for growth and supports gene activation in ER+ breast cancer cells.
a, Workflow depicting the depletion of MED1 by CRISPR/Cas9. Cas9 (FLAG-tagged) was stably expressed in cells and lentiviral infection was used to introduce plasmids expressing gRNAs targeting MED1 exons into pools of cells to deplete MED1. b, Immunoblotting confirming depletion of MED1 by targeting 3 independent regions (designated KO-1, 2, and 3) in exons of the MED1 gene (with a gRNA targeting the mouse Rosa26 locus as a non-targeting control) in MCF7 and T47D cells, as well as comparable ERα levels in control and KO cells. Immunoblots are representative of three independent experiments. c, Cell growth attenuated by MED1 depletion in either MCF7 or T47D cells, measured by CellTiter Blue assay. N = 3 (biological replicates) for each cell type. Data points report mean ± SEM. Two-sided unpaired t-test for each of KO cell type versus control for D6 data. d, Attenuated colony formation in MCF7 cells with MED1 KO compared with control, observed by staining with crystal violet for cells growing for 16 days after initial seeding.
Extended Data Fig. 2 Genomic analyses of gene regulation by MED1 in ER+ breast cancer cells.
a–c, Poly(A) RNA-seq for control and MED1 KO MCF7 cells. Two biological replicate samples were detected for control or each of KO cell types. a, A volcano plot showing the adjusted P value (two-sided) and fold-change of each detected gene (combined from KO-1, -2, and -3) where the cut-off to define DEGs (FC > 2, P adj < 0.05) is indicated. b, A clustered heatmap of Z-scores of genes with differential expression with P adj < 0.05 from MED1 KO (-1, -2 and -3) versus control cells. Colors in scale represent relative expression levels: blue (Z ≤ −2), white (Z = 0), and red (Z ≥ +2), with a linear gradient for intermediate values. c, Gene ontology analyses with adjusted P values with KEGG gene lists for up-regulated and down-regulated DEGs, respectively. d, Venn diagram showing overlap of genomic peaks of MED1, MED17 and Pol II. e–g, Analyses for peaks classified by occupancy of MED1, MED17 and Pol II. e, Pie diagrams showing constitution of each type of these peaks for types of regulatory elements. See Methods for the definition of regulatory elements. f, Genomic annotation of MED1+MED17+Pol II+ peaks that cannot be categorized into pre-defined types of regulatory elements. g, Pie diagrams showing constitution of each type of these peaks for association with up-, down-regulated genes or no-change-genes of MED1 KO versus control cells. h, Density of MED1 occupancy at active promoters associated with plotted genes with stratification of genes based on their fold-change values detected from RNA-seq of MED1 KO versus control cells (designated STRONG UP, MILD UP, NO-CHANGE, MILD DOWN, STRONG DOWN). Boxes span the 25th-75th percentiles (center line indicates median), whiskers extend to 1.5 X IQR, and points beyond represent outliers. Parentheses indicate sample sizes.
Extended Data Fig. 3 MED1 acetylation is regulated by p300/CBP and identification of acetylation sites in the MED1 IDR.
a, b, Detection of acetylation of FLAG-tagged Mediator subunits transiently expressed in HEK293T cells treated with Trichostatin A (TSA, 0.5 μM for 24 h) and nicotinamide (NAM, 5 mM for 3/6 h), referred to as T + N3 or T + N6 in the panel. Immunoblotting images (a) and densitometry quantification with AceK immunoblot signals normalized by the FLAG signals (b) are shown. c, Silver staining (left) and immunoblotting (right) confirming the yield and integrity of Mediator (presence of MED1 and MED17) purified from HeLa-S cells and the absence of p300 and SIRT1. d, Workflow for mass spectrometry identification of acetylation sites from immunoprecipitated endogenous MED1 from HEK293T cells with overexpression of p300 with or without A485. e, Immunoblotting confirming the overexpression of p300 and acetylation of MED1 in IP-Mass Spectrometry samples. Densitometry analysis was performed on immunoblotting images, and the normalized acetyl-lysine levels (relative to IP target protein, MED1) are shown. f, Analysis of MED1 protein sequence by VL-XT algorithm showing a disordered region (963-1320 aa) with high PONDR score. g, Multiple sequence alignments indicating that the 6 acetylation sites (arrows) in the MED1 IDR are conserved among species. h, Immunostaining with anti-FLAG antibodies combined with DAPI staining for MCF7 cells with ectopic expression of FLAG-tagged WT or 6KR MED1. Scale bar is 20 μm. For panels a (MED1 test), c, e, h, the results are representative of three independent experiments.
Extended Data Fig. 4 MED1 IDR deacetylation is regulated by SIRT1 under stress conditions and SIRT1 is associated with SEC.
a, Immunoblotting showing enhanced acetylation of FLAG-MED1 in HEK293T cells treated with Ex527. b, Immunoblotting showing enhanced acetylation of endogenous MED1 in MCF7 cells treated with Sirtinol. c, Immunoblotting showing reduced acetylation of FLAG-MED1 in MCF7 cells treated with CoCl2 (24 h). HIF-1α was detected to demonstrate the hypoxic response. d, Immunoblotting showing reduced acetylation of FLAG-MED1 in MCF7 cells under heat shock (42 °C, 2 h), while treatment of Ex527 (50 μM) prevented this effect. e, Immunoblotting showing elevated SIRT1 detected in association with endogenous AFF4 in T47D cells treated with DFOM (200 μM, 6 h). f, Immunoblotting showing elevated SIRT1 detected for association with endogenous CDK9 in MCF7 cells treated with DFOM (200 μM, 18 h), while equivalent association of Cyclin T1 with CDK9 with or without DFOM treatment was detected. g, Immunoblotting showing elevated SIRT1 detected in association with endogenous AFF4 in MCF7 cells challenged by heat shock (42゜C, 2 h), while equivalent association of CDK9 with AFF4 with or without heat shock was detected. h, Chart showing SEC components, SIRT1 and other AFF4-binding proteins detected by IP-Mass Spectrometry in each sample with area values and visualized with a color scale. The bait protein, AFF4, does not show a significant difference in levels among samples. i, IGV snapshots showing elevated occupancy of AFF4 and SIRT1 in promoter-proximal regions of representative genes in DFOM-treated (200 μM, 6 h) MCF7 cells detected by CUT&RUN-seq. For panels b and e–g, IgG was used as an IP control. For panels a–g, densitometry analysis was performed on immunoblotting images, and the normalized protein levels (relative to the IP target protein, FLAG tag of MED1 or endogenous MED1, AFF4 or CDK9) are shown, and these immunoblots are representative of three independent experiments.
Extended Data Fig. 5 Acetylation site mutant of MED1 alters gene expression in steady-state and stress conditions in ER+ breast cancer cells.
a, Workflow depicting the methods employed to replace endogenous MED1 with ectopic MED1 in MCF7 or T47D cells. Lentiviruses expressing gRNAs targeting MED1 were transfected into pools of cells expressing ectopic HA-tagged MED1 (WT or 6KR/Q mutant) with CRISPR-mutated target sequences. b, c, Immunoblotting confirming efficient depletion of MED1, expression of HA-tagged WT and 6KR/Q MED1 (after CRISPR KO) at levels comparable to that of endogenous MED1 in MCF7 (b) and T47D (c) cells. d–f, Poly(A) RNA-seq for 6KR/KO and WT/KO MCF7 cells in steady state. Three biological replicates were detected for each cell type. d, Volcano plot showing Fold-change versus Adjusted P value (two-sided) for each detected gene, with the cut-off for defining DEGs (FC > 2, P adj < 0.05) indicated. e, GSEA comparing the 6KR/KO versus WT/KO gene rank list with KEGG (left) and GOBP (right) gene lists. Adjusted P values are shown in addition to NES reported by bars. f, GSEA revealing that the up-regulated (upper) and down-regulated (lower) genes in MED1 KO versus control cells (strong or mild) are enriched, respectively, in the sets of down-regulated and up-regulated genes of 6KR/KO versus WT/KO cells. NES values are shown. g, Detection by qRT-PCR showing stronger induction of representative hypoxia-induced genes in 6KR/KO versus WT/KO T47D cells treated with 200 μM DFOM. For each of WT/KO or 6KR/KO cells, expression of the gene at each condition was normalized by its expression at control condition in this cell type. N = 3 (biological replicates) for each group. Bars report mean ± SEM. If < 0.05, the P values of two-sided unpaired t-test comparing cell types under specific conditions are shown. For panels b and c, immunoblots are representative of three independent experiments.
Extended Data Fig. 6 The acetylation-defective 6KR mutation of MED1 enhances chromatin occupancy of RNA Pol II which is also associated with endogenous acetylation level.
a, IGV snapshots of representative stress-induced genes with occupancy of HA (MED1) and Pol II (RPB1 S5P) shown for WT/KO and 6KR/KO cells. b, Comparable levels of representative Mediator subunits detected for association with immunoprecipitated endogenous MED17 in WT/KO, 6KR/KO and 6KQ/KO MCF7 cells. IgG was used as the IP control. c, Workflow depicting sequential ChIP-seq by anti-FLAG antibodies and pan-anti-acetyl-lysine antibodies to detect acetylation levels of FLAG-tagged MED1 (and its associated proteins) in genomic locations. d, Metaplots of occupancy of FLAG and acetylation for pre-defined MED1 peaks (from ChIP-seq in untreated MCF7) in High Ac, Mid Ac and Low Ac groups. e, Range-based heatmap of the occupancy of FLAG-MED1, acetyl-lysine (2nd ChIP), MED17, Pol II (RPB1) and p300 at pre-defined MED1 peaks (from ChIP-seq in untreated MCF7) that are stratified into 3 tiers based on relative acetyl-lysine density (lower 25%, middle 50% and upper 75% percentile, referred to as Low Ac, Mid Ac, High Ac). f, Metaplots of occupancy of MED17, Pol II (RPB1 S5P) and p300 for pre-defined MED1 peaks (from ChIP-seq in untreated MCF7) with comparison among High Ac, Mid Ac and Low Ac groups. g, h, Pie diagram showing the constitution of High/Mid/Low Ac groups of MED1 peaks for occupancy of MED17 and Pol II (g), and types of regulatory elements (h).
Extended Data Fig. 7 MED1 IDR acetylation mutation effects on LLPS and Pol II incorporation.
a, Colloidal blue staining for HIS-tagged EGFP-fused MED1 IDRs (6KR or 6KQ, 963-1320 aa) purified from E.coli. b, Reduced saturation concentration for 6KQ versus 6KR MED1 IDR. N = 4 (biological replicates). P value of two-sided unpaired t-test is shown. c, Colloidal blue staining for WT MED1 IDR fragment (fused to EGFP) and CBP (GST-tagged) purified from E.coli (left), and immunoblotting confirming the enforced acetylation of MED1 IDR (right). d, Droplets formed by WT MED1 IDR (fused with EGFP), with enhanced in-droplet partitioning of the acetylated versus control MED1 IDR. Representative images are shown (left) where the scale bars are 10 μm. The partition coefficient (inside versus outside droplets) is plotted, comparing control and the acetylated MED1 IDR (right). Bars report mean ± SEM. P value of two-sided unpaired t-test is shown. The parentheses show the numbers of droplets analyzed (across 3 biological replicates). e, Reduced saturation concentration for CBP-acetylated versus control WT MED1 IDR. N = 4 (biological replicates). P value of two-sided unpaired t-test is shown. f, g, Faster recovery of droplets formed by CBP-treated versus control WT MED1 IDR revealed by FRAP. Images for representative droplets (f) and recovery curves (g) are shown. Scale bar for images is 10 μm. Data points report mean ± SEM. Control and CBP-acetylated MED1 IDRs were tested for 6 and 7 droplets (across 3 biological replicates), respectively. P values (two-sided unpaired t-test) for each data point are shown. h, RNA Pol II CTD heptad sequences used in the Pol II co-condensation experiments (left) and colloidal blue staining of the mCherry-fused Pol II CTD heptads (right). i, j, Immunoblotting detection of Mediator subunits and Pol II (RPB1) remaining on beads after washing the purified Mediator complex from HeLa-S cells by buffers with increasing concentrations of salt (100/150/200/300 mM KCl) (i) or buffers with 1,6- or 2,5-Hexanediol (Hexn) at 2% or 10% concentration (j). For panels a, c, h–j, gel staining and immunoblots are representative of three independent experiments.
Extended Data Fig. 8 Acetylation-defective 6KR mutant MED1 promotes tumorigenesis and stress tolerance in culture.
a, More colonies formed by 6KR/KO versus WT/KO T47D cells stained with crystal violet following 25 days growth after initial seeding. b, c, Declined viability of 6KR/KO and WT/KO T47D cells treated for 48 h with various concentrations of DFOM (b) or paraquat (c) monitored by MTT assay showing tolerance of 6KR/KO. N = 4 (biological replicates) in each group. Bars report mean ± SEM. If < 0.05, the P values of two-sided unpaired t-test comparing cell types are shown. d, e, Enhanced migration of 6KR/KO versus WT/KO MCF7 cells revealed by wound healing assay. Representative images showing edges of adherent WT/KO or 6KR/KO MCF7 cells (d) and curves of distance of edge migration (e) are shown. Scale bar for images is 500 μm. N = 6 (biological replicates) for each cell type. Data points report mean ± SEM. P value of one-way ANOVA comparing cell types is indicated.
Extended Data Fig. 9 Acetylation-defective 6KR mutant MED1 promotes tumorigenesis and stress tolerance in a mouse model.
a, Measurement of tumor volume when mice bearing tumors of WT/KO or 6KR/KO MCF7 cells were sacrificed 133 or 95 days after implantation, respectively. Data are mean ± SEM. Number of tumors measured in each group is shown in parentheses. b, Immunoblotting showing comparable expression of HA-tagged (WT or 6KR) MED1 in WT/KO and 6KR/KO MCF7 tumors. Size-matched WT/KO and 6KR/KO MCF7 tumors were collected from NSG mice. Images with densitometry results (normalized by β-actin) are shown and plotted (right). c, Immunoblotting showing increased expression of NRF2 and NDRG1 in 6KR/KO versus WT/KO MCF7 tumors. Images with densitometry results (normalized by β-actin) are shown. d, e, Poly(A) RNA-seq for 6KR/KO and WT/KO MCF7 tumors developed in NSG mice. Three biological replicates were detected for each type of tumor. d, GSEA comparing the gene rank list with KEGG (left) and GOBP (right) pathway gene lists. Adjusted P values are noticed in addition to NES reported by bars. e, Fold change values of select stress-activated genes detected by RNA-seq. For b, c, each group has 3 tumors as biological replicates, the tests used the same samples, and the immunoblots are representative of three technical replicates. For a, b, the n.s. (no significance) indicates a p value > 0.05 (two-sided unpaired t-test).
Extended Data Fig. 10 Working model.
A model showing deacetylation of MED1 in promoter-proximal regions by SEC-associated SIRT1 under stress conditions and enhanced incorporation of Pol II through IDR-mediated interaction between MED1 IDR and Pol II CTD (shown in upper inset images). Thus, through increased RNA Pol II association, deacetylation of MED1 amplifies induction of cyto-protective genes and mitigates suppression of growth-supportive genes in stress conditions (shown in lower inset images). In parallel with the extensively-studied action of SEC/P-TEFb on Pol II pausing-release, we propose a SEC-SIRT1-MED1 axis that enables the sequential entry and action of Pol II to support survival and growth of stressed cells. This image was created with BioRender.
Supplementary information
Supplementary Information (download PDF )
Supplementary Tables 1 and 2, Figs. 1–8 and source data (uncropped scans) for supplementary figures.
Source data
Source Data Fig. 1 (download PDF )
Unprocessed western blots and/or gels.
Source Data Fig. 2 (download PDF )
Unprocessed western blots and/or gels.
Source Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Fig. 6 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 1 (download PDF )
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 3 (download PDF )
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 4 (download PDF )
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 5 (download PDF )
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 6 (download PDF )
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 6 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 7 (download PDF )
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 7 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 8 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 9 (download PDF )
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 9 (download XLSX )
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, R., Mo, Y., Barrows, D. et al. MED1 IDR deacetylation controls stress responsive genes through RNA Pol II recruitment. Nat Chem Biol 22, 501–513 (2026). https://doi.org/10.1038/s41589-025-02035-7
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41589-025-02035-7
