
Abstract
TRPM5 is a Ca2+-activated monovalent cation channel essential for taste perception, insulin secretion and gastrointestinal chemosensation. Canonical TRPM5 activation requires Ca2+ binding at two distinct sites: an agonist site within the lower vestibule of the S1–S4 pocket in the transmembrane domain (CaTMD) and a modulatory site in the intracellular domain (CaICD) that tunes voltage dependence and agonist sensitivity. Here we characterize CBTA as a noncalcium agonist that binds to the upper vestibule of the S1–S4 pocket, directly above CaTMD. CBTA alone mimics the dual role of CaTMD and CaICD, merging agonist activation with voltage modulation. CBTA also renders TRPM5 supersensitive to Ca2+, synergistically hyperactivating the channel even at near-resting Ca2+ levels. We further demonstrate that the inhibitor triphenylphosphine oxide binds the same site but stabilizes a nonconductive state. These opposing effects reveal the upper S1–S4 pocket as a multifunctional regulatory hub integrating activation, inhibition and modulation in TRPM5.

Similar content being viewed by others


Main
TRPM5 is a calcium-activated, nonselective monovalent cation channel that couples intracellular Ca2+ signals to membrane depolarization, driving critical physiological responses such as taste perception, insulin secretion and immune responses1,2,3,4,5,6,7,8,9,10,11. In taste receptor cells, it has a key role in detection of sweet, bitter and umami flavors4,5,12,13,14. Beyond chemosensation, TRPM5 also contributes to endocrine and metabolic functions: in pancreatic β cells, it amplifies depolarization in response to calcium oscillation, promoting insulin release3,6. Consistent with this role, loss of TRPM5 function has been shown to impair insulin secretion and glucose tolerance3,10, suggesting that pharmacological modulation of TRPM5 activity may offer a strategy for restoring β cell function in diabetic individuals15. However, the pharmacological landscape of TRPM5 remains limited because of a lack of TRPM5-specific small-molecules16.
Canonical TRPM5 activation is driven by Ca2+ binding at two discrete sites: a primary agonist site in the lower vestibule of S1–S4 pocket in the transmembrane domain (CaTMD) and a modulatory site in the intracellular domain (CaICD)17. The ICD site is known to regulate the voltage dependence and promote calcium binding at the CaTMD site, modulating sensitivity to physiological Ca2+ oscillations6,17. Until recently, Ca2+ was the only known agonist of TRPM5. The discovery of small-molecule compounds, such as 5-chloro-N-((5-chlorothiazol-2-yl)methyl)benzo[d]isothiazol-6-amine, hereafter referred to as CBTA, as the first noncalcium positive regulator for TRPM5 marks a major breakthrough18,19. Although it is still unclear whether CBTA activates TRPM5 independently of calcium, it opens avenues for the development of TRPM5-targeted therapies. In addition to agonists, TRPM5 is selectively inhibited by small-molecule inhibitors such as triphenylphosphine oxide (TPPO) and N′-(3,4-dimethoxybenzylidene)-2-(naphthalen-1-yl)acetohydrazide (NDNA)17,20, which provide tools for dissecting TRPM5 physiological function and potential leads for therapeutic development in settings of TRPM5 hyperactivity.
We previously showed that NDNA binds to the cleft between the S1–S4 domain and the S5–S6 pore domain, inhibiting TRPM5 by locking the TMD in an apo-like closed conformation17. In contrast, the precise binding sites and mechanisms of action for CBTA and TPPO remain unknown. Consequently, how these small molecules coordinate with the calcium binding sites in the TMD (CaTMD) and ICD (CaICD), which is essential for calcium or voltage-dependent channel activation, remains unclear17,21. Understanding these couplings is key to uncovering how TRPM5 integrates multiple regulatory inputs to achieving precise functional control and for drug development to treat TRPM5-linked diseases22.
Here, using cryo-electron microscopy (cryo-EM), electrophysiology and mutational analyses, our study establishes the upper S1–S4 pocket as a multifunctional regulatory site for channel activation, inhibition and modulation, providing insights into TRPM5 gating and proposing a unifying mechanism of TMD and ICD communication that underpins the polymodal regulation of TRPM channels.
Result
Direct activation of TRPM5 by CBTA and its synergy with Ca2+
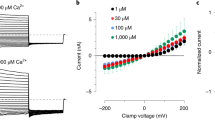
Although compound CBTA has been shown to evoke TRPM5 currents, it remains unclear whether CBTA acts as a direct agonist or as a modulator that potentiates calcium-activated TRPM5 currents, as previous studies did not test the effect of CBTA in the absence of intracellular calcium18. To address this, we chelated intracellular free calcium using EGTA and performed whole-cell patch-clamp experiments on zebrafish TRPM5. In the absence of CBTA, no TRPM5 currents were detected, confirming that calcium was effectively chelated by EGTA (Fig. 1a, black trace). Extracellular application of CBTA-activated TRPM5 at both positive and negative transmembrane voltages (Fig. 1a and Extended Data Fig. 1a,b). At low concentrations (≤1 µM), the CBTA-induced TRPM5 current was outwardly rectifying but became less rectifying at higher concentrations (≥3 µM) (Fig. 1a,b). This concentration-dependent voltage dependence is similar to that of the endogenous agonist calcium1,17. These findings establish compound CBTA as the first known noncalcium agonist of TRPM5.

a, Whole-cell recording of currents from cells overexpressing TRPM5, elicited by various concentrations of CBTA. The pipette solution contained 5 mM EGTA. Cells were held at −100 mV for 100 ms, followed by a voltage ramp from −100 mV to +100 mV over 200 ms. The pipette solution contained 5 mM EGTA. b, The ratio of TRPM5 current at +100 mV and −100 mV, indicating the level of voltage dependence (n = 5). Bars represent the mean; each dot corresponds to an independent cell measurement. Here and below, n represents the number of independent cells recorded in whole-cell patch-lamp experiments. c, Dose–response curves and EC50 estimates of TRPM5 activation by CBTA, measured with intracellular pipette solutions containing either 5 mM EGTA (n = 5) or 100 nM Ca2+ (n = 4). Circles represent the mean current and error bars represent the s.e.m.
To assess the potency of CBTA, we measured its concentration–response relationship and estimated its half-maximal effective concentration (EC50) in the micromolar range (Fig. 1c, red and pink traces). This result was surprising, as it was approximately 500 times less potent than previously reported18. A key difference in our experiments was the inclusion of EGTA to ensure a calcium-free intracellular solution, whereas the previous study maintained 1 µM free calcium, allowing for potential calcium interacting with TRPM5. This discrepancy prompted us to examine the effect of CBTA in the presence of calcium. Indeed, with just 100 nM free cytosolic calcium—a concentration of calcium that by itself does not activate TRPM5 (Extended Data Fig. 1c,d)—the EC50 of CBTA shifted to the nanomolar range (Fig. 1c, blue and cyan traces), similar to the previously reported value for human TRPM5 (ref. 18). Moreover, coapplication of 100 nM calcium and 100 nM CBTA robustly activated TRPM5 in a voltage-independent manner (Extended Data Fig. 1g,h). In contrast, neither 100 nM calcium nor 100 nM CBTA alone was sufficient to activate the channel (Extended Data Fig. 1g,h).
These findings demonstrate that CBTA can activate TRPM5 independently of calcium but also synergizes with calcium to dramatically enhance TRPM5 activity. While such a synergistic mechanism has not previously been described for TRPM5, TRPM4 or TRPM2, it is reminiscent of the cooperative action of calcium and icilin on TRPM8, where both ligands bind the voltage-sensing-like domain to drive channel activation17,21,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45.
CBTA binds to the upper S1–S4 pocket
To investigate the mechanism of CBTA agonism and its cooperativity with calcium, we determined the cryo-EM structures of zebrafish TRPM5 in the presence of CBTA alone and in combination with calcium. In the CBTA-only sample, 1 mM EDTA was included to chelate calcium and ensure a calcium-free condition, as established in our previous work17. The CBTA/EDTA–TRPM5 and CBTA/Ca2+–TRPM5 cryo-EM maps were refined to resolutions of 3.1 and 2.9 Å, respectively, with the TMD achieving a local resolution up to 2.6 Å (Supplementary Table 1 and Supplementary Fig. 1–2). The overall structures closely resemble the previously published TRPM5 structures, featuring the characteristic tetrameric assembly with a TMD and a large ICD consisting of MHR3/4 and MHR1/2 domains (Fig. 2a,b)17.

a, The cryo-EM map of CBTA/EDTA–TRPM5. One subunit of TRPM5 is colored in blue. The CBTA density is shown in yellow. The MHR1/2 and MHR3/4 domains in TRPM5 are also indicated. b,c, The cryo-EM map (b) and atomic model (c) of CBTA/Ca2+–TRPM5. One subunit of TRPM5 is colored in red. The CBTA density is shown in yellow. CaTMD and CaICD in CBTA/Ca2+–TRPM5 are also indicated using green spheres. d, A close-up view of the CBTA-binding site. Residues that form the CBTA-binding pocket are shown in stick representation. The cryo-EM density corresponding to the CBTA molecule is shown using a semitransparent surface. e, The effect of 20 µM CBTA on whole-cell currents—recorded at −100 mV and +100 mV—in nontransfected tsA201 cells (n = 4), TRPM5 WT (n = 5) and TRPM5 mutants F730A, L764A, R834A, R998A and N723A (n = 4 each), with 5 mM EGTA included in the recording pipette, presented as bar graphs (mean). f, Pore profiles of the open states identified in the CBTA/EDTA–TRPM5 and Ca2+/EDTA–TRPM5 datasets, compared to those of the apo closed state and Ca2+-bound open state. g, The distribution of open and closed states in the presence of different calcium concentrations. The CaTMD and CaICD are allosterically coupled as we were unable to identify conformations of TRPM5 with only CaTMD or CaICD bound.
Both CBTA/EDTA–TRPM5 and CBTA/Ca2+–TRPM5 structures revealed a well-defined density within the upper vestibule of the S1–S4 pocket of each subunit, positioned above the CaTMD binding site (Fig. 2c). This density is triangular, closely matching the shape of a CBTA molecule (Fig. 2d). Notably, this density was not observed in any previously reported TRPM5 structures, indicating a previously unrecognized ligand-binding site in TRPM5. However, other TRPM family members, such as TRPM3 and TRPM8, also harbor ligands within analogous regions of the S1–S4 pocket, suggesting that this site may represent a conserved ligand recognition hotspot across the family39,40,41,46,47,48. The CBTA-binding site is surrounded by several hydrophobic residues, such as L764, M726, Y727, F730, W760 and I801, which cluster around the upper portion of the pocket (Fig. 2d). In addition, several polar and charged residues, including R834, Y995, E768 and D797, engage the 5-chlorothiazole moiety of CBTA from the bottom of the site (Fig. 2d). Although calcium and CBTA bind in proximity, they interact with distinct sets of residues (Fig. 2c)17.
To validate the CBTA-binding site, we introduced alanine substitutions at key interacting residues and assessed their response to CBTA. The mutants showed negligible CBTA-induced currents (Fig. 2e and Extended Data Fig. 2a–c). As a control, calcium still activated these mutants, confirming that the loss of CBTA response was not because of protein misfolding or trafficking issues (Extended Data Fig. 2d–g).
CBTA renders TRPM5 supersensitive to calcium
The CBTA/Ca2+–TRPM5 structure adopts an open conformation, mirroring the open state previously observed with calcium alone (Fig. 2f)17. In contrast, the consensus map of CBTA/EDTA–TRPM5 revealed heterogeneity in the TMD and showed a weak and ambiguous signal at the CaTMD site, despite the presence of 1 mM EDTA, a condition known to eliminate calcium binding in TRPM5 (Extended Data Fig. 3a,b)17. To resolve this heterogeneity, we performed signal subtraction and focused classification (Methods and Supplementary Fig. 1). Strikingly, while most particles corresponded to a closed state with CBTA bound but no CaTMD density, a small fraction (~5%) adopted an open TMD conformation (Fig. 2f, Supplementary Table 1 and Extended Data Fig. 3c), closely resembling the TMD conformation in the CBTA/Ca2+–TRPM5 structure.
Surprisingly, in this minor open-state population, we observed robust Ca2+ density at the CaTMD site, whereas the CaICD site remained unoccupied (Extended Data Fig. 3c). This was unexpected given the stringent calcium-chelating conditions used. The most likely explanation is that the calcium signal originates from trace contaminants commonly found in buffer components, which can yield free calcium in the micromolar range. Remarkably, even under these conditions, CBTA-bound TRPM5 was able to compete with EDTA—a strong calcium chelator—to coordinate Ca2+ at the CaTMD site. This observation raises two possible explanations: either CaTMD intrinsically has higher affinity for calcium than CaICD or CBTA specifically enhances calcium binding at CaTMD. To distinguish between these possibilities, we conducted a calcium titration and determined TRPM5 structures across a range of concentrations—from saturating to subsaturating levels (Supplementary Figs. 3–5).
At high calcium concentrations (≥200 µM), both CaTMD and CaICD sites were consistently occupied and the channel adopted an open conformation (Fig. 2g and Extended Data Fig. 3d)17. At an intermediate concentration (6 µM), we observed a mixture of open and apo states, with either both sites occupied or neither17. At lower concentrations (2 or 1 µM), only the apo state was observed, with neither site occupied (Fig. 2g and Extended Data Fig. 3d). These results indicate that CaTMD and CaICD have comparable affinities to calcium and support the conclusion that CBTA specifically and dramatically enhances calcium binding at CaTMD. This CBTA-induced stabilization of CaTMD binding under otherwise nonpermissive conditions aligns with electrophysiological data showing enhanced calcium sensitivity (Extended Data Fig. 1i) and reveals a potent mechanism by which CBTA drives TRPM5 activation through synergistic engagement of the CaTMD site.
Synergetic interplay between CBTA and CaTMD
Our structural and functional data support a strong allosteric coupling between CBTA and calcium, which cooperatively drive TRPM5 hyperactivation. To understand how CBTA enhances calcium binding at CaTMD, we aimed to isolate its structural effect in the absence of calcium. Specifically, we asked whether CBTA binding alone could preconfigure the CaTMD site to favor calcium coordination, thereby explaining its apparent ability to recruit trace calcium even under chelating conditions. To address this, we used 5 mM EGTA, a more specific and potent calcium chelator than EDTA, and determined the cryo-EM structure of TRPM5 in the presence of CBTA and EGTA (Supplementary Table 2 and Supplementary Fig. 6). This condition yielded a true CBTA-only structure, in which CBTA occupied the same binding site as observed previously but no calcium density was observed at the CaTMD site, confirming effective calcium chelation (Fig. 3a).

a, The structure of CBTA/EGTA–TRPM5. Left: cryo-EM map of TRPM5 with one subunit colored in magenta. Right: a close-up view of the S1–S4 domain. The CBTA molecule is shown using stick representation and colored in yellow. The cryo-EM density for CBTA is shown using a semitransparent surface. b, A close-up view of the CaTMD site mutant D797A with 5 mM calcium. No CaTMD is observed. c, A close-up view of the CaTMD site mutant D797N with 5 mM calcium. While a strong CBTA density is present, no CaTMD is observed. d, The pore profile plot of Ca2+–TRPM5 WT and CBTA/Ca2+–TRPM5-D797N. Despite the presence of CBTA, the CBTA/Ca2+–TRPM5-D797N structure shows a closed state similar to the apo state.
Comparison of the apo and CBTA-only TRPM5 structures revealed a key mechanistic insight (Extended Data Fig. 3e–g); CBTA binding induces local rearrangements in the S1–S4 domain that remodel the CaTMD site. In the apo state, residue R834 forms salt bridges with E768 and D797, both of which are critical for calcium coordination17,49. Upon CBTA binding, R834 is displaced, disrupting these salt bridges and freeing E768 and D797. This reorganization primes the CaTMD site for calcium binding, providing a structural explanation for the dramatically enhanced calcium sensitivity observed in the presence of CBTA.
To further dissect this synergy, we examined TRPM5 mutants designed to disrupt either the CBTA or CaTMD binding site. Substitution of L764, a key hydrophobic residue in the CBTA pocket, abolished CBTA response but preserved calcium-dependent activation (Extended Data Fig. 2b–e). Strikingly, this mutant (L764A) showed no response to coapplication of low-dose CBTA and calcium (100 nM each), a combination that robustly activates wild-type (WT) TRPM5 (Extended Data Fig. 4a,b). Similarly, substitutions at the CaTMD site—D797A, D797N and Q771A—all abolished calcium-dependent activation (Extended Data Fig. 4e)17,49 and eliminated the synergistic response to CBTA and calcium (Extended Data Fig. 4f). These findings indicate that both binding sites must be intact to support the cooperative effect.
Structural analysis of the CaTMD mutants provided further insight into this coupling (Supplementary Tables 2 and 3 and Supplementary Figs. 7–9). In the D797A mutant, CBTA density was markedly weakened (Fig. 3b), likely because of the loss of a direct interaction between CBTA and the D797 side chain. By contrast, D797N preserved strong CBTA density (Fig. 3c), making it a suitable background to examine how the absence of calcium binding at CaTMD affects CBTA-induced channel gating. The structure of CBTA/Ca2+–TRPM5-D797N revealed a closed pore and no calcium density at CaTMD, closely resembling the CBTA-only structure rather than the open CBTA/Ca2+–TRPM5 structure (Fig. 3d). These findings reinforce a model in which CBTA and calcium engage distinct but structurally interdependent sites and their coordinated allosteric interactions are required to promote calcium binding and stabilize the open-channel conformation.
TPPO inhibits TRPM5 by targeting the upper S1–S4 pocket
The structure of CBTA-bound TRPM5 identified the upper S1–S4 pocket as a previously unrecognized agonist-binding site. In contrast, our earlier work showed that the antagonist NDNA binds at a distinct site, wedged between the S1–S4 domain and the S5–S6 pore domain17. These observations suggest that TRPM5 can be modulated by small molecules through multiple structural mechanisms. To further explore this concept, we turned to TPPO, a widely used TRPM5 antagonist20, whose binding site and mode of inhibition remained unknown.
TPPO potently inhibits human, mouse and zebrafish TRPM5 at 100 µM (Fig. 4a)20. We determined the cryo-EM structure of TRPM5 in the presence of calcium and TPPO at 3.0-Å resolution (Supplementary Table 3 and Supplementary Fig. 10). Including calcium enabled us to distinguish the specific inhibitory effect of TPPO from the apo closed state that occurs in the absence of agonist. The structure revealed a well-defined tripod-shaped TPPO density within the upper S1–S4 pocket, directly overlapping the CBTA-binding site (Fig. 4b,c). This finding shows that TPPO, despite being an inhibitor, engages the same allosteric site as the agonist CBTA.

a, I–V relationships of whole-cell current trace obtained using a voltage ramp protocol from −100 mV to +100 mV before and during application of 100 µM TPPO. b, The cryo-EM map of TPPO/Ca2+–TRPM5. One subunit of TRPM5 is colored in blue. The TPPO molecule is indicated in yellow. The CaTMD and CaICD are indicated using green spheres. c, Top: a close-up view of the TPPO-binding site in TPPO/Ca2+–TRPM5 structure. The cryo-EM density of TPPO and CaTMD is shown in surface representation and colored in yellow and green, respectively. Bottom: a 60° rotation view with some residues hidden for clarity. d, The relative inhibition of TPPO on WT and various binding-site mutants (WT, n =4; H837A, n = 4; I838A, n = 3; F730A, n = 4; R998A, n = 4). e, The pore domain structure of apo TRPM5, Ca2+–TRPM5 and TPPO/Ca2+–TRPM5. Bars represent the mean; each dot corresponds to an independent cell measurement. f, The allosteric effect of TPPO and CBTA to the pore domain. The S4–S6 helices of CBTA/Ca2+–TRPM5 (red) and TPPO/Ca2+–TRPM5 (blue) structures are shown with the S1–S4 domain of one subunit aligned. TPPO or CBTA binding affects the side-chain conformation of H837 and F839 in the S4 helix, which is further propagated to the S5 and S6 helices of adjacent subunit (indicated with a prime symbol). This eventually leads to different position of I966, which forms the gate of TRPM5. The CaTMD site at the bottom of CBTA/TPPO-binding site is also shown.
The binding of TPPO is mediated by both hydrophobic and polar interactions. Its three phenyl rings engage a hydrophobic pocket within the S1–S4 domain, while its oxygen atom forms a critical polar interaction with R998 (Fig. 4c). Consistent with this, substitution of R998 to alanine abolished TPPO inhibition (Fig. 4d and Extended Data Fig. 5a). Moreover, TPP, which lacks the oxygen atom of TPPO, exhibited markedly weaker inhibition, underscoring the importance of this polar interaction20. Additional substitutions in the TPPO-binding pocket also reduced the inhibitory effect, further supporting the functional relevance of the observed binding site (Fig. 4d and Extended Data Fig. 5b–e).
In the TPPO-bound structure, calcium densities at the CaTMD and CaICD sites remain well defined, indicating that TPPO does not interfere with calcium binding (Fig. 4c). Despite this, the pore remains nonconductive, with a narrowest pore radius of 1.0 Å (Fig. 4e). To understand why TPPO and CBTA, despite binding to the same S1–S4 pocket, exert opposite effects on TRPM5 gating, we compared the TPPO/Ca2+–TRPM5 closed state with the CBTA/Ca2+–TRPM5 open state. Structural alignment of their S1–S4 domains revealed that the distinct shapes and binding modes of CBTA and TPPO engage divergent allosteric pathways, with the V-shaped conformation of CBTA altering S4 dynamics in a manner opposite to the tripod-like TPPO. These ligands induce differential conformational shifts in the S4 helix, particularly at H837 and F839, with H837 facing the ligand-binding site and F839 oriented toward the S5 helix of the adjacent subunit. This intersubunit contact enables coupling that transmits conformational signals—whether triggered by agonist or antagonist binding at the S1–S4 domain—to the pore-lining S5–S6 helices and the gate residue I966 (Fig. 4f).
Together, these structures reveal that the upper S1–S4 pocket acts as a regulatory hub for TRPM5, capable of supporting either activation or inhibition depending on the chemical architecture of ligands. On the basis of these observations, we propose that TPPO inhibits TRPM5 by interfering with allosteric communication between the S1–S4 domain and the pore domain, thereby preventing the pore from fully opening. This mechanism contrasts sharply with that of NDNA, which locks the channel in an apo-like conformation with a fully closed pore, even in the presence of bound calcium17.
Bidirectional allosteric communication between ICD and TMD
Our previous findings showed that calcium-dependent TRPM5 activation requires calcium binding at both CaTMD and CaICD sites17. Disruption of the CaICD site, as in the E337A mutant, renders the channel voltage-dependent and inactive at physiological membrane potentials and reduces calcium affinity at CaTMD17. These findings highlight the critical role of ICD-to-TMD communication in channel activation.
Interestingly, our data show that CBTA can bypass this requirement, as a small subset of particles in the CBTA/EDTA dataset adopted an open conformation with CBTA bound but no CaICD density. However, because the ICD is relatively flexible and the map was obtained at more a moderate resolution (3.5 Å), we could not exclude the possibility that the apparent absence of CaICD was because of limited resolution. To address this, we turned to the E337A mutant, which eliminates CaICD binding by design. We found that CBTA robustly activates E337A with an EC50 comparable to WT (Fig. 5a,b and Extended Data Fig. 6a,b). By contrast, calcium alone fails to activate E337A under these conditions (Extended Data Fig. 6c–h)17. Consistent with the electrophysiological data, cryo-EM analysis of CBTA/Ca2+–TRPM5-E337A yielded a structure at substantially higher resolution (2.7 Å), unambiguously revealing a homogeneous open conformation despite the absence of CaICD density (Fig. 5c, Supplementary Table 3, Extended Data Fig. 7a,b and Supplementary Fig. 11), whereas Ca2+–TRPM5-E337A, lacking both CaICD and CBTA, remained closed (Fig. 5c)17. Structural comparison shows that CBTA induces movement of the TRP helix, a key structural element for channel gating by mediating the ICD–TMD communication (Fig. 5d), similar to that triggered by CaICD binding in the WT17. These findings suggest that CBTA functionally mimics CaICD by engaging the TRP helix, thereby replacing its role in ICD-to-TMD coupling.

a, Whole-cell recording of TRPM5-E337A elicited by various concentrations of CBTA. The pipette solution contained 5 mM EGTA. b, The dose–response curve and EC50 estimates of TRPM5-E337A to CBTA (n = 4). Circles represent the mean current and error bars represent the s.e.m. c, The pore profiles of apo TRPM5-E337A, Ca2+–TRPM5-E337A and CBTA/Ca2+–TRPM5-E337A. d, Structural comparison between CBTA/Ca2+–TRPM5(E337A) and Ca2+–TRPM5(E337A) (PDB 7MBU). The structures are shown in transparent cartoon representation, except for the TRP helix. CBTA is shown using yellow spheres. CBTA binding induced a motion of the TRP helix. e, The pore profiles of Ca2+–TRPM5-Q771A and CBTA/Ca2+–TRPM5-Q771A. f, The CaTMD site of CBTA/Ca2+–TRPM5-Q771A with the atomic model and cryo-EM density overlaid. The CBTA and CaTMD density is indicated. g, Bar graphs showing currents measured at +100 mV and −100 mV for Q771A (n = 4) and D797N (n = 4) mutants in the presence of 300 nM free calcium, without or with 1 μM CBTA.
To investigate whether the ICD–TMD coupling is bidirectional (that is, whether the TMD can in turn influence the ICD), we examined the structure of the CaTMD site mutant Q771A (Supplementary Table 4 and Supplementary Figs. 12 and 13), which abolishes Ca2+-induced channel activation. Although Ca2+–TRPM5-Q771A retains clear calcium density at CaTMD, unlike D797A/N (Extended Data Fig. 7c and Fig. 3b,c), it adopts a closed conformation with minimal S1–S4 rearrangement and TRP helix movement compared to Ca2+-bound WT (Fig. 5e and Extended Data Fig. 7d). Specifically, in WT TRPM5, CaTMD binding induces an outward tilt of the S2 helix, the S2–S3 linker and the S3 helix, repositioning the TRP helix to enable coordinated motion across the S1–S4 domain, the S5–S6 pore domain and the ICD (Extended Data Fig. 7d, left)17. In Q771A, the CaTMD-induced movements of the S1–S4 domain and TRP helix are negligible (Extended Data Fig. 7d, right). As a result, the pore domain of Ca2+–TRPM5-Q771A remains closed. In addition, the ICD adopts an intermediate conformation between the apo and calcium-bound WT states (Extended Data Fig. 7e), consistent with reduced CaICD occupancy (Extended Data Fig. 7f). Together, these analyses indicate that Q771 is dispensable for CaTMD coordination but required to couple CaTMD binding to conformational changes within the TMD (from the S1–S4 domain to the S5–S6 pore domain) and from the TMD to the ICD.
We then asked whether CBTA could restore this coupling. Electrophysiology experiments showed that CBTA and calcium synergistically hyperactivate Q771A (Fig. 5g and Extended Data Fig. 4e–h). The CBTA/Ca2+–TRPM5-Q771A structure exhibits strong CBTA and CaTMD densities and reveals an open pore with a radius of 2.7 Å (Fig. 5e,f and Supplementary Fig. 10). A structural comparison revealed that CBTA binding restores the S1–S4 domain conformation of Q771A seen in the open-state TRPM5 structure (Extended Data Fig. 7g) and focused classification showed full occupancy at the CaICD site (Extended Data Fig. 7h), in contrast to the calcium-alone condition. Together, these findings demonstrate that CBTA can restore impaired intra-TMD coupling (between the S1–S4 and S5–S6 pore domains) and TMD-to-ICD coupling by remodeling the S1–S4 domain and reestablishing allosteric communication across domains. CBTA, thus, acts as a bidirectional modulator, capable of bypassing or reconstituting defective coupling between ICD and TMD.
Discussion
Despite its critical roles in taste perception, insulin secretion, immune signaling and other physiological processes, the mechanisms underlying TRPM5 activation and pharmacological modulation have remained poorly defined. Here, we investigated CBTA, a potent synthetic agonist of TRPM5, and uncovered a previously unrecognized activation mechanism. Our structural and functional data reveal that CBTA binds a site in the upper vestibule of the S1–S4 pocket, activating TRPM5 independently of calcium (Fig. 6). Remarkably, CBTA also dramatically enhances calcium sensitivity at the CaTMD site and this synergy leads to robust TRPM5 hyperactivation (Fig. 6). These findings identify a druggable allosteric hotspot for the development of TRPM5-targeted therapeutics.

Cartoon illustrating how Ca2+, CBTA and TPPO differentially modulate TRPM5 activation. Canonical activation requires Ca2+ binding at the agonist site CaTMD and the modulatory site CaICD. CBTA, a noncalcium agonist, binds the upper vestibule of the S1–S4 pocket directly above CaTMD and mimics the dual roles of CaTMD and CaICD, merging agonist activation with voltage modulation. CBTA also renders TRPM5 supersensitive to Ca2+, synergistically hyperactivating the channel even at near-resting Ca2+ levels. In contrast, the inhibitor TPPO binds the same pocket but stabilizes a nonconductive conformation.
A central finding of this study is that the upper S1–S4 pocket serves as a multifunctional regulatory hub (Fig. 6)23. This site is adjacent to the TRP helix, a key structural element in channel gating. CBTA binding at this site triggers conformational changes across the protein similar to those induced by calcium, leading to channel opening. In contrast, TPPO, a TRPM5 antagonist, also binds to the same pocket but impairs the allosteric coupling between the binding site and pore domain, thereby preventing channel opening. These opposing outcomes—activation versus inhibition—from ligands engaging the same site underscore its versatility as a pharmacological switch. Notably, this mechanism echoes observations in other ion channels, including Cav1.1 and KCNQ2, where distinct ligands targeting an identical binding site can exert opposite effects on channel activity50,51.
This site also offers a conceptual contrast to the canonical activation mechanism of TRPM5, which requires calcium binding at two discrete binding sites (CaTMD and CaICD) to enable concerted gating transitions (Fig. 6)17,21. CBTA bypasses this dual-site requirement by driving a TMD-localized gating, which simplifies the activation pathway and provides a structural framework for drug targeting (Fig. 6).
Another key insight from this study is the discovery of bidirectional allosteric communication between the TMD and ICD. While our previous work established that calcium binding to the ICD modulatory site enhances agonist efficacy at the TMD—a well-recognized regulatory mechanism—our findings reveal that this relationship is reciprocal; agonist binding at the TMD, such as by CBTA or calcium, can in turn enhance calcium binding at the ICD site and promote conformational changes within the ICD17. This reciprocal interplay introduces a new conceptual layer to ion channel regulation, wherein spatially distinct agonist and modulatory binding sites communicate bidirectionally to coordinate gating.
Although our static cryo-EM structures reveal key ligand-binding modes, the dynamic processes and the associated free-energy landscape by which CBTA and TPPO influence channel gating remain incompletely understood. Future molecular dynamics simulations could complement our findings by visualizing the temporal sequence of conformational changes and allosteric coupling between the S1–S4 pocket and the pore, thereby deepening our mechanistic understanding of how agonists and antagonists exert opposing effects on TRPM5 function.
In summary, this work defines a shared allosteric pocket in TRPM5 that integrates activation, inhibition and interdomain coupling. We reveal how synthetic agonists can exploit this site to bypass or restore defective gating pathways. The modularity, accessibility and bidirectional signaling capacity of this regulatory hub make it an attractive platform for the rational design of selective TRPM5 modulators, with broad implications for targeting TRP channels in metabolic, sensory and neurological disorders.
Methods
Constructs and cloning
The genes encoding full-length TRPM5 from human (UniProt Q9NZQ8) and zebrafish (UniProt S5UH55) in pEG BacMam vector from our previous study were used17. The constructs included an N-terminal 8×His tag, an enhanced green fluorescent protein (GFP) and a thrombin cleavage site. Site-directed mutagenesis was carried out using either the QuikChange II site-directed mutagenesis kit (Qiagen) or the Q5 site-directed mutagenesis protocol (New England Biolabs), with all mutations verified by Sanger sequencing or whole-plasmid sequencing (Eurofins) (Supplementary Table 5).
Protein expression and purification
To produce baculovirus, each TRPM5 ortholog in the PEGN vector was transformed into DH10Bac cells (Gibco)52. P1 and P2 baculoviruses were then generated using Sf9 cells (Gibso, 12-659-017). P2 baculovirus (8%) was used to infect tsA201 cells (American Type Culture Collection, CRL-3216) grown in suspension culture with Freestyle 293 expression medium (Gibco). Following infection, the cells were incubated at 37 °C for 8–16 h before adding 10 mM sodium butyrate. The cultures were then shifted to 30 °C and agitated vigorously for 60 h. After 72 h of infection, cells were harvested by centrifugation at 4,000g for 30 min at 4 °C. The resulting cell pellets were washed with TBS buffer (150 mM NaCl and 20 mM Tris, pH 8.0) and stored at −80 °C.
Depending on the protein expression level, cell pellets from a 200–800-ml culture were thawed on ice and resuspended in TBS buffer supplemented with 1 mM PMSF, 0.8 μM aprotinin, 2 μg ml−1 leupeptin, 2 mM pepstatin A (protease inhibitors) and 1% GDN detergent (Anatrace). The membrane protein was extracted through whole-cell solubilization at 4 °C for 1.5 h with continuous rotation. Solubilized protein was clarified by centrifugation and the clear supernatant was incubated with 2 ml of TALON cobalt metal-affinity resin (Takara Bio) for 1 h. The resin was washed with 20 ml of TBS buffer containing 0.01% GDN and 15 mM imidazole. Protein was eluted using TBS buffer with 0.02% GDN and 250 mM imidazole. The eluate was concentrated to 500 μl and further purified by size-exclusion chromatography (SEC) using TBS buffer with 0.02% GDN. SEC fractions (0.3 ml each) containing TRPM5 were pooled and concentrated to >5 mg ml−1 for cryo-EM grid preparation.
EM sample preparation and data acquisition
Freshly purified TRPM5 protein in GDN detergent was mixed with designated amount of calcium, EDTA, EGTA, CBTA or TPPO before grid preparation (Extended Data Table 1). A 3-μl aliquot of the prepared sample was applied to a glow-discharged Quantifoil holey carbon grid (gold, 2-μm size, 1-μm hole, 300-mesh), blotted for 1.5 s under 100% humidity using a Vitrobot Mark III and plunge-frozen in liquid ethane cooled by liquid nitrogen. Grids were loaded into an FEI Titan Krios transmission EM instrument operated at 300 kV, with a nominal magnification of ×130,000 and a 20-eV slit-width energy filter. Data collections were conducted using a K3 direct electron detector in super-resolution mode, with a binned pixel size of 0.413 Å. K3 videos were dose-fractionated into 75 frames over 1.5 s, with a total dose of 49 e− per Å2. Automated image acquisition was performed using SerialEM53, with defocus values ranging from 0.9 μm to 2.1 μm.
Single-particle cryo-EM data analysis
The data-processing workflow is outlined in Supplementary Figs. 1–13. In summary, raw super-resolution TIFF video files from each dataset were motion-corrected and subjected to 2× binning using either MotionCor2 (version 1.4.1) or RELION (versions 4.0 or 5.0beta)54,55. Defocus values for individual micrographs were determined using CTFFIND (version 4.1.10)56. Particle picking was carried out with gautomatch or topaz (version 0.2.4)57. Two-dimensional classification and heterogeneous refinement by cryoSPARC (version 3.0.0) were used to exclude junk particles58. The remaining high-quality particles were used to construct an initial three-dimensional (3D) model through ab initio reconstruction, followed by homogeneous refinement with C4 symmetry. Further map resolution enhancements were achieved through iterative contrast transfer function refinement and Bayesian polishing in RELION55. We noticed obvious conformational heterogeneity in the TMD during consensus refinement, revealing notable flexibility in TRPM5, particularly within the extracellular pore loop region. To enhance the quality of the map, focused classification was conducted by subtracting TMD signals from the particles. Following TMD-focused classification, the map with the highest nominal resolution and a well-defined extracellular region was selected for atomic model construction. To identify heterogeneity of calcium binding at both TMD and ICD sites, we investigate the cryo-EM data of TRPM5 at the subunit level. Specifically, symmetry expansion was applied to the optimal particle set from the TMD-focused classification, followed by subtraction of single-subunit signals and 3D classification without image alignment. This approach revealed was instrumental for revealing the calcium occupancy in CaICD and CaTMD sites for Q771A and D797A mutant datasets.
The map resolution for all datasets was estimated using the gold-standard Fourier shell correlation criterion at 0.143 (ref. 59). Local map resolution was determined using RELION55. Cryo-EM maps were analyzed and visualized with UCSF ChimeraX, while PDB structures were displayed using either UCSF ChimeraX60 or PyMOL (Schrödinger).
Atomic model building
All the atomic models were generated from the TRPM5 WT (apo) (PDB 7MBP) and TRPM5 WT (Ca2+-bound) (PDB 7MBQ) that we published before17. Ligand models and restraints were generated using the Grade Web Server (http://grade.globalphasing.org). Substitutions were introduced using Coot and were subjected to real-space refinement61,62. The model statistics were calculated using phenix.molprobity and phenix.mtriage routines63,64.
Electrophysiology
Whole-cell patch-clamp recordings were conducted on tsA201 cells overexpressing GFP-tagged zebrafish TRPM5 channels, including both WT and mutant variants. Then, 1 day after transfection with Lipofectamine 2000, cells were detached using trypsin and replated onto glass coverslips precoated with 0.01% poly(l-lysine) (Sigma) to enhance attachment. Once the cells adhered, the coverslips were transferred to a recording chamber containing a bath solution (pH 7.4) composed of 150 mM NaCl, 3 mM KCl, 10 mM HEPES, 2 mM CaCl2, 1 mM MgCl2 and 12 mM mannitol. Patch pipettes were filled with internal solutions containing different free calcium concentrations (0–10 μM), each used in separate experiments. All solutions contained 150 mM NaCl, 1 mM MgCl2, 10 mM HEPES, 5 mM EGTA and CaCl2 adjusted accordingly (0–4.9 mM) to achieve the desired calcium levels (pH 7.3, 22 °C) (https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/CaEGTA-TS.htm). All experiments were carried out at room temperature.
During recordings, cells were held at −100 mV for 100 ms before applying a voltage ramp from −100 mV to +100 mV over 200 ms, repeating every 10 s (Clampex 11.3, Multiclamp 700B 2.2.2.2). For Extended Data Fig. 7c–h, an alternative protocol was used, stepping from −200 mV to +200 mV in 50-ms increments, with 20-mV step intervals. Membrane currents were digitally recorded at 10 kHz and filtered at 2 kHz. Current amplitudes were measured 5 ms before the end of the pulse at both +100 mV and −100 mV, with the values subtracted from the current at 0 mV to establish the baseline. Data were analyzed using custom Python scripts, including pyABF for loading and processing ABF electrophysiology files65. Electrophysiology files were parsed and analyzed in Python (version 3.11.8) executed in Spyder (version 6.0.7; Anaconda environment) on macOS 26.0.1 (ARM architecture). Data processing used pandas (version 2.2.2) and numerical operations used NumPy (v1.26.4). Statistical analyses were performed in statsmodels (version 0.14.5) using ordinary least squares and analysis of variance through anova_lm. Graphs were generated with Matplotlib (version 3.9.2) and finalized in GraphPad Prism (version 10.6.1). All custom analysis scripts used in this study are available on Zenodo (https://doi.org/10.5281/zenodo.17427642)65.
Electrophysiology experiments, as seen in Figs. 2a–c,f, 3d and 4a,b and Extended Data Figs. 2 and 4, involved the application of various concentrations of compound CBTA to a single cell. Only data from cells in which the current fully returned to baseline during washout were included for EC50 measurements. In the TPPO experiment, TRPM5 channels were activated using 0.3 μM free intracellular calcium and, after current stabilization for over 30 s, 100 μM TPPO was applied to assess its inhibitory effect.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM density maps of CBTA/EDTA–TRPM5(consensus), CBTA/EDTA–TRPM5(open), CBTA/Ca2+–TRPM5, 200 μM Ca2+–TRPM5, 1 μM Ca2+–TRPM5, 2 μM Ca2+–TRPM5, CBTA/EGTA–TRPM5, Ca2+–TRPM5-D797A, CBTA/Ca2+–TRPM5(D797A), CBTA/Ca2+–TRPM5-D797N, TPPO/Ca2+–TRPM5, CBTA/Ca2+–TRPM5-E337A, CBTA/Ca2+–TRPM5-Q771A and Ca2+–TRPM5-Q771A were deposited to the EM Data Bank under accession numbers EMD-71244, EMD-71245, EMD-71246, EMD-71247, EMD-71248, EMD-71249, EMD-71250, EMD-71251, EMD-71252, EMD-71253, EMD-71254, EMD-71255, EMD-71256 and EMD-71257, respectively. The structure models of EDTA/CBTA–TRPM5(open), Ca2+/CBTA–TRPM5, EGTA/CBTA–TRPM5, Ca2+–TRPM5-D797A, Ca2+/CBTA–TRPM5-D797A, Ca2+/CBTA–TRPM5-D797N, Ca2+/TPPO–TRPM5, Ca2+/CBTA–TRPM5-E337A, Ca2+/CBTA–TRPM5-Q771A and Ca2+–TRPM5-Q771A were deposited to the PDB under accession codes 9P3N, 9P3O, 9P3P, 9P3Q, 9P3R, 9P3S, 9P3T, 9P3U, 9P3V and 9P3W, respectively. Source data are provided with this paper.
Code availability
The custom analysis codes were deposited to Zenodo (https://doi.org/10.5281/zenodo.17427642)65.
References
Zhang, Z., Zhao, Z., Margolskee, R. & Liman, E. The transduction channel TRPM5 is gated by intracellular calcium in taste cells. J. Neurosci. 27, 5777–5786 (2007).
Howitt, M. R. et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 351, 1329–1333 (2016).
Brixel, L. R. et al. TRPM5 regulates glucose-stimulated insulin secretion. Pflug. Arch. 460, 69–76 (2010).
Perez, C. A. et al. A transient receptor potential channel expressed in taste receptor cells. Nat. Neurosci. 5, 1169–1176 (2002).
Zhang, Y. et al. Coding of sweet, bitter, and umami tastes: different receptor cells sharing similar signaling pathways. Cell 112, 293–301 (2003).
Colsoul, B. et al. Loss of high-frequency glucose-induced Ca2+ oscillations in pancreatic islets correlates with impaired glucose tolerance in Trpm5−/− mice. Proc. Natl Acad. Sci. USA 107, 5208–5213 (2010).
Bezencon, C. et al. Murine intestinal cells expressing Trpm5 are mostly brush cells and express markers of neuronal and inflammatory cells. J. Comp. Neurol. 509, 514–525 (2008).
Ketterer, C. et al. Genetic variation within the TRPM5 locus associates with prediabetic phenotypes in subjects at increased risk for type 2 diabetes. Metabolism 60, 1325–1333 (2011).
Tabur, S. et al. Role of the transient receptor potential (TRP) channel gene expressions and TRP melastatin (TRPM) channel gene polymorphisms in obesity-related metabolic syndrome. Eur. Rev. Med Pharm. Sci. 19, 1388–1397 (2015).
Philippaert, K. et al. Steviol glycosides enhance pancreatic beta-cell function and taste sensation by potentiation of TRPM5 channel activity. Nat. Commun. 8, 14733 (2017).
Colsoul, B. et al. Insulin downregulates the expression of the Ca2+-activated nonselective cation channel TRPM5 in pancreatic islets from leptin-deficient mouse models. Pflug. Arch. 466, 611–621 (2014).
Liu, D. & Liman, E. R. Intracellular Ca2+ and the phospholipid PIP2 regulate the taste transduction ion channel TRPM5. Proc. Natl Acad. Sci. USA 100, 15160–15165 (2003).
Damak, S. et al. Trpm5 null mice respond to bitter, sweet, and umami compounds. Chem. Senses 31, 253–264 (2006).
Liman, E. R. TRPM5 and taste transduction. In Transient Receptor Potential (TRP) Channels (eds Flockerzi, V. & Nilius, B.) (Springer, 2007).
Vennekens, R., Mesuere, M. & Philippaert, K. TRPM5 in the battle against diabetes and obesity. Acta Physiol. 222, e12949 (2018).
Virginio, C. et al. Identification of positive modulators of TRPM5 channel from a high-throughput screen using a fluorescent membrane potential assay. SLAS Discov. 27, 55–64 (2022).
Ruan, Z. et al. Structures of the TRPM5 channel elucidate mechanisms of activation and inhibition. Nat. Struct. Mol. Biol. 28, 604–613 (2021).
Barilli, A. et al. From high-throughput screening to target validation: benzo[d]isothiazoles as potent and selective agonists of human transient receptor potential cation channel subfamily M member 5 possessing in vivo gastrointestinal prokinetic activity in rodents. J. Med. Chem. 64, 5931–5955 (2021).
Sabat, M. et al. The discovery of (1R, 3R)-1-(3-chloro-5-fluorophenyl)-3-(hydroxymethyl)-1,2,3,4-tetrahydroisoquinoline-6-carbonitrile, a potent and selective agonist of human transient receptor potential cation channel subfamily m member 5 (TRPM5) and evaluation of as a potential gastrointestinal prokinetic agent. Bioorg. Med. Chem. 76, 117084 (2022).
Palmer, R. K. et al. Triphenylphosphine oxide is a potent and selective inhibitor of the transient receptor potential melastatin-5 ion channel. Assay. Drug Dev. Technol. 8, 703–713 (2010).
Karuppan, S., Schrag, L. G., Pastrano, C. M., Jara-Oseguera, A. & Zubcevic, L. Structural dynamics at cytosolic interprotomer interfaces control gating of a mammalian TRPM5 channel. Proc. Natl Acad. Sci. USA 121, e2403333121 (2024).
Chubanov, V., Kottgen, M., Touyz, R. M. & Gudermann, T. TRPM channels in health and disease. Nat. Rev. Nephrol. 20, 175–187 (2024).
Huang, Y., Fliegert, R., Guse, A. H., Lu, W. & Du, J. A structural overview of the ion channels of the TRPM family. Cell Calcium 85, 102111 (2020).
Guo, J. et al. Structures of the calcium-activated, non-selective cation channel TRPM4. Nature 552, 205–209 (2017).
Winkler, P. A., Huang, Y., Sun, W., Du, J. & Lu, W. Electron cryo-microscopy structure of a human TRPM4 channel. Nature 552, 200–204 (2017).
Autzen, H. E. et al. Structure of the human TRPM4 ion channel in a lipid nanodisc. Science 359, 228–232 (2018).
Duan, J. et al. Structure of full-length human TRPM4. Proc. Natl Acad. Sci. USA 115, 2377–2382 (2018).
Hu, J. et al. Physiological temperature drives TRPM4 ligand recognition and gating. Nature 630, 509–515 (2024).
Ekundayo, B. et al. Identification of a binding site for small molecule inhibitors targeting human TRPM4. Nat. Commun. 16, 833 (2025).
Zhang, Z., Tóth, B., Szollosi, A., Chen, J. & Csanády, L. Structure of a TRPM2 channel in complex with Ca2+ explains unique gating regulation. eLife 7, e36409 (2018).
Huang, Y., Winkler, P. A., Sun, W., Lu, W. & Du, J. Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nature 562, 145–149 (2018).
Wang, L. et al. Structures and gating mechanism of human TRPM2. Science 362, eaav4809 (2018).
Yin, Y. et al. Visualizing structural transitions of ligand-dependent gating of the TRPM2 channel. Nat. Commun. 10, 3740 (2019).
Huang, Y., Roth, B., Lu, W. & Du, J. Ligand recognition and gating mechanism through three ligand-binding sites of human TRPM2 channel. eLife 8, e50175 (2019).
Yu, X. et al. Structural and functional basis of the selectivity filter as a gate in human TRPM2 channel. Cell Rep. 37, 110025 (2021).
Huang, Y., Kumar, S., Lee, J., Lu, W. & Du, J. Coupling enzymatic activity and gating in an ancient TRPM chanzyme and its molecular evolution. Nat. Struct. Mol. Biol. 31, 1509–1521 (2024).
Toth, B., Jiang, Y., Szollosi, A., Zhang, Z. & Csanady, L. A conserved mechanism couples cytosolic domain movements to pore gating in the TRPM2 channel. Proc. Natl Acad. Sci. USA 121, e2415548121 (2024).
Yin, Y. et al. Structure of the cold- and menthol-sensing ion channel TRPM8. Science 359, 237–241 (2018).
Yin, Y. et al. Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 363, eaav9334 (2019).
Diver, M. M., Cheng, Y. & Julius, D. Structural insights into TRPM8 inhibition and desensitization. Science 365, 1434–1440 (2019).
Zhao, C. et al. Structures of a mammalian TRPM8 in closed state. Nat. Commun. 13, 3113 (2022).
Yin, Y. et al. Activation mechanism of the mouse cold-sensing TRPM8 channel by cooling agonist and PIP2. Science 378, eadd1268 (2022).
Palchevskyi, S. et al. Structure of human TRPM8 channel. Commun. Biol. 6, 1065 (2023).
Yin, Y. et al. Mechanisms of sensory adaptation and inhibition of the cold and menthol receptor TRPM8. Sci. Adv. 10, eadp2211 (2024).
Chuang, H. H., Neuhausser, W. M. & Julius, D. The super-cooling agent icilin reveals a mechanism of coincidence detection by a temperature-sensitive TRP channel. Neuron 43, 859–869 (2004).
Yin, Y. et al. Molecular basis of neurosteroid and anticonvulsant regulation of TRPM3. Nat. Struct. Mol. Biol. 32, 828–840 (2025).
Kumar, S. et al. Structural basis for agonist and heat activation of nociceptor TRPM3. Nat. Struct. Mol. Biol. https://doi.org/10.1038/s41594-025-01692-5 (2025).
Huffer, K., Denley, M. C. S., Oskoui, E. V. & Swartz, K. J. Conservation of the cooling agent binding pocket within the TRPM subfamily. eLife 13, RP99643 (2024).
Yamaguchi, S., Tanimoto, A., Iwasa, S. & Otsuguro, K. TRPM4 and TRPM5 channels share crucial amino acid residues for Ca2+ sensitivity but not significance of PI(4,5)P2. Int. J. Mol. Sci. 20, 2012 (2019).
Zhao, Y. et al. Molecular basis for ligand modulation of a mammalian voltage-gated Ca2+ channel. Cell 177, 1495–1506 (2019).
Li, J. et al. Small molecule inhibits KCNQ channels with a non-blocking mechanism. Nat. Chem. Biol. 21, 1100–1109 (2025).
Goehring, A. et al. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat. Protoc. 9, 2574–2585 (2014).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Kimanius, D., Dong, L., Sharov, G., Nakane, T. & Scheres, S. H. W. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem. J. 478, 4169–4185 (2021).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Henderson, R. et al. Outcome of the first electron microscopy validation task force meeting. Structure 20, 205–214 (2012).
Meng, E. C. et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 74, 531–544 (2018).
Davis, I. W. et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 (2007).
Afonine, P. V. et al. New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr. D Struct. Biol. 74, 814–840 (2018).
Lee, J. Custom Python scripts for TRPM5 electrophysiology analysis. Zenodo https://doi.org/10.5281/zenodo.17427642 (2025).
Acknowledgements
We thank members of the J.D. and W.L. Labs for thoughtful discussions. Cryo-EM data were collected at the David Van Andel Advanced Cryo-EM Suite. We appreciate the high-performance computing team of Van Andel Institute and the Structural Biology Facility and Northwestern IT Research Computing and Data Services (NUIT RCDS) at Northwestern University for computational support. W.L. is supported by National Institutes of Health (NIH) grants (R01HL153219, R01NS112363 and R35GM138321). J.D. is supported by a McKnight Scholar Award, a Klingenstein-Simon Scholar Award, a Sloan Research Fellowship in neuroscience, a Pew Scholar in the Biomedical Sciences award and NIH grants (R01NS111031 and R01NS129804). Z.R. is supported by an American Heart Association postdoctoral fellowship (20POST35120556) and an NIH grant (R00NS128258).
Author information
Authors and Affiliations
Contributions
W.L. and J.D. supervised the project. Z.R. carried out the protein purification, cryo-EM data collection and processing. Y.L. contributed to cryo-EM data processing. I.J.O. and J.L. performed the electrophysiological experiments. Z.R., J.D. and W.L. analyzed the structures and wrote the paper. All authors contributed to paper preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemical Biology thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Whole-cell recordings of CBTA-induced currents and dose-response relationships in TRPM5.
a, Representative plot showing currents measured at +100 and −100 mV during CBTA application and washout. b, Representative current-voltage relationships obtained using a ramp protocol from −100 to +100 mV during CBTA application. In a and b, whole-cell recordings were performed with pipette solution containing 5 mM EGTA. c, Representative whole-cell current traces recorded with intracellular solutions containing different free Ca2+ concentrations, using voltage steps of −100 mV (30 ms) followed by +100 mV (30 ms). d, Dose-response curve and EC50 estimates for intracellular free calcium (10 µM, n = 15; 3 µM, n = 10; 1.5 µM, n = 5; 1 µM, n = 15; 450 nM, n = 5; 300 nM, n = 6; 100 nM, n = 6; and 30 nM, n = 6). Circles represent mean, and error bars represent s.e.m. e, Representative plot showing currents measured at +100 and −100 mV during CBTA application and washout. f, Representative current-voltage relationships obtained using a ramp protocol from −100 to +100 mV during CBTA application. In e-f, whole-cell recordings were performed with pipette solution containing 100 nM free calcium. g, Representative whole-cell current traces recorded from three groups in drTRPM5-expressing cells: CBTA alone (100 nM), free Ca2+ alone (100 nM), and CBTA + free Ca2+ (100 nM each) using a voltage ramp from −100 to +100 mV. As a control, non-transfected tsA201 cells were recorded with CBTA + free Ca2+ (100 nM each). h, Quantification of current amplitudes at −100 and +100 mV for conditions in (g). Each group: n = 4. Bars represent mean; each dot corresponds to an independent cell measurement. i, Calcium-dependent activation of TRPM5 currents measured at +100 and −100 mV. Whole-cell currents were recorded at different intracellular free Ca2+ concentrations (1, 3, 10, 30, 100, 300, and 1000 nM; n = 6 cells each, except 1 nM: n = 5). Black ( + 100 mV) and gray (−100 mV) curves are measured without CBTA. Red ( + 100 mV) and light red (−100 mV) curves are measured with extracellular addition of 100 nM CBTA. Circles represent mean, and error bars represent s.e.m.
Extended Data Fig. 2 Whole-cell recordings of CBTA-binding site mutants of TRPM5.
a, Representative whole-cell current traces recorded using a ramp protocol from −100 to +100 mV in non-transfected tsA201 cells, cells transfected with wild-type TRPM5 and its mutants F730A, L764A, R834A, R998A, N723A. Black and red traces show baseline currents and currents with 20 μM CBTA, respectively. Pipette solution contains 5 mM EGTA. b, Estimated marginal means (EMMs) of CBTA-activated currents after adjusting for 1 µM free Ca2+-evoked currents using ANCOVA. c, Normalized adjusted CBTA responses. For both b and c, WT n = 5 and mutants n = 4 each. Circles represent mean, and error bars represent s.e.m. Method: Whole-cell currents evoked by 1 µM free Ca2+ served as a proxy for channel expression. Because Ca2+- and CBTA-activated currents were obtained from different cells, genotype-level median Ca2+ currents were assigned as expression values. Both CBTA and Ca2+ currents were log-transformed. An ANCOVA model (Python, statsmodels) was fitted with log(I_CBTA) as the dependent variable, log(I_Ca) as the covariate, and genotype as the fixed factor (WT as the reference). EMMs were extracted at the average Ca2+ expression level. d, Representative plot showing currents measured at +100 and −100 mV, with time 0 indicating immediately after whole-cell configuration. The pipette solution contains 1 μM free calcium. Peak and steady-state refer to the largest current and the stabilized current observed over 30 s, respectively. e, Peak currents measured at +100 and −100 mV for non-transfected tsA201 (n = 6), WT TRPM5 (n = 9), and mutants (F730A, n = 10; L764A, n = 10; R834A, n = 12; R998A, n = 10; N723A, n = 10). Bars represent mean; each dot corresponds to an independent cell measurement. f, Steady-state currents measured at +100 and −100 mV for non-transfected tsA201 cells (n = 6), WT TRPM5 (n = 9) and mutants (F730A, n = 10; L764A, n = 10; R834A, n = 12; R998A, n = 10; N723A, n = 10). Bars represent mean; each dot corresponds to an independent cell measurement. g, Representative traces of peak and steady-state currents recorded from non-transfected tsA201 cells, WT TRPM5 and mutants using a ramp protocol from −100 to +100 mV. The pipette solution contains 1 μM free calcium.
Extended Data Fig. 3 Structural analysis of TRPM5 with agonists calcium and CBTA.
a, A close-up view of the CaTMD site of TRPM5 in the presence of EDTA (EMDB-23740). The atomic model is overlaid with cryo-EM map (PDB: 7MBP). b, A close-up view of the CaTMD site of the consensus map of EDTA/CBTA–TRPM5 dataset. The atomic model is overlaid with cryo-EM map. The CBTA density is clearly defined (yellow stick), whereas a weak CaTMD density is seen (green sphere). c, The close-up view of CaTMD (left panel) and CaICD (right panel) sites of the open state identified in CBTA/EDTA–TRPM5 dataset. The CaTMD density is strong whereas the CaICD is absent. d, The structural titration experiment of TRPM5 in the presence of various calcium concentrations (200 μM, 6 μM, 2 μM, and 1 μM). The upper and lower panels showed the close-up view of the CaTMD and CaICD sites, respectively. At 6 μM calcium concentration, two conformations of TRPM5 are identified, including one with both CaTMD and CaICD occupied, and another one in the apo state. e, Comparison of the CBTA-induced conformational change with the apo state and calcium-bound open state of TRPM5. The left panel indicates the S1–S4 domain, whereas right panel shows the S5–S6 pore domain. f, Structural comparison between CBTA/EGTA–TRPM5 (magenta) and Apo–TRPM5 (yellow). Residues surrounding CBTA are shown in stick representation. g, The cation-π stacking between CBTA, R834 and H837 in CBTA/EGTA–TRPM5 structure.
Extended Data Fig. 4 Representative whole-cell current of CBTA/CaTMD binding site mutants.
a-d, Representative traces of whole-cell currents in CaTMD mutant (L764A, n = 4) and controls (non-transfected tsA201 cells, TRPM5-WT), measured either without CBTA (a and c) or with 100 nM CBTA (b and d). The pipette solution contains 100 nM free calcium in (a and b) and 300 nM free calcium in (c and d). Bar graphs show mean currents measured at +100 mV and −100 mV for each group (n = 4-5); each dot corresponds to an independent cell measurement. e-f, Representative traces of whole-cell currents in CaTMD mutants (Q771A, D797A, D797N) and controls (non-transfected tsA201 cells, TRPM5-WT), measured either without CBTA (e) or with 100 nM CBTA (f). The pipette solution contains 100 nM free calcium. Bar graphs show currents measured at +100 mV and −100 mV for each group (n = 4); each dot corresponds to an independent cell measurement. g-h, Representative traces of whole-cell currents in CaTMD mutants Q771A and D797N with 300 nM free calcium, measured either without CBTA (g) or with 1 μM CBTA (h).
Extended Data Fig. 5 Representative whole-cell current recordings and I–V relationships of WT TRPM5 and mutants of the TPPO binding site in response to extracellular 100 μM TPPO.
a–d, Raw current traces (left) and corresponding I–V plots (right) obtained from cells expressing R998A (a), F730A (b), I838A (c), and H837A (d). Currents were evoked using a voltage-ramp protocol from –100 mV to +100 mV, with 0.3 μM free intracellular Ca2+ in the pipette solution. 100 μM TPPO (red) was applied extracellularly via bath perfusion. I–V curves were generated from the current responses to the voltage ramp under bath (black) and TPPO (red) conditions. e, Representative time-course recordings of whole-cell TRPM5 currents measured at +100 mV and –100 mV from WT, R998A, F730A, H837A, and I838A. Currents were recorded with 0.3 μM free intracellular Ca2+, and 100 μM TPPO was applied extracellularly at the time indicated by the red bar.
Extended Data Fig. 6 Whole-cell current responses in TRPM5-WT and E337A: Effects of CBTA and varying intracellular calcium concentrations.
a, Representative plot showing currents measured at +100 mV and −100 mV during CBTA application (0.3 to 100 μM) and washout in TRPM5-E337A. b, Representative current-voltage relationships obtained using a ramp protocol from −100 mV to +100 mV during CBTA application (0.3 to 100 μM). In a and b, whole-cell recordings were performed with pipette solution containing 5 mM EGTA. c-h, Whole-cell current recordings in WT and E337A groups, with the pipette solution containing 1, 3, and 10 μM intracellular free calcium. Panels c, e, and g show the current-voltage relationships for TRPM5-WT(1 µM free Ca2+, n = 9; 3 µM free Ca2+, n = 11; 10 µM free Ca2+, n = 10) and E337A(1 µM free Ca2+, n = 6; 3 µM free Ca2+, n = 4; 10 µM free Ca2+, n = 4) obtained by applying step voltage protocols from −200 mV to +200 mV for 1 μM Ca2+ (c), 3 μM Ca2+ (e), and 10 μM Ca2+ (g) intracellular free calcium. Circles represent mean currents, and error bars represent s.e.m. Panels d, f, and h show representative current traces in response to step voltage protocols for 1 μM (d), 3 μM (f), and 10 μM (h) calcium groups in TRPM5-WT and E337A. 1 µM intracellular free calcium (WT: n = 9, E337A: n = 6), 3 µM (WT: n = 11, E337A: n = 4), and 10 µM (WT: n = 10, E337A: n = 4).
Extended Data Fig. 7 TMD-ICD coupling in TRPM5.
a and b, A close up view of the CaTMD (a) site and CaICD (b) site of CBTA/Ca2+–TRPM5(E337A) structure. The cryo-EM map is overlaid with atomic model with relevant residues shown in stick representation. The CBTA and CaTMD density are well defined, whereas CaICD is absent. c, A close-up view of the CaTMD (left panel) and CaICD (right panel) sites of Ca2+–TRPM5(Q771A) dataset. d, Subunit comparison of the EDTA/CBTA–TRPM5(WT) and CBTA/Ca2+–TRPM5(E337A) with the apo and open state of TRPM5. The root-mean-square-deviation (RMSD) of the intracellular domain (ICD) of the structures relative to the apo and open state is shown in the inlet. Both CBTA/EGTA–TRPM5(WT) and the CBTA/Ca2+–TRPM5(E337A) adopts an intermediate conformation between apo and open state. e, Comparison of the S1–S4 domain between Apo–TRPM5 and Ca2+–TRPM5 (left panel), and Ca2+–TRPM5(Q771A) and Apo–TRPM5(WT) (right panel). f, Comparison of the ICD of Ca2+–TRPM5(Q771A) with Apo–TRPM5 and Ca2+–TRPM5. The ICD of Ca2+–TRPM5(Q771A) adopts an intermediate conformation. g, Subunit-focused analysis identified classes with CaICD-bound (left panel) and -unbound (right panel) states for Ca2+–TRPM5(Q771A). h, Cryo-EM map comparison of the MHR1/2 domain between the CaICD-bound (cyan) and -unbound (magenta) subunits for the Ca2+–TRPM5(Q771A). h, Comparison of Ca2+/CBTA–TRPM5(Q771A) with the open state of TRPM5. i, The CaICD of Ca2+/CBTA–TRPM5(Q771A) is fully occupied, distinct from Ca2+–TRPM5(Q771A) as seen in (g). j, The CaICD of Ca2+/CBTA–TRPM5(Q771A) is fully occupied, distinct from the CaICD of Ca2+–TRPM5(Q771A) as seen in g.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–14 and Tables 1–5.
Source data
Source Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 6 (download XLSX )
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ruan, Z., Lee, J., Li, Y. et al. A single allosteric site merges activation, modulation and inhibition in TRPM5. Nat Chem Biol 22, 402–410 (2026). https://doi.org/10.1038/s41589-025-02097-7
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41589-025-02097-7
