
Abstract
The E3 ligase substrate adapter cereblon (CRBN), the primary target of clinical agents thalidomide and lenalidomide, recognizes endogenous substrates bearing the C-terminal cyclic imide modification. Although C-terminal cyclic imides can form spontaneously, an enzyme that regulates their formation and thereby promotes a biological pathway connecting substrates to CRBN is unknown. Here we report that protein carboxymethyltransferase (PCMT1) promotes formation of C-terminal cyclic imides on C-terminal asparagine residues of CRBN substrates. PCMT1 and CRBN coregulate the levels of metabolic enzymes including glutamine synthetase and inorganic pyrophosphatase 1 in vitro, in cells and in vivo, and this regulation is associated with the proepileptic phenotype of CRBN knockout mouse models. The discovery of an enzyme that regulates CRBN substrates through the C-terminal cyclic imide reveals a previously unknown biological pathway that is perturbed by thalidomide derivatives and provides a biochemical basis for the connection between multiple biological processes and CRBN.

This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

Data availability
All data are available in the main text and Supplementary Information. Proteomics data have been deposited to the PRIDE repository under the identifier PXD061730. The crystal structure of human CRBN–DDB1 in complex with GLUL-cN has been deposited to the PDB under the accession code 9NR3. Source data are provided with this paper.
Code availability
All code used in this study is commercially available or provided in a public repository.
References
Ito, T. et al. Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350 (2010).
Singhal, S. et al. Antitumor activity of thalidomide in refractory multiple myeloma. N. Engl. J. Med. 341, 1565–1571 (1999).
List, A. et al. Efficacy of lenalidomide in myelodysplastic syndromes. N. Engl. J. Med. 352, 549–557 (2005).
Kronke, J. et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305 (2014).
Lu, G. et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309 (2014).
Kronke, J. et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 523, 183–188 (2015).
Matyskiela, M. E. et al. A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature 535, 252–257 (2016).
Donovan, K. A. et al. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. eLife https://doi.org/10.7554/eLife.38430 (2018).
Matyskiela, M. E. et al. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Biol. 14, 981–987 (2018).
Ichikawa, S. et al. The E3 ligase adapter cereblon targets the C-terminal cyclic imide degron. Nature 610, 775–782 (2022).
Heim, C., Spring, A.-K., Kirchgäßner, S., Schwarzer, D. & Hartmann, M. D. Identification and structural basis of C-terminal cyclic imides as natural degrons for cereblon. Biochem. Biophys. Res. Commun. 637, 66–72 (2022).
Voorter, C. E., de Haard-Hoekman, W. A., van den Oetelaar, P. J., Bloemendal, H. & de Jong, W. W. Spontaneous peptide bond cleavage in aging alpha-crystallin through a succinimide intermediate. J. Biol. Chem. 263, 19020–19023 (1988).
Xu, W. et al. Genesis and regulation of C-terminal cyclic imides from protein damage. Proc. Natl Acad. Sci. USA 122, e2415976121 (2025).
Robinson, N. E. & Robinson, A. B. Deamidation of human proteins. Proc. Natl Acad. Sci. USA 98, 12409–12413 (2001).
O’Connor, C. M. 13 Protein L-isoaspartyl, D-aspartyl O-methyltransferases: catalysts for protein repair. Enzymes 24, 385–433 (2006).
Kim, E., Lowenson, J. D., MacLaren, D. C., Clarke, S. & Young, S. G. Deficiency of a protein-repair enzyme results in the accumulation of altered proteins, retardation of growth, and fatal seizures in mice. Proc. Natl Acad. Sci. USA 94, 6132–6137 (1997).
Lai, K. Y. et al. LanCLs add glutathione to dehydroamino acids generated at phosphorylated sites in the proteome. Cell 184, 2680–2695 (2021).
Higgins, J. J., Pucilowska, J., Lombardi, R. Q. & Rooney, J. P. A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology 63, 1927–1931 (2004).
Sheereen, A. et al. A missense mutation in the CRBN gene that segregates with intellectual disability and self-mutilating behaviour in a consanguineous Saudi family. J. Med. Genet. 54, 236–240 (2017).
Nguyen, T. V. et al. Glutamine triggers acetylation-dependent degradation of glutamine synthetase via the thalidomide receptor cereblon. Mol. Cell 61, 809–820 (2016).
Del Prete, D., Rice, R. C., Rajadhyaksha, A. M. & D’Adamio, L. Amyloid precursor protein (APP) may act as a substrate and a recognition unit for CRL4CRBN and Stub1 E3 ligases facilitating ubiquitination of proteins involved in presynaptic functions and neurodegeneration. J. Biol. Chem. 291, 17209–17227 (2016).
Kurihara, T., Asahi, T. & Sawamura, N. Cereblon-mediated degradation of the amyloid precursor protein via the ubiquitin-proteasome pathway. Biochem. Biophys. Res. Commun. 524, 236–241 (2020).
Fischer, E. S. et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53 (2014).
Lee, K. M., Jo, S., Kim, H., Lee, J. & Park, C. S. Functional modulation of AMP-activated protein kinase by cereblon. Biochim. Biophys. Acta 1813, 448–455 (2011).
Hohberger, B. & Enz, R. Cereblon is expressed in the retina and binds to voltage-gated chloride channels. FEBS Lett. 583, 633–637 (2009).
Hsiao, K., Zegzouti, H. & Goueli, S. A. Methyltransferase-Glo: a universal, bioluminescent and homogenous assay for monitoring all classes of methyltransferases. Epigenomics 8, 321–339 (2016).
Lowenson, J. D. & Clarke, S. Structural elements affecting the recognition of L-isoaspartyl residues by the L-isoaspartyl/D-aspartyl protein methyltransferase. Implications for the repair hypothesis. J. Biol. Chem. 266, 19396–19406 (1991).
Bennett, E. J. et al. Catalytic implications from the Drosophila protein L-isoaspartyl methyltransferase structure and site-directed mutagenesis. Biochemistry 42, 12844–12853 (2003).
Yang, H., Lowenson, J. D., Clarke, S. & Zubarev, R. A. Brain proteomics supports the role of glutamate metabolism and suggests other metabolic alterations in protein L-isoaspartyl methyltransferase (PIMT)-knockout mice. J. Proteome Res 12, 4566–4576 (2013).
Payne, N. C., Kalyakina, A. S., Singh, K., Tye, M. A. & Mazitschek, R. Bright and stable luminescent probes for target engagement profiling in live cells. Nat. Chem. Biol. 17, 1168–1177 (2021).
Ichikawa, S. et al. The cyclimids: degron-inspired cereblon binders for targeted protein degradation. Cell Chem. Biol. 31, 1162–1175 (2024).
Arad, G., Freikopf, A. & Kulka, R. G. Glutamine-stimulated modification and degradation of glutamine synthetase in hepatoma tissue culture cells. Cell 8, 95–101 (1976).
Lloyd, H. C. et al. A method for the detection and enrichment of endogenous cereblon substrates. Cell Chem. Biol. 32, 1028–1041 (2025).
Watson, E. R. et al. Molecular glue CELMoD compounds are regulators of cereblon conformation. Science 378, 549–553 (2022).
Petzold, G., Fischer, E. S. & Thoma, N. H. Structural basis of lenalidomide-induced CK1alpha degradation by the CRL4(CRBN) ubiquitin ligase. Nature 532, 127–130 (2016).
Costas-Insua, C. et al. The CB(1) receptor interacts with cereblon and drives cereblon deficiency-associated memory shortfalls. EMBO Mol. Med. 16, 755–783 (2024).
Ikegaya, Y. et al. Aberrant synaptic transmission in the hippocampal CA3 region and cognitive deterioration in protein-repair enzyme-deficient mice. Hippocampus 11, 287–298 (2001).
Eid, T. et al. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 363, 28–37 (2004).
Bird, C. M. & Burgess, N. The hippocampus and memory: insights from spatial processing. Nat. Rev. Neurosci. 9, 182–194 (2008).
Walker, M. C. Pathophysiology of status epilepticus. Neurosci. Lett. 667, 84–91 (2018).
Niu, H. et al. Crystallographic and modeling study of the human inorganic pyrophosphatase 1: a potential anti-cancer drug target. Proteins Struct. Funct. Bioinf. 89, 853–865 (2021).
Sarlo, G. L. & Holton, K. F. Brain concentrations of glutamate and GABA in human epilepsy: a review. Seizure 91, 213–227 (2021).
Eid, T., Lee, T. W., Patrylo, P. & Zaveri, H. P. Astrocytes and glutamine synthetase in epileptogenesis. J. Neurosci. Res. 97, 1345–1362 (2019).
Achleitner, M. T. et al. PPA1 deficiency causes a deranged galactose metabolism recognizable in neonatal screening. Metabolites 13, 1141 (2023).
Tezuka, Y. et al. Upregulation of inorganic pyrophosphatase 1 as a JNK phosphatase in hypothyroid embryonic chick cerebellum. Life Sci. 128, 94–100 (2015).
Schouten, M. et al. Multi-omics profile of the mouse dentate gyrus after kainic acid-induced status epilepticus. Sci. Data 3, 160068 (2016).
Racine, R. J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32, 281–294 (1972).
Yao, Z. et al. A high-resolution transcriptomic and spatial atlas of cell types in the whole mouse brain. Nature 624, 317–332 (2023).
Merino-Cacho, L. et al. Cullin-RING ligase BioE3 reveals molecular-glue-induced neosubstrates and rewiring of the endogenous cereblon ubiquitome. Cell Commun. Signal 23, 101 (2025).
Kroupova, A. et al. Design of a Cereblon construct for crystallographic and biophysical studies of protein degraders. Nat. Commun. 15, 8885 (2024).
Ran, F. A. et al. Genome engineering using the CRISPR–Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Batth, T. S., Francavilla, C. & Olsen, J. V. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J. Proteome Res. 13, 6176–6186 (2014).
Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Maroto, I. B. et al. Control of a hippocampal recurrent excitatory circuit by cannabinoid receptor-interacting protein Gap43. Nat. Commun. 14, 2303 (2023).
Acknowledgements
We thank S. Ichikawa, H. A. Flaxman, N. C. Payne, M. A. Leon-Duque and T. Long for helpful discussions, T. Wang for support with peptide synthesis, M. Chen and S. Trager from the Harvard University Mass Spectrometry and Proteomics Resource Laboratory for support with MS experiments, D. Cui and B. Tresco from the Harvard University Laukien-Purcell Instrumentation Center for support with peptide MS experiments, and J. A. Nelson and Z. T. Niziolek from the Harvard University Bauer Core for support with flow cytometry. His6-CRBN–DDB1 was a generous gift from Boehringer Ingelheim. Support from the National Institutes of Health (grant no. R01GM141406, C.M.W.), the Blavatnik Biomedical Accelerator at Harvard University (C.M.W.), Mark Foundation for Cancer Research (C.M.W.), Merkin Family Foundation (C.M.W.), the Starr Foundation (C.M.W.) and Spanish Ministerio de Ciencia, Innovación y Universidades (MICINU/FEDER; grant no. PID2021-125118OB-I00, M.G.) is gratefully acknowledged. N.Z. is supported by Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Contributions
Z.Z., W.X. and A.K.D.P. designed and synthesized peptides. E.Y.F., S.C., W.X., H.C.L. and A.K.D.P. generated recombinant and engineered proteins. Z.Z., W.X. and E.Y.F. performed in vitro enzyme activity and TR-FRET binding studies. S.C. and N.Z. designed and conducted structural, BLI and in vitro ubiquitination studies. Z.Z., W.X. and E.Y.F. performed cellular studies. H.C.L. developed cerebody methods. A.H.-L., P.P.-V. and M.G. designed and conducted in vivo measurements. E.Y.F. and W.X. analyzed mouse tissue samples. C.M.W. conceived of the project. C.M.W., Z.Z., W.X., E.Y.F. and S.C. drafted the paper. All authors reviewed and edited the paper.
Corresponding author
Ethics declarations
Competing interests
The Woo Laboratory, under the direction of C.M.W., receives or has received sponsored research support from Amgen, Ono Pharmaceuticals and Merck. N.Z. is one of the scientific cofounders and a shareholder of SEED Therapeutics, and serves as a member of the scientific advisory boards of SyntheX, Molecular Glue Labs, Cold Start Therapeutics and Differentiated Therapeutics, with financial interests. The other authors declare no competing interests.
Peer review
Peer review information
Nature Chemical Biology thanks Chul-Seung Park and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
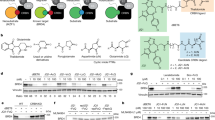
Extended Data Fig. 1 Formation of cyclic imides from asparagine.
(a) Scheme of protein damage events that occur through Asn residues and result in the C-terminal cyclic imide or isoaspartate post-translational modifications. (b) Chemical scheme and conversion of model peptides incubated in 20 mM NH4OAc (pH 7.4) buffer at 37 °C (n = 3). We detected two distinct hydrolysis products, which were constitutional isomers, but were unable to assign their identities. Data represent mean ± S.D. (c) Scheme of PCMT1 correction of isoaspartate modifications.
Extended Data Fig. 2 Efficiency of PCMT1-mediated C-terminal cyclic imide formation on various peptides.
(a) Time-course of peptides representing the C-termini of proteins previously linked to CRBN incubated with PCMT1 and SAM in 50 mM Tris-Cl (pH 7.4) buffer at 37 °C (n = 3). (b) Time-course incubation of Fmoc-FN and GGGFN with PCMT1 and SAM in 50 mM Tris-Cl (pH 7.4) buffer at 37 °C (n = 3). (c) Time-course incubation of synthetically methylated peptide PFQYKN(OMe) in 50 mM Tris-Cl (pH 7.4) buffer at 37 °C with or without PCMT1 and SAM. (d) Time-course incubation of Fmoc-FN and FQYKN with the catalytically inactive PCMT1 S60A mutant and SAM in 50 mM Tris-Cl (pH 7.4) buffer at 37 °C (n = 2). For all these data, note that the grey curves likely represent the sum of two species, the C-terminal Asn or Gln (that is, the starting material) or the C-terminal amide (a hydrolysis product), which are indistinguishable by mass spectrometry. Indicated sample sizes (n) represent biological replicates.
Extended Data Fig. 3 Sequence preference of PCMT1 activity and CRBN recognition.
(a) Structure of thalidomide-FITC, the TR-FRET tracer used to assess the engagement of ligands with His6-CRBN–DDB1 in this study. (b) Conversion efficiency and CRBN binding of GLUL C-terminal peptides with variable lengths after 14 h incubation with PCMT1 by mass spectrometry and TR-FRET assay, respectively (n = 3). (c) Conversion efficiency and CRBN binding of GLUL C-terminal peptides with variable –1 residue after 14 h incubation with PCMT1 by mass spectrometry and TR-FRET assay, respectively (n = 3). (d) Conversion efficiency and CRBN binding of GLUL C-terminal peptides with variable –2 residue after 14 h incubation with PCMT1 by mass spectrometry and TR-FRET assay, respectively (n = 3). Data represent mean ± S.D. in all graphs with error bars. Indicated sample sizes (n) represent biological replicates.
Extended Data Fig. 4 Full-length GLUL is recognized and regulated by PCMT1 and CRBN in cells.
(a) In vitro ubiquitination assay of Venus-GLUL with WT or catalytically inactive PCMT1 (n = 2). (b) Size exclusion chromatography of the GFP-GLUL protein before or after electroporation. The oligomerization state of the protein did not change upon electroporation. (c) Cerebody enrichment for APP from WT, PCMT1-KO, or CRBN-KO HEK293T cells after treatment with DMSO or 100 µM lenalidomide for 24 h (n = 3). Multiple bands are observed corresponding to different isoforms of APP with different degrees of PTMs. Enriched APP may stem from nonspecific binding. (d) Western blot of endogenous GLUL levels in sgCtrl, PCMT1-KO, or CRBN-KO HEK293T cells (n = 3). Quantified relative GLUL levels are shown. (e) Cycloheximide (CHX) chase assay to monitor the degradation of GLUL over 24 h in sgCtrl, PCMT1-KO, or CRBN-KO HEK293T cells (n = 2). Mean relative GLUL levels are shown. (f–g) Western blot of GLUL levels in sgCtrl, PCMT1-KO, or CRBN-KO HEK293T cells grown to low or high confluence, along with representative images of cells in culture from the same experiment (n = 3). Quantified relative GLUL levels, normalized to sgCtrl within each condition, are shown. (h) Western blot of GLUL levels in sgCtrl, PCMT1-KO, or CRBN-KO HEK293T cells treated with DMSO or 1 µM MLN4924 for 24 h (n = 3). Mean relative GLUL levels are shown. Indicated sample sizes (n) represent biological replicates.
Extended Data Fig. 5 Glutamine-triggered GLUL degradation is independent of PCMT1.
(a) Scheme of GLUL-catalyzed condensation of glutamate and ammonium into glutamine. (b) Time-course GLUL buildup in WT, sgCtrl, PCMT1-KO, or CRBN-KO HEK293T cells incubated in glutamine-free media (n = 3). (c) Time-course GLUL degradation of glutamine-starved sgCtrl, PCMT1-KO, or CRBN-KO HEK293T cells after refeeding with 4 mM glutamine (n = 3). Quantified relative GLUL levels are shown. (d) Normalized mRNA level of GLUL in HEK293T cells with or without glutamine starvation for 48 h (n = 3). Data represent mean ± S.D. Indicated sample sizes (n) represent biological replicates.
Extended Data Fig. 6 Impact of clinical agents on the GLUL-cN level in multiple cell lines.
(a) Cerebody enrichment for GLUL-cN from WT, PCMT1-KO, or CRBN-KO HEK293T cells after treatment with DMSO or 100 µM lenalidomide for 24 h (n = 3). (b) Cerebody enrichment for GLUL-cN from different human and mouse cell lines after treatment with DMSO or 100 µM lenalidomide for 24 h (n = 3). Elution for SK-N-SH and MEF was performed with 3× FLAG peptide, while elution for Neuro-2a was performed with 200 µM lenalidomide. Indicated sample sizes (n) represent biological replicates.
Extended Data Fig. 7 Time- and dose-dependence of clinical agents’ impact on GLUL-cN in cells.
(a) Cerebody enrichment for Glul-cN from Neuro-2a cells after treatment with varying doses of lenalidomide for 24 h (n = 3). (b) Cerebody enrichment for Glul-cN from Neuro-2a cells after treatment with 100 µM lenupalidomide for varying durations (n = 3). (c) Cerebody enrichment for Glul-cN from Neuro-2a cells after treatment with DMSO, 5 µM TAK-243, 1 µM MLN4924, or 10 µM MG132 for 6 h (n = 3). Indicated sample sizes (n) represent biological replicates.
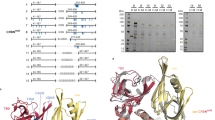
Extended Data Fig. 8 Characterization of GLUL-cN binding to CRBN.
(a) Close-up view of GLUL-cN in complex with CRBN. GLUL-cN is shown as a stick model, together with its positive Fo – Fc electron density (purple mesh) calculated and contoured at 2.5σ before it was built into the CRBN-DDB1 model. The C-terminal cN and the backbones of upstream residues were resolved with continuous density, along with clear density for the side chain of the tyrosine residue at the –2 position. CRBN is shown as an orange cartoon with the tri-Trp side chains highlighted. (b) GLUL-cN–CRBN aligned with CRBN–lenalidomide (PDB: 5FQD). For GLUL-cN–CRBN, CRBN is shown as an orange cartoon with the tri-Trp side chains highlighted. GLUL-cN is shown as sticks (carbon: green, oxygen: red, nitrogen: blue). For CRBN–lenalidomide, CRBN is shown as a purple cartoon, and lenalidomide (Len) is shown as sticks (carbon: purple, oxygen: red, nitrogen: blue). (c) GLUL-cN–CRBN aligned with MsCI4–QMQcN (PDB: 8BC6). CRBN is shown as an orange cartoon with the tri-Trp side chains highlighted. GLUL-cN is shown as sticks (carbon: green, oxygen: red, nitrogen: blue). MsCI4 is shown as a cyan cartoon, and the peptide (QMQcN) is shown as sticks (carbon: cyan, oxygen: red, nitrogen: blue, sulfur: yellow). (d–h) BLI measurements of GLUL-cN binding to CRBN WT or mutants (n = 2 biological replicates). (i) The C-terminal residues of GLUL across species from different classes of vertebrates. The conserved C-terminal N residues are highlighted in red.
Extended Data Fig. 9 Genetic knockout of Crbn leads to accumulation of Glul and Ppa1 and neurological phenotypes in mouse models.
(a) Volcano plot of tryptic peptides derived from protein C-termini that bear a C-terminal N or Q identified from the hippocampi of 4 individual WT or Crbn−/− mice (two male, two female). P-values for the abundance ratios were calculated using a one-way ANOVA with TukeyHSD post-hoc test. (b) Western blot of Glul and Ppa1 levels in WT or Crbn−/− mouse hippocampus (n = 3). (c) Normalized mRNA levels of Glul, Ppa1, Pcmt1, and Crbn in WT or Crbn−/− mouse hippocampus (n = 3). Comparisons were performed using unpaired two-tailed t-tests, and p-values are indicated. (d) The C-terminal residues of PPA1 across species from different classes of vertebrates. The conserved C-terminal N residues are highlighted in red. (e) Comparison of the time from KA injection to the first observation of convulsions (stage 3 of Racine scale) in WT (n = 19) and Crbn−/− (n = 16) mice. (f) Comparison of Racine scores of WT (n = 19) and Crbn−/− (n = 16) mice during each phase of the convulsion response. Comparisons were performed using unpaired two-tailed t-tests, and p-values are indicated. Data represent mean ± S.D. in all graphs with error bars. Indicated sample sizes (n) represent biological replicates.
Extended Data Fig. 10 PPA1 is a substrate of PCMT1 and CRBN.
(a) Time-course incubation of the peptide representing the C-terminus of human PPA1 with PCMT1 and SAM in 50 mM Tris-Cl (pH 7.4) buffer at 37 °C (n = 3). (b) Time-course incubation of the peptide representing the C-terminus of mouse Ppa1 with PCMT1 and SAM in 50 mM Tris-Cl (pH 7.4) buffer at 37 °C (n = 3). (c) TR-FRET assay of the human or mouse PPA1 C-terminal peptides after 14 h incubation with or without PCMT1 (n = 3). Data represent mean ± S.D. (d) Cerebody enrichment for PPA1-cN from different human and mouse cell lines after treatment with DMSO or 100 µM lenalidomide for 24 h (n = 3). Basal levels of enriched PPA1 may stem from nonspecific binding. Elution for HEK293T and Neuro-2a was performed with 200 µM lenalidomide, while elution for SK-N-SH was performed with 3× FLAG peptide. Indicated sample sizes (n) represent biological replicates.
Supplementary information
Supplementary Information
Supplementary Tables 1–4, Figs. 1 and 2, Notes 1 (materials and instrumentation) and 2 (synthetic procedures), NMR spectra, and LC–MS traces.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 3
Unprocessed western blots and/or gels.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 4
Unprocessed western blots and/or gels.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 6
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 4
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 5
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 6
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 7
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 9
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 10
Unprocessed western blots and/or gels.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhao, Z., Xu, W., Feng, E.Y. et al. PCMT1 generates the C-terminal cyclic imide degron on CRBN substrates. Nat Chem Biol (2025). https://doi.org/10.1038/s41589-025-02106-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41589-025-02106-9
This article is cited by
-
Cereblon induces G3BP2 neosubstrate degradation using molecular surface mimicry
Nature Structural & Molecular Biology (2026)
