
Abstract
Adeno-associated viruses (AAVs) are common vectors in gene therapy but can frequently cause liver complications in patients. The mechanisms underlying AAV-related liver toxicity remain poorly understood, posing challenges for effective prevention and intervention. Here we conducted a case study of a child with spinal muscular atrophy type 1 experiencing substantial hepatitis after receiving onasemnogene abeparvovec, undertaking long- and short-read metagenomic sequencing of liver tissue. We identified manufacturing plasmid sequences with complex structures and recombination. Vector genomes had extensive disruption and concatemerization as well as numerous vector–human fusion junctions. We also identified human betaherpesvirus 6B in the liver. Further work and investigation of more patients is needed to establish whether the presence of manufacturing plasmid sequences or helper viruses contribute to the formation of these complex concatemeric DNA structures in the liver, and whether these are a factor in the development of liver toxicity after AAV gene therapy.
Similar content being viewed by others


Main
Adeno-associated virus (AAV) gene therapies show promise for treating a variety of serious genetic conditions, including hemophilia1,2,3, muscular dystrophies4 and spinal muscular atrophy (SMA)5. As of 2025, there were seven AAV gene therapies approved by the US Food and Drug Administration6, with many more in clinical trials. The most common adverse effect of intravenously administered AAV gene therapies is hepatotoxicity, routinely treated with high dose steroids. Occasionally, liver toxicity is severe, and some patients have experienced fulminant liver failure7,8,9,10,11. Hepatotoxicity tends to be more severe in older patients with a higher body weight, who receive higher vector doses12,13.
The mechanisms underlying hepatotoxicity are incompletely understood, and it has been postulated to be caused by innate, humoral and cellular immune responses to the vector capsid, genome or transgene product14,15,16, by impurities within the vector preparation17,18 or from a direct toxic effect19,20. Acute sinusoidal endothelial injury resembling capillary leak syndrome has also been well documented in nonhuman primates using both empty capsids and therapeutic transgenes21.
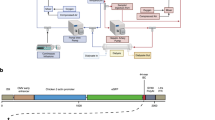
Onasemnogene abeparvovec (OA) is an AAV-vectored gene therapy for SMA, a neurodegenerative disease caused by deleterious variants in the survival motor neuron 1 (SMN1) gene22. OA is manufactured using three plasmids (Fig. 1): a vector plasmid (pSMN), which contains SMN and elements necessary for its expression; a packaging plasmid (pAAV2/9), which contains AAV2 replication (rep) and AAV9 capsid (cap) genes; and a helper plasmid (pHelper), which contains adenovirus (HAdV) genes necessary for AAV replication23,24. The resultant vector preparation contains therapeutic recombinant AAV (rAAV) particles that have an outer AAV9 capsid, containing a vector genome encoding human SMN. Manufacturing process-related impurities, including empty capsids, reverse-packaged plasmids, genome fragments and recombined products, are also present in rAAV preparations, even after good manufacturing practice procedures25,26,27. These manufacturing issues are complex to study and resolve, and the US Food and Drug Administration has released guidance on reporting and validating the steps in the manufacturing process28.

OA is produced by transfection of HEK293 cells with a vector plasmid (pSMN), containing SMN between AAV ITRs, an AAV plasmid containing AAV2 rep and AAV9 cap genes (pAAV2/9), and a helper plasmid containing HAdV genes such as E2A, E4 and VA RNA genes (pHelper)23,24. SMN, survival motor neuron; HAdV, human adenovirus; ssDNA, single-stranded DNA; dsDNA, double-stranded DNA. Created with BioRender.com.
We investigated a 7-year-old female patient treated with OA for SMA type 1 (homozygous deletion of exon 7 of SMN1, two copies of SMN2), whose clinical course has been reported previously (case 2, Finnegan et al.12). The patient weighed >20 kg at the time of infusion, and therefore required a high total vector dose of 2.2 × 1015 vector genomes. The patient experienced symptomatic hepatitis, with vomiting, jaundice, abdominal pain and dark urine. Serum hepatic markers, indicating liver injury, peaked 7 weeks after infusion (Extended Data Table 1). Liver injury was managed using steroids and tacrolimus. Tacrolimus was successfully withdrawn 7 months after infusion, and steroid treatment continued for 19 months.
A needle core liver biopsy, taken 7 weeks after infusion, showed mild perivenular and portal fibrosis and a single focus of porto-central necrosis. There was mild portal tract expansion, including a portal ductular reaction and periductal and intraepithelial neutrophils. There was a moderate portal inflammatory infiltrate composed predominantly of CD4- and CD8-positive T lymphocytes and occasional plasma cells, with mild interface inflammation and moderate lobular inflammation with foci of hepatocellular cholestasis (Fig. 2). Few CD20-positive B lymphocytes were detected. These histological features are consistent with those previously reported in children with hepatitis associated with wild-type AAV2 infection29,30 and in ‘indeterminate’ pediatric acute liver failure31. Adenovirus immunostaining was negative (Fig. 2h).

a, Liver biopsy of the patient shows marked periportal and lobular inflammation as well as interface inflammation (*); numerous hepatocytes with ballooning degeneration are present (**). b, High magnification of the box in a, with ballooning hepatocytes highlighting the swollen cytoplasm. c, Magnification of the ‘*’ region from a. d, Magnification of the ‘**’ region from a. e–g, Inflammation in the liver is shown by immunohistochemistry (IHC) detecting CD20 (e), CD8 (f) and CD4 (g). There was no noteworthy steatosis or spotty necrosis, and special stains did not show periportal diastase periodic acid-schiff (DPAS)-positive globules or iron deposition. h, Adenovirus immunostaining was negative. Scale bars, 400 µm (a), 60 µm (b and d), 100 µm (c) and 50 µm (e–h). All available tissue was stained, and representative images have been captured to illustrate the signal in the sample.
We conducted untargeted short-read metagenomic sequencing of DNA and RNA from the residual patient liver sample. In the DNA sequencing analysis, the initial assignment of nonhuman reads to the most likely microbial species identified multiple serotypes of AAV, primarily AAV2, as well as human mastadenovirus C (HAdV-C) and human betaherpesvirus 6B (HHV-6B) (Extended Data Table 2). Reads assigned to HHV-6B covered the breadth of the genome (Fig. 3b) and a specific polymerase chain reaction (PCR) for HHV-6B was positive (cycle threshold (CT) 26.2), indicating natural HHV-6B infection.

a, Genome coverage of wild-type (WT) AAV2 and HAdV-C from Illumina sequencing reads. Approximate locations of the genes present in the manufacturing plasmids are marked along the x axis. AAV2 alignment uses more stringent mapping parameters to more clearly differentiate between any AAV2- and AAV9-derived sequences (Methods). b, Alignment of Illumina sequencing reads to the HHV-6B genome shows reads cover the breadth of the genome. c, Alignment of Illumina sequencing reads to approximate manufacturing plasmid sequences shows the presence of plasmid sequences. CMV enhancer, cytomegalovirus enhancer; SV40 intron, simian virus 40 small intron; bGH poly(A), bovine growth hormone poly(A) signal. In the negative control, ten reads aligned to the pSMN sequence, while no reads aligned to the pAAV2/9 or pHelper sequences.
The incomplete genome coverage of AAV2 and HAdV suggested that the results did not derive from a wild-type infection (Fig. 3a). To investigate this further, we aligned the reads to the manufacturing plasmid sequences used in OA production. We found good coverage of the OA vector genome as expected, but also of the pSMN plasmid backbone and of pAAV2/9, and some reads mapping to pHelper (Fig. 3c). The reads originally classified as AAV2 or HAdV-C aligned only to sections of the viral genomes that are part of the OA manufacturing plasmids (AAV2 rep, HAdV E4, E2A, L4 and VA regions), suggesting the presence of plasmid sequences in the liver tissue rather than wild-type virus infection (Fig. 3a). A specific PCR for HAdV, targeting a region of the genome that is not present in the pHelper plasmid, was negative.
The presence of the pAAV2/9 plasmid sequences also potentially explains why multiple AAV serotypes, other than AAV2, including AAV4 and AAV8, were found in our initial classification. As there is not currently a RefSeq reference sequence for AAV9, it is not included in the metagenomics database. Therefore, reads from the AAV9-derived region in pAAV2/9 (AAV9 cap gene) were probably misclassified as other AAV serotypes in the initial analysis. We performed an alignment of reads from the liver to AAV1–9 genomes, finding the best alignment to the rep gene of AAV2 and the cap gene of AAV9 (Extended Data Fig. 1a and Supplementary Table 1), in keeping with the chimeric structure of pAAV2/9 (AAV2 rep gene and AAV9 cap gene). Some short regions of the pAAV2/9 plasmid sequence had no aligning reads (Fig. 3c), suggesting that the sequence we used was not fully identical to the plasmid sequence used in OA manufacture, which is proprietary. Analysis of long-read metagenomic DNA data yielded similar results: initial classification identified AAVs, HAdV and HHV-6B, but subsequent alignment revealed sequences corresponding to all three manufacturing plasmids (Extended Data Table 2).
Classification and alignment of the nonhuman RNA sequencing (RNA-seq) metagenomics data detected two reads corresponding to the AAV2 rep gene. Four further reads showed BLAST similarity to the AAV inverted terminal repeat (ITR) region but did not align. No RNA reads corresponding to pHelper, HAdV or HHV-6B were found. Previous published work has shown that our RNA-seq metagenomics protocol is as sensitive as targeted real-time PCR32. The low-level AAV RNA could result from transcription of the AAV2 rep gene; however, this signal is below the typical reporting cutoff of the metagenomics protocol and would require further validation. RNA-seq sequence alignment confirmed the presence of RNA transcripts corresponding to the OA vector genome, including SMN1 exon 7 (Extended Data Fig. 1b), suggesting successful expression of the therapeutic transgene.
Next, we performed in situ hybridization to confirm the presence and location of nucleic acid sequences derived from OA. A probe for human SMN confirmed successful vector transduction in the patient’s liver, with 28.5% of cells in the biopsy tissue showing a positive signal (control patients showed 0.4–1.5% positive cells; Fig. 4 and Extended Data Fig. 2). We observed both nuclear and cytoplasmic positive signals. To detect plasmid sequences, we designed probes complementary to regions of the manufacturing plasmids that are absent from both the therapeutic OA vector genome and the human genome. Analysis confirmed the presence of the bacterial origin of replication in pSMN, pHelper and pAAV2/9 plasmids in 5.1% of cells (probe vector-pHelper-C1, 0.2–1.1% positive in controls), as well as a sequence from the AAV9 cap gene present in pAAV2/9 in 5.8% of cells (probe AAV-HeB-T1-VP1-O1-C1, 0.2–1.1% positive in controls) (Fig. 4). The contaminant plasmid-specific sequences were found at lower levels than SMN, in agreement with the metagenomic sequencing.

a–c, In situ hybridization (ISH) for the detection of SMN1 in formalin-fixed paraffin-embedded (FFPE) liver tissue. a, A strong positive red signal was detected in the nucleus of ballooning hepatocytes separated by areas with severe immune cell infiltration (*). The box in a is magnified in b. b, Higher magnification of SMN1-positive hepatocytes next to the immune cell infiltrate (*). c, Dense nuclear signal for SMN1 and a mild-to-moderate, punctuated signal within the cytoplasm of hepatocytes and immune cells (*). d,e, ISH for the detection of manufacturing plasmid sequences in FFPE liver tissue: pSMN/pAAV2/9/pHelper ori (d) and pAAV2/9 cap (e). f, The percentage of positive cells versus two control liver tissues (control 1, explant liver tissue from a child affected by severe hepatitis in AAV2 outbreak; control 2, healthy adult liver). g, Schematic showing probe binding sites on manufacturing plasmid sequences. See Extended Data Fig. 2 for controls. Scale bars, 300 µm (a) and 100 µm (b–e). All available tissue was stained, and representative images have been captured to illustrate the signal in the sample.
We undertook detailed sequence analysis of individual reads from the long-read sequencing to determine the vector genome structures present in the liver. This showed high levels of vector genome concatemerization and complex genome structures with rearrangements (Fig. 5a–d and Extended Data Table 3). The concatemeric patterns observed, including head-to-head, head-to-tail and alternating repeats, showed similarities to those seen in replicating AAVs using rolling hairpin and rolling circle amplification33. Plasmid reads tended to not represent full-length plasmids but rather fragments of plasmid sequences in combination with the vector genome. The majority of pAAV2/9 reads also contained regions of the other manufacturing plasmids, indicating recombination between plasmids (Fig. 5 and Extended Data Table 3). Most of the complex structures and recombination events involved the vector genome, the rep–cap region of pAAV2/9 and the region of pHelper containing the HAdV-derived genes (Fig. 5 and Extended Data Table 3).

Alignment dot plots showing individual nanopore reads (x axis) aligning to representative sequences of the OA manufacturing plasmids (y axis). Red dots indicate alignment to the forward strand, and blue dots indicate alignment to the reverse strand. a, Explanation of dot plot format. b, Alignment against the vector region of the pSMN plasmid. c, Alignment against the entire pSMN plasmid. d, Alignment to the pAAV2/9 plasmid. e, Alignment to regions of all three plasmids—the vector region of pSMN, AAV rep and cap within pAAV2/9 and the HAdV gene region within pHelper. Representative images were selected; the number of reads in each category can be found in Extended Data Table 3, and diagrams for all reads are provided in the Supplementary Information. See the Supplementary Information and Methods for description of similar dot plots generated for human reads.
Rearranged sequences may derive from recombined plasmid contaminants outside vector particles, mispackaged recombined DNA from manufacture and/or recombination events after infusion. Many of the structures we observed were longer than the maximum packaging length of an AAV vector (up to 15 kb, while the packaging limit is approximately 5 kb (refs. 34,35)). Purification steps during manufacture are designed to remove nonpackaged DNA, and efficiency of uptake of any remaining DNA is likely to be low, suggesting that some recombination may have occurred in vivo, as previously described in nonhuman primate liver36.
We also identified numerous internal vector rearrangements at the DNA level from the short-read metagenomics. First, chimeric reads were identified, signifying read-through transcripts and noncanonical splice fusions at both DNA and RNA levels (Extended Data Fig. 3a). Mapping the reads to the vector plasmids revealed that most occurred between the AAV2 ITRs, with further junction points identified between the plasmid backbone and SMN transgene (Extended Data Fig. 3a). Without direct sequencing of the vector batch, we could not determine whether these rearrangements occurred during vector manufacture or within target cells, as investigated in previous studies36. Analysis of corresponding RNA reads showed substantially fewer chimeric transcripts, suggesting these rearranged DNA sequences generally did not produce stable transcripts (Extended Data Fig. 3).
Our study also revealed potential integration of AAV into the host genome. Analysis of chimeric DNA reads mapped to the pSMN plasmid revealed numerous vector–human junctions throughout the vector genome, including a small number of junctions in the plasmid backbone (Extended Data Fig. 4). However, only a subset of these junctions appeared in chimeric RNA reads (Extended Data Fig. 4b). Notably, we detected several chimeric RNA reads in the hybrid cytomegalovirus enhancer/chicken β-actin (CBA) promoter region. Analysis of the human portions of chimeric reads mapped to the human reference genome revealed no specific fusion hotspots at either DNA or RNA levels. Chimeric DNA reads predominantly localized within gene bodies, as determined by their positions relative to annotated gene loci (Supplementary Table 3). Chimeric RNA reads were detected at lower frequencies, also primarily within transcribed gene bodies without any discernible hotspots (Supplementary Table 4).
Random, low-frequency integration of rAAV vectors in patient tissue is now well recognized37,38,39,40, and AAV integrants in complex concatemers containing mixtures of rearranged and truncated vector genomes have been demonstrated in the liver tissue of nonhuman primates after intravenous administration of rAAV8 vectors36. Chimeric reads containing plasmid sequences and non-SMN human DNA were also identified by the long-read sequencing, but due to the use of a ligation library preparation kit, we were unable to verify that these were not sequencing artifacts. AAV vectors are expected to persist episomally in postmitotic cells, and therefore it is plausible that vectors and associated contaminating sequences are maintained even without integration.
In conclusion, our metagenomic sequencing approaches, together with in situ hybridization, provide evidence that sequences from all three manufacturing plasmids were present in the liver of a patient with severe hepatitis after treatment with OA, 7 weeks after infusion. Long-read sequencing also revealed extensive disruption and concatemerization of vector genomes and manufacturing plasmids, with evidence of recombination events. Complex structural rearrangements and concatemers of AAV vector genomes have previously been demonstrated in macaque liver after treatment with rAAVs36,41 and in human hepatocytes in a humanized mouse model42. Similar complex concatemeric structures have also been noted in liver samples from children with hepatitis associated with wild-type AAV2 infection29. It will be important to ascertain whether these genomic structures are also present in rAAV-treated patients without hepatitis.
The relevance of our finding of HHV-6B in the liver is unclear in this single case description. Although it is noteworthy that HHV-6 can act as a helper virus in wild-type AAV2 replication, we detected no HHV-6 RNA, suggesting no active viral replication at the time of biopsy. HHV-6 has also been found in liver tissue in a proportion of children with hepatitis associated with wild-type AAV2 infection, although also sometimes in controls29,30, and has been found in children with acute liver failure of unknown cause43,44.
The mechanism by which complex rAAV-derived genome structures are produced, and whether they arise solely during manufacture or within transduced liver cells, remains unclear. Unfortunately, we have been unable to the access the OA batch used to infuse this patient, and there is no obligation for it to be retained by the regulators. We postulate that presence of certain manufacturing plasmid sequences (such as AAV rep gene and HAdV helper regions) and/or helper viruses (such as HHV-6) could enable amplification of the vector genome within cells if expressed, giving rise to the complex concatemeric structures we observed. Formation of replication-competent rAAV particles due to nonhomologous recombination in the course of vector production has been described45. Alternatively, these large DNA concatemers may arise purely from ITR-driven intermolecular recombination of transduced rearranged vector genomes46,47.
Future work is needed to determine the frequency and pathological consequences of complex DNA structures in patient liver cells after rAAV gene therapy, whether they are episomal or integrated into the host genome, the putative role of contaminating plasmid sequences and their potential toxicity and/or immunogenicity, and how together these factors may relate to the hepatotoxicity of rAAV gene therapies. This may inform both the management of patients receiving gene therapies and the manufacture of rAAV vectors.
Methods
Ethics
The liver biopsy procedure was performed for diagnostic purposes. Liver biopsy was obtained under general anesthesia by the percutaneous route using a liver biopsy gun under ultrasound guidance. The biopsy was nontargeted from the right lobe and contained a 3–4-cm-long core of liver tissue. Written informed consent was obtained from the child’s parent for residual biopsy material to be analyzed in this study, with additional consent for research conducted under the International Severe Acute Respiratory and Emerging Infection Consortium (ISARIC) World Health Organization (WHO) Clinical Characterization Protocol UK (CCP-UK) (ISRCTN 66726260). Ethical approval for the ISARIC CCP-UK study was given by the South Central–Oxford Research Ethics Committee in England (13/SC/0149), the Scotland A Research Ethics Committee (20/SS/0028) and the WHO Ethics Review Committee (RPC571 and RPC572).
Short-read metagenomic sequencing
Untargeted Illumina metagenomic sequencing of the liver biopsy was carried out by the clinical metagenomics service at Great Ormond Street Hospital, according to the protocol previously described29,32. This is a clinical diagnostic virology laboratory and does not routinely work with plasmids, reducing the probability of contamination. A total of 44.1 million paired-end reads were obtained for DNA and 42.4 million for RNA. A negative control sample consisting of human DNA and RNA spiked with positive controls (cowpox DNA, and feline calicivirus and Escherichia phage MS2 RNA) was run in parallel, producing 44.3 and 46.3 million reads for DNA and RNA, respectively. Viruses were identified from the metagenomics data using Kraken248 and Bracken49 run through nf-core’s nextflow pipeline Taxprofiler50 with short-read quality control and host removal using hg38 enabled, as well as metaMix51 with the preprocessing pipeline previously described in ref. 29. A custom database based on all the complete bacterial, viral, fungal and protozoa genomes in RefSeq as of June 202352 was used for analysis.
Human-filtered reads from the metaMix pipeline (other than for alignment to pSMN, where raw reads were used) were aligned using Bowtie253 in very sensitive mode (apart from wild-type AAV2, where the parameters -score-min L,0,-0.1 -N 0 -L 22--mp 6,2--rdg 5,3--rfg 5,3 were used to provide more stringent mapping and help distinguish between the AAV2 and AAV9 cap sequences) to genome sequences of AAV2 (NC_001401), HHV-6B (NC_000898) and HAdV-C (NC_001405) obtained from RefSeq, as well as representative sequences of the plasmids used in OA manufacture (pSMN54, pAAV2/955, pHelper (pHGTI-Adeno1)56. A multi-fasta reference sequence consisting of AAV1–9 was also used (Supplementary Table 1). The sequence of the AMR gene region in the pSMN plasmid from the patent sequence did not match what was observed in the patient. This region was reconstructed using the long-read sequencing data, and it displayed over 99% similarity to publicly available KanR sequences (such as the KanR region of MH450172.1), suggesting that the AMR gene used in OA manufacture differs to the one in the relevant patent. The modified pSMN sequence was used in all alignments. PCR duplicates were removed from the resulting alignments using samtools markdup57, and alignments were plotted using a custom R script using tidyverse functions.
Long-read metagenomic sequencing
DNA from approximately 3 mg of liver was purified using the Qiagen DNeasy Blood & Tissue kit as per the manufacturer’s instructions. DNA was fragmented to an average size of 10 kb using a Megaruptor 3 (Diagenode) to reach an optimal molar concentration for library preparation. Quality control was performed using a Femto Pulse System (Agilent Technologies) and a Qubit fluorometer (Invitrogen). Samples were prepared for nanopore sequencing using the ligation sequencing kit SQK-LSK110. DNA was sequenced on a PromethION using R9.4.1 flowcells (Oxford Nanopore Technologies, ONT). Samples were run for 72 h, resulting in 14.1 million reads and 82.5 Gb with an N50 of 9,624 bp and a mean read quality score of 13.5. All library preparation and sequencing were performed by the UCL Long Read Sequencing facility.
Reads were trimmed using porechop58 with an adaptor threshold of 85 and were mapped to the human genome (ensemble GRCh38 v107) using minimap259 in map-ont mode. Unaligned reads were then aligned to the regions of the plasmids shown in the figures using minimap2, and the aligned reads were extracted using samtools57. A custom R script was used to filter reads that were over 1,000 bp in length, had a total alignment length of at least 80% of the total read length across all alignments and had a continuous stretch of matches/mismatches with no insertions or deletions of at least 100 bp. Alignment dot plots for these reads were created using redotable60 with a window size of 20. Representative examples are shown in the figures. Viruses were identified from the metagenomics data using Kraken2 and Bracken run through nf-core Taxprofiler50, with host removal with hg38 enabled.
Validation of alignment dot plots
To confirm that the concatemeric structures identified were not sequencing artifacts, we repeated the analysis using alignment to human genes other than SMN1. All the ONT reads were aligned to the whole human genome, and reads aligning to the GTF2H2 and ACTB genes were extracted. GTF2H2 was chosen because it is located close to endogenous SMN1 in the 5q13 region, and ACTB was chosen as a housekeeping gene on a different chromosome (Chr7). No evidence of complex concatemeric structures was found for these reads (Supplementary Fig. 2). Some duplex reads were identified, perhaps reflecting an ONT artifact where the complementary strand is sometimes sequenced directly after its pair. However, such duplex reads were excluded from the complex reads category in Extended Data Table 3 because they could result from the self-complementary vector (Supplementary Fig. 2 and Supplementary Table 2). There were also some reads that did not align completely to the targeted genes and surrounding regions, but instead partially aligned to another region of the human genome, usually on a different chromosome (Supplementary Fig. 2 and Supplementary Table 2). These could represent random ligation artifacts. However, both the frequency of these reads and the degree of concatemerization were much lower than those observed in the vector reads. Furthermore, in datasets that primarily consist of human reads, the probability of a ligation artifact arising between two human reads is likely to be much higher than the same between two vector or manufacturing plasmid reads, meaning that the human–human concatemers are more likely to have occurred by chance.
Chimeric read analysis of short-read metagenomics data
Processing of reads
Raw paired-end sequencing data were processed using fastp v0.23.261 for quality control and adapter removal. Read pairs were trimmed with a quality threshold of 20 (Phred score) and minimum length requirement of 50 bases. Adapter sequences and poly-G artifacts were automatically detected and removed using the paired-end detection algorithm. Overlapping paired-end reads were merged using PEAR v0.9.1162.
Mapping to custom reference genome
Chimeric reads were identified using STAR aligner63. A custom reference genome was prepared by adding the vector plasmid sequence (pSMN) as an additional chromosome to the human reference genome (hg38). This approach allowed simultaneous mapping to both the human genome and the vector sequence, facilitating the identification of vector–genome junctions. The STAR aligner index was generated using this modified reference with default parameters and four processing threads. The alignment was performed against the custom reference genome with minimum chimeric segment length of 12 nucleotides, minimum overhang for a chimeric junction of 12 nucleotides, and output of chimeric junctions and separate SAM files. Chimeric alignments were filtered with a minimum alignment score of 1, maximum score drop of 30 and score separation of 1. A maximum gap of 3 bases was allowed in chimeric segments. For spliced alignments, we specified a minimum overhang of 10 bases for splice junctions, and both mate gap and intron size were limited to 1,000,000 bases. The alignment was executed using four processing threads, and the output was generated as coordinate-sorted BAM files.
Analysis of chimeric vector reads
Chimeric junction data from STAR aligner output were parsed into a dataframe, filtering for fusion events involving the vector of interest (pSMN) by identifying chimeric reads where one fusion partner mapped to the vector sequence and the other to a genomic location. Chimeric junction data were processed to identify their proximity to endogenous genes using a custom Python script. Genomic coordinates from chimeric junctions were matched against gene annotations from GENCODE v3864. For each integration site, we identified the nearest gene and calculated the distance to its boundaries using a nearest-neighbor algorithm implemented in PyRanges65.
Vector coverage analysis
To evaluate read distribution and coverage patterns across the vector genome, sorted BAM files from STAR alignment were filtered using samtools (v1.15) with a BED file defining the vector regions of interest. For each sample, we generated position-specific coverage depth using the samtools depth command with the -a flag to report coverage at all positions, including those with zero coverage. Coverage profiles were visualized using a custom Python script with matplotlib. Visualizations were generated for chimeric reads that span vector–genome junctions to profile which positions in the vector genome were commonly implicated in fusions.
Mapping of internal vector rearrangements
To visualize the internal recombination events within the vector sequence, we developed a method to generate Circos plots using the pyCirclize66 Python package. Chimeric junction data were filtered to isolate vector-to-vector interactions (self-links), where both ends of a chimeric read mapped to different regions of the pSMN vector. A custom BED file was used to define the vector sequence boundaries. For each sample, vector self-interactions were represented as arcs connecting the respective start and end positions within the circular vector map. The positions were aligned against a circular representation of the parental pSMN map.
Specific pathogen PCRs
Human adenovirus (HAdV) and HHV-6 real-time PCRs were performed by the diagnostic Microbiology and Virology laboratory at Great Ormond Street Hospital, and are accredited by the UK Accreditation Service to ISO15189:2022 standards. The HAdV assay targets a 132-bp region of the HAdV hexon gene gene (forward primer: GCC ACS GTG GGG TTT CTA AAC TT, reverse primer: GCC CCA GTG GKC TTA CAT GCA CAT C, probe: TGC ACC AGA CCC GGR CTC AGG TAC TCC GA)67 and the HHV-6 assay targets a 74-bp region of the HHV-6 DNA polymerase gene (forward primer: GAA GCA GCA ATC GCA ACA CA, reverse primer: ACA ACA TGT AAC TCG GTG TAC GGT, probe: AAC CCG TGC GCC GCT CCC)68. Each target was multiplexed with an internal positive control targeting mouse (mus) DNA spiked into each sample during DNA purification, as described previously69 with detection of a noncoding sequence (forward primer: GGA CAC TAT GCC CCT CCT TAG A, reverse primer: AGC TCC AAA CTC CGT CTC TGT AA, probe: TTG GGA ACA AAA CAC CCA TGG AAG GA).
In brief, each 25-μl reaction consisted of 0.6 μM (HAdV) or 0.5 μM (HHV-6) of each primer with 0.12 μM of each mus primer, 0.4 μM (HAdV) or 0.3 μM (HHV-6) probe with 0.08 μM mus probe, and 12.5 μl Qiagen Quantifast Fast mastermix with 10 μl template DNA. PCR cycling was performed on an ABI 7500 Fast thermocycler (95 °C for 5 min followed by 45 cycles of 95 °C for 30 s and 60 °C for 30 s). Each PCR run included a no template control and a DNA-positive control for each target.
RNAscope in situ hybridization
Formalin-fixed paraffin-embedded liver sections were cut at 2–3 µm thickness and mounted on glass slides. According to the manufacturer’s instructions, RNAscope was performed with protease treatment and simmering in target solution (product codes 322360 and 322331, ACDBio) to detect the SMN gene (product code 553631, ACDBio, RNAscope Probe - Hs-SMN1-CDS - Homo sapiens survival of motor neuron 1 telomeric (SMN1) transcript variant d mRNA); the plasmid bacterial origin of replication in pSMN, pAAV2/9 and pHelper (product code 1261151-C1, ACDBio, RNAscope Probe - vector-pHelper-C1); and the AAV9 cap gene present in pAAV2/9 (product code 1261131-C1, ACDBio, RNAscope Probe - AAV-HeB-T1-VP1-O1-C1). As a positive control, a probe detecting ubiquitin (RNAscope Positive Control Probe - Hs-UBC, product code 310041, ACDBio) was used, and as a negative control, a probe for DapB (RNAscope Negative Control Probe - DapB, product code 310043, ACDBio) was used. Hematoxylin was used as a counterstain, and slides were digitized using the Leica Aperio 8 slide scanner.
To quantify the positive cells in the liver sections, the red signal of the ISH was detected using the deconvolution, cell segmentation and FISH module of the HALO-software (version 3.6, Indicalabs). A cell was considered positive if a red signal was detected in the cytoplasm and/or nucleus. Data were visualized using GraphPad Prism software (version 10).
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed paraffin-embedded tissue cut at a thickness of 3 µm, using the Ventana Benchmark ULTRA staining platform and Optiview DAB Detection kit, with a hematoxylin counterstain.
For CD4, CD8 and CD20, the positive control was tonsil. The following antibodies were used after heat-induced epitope removal (HIER) pretreatment: anti-CD4 (clone SP35, Roche, 790-4423), anti-CD8 (clone SP239, Roche, 790-7176) and anti-CD20 (clone L26, Dako (Agilent), M0755).
For adenovirus, the positive control was a known HAdV-positive gastrointestinal surgical case. A proteolytic-induced epitope removal (PIER) pretreatment with protease 1 for 4 min was used. Antibody incubation was carried out for 32 min (AdV clone 2/6 and 20/11, Roche, 760-4870, prediluted).
Statistics and reproducibility
This was a single case study, so no statistical analysis was performed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The full sequencing datasets cannot be shared because of their human genetic content, which could allow the patient to be identified. Human-filtered datasets are available from the corresponding author within 30 days of request.
Code availability
Code used for analysis in this study is available via GitHub at https://github.com/sarah-buddle/aav-gene-therapy.
References
Blair, H. A. Valoctocogene roxaparvovec: first approval. Drugs 82, 1505–1510 (2022).
Heo, Y.-A. Etranacogene dezaparvovec: first approval. Drugs 83, 347–352 (2023).
Dhillon, S. Fidanacogene elaparvovec: first approval. Drugs 84, 479–486 (2024).
Hoy, S. M. Delandistrogene moxeparvovec: first approval. Drugs 83, 1323–1329 (2023).
Hoy, S. M. Onasemnogene abeparvovec: first global approval. Drugs 79, 1255–1262 (2019).
Approved cellular and gene therapy products. FDA https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (2025).
Sarepta therapeutics shares safety update on ELEVIDYS. Sarepta Therapeutics https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-shares-safety-update-elevidys (2025).
Whiteley, L. O. An overview of nonclinical and clinical liver toxicity associated with AAV gene therapy. Toxicol. Pathol. https://doi.org/10.1177/01926233231201408 (2023).
Shieh, P. B. et al. Safety and efficacy of gene replacement therapy for X-linked myotubular myopathy (ASPIRO): a multinational, open-label, dose-escalation trial. Lancet Neurol. 22, 1125–1139 (2023).
Mullard, A. Gene therapy community grapples with toxicity issues, as pipeline matures. Nat. Rev. Drug Discov. 20, 804–805 (2021).
Chand, D. et al. Hepatotoxicity following administration of onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy. J. Hepatol. 74, 560–566 (2021).
Finnegan, R. et al. Risk-benefit profile of onasemnogene abeparvovec in older and heavier children with spinal muscular atrophy type 1. Neuromuscul. Disord. 42, 22–26 (2024).
Gowda, V. et al. Efficacy and safety of onasemnogene abeparvovec in children with spinal muscular atrophy type 1: real-world evidence from 6 infusion centres in the United Kingdom. Lancet Reg. Health Eur. https://doi.org/10.1016/j.lanepe.2023.100817 (2024).
Shirley, J. L., Jong, Y. P. de, Terhorst, C. & Herzog, R. W. Immune responses to viral gene therapy vectors. Mol. Ther. 28, 709–722 (2020).
Hösel, M. et al. Toll-like receptor 2–mediated innate immune response in human nonparenchymal liver cells toward adeno-associated viral vectors. Hepatology 55, 287–297 (2012).
Ashley, S. N., Somanathan, S., Giles, A. R. & Wilson, J. M. TLR9 signaling mediates adaptive immunity following systemic AAV gene therapy. Cell. Immunol. 346, 103997 (2019).
Larrey, D. et al. Drug-induced liver injury related to gene therapy: a new challenge to be managed. Liver Int. 44, 3121–3137 (2024).
Bucher, K. et al. Extra-viral DNA in adeno-associated viral vector preparations induces TLR9-dependent innate immune responses in human plasmacytoid dendritic cells. Sci. Rep. 13, 1890 (2023).
Hinderer, C. et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum. Gene Ther. 29, 285 (2018).
Audentes Therapeutics Inc (An Astellas Company). Comment on Docket FDA-2021-N-0651. FDA https://www.regulations.gov/comment/FDA-2021-N-0651-0013 (2021).
Hordeaux, J. et al. High-dose systemic adeno-associated virus vector administration causes liver and sinusoidal endothelial cell injury. Mol. Ther. 32, 952–968 (2024).
Groen, E. J. N., Talbot, K. & Gillingwater, T. H. Advances in therapy for spinal muscular atrophy: promises and challenges. Nat. Rev. Neurol. 14, 214–224 (2018).
Zolgensma. European Medicines Agency https://www.ema.europa.eu/en/medicines/human/EPAR/zolgensma (2020).
Wang, D., Tai, P. W. L. & Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 18, 358 (2019).
Wright, J. F. Product-related impurities in clinical-grade recombinant AAV vectors: characterization and risk assessment. Biomedicines 2, 80–97 (2014).
Srivastava, A., Mallela, K. M. G., Deorkar, N. & Brophy, G. Manufacturing challenges and rational formulation development for AAV viral vectors. J. Pharm. Sci. 110, 2609–2624 (2021).
Brimble, M. A. et al. Preventing packaging of translatable P5-associated DNA contaminants in recombinant AAV vector preps. Mol. Ther. Methods Clin. Dev. 24, 280–291 (2022).
Chemistry, manufacturing, and control (CMC) information for human gene therapy investigational new drug applications (INDs). FDA https://www.fda.gov/regulatory-information/search-fda-guidance-documents/chemistry-manufacturing-and-control-cmc-information-human-gene-therapy-investigational-new-drug (2020).
Morfopoulou, S. et al. Genomic investigations of unexplained acute hepatitis in children. Nature 617, 564–573 (2023).
Ho, A. et al. Adeno-associated virus 2 infection in children with non-A–E hepatitis. Nature 617, 555–563 (2023).
Chapin, C. A. et al. Activated CD8 T-cell hepatitis in children with indeterminate acute liver failure. J. Pediatr. Gastroenterol. Nutr. 71, 713–719 (2020).
Atkinson, L. et al. Untargeted metagenomics protocol for the diagnosis of infection from CSF and tissue from sterile sites. Heliyon 9, e19854 (2023).
Meier, A. F. et al. Herpes simplex virus co-infection facilitates rolling circle replication of the adeno-associated virus genome. PLoS Pathog. 17, e1009638 (2021).
Grieger, J. C. & Samulski, R. J. Packaging capacity of adeno-associated virus serotypes: impact of larger genomes on infectivity and postentry steps. J. Virol. 79, 9933–9944 (2005).
Dong, J.-Y., Fan, P.-D. & Frizzell, R. A. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther. 7, 2101–2112 (1996).
Greig, J. A. et al. Integrated vector genomes may contribute to long-term expression in primate liver after AAV administration. Nat. Biotechnol. 42, 1232–1242 (2024).
Gil-Farina, I. et al. Recombinant AAV integration is not associated with hepatic genotoxicity in nonhuman primates and patients. Mol. Ther. 24, 1100–1105 (2016).
Kaeppel, C. et al. A largely random AAV integration profile after LPLD gene therapy. Nat. Med. 19, 889–891 (2013).
Schmidt, M. et al. Molecular evaluation and vector integration analysis of HCC complicating AAV gene therapy for hemophilia B. Blood Adv. 7, 4966–4969 (2023).
Symington, E. et al. Long-term safety and efficacy outcomes of valoctocogene roxaparvovec gene transfer up to 6 years post-treatment. Haemophilia 30, 320–330 (2024).
Sun, X. et al. Molecular analysis of vector genome structures after liver transduction by conventional and self-complementary adeno-associated viral serotype vectors in murine and nonhuman primate models. Hum. Gene Ther. 21, 750–761 (2010).
Dalwadi, D. A. et al. AAV integration in human hepatocytes. Mol. Ther. 29, 2898–2909 (2021).
Warner, S., Brown, R. M., Reynolds, G. M., Stamataki, Z. & Kelly, D. A. Case report: acute liver failure in children and the human herpes virus 6-? A factor in the recent epidemic. Front Pediatr. 11, 1143051 (2023).
Yang, C. H. et al. Evaluating for human herpesvirus 6 in the liver explants of children with liver failure of unknown etiology. J. Infect. Dis. 220, 361–369 (2019).
Allen, J. M., Debelak, D. J., Reynolds, T. C. & Miller, A. D. Identification and elimination of replication-competent adeno-associated virus (AAV) that can arise by nonhomologous recombination during AAV vector production. J. Virol. 71, 6816–6822 (1997).
Yang, J. et al. Concatamerization of adeno-associated virus circular genomes occurs through intermolecular recombination. J. Virol. 73, 9468–9477 (1999).
Yan, Z., Zak, R., Zhang, Y. & Engelhardt, J. F. Inverted terminal repeat sequences are important for intermolecular recombination and circularization of adeno-associated virus genomes. J. Virol. 79, 364–379 (2005).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Lu, J., Breitwieser, F. P., Thielen, P. & Salzberg, S. L. Bracken: estimating species abundance in metagenomics data. PeerJ Comput. Sci. 3, e104 (2017).
taxprofiler: Introduction. nf-core https://nf-co.re/taxprofiler/1.0.1.html (2023).
Morfopoulou, S. & Plagnol, V. Bayesian mixture analysis for metagenomic community profiling. Bioinformatics 31, 2930–2938 (2015).
Buddle, S. et al. Evaluating metagenomics and targeted approaches for diagnosis and surveillance of viruses. Genome Med. 16, 111 (2024).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Kaspar, B. K., Burghes, A. & Porensky, P. Intrathecal delivery of recombinant adeno-associated virus 9. US patent US20190269798A1 (2022).
Gao, G., Wilson, J. & Alvira, M. Adeno-associated virus (AAV) serotype 9 sequences, vectors containing same, and uses therefore. US patent US7198951B2 (2005).
Gray, J. Molecule Information, pHGTI-Adeno1 (Harvard Gene Therapy Initiative, 2004).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078 (2009).
Wick, R. Porechop. GitHub https://github.com/rrwick/Porechop (2023).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Andrews, S. Redotable. GitHub https://github.com/s-andrews/redotable (2022).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Zhang, J., Kobert, K., Flouri, T. & Stamatakis, A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Frankish, A. et al. GENCODE 2021. Nucleic Acids Res. 49, D916–D923 (2021).
Stovner, E. B. & Sætrom, P. PyRanges: efficient comparison of genomic intervals in Python. Bioinformatics 36, 918–919 (2020).
Shimoyama, Y. pyCirclize. GitHub https://moshi4.github.io/pyCirclize/ (2025).
Brown, J. R., Shah, D. & Breuer, J. Viral gastrointestinal infections and norovirus genotypes in a paediatric UK hospital, 2014–2015. J. Clin. Virol. 84, 1–6 (2016).
Watzinger, F. et al. Real-time quantitative PCR assays for detection and monitoring of pathogenic human viruses in immunosuppressed pediatric patients. J. Clin. Microbiol. 42, 5189–5198 (2004).
Tann, C. J. et al. Prevalence of bloodstream pathogens is higher in neonatal encephalopathy cases vs. controls using a novel panel of real-time PCR assays. PLoS ONE 9, e97259 (2014).
Acknowledgements
S.B., O.T.M. and S.M. are funded by the National Institute for Health Research (NIHR) Blood and Transplant Research Unit for Genomics to Enhance Microbiology Screening (NIHR203338). L.-A.K.B. is funded by the NIHR Great Ormond Street Biomedical Research Centre (BRC). J.B. receives funding from the NIHR UCL/UCLH BRC. J.B. is an NIHR Senior Investigator. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. R.K. is funded by LifeArc P2020-0008 and P2023-0011, Great Ormond Street Hospital Children Charity and Dravet Syndrome UK Charity V4720 and V4919 and Therapeutic Acceleration Support (TAS), UCL. This work was supported by grants CRUSH MC_UU_00034/9 and Wellcome Trust 226141/Z/22/Z. The support of the GOSH and UCLH/ Institute of Neurology BRC to the Dubowitz Neuromuscular Centre Biobank is gratefully acknowledged. The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the paper. We thank the team of the Histology Research Service, University of Glasgow, for the excellent technical support. We also thank M. Deheragoda, King’s College Hospital NHS Foundation Trust Liver Pathology department, for her expert input.
Author information
Authors and Affiliations
Consortia
Contributions
S.B., L.-A.K.B., S.M., M.S., E.C.T., J.C., S.N.W., F.M. and J.B. designed the study. J.R.B., L.A., A.K., D.D., N. Storey, J.C., S.N.W. and S.B. performed and analyzed the short-read metagenomics. O.E.T.M., H.M., J.L. and S.B. performed and analyzed the long-read metagenomics. L.C. and N. Sebire performed and interpreted the histopathology. V.H., R.O., P.A. and G.I. performed and interpreted the in situ hybridization. H.B., M.G.S. and J.K.B. either obtained patient consent or were involved in maintaining ISARIC protocols. A.D., M.S. and G.B. provided clinical care for the patient and edited the paper. R.K. edited the paper. S.B. and L.-A.K.B. wrote the paper. All authors edited and approved the paper.
Corresponding author
Ethics declarations
Competing interests
G.B. is PI of clinical trials Sponsored by Roche, Novartis, Sarepta, Pfizer, NS Pharma, Reveragen, Percheron, Biomarin and Scholar Rock and has received speaker and/or consulting fees from Sarepta, PTC Therapeutics, Entrada Therapeutics, Pfizer, Biogen, Novartis Gene Therapies, Inc. (AveXis), and Roche, as well as grants from Sarepta, Roche and Novartis Gene Therapies. UCL has received funding from Sarepta, Roche, Pfizer, Italfarmaco, Santhera and Moderna. F.M. is the PI of the Novartis-sponsored trials in which OA was studied in the UK and is also involved in clinical trials sponsored by Biogen, Roche, Sarepta Therapeutics, Genethon, PTC therapeutics and Solid Bioscience. He has received consulting fees from Pfizer, Sarepta, Roche, Biogen, Novartis, Solid, Dyne Therapeutics, Entrada, PTC and Edgewise. M.S. is the sub-I of the Novartis sponsored trials in which OA was studied in the UK and is also involved in clinical trials sponsored by Biogen, Roche and Dyne. She has received consulting fees from Roche, Biogen and Novartis. The other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Eugenio Montini, Simone Spuler and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Anna Maria Ranzoni, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Additional alignments of metagenomics data.
Alignment of short-read metagenomics data to A: AAV1-9 (DNA reads) and B: pSMN plasmid (RNA reads). Exon 7 is highlighted in grey. A full reference genome is not available for AAV9, so the sequence used is the closest full-genome BLAST match to the AAV9 cap sequence in the pAAV2/9 plasmid. The AAV9 rep sequence (shown in grey) is not verified and should be treated with caution, although in this sequence it closely resembles AAV2 rep. AAV accession numbers can be found in Supplementary Table 1.
Extended Data Fig. 2 In situ hybridization controls.
A) Abundant ubiquitin (RNA integrity, reference gene) positive signal detected. B) DapB (negative control, bacterial gene) no signal detected. Scale bars = 100 µm.
Extended Data Fig. 3 Internal vector rearrangements within pSMN plasmid.
Circular representation of internal vector rearrangements for A) DNA and B) RNA reads. Genomic tracks were drawn using pyCirclize software. The vector reference map was created using SnapGene software.
Extended Data Fig. 4 Mapping of human-AAV chimeric reads against the vector plasmid.
A) DNA and B) RNA chimeric read alignment showing sequence coverage plotted as the frequency each nucleotide position appears in chimeric reads along the vector reference plasmid (x-axis). The plasmid map above each graph illustrates the corresponding functional elements. Key components include: M13 ori (bacteriophage origin of replication), ITR (inverted terminal repeat), CMV (cytomegalovirus enhancer), CBA (chicken beta-actin core promoter), SV40 intron (simian virus 40 small intron), SMN (spinal motor neuron coding sequence), SMN 3’UTR (noncoding region from endogenous SMN locus), bGH polyA (bovine growth hormone polyA signal), AmpR promoter (bacterial promoter for ampicillin resistance), KanR (kanamycin resistance marker), and ori (bacterial origin of replication).
Supplementary information
Supplementary Information (download PDF )
Supplementary Tables 1–4 and Figs. 1 and 2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Buddle, S., Brown, LA.K., Morfopoulou, S. et al. Contaminating plasmid sequences and disrupted vector genomes in the liver following adeno-associated virus gene therapy. Nat Med 32, 472–480 (2026). https://doi.org/10.1038/s41591-025-04073-z
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41591-025-04073-z
