
Abstract
To understand gene function, it is necessary to compare cells carrying the mutated target gene with normal cells. In most biomedical studies, the cells being compared are in different mutant and control animals and, therefore, do not experience the same epigenetic changes and tissue microenvironment. The experimental induction of genetic mosaics is essential to determine a gene cell-autonomous function and to model the etiology of diseases caused by somatic mutations. Current technologies used to induce genetic mosaics in mice lack either accuracy, throughput or barcoding diversity. Here we present the iFlpMosaics toolkit comprising a large set of new genetic tools and mouse lines that enable recombinase-dependent ratiometric induction and single-cell clonal tracking of multiple fluorescently labeled wild-type and Cre-mutant cells within the same time window and tissue microenvironment. The labeled cells can be profiled by multispectral imaging or by fluorescence-activated flow cytometry and single-cell RNA sequencing. iFlpMosaics facilitate the induction and analysis of genetic mosaics in any quiescent or progenitor cell, and for any given single or combination of floxed genes, thus enabling a more accurate understanding of how induced genetic mutations affect the biology of single cells during tissue development, homeostasis and disease.
Similar content being viewed by others
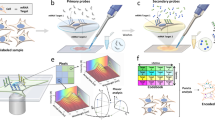

Main
The induction of a gene deletion or mutation can substantially alter a cell’s phenotype, and over time, it can also affect the surrounding tissue’s phenotype. Scientists often analyze mutant cells or tissues that have carried genetic mutations for several days, months or even years and compare their phenotype with that of independent control cells or tissues from distinct nonmutant animals. During the process from gene mutation to phenotypic manifestation and analysis, the biology of the targeted and surrounding tissue often undergoes significant changes. Since the wild-type cells surrounding mutant cells are themselves a source of biochemical factors, any alteration to their development or function by the mutant cells will trigger changes in a key tissue feedback mechanism, and any such changes will impact the phenotype of the mutant cells in a noncell-autonomous manner, that is, not directly dependent of the initially induced genetic mutation itself1,2,3. Over time, this phenomenon often generates secondary mutant tissue phenotypes that can confound interpretation of the primary impact of a gene mutation on a cell’s phenotype.
Genetic mosaics are a powerful research tool because they allow the study of cell-autonomous gene function when mutant and wild-type cells originate from the same progenitor cells. In this scenario, the only difference between the cells being compared is the induced mutation, in an otherwise identical organism, genetic background and tissue microenvironment. Mouse models that allow the timed induction of somatic genetic mosaics are essential to accurately understand a gene function and model biological or disease processes caused by sporadic somatic mutations.
In Drosophila, interchromosomal mitotic recombination associated with distinct tissue markers has been widely used to induce and track genetic mosaics. A similar approach, called mosaic analysis with double markers (MADM), has been developed in mice to allow the labeling of control and mutant cells with different fluorescent markers4,5,6. This method allows a full correlation between the expression of a given fluorescent marker and the genetic status (wildtype, heterozygous or knockout (KO)) of the locus carrying the mutation. However, MADM relies on rare interchromosomal Cre-dependent recombination events and cannot be efficiently induced with tamoxifen-inducible CreERT2 lines, which are weaker and only transiently active. Moreover, this technology requires chromosomal genetic linkage between the engineered MADM elements and another gene mutation, requiring the use of a different mouse line for each of the 20 pairs of mouse chromosomes, limiting its applicability. MADM is also difficult to use in epistasis analyses that require simultaneous loss-of-function of multiple genes present in distinct chromosomes and cannot be induced in quiescent cells.
Given the limitations of MADM, the most widely used method for generating conditional somatic genetic mosaics in the mouse remains the much simpler CreERT2 recombinase-dependent mosaic induction of a floxed-gene (flanked by loxP sites) deletion. With this method, the location, timing and frequency of the recombination events can be regulated by restricting CreERT2 expression to a specific tissue and by varying the timing and dose of treatment with the CreERT2 activating ligand tamoxifen. This simpler method requires the use of independent fluorescent reporters of Cre recombination to detect cells with CreERT2 activity. However, several studies have shown that Cre-activity reporter alleles only accurately report recombination of themselves and cannot be used to reliably report the mosaic recombination of other floxed genes7,8,9. Thus, though much easier to implement than MADM, CreERT2-dependent genetic mosaic approaches generate a high frequency of false positives and false negatives and should not be used for mosaic functional genetics.
To overcome the limitations of current methods for the generation of genetic mosaics in mice, we have developed iFlpMosaics, a compendium of new genetic tools and mouse lines that enable Flp recombinase-dependent-induction of multispectral and ratiometric genetic mosaics of both wild-type and mutant cells in any tissue. iFlpMosaics provide a high degree of fluorescent barcoding and clonal resolution of both mutant and wild-type cells and tighter control of the frequency of genetic mosaicism over time. This technology is compatible with all existing floxed alleles and can be induced in any quiescent or proliferating cell. The combination of iFlpMosaics with an additional iDre/FlpProgenitor allele enables its additional induction by any DreERT2 line and the generation of twin-spot clones derived from a single-progenitor cell. The ratiometric mutant and wild-type cell profiling and comparative analysis can be done by in situ multispectral imaging or ex situ by fluorescence-activated flow cytometry (FACS) or single-cell RNA sequencing (scRNA-seq). iFlpMosaics will enable the accurate and high-throughput quantitative analysis how an individual or combinatorial set of genetic mutations affects the biology of single cells during tissue development, regeneration or disease.
Results
Design and validation of iFlpMosaic mice
Several studies have shown that the induction of genetic mosaics in mice using CreERT2 is prone to the induction of false positives and false negatives at high frequency7,8. We confirmed that the efficiency of Cre-dependent recombination of different reporters and floxed alleles varies widely (Extended Data Fig. 1a,b) and that the frequency of cells having the desired recombination of two independent floxed alleles (true positives) is very low, particularly at the relatively low doses of tamoxifen needed to induce genetic mosaics with CreERT2 (Extended Data Fig. 1c,d). These data show that most functional genetic studies using CreERT2-dependent mosaic genetics will be unable to uncover the real effect of a deleted floxed gene on single-cell biology with high accuracy and statistical power. In addition, unlike MADM, CreERT2-dependent genetics only induce the labeling of pseudomutant cells, not the wild-type cells in the same tissue microenvironment, which substantially limits the comparative analysis.
To overcome the caveats of existing technologies used to induce genetic mosaics in mice, we developed a toolbox consisting of several mouse lines that allow the FlpO/FlpO-ERT2-recombinase-dependent induction of ratiometric fluorescent mosaics of bona fide mutant (gene KO) and wild-type cells (Fig. 1a,b and Extended Data Fig. 2a,b). We call these new mouse alleles iFlpMosaics (R26-iFlpMTomato-Cre/MYFP and Tg-iFlpMTomato-H2B-GFP-Cre/MYFP-H2B-Cherry-FlpO). They enable mosaic multispectral labeling and fate mapping of different mutant cells expressing the fluorescent membrane-tagged tomato protein (MTomato) and Cre, as well as of wild-type cells expressing membrane-tagged YFP (MYFP), in any tissue expressing FlpO or FlpO-ERT2. The two iFlpMosaic lines differ either in the genomic location, ROSA26 (R26) versus random transgene (Tg) or in the expression of additional chromatin-bound fluorescent proteins (H2B-tag) for increased cell resolution and nuclei segmentation.

a, Flp/FlpO-ERT2-induced recombination of the iFlpMosaic allele leads to one of two possible outcomes: cells expressing MTomato and Cre (mutant) or cells expressing MYFP (wildtype) (Extended Data Fig. 2). A confocal micrograph shows some labeled retina astrocytes (AC), endothelial cells (EC, ERG+ nuclei) and blood cells (BC). b, The Tg-iFlpMTomato-H2B-GFP-Cre/MYFP-H2B-Cherry-FlpO allele and confocal micrographs showing the full spectral separation of the four fluorescent labels expressed in mutant and wild-type retina cells (ECs and few ACs). c, The proportion of MTomato+ and MYFP+ cells detected by FACS. n.s., not significant. d, A comparison of MTomato intensity and Cre expression. e, Recombination of the Cre-reporter Rosa26-LSL-YFP in MTomato+ cells (white arrowheads) after induction of the R26-iFlpMTomato-Cre/MYFP allele by Apln-FlpO. There are few false positives (red arrows, MTomato+). f, qRT-PCR assessing gene deletion efficiency. P values are indicated on top of the horizontal lines. g, Induction of R26-iFlpMTomato-Cre/MYFP in mice carrying a floxed allele for Rbpj, an essential gene for arterial development, results in only 3.5% MTomato-2A-Cre+ cells in arteries (A), versus 96% in veins (V) and capillaries. h, Immunostaining for VEGFR2 and quantification of gene deletion in P7 heart sections ECs (Apln+). i, Immunostaining for RBPJ and quantification of gene deletion in adult lung epithelium (induced with FlpO-ERT2). j, Immunostaining for MYC and MYCN and quantification of genetic deletion in P7 retina ECs (Apln+). k, Immunostaining for DLL4 and quantification of gene deletion in adult lung epithelium (induced with FlpO-ERT2). The red arrows indicate the cells with MTomato-2A-Cre expression, the green arrows indicate the cells with MYFP expression and the gray arrows indicate the nonfluorescent/wild-type cells. The data are presented as mean values ± standard deviation. Scale bars, 50 μm for b, e, g, h and j and 10 μm for i and k.
Transfection of iFlpMosaic embryonic stem (ES) cells with FlpO-expressing plasmids or induction of these mosaics in mice carrying FlpO/FlpO-ERT2 alleles induced the expected recombination between the pair of FRTF3 or FRT5T2 sites but not among the different sites, which enables the induction of ratiometric mosaics of MTomato+ and MYFP+ cells, with very few cells being double positive due to recombination during the G2/M phase of the cycle (Fig. 1a and Extended Data Fig. 2c–g). The observed higher frequency of MTomato+ cells is consistent with the shorter genetic distance between the FRTF3 sites than between the FRT5T2 sites (Fig. 1a,b and Extended Data Fig. 2a,b,e).
In ES cells and mice, the initially induced cell ratios did not change after prolonged culture or during the entire embryo development in vivo (Fig. 1c and Extended Data Fig. 2h), showing that expression of MTomato-2A-Cre (or MYFP) is not deleterious to cells. Notably, we found on average ten times lower expression of the MTomato-2A-Cre cassette in iFlpMosaic mice than in the previously generated iSuRe-Cre mice (Fig. 1d), which substantially reduces the chance of Cre toxicity8.
In previous reports, inducible Cre-expressing alleles located in the Rosa26 locus were found to be leaky and to induce germline recombination, even in the absence of FlpO/CreERT2 activity8,10. None of the analyzed iFlpMosaic mice had leaky expression of the fluorescent proteins contained in their constructs in the absence of an extra allele expressing FlpO (Extended Data Fig. 2i). When combined with other sensitive Cre reporters, the Tg-iFlpMTomato-H2B-GFP-Cre/MYFP-H2B-Cherry-FlpO mouse line had very low nonself Cre leakiness, lower than standard CreERT2 lines (Extended Data Fig. 2j,k). The R26-iFlpMTomato-Cre/MYFP line had no leakiness (Extended Data Fig. 2i).
In iFlpMosaic mice, MTomato-2A-Cre+ cells efficiently recombined other Cre-reporter alleles and all the six floxed genes tested (Fig. 1e–k). These data show that unlike the commonly used CreERT2-dependent mosaic genetics (Extended Data Fig. 1), iFlpMosaics enable the induction and labeling of bona fide mutant and wild-type cells, ensuring high genetic reliability in the analysis of mosaic and single-cell phenotypes.
Generation of ubiquitous and tissue-specific FlpO-ERT2 mice
Multispectral genetic mosaics are a particularly valuable research tool if they can be induced at a specific timepoint and in a substantial number of cells, something that is not possible with the MADM system4,5,6,11. To obtain temporal control over Flp activity or induction, previous studies generated mouse lines that enabled the expression of Flpe-ERT2 or the mammalian codon-optimized FlpO-ERT210,12. These studies showed that FlpO-ERT2 activity is substantially lower than CreERT2, and very few cells in each organ were shown to recombine10. In agreement with this, we found that the published R26CAG-FlpO-ERT2 line recombined the R26-iFlpMTomato-Cre/MYFP allele in only ~0.2% of embryonic cells and was also weak in postnatal and adult organs (Fig. 2a).

a, Recombination of the R26-iFlpMTomato-Cre/MYFP allele induced by the published R26CAG-FlpO-ERT2 allele after a single dose of tamoxifen. b, A schematic of novel Tg(Ins-CAG-FlpO-ERT2) alleles containing DNA elements to enhance FlpO-ERT2 expression (Extended Data Fig. 2). c, A comparison of FlpO copy number by genomic DNA qRT-PCR of founders (F) 1–5. d–g, A comparison by FACS and histology of the recombination efficiency of the different FlpO-ERT2-expressing mouse lines/founders at embryonic, postnatal and adult stages. The fold change is indicated for the most substantial changes. CardioM, cardiomyocytes; LSK+, Lin−Sca1+c-Kit+ cells; MP, myeloid progenitors. h, CRISPR–Cas9 targeting with a guide RNA (gRNA) and a donor DNA for the generation of novel endothelial-specific Cdh5-FlpO-ERT2-expressing mouse lines from the published Cdh5-CreERT2 mouse line. i, A comparison of FlpO and Cre copy number by genomic DNA qRT-PCR. j, Recombination efficiency of founder number 1 of the newly generated Tg(Cdh5-FlpO-ERT2) mouse line in embryonic, postnatal and adult tissues and some representative confocal micrographs. White box delimits the magnified area shown to the right. The data are presented as mean values ± standard deviation. Scale bars, 100 μm.
To enhance FlpO-ERT2 expression, we, therefore, sought to develop new constructs containing optimized DNA expression elements13,14 (Fig. 2b). We generated and screened five transgenic ubiquitous FlpO-ERT2 mouse lines with variable copy number (Fig. 2c). Compared with the published R26CAG-FlpO-ERT2 line, the best of these new Tg(Ins-CAG-FlpO-ERT2) lines (founder no. 2) induced several-fold higher recombination of the iFlpMosaic allele in embryos and different organs and cell types (Fig. 2d–g and Extended Data Fig. 3a–c). With this higher recombination frequency, we frequently observed mutant (MTomato+) and wild-type (MYFP+) cells close to each other, enabling direct comparison of cells experiencing the same tissue microenvironment (Fig. 2f,g and Extended Data Fig. 3d). We also confirmed that the ubiquitous and strong FlpO-ERT2 expression is not leaky in any organ (Extended Data Fig. 3e). Besides generating ubiquitous FlpO-ERT2 lines of general relevance, we also developed a method to easily target and modify preexisting and prevalidated CreERT2-expressing transgenic alleles to achieve tissue-specific expression of FlpO-ERT2 (Fig. 2h). Using this method, we generated the Cdh5-FlpO-ERT2 mouse line, that induced the specific recombination of iFlpMosaics in endothelial cells (Fig. 2h–j).
Ratiometric functional genetics with iFlpMosaic mice
Unlike classical Cre reporters, iFlpMosaics were designed to be ratiometric. This allows quantification by histology or FACS of the relative frequency (ratio) of mutant (MTomato-2A-Cre+) and wild-type (MYFP+) cell populations induced in the same tissue and experiencing the same microenvironment throughout the pulse–chase period. We envisioned that this property would allow high-throughput and accurate analysis of the cell-autonomous function of genes in virtually all cell types and over long periods. Such an analysis is not possible with classical Cre/CreERT2 mosaic genetics because of the high frequency of false positives and negatives (Extended Data Fig. 1c,d) and the lack of simultaneous labeling of wild-type cells, which provide an important internal control for comparative mosaic analysis in the same tissue.
To demonstrate the utility of iFlpMosaic mice to reveal the effect of induced mutations in multiple cell lineages, we intercrossed them with mice containing either the ACTB:FlpE allele15 (recombines early embryo progenitor cells), the Apln-FlpO allele16 (recombines endothelial and derived hematopoietic cells) or the new inducible Tg(Ins-CAG-FlpO-ERT2) allele.
When the R26-iFlpMTomato-Cre/MYFP allele was combined with the ACTB:FlpE or Apln-FlpO alleles and the Rbpj, Myc and Foxo floxed alleles, mosaic mutant animals survived until adult stages, unlike embryos carrying mutations of these genes in all cells or in endothelial cells (ECs)17,18. We first conducted a high-throughput FACS analysis to score the relative ratios of mutant cells (MTomato-Cre+) and wild-type cells (MYFP+) in major organs of postnatal day (P)7 animals. The assumption was that if the targeted gene is important for a given cell type or organ, its deletion would change the proliferation or differentiation rate of the mutant cells in relation to wild-type cells, and this would be noticed in the final MTomato+/MYFP+ log ratio.
MYC loss has previously been linked to a general cell competitive disadvantage19 and a lack of proliferation by ECs20. However, we found that mosaic Myc deletion had a relatively minor effect on EC clonal expansion throughout embryonic development (Fig. 3a). Instead, the highest sensitivity to the mosaic loss of MYC was observed in the hematopoietic lineage (Fig. 3b and Extended Data Fig. 3f,g). Unlike liver and lung, heart CD31−CD45− cells were not significantly affected by Myc deletion (Fig. 3c), probably due to compensation by the homologous Mycn gene in cardiomyocytes as previously reported21.

a,b, R26-iFlpMTomato-Cre/MYFP mosaics were induced with Apln-FlpO (recombines endothelial and derived hematopoietic cells at E9.5) on control (wildtype) and Myc-floxed backgrounds, and the tissues were analyzed at P7. The chart and FACS data correspond to CD31+ (ECs, in a) and CD45+ (blood, in b) cells. The absolute fold change of the log2 ratio is indicated in red and P values in black. c, Similar FACS analysis of the indicated organs from R26-iFlpMTomato-Cre/MYFP mice carrying the ACTB-FlpE allele. d, iFlpMosaic deletion of Foxo1/3/4 from P1 to P15 increases the FACS frequency of MTomato+/mutant cells in the heart but not in the liver or lungs. e, Confocal micrographs of control and Foxo1/3/4 mosaic P6 and P15 retinas from animals induced at P1. Dense clusters of ERG+ ECs are indicated with yellow circles. The chart shows ratiometric analysis of retinal immunostainings for MTomato, MYFP and ERG (labels EC nuclei). f, Confocal micrographs of P6 retinas showing FOXO1 immunostaining and deletion exclusively in MTomato+ cells. The data are presented as mean values ± standard deviation. Scale bars, 100 μm.
We also confirmed the ability of the system to support mosaic epistasis analysis of mutant cells having multiple deleted genes, in this case, three Foxo genes (six floxed alleles), present in distinct chromosomes. Foxo genes partially compensate each other’s function in several cell types, and together, they are considered important negative regulators of cell proliferation and metabolism downstream of AKT signaling, preventing uncontrolled cell growth20,22,23. Surprisingly, our analysis showed that mosaic deletion of three Foxo genes (Foxo1/3/4KO) had relatively minor consequences for cell expansion in the liver and lung but especially affected heart cells (Fig. 3d). This suggests cell-type and organ-specific functions for Foxo genes. The data also show that postnatal retinal ECs are especially responsive to the loss of Foxo1/3/4. These cells were much more proliferative, formed dense clusters and outcompeted wild-type cells over time (Fig. 3e,f).
Finally, when iFlpMosaics were combined with the Rbpj-floxed allele, we could find that single RbpjKO ECs proliferate normally but mobilize preferentially to the leading edge of the vessels, being absent in arteries (Extended Data Fig. 4).
Overall, these results show the utility of the iFlpMosaics technology for the accurate modeling and high-throughput analysis via FACS or direct tissue imaging of the impact of single or multiple somatic gene mutations on cellular expansion and mobilization.
Single-cell functional mosaic genetics with iFlpMosaics
Both MADM and iFlpMosaics label mutant and wild-type cells with two different fluorescent proteins; however, for single-cell clonal analysis at high resolution, higher cellular barcoding is needed. To obtain higher clonal resolution of both mutant and wild-type cells, we developed the iFlpH2B-Cherry/GFP/Cerulean allele, which enables the chromatin/nuclei labeling of a fraction of the membrane-labeled mutant and wild-type cells (Extended Data Fig. 5a–d), which is ideal for the analysis of the clonal dynamics of single-cells. For even higher multispectral barcoding, it is possible to combine iFlpMosaics with the iCreMYFP/Tomato/MTFP1 allele13 and, in this way, obtain up to 12 different fluorescent barcodes for mutant cells and 7 for wild-type cells (Extended Data Fig. 5e,f).
In the liver, a high percentage of neonatal hepatocytes are in the S phase (EdU+) or in cycle (Ki67+) at P1 and P7, and most stop dividing before P20 (Fig. 4a). However, the hepatocyte single-cell clonal expansion dynamics are still unknown. iFlpMosaic × iFlpH2B-Cherry/GFP/Cerulean mice were induced at P1, and livers were collected at P20 for multispectral imaging and single-cell clonal analysis. Multispectral confocal scanning and spatial mapping of clones revealed that hepatocyte proliferation (clones with two cells or more) was dispersed throughout the postnatal liver (Fig. 4b and Extended Data Fig. 6a,b). The average clone size obtained was similar at both low and high iFlpMosaic recombination frequencies (Fig. 4c), confirming that there is enough multispectral clonal and quantitative resolution even at high induction rates.

a, Quantification of Ki67+ cells (in cycle) and EdU+ cells (S phase) in livers. b, Left: a schematic of the alleles and how they can be used to fluorescently barcode and track the clonal expansion of single mutant/MTomato+ and wild-type (WT)/MYFP+ cells from P1 to P20. Right: representative confocal micrographs with magnified insets (white boxes) of P20 livers (from animals induced with 4-OHT at P1) with different recombination frequencies (Extended Data Fig. 6). c, The clone size distributions of hepatocytes at both low and high recombination rates. d, The whole-liver weight at P1 and P20. e, A quantification of MYFP+ (MycWT) and MTomato+ (MycKO) clone size frequency. f, A chart depicting the normalized expansion of MycWT and MycKO hepatocytes. g, The representative confocal micrographs of P20 pancreas from control and Myc-floxed mice carrying the indicated iFlpMosaic alleles. Right: inverted LUT images depicting the clone mapping (Fiji image analysis scripts) according to their dual color code. h, The quantification of clone size frequency. i, A chart depicting the normalized expansion of MycWT and MycKO pancreatic cells. j, log2 ratio of MTomato+ cells to MYFP+ cells in control and Myc-floxed pancreas, showing a 3.7-fold decrease of MTomato+ (MycKO) cells over 20 days. k, Representative confocal micrographs of adult livers from control and Myc-floxed mice in which the indicated alleles were induced 14 days before tissue collection. Right: a semiautomatic mapping of clones according to their fluorescent barcode. Note the loss of MTomato+ cells in the Myc-floxed background. l, The clone size, showing that WT hepatocytes rarely divide or expand over a 2 week period. m, The clone frequency, showing the lack of impact of Cre expression in WT MTomato+ cells. n, The percentages of proliferative (composed of more than one cell) and nonproliferative clones. o, log2 ratio of the MTomato+ and MYFP+ cell frequency in control and adult Myc-floxed livers. p, The clone frequency MycWT and MycKO hepatocytes. q, The percentages of proliferative and nonproliferative MycWT and MycKO clones. The data are presented as mean values ± standard deviation. Scale bars, 100 μm.
Analysis of iFlpMosaics revealed that 17.2% of neonatal hepatocytes did not expand clonally, with most hepatocytes (69%) dividing only once or twice in 20 days. Only 2.4% of single hepatocytes yielded clones with eight or more cells. The average clone size for wild-type cells was 2.9, in agreement with the observed 2.9-fold liver weight increase from P1 to 20 (Fig. 4c,d). A comparison of clonal expansion between wild-type (MYFP+) and mutant MycKO (MTomato-2A-Cre+) hepatocytes revealed that MYC is necessary for optimal postnatal hepatocyte clonal expansion. Mean MycKO hepatocyte clone size was 2.25, and these clones generated only 125% more hepatocytes (125 new hepatocytes for every 100) over 20 days, whereas wild-type-cell clone size was 2.87, and these clones generated 187% more hepatocytes over the same period (Fig. 4e,f).
The analysis of postnatal pancreatic cells revealed similar Ki67+ and EdU+ frequencies to those of liver cells (see Fig. 4a and Extended Data Fig. 6c for a comparison). However, iFlpMosaics analysis revealed that wild-type pancreatic cells expand substantially more than wild-type hepatocytes until P20 (Fig. 4g), with a mean clone size of 3.60 instead of 2.87 (compare wild-type values of Fig. 4h,i with Fig. 4e,f). As in the liver, MycKO cells in the pancreas also expanded substantially less than wild-type cells (Fig. 4h,i) and were 3.7-fold less frequent than neighboring wild-type cells at the end of the analysis (Fig. 4j).
Evidence from recent studies suggested the existence of a highly proliferative hepatocyte population in adult liver, based on a new Dre-inducible Ki67-Cre (ProTracer) allele24 or CreERT2-based single-cell lineage tracing of a unicolor reporter25. Ki67 immunostaining showed that only 1.87% of adult liver hepatocytes are in cycle at any given moment (Extended Data Fig. 6d). Ki67 can be expressed in metabolically activated (primed and in cycle) or DNA-replicating (S phase) or in polyploid cells, but many Ki67+ cells will not undergo productive cell division or will die after being in cycle. The iFlpMosaics system can measure both productive cell expansion and cell survival. Using the iFlpMosaic mice with the highest number of multispectral combinations and, therefore, the highest single-cell clonal resolution, we observed very limited proliferation or clonal expansion of single mononucleated or binucleated hepatocytes during the 2 weeks after induction. The mean size of adult hepatocyte clones was 1.10 cells after 2 weeks (Fig. 4k,l), notably lower than the 1.38 cells after only 1 week of pulsing reported for a simpler unicolor reporter25. During the 2 weeks after induction, only 9.5% of single mononucleated or binucleated diploid hepatocytes underwent productive division or cytokinesis from binucleated cells to form a two-cell clone (Fig. 4m,n). Proliferation frequency was also low in polyploid hepatocytes (4.9%).
Despite the absence of substantial hepatocyte proliferation in homeostasis, when we induced iFlp ratiometric mosaics in adult Mycflox livers, we still observed a very significant loss of mutant MycKO cells (4.2-fold decrease) only 2 weeks after induction (Fig. 4o). Since there is limited clonal expansion of wild-type cells in adult liver (mean clone size 1.10, meaning that only 10 new hepatocytes were generated for every 100 initial hepatocytes over 2 weeks; Fig. 4l), these data suggest that MYC loss induces a significant decrease in adult hepatocyte survival. Interestingly, wild-type hepatocytes expanded more when surrounded by MycKO hepatocytes than when surrounded by wild-type hepatocytes (see Fig. 4m,n,p,q and Extended Data Fig. 6e for a comparison), probably to compensate for the loss of MycKO hepatocytes. These data suggest the existence of MYC-dependent cell competition among adult hepatocytes, in which cells loosing MYC are less metabolically fit and are gradually excluded, as previously shown to occur during early embryonic development19.
iDre/Flp Progenitor enables the induction of twin-spot clones
The results above show that there is substantial intercellular clonal variability when different progenitor cells, occupying different tissue locations, are induced. The iFlpMosaics technology shown above can only be induced in distinct progenitor cells, and therefore, it requires averaging of the clonal expansion data from many different independently labeled progenitor cells. By contrast, The MADM approach allows the generation of labeled wild-type and mutant cells from the same progenitor cell (twin-spot clones) and, in this way, gives a very precise estimate of how a gene mutation impacts the mobilization and proliferation phenotypes of a single-cell derived progeny. However, as mentioned above, MADM is a cumbersome method and cannot be effectively induced at a specific timepoint with CreERT2.
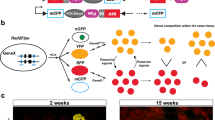
To overcome the limitations of current approaches to understanding the role of genes in single-progenitor cell biology, we designed the iDre/FlpProgenitor allele (Fig. 5a). This allele can be induced by both DreERT2 or FlpO-ERT2. Therefore, we also generated a new Tg(Ins-CAG-DreERT2) allele by using clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) assisted targeting into the preexisting and screened Tg(Ins-CAG-FlpO-ERT2) allele (Fig. 5b), which allowed us to induce much higher frequencies of recombination (Fig. 5c). The iDre/FlpProgenitor allele also enabled us to increase the sensitivity and recombination efficiency of all FlpO-ERT2 lines. With the Cdh5-FlpO-ERT2 line, we increased EC-recombination efficiency 14-fold, recombining 51% of all ECs (Fig. 5d). Recombination efficiency after combining iDre/FlpProgenitor with the Tg(Ins-CAG-FlpO-ERT2) allele was increased 25-fold (Extended Data Fig. 7a).

a, A schematic of the iDre/FlpProgenitor DNA construct used to generate transgenic mice. b, A strategy for the generation of the Tg(Ins-CAG-DreERT2) allele. c,d, Retinal confocal micrographs showing that the iDre/FlpProgenitor allele increases the recombination frequency of the iFlpMosaic allele, when induced either by the CAG-DreERT2 (c) or Cdh5-FlpOERT2 (d) alleles. H2B-V5 labels the nuclei of cells expressing the iDre/FlpProgenitor allele. e, A schematic of the genetic recombination cascade and cellular markers in the progenitor and daughter cells. f, The kinetics of the induced recombination cascade determined by FACS of in vitro cultured fibroblasts. Top: plots and chart show the hCD2 reporter signals when contrasted with the autofluorescence control PerCP dye channel. n.s., not significant. Bottom: FACS plots show the recombination frequency and expression of the iFlpMosaic allele reporters over time, within hCD2+ cells. g, A confocal analysis of the P6 pancreas, 3 days after a very low dose of 4-OHT induction, showing twin-spot clones. Only H2B-V5+ (iDre/FlpProgenitor+) cells give rise to twin-spot clones having both Tomato+ and YFP+ cells (orange bar). h, Induction with a single dose of 4-OHT (40 mg kg−1) 5 days before collecting the tissues shows that the new Ki67-2A-DreERT2 allele (Extended Data Fig. 7f) drives recombination in Ki67+ progenitor cells of the indicated organs, generating twin-spot clones. Background immuno, background or noise signal from immunostaining with anti-GFP/YFP; Exp., expected frequencies of recombination. The data are presented as mean values ± standard deviation. Scale bars, 24 μm in g and h, and 500 μm in c and d.
We reasoned that the combination of iFlpMosaics with the iDre/FlpProgenitor allele would allow tamoxifen induction of a genetic cascade culminating in the generation of genetic mosaics of mutant and wild-type cells derived from the same progenitor cells. After tamoxifen induction of FlpO-ERT2 or DreERT2, the iDre/FlpProgenitor allele is first recombined, and this triggers the permanent expression of FlpO, H2B-V5 and the cell-surface marker hCD2 (Fig. 5e). FlpO then effectively recombines the R26-iFlpMTomato-Cre/MYFP allele in H2B-V5+/hCD2+ cells 24–48 h after the 4-hydroxytamoxifen (4-OHT) induction, generating both gene KO (MTomato+) and wild-type (MYFP+ ) cells derived from the same progenitor cells (Fig. 5f).
In the pancreas of newborns containing the iDre/FlpProgenitor allele, anti-V5 immunostaining enabled the detection of the nuclei of the progenitor cell and its progeny (Fig. 5g). Even when very sparse labeling was used, V5+MTomato+ and V5+MYFP+ cells were frequently found located close to each other (25% of all clones), confirming that they arose from the same single-progenitor cell, whereas H2B-V5− cells of the same animals did not show any clones containing both MTomato+ and MYFP+ cells close to each other (Fig. 5g). In angiogenic retina vessels, endothelial cells migrate extensively as they proliferate, making it more difficult to track single-cell-derived clones. However, in animals without the iDre/FlpProgenitor allele, MYFP+ wild-type cells were rarely intermixed with mutant MTomato+ cells (Extended Data Fig. 7b). In animals or cells with the iDre/FlpProgenitor allele, V5+MTomato+ and V5+MYFP+ cells were frequently located close to each other, suggesting that they arose from the same progenitor cells (Extended Data Fig. 7c,d). Time-lapse imaging of mouse ES cells confirmed that single hCD2+ cells divide and form twin-spot clones (Extended Data Fig. 7e).
We also generated a new Ki67-2A-DreERT2 line to recombine iDre/FlpMosaic alleles specifically in dividing Ki67+ progenitor cells. This allowed us to induce twin-spot clones in any proliferating cell of different organs (Fig. 5h and Extended Data Fig. 7f).
scRNA-seq of ratiometric iFlpMosaics
We have shown that iFlpMosaics allow the reliable induction and isolation by FACS of bona fide mutant cells and wild-type cells from the same tissue (Fig. 1), unlike CreERT2-dependent mosaic genetics (Extended Data Fig. 1). Therefore, we combined iFlpMosaics with single-cell RNA sequencing (scRNA-seq) to achieve high-throughput scoring of gene function in the proliferation and differentiation of all early embryo cell types. For this analysis, we first selected to induce the mosaic deletion of Rbpj, which encodes a transcription factor essential for Notch signaling, one of the most important pathways for cell proliferation and differentiation during embryonic development26. We pulsed embryos with 4-OHT at embryonic day (E)9.5 and collected MTomato+ and MYFP+ cells by FACS at E13.5 (Fig. 6a). Based on their transcriptome and known marker genes, we identified 16 major cell types (Fig. 6b). In MTomato-2A-Cre+ cells, deletion of Rbpj exons 6 and 7 generates a less stable but still detectable 3′ messenger RNA27, together with a decrease in the expression of the main canonical downstream target Hes1 (Fig. 6c), whose expression can also be regulated by other pathways.

a, A schematic of the induction and analysis stages (four embryos were used). b, Uniform manifold approximation and projections (UMAPs) showing RbpjKO (MTomato+) and RbpjWt (MYFP+) cells and dot plots with the top markers used to identify the major cell types. CNS, central nervous system; PNS, peripheral nervous system. c, A dot plot showing the expression for Rbpj whole mRNA (mostly the 3′ undeleted mRNA) and the deleted floxed exons 6–7. Bottom: the expression of the canonical NOTCH/RBPJ signaling target Hes1 (regulated also by other pathways) and MKi67. d,Histobars with the proportions of the different clusters identified in c. e, Comparative UMAP showing that mosaic deletion of Rbpj changes the relative proportion of different cell types. f, Analysis within the endothelial cell cluster, including general embryo, brain and liver ECs, with some cells undergoing endothelial-to-mesenchymal transition. g,h, RbpjKO mutant cells present a deregulation of arterial (Gja4 and Gja5) (g) and tip cell (Esm1, Kcne3 and Apln) marker genes (h). i, Gene set enrichment analysis (GSEA) comparing RbpjWt with RbpjKO cells (see Extended Data Fig. 8 for the analysis of all other cell clusters). NES, normalized enrichment score. j, UMAPs showing the differential distribution of the Notch1/2/3KO (MTomato+) and Notch1/2/3Wt (MYFP+) cells (collected from four embryos) and the different clusters they form. k, A dot plot showing the frequency and amplitude of expression for the three Notch receptors whole mRNA (mostly the 3′ undeleted mRNA in the case of Notch2 and Notch3) and their deleted floxed exons. Bottom: the expression of the Notch receptors (whole mRNA) and their canonical signaling targets Hes1/Hey1/Hey2 (regulated also by other pathways) and the G2/M phase marker MKi67. l, The histobars and UMAP showing that mosaic deletion of Notch1/2/3 (MTomato+ cells) changes the relative proportion of different cell types, particularly neural cells (Extended Data Figs. 9 and 10).
Global iFlpMosaic-driven Rbpj deletion at E9.5 compromised the differentiation and expansion of peripheral neurons, ECs, cardiomyocytes, erythrocytes, hepatocytes and myocytes, whereas the frequency of epithelial and mesenchymal cells increased (Fig. 6d,e). RBPJ loss also changed the transcriptional programs of these cells. As an example, mosaic Rbpj deletion in ECs led to the cell-autonomous decrease in expression of the arterial marker genes Gja5 and Gja4 and upregulation of the tip-cell genes Esm1, Kcne3, Apln and Cdkn1a (Fig.6f–i). This correlates with the essential cell-autonomous role of RBPJ in the specification of arterial ECs and the inhibition of tip ECs16,28.RBPJ loss also changed the relative frequency or genetic programs of many other embryo cell types (Extended Data Fig. 8).
In addition to Rbpj, we also used iFlpMosaics to induce the mosaic and simultaneous deletion of the three main Notch receptors in embryo single cells (Fig. 6j,k), something not achievable with CreERT2 or MADM genetics. In contrast to deletion of Rbpj from E9.5 to E13.5, deletion of the three Notch receptors (six floxed alleles) from E7.5 to E13.5 led to a very substantial loss of all neuronal cell types (Fig. 6j), confirming the essential role for Notch in the maintenance of an early pool of neuronal progenitor cells and the prevention of their premature differentiation and exhaustion26. Notch receptor deletion also led to an increase in the frequency of epithelial cells, lymphatic and blood ECs and some mesenchymal cells (Fig. 6l). In addition, it deregulated many cell types differentiation and transcriptional programs (Extended Data Figs. 9 and 10).
These scRNA-seq data provide an important resource for further exploring the role of RBPJ and NOTCH signaling in the early expansion and differentiation of embryonic lineages. This analysis also exemplifies how the combination of ratiometric iFlpMosaics with scRNA-seq analysis is a very powerful tool for the high-throughput determination of cell-autonomous gene functions in all cell types during development or disease.
Discussion
Genetic mosaics are essential tools for determining the cell-autonomous function of a gene and avoid the confounding effects frequently associated with classical whole-animal or tissue-specific conditional genetics1,3. With inducible mosaic genetics, the mutant and wild-type cells share the same microenvironment and developmental or physiological history, with the only difference between them being the deleted gene. This provides a much more accurate way to study and understand cell-autonomous gene function during tissue development, homeostasis or disease.
CreERT2-dependent genetic mosaics are easy to induce but are unreliable7,8,9. They also lack the coinduction of wild-type cells labeling required for direct phenotypic comparison in the same tissue. MADM enables the colabeling of both mutant and wild-type cells and is genetically very reliable but is a technically cumbersome method4,5,6. In addition, MADM interchromosomal recombination events are difficult to induce with CreERT2, resulting in very few labeled cells per animal11.
The set of new genetic tools we describe here substantially improves on both of these technologies. Unlike CreERT2 genetics, iFlpMosaics provide clearly distinguishable and reliable fluorescent labels for both true mutant cells (MTomato+) and wild-type cells (MYFP+), which enable the accurate measurement and comparison of the proliferation, differentiation, migration and survival of these populations over time within the same animal. Unlike MADM, iFlpMosaics provide a high degree of inducibility at any specific timepoint and in any cell type, including quiescent cells present in adult mice, which will be very important for modeling the etiology of diseases caused by sporadic somatic mutations, or the consequences of CRISPR-based gene editing, which are by their nature also mosaic. In addition, iFlpMosaics are compatible with all existing floxed mouse alleles, enabling mosaic deletion of one or multiple floxed genes in cis (we showed Myc/Mycn or Notch1/2/3 or Foxo1/3/4), which is essential to perform functional genetic epistasis with single-cell resolution. With iFlpMosaics, instead of only 1 cell barcode (as in standard CreERT2 or MADM), up to 12 different multispectral barcodes are possible for mutant cells and up to seven for wild-type cells. This substantially higher spectral barcode diversity and dual labeling (membrane and nuclei) further increases single-cell and clonal quantitative resolution during a pulse–chase genetic experiment.
iFlpMosaics allowed us to substantially extend and refine previous findings on the roles of several genes (Myc, Foxo1/3/4, Rbpj or Notch1/2/3) in the development and differentiation of most embryonic lineages, the single-cell clonal expansion of postnatal tissues or the competitive survival of cells during the normal homeostasis and turnover of adult tissues. The ability to induce ratiometric genetic mosaics in multiple cell types simultaneously and at both low and high frequencies increases the ease and throughput of analyzing gene cell-autonomous functions. With iFlpMosaics, determining a gene cell-autonomous function no longer requires the crossing of mice carrying the floxed gene with multiple independent CreERT2 lines (one for each cell type) or with multiple MADM alleles (one for each chromosome carrying the mutant allele)4.
Single-cell biology during tissue development is highly heterogeneous, and it is, therefore, important to be able to reliably induce and analyze genetic mosaics of mutant and wild-type cells derived from the same single-progenitor cell and with a negligible level of false positives and false negatives. The established MADM technology is ideal for this4,5,6 but either lacks temporal inducibility with Cre or efficiency with CreERT2. iFlpMosaics are inducible by FlpOERT2, which is a much weaker recombinase than the established CreERT2. For this reason, few tissue-specific FlpOERT2 lines exist, and this may be a disadvantage of iFlpMosaics when cell-type specificity is required. However, with the new iDre/FlpProgenitor allele, we were able to substantially increase the inducibility and specificity of iFlpMosaics. This allele is much more sensitive to both DreERT2 or FlpO-ERT2 activity, which enables the use of weaker and cell/tissue-specific FlpOERT2 or DreERT2 lines, such as the generated Cdh5-FlpOERT2 and Ki67-2A-DreERT2 lines. In addition, the iDre/FlpProgenitor allele results in a genetic cascade that includes a delay in the expression of FlpO. This delay enables the generation of different mutant and wild-type cells within the progeny of a single starting progenitor cell, something previously achievable only with MADM. With the generated Ki67-2A-DreERT2 allele, iFlpMosaics and twin-spot clones can be induced, specifically in the progeny of any proliferating cell.
Given the limitations of current technologies, iFlpMosaics will greatly facilitate the induction and high-throughput functional analysis of genetic mosaics. It will be crucial for understanding how individual or combined genetic mutations affect the biology of single cells during tissue development, regeneration or disease.
Methods
Mice
All mouse husbandry and experimentation were conducted using protocols approved by local animal ethics committees and authorities (Comunidad Autónoma de Madrid and Universidad Autónoma de Madrid CAM-PROEX 177/14, CAM-PROEX 167/17, CAM-PROEX 164.8/20 and PROEX 293.1/22). The mouse colonies were maintained in racked individual ventilation cages according to current national legislation. The mice had dust and pathogen-free bedding and sufficient nesting and environmental enrichment material for the development of species-specific behavior. All mice had ad libitum access to food and water in environmental conditions of 45–65% relative humidity, temperatures of 21–24 °C and a 12–12 h light–dark cycle. To preserve animal welfare, mouse health was monitored with an animal health surveillance program that followed the Federation of European Laboratory Animal Science Associations (FELASA) recommendations for specific pathogen-free facilities.
We generated and used Mus musculus lines on the C57BL6 or C57BL6 × 129SV or B6CBAF1 or C57BL6 × DBA genetic backgrounds. All mice were backcrossed to C57Bl6 for several generations. To generate mice for analysis, we intercrossed mice aged between 7 and 30 weeks. We analys¡zed mice of both sexes. We do not anticipate any influence on our data of mouse sex. The following mouse lines were used and intercrossed: Tg(Cdh5-CreERT2)29, Tg(iSuRe-Cre)8, Notch1flox30, Notch2flox31, Notch3flox32, Rbpjflox33, Mycflox34, Mycnflox35, Dll4flox36, Kdrflox37, Foxo1/3/4flox23, Rosa26-EYFP38, Rosa26-iChr2-Mosaic13, Tg-iMb2-Mosaic or iCreMYFP/MTomato/MTFP113, Gt(Rosa)26tm3(CAG-FlpO-ERT2)Ali10, Gt(Rosa)26Sortm14(CAG-LSL-tdTomato)Hze39, R26-iFlpMTomato-2A-Cre/MYFP-2A-FlpO, ACTB:FlpE allele15 and Apln-FlpO16. The following mouse lines were produced in this study: R26-iFlpMTomato-Cre/MYFP, Tg-iFlpMTomato-H2B-GFP-Cre/MYFP-H2B-Cherry-FlpO, Tg(INS-CAG-FlpO-ERT2), Tg(INS-CAG-DreERT2), Tg(Cdh5-FlpO-ERT2), Tg-iFlpH2B-Cherry/GFP/Cerulean, Tg-iDre/FlpProgenitor and MKi67-2A-DreERT2.
The Tg-iFlpMTomato-H2B-GFP-Cre/MYFP-H2B-Cherry-FlpO, Tg(INS-CAG-FlpO-ERT2), Tg-iFlpH2B-Cherry/GFP/Cerulean and Tg-iDre/FlpProgenitor mouse alleles were generated by standard DNA injection into mouse eggs and screening of several transgenic founders, as indicated in the main text or figures. The chromosomal position of these transgenic alleles was mapped by targeted locus amplification and sequencing (Cergentis). The Tg-iFlpMTomato-H2B-GFP-Cre/MYFP-H2B-Cherry-FlpO allele is in chromossome 3:44,078,483, the Tg-iFlpH2B-Cherry/GFP/Cerulean allele is in chromossome 16:66,008,291 and the Tg-iDre/FlpProgenitor allele is in chromosome 13:90,390,612. The Cergentis report also indicated that there are no genes in the integration sites for all these three alleles. The R26-iFlpMTomato-2A-Cre/MYFP-2A-FlpO allele was generated by CRISPR-assisted gene targeting of the Rosa26 locus in G4 ES cells, using the guide GACTGGAGTTGCAGATCACGA_GGG (IDT DNA) and a donor DNA, as previously described16. The targeted ES cells were used for mice production. After eggs from these mice were injected with guide RNAs recognizing the sequence CCTGTGCAAGACCCCCCCCA_AGG and GGCGGATCTGATAAGCTCGA_GGG and a donor DNA coding for YFP-WPRE-pA to delete the 2A-FlpO cassette and obtain the R26-iFlpMTomato-Cre/MYFP allele. The Tg(Cdh5-FlpO-ERT2) line was generated by CRISPR–Cas9-assisted gene targeting of the existing Tg(Cdh5-CreERT2) line29 in mouse eggs, using the guide AAGCTTATCGATACCGTCGA_CGG and a donor DNA containing the Cdh5-FlpO-ERT2 sequence. Tg(INS-CAG-DreERT2) mice were generated by CRISPR–Cas9 using guide RNAs (GATGTCGAACTGGCTCATGG_TGG and AACAGGCGGATCTGCGTACG_CGG) targeting the existing FlpO sequence contained in the prescreened Tg(INS-CAG-FlpO-ERT2) transgene and a donor DNA containing the CAG-DreERT2 sequence. The Mki67-2A-DreERT2 line was generated by CRISPR–Cas9 using guide RNA ACTGGAGGTGAAAACCACAC_TGG targeting a sequence after the stop codon of the Mki67 gene and a donor DNA fragment containing the sequence MKi67-2A-DreERT2-WPRE-sv40pA-MKi67 (Extended Data Fig. 7f). The injection mixtures consisted of 0.305 mM of the described crRNAs (IDT) and tracrRNA (IDT, catalog 1072533) and 20 ng µl−1 Cas9 nuclease (Alt-R Streptococcus pyogenes HiFi Cas9 Nuclease V3, 100 µg, catalog 1081060). The founders were initially screened by polymerase chain reaction (PCR) with the primers described in Supplementary Table 1.
To induce CreERT2, FlpO-ERT2 or DreERT2 activity in adult mice, 1 g tamoxifen (Sigma-Aldrich, P5648_1G) was dissolved in 50 ml corn oil (stock tamoxifen concentration, 20 mg ml−1), and aliquots were stored at −20 °C. The animals received intraperitoneal injections of 50–150 µl of this stock solution (total dose, 1–3 mg tamoxifen per animal at 40–120 mg kg−1), as indicated in the figures. For treatment of pregnant females, the tamoxifen was dissolved together with progesterone to reduce miscarriage (2 mg tamoxifen and 1 mg progesterone per mouse). To activate recombination in pups, 4-OHT was injected at the indicated stages at a dose of 40 mg kg−1 or 4 mg kg−1 body weight, as indicated in the figures. All mouse lines and primer sequences required for genotyping are provided in Supplementary Table 1.
Immunofluorescence on tissue cryosections
For multispectral iFlpMosaics tissue collecting, the mice were killed in CO2 chambers, 10 ml of 50 mM KCl were injected into the left ventricles and whole mice were perfused with 3.7–4% formaldehyde (ITW Reagents, 252931) at pH 7. The explanted tissues were postfixed for 2 h in 4% paraformaldehyde (PFA) (Thermoscientific, 043368.9 M) in phosphate-buffered saline (PBS) at 4 °C with gentle rotation. After three washes in PBS for 10 min each, the organs were stored overnight in 30% sucrose (Sigma) in PBS. The organs were then embedded in optimal cutting temperature coumpound (Sakura) and frozen at −80 °C. The cryosections (35 µm) were cut on a cryostat (Leica), washed three times for 10 min each in PBS and blocked and permeabilized in PBS containing 10% donkey serum (Millipore), 10% fetal bovine serum (FBS) and 1% Triton X-100. The primary antibodies (Supplementary Table 2) were diluted in the same buffer and incubated with sections overnight at 4 °C. This step was followed by three 10 min washes in PBS and incubation for 2 h with conjugated secondary antibodies (Supplementary Table 2) and 4,6-diamidino-2-phenylindole (DAPI) in PBS at room temperature. After three washes in PBS, the sections were mounted with Fluoromount-G (SouthernBiotech). To detect the DLL4 protein, we had to use streptavidin/tyramide-based signal amplification. Briefly, BLOXALL endogenous blocking solution (Vector Laboratories, SP-6000) was used to quench endogenous peroxidase activity. After adding the primary goat anti-Dll4 and secondary antibody biotin-donkey anti-goat (as mentioned above), the slides were washed three times for 10 min in Tris-buffer with sodium and tween (TBST) (0.05 M Tris–HCl pH 7.5, 0.3 M NaCl and 0.1% Tween20) and then incubated with ABC reagent (Vectastain Elite ABC-HRP Kit, PK-6100) for 1 h at room temperature. After three washes with TBST buffer, the slides were incubated 3 min with tyramide signal amplification (TSA) fluorescein (PerkinElmer NEL701A) to amplify the signal.
Immunofluorescence on paraffin sections
The RBPJ protein was detected with the TSA kit (NEL701A) procedure in paraffin sections after antigen retrieval. In brief, the sections were dewaxed and rehydrated, followed by antigen retrieval in sub-boiling sodium citrate buffer (10 mM, pH 6.0) for 30 min. The slides were cooled down to room temperature for 30 min, followed by incubation for 10 min in BLOXALL Endogenous Blocking Solution (Vector Laboratories, SP-6000) to quench endogenous peroxidase activity. Next, the slides were washed twice for 5 min each in TBST buffer (0.3 M NaCl, 0.05 M Tris–HCl pH 7.5 and 0.1% Tween20), followed by blocking for 1 h in PBS containing 10% donkey serum, 10% FBS and 0.3% Triton. The sections were then incubated with primary antibodies (rat anti-RBPj, rabbit anti-RFP-594 and goat anti-GFP (green fluorescent protein); Supplementary Table 2) at 4 °C in TBST buffer containing 5% donkey serum and 1% FBS. The slides were washed three times for 10 min in TBST buffer and then incubated for 1 h at room temperature with biotin-SP-donkey anti-rat and donkey anti-goat AF680 antibodies. The slides were washed three times for 10 min in TBST buffer and then incubated with ABC reagent (Vectastain Elite ABC-HRP Kit, PK-6100) for 1 h at room temperature. After three washes with TBST buffer, the slides were incubated for 3 min with TSA fluorescein (PerkinElmer NEL701A). The slides were then washed three times for 10 min in TBST buffer and stained with DAPI before mounting with Fluoromount-G (SouthernBiotech).
Whole-mount immunofluorescence of retinas
For postnatal mouse retina immunostaining, eyes were collected and fixed in ice-cold 4% PFA in PBS for 10 min. The eyes were then incubated in the same solution for a further 15 min at room temperature, washed once in PBS and kept on ice. After microdissection with spring scissors (FST), the retinas were fixed in 4% PFA for an additional 45 min at room temperature, followed by two PBS washes of 10 min each. The retinas were blocked and permeabilized for 1 h in PBTS buffer (0.3% Triton X-100, 3% FBS and 3% donkey serum). The samples were then incubated overnight at 4 °C with biotinylated isolectin B4 (Vector Labs, B-1205, diluted 1:50) and primary antibodies (Supplementary Table 2) diluted in PBTS buffer. After five washes (20 min each) in PBTS buffer diluted 1:2, the samples were incubated for 2 h at room temperature with Alexa-conjugated secondary antibodies (Supplementary Table 2). After three washes of 30 min each in PBTS buffer (diluted 1:2) and two washes of 15 min each in PBS, the retinas were mounted with Fluoromount-G (SouthernBiotech).
In vivo EdU labeling and detection of EC proliferation
To detect EC proliferation in postnatal liver and pancreas, 20 μg g−1 body weight EdU (Invitrogen, A10044) was injected intraperitoneally into P1, P7 or P20 mice 4 h before dissection. Livers and pancreases were fixed in 4% PFA and processed for cryosectioning as described above. The EdU signals were detected with the Click-it EdU Alexa Fluor 647 or 488 Imaging Kit (Invitrogen, C10340 or C10337). In brief, after all other primary and secondary antibody incubations, the samples were washed according to the immunofluorescence staining procedure and then incubated with Click-iT EdU reaction cocktail for 40 min, followed by DAPI counterstaining.
Isolation of lung fibroblasts from adult mice
To establish primary cell cultures of lung fibroblasts we followed a published protocol40, with some modifications. Briefly, lungs were dissected from mice under sterile conditions and placed in sterile PBS. In a cell culture hood, the lungs were removed from PBS and then chopped into small fragments with scissors and placed in 10 ml of digestion buffer containing Dulbecco’s phosphate-buffered saline (DPBS, Thermofisher 14040141) with Liberase TL (0.14 Wunsch units per milliliters; Merck 5401020001) and 1× antibiotic/antimycotic (Thermofisher 15240096). The tissue in digestion buffer was incubated in a water bath at 37 °C for 30 min, with mixing every 2–3 min. After this period, the solution was pipetted up and down to break clumps and after 10 ml of culture medium containing DMEM buffer/F12 (Thermofisher 11320033), 15% FBS and 2× antibiotic/antimycotic solution was added. This cell suspension was centrifuged at 500g for 5 min. The cell pellet was washed two times with culture medium to remove liberase completely. At the end, the cells were resuspended in 12 ml of culture medium and seeded in a p100 dish and incubated at 37 °C, 5% CO2 and 3% O2. The medium was changed after 3–5 days, when fibroblasts had crawled out of tissue fragments. The fibroblasts were then expanded and maintained in full medium at 37 °C, 5% CO2 and 3% O2 for expansion and only transferred to normoxic conditions to carry out experiments.
To induce recombination, the cells were trypsinized, resuspended and plated in culture medium containing 0.2 μM of 4-OH tamoxifen for 4 h and washed after in culture medium. The cells were then trypsinized at different timepoints after induction (24, 48, 72, 96 and 120 h), stained with DAPI and analyzed or sorted in a FACS machine. A minimum of 20,000 cells per experimental group were sorted into 300 μl of a solution containing DPBS and 10% FBS. At the end, the cells were centrifuged at 500g for 5 min at 4 °C, and the supernatants removed. The cell pellets were stored at −80 °C until all the samples were ready to be further processed. Genomic DNA was extracted from cell pellets by incubating each pellet in 40 μl of DirectPCR Lysis Reagent (Viagen 301-C) supplemented with proteinase K (0.33 mg ml−1) and incubated at 55 °C overnight. Proteinase K was inactivated by incubating samples at 85 °C for 45 min. A total of 1 μl of each sample was directly used for quantitative real-time PCR (qRT-PCR) using Taqman universal master mix and an Applied Biosystems QuantStudio5 machine.
Derivation and live imaging of mouse embryonic stem cells
To derive genetically modified mouse ES cells, we intercrossed mice containing the desired alleles, and the pluripotent cells unit at CNIC (centro nacional de investigaciones cardiovasculares) expanded in vitro their blastocysts according to established protocols41. Briefly, the blastocysts were transferred individually to a 24-well plate containing feeder layers of freshly inactivated MEFs and in ES-2i medium (DMEM/Glutamax, GIBCO 31966-021; NEAA, β-mercaptoethanol, LIF, 20% serum replacement, 3 µM CHIR and 1 µM PD). The blastocysts were cultured without disturbance for 3 days. From day 4, the medium was changed every other day, and when each embryo inner cell mass had grown sufficiently, it was disaggregated by gently trypsinization and individually passaged to a new 24-well plate. Following several passages, the independent cell lines were genotyped, expanded and frozen. A selected ES clone carrying the alleles CAG-FlpO-ERT2, iDre/FlpProgenitor and R26-iFlpMTomato-Cre/MYFP was plated on matrigel and induced with 1 μM of 4-OHT for 3 h and incubated for 22 h more with ES-2i medium. The antibody hCD2-APC was added 2 h before starting the time-lapse live imaging (24 h after 4-OHT induction) on a Leica SP8 Navigator with a chamber set at 37 C and 5% CO2.
Image acquisition and analysis
For confocal scanning, the immunostained organ sections or whole-mount retinas were imaged at high resolution with a Leica SP8 Navigator confocal microscope fitted with a 10×, 20× or 40× objective. The individual fields or tiles of large areas were acquired. All the images shown are representative of the results obtained for each group and experiment. The animals were dissected and processed under exactly the same conditions. Comparisons of phenotypes or signal intensities were made using images obtained with the same laser excitation and confocal scanner detection settings. ImageJ/FIJI v1.53c was used to threshold, select and quantify the objects in confocal micrographs. Manual and automatic ImageJ public plugins and custom-made Fiji macros (file ‘Script_Reporter_Marker detection_FINAL.ijm’ for quantifying ERG (EC nuclei marker) or DAPI colocalization with reporters and other markers; Figs. 1e–k, 2f–h and 3e and Extended Data Figs. 1d, 3d, 4 and 6c,d) and R scripts were used for quantification: ‘Script_PostnatalLiver_CloneOutput’ (for postnatal liver clonality analysis; Fig. 4b,e), ‘Script_PostnatalPancreas_CloneOutput’ (for postnatal pancreas clonality analysis; Fig. 4g–i), ‘Script_AdultLiver_CloneOutput’ (for adult liver clonality analysis; Fig. 4k–p and Extended Data Fig. 6e) and ‘RMacroForClustering_iProgenitor’ (for clustering analysis of iProgenitor mouse line; Extended Data Fig. 7b,c). Adobe Photoshop CC 19.1.5 and Adobe Illustrator CC v22.1 were used for downstream image processing, analysis and illustration.
FACS and sorting
Embryonic, postnatal or adult tissues were dissociated before FACS. The embryonic tissues were dissociated using the Miltenyi Biotec Tissue Dissociation Kit 2 (130-110-203). Postnatal and adult mouse tissues were first digested for 20 min at 37 °C with 2.5 mg ml−1 collagenase type I (Thermofisher), 2.5 mg ml−1 dispase II (Thermofisher) and 50 μg ml−1 DNAseI (Roche).
The dissociated samples were filtered through a 70 μm cell strainer, and the cells were centrifuged (400g, 4 °C for 5 min). The cell pellets were gently resuspended in blood lysis buffer (0.15 M NH4Cl, 0.01 M KHCO3 and 0.01 M EDTA buffer in distilled water) and incubated for 10 min on ice to remove erythroid cells. The cells were centrifuged (400g at, 4 °C for 5 min), and the cell pellets were gently resuspended in blocking solution (PBS without Ca2+ or Mg2+ and containing 3% dialyzed FBS (Termofisher)) and incubated at 4 °C with shaking for 20 min. The cells were centrifuged (300g at 4 °C for 5 min), resuspended and incubated for 30 min at 4 °C with APC rat anti-mouse CD31 and APC-CY7 anti-CD45 (Supplementary Table 2). The cells were then centrifuged (400g, 4 °C for 5 min), resuspended, washed in PBS without Ca2+ or Mg2+ and centrifuged again, and the cell pellets were resuspended in blocking solution. The cells were kept on ice until used for FACS. DAPI (5 mg ml−1) was added to the cells immediately before analysis. The cells were routinely analyzed with a LSRFortessa cell analyzer or sorted in a FACS Aria Cell Sorter (BD Biosciences) (see Supplementary Fig. 2 for an example of the FACS gating strategy). BD FACS Diva V8.0.1 and Flow JO v10 were utilized for FACS data collection and analysis.
Cell isolation for transcriptomic analysis
For qRT-PCR analysis, approximately 20,000 DAPI-negative MTomato+ or MYFP+ cells from dissociated tissues were sorted directly to RLT buffer (RNAeasy Micro kit, Qiagen), and RNA was extracted according to the manufacturer’s instructions and stored at −80 °C. This RNA was later used for qRT-PCR or RNA sequencing analysis. For qRT-PCR, the total RNA was retrotranscribed with the High Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Thermo fisher, 4368814). Complementary DNA (cDNA) was preamplified with Taqman PreAmp Master Mix containing a Taqman Assay-based preamplification pool composed of a mix of the Taqman assays indicated in Supplementary Table 3. The preamplified cDNA was used for qRT-PCR using the same gene-specific Taqman Assays and Taqman Universal Master Mix in an AB7900 thermocycler (Applied Biosystems). The data were retrieved and analyzed with AB7900 software.
For RNA-seq analysis (Fig. 6), the embryos were dissociated using Miltenyi Biotec Tissue Dissociation Kit 2 (130-110-203). A 1.2 ml volume of dissociation solution was placed together with a single embryo in a 2 ml round-bottom tube. Following a 5 min incubation in a 37 °C water bath, the tubes were placed inside a prewarmed 50 ml falcon tube and incubated for 25 min on a MACSmix Tube Rotor. The cells were then resuspended with 30 strokes with a Gilson P1000 pipette, starting slow and increasing speed gradually. To the 1.2 ml cell suspension 3 ml of sorting buffer was added (10% FBS in Ca2- and Mg2-free PBS), and the resulting solution was transferred to a 5 ml syringe. This volume was pressed slowly through a small 70 µm strainer to remove clumps. Each embryo yielded around 30 million cells at this stage and was processed independently. The cell suspensions were spun at 400g for 6 min, and the pellets were detached and resuspended in 300 µl sorting buffer containing DAPI. DAPI-negative, single and live MTomato+ and MYFP+ cells were sorted to eppendorf tubes containing 300 µl of sorting buffer. A total of 66,000 MTomato+ cells and 30,000 MYFP+ cells were sorted using a 100 µm nozzle, 20 PSI, and high purity scale and relatively low flow rate (less than 3,000 events per second) to preserve cell viability and decrease contamination. The sorted cells were spun at 500g for 5 min and resuspended in 30–40 µL cell-capture buffer (Ca2- and Mg2-free PBS supplemented with 0.04% bovine serum albumin). After counting the cells in a Countess 3 Automated Cell Counter (Thermo Fisher Scientific), two independent 10x Genomics ports were loaded with either 16,000 MTomato+ cells (90% viability) or 16,000 MYFP+ cells (88% viability).
Next-generation sequencing sample and library preparation
Next-generation sequencing experiments were performed in the CNIC Genomics Unit. For scRNA-seq experiments, the single cells were encapsulated in emulsion droplets using the Chromium Controller instrument (10x Genomics). scRNA-seq libraries were prepared according to the manufacturer’s instructions. The aimed target cell recovery for each port was 10,000 cells. The generated libraries were sequenced in a HiSeq4000 or NextSeq2000 system (Illumina).
Transcriptomic data analysis
Transcriptomic data were analyzed by the CNIC Bioinformatics Unit, Alvaro Regano and Irene Garcia.
The following pipeline was followed for scRNA-seq data processing and in silico cell-type selection. For alignment and quantification of gene expression, the reference transcriptome was built using mouse genome GRCm38 and Ensembl gene build v98 (sep2019.archive.ensembl.org). The WPRE-sv40pa sequences expressed in the MTomato+ and MYFP+ samples were added to the reference. The gene metadata were obtained from the corresponding Ensembl BioMart archive. The reads from transcripts were processed, aligned and quantified using the Cell Ranger v6.1.2 for the Rbpj wild-type versus KO samples and Cell Ranger v7.1.0 for the Notch1/2/3 wild-type versus KO samples. The single-cell analysis was based on the Seurat v4.1.3 (https://satijalab.org/seurat/)42 and DoubletFinder R packages. Low-quality cells were filtered out using the following criteria: total counts, >1,000 and <55,000; genes detected, >500 and <7,500; mitochondrial transcripts content, <15%; hemoglobin transcripts, <1%; and ribosomal transcripts, <35. The counts were log-normalized and scaled, followed by a principal component analysis and clustering using the shared nearest-neighbors algorithm and Louvain clustering (settings as defaults except for the 2,000 most variable genes, 24 principal components and a resolution of 0.35). The clusters and cells were classified on the basis of the SingleR function from the SingleR package (v2.0)43. Spearman correlation coefficients with an 80th percentile threshold were taken into consideration for correct cell type assignment using the following three datasets as reference for the label transfer: Blueprint ENCODE, the Human Primary Cell Atlas cell-type profile collection43 and a scRNA-seq mouse dataset from the celldex package (https://bioconductor.org/packages/release/data/experiment/vignettes/celldex/inst/doc/userguide.html). This identification was used to predefine the different cell types present in the dataset for the analysis. The doublets were removed using DoubletFinder, using a first annotation with 24 PCs (principal components), number of artificial doublets (pN) 0.25 and a neighbourhood size (pK) of 0.05, and the second annotation with 10 PCs, pN 0.25 and pK 0.05. The cells classified as singlets were preserved. The singlets were then reclustered using 19 principal component analyses and a clustering resolution of 0.35. A manual curation of the identified clusters was applied to confirm and finetune the identity of the clusters based on available scRNA-seq bibliography44,45. R scripts used to analyze RNA sequencing data, ‘Script_Embryos_Jan2023’ and ‘ToGenerateGraphsOnly’ (Fig. 6 and Extended Data Figs. 8–10), are available at GitHub (https://github.com/RuiBenedito/Benedito_Lab/tree/main/iFlpMosaics).
To evaluate the rate of exon deletion (for Rbpj, Notch1, Notch2 and Notch3) in scRNA-seq data obtained from the different iFlpMosaic experiments (Fig. 6), we evaluated the number of reads with unique molecule identifiers aligning with each of the exons (included the deleted/floxed exons) and the last 3’ exon–UTR sequence, the latter usually overrepresented in 3′ mRNA 10x Chromium sequencing data. A valid cell barcode, unique molecule identifier and compatible strand was considered in the quantification. The exon read data were incorporated into the Seurat object as metadata and normalized to the total RNA counts per cell. If more than one exon was flanked by loxP sites, the sum of their reads was aggregated as a single variable to quantify the deletion. Subsequently, we visualized the data using a dot plot analysis showing the percentage of cells and average expression of the whole mRNA (which includes the last 3′ UTR exon that is overrepresented) or the specific gene floxed exons, expected to be deleted by Cre in MTomato+ (mutant) cells.
Copy number assays
To determine the copy number of FlpO- or Cre-expressing cassettes after transgenesis, genomic DNA was extracted from mouse tail biopsies. Briefly, the blood samples were digested overnight in 500 μl proteinase K solution (10 mM Tris–HCl (pH 8.0), 100 mM NaCl, 10 mM EDTA (pH 8.0), 0.5% sodium dodecyl sulfate and 0.25 mg ml−1 proteinase K) with occasional vortexing. To this solution, 250 μl each of phenol and chloroform was added, followed by vigorous vortexing to ensure thorough mixing. The samples were immediately microcentrifuged at maximum speed for 5 min to separate the aqueous and organic layers. The upper aqueous layer (300 μl) was removed, with special care taken not to disturb the interface. To 300 μl solution, 30 μl (0.1× volume) of 3 M NaAc and 825 μl (2.5× volume) of 100% EtOH were added. The tubes were then shaken ten times to precipitate the DNA. The samples were spun down at maximum speed for at least 45 min at 4 °C. The supernatant was removed and pelleted DNA was washed with 500 μl 70% ethanol, followed by centrifuging at max speed for 5 min. The washed pellets were resuspended overnight in 100 μl Tris-EDTA solution (Tris 10 mM and 0.1 mM EDTA, pH 8). The next day, the DNA concentrations were measured in a NanoDrop microvolume spectrophotometer (Thermo Fisher Scientific) and diluted to a final concentration of 10 ng μl−1.
For the copy number assays, we performed qRT-PCR with TaqMan Universal Master Mix (TaqMan, 4440049) and the following probes: Tert TaqMan Copy Number Reference Assay (TaqMan probe, 4458369, with dye VIC: 2'-chloro-7'-phenyl-1,4-dichloro-6-carboxyfluorescein), and probes and primer sets with the fluorophore fluorescein amidite (FAM) designed in the lab and synthetized by integrated DNA technologies (idtdna.com). To detect FlpO (qPCR FlpO probe: TCTTGATGTCGCTGAACCTGCCG with primers FlpO-qPCR-F: CTGTACCAGTTCCTGTTCCTG and FlpO-qPCR-R: CTTGTCTTGGTCTCGGTCAC) and a predesigned Cre assay to detect the Cre gene (Mr00635245_cn, ThermoFisher).
The relative number of copies of the genes of interest was determined using the Tert probe as a reference. The Tert and FlpO/Cre probes were combined in the same reaction since they emit different fluorophores, VIC and FAM, respectively. The qRT-PCR reactions were run in an AB7900 thermocycler (Applied Biosystems).
Statistics and reproducibility
The numerical data was first processed with Microsoft Excel 2016 and after analyzed and plotted with Graphpad Prism v7.03. All bar graphs show mean ± standard deviation. The experiments were repeated with independent animals, as stated in the source data file or figure legends. The comparisons between two sample groups with a Gaussian distribution were by unpaired two-tailed Student t-tests. The comparisons among more than two groups were by one-way analysis of variance followed by multiple comparison tests. Datapoints were analyzed and plotted with GraphPad Prism. No randomization or blinding was used, and the animals or tissues were selected for analysis based on their genotype, the detected Cre/FlpO/Dre-dependent recombination frequency and the quality of multiplex immunostaining. The sample sizes were chosen according to the observed statistical variation and published protocols.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq data can be viewed at the Gene Expression Omnibus under accession number GSE257723. The instructions and code to reproduce all scRNA-seq or image analysis results can be found at GitHub via https://github.com/RuiBenedito/Benedito_Lab/tree/main/iFlpMosaics. The unprocessed FACS raw data files or original microscopy images of the data are available upon request. All other data supporting the findings in this study are included in the main article and associated files. Source data are provided with this paper.
References
Hansen, A. H. et al. Tissue-wide effects override cell-intrinsic gene function in radial neuron migration. Oxford Open Neurosci. https://doi.org/10.1093/oons/kvac009 (2022).
Hansen, A. H. & Hippenmeyer, S. Non-cell-autonomous mechanisms in radial projection neuron migration in the developing cerebral cortex. Front. Cell Dev. Biol. 8, 574382 (2020).
Vazquez-Liebanas, E. et al. Mosaic deletion of claudin-5 reveals rapid non-cell-autonomous consequences of blood–brain barrier leakage. Cell Rep. 43, 113911 (2024).
Contreras, X. et al. A genome-wide library of MADM mice for single-cell genetic mosaic analysis. Cell Rep. 35, 109274 (2021).
Zong, H. Generation and applications of MADM-based mouse genetic mosaic system. Methods Mol. Biol. 1194, 187–201 (2014).
Zong, H., Espinosa, J. S., Su, H. H., Muzumdar, M. D. & Luo, L. Mosaic analysis with double markers in mice. Cell 121, 479–492 (2005).
Liu, J. et al. Non-parallel recombination limits cre-loxP-based reporters as precise indicators of conditional genetic manipulation. Genesis 51, 436–442 (2013).
Fernandez-Chacon, M. et al. iSuRe-Cre is a genetic tool to reliably induce and report Cre-dependent genetic modifications. Nat. Commun. 10, 2262 (2019).
Schmidt-Supprian, M. & Rajewsky, K. Vagaries of conditional gene targeting. Nat. Immunol. 8, 665–668 (2007).
Lao, Z., Raju, G. P., Bai, C. B. & Joyner, A. L. MASTR: a technique for mosaic mutant analysis with spatial and temporal control of recombination using conditional floxed alleles in mice. Cell Rep. 2, 386–396 (2012).
Cheung, G. et al. Multipotent progenitors instruct ontogeny of the superior colliculus. Neuron 112, 230–246 e211 (2024).
Hunter, N. L., Awatramani, R. B., Farley, F. W. & Dymecki, S. M. Ligand-activated Flpe for temporally regulated gene modifications. Genesis 41, 99–109 (2005).
Pontes-Quero, S. et al. Dual ifgMosaic: a versatile method for multispectral and combinatorial mosaic gene-function analysis. Cell 170, 800–814 e818 (2017).
Cai, D., Cohen, K. B., Luo, T., Lichtman, J. W. & Sanes, J. R. Improved tools for the Brainbow toolbox. Nat. Methods 10, 540–547 (2013).
Rodriguez, C. I. et al. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat. Genet. 25, 139–140 (2000).
Luo, W. et al. Arterialization requires the timely suppression of cell growth. Nature 589, 437–441 (2021).
Dharaneeswaran, H. et al. FOXO1-mediated activation of Akt plays a critical role in vascular homeostasis. Circ. Res. 115, 238–251 (2014).
He, C. et al. c-myc in the hematopoietic lineage is crucial for its angiogenic function in the mouse embryo. Development 135, 2467–2477 (2008).
Claveria, C., Giovinazzo, G., Sierra, R. & Torres, M. Myc-driven endogenous cell competition in the early mammalian embryo. Nature 500, 39–44 (2013).
Wilhelm, K. et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 529, 216–220 (2016).
Munoz-Martin, N., Sierra, R., Schimmang, T., Villa Del Campo, C. & Torres, M. Myc is dispensable for cardiomyocyte development but rescues Mycn-deficient hearts through functional replacement and cell competition. Development 146, dev170753 (2019).
Eijkelenboom, A. & Burgering, B. M. FOXOs: signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 14, 83–97 (2013).
Paik, J. H. et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 128, 309–323 (2007).
He, L. et al. Proliferation tracing reveals regional hepatocyte generation in liver homeostasis and repair. Science 371, eabc4346 (2021).
Wei, Y. et al. Liver homeostasis is maintained by midlobular zone 2 hepatocytes. Science 371, eabb1625 (2021).
Siebel, C. & Lendahl, U. Notch signaling in development, tissue homeostasis, and disease. Physiol. Rev. 97, 1235–1294 (2017).
Fernández-Chacón, M. et al. Incongruence between transcriptional and vascular pathophysiological cell states. Nat. Cardiovasc. Res. 2, 530–549 (2023).
Pontes-Quero, S. et al. High mitogenic stimulation arrests angiogenesis. Nat. Commun. 10, 2016 (2019).
Wang, Y. et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486 (2010).
Radtke, F. et al. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity 10, 547–558 (1999).
McCright, B., Lozier, J. & Gridley, T. Generation of new Notch2 mutant alleles. Genesis 44, 29–33 (2006).
Garcia-Gonzalez, I. et al. iSuRe-HadCre is an essential tool for effective conditional genetics. Nucleic Acids Res. https://doi.org/10.1093/nar/gkae472 (2024).
Han, H. et al. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int Immunol 14, 637–645 (2002).
de Alboran, I. M. et al. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity 14, 45–55 (2001).
Knoepfler, P. S., Cheng, P. F. & Eisenman, R. N. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev 16, 2699–2712 (2002).
Koch, U. et al. Delta-like 4 is the essential, nonredundant ligand for Notch1 during thymic T cell lineage commitment. J. Exp. Med. 205, 2515–2523 (2008).
Haigh, J. J. et al. Cortical and retinal defects caused by dosage-dependent reductions in VEGF-A paracrine signaling. Dev. Biol. 262, 225–241 (2003).
Srinivas, S. et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol 1, 4 (2001).
Madisen, L. et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010).
Seluanov, A., Vaidya, A. & Gorbunova, V. Establishing primary adult fibroblast cultures from rodents. J. Vis. Exp. https://doi.org/10.3791/2033 (2010).
Bryja, V., Bonilla, S. & Arenas, E. Derivation of mouse embryonic stem cells. Nat. Protoc. 1, 2082–2087 (2006).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529 (2021).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
He, P. et al. The changing mouse embryo transcriptome at whole tissue and single-cell resolution. Nature 583, 760–767 (2020).
Acknowledgements
The research in Rui Benedito laboratory was supported by the European Research Council Starting Grant AngioGenesHD (638028), the European Research Council Consolidator Grant AngioUnrestUHD (101001814), the Ministerio de Ciencia, Innovación y Universidades (SAF2017-89299-P and PID2020-120252RB-I00) and ‘la Caixa’ Banking Foundation (project code HR19-00120 and HR22-00316 AngioHeart) awarded to R.B. The CNIC is supported by the Instituto de Salud Carlos III, the Ministerio de Ciencia, Innovación y Universidades (MICIU) and the Pro CNIC Foundation and is a Severo Ochoa Center of Excellence (grant CEX2020-001041-S funded by MICIU/AEI/10.13039/501100011033). The microscopy experiments were performed in the CNIC Microscopy and Dynamic Image Unit, an ICTS-ReDib co-funded by MCIN (/AEI/10.13039/501100011033) and the EDRF ‘A Way to Build Europe’ (number ICTS-2018-04-CNIC-16). I.G.-G. was supported by a PhD fellowship from Fundación La Caixa (CX-SO-16-1). A.R. was supported by The Youth Employment Initiative PEJD-2019-PRE/BMD-16990. L.G.-O. was supported by the Spanish Ministry of Economy and Competitiveness (PRE2018-085283). S.G. was supported by a Juan de la Cierva Fellowship (FJC2020-044237-I). M.F.-C. was supported by PhD fellowships from Fundación La Caixa (CX_E-2015-01). We thank S. Bartlett (CNIC) for English editing and the members of the CNIC transgenesis, microscopy, genomics, cytometry and bioinformatics units. We also thank F. Alt (Boston Children’s Hospital, Harvard Medical School), T. Honjo (Kyoto University Institute for Advanced Studies), F. Radtke (Swiss Institute for Experimental Cancer Research), R. H. Adams (Max Planck Institute for Molecular Biomedicine) and R. De Pinho (MD Anderson Cancer Center) for sharing the Mycfloxed, Rbpjfloxed, Notch1floxed, Cdh5(PAC)-creER and Foxos1/3/4floxed mice, respectively. We also thank Master’s students J. M. Arbonés and A. R. Sánchez for their work on iFlpMosaics.
Author information
Authors and Affiliations
Contributions
I.G.-G. and R.B. designed most of the experiments, interpreted results, assembled the figures and wrote the paper. R.B. designed all DNA vectors used for transgenesis. I.G.-G. did most of the DNA engineering (cloning), CRISPR–Cas9 genome targeting, animal experiments, confocal microscopy, FACS and scRNA-seq analysis. The mice were generated by the CNIC transgenesis unit. S.G. developed new methods for iFlpMosaics induction, tissue immunostaining and multispectral microscopy imaging, performed animal experiments and image analysis and interpreted results. S.F.R. performed immunostainings, microscopy, qRT-PCR and FACS analysis, image quantifications in Fiji and GraphPad, edited text and figures and assembled figures. L.G.-O, W.L., I.Z., M.D.-L. and M.F.-C. performed animal experiments, FACS, histology and confocal imaging. A.R. cloned and validated the iDre/FlpProgenitor allele. I.G.-G., A.R., C.T. and F.S.-C. analyzed the scRNA-seq data. M.L., M.S.S.-M., A.G.-C., F.L. and V.C.-G. gave general technical assistance with experiments and genotyped the mouse colonies. All authors approved the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Madhura Mukhopadhyay, in collaboration with the Nature Methods team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 False positives and false negatives with Cre-dependent mosaic genetics.
a, Schematic showing how a standard Cre-reporter can recombine and label cells without the deletion of any floxed gene (G) (false positives) and how the floxed gene can be deleted in non-Cre-recombined cells (false negatives). b, Different recombination efficiencies of different Rosa26 Cre-reporters in postnatal day 7 mice having the Cdh5-CreERT2 allele, despite all localizing to the Rosa26 locus and having a similar genetic distance between LoxP sites. c,d, Analysis of FACS or immunohistochemistry data reveals that Cre-reporters only accurately report recombination of themselves, not that of other floxed alleles (reporters), particularly at low tamoxifen doses. Data are presented as mean values +/− SD.
Extended Data Fig. 2 iFlpMosaics are neither toxic nor leaky.
a, b, Schematic diagrams of the novel Rosa26-iFlpMTomato-Cre/MYFP and Tg-iFlpMTomato-H2B-GFP-Cre/MYFP-H2B-Cherry-FlpO alleles, showing the genetic distances between the mutually exclusive FRT site pairs and the expected outcomes after FlpO/FlpO-ERT2 recombination. c, d Confocal microscopy and FACS analysis of mouse ES cells used to generate Rosa26-iFlpMTomato-Cre/MYFP mice 3 days after transfection with FlpO-expressing plasmids. e-g, Frequency of recombination and expression detected by microscopy in ES cells and ECs derived from embryoid bodies (EBs). h, Relative frequency of MTomato-2A-Cre+ and MYFP+ cells in ES cells (in vitro) and in blood (in vivo); the absence of change over time shows that permanent expression of Cre is non-toxic to cells. i, j FACS analysis of FlpO versus Cre-dependent leakiness in adult organs of mice carrying the standard Rosa26-LSL-YFP allele in combination with iFlpMosaics alleles. FlpO leakiness was not detected (0% MTomato+ cells), and Cre-non-self-leakiness (only detectable with the additional Rosa26-LSL-YFP allele) was observed only in a very small fraction of cells from animals carrying the Tg-iFlpMTomato-H2B-GFP-Cre/MYFP-H2B-Cherry-FlpO allele and not at all in animals carrying the Rosa26-iFlpMTomato-Cre/MYFP allele. k, CreERT2 alleles (that is Cdh5-CreERT2) can be leakier than iFlpMosaics. Scale bars 50μm (c and g left) and 200μm in f and g center and right. Data are presented as mean values +/− SD. For statistics see Source Data file 1.
Extended Data Fig. 3 Comparative analysis of recombination efficiency in FlpO-ERT2 mouse lines.
a-c, Representative FACs plots of different adult organs (see quantitative data in Fig. 2g), showing MYFP+ and MTomato+ cell frequencies in mice carrying the iFlpMTomato-Cre/MYFP allele in combination with the published R26CAG-FlpO-ERT2 or the newly generated Tg(Ins-CAG-FlpO-ERT2)F2 allele. Induction was 3 daily consecutive injections of tamoxifen (60mg/kg) and tissues collected 7 days after. d, Confocal micrograph of a flat-mounted adult mouse aorta 2 weeks after induction. e, FACS analysis showing the lack of leakiness of the Tg(Ins-CAG-FlpO-ERT2)F2 and iFlpMosaic alleles in the absence of induction/tamoxifen. f, g, Representative FACS plots showing that in both the heart and the lung CD45+ blood cells are much more affected by Myc deletion than CD31+ ECs. Scale bar 100μm.
Extended Data Fig. 4 Validation of iFlpMosaics for ratiometric quantitative gene function analysis in cell migration and proliferation.
a, Representative confocal micrographs of a P6 retina from a Rbpj-floxed mouse carrying the iFlpMTomato-Cre/MYFP and Apln-FlpO alleles. Apln is an X-chromosomal gene (mosaically expressed in females) expressed at the angiogenic front of growing vessels, recombining in angiogenic front ECs that later form the entire vasculature. Apln also recombines in non-endothelial (ERG-negative) cells in the retina. The right micrograph shows ERG-signal segmentation (EC nuclei), with nuclei of MTomato+ (RbpjKO) cells in red and nuclei of MYFP+ (RbpjWT) cells in green. Grey nuclei are in non-recombined ERG+ cells. Dashed white lines demark the leading edge (LE), angiogenic front (AF), and mature area (MA). A, arteries; V, veins. The chart shows the log2 ratios of MTomato+ to MYFP+ ECs in each retinal region, showing the enrichment of RbpjKO cells in the LE and AF, which suggests that loss of this gene induces cell migration to the front of the plexus. b, c Representative confocal micrographs of a P6 retina from Rbpj-floxed mouse carrying the iFlpMTomato-H2BGFP-Cre/MYFP-H2BCherry and Apln-FlpO alleles. This allows the nuclear labeling of RbpjKO (H2B-GFP+) and RbpjWT (H2B-Cherry+) ECs and blood cells, which is more convenient for cell object segmentation and quantification of nuclear-specific proteins, such as the proliferation and cell-cycle arrest markers Ki67 and p21. Nuclear segmentation outlines: red, Cherry+/Ki67- or p21-; pink, Cherry+/Ki67+ or p21+; green, GFP+/Ki67- or p21-; cyan, GFP+/Ki67+ or p21+. Data are presented as mean values +/− SD. For statistics see Source Data file 1. Scale bars, 150μm.
Extended Data Fig. 5 Combining iFlpMosaic with iFlpChromatin yields higher single-cell clonal resolution.
a, Representative confocal micrograph of adult liver sections from mice carrying the indicated alleles. Combination of these membrane and nuclear reporter alleles allows up to 3 mutant and 1 wildtype cell barcodes (labeling). Note that binucleated (2x2n) or polyploid (4n) hepatocytes can have more multispectral barcodes. b, Representative confocal micrograph of a P7 retina from an animal containing the indicated alleles and induced at P1 with 4-OHT. The bar shows the ratiometric frequency of each nuclear/H2B+ fluorescent marker from the iFlpChromatin allele. c, Representative confocal micrograph of the indicated adult organ sections. Mononucleated diploid cells (kidney and lung) express only 1 of 3 possible nuclear/chromatin markers, whereas multinucleated or polyploid cells (hepatocytes and cardiomyocytes) can express a combination of 2 or 3 possible markers (6 possible chromatin barcodes in total). d, Recombination and cell barcoding possibilities in animals carrying the indicated alleles. The representative confocal micrographs of a P7 retina show that a fraction of wildtype and mutant cells can both be labeled in 4 different ways, substantially increasing single-cell clonal resolution. The panel to the right illustrates how a Fiji image analysis script automatically detects, segments, and pseudocolors the nuclei of dual-labeled cells (cells with both nuclei and membrane labelled). e, Recombination and barcoding possibilities in a mononucleated diploid cell in animals carrying the indicated alleles. f, Representative confocal micrographs of an adult liver, collected 2 weeks after tamoxifen induction, show that mutant hepatocytes can be labeled with up to 16 different fluorescent barcodes, depending on whether the cells are mononucleated (diploid) or multinucleated (polyploid). Wildtype hepatocytes can have up to 8 different fluorescent barcodes (only 6 shown). Scale bars, 100μm.
Extended Data Fig. 6 Mapping the clonal expansion of single mutant and wildtype cells with iFlpMosaics.
a,b, Representative confocal images (low magnification large tile scans) of entire P20 liver sections from mice carrying the indicated alleles. Recombination of the reporter alleles was induced at P1 and resulted in different recombination frequencies. Right panels show the results of automatic image processing with Fiji scripts, which generate spatial maps of dual-labeled clones of cells (with membrane and nuclear labeling). The bigger the circle, the bigger the clone. c, Quantification and representative confocal micrographs of proliferative pancreatic cells (Ki67+ in cycle or EdU+ in S-phase) at different postnatal stages. d, Quantification and representative confocal micrograph of Ki67+/DAPI+ cells in the adult liver. e, Representative confocal micrographs of adult liver sections from control and Myc-floxed mice carrying the indicated alleles. Left panels show endogenous fluorescent signals from the reporter alleles. Center panels show sections from the same mice immunostained with antibodies to DsRed (detects MTomato and H2B-Cherry), GFP (detects MYFP and H2B-GFP), or the HA epitope (detects MTFP1 and H2B-Cerulean) together with Fiji-script-generated spatial maps of dual-labeled clones (with membrane and nuclear labeling) reveal hepatocyte ploidy and clone size. Graphs show the percentages of mononucleated cells, binucleated cells, and 2-cell mononuclear clones. In control mice, MYFP+ (WT) and MTomato+ (WT) diploid cells occur in similar proportions, whereas in Myc-floxed mutants MTomato+ (MycKO) diploid cells are more frequently binucleated and give rise to fewer 2-cell mononucleated clones than MYFP+ (MycWT) cells. Scale bars, 50μm (c, d), the rest 300μm. Data are presented as mean values +/− SD. For numerical data see Source Data file 1.
Extended Data Fig. 7 iDre/FlpProgenitor enables the effective induction of genetic mosaics from single progenitor cells.
a, FACS plots showing that the iDre/FlpProgenitor allele significantly increases the recombination frequency of the iFlpMTomato-Cre/MYFP allele when combined with the Tg(Ins-CAG-FlpO-ERT2) allele, because it is much more sensitive to FlpO-ERT2 activity. b,c, In animals lacking the iDre/FlpProgenitor allele, most single-cell derived clones are formed by just one type of cell (MTomato+ or MYFP+). With the iDre/FlpProgenitor allele, it is possible to induce clones formed by mutant (MTomato+) and wildtype (MYFP+) cells derived from the same single progenitor cells. d, Representative confocal micrograph of a P7 retina from an animal carrying the indicated alleles and induced with a very low dose of 4-OHT (0,04 mg/kg) at P3. After induction of the iDre/FlpProgenitor allele, cells express the nuclear marker H2B-V5 and FlpO. After cell division, FlpO will recombine the iFlpMTomato-Cre/MYFP allele, generating MYFP+ and MTomato+ progeny cells derived from a single initial recombination event. Cells at the angiogenic front disperse/migrate more after division than cells in the mature plexus. e, Time-lapse confocal microscopy of mouse ES cells derived from a mouse with the indicated genotype. 24h after administration of 4-OHT (1μM), ES cells first co-express the marker hCD2 at their surface (labelled with anti-hCD2-APC) and FlpO. During the subsequent hours, the iFlpMosaic-recombined progeny of these single progenitor ES cells express either MTomato-2A-Cre+ or MYFP+ (twin clones). f, Through the use of CRISPR-Cas9 mediated gene targeting, we generated several founder mice carrying the Mki67-2A-DreERT2 knock-in allele detected with primers P1 to P3. On the right a representative confocal micrograph from a growing hair follicle (see also Fig. 5h) containing many labelled cells 5 days after induction. Scale bars 500 μm in b, c, 100μm in d, 20μm in e, and 25μm in f. Data are presented as mean values +/− SD. For statistics see Source Data file 1.
Extended Data Fig. 8 Single cell RNA-seq analysis combined with ratiometric iFlpMosaics uncovers the cell-autonomous Rbpj gene function in diverse cell types.
a, Umaps and barplots showing the identified clusters and their frequencies among MYFP+ (RbpjWT) and MTomato+ (RbpjKO) cells. Dot plots show the frequency (size) and expression level (color intensity) for the top cluster marker genes. b, Top differentially expressed genes per cluster between MYFP+ (RbpjWT) and MTomato+ (RbpjKO) cells. Most of the charts also show the expression of Rbpj, its canonical target Hes1, and the proliferation markers Ki67 (cells in G2/M of the cell-cycle) and Cdkn1a (likely arrested cells). c, Gene set enrichment analysis (GSEA) pathways with their normalized enrichment score.
Extended Data Fig. 9 Single cell RNA-seq analysis combined with ratiometric iFlpMosaics uncovers the combined NOTCH1/2/3 receptors cell-autonomous function in diverse embryo cell types.
a, Umaps and barplots showing the identified clusters and their frequencies among MYFP+ (Notch1/2/3WT) and MTomato+ (Notch1/2/3KO) cells. Dot plots showing the frequency (size) and expression level (color intensity) for the top cluster marker genes. b, Top differentially expressed genes per cluster between MYFP+ (Notch1/2/3WT) and MTomato+ (Notch1/2/3KO) cells. Dot plots also show the expression of Notch1 (note that Notch2 and Notch3 3′mRNA is still expressed after deletion and detected by scRNAseq, as shown in Fig. 6k), its canonical target genes (Hes and Hey, these are also regulated by other pathways), and the proliferation markers Ki67 (labels cells in G2/M of the cell-cycle) and Cdkn1a (likely arrested cells). c, Gene set enrichment analysis (GSEA) pathways with their normalized enrichment score.
Extended Data Fig. 10 Single cell RNA-seq analysis combined with ratiometric iFlpMosaics uncovers the combined NOTCH1/2/3 receptors cell-autonomous function in diverse embryo cell types.
a, Umaps and barplots showing the identified clusters and their frequencies among MYFP+ (Notch1/2/3WT) and MTomato+ (Notch1/2/3KO) cells. Dot plots showing the frequency (size) and expression level (color intensity) for the top cluster marker genes. b, Top differentially expressed genes per cluster between MYFP+ (Notch1/2/3WT) and MTomato+ (Notch1/2/3KO) cells. Dot plots also show the expression of Notch1 (note that Notch2 and Notch3 3′mRNA is still expressed after deletion and detected by scRNAseq, as shown in Fig. 6k), its canonical target genes (Hes and Hey, these are also regulated by other pathways), and the proliferation markers Ki67 (labels cells in G2/M of the cell-cycle) and Cdkn1a (likely arrested cells). c, Gene set enrichment analysis (GSEA) pathways with their normalized enrichment score.
Supplementary information
Supplementary Table 1 (download XLSX )
Genotyping primers.
Supplementary Table 2 (download XLSX )
Antibodies list.
Supplementary Table 3 (download XLSX )
Taqman assays.
Source data
Source data (download XLSX )
All numerical and statistical data corresponding to Figs. 1–5 and Extended Data Figs 1–10.
Source Data Fig. 2 (download PDF )
A gating strategy example for one of the samples used in Fig. 2, as requested in Reporting summary.
Source Data Extended Data Fig. 7 (download PDF )
Unmodified gels are shown in Extended Data Fig. 7f.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Garcia-Gonzalez, I., Gambera, S., Rocha, S.F. et al. iFlpMosaics enable the multispectral barcoding and high-throughput comparative analysis of mutant and wild-type cells. Nat Methods 22, 323–334 (2025). https://doi.org/10.1038/s41592-024-02534-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41592-024-02534-w
