
Abstract
Large yellow croaker (Larimichthys crocea) has been demonstrated to be divided into three geographical stocks from south to north along the coast of China, including Nanhai, Mindong, and Daiqu. Although multiple versions of L. crocea have been published, no high-quality Nanhai and Daiqu genomes have been assembled, hampering the assessment of the fine-scale genetic structure and adversely affecting wild stock conservation, fishery management, and germplasm exploitation of large yellow croaker. To fill the gap, we sequenced the genomes of three L. crocea stocks using a combination of PacBio and Hi-C technologies. We assembled each genome (~712 Mb) into 24 chromosomes with a contig N50 of 19.46–29.71 Mb and an integration efficiency of 88.13–92.80%. Furthermore, 26,851–28,133 protein-coding genes were predicted. The reference genomes of three geographical stocks of L. crocea provide vital resources for future research on the conservation and utilization of genetic diversity.
Similar content being viewed by others
Background & Summary
Large yellow croaker (Larimichthys crocea) is a marine fish that inhabits the nearshore seas and estuaries of the northwestern Pacific Ocean, typically in temperate areas. The Chinese mariculture industry currently yields over 281,000 tons of large yellow croaker, indicating the important economic value of this fish1. Abundant genomic resources have been established for this species, comprising six genetic maps2,3,4,5,6,7, three draft genomes utilizing Illumina technology8,9,10, a draft genome using the combination of Illumina and PacBio sequencing technologies11, and a chromosome-level reference genome generated based on PacBio and Hi-C technologies12.
In the 1960s, some studies divided the large yellow croaker along the Chinese coastline into three stocks based on morphological data: Naozhou stock (NZ, located in the west South China Sea), Min-Yuedong stock (MYD, located in the eastern South China Sea and Taiwan Strait), and Daiqu stock (DQ, located in the East China Sea)13,14. In recent years, with the application of molecular genetic markers, more studies have reported the analysis of the population structure of large yellow croaker. Lin et al. have utilized a UPGMA tree based on eight strictly chosen simple sequence repeats (SSRs) to classify the species into NZ, MY (i.e. MYD), and DQ stocks15. Single nucleotide polymorphism (SNP), as a new generation of genetic markers, has also been used to evaluate the genetic structure of populations. Principal component analysis (PCA) based on SNP data reveals that large yellow croaker populations can be divided into three stocks, i.e., Nanhai (NH, distributed in the South China Sea and the Taiwan Strait), Mindong (MD, distributed near the Taiwan Strait, i.e. MYD or MY) and Daiqu (DQ, distributed in the East China Sea)16. Chen et al. also reported that climate change drove the boundary between Naozhou (i.e. Nanhai) stock and Min-Yuedong (i.e. Mindong) stock might have moved northwards from the Pearl River Estuary to the northern part of the Taiwan Strait, accompanied by highly asymmetric introgression16. A series of studies have shown that there are differences in genetic information among the three geographical stocks of large yellow croaker. However, so far, only the chromosome-level reference genome of Mindong stock has been reported12. The lack of high-quality reference genomes of the other two stocks hinders the assessment of the fine-scale genetic structure of large yellow croaker and adversely affects the population genetic and evolutionary studies, wild stock conservation, fishery management, and germplasm exploitation.
To fill the gap, we sequenced and constructed reference genomes for L. crocea from three geographical stocks of Nanhai (NH), Mindong (MD), and Daiqu (DQ). Using a combination of the PacBio single-molecule real-time sequencing technique (SMRT) and high-through chromosome conformation capture (Hi-C) technologies, we assembled each genome at the chromosome level with a total length of 706.71–722.73 Mb, a contig N50 of 19.46–29.71 Mb, a scaffold N50 of 27.00–27.83 Mb, and a complete BUSCO value of 98.5%-98.7%. To supply materials for gene annotation and conduct functional analysis, we additionally sequenced the transcriptomes of 4 tissues for each of the three stocks of L. crocea. A total of 209.84–219.64 Mb (29.69%-30.39% of the assemblies) of repeat content, 26,851–28,133 protein-coding genes, and 21,156–33,583 ncRNAs were identified. In conclusion, this study reports high-quality chromosome-level reference genomes of different geographical stocks of large yellow croaker for the first time, serving as valuable genomic resources for large yellow croaker and providing a vital reference for future research on the conservation and utilization of genetic diversity.
Methods
Sample collection and nucleic acid extraction
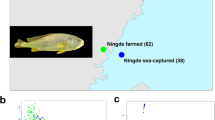
Three healthy female L. crocea from NH, MD, and DQ stocks were obtained from Dongshan Sea (Huizhou, Guangdong), Sandu Bay (Ningde, Fujian), Xiangshan Bay (Ningbo, Zhejiang) in China, respectively (indicated in Fig. 1). Muscle, brain, liver, and spleen were sampled from each fish. All samples were snap-frozen in liquid nitrogen and stored at −80 °C to preserve nucleic acid integrity. Genomic DNA (gDNA) of L. crocea was extracted from muscle tissues using an SDS-based DNA extraction method17, while total RNA was extracted from the brain, spleen, liver, and muscle by a TRIzoL kit (Invitrogen, CA, USA) and mixed at an equal concentration for transcriptome sequencing. The quality of gDNA was assessed by 1.5% agarose gel electrophoresis and DNA was quantified by a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA). The quality of RNA was assessed by the Fragment Analyzer 5400 (Agilent Technologies, CA, USA).

Geographical distribution of three large yellow croaker stocks. The sampling points of the three fish sequenced in this paper are marked as red dots. NH, Nanhai; MD, Mindong; DQ, Daiqu.
Library construction and sequencing
For HiFi read generation, high-molecular-weight (HMW) gDNA was sheared to 15,000–20,000 bp, and the PacBio HiFi library was constructed using the SMRTbell Express Template Prep Kit 2.0 (Pacbio, USA). The genomic library was sequenced in CCS mode on the PacBio Sequel II system at Novogene (Tianjin, China). A total of 52.18–56.59 Gb of CCS clean reads were obtained with an average read length of 7,947–8,284 bp, resulting in 73.84–79.94-fold coverage of the three L. crocea genomes. For Hi-C library construction, the MboI restriction enzyme was used to digest the cross-linked high-molecular-weight (HMW) gDNA. DNA was purified by the phenol-chloroform extraction and randomly sheared into 300–500 bp fragments. After the addition of A-tails to the fragment ends and the subsequent ligation by the Illumina paired-end (PE) sequencing adapters, Hi-C sequencing libraries were amplified by PCR (12–14 cycles) and sequenced on the Illumina platform to obtain PE150 reads. Finally, 43.16–45.78 Gb of paired-end clean reads were generated from the Hi-C library. The RNA-seq library was constructed using the NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB, USA) following the manufacturer’s recommendations and sequenced on the Illumina Novaseq 6000 platform. A total of 24.65–25.58 Gb of paired-end clean reads were generated from the RNA-seq library. For the PacBio Iso-Seq library, RNA was converted into cDNA using the SMARTer PCR cDNA Synthesis Kit (Clontech, USA). The library was then sequenced on the PacBio Sequel II system at Novogene (Tianjin). As shown in Table 1, 30.10–60.44 Gb of long reads were obtained from the sequencing of Iso-Seq libraries.
Genome assembly
HiFiasm18 (v0.18.5) was used to generate contig-level genomes based on the HiFi long reads with default parameters, resulting in three preliminary assemblies of each of the three stocks. For NH stock, the assembly contained 100 contigs, with a total length of 722.73 Mb and a contig N50 of 29.71 Mb. For MD stock, the assembled genome size was 707.86 Mb, including 87 contigs, with a contig N50 of 22.39 Mb. For DQ stock, we gained the preliminary assembly with a total length of 706.71 Mb, including 101 contigs, with a contig N50 of 18.46 Mb. The complete BUSCO value ranged from 98.5% to 98.7%, indicating eximious assembly integrity (Table 2).
Hi-C sequencing data was provided to Juicer19 (v1.11.08) and 3D-DNA pipeline20 for chromosome-level genome assemblies of the three L. crocea stocks. We mapped Hi-C clean reads to the preliminary genomes by Juicer. Then, the genomic proximity signal in the Hi-C datasets was used to obtain the chromosome-level scaffolds. Subsequently, the 3D-DNA pipeline was used for scaffolding the genomes. Ultimately, chromosome-level genomes were finalized by using Juicebox21 to adjust misjoins, translocations, inversions, and chromosome boundaries. The size of each chromosome-level genome assembly was estimated to be 636.97 Mb (NH stock, 88.13% of the total length of contigs), 656.91 Mb (MD stock, 92.80% of the total length of contigs), and 644.04 Mb (DQ stock, 91.13% of the total length of contigs). All three assemblies contained 24 chromosomes, with an average chromosome length of 26.54 Mb, 27.37 Mb, and 26.83 Mb, respectively (Table 3).
Annotation of repetitive sequences
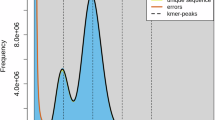
The repetitive sequences of three L. crocea genomes were identified using both homology-based and de novo strategies. First, RepeatModeler22 (v2.0.1) was utilized to detect repetitive sequences and generate a de novo repeat library. Then, unknown repeats were classified by TEclassTest.pl in TEclass23 (v2.1.3). In another way, repeat elements were forecasted based on the library from RepeatMasker24 (v4.1.2). Finally, all repetitive regions were integrated and masked. Totally, 219.63 Mb (30.39% of the assembled genome), 211.82 Mb (29.92%), and 209.84 Mb (29.69%) of consistent and non-redundant repeat sequences were obtained from genomes of NH, MD, and DQ stocks, respectively. The most abundant repetitive elements for three genomes were DNA transposons. Notably, 47.74 Mb LTRs (6.6% of the assembled genome) were identified from the genome of NH stock, which was higher than the other two stocks (Table 4, Fig. 2A–C).

Gene and repetitive element annotations of three L. crocea genomes. A–C: Distribution of divergence rate for TEs in Nanhai (NH) genome (A), Mindong (MD) genome (B), and Daiqu (DQ) genome (C). D–F: Venn diagrams of functional annotation based on different databases in NH genome (D), MD genome (E), and DQ genome (F).
Genome annotation
For noncoding RNA (ncRNA) annotation, Infernal25 (v1.1.4) was utilized based on the Rfam database (http://eggnogdb.embl.de/). Five types of ncRNA were identified from the L. crocea genomes. For NH stock, 7,110 tRNAs, 1,699 miRNAs, 23,465 rRNAs, 1,305 snRNAs, and 4 lncRNAs were identified. For MD stock, there were 6,206 tRNAs, 2,032 miRNAs, 11,805 rRNAs, 1,109 snRNAs and 4 lncRNAs. For DQ stock, 6,331 tRNAs, 2,085 miRNAs, 15,114 rRNAs, 960 snRNAs, and 3 lncRNAs were identified (Table 5, Fig. 3).

Circos plot of the reference genomes of three geographical stocks of L. crocea. Circos plot of 24 chromosome-level scaffolds, representing annotation results of genes and ncRNA of (A) Nanhai (NH) stock, (B) Mindong (MD) stock, (C) Daiqu (DQ) stock. The tracks from inside to outside are ncRNA abundance of the positive strand, ncRNA abundance of the negative strand, GC content, gene abundance of the positive strand, gene abundance of the negative strand, and 24 chromosome-level scaffolds.
For gene structure annotation, both ab intio and transcriptome-based strategies were used for gene prediction in the genome of each stock after soft-masking all repeat regions. RNA-seq data was assembled into transcripts using Trinity26 (v2.8.5). For the ab intio approach, BRAKER227 was executed based on the transcripts assembled from RNA-seq and known genes of L. crocea and Cyprinus carpio. For the transcriptome-assisted approach, RNA-seq data was aligned to the genomes to assemble into transcriptome by HISAT228 (v2.2.1) and StringTie29 (v2.1.4). After that, the open reading frame (ORF) was predicted via TransDecoder (https://github.com/TransDecoder/TransDecoder) (v5.5.0). Eventually, EvidenceModeler (v1.1.1) was adopted to produce comprehensive gene sets, which was further annotated for protein-coding gene structure by PASA30 (v2.4.1). As a result, we predicted 36,151 (NH stock), 36,151 (MD stock), and 33,013 (DQ stock) protein-coding genes, respectively, which were subsequently used for functional annotation (Table 6).
For functional annotation of protein-coding genes, Diamond31 (v2.0.6) was applied to align protein-coding genes to the NCBI nr, TrEMBL (http://www.uniprot.org/), and Swiss-Prot (http://www.uniprot.org/) protein databases with the threshold of E-values less than 1 × 10−5. The annotation of GO and KEGG pathways was performed using InterProScan32 (v5.53) and the online website KEGG Automatic Annotation Server33 (KAAS, https://www.genome.jp/tools/kaas/). After integration and de-redundancy, a total of 28,133 (NH stock), 26,851 (MD stock), and 27,279 (DQ stock) protein-coding genes were annotated (Table 6, Figs. 2D–F, 3).
Data Records
All the PacBio long DNA reads, Hi-C reads, Illumina short RNA reads, and PacBio long RNA reads are available from NCBI via the accession numbers SRR29302595-SRR2930260634. The assembled genomes of NH, MD, and DQ have been deposited at Genbank under the accession numbers of JBEDUZ00000000035, JBEDUY00000000036, and JBEDUX00000000037, respectively. Moreover, the assembled genomes and genome annotation are available on Figshare38,39,40.
Technical Validation
Evaluation of genome assemblies and annotation
To ensure the accuracy and integrity of the assemblies, we assessed the completeness of the final genome assemblies using Benchmarking Universal Single-Copy Orthologues (BUSCO)41 with the Actinopterygii_odb10 lineage database. Out of 3,640 single-copy orthologues, all of the three assemblies have > 98.5% BUSCO completeness, which are comparable to those of NH (98.7%), MD (98.5%), DQ (98.6%) (Supplementary Table 1). We additionally used merqury42 (version 1.3) to assess the quality of the three assemblies. The results revealed exceptionally low error rates across all three genomes, with QV values exceeding 60 (Supplementary Table 2), which is sufficient to indicate that all three assemblies are of good quality.
We plotted the Hi-C interaction heatmaps of the chromosomes in each of the three assemblies (Supplementary Fig. 1). The contigs were anchored on 24 chromosomes in all three genomes, which formed squared boxes along the main diagonal of the heatmap matrix. The assembled chromosomes in each of the three assembled genomes display high collinearity with those of the large yellow croaker reference genome published in 201912, indicating that the structures of the assembled genomes are consistent. Meanwhile, for all three genomes, the number of gaps in the assembled chromosomes was significantly reduced compared with the genome released in 201912 (Supplementary Fig. 2). No gap was observed in chr9 and chr10 of the NH genome and chr6 of the MD genome. These results demonstrate improvements in the assembly and anchoring completeness of the new genomes.
Moreover, we successfully obtained a total of 36,151, 36,151, and 33,013 protein-coding genes by combining ab initio strategies and transcriptome-assisted approaches in NH, MD, and DQ genomes, respectively. A total of 28,133, 26,851, and 27,279 genes were functionally annotated in at least one of these databases in three genomes (Fig. 2D–F, Table 6). Taken together, these results suggest that the three assembled L. crocea genomes were of superior quality.
Code availability
The software settings and parameters used in this study are as follows:
Genome assembly:
Hifiasm: all parameters were set as default.
Genome annotation:
(1) RepeatModeler: parameters: -engine ncbi.
(2) TEclass: all parameters were set as default.
(3) RepeatMasker: parameters: -e ncbi -no_is -nolow -norna -gff -poly -html -a.
(4) BRAKER2: all parameters were set as default.
(5) HISAT2: parameters: --dta.
(6) EvidenceModeler: parameters: all parameters were set as default.
(7) PASA: --ALIGNERS blat.
(8) InterProScan: parameters: -appl Pfam -goterms -iprlookup –pa.
No custom code was used during this study for the curation and validation of the dataset.
References
China MOaaRaOTPSRO, C. N., Fisheries CSO. China Fishery Statistical Yearbook 2024 (China Agriculture Press, 2024).
Ye, H., Liu, Y., Liu, X., Wang, X. & Wang, Z. Genetic mapping and QTL analysis of growth traits in the large yellow croaker Larimichthys crocea. Marine biotechnology 16, 729–738 (2014).
Ning, Y. et al. A genetic map of large yellow croaker Pseudosciaena crocea. Aquaculture 264, 16–26 (2007).
Xiao, S. et al. Gene map of large yellow croaker (Larimichthys crocea) provides insights into teleost genome evolution and conserved regions associated with growth. Scientific reports 5, 18661 (2015).
Ao, J. et al. Construction of the high-density genetic linkage map and chromosome map of large yellow croaker (Larimichthys crocea). International Journal of Molecular Sciences 16, 26237–26248 (2015).
Kong, S. et al. Constructing a high-density genetic linkage map for large yellow croaker (Larimichthys crocea) and mapping resistance trait against ciliate parasite Cryptocaryon irritans. Marine Biotechnology 21, 262–275 (2019).
Yu, X., Joshi, R., Gjøen, H. M., Lv, Z. & Kent, M. Construction of genetic linkage maps from a hybrid family of large yellow croaker (Larimichthys crocea). Frontiers in Genetics 12, 792666 (2022).
Wu, C. et al. The draft genome of the large yellow croaker reveals well-developed innate immunity. Nature communications 5, 5227 (2014).
Wang, Z. et al. Proto-sex locus in large yellow croaker provides insights into early evolution of the sex chromosome. Biorxiv, 2020.2006. 2023.166249 (2020).
Ao, J. et al. Genome sequencing of the perciform fish Larimichthys crocea provides insights into molecular and genetic mechanisms of stress adaptation. PLoS genetics 11, e1005118 (2015).
Mu, Y. et al. An improved genome assembly for Larimichthys crocea reveals hepcidin gene expansion with diversified regulation and function. Communications biology 1, 195 (2018).
Chen, B. et al. The sequencing and de novo assembly of the Larimichthys crocea genome using PacBio and Hi-C technologies. Scientific data 6, 1–10 (2019).
Tian, M., Xu, G. & Yu, R. Geographic variation and population of morphological characteristics of Pseudosciaena crocea (Richardson). Studia Mar Sinica 2, 79–97 (1962).
Xu, G., Tian, M. & Zheng, W. The stocks of Pseudosciaena crocea (Richardson). Proceeding the 4th Plenum the comm fish res the west part the Pacific Ocean. Science Press, Beijing, 39–46 (1963).
Lin, N., Su, Y., Ding, S. & Wang, J. Genetic analysis of large yellow croaker (Pseudosciaena crocea) stocks using polymorphic microsatellite DNA. Fujian Journal of Agricultural Sciences 27, 661–666 (2012).
Chen, B. et al. Population structure and genome-wide evolutionary signatures reveal putative climate-driven habitat change and local adaptation in the large yellow croaker. Marine Life Science & Technology 5, 141–154 (2023).
Goldenberger, D., Perschil, I., Ritzler, M. & Altwegg, M. A simple” universal” DNA extraction procedure using SDS and proteinase K is compatible with direct PCR amplification. Genome Research 4, 368–370 (1995).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature methods 18, 170–175 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell systems 3, 99–101 (2016).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
Abrusán, G., Grundmann, N., DeMester, L. & Makalowski, W. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330 (2009).
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 5, 4.10. 11–14.10. 14 (2004).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nature biotechnology 29, 644 (2011).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR genomics and bioinformatics 3, lqaa108 (2021).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature biotechnology 33, 290–295 (2015).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome biology 9, 1–22 (2008).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nature methods 18, 366–368 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic acids research 35, W182–W185 (2007).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP512089 (2024).
Genbank https://identifiers.org/insdc:JBEDUZ000000000 (2024).
Genbank https://identifiers.org/insdc:JBEDUY000000000 (2024).
Genbank https://identifiers.org/insdc:JBEDUX000000000 (2024).
Chen, X. The genome of Larimichthys crocea (MD stock). figshare https://doi.org/10.6084/m9.figshare.25981759.v1 (2024).
Chen, X. The genome of Larimichthys crocea (NH stock). figshare https://doi.org/10.6084/m9.figshare.25980427.v1 (2024).
Chen, X. The genome of Larimichthys crocea (DQ stock). figshare https://doi.org/10.6084/m9.figshare.25981936.v1 (2024).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome biology 21, 1–27 (2020).
Acknowledgements
This work was supported by the Research on breeding technology of candidate species for Guangdong modern marine ranching (2024-MRB-00-001), the National Science Fund for Distinguished Young Scholars (32225049), the “Science and Technology Innovation 2025” Major Special Project of Ningbo City (2021Z002), and the Fundamental Research Funds for the Central Universities (20720240107).
Author information
Authors and Affiliations
Contributions
P.X. conceived and supervised the study. Q.H., Q.Z.K. and P.X. collected the samples. X.T.C., L.W.M. and Q.H. performed bioinformatics analysis. X.T.C. drafted the manuscript. F.P. helped with manuscript preparation. P.X., T.Z. and N.L. revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, X., Miao, L., He, Q. et al. Chromosome-level genome assembly for three geographical stocks of large yellow croaker (Larimichthys crocea). Sci Data 11, 1364 (2024). https://doi.org/10.1038/s41597-024-04126-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-04126-x
