
Abstract
Malus niedzwetzkyana (MN) is the mother of Rosybloom hybrids, ornamental crabapples with red and purple flowers. We present a high-quality chromosome-scale genome for MN with a size of 672.64 Mb, anchored to 17 chromosomes and with a high BUSCO completeness score of 98.6%, reaching the ‘gold standard’ level. Moreover, our assembly has captured 28 telomeres. A total of 43,813 protein-coding genes was annotated in the MN genome. The assembled high quality provides a valuable opportunity to enhance our understanding of the genetic basis of flower colour and other ornamental traits in crabapples, thereby advancing the field of genetics and breeding.
Similar content being viewed by others


Background & Summary
The ornamental crabapples (Malus spp., Rosaceae) are woody plants with fruit diameters of less than 5 cm, whose flowers, leaves, fruits and other traits have significant ornamental value1. Interspecific hybridization plays a pivotal role in the breeding of new varieties of ornamental crabapple. For a considerable period, the predominant colours observed in crabapple flowers were white and pink. Following the discovery of Malus niedzwetzkyana (MN), there was a notable increase in the intensity of the colours red and purple, which significantly enhanced the ornamental value of crabapples. The Rosybloom hybrids, which are descendants of MN, exhibit flowers that are dark, deep rose, red, and purple in colour1. They have been widely used in garden landscapes around the world. Previous studies have demonstrated that the colour of flowers, as well as foliage and fruit is primarily influenced by anthocyanins, a category of flavonoid compounds2,3,4. The genetic analysis of MN at the molecular level provides a valuable opportunity to enhance our understanding of the genetic basis of flower colour and other ornamental traits in crabapples, thereby advancing the field of genetics and breeding.
De novo genome assembly represents a foundational and efficacious instrument employed in the field of molecular genetics research. Moreover, the development of third-generation PacBio HiFi sequencing and Hi-C techniques has markedly enhanced the completeness of genome assembly. Several chromosome-level genomes of crabapples are now accessible in public databases, including the European crabapple (M. sylvestris)5, the Pacific crabapple (M. fusca)6, M. prunifolia7, and two ornamental crabapple cultivars ‘Royalty’ and ‘Flame’4. The advent of chromosome-level genome sequences has opened new avenues of research, offering researchers the opportunity to investigate the functional, regulatory, and evolutionary aspects of the genome in the Malus genus, and to gain a more nuanced understanding of its significant characteristics.
This study presents a high-quality genome for MN, generated through an integrated approach utilising Illumina short-read, PacBio high-fidelity (HiFi) long-read, and high-throughput chromosome conformation capture (Hi-C) sequencing data. In total, we generated 40.02 Gb (~59 × coverage) of clean short reads, 29.42 Gb (~43 × coverage) of PacBio HiFi CCS reads with an N50 of 17.97 kb, and 63.95 Gb of clean Hi-C data (~94 × coverage) (Table 1). The estimated genome size, heterozygosity, and repetitive content were 678.26 Mb, 0.57% and 33.74%, respectively, using 17-mer analysis. The final assembled genome size was 672.64 Mb with a contig N50 of 36.45 Mb (Table 2). The assembled contigs were then anchored onto 17 pseudochromosomes, with an anchor rate of 98.38% (Fig. 1). The quality and completeness of the assembly were validated by employing four distinct approaches. First, the clean short reads and the PacBio HiFi CCS reads were mapped to the assembly, yielding a mapping ratio of 99.32% and 99.95%, respectively (Table 3). Secondly, two telomeres were identified at both ends of the twelve chromosomes, one telomere was identified at one end of four chromosomes (chr1, chr5, chr13, and chr16), and only chromosome 15 did not have a telomere identified (Fig. 2). Thirdly, the BUSCO results showed that 1590 (98.6%) genes could be compared with the lineal homologous database, of which 1058 (65.6%) were single-copy and 532 (33.0%) were duplicates (Table 4). Finally, the long terminal repeat (LTR) assembly index (LAI) score of the MN genome assembly was 21.99, reaching the ‘gold standard’ level (LAI > 20).

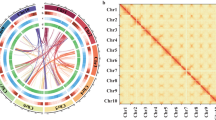
Hi-C interaction analysis and circos map. (a) Hi-C interaction heatmap of MN. (b) The circos map of MN. For the circos map, the tracks from inside to outside are chromosome ID and length (I), GC content (II), density of protein-coding genes (III), density of LTR elements (IV).

Overview of the near T2T and gap-free MN genome. The triangle and block represent the telomere regions and gaps, respectively.
Repeat sequences accounted for 476.89 Mb, representing 70.90% of the assembly (Table 5). The retransposon LTR was the most abundant component among the repetitive elements, accounting for 52.82% (Table 6). Gene annotation identified a total of 43,813 protein-coding genes (Table 2). The predicted proteins attained a complete BUSCO score of approximately 98.30%, which indicates a high quality of annotation (Table 4). A total of 42,972 (98.08%) protein-coding genes were successfully annotated in various databases, including Interpro, Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), SwissProt, Translation of European Molecular Biology Laboratory (Trembl), and NCBI non-redundant database (NR) (Table 7). A total of 4,351 non-coding RNA genes (i.e., 152 microRNAs [miRNAs], 770 transfer RNAs [tRNAs], 3074 ribosomal RNAs [rRNAs], and 355 small nuclear RNAs [snRNAs]) in the genome (Table 8).
Methods
Sample collection, DNA and RNA extractions
We collected fresh leaves, flowers, bark on tender branches and young fruits from a mature MN tree growing in the China National Botanical Garden (Beijing, China). The leaf samples were subjected to DNA isolation via a modified cetyltrimethylammonium bromide (CTAB) method, and all samples collected were subjected to RNA isolation using Trizol reagent (Invitrogen, CA, USA). The quality of DNA and RNA were examined using a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and agarose gel electrophoresis. DNA and RNA quantity was determined using the Qubit dsDNA HS Assay Kit on a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and a Bioanalyzer 2100 system (Agilent Technologies, CA, USA), respectively.
Preparation and sequencing of short insert libraries
For the DNA short insert libraries, a starting material of 1 μg DNA was used and sequencing libraries were constructed using the VAHTS Universal DNA Library Prep Kit for MGI (Vazyme, Nanjing, China). For the RNA-seq libraries, 1 μg RNA per sample served as the input material, with mRNA being isolated from the total RNA magnetic beads equipped with poly-T. Sequencing libraries were created using the VAHTS Universal V6 RNA-seq Library Kit for MGI (Vazyme, Nanjing, China). Unique index codes were incorporated to differentiate sequences among various samples. The quantification and sizing of these libraries were assessed with the Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Finally, both DNA and RNA libraries were sequenced on a MGI-SEQ. 2000 platform to generate 150-bp paired-end reads.
Preparation and sequencing of long insert libraries
For the DNA PacBio long insert sequencing, a SMRTbell library was constructed following the manufacturer’s protocol using the SMRTbell Express Template Prep kit 2.0 (Pacific Biosciences). Initially, 15 μg of genomic DNA was processed in the first enzymatic step to eliminate single-stranded overhangs, followed by application of repair enzymes to mend any damage along the DNA backbone. The ends of the double-stranded DNA fragments were smoothed and then extended to create an A-overhang. The ligation with T-overhang SMRTbell adapters was conducted at 20 °C for 60 minutes. The SMRTbell library was cleaned with 1 × AMPure PB beads. The library’s size distribution and concentration were evaluated with the FEMTO Pulse automated pulsed-field capillary electrophoresis device (Agilent Technologies, Wilmington, DE) and the Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). Following library characterization, 3 μg DNA underwent size selection with the BluePippin system (Sage Science, Beverly, MA), to eliminate SMRTbells shorter than 15 kb. The library was again purified with 1 × AMPure PB beads. The library’s size and quantity were reassessed using the FEMTO Pulse and the Qubit dsDNA HS reagents Assay kit. The sequencing primer and Sequel II DNA Polymerase were annealed and bound to the final SMRTbell library, respectively. The library was loaded at an on-plate concentration of 120 pM via diffusion loading. SMRT sequencing was executed using a single 8 M SMRT Cell on the Sequel II System, using the Sequel II Sequencing Kit and 1800-minute movies.
Hi-C Library construction and sequencing process
The Hi-C library was prepared according to a previous report8. In summary, samples were fixed by vacuum infiltration with 3% formaldehyde for 30 min at 4°C and then treated with 0.375 M glycine solution to quench the action for 5 min. The fixed samples were lysed, and endogenous nucleases were neutralized with 0.3% SDS. Chromatin DNA was digested using 100 U of MboI (NEB), labeled with biotin-14-dCTP (Invitrogen) and subsequently ligated with 50 U T4 DNA ligase (NEB). After reversing the cross-links, the ligated DNA was purified using the QIAamp DNA Mini Kit (Qiagen) as per the manufacturer’s guidelines. The extracted DNA was fragmented to 300–500 bp pieces, subjected to blunt-end repair, A-tailing and adaptor ligation, followed by purification via biotin-streptavidin pull-down and PCR amplification. The Hi-C libraries were then quantified and sequenced using the Illumina Nova-seq platform (San Diego, CA, USA).
Genome survey and analysis
After quality assessment and filtering, 40.02 Gb of clean DNA short reads were obtained for genome survey. The genome size, heterozygous ratio, and percentage of repetitive sequence were estimated using the GCE (v1.0.2)9 software with K-mer size of 17 bp and cleaned short reads.
Genome assembly
The yielding 29.42 Gb PacBio long high-fidelity (HiFi) reads were assembled to construct the draft genome using hifiasm (v0.16.1)10 with default parameters. The Hi-C reads were applied to assemble the contigs into 17 chromosomes using Juicer tools (v.1.6)11 and 3D-DNA12 based on Hi-C interaction data (70.74 Gb) (Fig. 1). Finally, JucieBox (v1.11.08)13 was used to manually adjusted the assembled genome. The BUSCO (Benchmarking Universal Single-Copy Orthologs) pipeline (v5.2.2)14 was used to assess the coverage of highly conserved genes to validate the completeness of the genome assembly with the embryophyte_odb10 dataset, which contains 1614 BUSCO gene sets.
Repetitive sequences annotation and telomeres identification
We used two methods to identify the repeat contents in the MN genome, combining de novo and homology-based prediction. For the homology-based analysis, we utilized RepeatMasker (v4.1.2; http://repeatmasker.org) with the Repbase TE library15 to detect known transposable elements (TEs) within the genome. Additionally, RepeatProteinMask (v4.1.2) was employed to search against the TE protein database. In terms of de novo prediction, we generated a custom repeat library for the genome using RepeatModeler (v2.0.2a; http://www.repeatmasker.org/RepeatModeler/), which automates the execute of RECON (v1.08)16 and RepeatScout (v1.0.5)17, two key tools for identifying, refining, and classifying potential interspersed repeats. Furthermore, LTR_FINDER (v1.0.7)18 was deployed for a de novo search of long terminal repeat (LTR) retrotransposons within the genome sequences. Tandem repeats were identified with the Tandem Repeat Finder (v4.10.0)19 and non-interspersed repeat sequences, such as low-complexity repeats, satellites and simple repeats, were detected using RepeatMasker. The repeat libraries from both methods were then integrated to ascertain the repeat content. Telomeric sequences in MN genome assembly were pinpointed using quarTeT (v1.0.3)20 with the “-c plant” option, revealing the telomere repeat monomer as TTTAGGG.
Prediction and functional annotation of protein-coding genes
We used three strategies to predict genes, including ab initio prediction, homology-based prediction, and transcriptome-based prediction, all applied to the repeat-masked, chromosome-scale genome. First, ab initio prediction was performed with Augustus (v3.3.1)21 and Genescan (v1.0)22 utilizing models trained on a curated set of high-quality proteins derived from RNA-Seq data. Second, the genome sequences were aligned with the protein sequences of seven plants, M. domestica, M. sieversii, M. sylvestris, M. prunifolia, Pyrus pyrifolia, Crataegus pinnatifida, and Prunus persica, and gene structures were predicted with Exonerate (v2.2.0)23 with default parameters. Third, for transcriptome-based gene prediction, the gene structure was built using PASA (v2.4.1)24. For Iso-Seq-based gene prediction, the Iso-seq reads were aligned to scaffolds using GMAP (v2017-11-15)25. The transcripts were used to predict Open Reading Frames (ORFs) using PASA, and full-length cDNA was screened as a training set. Finally, Maker (v3.00)26 synthesized the predictions from the three approaches to generate gene models. The output was a coherent and non-overlapping set of sequence assemblies that delineated the gene structures. Gene functions were assigned based on the highest scoring matches of the alignments to the National Center for Biotechnology Information (NCBI) Non-Redundant (NR), TrEMBL27, InterPro28 and Swiss-Prot27 protein databases using BLASTP (v2.6.0+)29 and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database30, all with an E-value threshold of 1E-5. Protein domains were annotated with InterProScan (v5.3574.0)31 based on InterPro protein databases. Pfam database32 was used to identify the motifs and domains within gene models. IDs of Gene Ontology (GO)33 were assigned to each gene by Blast2GO34. Non-coding RNA, including miRNA, tRNA, rRNA, and snRNA, were annotated using the following methods: tRNA were predicted using tRNAscan-SE (v1.3.1)35 with default parameters; rRNA were identified by mapping Arabidopsis thaliana rRNA sequences to the MN genome using BLASTN-short v2.2.28; miRNA and snRNA were analyzed using INFERNAL (v1.1.3)36 against the Rfam database with default parameters.
Data Records
The DNA sequence reads of MN (Experiments of DNA sequencing data from PacBio HiFi library: SRR3012738137; Experiments of DNA sequencing data from Hi-C library: SRR3014070838) have been deposited in the Sequence Read Archive (SRA) under BioProject accession PRJNA1143952. The genome assembly have been deposited in the GenBank database under the accession number JBHDYX00000000039. The files of the gene structure annotation, repeat predictions and gene functional annotation have been deposited at Figshare database (https://doi.org/10.6084/m9.figshare.26962936)40.
Technical Validation
We utilized a variety of methods to ascertain the accuracy and completeness of the MN genome assembly. First, the Hi-C heatmap demonstrated the accuracy of the genome assembly, with distinct Hi-C signals between the 17 pseudo-chromosomes, indicating their relative independence (Fig. 1). Second, the completeness and accuracy of the assembled genome was further substantiated by the benchmarking universal single-copy orthologues (BUSCO) analysis, which revealed that 1,590 complete plant orthologues (98.6%) were identified (Table 4). Third, the long terminal repeat (LTR) assembly index (LAI) score of the present assembly was 21.99, reaching the ‘gold standard’ level (LAI > 20). The accuracy was further corroborated by the very high mapping rates of two types of sequences to the MN assembly, with 99.32% of short insert reads and 99.95% of HiFi reads mapping to the MN assembly (Table 2). Finally, the chromosome telomere location map showed that the assembled genome extended to the telomeres, with the exception of chromosome 15, and that the majority of the chromosomes were assembled into double-ended telomeres (Fig. 2).
Code availability
There is no custom code was used during this study. All software and pipelines were executed according to the manual and protocols of the published bioinformatics tools. The version and code/parameters of software have been detailed and described in Methods.
References
Fiala, J. Flowering crabapples: the genus Malus. (Timber Press, Incorporated, 2003).
Lawrence, W. J. C. & Price, J. R. The genetics and chemistry of flower colour variation. Biol. Rev. 15, 35–57 (1940).
Glover, B. J. & Martin, C. Anthocyanins. Curr. Biol. 22, R147–R150 (2012).
Li, H. et al. Chromosomal level genome assemblies of two Malus crabapple cultivars Flame and Royalty. Sci. Data 11, 201 (2024).
Sun, X. et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nat. Genet. 52, 1423–1432 (2020).
Mansfeld, B. N. et al. A haplotype resolved chromosome‐scale assembly of North American wild apple Malus fusca and comparative genomics of the fire blight Mfu10 locus. Plant J. 116, 989–1002 (2023).
Li, Z. et al. Chromosome-scale reference genome provides insights into the genetic origin and grafting-mediated stress tolerance of Malus prunifolia. Plant Biotechnol. J. 20, 1015–1017 (2022).
Padmarasu, S., Himmelbach, A., Mascher, M. & Stein, N. In situ Hi-C for plants: an improved method to detect long-range chromatin interactions. in Plant long non-coding RNAs (eds. Chekanova, J. A. & Wang, H.-L. V.) 441–472 (Springer New York, New York, NY, 2019).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. (2013).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Robinson, J. T. et al. Juicebox.js provides a cloud-based visualization system for Hi-C data. Cell Syst. 6, 256–258.e1 (2018).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6 (2015).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 13, 1269–1276 (2003).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Hortic. Res. 26–28 (2023).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997).
Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6 (2005).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9 (2008).
Wu, T. D. & Watanabe, C. K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005).
Cantarel, B. L. et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370 (2003).
Mitchell, A. et al. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 43, D213–D221 (2015).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40 (2012).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Dwight, J. T. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Conesa, A. & Gotz, S. Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008, 1–12 (2008).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods in Mol. Biol. 1962, 1–14 (2019).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30127381 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30140708 (2024).
Wang, R. et al. Malus niedzwetzkyana isolate LD-2024a, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JBHDYX000000000 (2024).
Wang, R. Chromosome-level genome assembly of Malus niedzwetzkyana, the mother of Rosybloom crabapple. figshare https://doi.org/10.6084/m9.figshare.26962936 (2024).
Acknowledgements
This work was supported by the Beijing Natural Science Foundation (6224061), Beijing Municipal Administration Center of Parks (kjxx2021004, kjxx2023006, zx2022010) and the Open Fund of State Key Laboratory of Tree Genetics and Breeding (Chinese Academy of Forestry) (TGB-KF-2024-001).
Author information
Authors and Affiliations
Contributions
Ran He, Ling Guo, and Leiming Dong supervised the project. Hengxing Liu prepared the plant material. Ruizhen Wang, Jian Quan and Boyang Liu drafted the manuscript. Yu Wei prepared all figures and revised the manuscript. Ran He, Ling Guo, and Leiming Dong contributed to the discussions and improvement of the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, R., Quan, J., Liu, B. et al. Chromosome-level genome assembly of Malus niedzwetzkyana, the mother of Rosybloom crabapple. Sci Data 12, 211 (2025). https://doi.org/10.1038/s41597-024-04221-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-04221-z
This article is cited by
-
Chromosome-scale genome assembly of Flemingia macrophylla
Scientific Data (2025)
