
Abstracts
The large-scale loach (Paramisgurnus dabryanus; Cypriniformes: Cobitidae) is primarily distributed in East Asia. It is an important economic fish species characterized by fast growth, temperature-dependent sex determination and the ability to breathe air. Currently, molecular mechanism studies related to some aspects such as sex determination, toxicology, feed nutrition, growth and genetic evolution have been conducted. However, the lack of a high-quality reference genome has hindered further research. In this study, we performed PacBio HiFi and Hi-C sequencing on a female (ZW) specimen and assembled the first high-quality chromosome-level genome of the large-scale loach. The assembled genome comprises 24 chromosomes, with a total length of 1.04 Gb, a scaffold N50 of 41.7 Mb, and a BUSCO completeness of 95.8%, including 28,311 protein-coding genes. These findings not only provide new insights into the genome structure of the large-scale loach but also establish a crucial reference point for omics studies and serve as an essential genomic resource for breeding programs in this species.
Similar content being viewed by others


Background & Summary
The large-scale loach (Paramisgurnus dabryanus), belonging to the order Cypriniformes, family Cobitidae, and subfamily Cobitinae, is a small, benthic freshwater fish primarily distributed in East Asia. Highly enriched with protein, fats, minerals, and various vitamins, it is considered one of the most important aquaculture species due to its high nutritive and economical values1,2,3.
Karyotype analysis has shown that the sex chromosomes of large-scale loach follow a ZZ/ZW system, with females being heterogametic (ZW)4,5. This species exhibits sexual dimorphism in growth phenotypes, with females being larger and growing faster than males. Gonadal differentiation occurs 30 days post-fertilization in males and 45 days in females, with sexual maturity reached within six months. In addition to genotypic factors, temperature plays a crucial role in the sex determination of large-scale loach. Studies have shown that at 20 °C, the sex ratio remains 1:1, but at 25 °C, the proportion of males exceeds 70%, with this ratio gradually increasing as the temperature rises further, making large-scale loach an ideal model for studying temperature-dependent sex regulation6,7. Current research on sex determination focuses on the development of sex markers and the molecular mechanisms of temperature’s effect on sex determination. However, the lack of an available reference genome has hindered the identification of universal sex-specific molecular markers8, and research on the impact of temperature on sex determination remains in its early stages9,10. Although the mitochondrial genome is frequently employed in phylogenetic analysis11, evolutionary genomics12, and species identification13 of large-scale loach, its restricted genetic renders it unsuitable for the investigation of any economic trait. Assembling a chromosome-level reference genome of large-scale loach will not only aid in the detailed study of chromosomal structure but also provide a foundation for investigating the mechanisms underlying sex determination in this species.
In recent years, various omics technologies have played an important role in unraveling the complex molecular and genetic mechanisms of large-scale loach. However, due to the lack of a reference genome, researchers have often relied on de novo strategies, which focus on only a limited number of genes and cannot fully explore related mechanisms or reveal the species’ complex biological characteristics. For example, de novo transcriptomics has been used to explore the ammonia tolerance and detoxification mechanisms of this species14,15,16, identifying key regulatory genes that may be used in fish breeding17. Additionally, combined metabolomics and de novo transcriptomics analyses have preliminarily examined the effects of different protein and lipid ratios on growth performance and their molecular mechanisms18. Without genome support, these studies have been unable to fully elucidate the regulatory mechanisms involved. Furthermore, omics tools have been increasingly applied in studies related to growth1,19, pathogen infection and prevention20, environmental toxicology21,22,23, and interspecies hybridization24,25 of large-scale loach.
The publication of the genome will significantly advance omics studies of large-scale loach, providing a more comprehensive genetic background for identifying new gene functions and biological pathways, and contributing to future studies on systematics, evolutionary analysis, and genetic breeding.
In this study, we employed PacBio sequencing and Hi-C assembly technologies to construct a chromosome-level reference genome for large-scale loach. Based on the karyotype (2n = 48), 99.56% of the assembled contig sequences were successfully anchored to 24 chromosomes. The total genome assembly length was 1.04 Gb, with a scaffold N50 of 41.7 Mb and a BUSCO completeness score of 95.8%, containing 28,311 protein-coding genes, 97.49% of which were annotated. This genome provides crucial data for the development of molecular markers related to economically important traits such as growth and sex-specific traits, as well as a foundation for identifying key genes and elucidating molecular mechanisms.
Methods
Ethics statement
All experiments and animal treatments were carried out according to the principles of the Animal Care and Use Committee of the Institute of Hydrobiology, Chinese Academy of Sciences.
Sample collection and gynogenesis
A set of large-scale loaches was sourced from the Baishazhou Aquatic Product Market in Wuhan, Hubei Province, China. Jellyfish (v2.2.10)26 and GenomeScope (v2.0)27 were employed to analyze the frequency distribution of 21-kmer depths, estimating the average heterozygosity of the large-scale loach genome via ten randomly selected female samples. A sexually mature female with a heterozygosity of 1.44% was selected as the parent for gynogenetic induction through heat shock. This method inhibited the extrusion of the second polar body, resulting in gynogenetic progeny with lower heterozygosity. One female individual from the progeny was subsequently selected for further analysis (Fig. 1A). After anesthetization with MS222 (0.05%), blood and muscle tissue samples were immediately collected, flash-frozen in liquid nitrogen and stored at −80 °C for future analysis.

Genome survey and morphological characterization of gynogenetic offspring of the large-scale loach for genome assembly. (A) Dorsal view of the gynogenetic offspring of the large-scale loach. (B) The frequency distribution of 21-kmer depths.
Illumina sequencing and genome survey
Genomic DNA was extracted from both the gynogenetic parent and offspring using a modified cetyltrimethyl ammonium bromide (CTAB) protocol. Whole-genome resequencing was conducted using the paired-end (PE-150) strategy on the Illumina DNBSEQ-T7 platform. Additionally, whole-genome sequence data from ten female large-scale loaches from our previous study28 were incorporated for comparative genome survey analysis. GenomeScope (v2.0) was employed to analyze the data from the gynogenetic parent, offspring, and the ten female loaches. The results of the resequencing and genome survey analyses indicated a notable reduction in heterozygosity to 0.98% in the gynogenetic offspring, a figure that is significantly lower than that observed in the parent (1.44%) and the wild population (1.69%) (Table 1). The genome size of the large-scale loach was estimated at 0.94 Gb, based on 59.42 GB of clean sequencing data, closely aligning with the 1.04 Gb size obtained from the assembly (Fig. 1B).
Hi-C based and PacBio sequencing
Hi-C libraries were constructed following established protocols29. In brief, samples were cross-linked with 1% formaldehyde for 10 minutes at room temperature, then cool down with 0.125 M glycine for 5 minutes. The cross-linked cells were lysed and endogenous nucleases were inactivated with 0.3% SDS. Chromatin DNA was digested with 100U MboI (NEB), labeled with biotin-14-dCTP (Invitrogen), and ligated using 50U T4 DNA ligase (NEB). After reversing the cross-links, the ligated DNA was extracted using the QIAamp DNA Mini Kit (Qiagen) following the manufacturer’s instructions. The purified DNA was cut into 300–500 bp fragments, blunt-end repaired, A-tailed, and adaptors were added. Biotin-streptavidin pull-down followed by PCR amplification was then performed. The Hi-C libraries were quantified and sequenced on the MGISEQ-2000 platform (BGI, China).
For PacBio sequencing, a genomic library with inserts ranging from approximately 15 K to 20 K bp was prepared using the SMRTbell Express Template Prep Kit 3.0 (Pacific Biosciences, Menlo Park, CA, USA). Sequencing was conducted on the PacBio Sequel II platform (Pacific Biosciences, Menlo Park, CA, USA), following the manufacturer’s protocol. To process raw sequencing reads, default parameters of min passes = 3 and min RQ = 0.99 in the CCS software (https://github.com/PacificBiosciences/ccs) were applied to generate high-fidelity (HiFi) reads with quality exceeding Q20. After quality control and filtering, the final dataset comprised 65.55 GB of high-quality long reads (Table 2).
Genome assembly
We used HiFi data for contig assembly with hifiasm30 (v 0.15.2, parameters: --h1 --h2 -s 1 -10), resulting in haplotype A and haplotype B. Haplotype A assembly contained 1.05 Gb in 501 contigs with a contig N50 of 40.57 Mb, while haplotype B assembly contained 1.04 Gb in 374 contigs with a contig N50 of 45.64 Mb. Both haplotypes were scaffolded using Lachesis31 based on valid Hi-C data processed via HiC-Pro32 (v2.9.0), producing two chromosome-level haploid genomes, Haplotype A and Haplotype B. Haplotype B had a slightly higher mapping rate of short reads from second-generation sequencing compared to Haplotype A (Supplementary Table S1) and demonstrated higher completeness, making it the reference genome used for structural and functional annotation. Based on karyotype analysis, the assembled contigs were anchored to 24 chromosomes with a length of 1,038,659,332 bp, representing 99.56% of the total genome length. The final genome assembly was 1,043,469,091 bp in length, comprising 219 contigs, including both chromosomes and unanchored fragments (Table 3).
Repeat sequence annotation
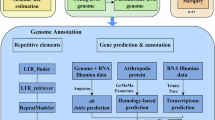
We identified repeat sequences and transposable elements (TEs) using the methods of de novo assembly33 and homologous prediction. First, we used RepeatModeler34 (v2.0.2) to predict the repeat sequence with default parameters. Then, RepBase database35 and RepeatMasker36 (v4.1.2) were used to annotate the sequence homologs. The results showed that 615.59 Mb are repeat sequences, accounting for 58.99% of the large-scale loach genome. Among these repeat sequences, most (37.74%) are DNA transposons, followed by 8.72% of unclassified elements and 3.86% of long terminal repeats (LTRs) (Table 4).
Coding gene prediction
We used three complementary approaches for coding gene prediction: (1) de novo prediction, homology-based prediction, and transcriptome-based prediction. De novo prediction: Augustus (v3.1.0, parameters: --species=zebrafish --gff3=on --strand=both)37, SnapGene (v2006-07-28)38, GeneID (v1.4), GlimmerHMM (v3.0.1), and GeneMarkES (v4.48)39 were employed. (2) Homology-based prediction: GeMoMa (v1.9)40 was used to predict homologous genes from zebrafish, common carp, and loach species. (3) Transcriptome-based prediction: We collected publicly available transcriptomic data (PRJNA26673941 and PRJNA62318919) along with self-generated data. Two assembly methods were employed: reference-guided assembly using Hisat2 (v2.1.0)42 and StringTie (v2.1.4)43, and de novo assembly using Trinity 40 (v2.1.1)44, followed by PASA (v2.4.1)45 to align the assembled transcripts back to the reference genome for gene prediction.
Finally, we integrated the predictions from these three methods using EVM (v1.1.1)46 and refined them with PASA, resulting in the identification of 28,311 genes (Table 5). Based on the annotation results, syntenic blocks across the 24 chromosomes were identified using MCScanX (https://github.com/wyp1125/MCScanx) with parameters set to -e 1e-10 and -s 5. A circular plot illustrating the distribution of gene and repeat density, GC content, and synteny within the genome was generated using TBtools-II47 (Fig. 2A).

The profiles of the genome structure and interactions among genes. (A) The circos plot showing the features of the large-scale loach genome. Tracks from outer to inner layers represent the 24 chromosomes, repeat elements density, gene density, GC content, and links of intragenomic syntenic blocks within 100 K bp sliding windows. (B) Hi-C interaction heatmap indicating the interactions among chromosomes.
Noncoding RNAs annotation
Non-coding RNAs, including tRNAs, rRNAs, miRNAs, snoRNAs, and snRNAs, were annotated using established tools. tRNAs were identified based on structural features using tRNAscan-SE (v2.0.12)48 with default parameters. rRNAs were predicted using RNAmmer (v1.2)49 (parameters: -S euk -m tsu, lsu, ssu) based on structural characteristics. miRNAs, snRNAs, and snoRNAs were predicted using covariance models from Rfam (v14.1)50 through INFERNAL (v1.1.4, parameters: -cut_ga -rfam -nohmmonly -fmt 6)51. In total, 2,660 miRNAs, 23,781 tRNAs, 10,126 rRNAs, 1,457 snRNAs, and 387 snoRNAs were annotated (Table 6).
Gene function annotation
Gene functions were annotated through comparisons with public databases, including UniProt (Swiss-Prot and TrEMBL), Pfam (The Protein Families Database), NR (Non-Redundant Protein Database), EggNOG-mapper, KEGG (Kyoto Encyclopedia of Genes and Genomes), KOG (Eukaryotic Orthologous Groups), and GO (Gene Ontology). Diamond (v2.1.8.162)52 was used to align the protein sequences predicted by EvidenceModeler with these databases, using an E-value cutoff of 1e−05. The best hits were retained and results from the seven databases were integrated. A total of 27,600 genes (97.49% of the 28,311 predicted protein-coding genes) were annotated, with 97.15%, 84.27%, 81.22%, 88.22%, 84.02%, 18.67%, and 20.23% of genes found in NR, UniProt, Pfam, EggNOG, KOG, KEGG, and GO, respectively (Table 7).
Chromosomal synteny analysis
To accurately evaluate genome structural features and validate assembly quality, we performed synteny analysis between the large-scale loach and two other loach species, the pond loach and plateau loach, both of which have available chromosome-level genomes. Using Last (v1559)53 and JCVI (v1.3.8)54, we identified syntenic gene pairs and homologous regions between each species’ genomes and visualized the results. The analysis revealed significant synteny among the three species’ genomes, with a high degree of chromosomal structural consistency between large-scale loach and both pond loach and plateau loach, further validating the quality of our genome assembly and annotation (Fig. 3).

Genome Synteny among Large-scale loach, Pond loach, and Plateau loach. The 17th chromosome, hypothesized to be the sex chromosome, is displayed as the final chromosome in the figure. The female-specific molecular marker Pd-WM1, located on the putative Z and W chromosomes, is shown on the right.
We used GeMoMa (v1.9) to align homologous genes between Haplotype A and Haplotype B, using Haplotype B as the reference. The two haplotypes exhibited good synteny (Supplementary Figure S1). In our previous studies, we identified a 383 bp sex-specific molecular marker (Pd-WM1) present only in females (NCBI accession no. PQ346371). Using primers designed from this sequence, we observed that in addition to the 383 bp band in females, both males and females exhibited a 544 bp band. Sequence alignment revealed that the longer band contained an insertion compared to the shorter band. BLAST alignment localized the two sequences uniquely to chromosome 17 of Haplotype A and Haplotype B, respectively, suggesting that this chromosome is the sex chromosome. Given the good synteny between the two chromosomes, we designated chromosome 17 of Haplotype B as the W chromosome and that of Haplotype A as the Z chromosome, consistent with the marker length characteristics (Fig. 3).
Data Records
Raw llumina short read, PacBio long read, and Hi-C sequencing data for generating genome assembly of the gynogenetic offspring and raw llumina short read of the gynogenetic female parent have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2024), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences55,56 under the accession number CRA01930557. RNA-seq data for annotating the large-scale loach assembly are available at Genome Sequence Archive (GSA) of NGDC the accession number CRA01889758. The chromosome-level genome assembly, based on Haplotype B, is available in NCBI under accession number GCA_030506205.259. Genome annotation files and Haplotype A assembly results have been deposited in the Figshare database60. All files related to assembly and annotation are also available at Aquatic Bioinformatics Archive61 .
Technical Validation
Quality evaluation of the genome assembly and annotation
The genome was evaluated for quality by mapping second-generation sequencing reads to the assembly using Bowtie2 (v2.1.0)62, yielding a mapping rate of 97.16%. For chromosome-level quality assessment, the Hi-C heatmap revealed strong interaction signals along the diagonal without significant noise in other regions (Fig. 2B), indicating high accuracy in chromosome assembly. Genome completeness was evaluated using BUSCO (v5.2.2, parameters: -c 10 -long -f)63 with the actinopterygii_odb10 database. The genome showed a BUSCO completeness score of 95.8%, including 94.2% single-copy and 1.6% duplicated genes (Table 8), which is slightly higher than those of the other two loach species, confirming the high completeness of our assembled genome.
Code availability
All commands and pipelines used for data processing followed the official manuals and protocols of the respective bioinformatics tools. No custom scripts or code were used. Specific parameters for each tool are provided in the Methods section, and default parameters were applied where not specified.
References
Wei, J., Chen, Y. & Wang, W. A high-density genetic linkage map and QTL mapping for sex and growth-related traits of large-scale loach (Paramisgurnus dabryanus). Front Genet 10, 1023, https://doi.org/10.3389/fgene.2019.01023 (2019).
You, C., Yu, X. & Tong, J. Detection of hybridization between two loach species (Paramisgurnus dabryanus and Misgurnus anguillicaudatus) in wild populations. Environ Biol Fishes 86, 65–71 (2009).
Li, K., Li, Y. C. & Zhou, D. A comparative study of the karyotypes in two species of mud loaches. Zool Res 4, 75–81 (1983).
Chang, Z. J. & Yu, Q. X. The cytogenetic evidences of ZZ/ZW sex determination in Paramisgurnus dabryanus. Hereditas 19, 17–19, https://doi.org/10.16288/j.yczz.1997.03.005 (1997).
You, C., Yu, X., Tan, D. & Tong, J. Gynogenesis and sex determination in large-scale loach Paramisgurnus dabryanus (Sauvage). Aquac Int 16, 203–214, https://doi.org/10.1007/s10499-007-9138-6 (2007).
Nomura, T., Arai, K., Hayashi, T. & Suzuki, R. Effect of temperature on sex rations of normal and gynogenetic diploid loach. Fish Sci 64, 753–758, https://doi.org/10.2331/fishsci.64.753 (1998).
Nan, P., Du, Q., Yan, S. & Chang, Z. Effects of temperature on sex differentiation of gonads and the cloning and expression of CYP19a in two species of loaches. Journal of Fishery Sciences of China 12, 407–413 (2005).
Xia, X., Zhao, J., Du, Q., Zhi, J. & Chang, Z. Cloning and identification of a female-specific DNA marker in Paramisgurnus dabryanus. Fish Physiol Biochem 37, 53–59, https://doi.org/10.1007/s10695-010-9415-6 (2011).
Xia, X. et al. Molecular cloning, characterization, and expression profiles of the Sox3 gene in Chinese loach Paramisgurnus dabryanus. Evol Dev 20, 108–118, https://doi.org/10.1111/ede.12252 (2018).
Xia, X. et al. Molecular cloning and mRNA expression pattern of Sox4 in Paramisgurnus dabryanus. Gene Expr Patterns 25–26, 109–117, https://doi.org/10.1016/j.gep.2017.06.008 (2017).
Zhu, M. & Liu, F. Mitochondrial genome sequence of Paramisgurnus dabryanus from the Yellow River estuary: implication for Cobitidae phylogeny. Mitochondrial DNA A DNA Mapp Seq Anal 27, 3039–3040, https://doi.org/10.3109/19401736.2015.1063124 (2016).
Dai, L. et al. Complete mitochondrial genome of Paramisgurnus dabryanus. Mitochondrial DNA A DNA Mapp Seq Anal 27, 1257–1259, https://doi.org/10.3109/19401736.2014.945545 (2016).
Liu, Y., Hou, J., Wang, G., Zhang, X. & Liu, H. A species-specific primer pair for distinguishing between Paramisgurnus dabryanus and Misgurnus anguillicaudatus based on mitochondrial DNA polymorphisms. Mitochondrial DNA A DNA Mapp Seq Anal 27, 2326–2329, https://doi.org/10.3109/19401736.2015.1022764 (2016).
Zhang, Y. L., Zhang, H. L., Wang, L. Y., Gu, B. Y. & Fan, Q. X. Changes of ammonia, urea contents and transaminase activity in the body during aerial exposure and ammonia loading in Chinese loach Paramisgurnus dabryanus. Fish Physiol Biochem 43, 631–640, https://doi.org/10.1007/s10695-016-0317-0 (2017).
Zhang, Y. L. et al. Partial Amino Acid Metabolism and Glutamine Synthesis as the Ammonia Defensive Strategies During Aerial Exposure in Chinese Loach Paramisgurnus dabryanus. Front Physiol 10, 14, https://doi.org/10.3389/fphys.2019.00014 (2019).
Shang, Z. H. et al. Transcriptomic analyses of the acute aerial and ammonia stress response in the gill and liver of large-scale loach (Paramisgurnus dabryanus). Comp Biochem Physiol C Toxicol Pharmacol 250, 109185, https://doi.org/10.1016/j.cbpc.2021.109185 (2021).
Huang, M., Shang, Z. H., Wu, M. X., Zhang, L. J. & Zhang, Y. L. Regulation of Rhesus glycoprotein-related genes in large-scale loach Paramisgurnus dabryanus during ammonia loading. Ecotoxicol Environ Saf 244, 114077, https://doi.org/10.1016/j.ecoenv.2022.114077 (2022).
Wang, Z. R. et al. Metabolomic and transcriptomic profiling reveals the effect of dietary protein and lipid levels on growth performance in loach (Paramisgurnus dabryanus). Front Immunol 14, 1236812, https://doi.org/10.3389/fimmu.2023.1236812 (2023).
Zhao, L. et al. Comparative transcriptome profiles of large and small bodied large-scale loaches cultivated in paddy fields. Sci Rep 11, 4936, https://doi.org/10.1038/s41598-021-84519-9 (2021).
Luo, Z. et al. First report of a disease caused by Bacillus cereus in cultured loach Paramisgurnus dabryanus. Dis Aquat Organ 147, 87–95, https://doi.org/10.3354/dao03629 (2021).
Wang, X. et al. Enrichment of polystyrene microplastics induces histological damage, oxidative stress, Keap1-Nrf2 signaling pathway-related gene expression in loach juveniles (Paramisgurnus dabryanus). Ecotoxicol Environ Saf 237, 113540, https://doi.org/10.1016/j.ecoenv.2022.113540 (2022).
Xia, X. et al. Reproductive toxicity and cross-generational effect of polyethylene microplastics in Paramisgurnus dabryanus. Chemosphere 313, 137440, https://doi.org/10.1016/j.chemosphere.2022.137440 (2023).
Xia, X. et al. Damage of polyethylene microplastics on the intestine multilayer barrier, blood cell immune function and the repair effect of Leuconostoc mesenteroides DH in the large-scale loach (Paramisgurnus dabryanus). Fish Shellfish Immunol 147, 109460, https://doi.org/10.1016/j.fsi.2024.109460 (2024).
Huang, S., Cao, X., Wang, W. & Nasr, M. Fertility and ploidy of gametes of allodiploid and allotriploid loaches produced by diploid Misgurnus anguillicaudatus females and Paramisgurnus dabryanus males. Fish Physiol Biochem 44, 13–20, https://doi.org/10.1007/s10695-017-0409-5 (2018).
Chen, Y. et al. Identification, expression and bioactivity of Paramisgurnus dabryanus β-defensin that might be involved in immune defense against bacterial infection. Fish Shellfish Immunol 35, 399–406, https://doi.org/10.1016/j.fsi.2013.04.049 (2013).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Zhang, L. et al. FishSNP: a high quality cross-species SNP database of fishes. Sci Data 11, 286, https://doi.org/10.1038/s41597-024-03111-8 (2024).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680, https://doi.org/10.1016/j.cell.2014.11.021 (2014).
Cheng, H. Y., Concepcion, G. T., Feng, X. W., Zhang, H. W. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 31, 1119–1125, https://doi.org/10.1038/nbt.2727 (2013).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16, 1–11, https://doi.org/10.1186/s13059-015-0831-x (2015).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile Dna 6, 1–6, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 25, 4.10. 11–14.10. 14, https://doi.org/10.1002/0471250953.bi0410s05 (2009).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Korf, I. Gene finding in novel genomes. BMC bioinformatics 5, 1–9, https://doi.org/10.1186/1471-2105-5-59 (2004).
Lomsadze, A., Burns, P. D. & Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res 42, e119–e119, https://doi.org/10.1093/nar/gku557 (2014).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol, 161-177, https://doi.org/10.1007/978-1-4939-9173-0_9 (2019).
Chen, J. et al. Analysis of multiplicity of hypoxia-inducible factors in the evolution of Triplophysa fish (Osteichthyes: Nemacheilinae) reveals hypoxic environments adaptation to Tibetan plateau. Front Genet 11, 433, https://doi.org/10.3389/fgene.2020.00433 (2020).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20, 1–13, https://doi.org/10.1186/s13059-019-1910-1 (2019).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652, https://doi.org/10.1038/nbt.1883 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, 1–22, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Chen, C. et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol Plant 16, 1733–1742, https://doi.org/10.1016/j.molp.2023.09.010 (2023).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res 49, 9077–9096, https://doi.org/10.1093/nar/gkab688 (2021).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35, 3100–3108, https://doi.org/10.1093/nar/gkm160 (2007).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res 49, D192–D200, https://doi.org/10.1093/nar/gkaa1047 (2021).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nature methods 18, 366–368, https://doi.org/10.1038/s41592-021-01101-x (2021).
Frith, M. C., Hamada, M. & Horton, P. Parameters for accurate genome alignment. BMC bioinformatics 11, 1–14, https://doi.org/10.1186/1471-2105-11-80 (2010).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488, https://doi.org/10.1126/science.1153917 (2008).
Chen, T. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genomics Proteomics Bioinformatics, https://doi.org/10.1016/j.gpb.2021.08.001 (2021).
Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2024. Nucleic Acids Res 52, D18-d32, https://doi.org/10.1093/nar/gkad1078 (2024).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA019305 (2024).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA018897 (2024).
Zhang, W. NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_030506205.2 (2024).
Zhang, L., Zhang, W. T., Shi, M. J. & Xia, X. Q. Chromosome-level genome assembly and annotation of the gynogenetic large-scale loach (Paramisgurnus dabryanus). figshare https://doi.org/10.6084/m9.figshare.27130323 (2024).
Aquatic Bioinformatics Archive, https://bioinfo.ihb.ac.cn/archive/PD_genome_v2 (2025).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, https://doi.org/10.1038/nmeth.1923 (2012).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molecular Biology and Evolution 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Acknowledgements
This work was supported by the grants from the Strategic Priority Research Program of the Chinese Academy of Sciences (Precision Seed Design and Breeding (XDA24010206), the National Key R&D Program of China (2021YFD1200804, 2018YFD0901201), the National Natural Science Foundation of China (31801055), the National Key R&D Program of China (2023YFD2401603), the Key R&D Program of Shandong Province, China (No.2023LZGC020) and the Foundation of State Key Laboratory of Mariculture Biobreeding and Sustainable Goods (No.BRESG202306).
Author information
Authors and Affiliations
Contributions
Xiao-Qin Xia, Mijuan Shi, and Lei Zhang conceived and designed the study. Lei Zhang and Wanting Zhang assembled the genome and conducted data analysis, and Lei Zhang drafted the manuscript. Yingyin Cheng, Yutong Fang, Xin Guan, Ao Gong, Yanxin Jiang,You Duan and Lei Huang prepared the fish samples. Waqar Younas and Yaping Wang reviewed and revised the manuscript. All authors contributed to editing and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, L., Zhang, W., Cheng, Y. et al. Chromosome-level genome assembly and annotation of the gynogenetic large-scale loach (Paramisgurnus dabryanus). Sci Data 12, 155 (2025). https://doi.org/10.1038/s41597-025-04498-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04498-8
