
Abstract
Yanbian cattle are a native cattle breed originating from the Yanbian Korean Autonomous Prefecture in Jilin Province, northeastern China. Developed over centuries through natural and artificial selection under cold climatic conditions, this breed is highly adapted to harsh environments, including low temperatures, rugged terrain, and sparse forage. Here, we constructed a high-quality chromosome-level genome assembly for Yanbian cattle using HiFi, Hi-C and RNA-seq data. The genome sequence was anchored to 30 chromosomes, with a total genome length of 2.8 Gb, a contig N50 of 86.41 Mb and a scaffold N50 of 111.08 Mb. Short-read data showed the average 99.56% mapping ratio to the assembly, validating base-level accuracy. Also, 93% complete BUSCOs verified the integrity of the assembled genome. 51.94% repetitive elements of the genome and 20,421 protein-coding genes were annotated. This Yanbian cattle genome serves as an indispensable resource for bovine genomic studies and local breed conservation, enabling both evolutionary insights and genetic characteristic analyses.
Similar content being viewed by others

Background & Summary
Yanbian cattle (Bos taurus), locally referred to as Yanbian yellow cattle, represent one of China’s five premier indigenous cattle breeds. Native to northeastern China, this breed exhibits exceptional adaptive traits including cold tolerance, disease resistance, and remarkable roughage utilization efficiency1,2. In recent years, Yanbian cattle have gained increasing agricultural and commercial importance due to their superior meat quality characteristics - particularly their highly marbled, tender beef with distinctive flavor profiles - meeting the growing demand for premium beef products in China’s evolving market3.
Current research has identified several candidate genes associated with the breed’s distinctive phenotypes. Studies have revealed key genetic factors (such as CORT, FGF5, and CD36) contributing to cold climate adaptation2, while polymorphisms in CAPN1 have been linked to meat quality traits4. However, existing investigations have primarily relied on the Hereford cattle reference genome5, leaving significant gaps in our understanding of Yanbian-specific genetic variations and their molecular mechanisms. The absence of a Yanbian-specific cattle reference genome substantially limits comprehensive genomic studies. Therefore, a complete, high-quality Yanbian cattle genome assembly would be essential, enabling precise identification of breed-specific genes, regulatory elements, and functional DNA regions, thereby facilitating deeper investigation into the genetic basis of its valuable traits.
In this study, we presented the first high-quality chromosome-level genome assembly of Yanbian cattle, generated through PacBio HiFi circular consensus sequencing (106.7 Gb) and high-throughput chromosome conformation capture (Hi-C) scaffolding (341.51 Gb raw data) combined with RNA-seq short reads6,7,8. The final assembly comprises 30 chromosomes (scaffold N50 ~111.08 Mb), represented by 59 contigs (contig N50 ~86.41 Mb), covering 97.45% of the original genome length. Notably, 17 chromosomes were completely assembled without gaps, demonstrating exceptional assembly continuity. The assembly also exhibits high completeness, with 93% of BUSCO genes identified and minimal missing (5.1%) or fragmented (2%) sequences. In summary, the first Yanbian cattle assembly (YB_JAAS) provides new insights into mining the specific genetic variation information of Yanbian cattle and in-depth understanding of its origin, domestication and development.
Methods
Blood samples and tissues collection
Blood samples were collected from a 10-year-old male Yanbian cattle from Yanbian Livestock Development Corporation, in Yanji city, Jilin Province for HiFi genome and HiC sequencing, supplemented by whole-genome resequencing of five 20-month-old Yanbian cattle from the same region. Tissue samples (heart, liver, spleen, lung, kidney, muscle, small intestine, and rumen) from one 4-year-old Yanbian cattle were snap-frozen in liquid nitrogen and processed by Wuhan Frasergen Bioinformatics Co., Ltd for RNA-seq.
DNA and RNA extraction
High-quality genomic DNA was extracted using a modified CTAB method9. Briefly, whole blood samples were lysed in 4 × CTAB buffer containing β-mercaptoethanol, incubated at 65 °C for 1.5 h, and then cooled to room temperature. DNA was extracted with chloroform-isoamyl alcohol (24:1) and precipitated with ethanol. Total RNA was extracted using Trizol reagent (Invitrogen, CA, USA). RNA purity and integrity were assessed using a NanoDrop spectrophotometer and an Agilent 2100 Bioanalyzer, while degradation was evaluated by 1.5% agarose gel electrophoresis.
Genome sequencing
The HiFi sequencing was conducted on the PacBio Revio platform, leveraging the SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences) for library construction. Original sequencing data were processed via SMRTlink and ccs tools to split subreads and generate HiFi reads, which referred to those sequences with full passes the same ZMW (Zero-Mode Waveguide) >3 and an accuracy consensus sequences >99%. The final output consisted of 6,503,032 high-quality reads, accumulating to 106.7 Gb of sequence data at an average length of 16.41 kb. (Table 1).
To resolve scaffolding ambiguities and improve assembly quality, we employed a combined approach integrating Hi-C data with PacBio HiFi long-read sequencing. Hi-C libraries were prepared using conventional methods10. Chromatin was cross-linked with 1% formaldehyde (10 min, RT), quenched with 0.125 M glycine, and lysed. After SDS treatment (0.3%) to inactivate nucleases, chromatin was digested with MboI (100U), biotin-labeled, and ligated using T4 DNA ligase (50U). Cross-links were reversed and DNA purified (QIAamp DNA Mini Kit). The DNA was sheared (300–500 bp), end-repaired, A-tailed, and adapter-ligated. Biotinylated fragments were enriched via streptavidin pull-down, PCR-amplified, and sequenced (BGI PE150). Finally, 341.51 Gb Hi-C raw data were generated. After trimming adaptors and filtering low-quality reads using Trimmomatic11, we obtained 330.78 Gb clean reads (Table 2). Subsequent bioinformatic analyses were based on clean data.
For genome annotation support, polyadenylated mRNA was enriched from 1 μg total RNA using oligo(dT) magnetic beads. RNA-seq libraries were prepared with the VAHTS Universal V6 kit and sequenced in paired-end mode on a DNBSEQ-T7 system. Finally, the RNA-seq experiment yielded 26.23 Gb of high-quality paired-end reads for further genome annotation.
Genome assembly
Filtered HiFi reads were assembled into contigs using hifiasm v0.19.8-r603, yielding a contig-level genome12. 2,215,797,704 clean Hi-C reads were aligned to the ARS-UCD2.0 reference genome using Juicer with default parameters to construct scaffold-level assembly (Table 2)13,14. Heterozygous and contaminated contigs were filtered out using Juicer13,15. We finally anchored the scaffolds on 30 chromosomes (29 autosomes plus X) and successfully gained a Yanbian cattle assembly with total length up to 2,849,678,991 bp (Fig. 1, Table 3). The contig N50 and scaffold N50 are 86.41 Mb and 111.08 Mb respectively. The Hi-C assistant assembly result was shown in Table 3. Notably, most chromosomes had only 1 contig, meaning that the assembled genome has good integrity with lots of 0 gaps (Table 3)16.

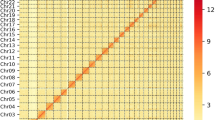
Genome-wide Hi-C interaction map with 500k resolution. The colors in the figure, from light to dark, indicate the increase in the intensity of the interaction. The darker the color, the stronger the interaction. The horizontal and vertical coordinates represent its N*bin position on the genome. The first 30 squares in the picture represent the 30 chromosomes of cattle.
Assembly quality assessment
The assembled genome was assessed for completeness through BUSCO analysis with OrthoDB as the reference17. Overall, 93.0% complete BUSCOs were identified in YB assembly (Table 4). In addition, the genome sequences mounted by Hi-C were sorted according to the reference genome sequences using Mummer to perform a collinearity analysis. The result indicated good collinearity with the reference genome (Fig. 2). Moreover, we mapped the whole genome resequencing data of 5 Yanbian cattle to YB_JAAS genome. This analysis illustrated the high mapping ratio with an average mapping ratio of 99.56% and an average properly mapping ratio of 98.22% (Table 5). All the evidence confirmed the high quality of our assembled genome (YB_JAAS).

The mapping result of collinearity analysis using Mummer. The horizontal axis represents the reference genome, and the vertical axis represents the genome after Hi-C mounting. The red line indicates the forward matching of the sequence, and the blue line indicates the reverse complementary matching.
Annotation of repetitive sequences
To comprehensively characterize the repetitive elements in the YB_JAAS genome, we employed an integrated approach combining homology-based and de novo prediction strategies. For homology-based identification, known transposable elements (TEs) were annotated using RepeatMasker (version 4.1.2) with the Repbase TE database as a reference library18,19,20. Additionally, RepeatProteinMask (Revision 1.36) was implemented to detect repetitive elements using a curated TE protein database19. For de novo prediction, RepeatModeler was employed to build a de novo genome-specific (YB_JAAS) repeat library, which captured both known and novel repetitive elements including TEs, low-complexity regions, and unclassified repeats by integrating two complementary algorithms RECON21 and RepeatScout22. To further improve LTR retrotransposon detection, we performed a specialized search using LTR_FINDER (v1.0.7)22,23. Tandem repeats were identified using the Tandem Repeat Finder (TRF)24, while low-complexity repeats, satellites and simple repeats were annotated via RepeatMasker20. Finally, after integrating the libraries derived from both homology-based and de novo approaches and performing a comprehensive repeat annotation, 51.94% of the whole genome sequences were defined as repetitive sequences, achieving a complete characterization of repetitive elements in the YB_JAAS genome (Table 6). A total of 1.32 Gb sequences were identified as combined TEs, accounting for 45.22% of the whole genome length (Table 7). Of all the types in TEs, LINE sequences were annotated most, reaching 31.91% of the whole genome (Fig. 3).

The classification statistics of TEs.
Gene annotation
To comprehensively identify protein-coding genes, we applied three prediction methods to the YB_JAAS genome: ab initio modeling, homology-based alignment and RNA-Seq-guided annotation. For homology-based prediction, we use tblastn to align the sequences of proteins encoded by five known related species including wild yak (Bos mutus), zebu (Bos indicus), water buffalo (Bubalus bubalis), American bison (Bison bison) and Banteng (Bos javanicus) to the YB_JAAS genome25, and utilize Exonerate to predict the gene structure26. Comparative analysis revealed conserved GC content distributions in both genes and coding sequences relative to these species, while demonstrating increased exon and introns number in the YB_JAAS genome (Fig. 4). Augustus (v3.3.1) and Genescan were adopted to perform ab initio gene prediction27,28. To accurately determine the splicing sites and exon regions, we first assembled clean RNA-Seq reads into transcripts using trinity29, and the gene structure were formed using PASA30. Lastly, MAKER (v3.00) was used to integrate the gene set predicted by various methods into a non-redundant and more complete gene set31. Then PASA combined with transcriptome data was used to update the gene structure30. The gene prediction results obtained by different kinds of software are elaborated in Table 8. Finally, the genome annotation identified 20,421 protein-coding genes, with a median gene length of 48.43 kb. Exon-intron structure analysis revealed an average of 9.85 exons per gene, with exon lengths averaging 322.13 bp and introns spanning 5,040.47 bp on average (Table 8). The average CDS length is 1,688.59 bp (Table 8).

The comparison results of gene characteristics between YB_JAAS (Bos_taurus) with wild relatives.
Functional annotation of protein-coding genes
Functional predictions were derived from sequence similarity analyses using BLASTP (v2.6.0 + )32,33 against curated protein databases (NCBI NR, TrEMBL34, Swiss-Prot34) and domain databases (InterPro35), supplemented with KEGG36 pathway mapping, applying a conservative e-value threshold of 1e-5 for all alignments. Gene Ontology (GO) IDs for each gene were obtained from Blast2GO37. In total, approximately 19,880 (about 97.35%) of the predicted protein-coding genes of YB_JAAS genome could be functionally annotated (Table 9) with InterPro, GO, KEGG, SwissProt, TrEMBL, and NR databases covering 85.08%, 84.25%, 96.42%, 93.99%, 95.54%, and 97.24%, respectively. Within the GO database, the cellular component category contained the highest number of annotations (91,115 terms) and genes (11,567 genes), followed by molecular function (82,501 terms, 11,147 genes) and biological process (56,524 terms, 9,543 genes) (Table S1). The KEGG database annotated 56.54% of genes into Brite Hierarchies, primarily associated with genetic information processing, metabolism, and signaling and cellular processes (Table S2). Among pathways, Human Diseases represented the largest proportion (16.21%), followed by Organismal Systems (15.16%). Other categories such as Environmental Information Processing, Cellular Processes, Metabolism and Genetic Information Processing comprised ~10% each (Table S2).
Annotation of non-coding RNA genes
According to the structural characteristics of tRNA, tRNAscan-SE was used to find the tRNA sequences in the genome38. RNAmmer (version 1.2) was carried out to predict rRNA in the genome39. In addition, the covariance model of Rfam (http://xfam.org/) family and the INFERNAL software provided by Rfam could be used to predict miRNA and snRNA sequence information on the genome40,41. Finally, we identified a total of 269,330 tRNAs, 2,946 rRNAs (including 14 18S, 14 28S and 2,9185S), 982 miRNAs and 1,795 snRNAs (including 199 CD-box, 364 HACA-box, 1,201 splicing and 31 scaRNA). The annotation results of various types of non-coding RNAs were summarized in Table 10. Among these types, tRNA accounted for the largest proportion, up to 67.49% of the whole genome (Table 10).
Ethics statement
The blood and tissues samples were obtained from Molecular Breeding Research Laboratory, Institute of Animal Husbandry and Veterinary Medicine, JAAS, China. This study has been approved by the Institutional Animal Ethics Committee of Jilin Academy of Agricultural Sciences (JAAS). The Number of Permit is JNK20221218-02.
Data Records
The whole genome sequence data in this paper have been deposited in the Genome Warehouse in National Genomics Data Center42,43, Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation, under accession number (SAMC4847615, PRJCA037127) that is publicly accessible at NGDC Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/92360/show (2025)44, also presented at DDBJ/ENA/GenBank under the accession JBPCCZ00000000045. The raw resequencing and RNA-seq data reported in this paper have been deposited in the Genome Sequence Archive46 in National Genomics Data Center43 China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA023772) that are publicly accessible at NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA023772 (2025)47 and SRA database in NCBI with accession SRP591175 (https://www.ncbi.nlm.nih.gov/sra/SRP591175)48. The genome annotation file has been shown in Figshare database (https://doi.org/10.6084/m9.figshare.29310821)49.
Technical Validation
Based on the single copy homologous gene set in OrthoDB, BUSCO was used to predict these genes and calculate their integrity, fragmentation, and possible loss rate. Thus, the integrity of gene regions in the overall assembly result was assessed. Our genome assembly (YB_JAAS) achieved a BUSCO completeness score of 93% (vertebrata_odb10), with 91.4% single-copy and 1.6% duplicated genes. The fragmented BUSCOs genes and missing BUSCOs genes were 66 and 170, respectively (Table 4). In addition, the mapped results using the whole genome sequences demonstrated extremely high mapping ratio, averaging 99.56% (Table 5). Functional annotations covered ~97.35% of genes by six databases (Table 9). These analyses all confirmed the high continuity and completeness of YB_JAAS.
Code availability
The software including the version number, parameters and source website used in this study are listed in the supplementary table (Table S3).
References
Nawaz, A. et al. Fatty acid profiles unveiled: gene expression in Yanbian yellow cattle adipose tissues offers new insights into lipid metabolism. Arch Anim Breed 67(4), 469–480 (2024).
Shen, J. et al. Whole Genome Scan and Selection Signatures for Climate Adaption in Yanbian Cattle. Front Genet 11, 94 (2020).
Ji, S. et al. Differential Expression of PPARgamma, FASN, and ACADM Genes in Various Adipose Tissues and Longissimus dorsi Muscle from Yanbian Yellow Cattle and Yan Yellow Cattle. Asian-Australas J Anim Sci 27(1), 10–18 (2014).
Xin J. et al. Association of Polymorphisms in the Calpain I Gene with Meat Quality Traits in Yanbian Yellow Cattle of China. Asian-Australasian Journal of Animal Sciences 2011, 24.
Rosen B. D. et al. De novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience 2020, 9(3).
Burton J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 2013, 31(12):1119-1125.
Wei Z. et al. Chromosome-level genome assembly and annotation of the Yunling cattle with PacBio and Hi-C sequencing data. Scientific Data 2024, 11(1).
Qiu S., Wu N., Sun X., Xue Y., Xia J.: Chromosome-level genome assembly of soybean aphid. Scientific Data 2025, 12(1).
Porebski, S., Bailey, L. G. & Baum, B. R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Molecular Biology Reporter 15(1), 8–15 (1997).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159(7), 1665–1680 (2014).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15), 2114–2120 (2014).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18(2), 170–175 (2021).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Systems 3(1), 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356(6333), 92–95 (2017).
Forcato, M. et al. Comparison of computational methods for Hi-C data analysis. Nature Methods 14(7), 679–685 (2017).
Ben Zouari, Y., Molitor, A. M., Sikorska, N., Pancaldi, V. & Sexton, T. ChiCMaxima: a robust and simple pipeline for detection and visualization of chromatin looping in Capture Hi-C. Genome Biology 20(1), 102 (2019).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol Biol 1962, 227–245 (2019).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and Genome Research 110(1-4), 462–467 (2005).
Saha, S., Bridges, S., Magbanua, Z. V. & Peterson, D. G. Computational Approaches and Tools Used in Identification of Dispersed Repetitive DNA Sequences. Tropical Plant Biology 1(1), 85–96 (2008).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics Chapter 4, 4.10.11–14.10.14 (2009).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res 12(8), 1269–1276 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), i351–358 (2005).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35(suppl_2), W265–W268 (2007).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27(2), 573–580 (1999).
Gertz, E. M., Yu, Y.-K., Agarwala, R., Schäffer, A. A. & Altschul, S. F. Composition-based statistics and translated nucleotide searches: Improving the TBLASTN module of BLAST. BMC Biology 4(1), 41 (2006).
Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6(1), 31 (2005).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research 34(suppl_2), W435–W439 (2006).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. Journal of molecular biology 268(1), 78–94 (1997).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29(7), 644–652 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31(19), 5654–5666 (2003).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18(1), 188–196 (2008).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17), 3389–3402 (1997).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10(1), 421 (2009).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31(1), 365–370 (2003).
Mitchell, A. et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res 43(Database issue), D213–221 (2015).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40(Database issue), D109–114 (2012).
Conesa, A. & Götz, S. Blast2GO: A Comprehensive Suite for Functional Analysis in Plant Genomics. International Journal of Plant Genomics 2008(1), 619832 (2008).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25(5), 955–964 (1997).
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H. & Rognes, T. Ussery DW: RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35(9), 3100–3108 (2007).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Research 33(suppl_1), D121–D124 (2005).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29(22), 2933–2935 (2013).
Ma Y. et al. The Updated Genome Warehouse: Enhancing Data Value, Security, and Usability to Address Data Expansion. Genomics, proteomics & bioinformatics 2025.
Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res 2022, 50(D1):D27-d38.
National Genomics Data Center (NGDC). Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/92360/show (2025).
NCBI GenBank http://identifiers.org/ncbi/insdc:JBPCCZ000000000 (2025).
Chen, T. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genomics, proteomics & bioinformatics 19(4), 578–583 (2021).
National Genomics Data Center (NGDC). Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA023772 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP591175 (2025).
Fang, W. et al. De novo genome assembly of Yanbian cattle using PacBio HiFi and Hi-C combined with RNA-seq data. YB_JAAS. figshare. https://doi.org/10.6084/m9.figshare.29310821 (2025).
Acknowledgements
The authors acknowledge the funding support received from Agricultural Science and Technology Innovation Project of Jilin Province (CXGC2023TD103) and National Beef Cattle and Yak Industrial Technology System (CARS-37).
Author information
Authors and Affiliations
Contributions
Y. Cao, Y. Zhao and W. Fang conceived the project; Y. Liu, J. Wu, L. Miao, C. Xiao, X. Li and X. Huang collected the samples; W. Fang and Y. Cao analyzed the data; W. Fang finished the original manuscript. Y. Cao and Y. Zhao revised the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fang, W., Cao, Y., Liu, Y. et al. De novo genome assembly of Yanbian cattle using PacBio HiFi and Hi-C combined with RNA-seq data. Sci Data 12, 1399 (2025). https://doi.org/10.1038/s41597-025-05689-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05689-z
