
Abstract
Micro RNAs (miRNAs, miRs) and relevant networks might exert crucial functions during differential host cell infection by the different Leishmania species. Thus, a bioinformatic analysis of microarray datasets was developed to identify pivotal shared biomarkers and miRNA-based regulatory networks for Leishmaniasis. A transcriptomic analysis by employing a comprehensive set of gene expression profiling microarrays was conducted to identify the key genes and miRNAs relevant for Leishmania spp. infections. Accordingly, the gene expression profiles of healthy human controls were compared with those of individuals infected with Leishmania mexicana, L. major, L. donovani, and L. braziliensis. The enrichment analysis for datasets was conducted by utilizing EnrichR database, and Protein–Protein Interaction (PPI) network to identify the hub genes. The prognostic value of hub genes was assessed by using receiver operating characteristic (ROC) curves. Finally, the miRNAs that interact with the hub genes were identified using miRTarBase, miRWalk, TargetScan, and miRNet. Differentially expressed genes were identified between the groups compared in this study. These genes were significantly enriched in inflammatory responses, cytokine-mediated signaling pathways and granulocyte and neutrophil chemotaxis responses. The identification of hub genes of recruited datasets suggested that TNF, SOCS3, JUN, TNFAIP3, and CXCL9 may serve as potential infection biomarkers and could deserve value as prognostic biomarkers for leishmaniasis. Additionally, inferred data from miRWalk revealed a significant degree of interaction of a number of miRNAs (hsa-miR-8085, hsa-miR-4673, hsa-miR-4743-3p, hsa-miR-892c-3p, hsa-miR-4644, hsa-miR-671-5p, hsa-miR-7106-5p, hsa-miR-4267, hsa-miR-5196-5p, and hsa-miR-4252) with the majority of the hub genes, suggesting such miRNAs play a crucial role afterwards parasite infection. The hub genes and hub miRNAs identified in this study could be potentially suggested as therapeutic targets or biomarkers for the management of leishmaniasis.
Similar content being viewed by others


Introduction
Leishmaniasis is a widespread tropical and subtropical disease, caused by various species of Leishmania spp., which are transmitted through the bite of the female Phlebotomine sandflies1,2,3. The estimated epidemiological range of 12 million cases with an addition of 2 million new annual cases, poses a substantial threat of leishmaniasis to public health. It is noteworthy that the reported leishmaniasis cases are concentrated in more than 98 countries across all continents4. Leishmaniasis presents with a diverse range of clinical manifestations, encompassing cutaneous leishmaniasis (CL), causing non-lethal skin lesions, and visceral leishmaniasis (VL), commonly called kala-azar, affecting internal organs, leading to pancytopenia, hepatomegaly, and splenomegaly5. Leishmania major, L. tropica, L. aethiopica, and L. donovani, L. infantum have been recognized as the main causative agents of CL and VL, respectively6.
Leishmania protozoan parasites employ different and sophisticated evasion strategies dealing with the host’s immune responses including the inhibition of complement system and modulating anti-parasitic activity of immune T-cells. Furthermore, these intracellular parasites by expressing crucial parasite proteins are able to affect downstream proteins and consequently affect and change the host cell signaling pathways and responses to favor the parasite survival and pathogenesis persistence7,8. In this sense, small and long non-coding RNAs are considered as biomolecules involved in many cell regulatory networks (through regulating gene expression) during parasite infections9,10. Thus, such pivotal biomolecules can impact key biological processes including immunity and pathogenesis processes of different parasitic diseases such as leishmaniasis11.
microRNAs (miRNAs, miRs) are small endogenous ncRNAs implicated in post-transcriptional regulation by binding to the 3’ UTR of target mRNA, leading to mRNA degradation or translational inhibition12. They exert a modulatory influence on diverse biological processes including cell proliferation, differentiation, and immune response regulation13,14. Consequently, expression and modulation of miRNAs can be implicated in the development of pathogenesis processes of several human diseases including parasitic infections. Therefore, the identification of such biomarkers could provide novel therapeutic and diagnostic prospects for the management of leishmaniasis15. Furthermore, unique miRNAs are directly implicated in spike glycoprotein production upon coronavirus (COVID) vaccination which might also open new avenues to promote protective signals during nano-vaccine development in parasitic diseases such as leishmaniasis16. There is a great deal of studies that identified various and crucial miRNAs (miRNAs expressed in both parasite and infected-host cells) during leishmaniasis infection17,18.
Given the significance of accurate molecular diagnosis in the context of leishmaniasis, this study aims to discover the crucial genes and miRNAs linked to Leishmania spp. infection by conducting a meta-analysis of expression microarray datasets using bioinformatic techniques. Additionally, the interaction of the miRNA-hub genes implicated in leishmaniasis along with their associated signaling pathways will also be investigated.
Materials and methods
Collection and selection of eligible gene expression datasets
In the discovery step, we identified first microarray datasets that compared gene expression between leishmaniasis patients and healthy controls in different tissues and cell types. Microarray data were retrieved from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) database with “Leishmania”, “Expression profiling by array”, “microarrays”, and “Homo sapiens” as keywords. During the second step, the selection process for qualified studies and datasets, we followed a number of strict criteria including human case–control studies, gene expression profiling analysis, comparable test conditions, and the downloadable of complete raw and processed microarray data. Other clinical covariates, including age, sex, and therapeutic status were not available for all samples, therefore, to avoid the introduction of false positives by imputation, have not been included. Disqualifying factors for studies included the use of cell lines, exclusive reliance on Real-Time PCR (RT-PCR) profiling, absence of case–control design, and investigation of specific factors' impact on leishmaniasis. The datasets and references that met the aforementioned requirements were all manually reviewed14,19.
Data extraction and processing
For each dataset, the series matrix file was downloaded and processed in several steps including background correction, log2 transformation, and quantile normalization which were performed using R language v4.2.2 and GEO2R analysis software. We used the R packages including Limma, GEOquery, BiocGenerics, Biobase, parallel, reshape, reshape2, ggplot2, grid, plyr, dplyr, data.table, sva, and affy from Bioconductor to process the data. After computation, all datasets related to Leishmania infections were merged. During this step, the expression data were mean-centered and reduced to the number of common probes across all data sets20. The gene expression data of the merged datasets were batch-adjusted using the ComBat method, implemented in the sva package v3. Assessing the success of batch correction was confirmed by boxplots.
Differential gene expression screening
For every dataset, analysis to identify differentially expressed genes (DEGs) were developed. Limma package (https://bioconductor.org/packages/release/bioc/html/limma.html), which applies linear models to examine the expression patterns of individual genes, was used to obtain the required tools to analyze DEGs with T test. False discovery rate (FDR) was calculated using Benjamini & Hochberg method. The FDR cut-off value for DEGs was below 0.01. By calculating adjusted p values and fold changes (FC), we identified genes which exhibited differential expression, defined as those with an adjusted p value less than 0.05 and |log FC|> 1.5. Additionally, Heatmap was generated for each dataset using the Python packages (version 3.11) to represent significant DEGs.
Enriched gene ontology (GO) and pathways analysis
Gene ontology servers as a comprehensive resource for the high-throughput annotation of biological functions. This ontology framework encompasses three fundamental biological dimensions, including the biological process (BP), molecular function (MF), and cellular component (CC). Here, GO enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of DEGs were conducted using the Enrich database (https://maayanlab.cloud/Enrichr/) (www.kegg.jp/kegg/kegg1.html)21. To ensure the statistical validity, selection criteria were utilized, which included the presence of at least three genes within a cluster with a GO tree interval ranging from 3 to 8 and a kappa score of 0.4 for pathway network connectivity22. Consequently, any P value less than 0.05 was considered to possess statistical significance.
Clustering gene expression data
Python libraries including matplotlib, and seaborn were imported to cluster DEGs and visualize the results. We used hierarchical clustering, a potent technique for examining high throughput expression data. Python determined the degree of similarity among the genes in each set of data, displayed the expression value using colors, and then clustered the genes.
Protein–protein interaction (PPI) network construction, cluster networks, and identification of hub genes
The Search Tool for the Retrieval of Interacting Genes (STRING) database, available at (http://string-db.org), was utilized to establish a Protein–Protein Interaction (PPI) network. Gene co-expression analysis was subsequently performed on the network, employing a confidence score threshold of more than 0.4 for GSE42088, GSE43661, GSE63931, GSE64610, and GSE69252 datasets and 0.7 for GSE55664 dataset23. In order to identify hub genes, a protein–protein interaction (PPI) network was constructed using the software Cytoscape 3.7.1 (http://www.cytoscape.org). Genes that met the following cut-off thresholds were identified as hub-genes: Degree > 10, K core > 2, and max depth > 100. The top 10 hub genes were determined using cytoHubba, employing four algorithms based on closeness, betweenness, and connectivity (degree) methods14. The final selection of hub genes was made by finding the intersection between the sets of hub genes obtained from all three algorithms. In addition, the Molecular Complex Detection (MCODE) tool was used to visually examine clusters within the PPI network. The MCODE analysis involved specific parameter settings, including a degree cutoff of 2, a node score cutoff of 0.2, a k-core value of 2, and a maximum depth of 100. Genes exhibiting the highest MCODE scores were identified as potential hub genes. In order to determine the hub genes that were shared among different methods and analysis, a Venn diagram was employed (http://bioinformatics.psb.ugent.be/webtools/Venn/). This Venn diagram facilitated the identification of common hub genes across various approaches.
Hub gene validation
The predictive effects of hub genes on infection establishment were evaluated by employing receiver operating characteristic (ROC) curve analysis. The diagnostic value of the hub genes was compared by calculating the area under the ROC curve (AUC). The MedCalc version 22 was used to perform this analysis24.
Evaluation of miRNAs-hub genes interaction network
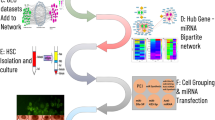
In order to determine the specific miRNAs associated with the hub genes, we utilized various resources including the miRTarBase database (https://www.mirtarbase.cuhk.edu.cn), miRWalk (https://mirwalk.umm.uni-heidelberg.de/), and TargetScan (https://www.targetscan.org/). In addition to evaluating miRNAs through gene-miRNAs interaction, a comprehensive review of literature studies was also conducted. Furthermore, the miRNet database (https://www.mirnet.ca/) was employed to generate a graphical depiction of the interactions between miRNAs and the hub genes. The steps of microarray analysis approach are outlined in Fig. 1, providing a visual representation of the entire pipeline.

The flowchart of microarray analysis approach towards biomarker discovery in Leishmania infections. Six gene expression datasets were downloaded from GEO, and the differentially expressed genes (DEGs) in leishmaniasis patients and healthy controls with an adjusted P value < 0.05 and a |log fold change (FC)|> 1.5 were first identified by GEO2R or R language version 4.2.2. Next, Gene Ontology (GO) and Kyoto Encyclopaedia of Genes and Genomes (KEGG) analysis (www.kegg.jp/kegg/kegg1.html) were performed for enrichment analysis of these DEGs. Then, the hub genes were identified by the cytoHubba plugin and the other bioinformatics approaches including protein–protein interaction (PPI) network analysis, and miRNA-hub gene network construction was also performed. This approach promotes a comprehensive understanding of parasite infection and for biomarker discovery useful for early diagnosis.
Results
Dataset’s characteristics
Our prespecified criteria led to the identification of six datasets, namely GSE69252 and GSE43661 datasets which were based on the GPL6244 platform (Affymetrix Human Gene 1.0 ST Array), while GSE42088 was analyzed using the GPL70 platform (Affymetrix Human Genome U133 Plus 2.0 Array), GSE64610 was based on the GPL16025 platform (NimbleGen Homo Sapiens Expression Array), GSE63931 was evaluated by GPL17077 platform (Agilent-039494 SurePrint G3 Human GE v2 8 × 60K Microarray 039,381), and GSE55664 was related to GPL10558 platform (Illumina HumanHT-12 V4.0 expression beadchip). Table 1 displays the fundamental characteristics of the datasets that were assimilated. Moreover, the microarray analysis employed diverse types of cell sorts which clearly depicted in Fig. 2.

Details of the recruited datasets related to each species of Leishmania along with the sort of the studied cells. Datasets belong to different cells of the different species of Leishmania.
Identification of DEGs
The DEGs were attained through the examination of six microarray datasets in correlation with four distinct Leishmania species. The results of our mRNA analysis represent a total of 528 DEGs (475 upregulated, 53 downregulated) for GSE55664, 107 DEGs (70 upregulated, 37 downregulated) for GSE42088, 187 DEGs (63 upregulated, 124 downregulated) for GSE43661, 71 DEGs (58 upregulated,13 downregulated) for GSE63931, 118 DEGs (108 upregulated, 10 downregulated) for GSE64610, and 67 DEGs (63 upregulated, 4 downregulated) for GSE69252. Table 2 presents a categorical arrangement of the genes that have been up-regulated and down-regulated, which were detected as remarkably significant in the microarray examination.
Gene ontology
The current investigation performed a GO enrichment analysis on DEGs to uncover the fundamental biological pathways of leishmaniasis caused by different Leishmania spp. The findings of GO clarified that, DEGs are significantly enriched in immunological process, functions and, pathways such as inflammatory response, cytokine-mediated signaling pathway, granulocyte and neutrophil chemotaxis, etc. which is demonstrated in Table 3. The results of enrichment showed that most of the DEGs were involved in cellular response to cytokine stimulus (GO:0071345), cellular response to interferon-gamma (GO:0071346), inflammatory response (GO:0006954), cytokine-mediated signaling pathway (GO:0019221), abnormal macrophage physiology (MP:0002451), decreased CD8-positive, alpha–beta T cell number (MP:0008079), increased CD4-positive, alpha beta T cell number (MP:0008074), and also pathways including rheumatoid arthritis, cytokine–cytokine receptor interaction and, chemokine signaling pathway (Fig. 3).

Enrichment analysis associated with DEGs obtained from the Enricher database. Green nodes represent the genes while pink nodes display gene ontology terms, purple nodes show pathways and mammalian phenotype data displayed by orange node.
Construction of PPI network and identification of central hub genes
PPI networks serve as numerical depictions of the actual physical interactions that occur among proteins within the cell. The results of the number of nodes and edges of the examined datasets are listed in Table 4. The ultimate selection of hub genes was established by identifying the intersection between the sets of hub genes obtained from three algorithms (Betweenness, closeness, and degree) and the resultant of MCODE25. The findings indicated that SOCS3, TNFAIP3, JUN and, TNF were the hub genes identified for GSE42088. In addition, CCR7 and IDO1 were recognized as the hub genes that were identified for GSE69252. Our results shown that CXCL8, CXCL9, and CXCL10 are hub genes in GSE55664. Additionally, CXCL9, CXCL10, and FCGR3A represent the hub genes identified in relation to GSE63931. Moreover, VEGFA, IL1B and TNF were found as hub genes for GSE64610. Finally, LRRK2, HSPA1B, CXCL9, and, RPL13 recognized to be the hub genes for GSE43661 dataset (Fig. 4 and Table 5).

The overlap of hub genes between all analyzed datasets is shown by Venn diagram. Accordingly, there are no common genes between all of these datasets.
Validation of hub genes
By utilizing the ROC curve (AUC > 0.6) to assess the prognostic value of hub genes in the PPI network26, 5 out of 14 hub genes, namely, TNF, SOCS3, JUN, TNFAIP3, and CXCL9 showed potential indications as potential infection biomarkers (Table 6). However, the remaining genes, were not validated, probably owing to the limited number of samples. To ensure the accuracy and reliability of these genes as potential biomarkers, conducting further investigations using larger sample groups is highly recommended. The area under the ROC curve (AUC) reflects diagnostic value of the test (Fig. 5).

This figure showed the evaluation of sensitivity and specificity of hub genes in the diagnostic of the infection.
Identification of miRNA-hub genes interaction
Based on the result obtained from miRWalk, hsa-miR-8085, hsa-miR-4673, hsa-miR-4743-3p, hsa-miR-892c-3p, hsa-miR-4644, hsa-miR-671-5p, hsa-miR-7106-5p, hsa-miR-4267, hsa-miR-5196-5p, hsa-miR-4252 display a substantial degree of interaction with the majority hub genes which candidate them as principal miRNA signatures of cell specific infections (Fig. 6 and Table 7).

The interaction network between miRNAs and hub genes in miRWalk. Blue nodes represent miRNAs, while orang node displays hub genes.
In this study, in addition to evaluating miRNAs through gene-miRNAs interaction, we also conducted a comprehensive review of literature studies. The reviewed studies are presented in Table 8. Based on the comparison between the results of the present study and previous ones, some miRNA hubs play an important role during Leishmania spp. infection and probably during disease progression.
Discussion and conclusion
Leishmaniasis, a parasitic illness caused by protozoan parasites, is commonly found in several regions of the tropics, subtropics, and southern Europe32. It is an infectious disease caused by Leishmania parasites and transmitted through the bite of phlebotomine sand flies, is classified as a neglected tropical disease (NTD) with a number of associated risk factors33. The diagnosis and treatment of this particular parasitic infection, which is caused by a variety of Leishmania parasite species, presents considerable challenges34. Consequently, the discovery of reliable biomarkers or molecular targets if including therapeutics offers enormous potential for improving disease management and patient outcomes35.
In this article, we explored the gene expression profile of individuals infected with various Leishmania spp. through analyzing multiple microarray datasets, in order to uncover potential biomarkers such as genes and miRNAs for Leishmania infections, highlighting their implications for diagnosis, treatment monitoring, and future research directions. Accordingly, the abnormal expression of miRNAs could serve as effective and non-invasive biomarkers for diagnosing and predicting various disorders, including infectious diseases18.
Since the raw data of GSE64610 dataset included the total data of both species, we reported the total number of genes. Although in our study the number of infected (97) and normal (54) samples were not in numerical balance, this issue could not affect the final statistical balance and we obtained acceptable data concerning DEG's. In addition, we rigorously filtered the obtained DEG's and determined a limited number of final hub genes. Accordingly, other studies have also recruited the same conditions. For example, in Li et al.’s study36, the number of infected samples (33) and normal (18) samples were not in numerical balance. Hence, the obtained DEGs (108 upregulated, 10 downregulated) are actually the sum of the DEGs in samples infected with L. donovani and L. major. Some of these genes have had expression changes in L. donovani and some in L. major.
Our study has identified the TNF gene as a potential hub gene in the context of differential Leishmania infections. This gene has a significant impact on the immune response during Leishmania infection, mainly by controlling the production and activation of pro-inflammatory cytokines, including TNF-α37. Evidences demonstrated a notable increase in TNF-α concentration in the serum of patients across all age groups with CL when compared to their respective control groups38,39. The function of TNF-α in Leishmania infections is a subject of debate, but many studies suggest that moderate amounts of TNF-α can aid in the clinical outcome of Leishmania infection by stimulating T-helper 1 (Th1) response40. Nevertheless, high levels of this cytokine can lead to tissue damage and the formation of persistent lesions. Despite the existence of numerous polymorphisms within the TNF gene locus, it is not yet certain whether these polymorphisms have a direct impact on TNF-α levels41. This suggests that the TNF gene might be linked to how susceptible people are to leishmaniasis. However, further research is needed to fully understand the role of the TNF gene in leishmaniasis42.
Additionally, SOCS3 was another potential hub gene in this context. SOCS3, a gene known for regulating the immune responses in various infections, plays a crucial role in L. major infection. Mathematical modeling revealed that the critical role of the SOCS1/SOCS3 ratio in bolstering the early immune response43. This ratio was quantified both computationally and experimentally, signified an essential immune axis that regulates macrophage phenotypes during L. major infection. Notably, SOCS1's ability to inhibit the JAK/STAT1 signaling pathway, leading to the suppression of pro-inflammatory cytokine expression44, positions it as a promising candidate for therapeutic intervention in leishmaniasis. Studies have demonstrated that transgenic mice with increased levels of SOCS3 gene expression exhibit heightened airway responsiveness, indicating that the expression of SOCS3 in CD4+ T cells encourages Th2-dependent responses like allergic reactions45. Additionally, it has been discovered that excessive SOCS3 expression in T cells also contributes to the advancement of leishmaniasis by promoting a dominant IL-4 response. Furthermore, abnormal IL-4 production in the initial stages of infection is responsible for the disease progression in transgenic SOCS3 mice46.
JUN as another potential hub gene, is known to play a pivotal role in regulating immune responses to bacterial infections by modulating the expression of critical cytokines and chemokines47. While there is no direct evidence of JUN ‘s role in the immune response to leishmaniasis, its involvement in the regulation of the immune response to other infections suggests it might also affect how our body responds to leishmaniasis48. In addition, in our study, there is evidence to suggest that the vascular endothelial growth factor (VEGF) plays a role in the pathogenesis of Leishmania infection specially during lymphangiogenesis, or the formation of new blood vessels. It is also involved in the regulation of immune responses and has been implicated in the development and progression of several diseases, including cancer and infectious diseases49. Several studies have investigated the relationship between VEGF gene and Leishmania infection50. Tiffani et al. have shown that macrophages are the predominant cell type expressing VEGF-A during L. major infection. Given that Leishmania parasites activate hypoxia-inducible factor 1α (HIF-1α) and this transcription factor can drive VEGF-A expression51. These findings also suggest that targeting VEGF signaling may be a potential therapeutic strategy for the treatment of leishmaniasis52.
The immune response to Leishmania infection is complex and involves a variety of immune cells and biomolecules. One important differentially activated immune gene in this response is CXCL9, a chemokine codifying gene that is expressed by infected macrophages leading to the recruitment of T cells to the site of infection and inflammation been thus implicated in a number of disease conditions like in severe diseases and also in favoring M1 macrophage polarization upon infections53. In laboratory studies on visceral leishmaniasis, infected kupffer cells quickly release CCL2, CCL3, and CXCL10. These molecules attract inflammatory monocytes and T cells, leading to the formation of a granuloma54. There is a direct relationship between the severity of splenic damage and the levels of CXCL9, CXCL10, IFN-γ, and IL-10 in the blood during VL. This suggests that the immune system responds to high levels of parasitic activity by producing both pro-inflammatory and regulatory molecules to control parasitemia. The host may use this strategy to limit parasite growth during VL55. This process plays a crucial role in initiating an immune response against the parasites. Several studies have investigated the role of CXCL9 in leishmaniasis. In a study by Gomes et al., CXCL9 levels were found to be elevated in individuals with cutaneous leishmaniasis, suggesting that it may be a useful biomarker for monitoring disease progression and treatment efficacy56. Similarly, a study by de Brito et al. found that CXCL9 levels were elevated in individuals with cutaneous leishmaniasis, and that treatment with pentoxifylline drugs led to a decrease in CXCL9 levels57. Overall, the literature suggests that CXCL9 plays an important role in the immune response against leishmaniasis, and may be a useful biomarker for monitoring disease progression and treatment efficacy58. In addition, targeting this molecule for macrophage polarization modulation might be a possible therapeutic strategy.
TNFAIP3, also known as A20, was identified as a gene whose expression is rapidly induced by the Tumor Necrosis Factor (TNF). This gene encodes a ubiquitin-editing enzyme, which inhibit NF-kappa B activation as well as TNF-mediated apoptosis. TNFAIP3 protein is involved in the cytokine-mediated immune and inflammatory responses59,60. According to a previous investigation, Leishmania species inhibit the expression of NLRP3 inflammasome, which is a multiprotein signaling platform. This, in turn, inhibits the activation of caspase-1 and the maturation of IL-1β. The reduction of NLRP3 and pro-IL-1β during infection is due to a decrease in NF-κB activity. This decrease in NF-κB activity is linked to an increase in the expression of TNFAIP1, which is a negative regulator of NF-κB signaling61. In another study, increased transcript abundance was observed in L. amazonensis infection for TNFAIP362. Overall, RG1 was highly upregulated in VL (L. donovani) cases, and suppressed TLR-triggered proinflammatory responses through elevated reactive oxygen species stimulating TNFAIP363. In line with these studies, our study also showed a significant increase in the expression of the TNFAIP3 gene in patients compared to the control groups.
miRNAs are a type of non-coding RNA that have a vital role in regulating various cellular functions64. Their modulation and abnormal expression have been associated with the development of pathogenesis processes of several disorders, including infectious diseases such as leishmaniasis. Therefore, gaining a thorough understanding of how miRNAs interact with their targets could provide valuable insights into the underlying mechanisms of these diseases15.
Some research demonstrated a notable upregulation of miR-302b-3p, miR-372-3p, miR-373-3p, and miR-607 in THP-1 cells in response to intracellular parasitism compared to the healthy controls31. Additionally, differential miRNA expression profiles were observed in L. (V.) braziliensis promastigote-infected THP-1 cells, including let7i-5p, miR-30e-5p, miR-302a-3p, miR-302b-3p, and miR-34c-5p30. Furthermore, based on some studies, the upregulation of hsa-miR-146, miR-106, miR-324, miR-221, miR-9, and miR-155 in infected patients have the potential to downregulate the IFN-γ signaling pathway, contributing to disease progression during Post-Kala-Azar Dermal Leishmaniasis (PKDL)65. Our meta-analysis employing accessible datasets was able to clarify and identify putative biomarkers related to infection. As a result, the identification of five prospective gene biomarkers, such as TNF, SOCS3, JUN, TNFAIP3, and CXCL9, that are differently expressed in Leishmania infected cells, offers possibilities as clinically diagnostic biomarkers for an accurate identification of Leishmania infected individuals. Furthermore, protein interaction results revealed dysregulated biochemical pathways (cellular response to immunologic stimulus and response to inflammatory stimulus) that might be useful as therapeutic targets. The validation of these biomarkers and metabolic pathways by experimental and functional researches might provide future interesting prospects for the management of leishmaniasis.
We evaluated gene enrichment analysis to find the relevant pathways associated with Leishmania infection. Leishmaniasis is recognized for triggering an intricate immune reaction that encompasses both innate and adaptive immune responses. We have discovered that DEGs are enriched in pathways connected to the regulation of immune response, such as signaling pathways for cytokines (such as TNF, IL-12, IFN-gamma), NF-kappa B signaling, toll-like receptors and IL-17 signaling pathways. Hints of the immune system having an important function in fighting against leishmaniasis infection66. IL-17 is a cytokine with proinflammatory properties, primarily secreted by activated T cells, specifically CD4+ cells over CD8+ cells. This cytokine has been implicated in various inflammatory human diseases, such as rheumatoid arthritis and psoriasis67. In addition to T cells, IL-17 also stimulates other cell types, such as macrophages, to generate inflammatory mediators like TNFα, IL-1, and chemokines. These events ultimately result in the recruitment of neutrophils68. Previous studies have demonstrated that the increased recruitment of neutrophils, dependent on IL-17, into lesions of BALB/c mice infected with L. major, significantly contributes to the outcome of the disease69. Also increased levels of IL-17A in BALB/c mice infected with L. major, were linked to higher production of IL-23 by infected DC70.
Furthermore, we found that DEGs significantly enriched in pathways such as neutrophil/ granulocyte chemotaxis and migration. Neutrophils serve as the initial defense against infection and are promptly summoned to the infection site once the parasite infiltrates the host. While they possess the capability to engulf the Leishmania parasites, their capacity to eliminate the parasites remains limited. Besides, neutrophils release antimicrobial peptides and reactive oxygen species to aid in the management of the infection. Neutrophil and macrophages both utilize autophagy to degrade and recycle cellular component, including pathogens71. Furthermore, the enrichment of cellular components such as “Azurophil granule”, “Lysosome”, and “Cytolytic granule” may indicate an enhanced neutrophil response to the infection, highlighting the importance of these cells in the immune defense against Leishmania.
In conclusion, our meta-analysis employing accessible datasets was able to clarify and identify putative biomarkers related to cell specific Leishmania infections. As a result, since key immune responses can be considered as potential correlates of immunity against infectious pathogens thus having potential diagnostic value, the identification of five prospective immune response genes that are differently expressed in the infected cells, such as TNF, SOCS3, JUN, TNFAIP3, and CXCL9, offers possibilities to serve as biomarkers to identify Leishmania infected individuals. Furthermore, protein interaction research revealed major pathways linked to probable biological events such as various dysregulated biochemical pathways (cellular response to immunologic stimulus and response to inflammatory stimulus). Whit the aim to select key genes and miRNAs regulating the pathogen-host interactions and thus potentially useful for disease management particularly at the early stages of inflammatory responses, we have here approached in silico tools that predict the biological roles of human genes and miRNAs. Of course, all these predictions have to be well validated as in many (it not almost all) initial in silico studies. A thorough functional driven investigation of these biomarkers with well controlled samples including different cells and Leishmania spp. will provide new interesting research papers to improve disease management. As a summary, it seems that we need to go deeper, particularly in the study of microRNAs. The validation of these biomarkers and metabolic pathways by experimental and functional research might fully confirm the current study’s findings.
Data availability
The authors confirm that the data supporting the findings of this study is available within the article.
References
Scott, P. & Novais, F. O. Cutaneous leishmaniasis: immune responses in protection and pathogenesis. Nat. Rev. Immunol. 16, 581–592 (2016).
dos Santos Meira, C. & Gedamu, L. Protective or detrimental? Understanding the role of host immunity in leishmaniasis. Microorganisms 7, 695 (2019).
Mann, S. et al. A review of leishmaniasis: Current knowledge and future directions. Curr. Trop. Med. Rep 8, 121–132 (2021).
Nagle, A. S. et al. Recent developments in drug discovery for leishmaniasis and human African trypanosomiasis. Chem. Rev. 114, 11305–11347 (2014).
Torres-Guerrero, E., Quintanilla-Cedillo, M. R., Ruiz-Esmenjaud, J. & Arenas, R. Leishmaniasis: A review. F1000Res 6, 750 (2017).
Roatt, B. M. et al. Recent advances and new strategies on leishmaniasis treatment. Appl. Microbiol. Biotechnol. 104, 8965–8977 (2020).
Gurung, P. & Kanneganti, T. D. Innate immunity against Leishmania infections. Cell. Microbiol. 17, 1286–1294 (2015).
de Freitas e Silva, R. & Von Stebut, E. Unraveling the role of immune checkpoints in leishmaniasis. Front. Immunol. 12, 620144 (2021)
Bayer-Santos, E., Marini, M. M. & da Silveira, J. F. Non-coding RNAs in host-pathogen interactions: subversion of mammalian cell functions by protozoan parasites. Front. Microbiol. 8, 474 (2017).
Rashidi, S. et al. miRNAs in the regulation of mTOR signaling and host immune responses: the case of Leishmania infections. Acta Trop. 231, 106431 (2022).
Kataria, P., Surela, N., Chaudhary, A. & Das, J. MiRNA: Biological regulator in host-parasite interaction during malaria infection. Int. J. Environ. Res. Public Health 19, 2395 (2022).
Riolo, G., Cantara, S., Marzocchi, C. & Ricci, C. miRNA targets: From prediction tools to experimental validation. Methods Protoc. 4, 1 (2020).
Vafadar, A. et al. Long non-coding RNAs as epigenetic regulators in cancer. Curr. Pharm. Des. 25, 3563–3577 (2019).
Hashemi, K. S., Aliabadi, M. K., Hemmati, A. A. & Alashti, S. K. A meta-analysis of microarray datasets to identify biological regulatory networks in alzheimer’s disease. Front. Genet. 14, 1225196 (2023).
Paul, S. et al. Human microRNAs in host-parasite interaction: A review. 3 Biotech 10, 510 (2020).
Seneff, S., Nigh, G., Kyriakopoulos, A. M. & McCullough, P. A. Innate immune suppression by SARS-CoV-2 mRNA vaccinations: The role of G-quadruplexes, exosomes, and MicroRNAs. Food Chem. Toxicol. 164, 113008 (2022).
Acuña, S. M., Floeter-Winter, L. M. & Muxel, S. M. MicroRNAs: Biological regulators in pathogen-host interactions. Cells 9, 113 (2020).
Ghalehnoei, H., Bagheri, A., Fakhar, M. & Mishan, M. A. Circulatory microRNAs: Promising non-invasive prognostic and diagnostic biomarkers for parasitic infections. Eur. J. Clin. Microbiol. Infect. Dis. 39, 395–402 (2020).
Muka, T. et al. A 24-step guide on how to design, conduct, and successfully publish a systematic review and meta-analysis in medical research. Eur. J. Eepidemiol. 35, 49–60 (2020).
Zetterberg, H., Andreasen, N. & Blennow, K. Increased cerebrospinal fluid levels of transforming growth factor-β1 in Alzheimer’s disease. Neurosci. Lett. 367, 194–196 (2004).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
Bindea, G. et al. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 (2009).
Yu, J. & Finley, R. L. Jr. Combining multiple positive training sets to generate confidence scores for protein-protein interactions. Bioinformatics 25, 105–111 (2009).
Hajian-Tilaki, K. Receiver operating characteristic (ROC) curve analysis for medical diagnostic test evaluation. Caspian J. Intern. Med. 4, 627 (2013).
Bader, G. D. & Hogue, C. W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 4, 2 (2003).
Tang, Y. et al. Hub genes associated with the diagnosis of diabetic retinopathy. Int. J. Gen. Med. 1739–1750 (2021).
Rashidi, S. et al. Highlighting the interplay of microRNAs from Leishmania parasites and infected-host cells. Parasitology 148, 1434–1446 (2021).
Kumar, A. et al. Differential regulation of miRNA profiles of human cells experimentally infected by Leishmania donovani isolated from Indian visceral leishmaniasis and post-Kala-Azar dermal leishmaniasis. Front. Microbiol. 11, 1716 (2020).
Varikuti, S., Verma, C., Natarajan, G., Oghumu, S. & Satoskar, A. R. MicroRNA155 plays a critical role in the pathogenesis of cutaneous Leishmania major infection by promoting a Th2 response and attenuating dendritic cell activity. Am. J. Pathol. 191, 809–816 (2021).
Souza, Md. A. et al. miR-548d-3p alters parasite growth and inflammation in Leishmania (Viannia) braziliensis infection. Front. Cell. Infect. Microbiol. 11, 687647 (2021).
Ramos-Sanchez, E. M. et al. miR-548d-3p is up-regulated in human visceral leishmaniasis and suppresses parasite growth in macrophages. Front. Cell. Infect. Microbiol. 12, 110 (2022).
Ready, P. Leishmaniasis emergence in Europe. Euro Surveill. 15, 19505 (2010).
Cecílio, P., Cordeiro-da-Silva, A. & Oliveira, F. Sand flies: Basic information on the vectors of leishmaniasis and their interactions with Leishmania parasites. Commun. Biol. 5, 305 (2022).
Kobets, T., Grekov, I. & Lipoldova, M. Leishmaniasis: Prevention, parasite detection and treatment. Curr. Med. Chem. 19, 1443–1474 (2012).
Rostami, M. N. & Khamesipour, A. Potential biomarkers of immune protection in human leishmaniasis. Med. Microbiol. Immunol. 210, 81–100 (2021).
Li, J.-X. et al. Screening of potential hub genes involved in Cutaneous Leishmaniasis infection via bioinformatics analysis. Acta Trop. 236, 106645 (2022).
Bosch-Nicolau, P. et al. Leishmaniasis and tumor necrosis factor alpha antagonists in the Mediterranean basin. A switch in clinical expression. PLoS Negl. Trop. Dis. 13, 0007708 (2019).
Khudhur, H. R. & Alomashi, G. B. Effect of NRAMP1 gene polymorphism on levels of (TNF-α1and IL-1β) cytokines in cutaneous Leishmaniasis patients in Iraq. J. Immunol. Clin. Microbiol. 3, 15–22 (2018).
Oliveira, F. et al. Lesion size correlates with Leishmania antigen-stimulated TNF-levels in human cutaneous leishmaniasis. Am. J. Trop. Med. Hyg. 85, 70–73 (2011).
Mirzaei, A., Maleki, M., Masoumi, E. & Maspi, N. A historical review of the role of cytokines involved in leishmaniasis. Cytokine 145, 155297 (2021).
Kirik, F. E., Ülger, M., Tezcan Ülger, S. & Aslan, G. Association of cytokine gene polymorphisms with susceptibility to cutaneous leishmaniasis in a Turkish population. Parasite Immunol. 42, e12775 (2020).
Sette, A., Sidney, J. & Crotty, S. T cell responses to SARS-CoV-2. Annu. Rev. Immunol. 41, 343–373 (2023).
Veras, P. S. T., Ramos, P. I. P. & De Menezes, J. P. B. In search of biomarkers for pathogenesis and control of leishmaniasis by global analyses of Leishmania-infected macrophages. Front. Cell. Infect. Microbiol. 8, 326 (2018).
Liau, N. P. et al. The molecular basis of JAK/STAT inhibition by SOCS1. Nat. Commun. 9, 1558 (2018).
Seki, Y.-I. et al. SOCS-3 regulates onset and maintenance of TH2-mediated allergic responses. Nat. Med. 9, 1047–1054 (2003).
Nakaya, M. et al. Aberrant IL-4 production by SOCS3-over-expressing T cells during infection with Leishmania major exacerbates disease manifestations. Int. Immunol. 23, 195–202 (2011).
Salih, M. et al. Expression profiling of Sudanese visceral leishmaniasis patients pre-and post-treatment with sodium stibogluconate. Parasite Immunol. 39, e12431 (2017).
Atayde, V. D. et al. Leishmania exosomes and other virulence factors: Impact on innate immune response and macrophage functions. Cell. Immunol. 309, 7–18 (2016).
Weinkopff, T. et al. Leishmania major infection-induced VEGF-A/VEGFR-2 signaling promotes lymphangiogenesis that controls disease. J. Immunol. 197, 1823–1831 (2016).
Fraga, C. A. C. et al. Immunohistochemical profile of HIF-1α, VEGF-A, VEGFR2 and MMP9 proteins in tegumentary leishmaniasis. An. Bras. Dermatol. 87, 709–713 (2012).
Weinkopff, T., Roys, H., Bowlin, A. & Scott, P. Leishmania infection induces macrophage vascular endothelial growth factor A production in an ARNT/HIF-dependent manner. Infect. Immun. 87, e00088-e119 (2019).
Gioseffi, A. et al. Leishmania-infected macrophages release extracellular vesicles that can promote lesion development. Life Sci. Alliance 3, e202000742 (2020).
Laidlaw, B. J., Craft, J. E. & Kaech, S. M. The multifaceted role of CD4+ T cells in CD8+ T cell memory. Nat. Rev. Immunol. 16, 102–111 (2016).
Stanley, A. C. & Engwerda, C. R. Balancing immunity and pathology in visceral leishmaniasis. Immunol. Cell Biol. 85, 138–147 (2007).
Singh, N. & Sundar, S. Inflammatory chemokines and their receptors in human visceral leishmaniasis: Gene expression profile in peripheral blood, splenic cellular sources and their impact on trafficking of inflammatory cells. Mol. Immunol. 85, 111–119 (2017).
Gomes, R. & Oliveira, F. The immune response to sand fly salivary proteins and its influence on Leishmania immunity. Front. Immunol. 3, 110 (2012).
Brito, G. et al. Clinical and immunological outcome in cutaneous leishmaniasis patients treated with pentoxifylline. Am. J. Trop. Med. Hyg. 90, 617 (2014).
de Araújo, F. F. et al. Chemokines in Leishmaniasis: Map of cell movements highlights the landscape of infection and pathogenesis. Cytokine 147, 155339 (2021).
Lee, E. G. et al. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science 289, 2350–2354 (2000).
Opipari, A., Boguski, M. & Dixit, V. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J. Biol. Chem. 265, 14705–14708 (1990).
Gupta, A. K. et al. Leishmania donovani inhibits inflammasome-dependent macrophage activation by exploiting the negative regulatory proteins A20 and UCP2. FASEB J. 31, 5087–5101 (2017).
Lecoeur, H. et al. Leishmania amazonensis subverts the transcription factor landscape in dendritic cells to avoid inflammasome activation and stall maturation. Front. Immunol. 11, 1098 (2020).
Blackwell, J. M., Fakiola, M. & Singh, O. P. Genetics, transcriptomics and meta-taxonomics in visceral leishmaniasis. Front. Cell. Infect. Microbiol. 10, 590888 (2020).
Zhang, X. et al. Mechanisms and functions of long non-coding RNAs at multiple regulatory levels. Int. J. Mol. Sci. 20, 5573 (2019).
Kumar, A. et al. Differential regulation of miRNA profiles of human cells experimentally infected by Leishmania donovani isolated from Indian visceral leishmaniasis and post-kala-azar dermal leishmaniasis. Front. Microbiol. 11, 536265 (2020).
Rodrigues, V., Cordeiro-da-Silva, A., Laforge, M., Silvestre, R. & Estaquier, J. Regulation of immunity during visceral Leishmania infection. Parasit. Vectors 9, 118 (2016).
Park, H. et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 6, 1133–1141 (2005).
Griffin, G. K. et al. IL-17 and TNF-α sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J. Immunol. 188, 6287–6299 (2012).
Ribeiro-Gomes, F. L. et al. Macrophage interactions with neutrophils regulate Leishmania major infection. J. Immunol. 172, 4454–4462 (2004).
Lopez Kostka, S. et al. IL-17 promotes progression of cutaneous leishmaniasis in susceptible mice. J. Immunol. 182, 3039–3046. https://doi.org/10.4049/jimmunol.0713598 (2009).
Takele, Y. et al. Following successful anti-leishmanial treatment, neutrophil counts, CD10 expression and phagocytic capacity remain reduced in visceral leishmaniasis patients co-infected with HIV. PLOS Neglected Trop. Dis. 16, e0010681 (2022).
Funding
This work was supported by the Khomein University of Medical Sciences [grant number: 401000021, ethical code: IR.KHOMEIN.REC.1402.009].
Author information
Authors and Affiliations
Contributions
S.R and R.M.R. conceptualized and designed the paper. R.M.R, S.R, A.S, S.K.A, A.V, M.S, M.B, J.K and K.S.H wrote original draft and analyzed bioinformatic data. A.M revised and reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Savardashtaki, A., Khalili Alashti, S., Vafadar, A. et al. An integrated bioinformatic analysis of microarray datasets to identify biomarkers and miRNA-based regulatory networks in leishmaniasis. Sci Rep 14, 12981 (2024). https://doi.org/10.1038/s41598-024-63462-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-63462-5
Keywords
This article is cited by
-
Meta-analysis of microarray data to identify potential signature genes and MiRNAs associated with the pathogenesis of asthma
Journal of Translational Medicine (2025)
-
Antibacterial and antibiofilm activity of curcumin against multidrug-resistant bacteria: insights into FtsZ inhibition and mechanistic pathway
Proceedings of the Indian National Science Academy (2025)
