
Abstract
Urate nephropathy, a common complication of hyperuricemia, has garnered increasing attention worldwide. However, the exact pathogenesis of this condition remains unclear. Currently, inflammation is widely accepted as the key factor in urate nephropathy. Therefore, the aim of this study was to elucidate the interaction of lincRNA-p21/AIF-1/CMPK2/NLRP3 via exosomes in urate nephropathy. This study evaluated the effect of lincRNA-p21/AIF-1/CMPK2/NLRP3 using clinical data collected from patients with urate nephropathy and human renal tubular epithelial cells (HK2) cultured with different concentrations of urate. In clinical research section, the level of lincRNA-p21/AIF-1 in exosomes of urine in patients with hyperuricemia or urate nephropathy was found to be increased, particularly in patients with urate nephropathy. In vitro study section, the level of exosomes, inflammation, autophagy, and apoptosis was increased in HK2 cells induced by urate. Additionally, the expression of lincRNA-p21, AIF-1, CMPK2, and NLRP3 was upregulated in exosomes and HK2 cells. Furthermore, manipulating the activity of lincRNA-p21, AIF-1, CMPK2, and NLRP3 through overexpression or interference vectors regulated the level of inflammation, autophagy, and apoptosis in HK2 cells. In conclusion, the pathway of lincRNA-p21/AIF-1/CMPK2/NLRP3 contributed to inflammation, autophagy, and apoptosis of human renal tubular epithelial cell induced by urate via exosomes. Additionally, the specific exosomes in urine might serve as novel biomarkers for urate nephropathy.
Similar content being viewed by others

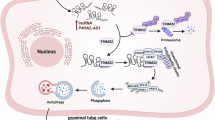
Introduction
Increasing evidence has confirmed that hyperuricemia is a global metabolic disease and an independent risk factor for chronic kidney disease1. The accumulation of urate in the kidneys is known as urate nephropathy2. Retrospective analyses have indicated that the risk of renal injury increases by 7–11%, and the risk of renal deterioration increases by 14% with every 1 mg/dl increase in serum uric acid concentration3,4,5. In recent decades, it has become evident that inflammation plays a central role in urate nephropathy6,7. Studies using human kidney biopsy samples have demonstrated a positive association between serum uric acid concentration and inflammation in tubulointerstitium8,9. Therefore, inflammation is recognized as the fundamental pathological process in urate nephropathy. However, the inflammatory signals which are activated, regulated, and transmitted among different renal cells have been no breakthroughs. Based on our preliminary experiment, long intergenic non-coding RNA p21 (lincRNA-p21), allograft inflammatory factor 1 (AIF-1), mitochondrial nucleotide phosphokinase 2 (CMPK2), and NLR family pyrin domain containing 3 (NLRP3) associated with inflammation, were upregulated in human renal tubular cells cultured with urate. However, the exact roles in urate nephropathy remain unclear.
LincRNA-p21, as a non-coding RNA, located approximately 15 KB upstream of the cell cycle regulation gene P21, regulates proliferation, apoptosis, and DNA damage progression through bind to specific regulatory regions of genome. Numerous studies have demonstrated the effect of lincRNA-p21 in inflammatory diseases. For example, lincRNA-p21 promoted hepatocyte apoptosis and fibrosis10, and vascular smooth muscle cell apoptosis in thoracic aortic aneurysms11. Down-regulation of lincRNA-p21 has been shown to alleviate extracellular matrix in diabetic nephropathy12. Deletion of lincRNA-p21 in renal tubular epithelial cells attenuated kidney injury caused by persistent obesity and hyperlipidemia through inflammation, apoptosis, and endoplasmic reticulum stress13. Spurlock et al. also have confirmed that administration of methotrexate upregulates the expression of lincRNA-p21 and inhibits the activity of NF-κB14. It is necessary to clarify the effect of lincRNA-p21 in inflammation of urate nephropathy. AIF-1 is also involved in inflammatory diseases, including atherosclerosis, rheumatoid arthritis, renal tubulointerstitial fibrosis, vascular calcification, and diabetic kidney disease15,16,17,18,19. However, the upstream and downstream regulatory mechanisms of AIF-1 are still unknown. Furthermore, NLRP3 as a best-characterized inflammasomes has been shown to activate inflammation20. It has suggested NLRP3 serving as a major sensor of sterile inflammatory signaling induces various inflammatory diseases21, including autoimmune diseases22, metabolic disorders23, particularly diseases caused by crystal formation24,25. Crystal could induce inflammatory responses through the NLRP3 pathway, such as gout attacks triggered by urate crystals26,27, atherosclerosis caused by cholesterol crystals28, and renal function decline associated with calcium oxalate crystals29. CMPK2 predominantly located in cell mitochondria is known as an upstream factor to NLRP3 activation30. Luo et al. proposed that CMPK2 is essential for mitochondrial DNA synthesis and NLRP3 activity, promoting a series of inflammatory responses in liver ischemia and reperfusion injury31. The axis of CMPK2/NLRP3 directly plays an important role to inflammation. Increasing evidence suggests inflammation plays an important role in pathophysiology of kidney diseases. Autophagy plays multiple roles in inflammation, and protects kidney from several inflammatory insults. Regulation of inflammation by autophagy has great potential as a therapeutic target for kidney disease. It is well-known that urate becomes oversaturated in proximal tubules, leading to the formation of urate crystals, which are endocytosed and causes lysosomal rupture inducing inflammatory injuries32,33,34. Autophagy engulfs damaged lysosomes and sequesters them from the cytosol to prevent cellular injury. Through this process, autophagy also restores lysosomal functions and biogenesis. It has confirmed that autophagy also plays a functional role in hyperuricemia-induced inflammation34,35. Activation of autophagy may inhibit inflammasome activity36 and downstream inflammatory responses37,38 induced by hyperuricemia.
Exosomes as an intercellular information carrier play a crucial role to regulate different cellular functions, which released by living cells39. Whether the renal exosomes might mediate the function of lincRNA-p21, AIF-1, CMPK2, and NLRP3 in inflammation induced by urate need further study. Elucidating the pathogenesis of exosomes holds promise for identifying new biomarkers and therapeutic target of urate nephropathy. Consequently, we hypothesized that lincRNA-p21, AIF-1, CMPK2, and NLRP3 regulated the pathogenesis of inflammation induced by urate via exosomes as shown in Fig. 1. The purpose of this study was to elucidate the mechanisms underlying the roles of lincRNA-p21, AIF-1, CMPK2, and NLRP3 via exosomes in urate nephropathy.

The possible mechanism of lincRNA-p21/AIF/CMPK2/NLRP3 pathway through exosomes in inflammation, autophagy, and cell death of renal tubular epithelial cells induced by urate.
Materials and methods
Patients
In accordance with the clinical study program approved by the ethics committee of Southern University of Science and Technology Hospital and the ethical standards of the 1964 Helsinki Declaration, 20 asymptomatic hyperuricemia patients who were treated in Southern University of Science and Technology Hospital during the period from January 1, 2022 to December 31, 2023 were selected. The patients were divided to group 1 (asymptomatic hyperuricemia with renal injury) and group 2 (asymptomatic hyperuricemia only). The inclusion criteria of group 1 were used as follows: ⑴ age between 18 and 40 years old; ⑵ serum uric acid ≥ 6 mg/dl; ⑶ with markers of tubulointerstitial lesion (KIM-1, NAG enzyme and β2-microglobulin in urine) above normal values; ⑷ serum creatinine ≥ 1.1 mg/dl, eGFR ≤ 90 ml/min, serum cystatin C ≤ 1.07 mg/l, uACR ≤ 30 mg/g; ⑸ without any glomerular or tubulointerstitial diseases; ⑹ without any chronic underlying diseases such as hypertension, diabetes, coronary heart diseases and ischemic cerebrovascular diseases and so on; ⑺ without abnormal physiological and biochemical indexes; no history of renal damaging drugs treatment such as nonsteroidal anti-inflammatory drugs and so on. However, the inclusion criteria of group 2 were the same as group 1 except ⑶ and ⑷ which were exclusion criterion to group 1. In addition, 10 healthy people without any underlying disease served as the control group. 100 mL urine specimens were collected, centrifugated and stored at – 80 ℃ for further examination. Serum uric acid ≥ 6 mg/dl was defined as hyperuricemia. What’s more, urate nephropathy was defined as hyperuricemia with serum creatinine ≥ 1.1 mg/dl, eGFR ≤ 90 ml/min, serum cystatin C ≤ 1.07 mg/l, abnormal markers of tubulointerstitial lesion, normal markers of glomerular injury excluding other kidney diseases. All volunteers had signed the consent for use of urine specimens and biochemical data.
Clinical index measurement
General biochemical indicators of all volunteers were collected from medical records. The levels of uric acid, creatinine, and cystatin C in serum were measured by commercial assay kits (Roche Diagnostics, Basel, Switzerland) on an automatic blood chemistry analyzer (Roche-Hitachi 7180, Roche Diagnostics, Basel, Switzerland). Estimated glomerular infiltration rate (eGFR) was calculated using the 2009 Chronic Kidney Disease Epidemiology Collaboration Equation. In addition, the levels of creatine, albumin, β2-microglobulin, specific gravity, kidney injury molecule-1, N-acetyl-beta-d-glucosaminidase, in urine were measured by automatic biochemical analyser or immunonephelometric assay. Moreover, exosomes extracted from urine samples were quantitated and detected. The levels of lincRNA-p21 and AIF-1 in urinary exosomes were detected by qPCR and ELISA. The final results were standardized with urinary creatinine.
Cell culture
Human renal tubular epithelial cell lines (HK2) were purchased from ATCC and used in all cell experiments. After recovery, the cells were cultured with DMEM/F12 medium containing 10% fetal bovine serum at 37 °C and 5% CO2. The medium was changed every 2—3 days. When cells were 80—90% fusion, trypsin was added for digestion and passage. The cells were used for further experiment after 12 h synchronization cultured with serum-free DMEM/F12 medium. First, HK2 cells were cultured with different concentrations of urate (300 μM, 500 μM and 700 μM) for 24 h, and collected for follow-up experiments. Second, HK-2 cells were transfected with lincRNA-p21, AIF-1 and CMPK2 overexpression or interference vector. After transfection for 6 h—8 h, cells were treated with 500 μM urate for 24 h, and collected for follow-up experiments. Third, HK2 cells were treated with 10 μM INF39 or Nigericin. After treatment for 12 h, the cells were treated with 500 μM urate for 24 h, and collected for follow-up experiments. Finally, the morphological changes of cells were observed under an inverted microscope. Moreover, the levels of inflammation and autophagy in cells and exosomes were detected.
Plasmid construction
In order to construct an overexpressed plasmid, the sequences of lincRNA-p21 (GenBank: KU881769.1, 2882 bp), AIF-1 (NM_001318970.2, CDS 282 bp) or CMPK2 (NM_207315.4, CDS 1350 bp) were amplified and connected to pcDNA3.1 (generalbiol, China). The cleavage site is BamHI/HindII. siRNA of lincRNA-p21, AIF-1 or CMPK2 was designed and synthesized by JST biological technology co, China. Each siRNA was designed with three types as Table 1.
Cell transfection
According to experimental groups, the cells were inoculated into 6-well plates, 4 × 105 per well, cultured in an incubator at 37 ℃ and 5% CO2, and transfected 24 h later. To prepare the transfection reagent, taken one of the 6-well plates as an example: solution 1, 125 μl of opti-MEM medium (Thermo Scientific) + 7.5 μl of lipo3000 (Invitrogen); solution 2, 125 μl of opti-MEM medium (Thermo Scientific) + 2.5 μg plasmid + 5 μl P3000 (auxiliary transfection reagent, Invitrogen) or 75 pmol sequence. Solution 1 and 2 were mixed and placed at room temperature for 15 min.
The mixture of solution 1 and solution 2 was added into 6-well plates, which were gently shaken and mixed, and continued to culture for 24 h.
Fluorescence in situ hybridization (FISH)
The expression of linRNA-p21 was detected by RNA FISH kit (Shanghai GenePharma Co, China). According to the manufacturer’s operation, the process was described as follows. Experimental cells were fixed with 4% paraformaldehyde for 10 min, and soaked with PBS for 5 min × 3 times. Added triton X-100 to completely cover cells and incubate at 4 ℃ for 15 min, and soaked with PBS for 5 min × 3 times. Added 100 μl sealing solution per well and incubate at 37 ℃ for 30 min, 100 μl 2 × Buffer C at 37 ºC for 30 min. Preparation of probe working liquid: for example, in a 10 μl system, 1 μmol/L biotin-probe 1 μl + 1 μmol/L SA-Cy3 1 μl + PBS 8 μl, and incubated at 37 ºC for 30 min. Mix the 10 μl probe working solution with 90 μl Buffer E. 100 μl denatured probe mixture was added to each well, and hybridized in a culture box at 37 ºC overnight after taking measures to avoid light. On the next day of hybridization, samples were removed from the incubator at 37 ºC, the probe mixture was sucked away, and 100 μl 0.1% Buffer F was added to each well and washed at 37 ºC for 10 min. Removed 0.1% Buffer F, added 100 μl of 2 × Buffer C to each well, and washed at 60 ºC for 10 min × 3 times, at 37 ℃ for 10 min × 3 times. Removed 2 × Buffer C, added 100 μl diluted DAPI working liquid to each well, and stained away from light for 5 min. Removed DAPI working liquid, and washed with PBS twice, 5 min each time. Added anti-fluorescence attenuation sealer, covered with the cover glass, and observed the sealer under a fluorescence microscope. The staining effect was observed and taken pictures under fluorescence microscope.
Extraction of exosomes
Exosomes of urine or culture supernate were extracted by ultrafast centrifugation method. Samples were melted at 37℃, moved to a new centrifuge tube and centrifuged at 4℃ and 2000 rpm for 30 min. The supernatant was carefully moved into a new centrifuge tube and centrifuged again at 4 ℃ 10,000 rpm for 45 min to remove large vesicles. Taken the supernatant and filtered by 0.45 μm filter membrane. And the filtrate was centrifuged at 100,000 rpm for 70 min. After the supernatant was removed and re-suspended with 10 mL pre-cooled PBS, and centrifuged again at 100,000 rpm for 70 min. After supernatant was removed, the exosomes were re-suspended with 100 μL pre-cooled PBS preserved at − 80 ℃.
Transmission electron microscopy
Exosome samples were observed by transmission electron microscopy. Taken 10 μL of exosomes and added into the copper net to precipitate for 1 min. 10 μL of uranyl acetate added into the copper net and precipitate for 1 min. The float was absorbed by filter paper. Exosome samples was dried at room temperature for a few minutes, examined and taken pictures by transmission microscopy (HT-7700, Hitachi) at 100 kv.
Particle size analysis of exosome
The concentration of exosomes was detected by nanoflow meter, and the purity of exosomes was detected by nanoparticle tracking analyzer (NTA). 10μL of exosome samples were diluted to 30 μL. After the instrument performance test with standard products is qualified, the exosome samples were loaded. It should be noted that gradient dilution should be carried out to avoid sample clogging of the injection needle. The particle size and concentration information of exosomes were detected by nanoparticle tracking analyzer (N30E, NanoFCM).
qPCR analysis (SYBR method)
Total RNAs of urine or HK2 were extracted using the total RNAs Isolation Kit, and the concentration of RNA was determined using ultraviolet spectrophotometer NANO 2000. Obtained RNA samples were reversely transcribed synthesized to cDNA by MMLV reverse transcription kit, according to the manufacturer’s protocols. 20 μl cDNA sample was stored at -20℃ or used for downstream experiments. The relative expression of RNA was amplified and determined by Exicycler 96 (BIONEER, South Korea), as described by the manufacturer. The primer sequences were used as Table 2. β-actin was used as an endogenous reference gene for normalization of the data. The reaction system was 20 μL (cDNA template 1 μL, SYBR GREEN mastermix 10 μL, upstream primers 0.5 μL, downstream primers 0.5 μL, ddH2O 8 μL). And the reaction conditions were as follows: incubated at 95℃ for 5 min, 95 ℃ for 10 s, 60 ℃ for 10 s, 72 ℃ for 15 s, after then went to line 2 for 40 cycles: incubated at 72 ℃ for 90 s, incubate at 40 ℃ for 1 min, melted from 60 ℃ to 94 ℃, every 1 ℃ for 1 s, incubated at 25 ℃ for 1 min. The method of 2−△△CT was used in this experiment.
Apoptosis analysis
Apoptotic cells were detected and quantified by flow cytometry using Annexin V-FITC/PI apoptosis Kit (wanleibio, China). The specific detection process was as follows. Experimental cells were collected and washed twice with PBS. And then the cells were suspended with 500 μL binding buffer to adjust the concentration to 106/ml. 5 μL AnnexinV-FITC and 10 μL Propidium Iodide was added and mixed which was incubated at room temperature for 15 min away from light. Cells suspension was filtered by mesh screen, and detected by flow cytometry (NovoCyte, Agilent, USA). In the two-parameter scatter plot, the living cells were shown in the lower left quadrant (Annexin V FITC-/PI-); the non-living cells namely the late apoptotic cells were shown in the upper right quadrant (Annexin V FITC+/PI+); the early apoptotic cells were shown in the lower right quadrant (Annexin V FITC+/PI-).
Western blot analyses
Total protein was extracted with total protein extraction kit (wanleibio, China). The protein concentration was detected by the BCA protein concentration determination kit (wanleibio, China). For electrophoresis, 8%, 12% or 15% SDS-PAGE gels were made respectively. 40 μg of total protein were loaded onto SDS-PAGE gels and separated by double vertical protein electrophoresis apparatus (DYCZ-24DN, Liuyi Biotechnology, China). After electrophoresis, the separated proteins were transferred onto a polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA). The membrane was blocked in Western Blocking Buffer for 1 h at room temperature and then incubated with primary antibodies against AIF-1(1:500, wanleibio), CMPK2 (1:3000, Proteintech), NLRP3 (1:500, wanleibio), p53 (1:500, wanleibio), p62 (1:500, wanleibio), LC3BII/I (1:500, wanleibio), β-actin (1:1000, wanleibio) and CD63 (1:500, Affinity Biosciences) for at 4℃ overnight. Membranes were then incubated with goat anti-rabbit IgG-HRP (1:5000, wanleibio) at 37 ℃ for 45 min. After incubation with second antibody, the PVDF membrane was immersed in TBST, shaken for 5 min, 6 times. When the water of PVDF was drained with filter paper, and evenly sprinkled the ECL luminescent solution, at room temperature for 5 min. Finally, PVDF was transferred to a dark box, and exposed in the dark room. The films were scanned and the optical density value of the target strip was analyzed with the Gel image processing system (GEL-Pro-Analyzer software, China). Protein levels were quantified and normalized to β-actin level.
Ethical approval
The clinical study program was approved by the ethics committee of Southern University of Science and Technology Hospital (2021–080). All volunteers also signed the consent for use of urine specimens and biochemical data in this study.
Results
The level of lincRNA-p21/AIF-1 in exosome of urine was related to urate nephropathy
The study analyzed the levels of lincRNA-p21 and AIF-1 in urinary exosomes of individuals with urate nephropathy, hyperuricemia, and healthy individuals. The study included 30 male volunteers, with 10 individuals in each group. Several biochemical indicators were measured, including age, eGFR (estimated glomerular filtration rate), serum cystatin C, uACR (urine albumin-to-creatinine ratio), urine specific gravity, serum uric acid, serum creatinine, uβ2-MG (urine β2-microglobulin), uKim-1 (urine kidney injury molecule-1), and uNAG (urine N-acetyl-β-D-glucosaminidase). The results as shown in Table 3 indicated that there were no significant differences in age, eGFR, serum cystatin C, uACR, and urine specific gravity between the healthy individuals, hyperuricemia group, and urate nephropathy group. However, patients with urate nephropathy had significantly increased levels of serum uric acid, serum creatinine, uβ2-MG, uKim-1, and uNAG compared to the other two groups. Furthermore, the levels of lincRNA-p21 and AIF-1 in urine exosomes were also increased in both the hyperuricemia and urate nephropathy groups. This suggests that urine exosomal lincRNA-p21/AIF-1 may be involved in the kidney injury induced by urate.
Urate induced inflammation, autophagy and apoptosis of HK2 cells
It is well-known that tubulointerstitial fibrosis is a major complication of hyperuricemia. Therefore, it is crucial to investigate the mechanism of renal tubular epithelial cell injury induced by urate in order to understand urate nephropathy. In this study, HK2 cells were cultured with different concentrations of urate for 24 h. The results shown in Fig. 2 indicated that compared to the control group, there were significant differences in the expression of lincRNA-p21, inflammatory proteins (AIF-1, CMPK2, and NLRP3), autophagy-related proteins (p53, p62, and LC3BII/I), and the level of apoptosis. These differences were positively correlated with the concentration of urate, with the most significant variation observed in the 700 μM urate group. However, it was worth noting that the apoptosis of HK2 cells was also highest in the 700 μM urate group. Therefore, a concentration of 500 μM urate was chosen for subsequent experiments.

The level of lincRNA-p21, inflammation, autophagy and apoptosis were increased in HK2 cells cultured with urate. (A and B) The expression of lincRNA-p21 in HK2 cells was detected by FISH and qPCR. (C and D) The expression of AIF-1, CMPK2, NLRP3, p53, p62, and LC3BII/I in HK2 cells were detected by western blot. (E and F) The apoptosis of HK2 cells was detected by Annexin V-FITC/PI. Versus control group, *p < 0.05, **p < 0.01, ***p < 0.001.
Urate increased the level of exosome in HK2 cells
Exosomes play a crucial role in kidney diseases by serving as carriers for intercellular communication within the kidney. To investigate the impact of exosomes on renal tubular epithelial cell injury induced by urate, exosomes were extracted and analyzed from the cell supernatant of HK2 cells. As depicted in Fig. 3, the average particle size of the exosomes was measured to be 80 nm, and their concentration was found to increase in a concentration-dependent manner in response to urate stimulation. Furthermore, compared to the control group, the levels of inflammatory proteins (AIF-1, CMPK2, and NLRP3) and autophagy-related proteins (p53, p62, and LC3BII/I) within the exosomes were significantly elevated. These findings provide compelling evidence that exosomes are involved in the inflammation and autophagy processes in HK2 cells induced by urate.

The level of exosome was increased in HK2 cells cultured with urate. (A) Exosome morphology was detected by electron microscopy. (B, C, D and E) The particle size and concentration information of exosomes were detected by nanoparticle tracking analyzer. (F) The expression of lincRNA-p21 in exosome was detected by qPCR. (G and H) The expression of AIF-1, CMPK2, NLRP3, p53, p62, and LC3BII/I in exosome were detected by western blot. Versus control group, *p < 0.05, **p < 0.01, ***p < 0.001.
LincRNA-p21 contributed to inflammation, autophagy and apoptosis of HK2 cells induced by urate
LincRNA-p21, a non-coding RNA, plays a role in regulating inflammation and other cellular processes. To assess the impact of lincRNA-p21 on inflammation, autophagy, and apoptosis in HK2 cells induced by urate, overexpression or interference vectors of lincRNA-p21 were transfected into HK2 cells. As depicted in Fig. 4, the expression of lincRNA-p21 was significantly increased in the overexpression group and significantly decreased in the interference group. Compared to the control group, lincRNA-p21 notably enhanced the expression of inflammatory proteins (AIF-1, CMPK2, and NLRP3) and autophagy-related proteins (p53, p62, and LC3BII/I) in both HK2 cells and exosomes. Additionally, lincRNA-p21 induced apoptosis in HK2 cells. Conversely, the siRNA targeting lincRNA-p21 had the opposite effects. Based on these research findings, it could be concluded that lincRNA-p21 promoted autophagy and apoptosis in HK2 cells induced by urate via the AIF-1/CMPK2/NLRP3 pathway.

LincRNA-p21 contributed to inflammation, autophagy and apoptosis of HK2 cells cultured with urate. (A) The expression of lincRNA-p21 in HK2 cells was detected by qPCR. (B) The apoptosis of HK2 cells was detected by Annexin V-FITC/PI. (C and D) The expression of AIF-1, CMPK2, NLRP3, p53, p62, and LC3BII/I in HK2 cells were detected by western blot. (E and F) The expression of AIF-1, CMPK2, NLRP3, p53, p62, and LC3BII/I in exosome were detected by western blot. Versus normal control group, *p < 0.05, **p < 0.01, ***p < 0.001.
AIF-1 contributed to autophagy and apoptosis of HK2 cells induced by urate via CMPK2/NLRP3
Numerous studies have provided evidence supporting the involvement of AIF-1 in renal tubulointerstitial inflammation and fibrosis. To investigate the impact of AIF-1 on inflammation, autophagy, and apoptosis in HK2 cells induced by urate, overexpression or interference vectors of AIF-1 were transfected into HK2 cells. As depicted in Fig. 5, the expression of AIF-1 mRNA and protein was significantly increased in the overexpression group and significantly decreased in the siRNA group. Compared to the control group, the expression of lincRNA-p21 did not show a significant effect in either the AIF-1 overexpression or interference group. However, AIF-1 significantly promoted cell apoptosis and the expression of inflammatory proteins (CMPK2 and NLRP3) and autophagy-related proteins (p53, p62, and LC3BII/I) in both HK2 cells and exosomes. Conversely, the siRNA targeting AIF-1 had the opposite effects. Therefore, these results suggested that, in addition to lincRNA-p21, AIF-1 could promote autophagy and apoptosis in HK2 cells induced by urate via the CMPK2/NLRP3 pathway.

AIF-1 contributed to inflammation, autophagy and apoptosis of HK2 cells cultured with urate. (A and B) The expression of AIF-1 in HK2 cells was detected by qPCR and western blot. (C) The expression of lincRNA-p21 in HK2 cells was detected by qPCR. (D and E) The expression of CMPK2, NLRP3, p53, p62, and LC3BII/I in HK2 cells were detected by western blot. (F and G) The expression of CMPK2, NLRP3, p53, p62, and LC3BII/I in exosome were detected by western blot. (H) The expression of lincRNA-p21 in exosome was detected by qPCR. (I) The apoptosis of HK2 cells was detected by Annexin V-FITC/PI. Versus normal control group, *p < 0.05, **p < 0.01, ***p < 0.001.
The effect of CMPK2 on inflammation, autophagy and apoptosis of HK2 cells induced by urate
To investigate the impact of CMPK2 on inflammation, autophagy, and apoptosis in HK2 cells induced by urate, overexpression or interference vectors of CMPK2 were transfected into HK2 cells. As depicted in Fig. 6, the expression of CMPK2 mRNA and protein was significantly increased in the overexpression group and significantly decreased in the siRNA group. Compared to the control group, CMPK2 overexpression significantly promoted cell apoptosis and the expression of inflammatory proteins (NLRP3) and autophagy-related proteins (p53, p62, and LC3BII/I) in both HK2 cells and exosomes. Conversely, the siRNA targeting CMPK2 had the opposite effects. Therefore, these results suggested that CMPK2 can promote autophagy and apoptosis in HK2 cells induced by urate via the NLRP3 pathway.

CMPK2 contributed to inflammation, autophagy and apoptosis of HK2 cells cultured with urate. (A and B) The expression of CMPK2 in HK2 cells was detected by qPCR and western blot. (C) The expression of lincRNA-p21 and AIF-1 in HK2 cells was detected by qPCR. (D and E) The expression of NLRP3, p53, p62, and LC3BII/I in HK2 cells were detected by western blot. (F and G) The expression of NLRP3, p53, p62, and LC3BII/I in exosome were detected by western blot. (H) The apoptosis of HK2 cells was detected by Annexin V-FITC/PI. Versus normal control group, *p < 0.05, **p < 0.01, ***p < 0.001.
The effect of NLPR3 on inflammation, autophagy and apoptosis of HK2 cells induced by urate
To investigate the effect of NLRP3 on inflammation, autophagy, and apoptosis in HK2 cells induced by urate, HK2 cells were treated with an activator or inhibitor of NLRP3. As shown in Fig. 7, compared to the control group, the NLRP3 activator (Nigericin) significantly promoted cell apoptosis and the expression of inflammatory proteins (CMPK2) and autophagy-related proteins (p53, p62, and LC3BII/I) in both HK2 cells and exosomes. On the other hand, the NLRP3 inhibitor (INF39) had the opposite effects. Therefore, these results suggested that NLRP3 interacts with CMPK2, leading to the promotion of autophagy and apoptosis in HK2 cells via inflammation.

NLRP3 contributed to inflammation, autophagy and apoptosis of HK2 cells cultured with urate. (A and B) The expression of CMPK2, p53, p62, and LC3BII/I in HK2 cells was detected by western blot. (C and D) The expression of CMPK2, p53, p62, and LC3BII/I in exosome was detected by western blot. (E) The apoptosis of HK2 cells was detected by Annexin V-FITC/PI. Versus normal control group, *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Urate nephropathy, the main complication of hyperuricemia, contributes to chronic kidney disease (CKD)40,41. The pathogenesis of urate nephropathy involves crystal deposition of monosodium urate, oxidative stress, endothelial dysfunction, renal tubule interstitial fibrosis, and inflammation42,43. Inflammatory and fibrotic lesions are the primary features of urate nephropathy, as indicated by renal biopsy findings. The main manifestation is the disappearance of normal renal tubular epithelial cells in the kidney and replacement by inflammatory cells and fibrous tissue in the renal interstitium, leading to the loss of physiological function. It is well known that under pathological stimuli, cells could undergo various modes of death, including autophagy, apoptosis, necrosis, pyroptosis, and ferroptosis and so on. Different cell death processes involve overlapping molecular mechanisms and may be co-activated in some cases, leading to the coexistence of multiple cell death modes in the pathological process. Therefore, research on cell death may require a comprehensive view of different types of cell death44,45. Our preliminary experimental data show that differential expression of lincRNA-p21, AIF-1, CMPK2, and NLRP3 might be involved in urate nephropathy, but the exact mechanism remains unclear. In this study, we explored the interaction mechanisms of lincRNA-p21, AIF-1, CMPK2, and NLRP3 in uric acid-induced inflammation and cell death of human renal tubular epithelial cells, by modulating gene expression in vitro and analyzing clinical urine samples. The data of the study suggested that lincRNA-p21, AIF-1, CMPK2, and NLRP3 co-acted and activated autophagy, and apoptosis via exosome in human tubular epithelial cells induced by urate.
LincRNA-p21, as a non-coding RNA, is a crucial regulator of cell proliferation, inflammation, apoptosis, and DNA damage responses, which plays pro-inflammatory roles in various diseases. For example, it has been observed that overexpression of lincRNA-p21 promotes the proinflammatory phenotype transformation of microglia in neuroinflammatory models46. Wu et al. confirmed that lincRNA-p21 inhibited cell proliferation and induced apoptosis in vascular smooth muscle cells and macrophages in vitro and in vivo via the p53 pathway47. Additionally, Zhou et al. suggested that lincRNA-p21 promoted the proliferation of lung fibroblasts, leading to pulmonary fibrosis48. Li B et al. observed that the deletion of lincRNA-p21 in renal tubule cell attenuated oxidative stress, inflammation, apoptosis, and endoplasmic reticulum stress via p53-dependent mechanism, leading to reduction of histological and functional kidney injury despite persistent obesity and hyperlipidemia13. Be consistent with the above studies, and we also found that lincRNA-p21 was involved in inflammation, autophagy, and apoptosis in renal tubular epithelial cells induced by urate. We speculated that lincRNA-p21 served as an initiator of inflammation and death of renal tubular epithelial cells induced by urate. LincRNA-p21 is a non-coding RNA that is not translated into protein but mainly plays a role in regulating gene transcription and expression. AIF-1 is a calcium-binding protein produced by monocytes, macrophages, and lymphocytes and so on. It has confirmed that AIF-1 contributes to several kidney diseases, such as diabetic nephropathy, renal tubulointerstitial inflammation and fibrosis19,49. In this study, we firstly approved that lincRNA-p21 promoted inflammation, autophagy and apoptosis induced by urate via AIF-1. And further clarified AIF-1 played a pathogenic role through CMPK2/NLRP3 pathway.
The axis of CMPK2/NLRP3 plays a crucial role in inflammation, and CMPK2 induces inflammation through NLRP3, which is a critical process of inflammatory reaction50,51. Previous studies suggest that CMPK2 induces pyroptosis resulting in inflammation by NLRP3 pathway52,53,54,55. However, a recent study shows that NLRP3 inflammasome activation not only leads to pyroptosis, but also to other types of cell death, including apoptosis, necrotic apoptosis, and iron death56,57. In addition, various effectors of cell death have been reported to regulate NLRP3 inflammasome activation, suggesting that cell death is closely related to NLRP358. In this study, we also confirmed that CMPK2/NLRP3 contributed to inflammation, autophagy, and apoptosis of renal tubular epithelial cells induced by urate. What’s more, we observed that the inhibitor or agonist of NLRP3 could affect the expression of CMPK2. However, no one has studied the effect of NLRP3 on CMPK2 in reverse. The results of this study showed CMPK2 increased after using the NLRP3 agonist and decreased after using the NLRP3 inhibitor. The possible reason for this situation was that the NLRP3 agonist or inhibitor works through CMPK2. Zhong Z et al. have confirmed that CMPK2 is necessary for the production of oxidized mtDNA fragments after exposure to NLRP3 activators. In addition, cytosolic oxidized mtDNA is required for NLRP3 inflammasome complex and activation30. Therefore, the specific mechanism of interaction between NLRP3 and CMPK2 required to be further studied to clarify.
Exosomes, as a carrier of information transfer between different cells, are involved in the pathogenesis of various diseases. In this study, we had demonstrated that the levels of lincRNA-p21, AIF-1, CMPK2, and NLRP3 in exosomes was corresponding to intracellular levels. LincRNA-p21, AIF-1, CMPK2, and NLRP3 entered exosomes, and were transmitted among the surrounding cells. Exosomes protect the contents from destruction and degradation, and facilitate the interaction between different cells, potentially serving as markers of pathological processes59,60. Chen et al. proposed that urinary exosome tsRNAs served as biomarkers for the diagnosis and prediction of nephritis in systemic lupus erythematosus61. In according with the results of in vitro, we also observed that there was high level of urinary exosome which contained lincRNA-p21, AIF-1, CMPK2, and NLRP3 in patients with hyperuricemia. Therefore, we suggested that lincRNA-p21, AIF-1, CMPK2, and NLRP3 in urinary exosome might be novel biomarkers for renal tubulointerstitial inflammation and fibrosis.
In conclusion, based on the aforementioned results, we proposed that the pathway of lincRNA-p21/AIF-1/CMPK2/NLRP3 contributed to inflammation, autophagy, and apoptosis of human renal tubular epithelial cell induced by urate via exosomes. Additionally, the specific exosomes in urine might serve as novel biomarkers for urate nephropathy. Although this study had experimentally confirmed the interaction among lincRNA-p21, AIF-1, CMPK2, and NLRP3 through in vitro activation or inhibition, the results required further validation in vitro and in vivo due to the complexity and cross-talk of interactions among various biological molecules. Additionally, the limited number of clinical samples in this study had resulted in poor stability and persuasiveness of the research findings, necessitating an expansion of the sample size for validation (Supplementary Information).
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author.
References
Wang, Y. et al. Effect of salt intake on plasma and urinary uric acid levels in Chinese adults: An interventional trial. Sci. Rep. 8(1), 1434 (2018).
Chinese guidelines for the diagnosis and treatment of hyperuricemia and gout (2019) [J]. Chinese Journal of Endocrinology and Metabolism. 2020(01):1–13.
Wang, Y. et al. Association between urinary sodium excretion and uric acid, and its interaction on the risk of prehypertension among Chinese young adults. Sci. Rep. 8(1), 7749 (2018).
Domrongkitchaiporn, S. et al. Risk factors for development of decreased kidney function in a Southeast Asian population: A 12-year cohort study. J. Am. Soc. Nephrol. 16(3), 791–799 (2005).
De Cosmo, S. et al. Serum uric acid and risk of CKD in type 2 diabetes. Clin. J. Am. Soc. Nephrol. 10(11), 1921–1929 (2015).
Kang, D. H. et al. A role for uric acid in the progression of renal disease. J. Am. Soc. Nephrol. 13(12), 2888–2897 (2002).
Mazzali, M. et al. Hyperuricemia exacerbates chronic cyclosporine nephropathy. Transplantation 71(7), 900–905 (2001).
Fan, S. et al. Hyperuricemia and its related histopathological features on renal biopsy. BMC Nephrol. 20(1), 95 (2019).
Zhou, J. et al. Plasma uric acid level indicates tubular interstitial leisions at early stage of IgA nephropathy. BMC Nephrol. 15, 11 (2014).
Yang, L. et al. Tβ4 suppresses lincRNA-p21-mediated hepatic apoptosis and fibrosis by inhibiting PI3K-AKT-NF-κB pathway. Gene 758, 144946 (2020).
Hu, W. et al. Upregulation of lincRNA-p21 in thoracic aortic aneurysms is involved in the regulation of proliferation and apoptosis of vascular smooth muscle cells by activating TGF-β1 signaling pathway. J. Cell. Biochem. 120(3), 4113–4120 (2019).
Zhang, J. et al. LincRNA-p21 sponges miR-18b to promote the progression of diabetic nephropathy. Am. J. Transl. Res. 10(5), 1481–1489 (2018).
Li, B. et al. Tubule-specific deletion of LincRNA-p21ameliorates lipotoxic kidney injury. Mol. Ther. Nucl. Acids 26, 1280–1290 (2021).
Spurlock, C. F. et al. Methotrexate inhibits NF-κB activity via long intergenic (noncoding) RNA-p21 induction. Arthritis Rheumatol. 66(11), 2947–2957 (2014).
Sikora, M. et al. Role of allograft inflammatory factor-1 in pathogenesis of diseases. Immunol. Lett. 218, 1–4 (2020).
Zhang, Y., Tedgui, A. & Ait-Oufella, H. Allograft inflammatory factor-1, a multi-target regulator of atherosclerosis. Atherosclerosis 289, 179–180 (2019).
Piotrowska, K. et al. Over-expression of allograft inflammatory factor-1 (AIF-1) in patients with rheumatoid arthritis. Biomolecules 10(7), 1064 (2020).
Hao, J. B. et al. The crosstalk between calcium ions and aldosterone contributes to inflammation, apoptosis, and calcification of VSMC via the AIF-1/NF-κB pathway in Uremia. Oxid. Med. Cell Longev. 2020, 3431597 (2020).
Hao, J. B. et al. The effect of allograft inflammatory factor-1 on inflammation, oxidative stress, and autophagy via miR-34a/ATG4B pathway in diabetic kidney disease. Oxid. Med. Cell. Longev. 2022(29), 1668000 (2022).
Liston, A. & Masters, S. L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 17(3), 208–214 (2017).
Misawa, T. et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 14, 454–460 (2013).
Vande Walle, L. et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 512(7512), 69–73 (2014).
Martinon, F. et al. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440(7081), 237–241 (2006).
Goldberg, E. L. et al. β-Hydroxybutyrate deactivates neutrophil NLRP3 Inflammasome to relieve gout flares. Cell Rep. 18(9), 2077–2087 (2017).
Ludwig-Portugall, I. et al. An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int. 90(3), 525–539 (2016).
Crișan, T. O. et al. Soluble uric acid primes TLR-induced proinflammatory cytokine production by human primary cells via inhibition of IL-1Ra. Ann. Rheum. Dis. 75(4), 755–762 (2016).
Kim, T. W. et al. The critical role of IL-1 receptor-associated kinase 4-mediated NF-κB activation in modified low-density lipoprotein-induced inflammatory gene expression and atherosclerosis. J. Immunol. 186(5), 2871–2880 (2011).
Duewell, P. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464(7293), 1357–1361 (2010).
Mulay, S. R. et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J. Clin. Invest. 123(1), 236–246 (2013).
Zhong, Z. et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560(7717), 198–203 (2018).
Luo, Y. et al. CMPK2 accelerates liver ischemia/reperfusion injury via the NLRP3 signaling pathway. Exp. Ther. Med. 22(6), 1358 (2021).
Isaka, Y. et al. Hyperuricemia-induced inflammasome and kidney diseases. Nephrol. Dial. Transplant. 31, 890 (2015).
Kimura, T., Isaka, Y. & Yoshimori, T. Autophagy and kidney inflammation. Autophagy 13(6), 997–1003 (2017).
Maejima, I. et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 32(17), 2336–2347 (2013).
Saitoh, T. et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456(7219), 264–268 (2008).
Shi, C. S. et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13(3), 255–263 (2012).
Choe, J. Y. et al. Enhanced p62 expression through impaired proteasomal degradation is involved in caspase-1 activation in monosodium urate crystal-induced interleukin-1b expression. Rheumatology (Oxford) 53(6), 1043–1053 (2014).
Wu, M. et al. Hyperuricemia causes kidney damage by promoting autophagy and NLRP3-mediated inflammation in rats with urate oxidase deficiency. Dis. Model Mech. 14(3), dmm048041 (2021).
Wang, J. et al. Exosomes: A novel strategy for treatment and prevention of diseases. Front. Pharmacol. 8, 300 (2017).
Vargas-Santos, A. B. & Neogi, T. Management of gout and hyperuricemia in CKD. Am. J. Kidney Dis. 70(3), 422–439 (2017).
Tsao, H. M. et al. Serum urate and risk of chronic kidney disease: A mendelian randomization study using Taiwan biobank. Mayo Clin. Proc. 98(4), 513–521 (2023).
Johnson, R. J. et al. Hyperuricemia, acute and chronic kidney disease, hypertension, and cardiovascular disease: Report of a scientific workshop organized by the national kidney foundation. Am. J. Kidney Dis. 71(6), 851–865 (2018).
Mauer, M. & Doria, A. Uric acid and diabetic nephropathy risk. Contrib. Nephrol. 192, 103–109 (2018).
Park, W. et al. Diversity and complexity of cell death: A historical review. Exp. Mol. Med. 55(8), 1573–1594 (2023).
Kayagaki, N., Webster, J. D. & Newton, K. Control of cell death in health and disease. Annu. Rev. Pathol. 19, 157–180 (2024).
Ye, Y. et al. A lincRNA-p21/miR-181 family feedback loop regulates microglial activation during systemic LPS- and MPTP- induced neuroinflammation. Cell Death Dis. 9(8), 803 (2018).
Wu, G. et al. LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation 130(17), 1452–1465 (2014).
Zhou, W. Q. et al. Lipopolysaccharide promotes pulmonary fibrosis in acute respiratory distress syndrome (ARDS) via lincRNA-p21 induced inhibition of Thy-1 expression. Mol. Cell. Biochem. 419(1–2), 19–28 (2016).
Yuan, X. et al. Aldosterone promotes renal interstitial fibrosis via the AIF-1/AKT/mTOR signaling pathway. Mol. Med. Rep. 20(5), 4033–4044 (2019).
Wu, W. et al. Endothelial Gata6 deletion reduces monocyte recruitment and proinflammatory macrophage formation and attenuates atherosclerosis through Cmpk2-Nlrp3 pathways. Redox Biol. 64, 102775 (2023).
Xian, H. et al. Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity 54(7), 1463-1477.e11 (2021).
Tang, Z. et al. Drugs targeting CMPK2 inhibit pyroptosis to alleviate severe pneumonia caused by multiple respiratory viruses. J. Med. Virol. 96(5), e29643 (2024).
Zhang, X., Zhang, K. & Zhang, Y. Pigment epithelium-derived factor facilitates NLRP3 inflammasome activation through downregulating cytidine monophosphate kinase 2: A potential treatment strategy for missed abortion. Int. J. Mol. Med. 45(5), 1436–1446 (2020).
Guan, X. et al. Microglial CMPK2 promotes neuroinflammation and brain injury after ischemic stroke. Cell Rep. Med. 5(5), 101522 (2024).
Wang, H., Wang, S. & Huang, S. MiR-494-3p alleviates acute lung injury through regulating NLRP3 activation by targeting CMPK2. Biochem. Cell Biol. 99(3), 286–295 (2021).
Huang, Y., Xu, W. & Zhou, R. NLRP3 inflammasome activation and cell death. Cell. Mol. Immunol. 18(9), 2114–2127 (2021).
Malireddi, R. K. S., Kesavardhana, S. & Kanneganti, T. D. ZBP1 and TAK1: Master regulators of NLRP3 Inflammasome/Pyroptosis, apoptosis, and necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 9, 406 (2019).
Barnett, K. C. et al. A 360° view of the inflammasome: Mechanisms of activation, cell death, and diseases. Cell 186(11), 2288–2312 (2023).
Barile, L. & Vassalli, G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacol. Ther. 174, 63–78 (2017).
Song, J. et al. Clinical and pathological correlation between urinary exosome miR-223 and IgAN patients. Clin. Nephrol. 100(5), 209–215 (2023).
Chen, S. et al. Urinary exosome tsRNAs as novel markers for diagnosis and prediction of lupus nephritis. Front. Immunol. 14, 1077645 (2023).
Acknowledgements
We would also like to thank the relevant institutions and teams of Southern University of Science and Technology Hospital for their support and collaboration. Finally, we extend our gratitude to all individuals who have contributed to this study, including members of the research team and volunteers involved in the experiments. Their efforts and dedication have been crucial to the success of this research.
Funding
The study was received funding from the Key Project of Sub-item Funding for Technology Research and Creative Design in Nanshan District (Project No. NS006), the High-level Project A Cultivation Program of the Chief Research Fund of the Southern University of Science and Technology Hospital (Project No. 2021-A05), Shenzhen Science and Technology Program (Project No. JCYJ20220530112606015), and the Sanming Project of Medicine in Nanshan Shenzhen (Project No. SZSM202103002).
Author information
Authors and Affiliations
Contributions
The study’s overall design and research objectives were conceptualized by the HJB and HLR. WSY, GXY, GXJ, YK and CRH collected clinical data from patients with urate nephropathy and conducted experiments. HJB, GXY and HLR performed data analysis, including statistical analysis and interpretation of the results obtained from the clinical data and in vitro experiments. HJB, WSY, GXY, GXJ, YK and CRH wrote the manuscript, including the introduction, methods, results, and discussion sections, based on the study findings and relevant literature. HJB and HLR. WSY, GXY, GXJ, YK and CRH conducted experiments in the laboratory. HJB and HLR interpreted the experimental results and discussed their implications in the context of urate nephropathy pathogenesis. HJB and HLR critically reviewed and revised the manuscript based on feedback from co-authors and reviewers, ensuring the accuracy and clarity of the scientific content.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hao, J., Guo, X., Wang, S. et al. LincRNA-p21/AIF-1/CMPK2/NLRP3 pathway promoted inflammation, autophagy and apoptosis of human tubular epithelial cell induced by urate via exosomes. Sci Rep 14, 18146 (2024). https://doi.org/10.1038/s41598-024-69323-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-69323-5
