
Abstract
Lumbar disc herniation (LDH) is a common clinical spinal disorder, yet its etiology remains unclear. We aimed to explore the role of cuproptosis-related genes (CRGs) and identify potential diagnostic biomarkers. Our analysis involved interrogating the GSE124272 and GSE150408 datasets for differential gene expression profiles associated with CRGs and immune characteristics. Molecular clustering was performed on LDH samples, followed by expression and immune infiltration analyses. Using the WGCNA algorithm, specific genes within CRG clusters were identified. After selecting the most predictive genes from the optimal model, four machine learning models were constructed and validated. This study identified nine CRGs associated with copper-regulated cell death. Two copper-containing molecular clusters linked to death were detected in LDH samples. Elevated expression and immune infiltration levels were found in LDH patients, particularly in CRG cluster C2. Utilizing XGB, five genes were identified for constructing a diagnostic model, achieving an area under the curve values of 0.715. In conclusion, this research provides valuable insights into the association between LDH and copper-regulated cell death, alongside proposing a promising predictive model.
Similar content being viewed by others
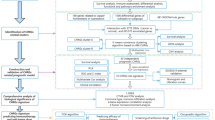

Introduction
Lumbar disc herniation (LDH) is a common orthopedic condition characterized by back pain, weakness, numbness, leg or foot pain, and tingling1,2. These symptoms result from mechanical compression, chemical radiculitis, and autoimmune responses. The recurrence rate of LDH ranges from 5 to 25%, posing challenges to clinical treatment3. Based on available reports4, LDH affects an estimated 9% of the global population. The presence of symptomatic LDH and associated back pain significantly impacts the quality of life of affected individuals. While the precise etiology of LDH remains uncertain, clinical observations suggest a close association with factors such as gender, age, height, weight, occupation, exercise, trauma, and other medical conditions5. LDH is influenced by inflammatory cell infiltration and the release of atypical cytokines, which exacerbate inflammation and pain symptoms6. Currently, no effective drug treatment exists for LDH. Therefore, understanding the immunological characteristics of LDH is essential for developing effective treatment strategies.
Copper is an essential trace element that plays a pivotal role in a multitude of critical biological processes in eukaryotic organisms, including mitochondrial respiration, iron absorption, kinase signaling, autophagy, protein quality control, and antioxidant/detoxification mechanisms7. Recent research has unveiled copper death as an emerging type of cell demise instigated by copper. Noteworthy genes associated with this phenomenon comprise SLC31A1, PDHB, PDHA1, LIPT1, FDX1, DLD, DLST, DBT, LIAS, DLAT, GCSH, ATP7A, and ATP7B8,9. The proposed mechanism involves the direct binding of copper ions to key proteins, such as acyl-CoA dehydrogenase, which is involved in the tricarboxylic acid (TCA) cycle within mitochondria. This interaction results in the depletion of iron-sulfur cluster proteins, ultimately triggering cellular apoptosis. Copper predominantly exists in the form of cytochrome C oxidase (COX) and superoxide dismutase 1 (SOD1) within mitochondria, exhibiting a close association with mitochondrial regulation10,11,12. Furthermore, several studies have indicated that copper exerts a substantial influence on the immune regulation mechanism within the human body13. Under physiological conditions, the intracellular concentration of copper ions is meticulously regulated through active homeostasis mechanisms. However, when the levels of copper ions gradually surpass the threshold, the excess ions can induce excessive cellular respiration, leading to cytotoxicity and potentially resulting in a form of cell death associated with copper toxicity14. Genetic factors, nutritional imbalances, and liver dysfunction are significant contributors to the dysregulation of copper levels in the body, which can have substantial health implications. Dyshomeostasis of copper is linked to various pathological conditions, including Menkes disease (MD), Wilson's disease (WD), neurodegenerative diseases, cancer, and cardiovascular disease15. Notably, both mitochondrial function and immune regulation mechanisms play crucial roles in the initiation and progression of LDH6,16. Consequently, it is plausible to hypothesize that a correlation exists between the onset and progression of LDH and copper depletion.
This study presents the first analysis of the differential expression and immune characteristics of cuproptosis-related genes (CRGs) in individuals with LDH and those without the condition. The primary objective was to explore the pathogenesis of LDH and the potential role of copper dysregulation in its development. To accomplish this, microarray datasets comprising samples from both LDH and non-LDH individuals were sourced from the Gene Expression Omnibus (GEO) database. Patients with LDH were then stratified into two gene clusters based on their expression profiles of CRGs, which were hypothesized to be related to copper-mediated cell death mechanisms. A thorough examination of the expression differences and the immune cell landscape within these clusters was conducted.
Materials and methods
Download and analyze the data of LDH and cuproptosis-related genes
To conduct an analysis of genes associated with copper-induced cell death in the context of Lumbar Disc Herniation (LDH), it is necessary to obtain and examine relevant microarray data. The Gene Expression Omnibus (GEO) (www.ncbi.nlm.nih.gov/geo) provides access to the required LDH microarray data17. Specifically, the datasets GSE124272 and GSE150408, as reported by Wang et al.18, contain transcriptomic data from 8 LDH patients and 8 non-LDH patients, and 17 LDH patients and 17 non-LDH patients, respectively. To address potential heterogeneity resulting from variations in experimental batches and platforms, the R software package “sva” installed on Bioconductor should be utilized (https://bioconductor.org/). The source of the cuproptosis-related genes (CRGs) is Tsvetkov et al. A total of 20 CRGs were identified19. To address potential heterogeneity resulting from variations in experimental batches and platforms, the R software package “sva” installed on Bioconductor should be utilized (https://bioconductor.org/). The source of the cuproptosis-related genes (CRGs) is Tsvetkov et al. A total of 20 CRGs were identified19. The details of these microarray data are listed in the Table 1.
The criteria of sample collections
For the dataset GSE124272, 8 LDH patients (4 males and 4 females, aged 33–60 years) and 8 controls (4 males and 4 females, aged 19–23 years) were recruited. The inclusion criteria for LDH patients were as follows: age between 35 and 60 years, severe lower back pain and sciatica within the past 4 weeks, and LDH confirmed by magnetic resonance imaging (MRI). The control group consisted of individuals aged 18–25 years with no history of lower back pain or sciatica.
The dataset GSE150408 included 17 LDH patients aged 19–54 years (average age of 40 years) and 17 controls without LDH, aged 19–30 years (average age of 23 years). Eligible LDH patients experienced sciatica confirmed by MRI, demonstrating single-level lumbar disc herniation at the L4/5 or L5/S1 level, resulting in nerve root compression. Patients with additional neuropathies, spinal diseases, infections, rheumatism, cardiovascular diseases, metabolic diseases, dementia, mental health disorders, a history of surgery, congenital diseases, tuberculosis, tumors, pregnancy, lactation, or recent medication use were excluded from the study.
CRGs for correlation and differential expression analysis
The limma software package was employed to filter differential CRGs by comparing the expression values of CRGs between LDH and non-LDH samples. The selection criteria for CRGs were based on a log2-fold change (FC) greater than 2 or less than -2, and an adjusted p-value of less than 0.05. Bar plots were generated to visualize these differences, and the correlation coefficients of CRGs expression across the samples were analyzed using the “corrplot” R package.
Evaluation of immune cell infiltration
Gene expression data from the datasets GSE124272 and GSE150408 were utilized to evaluate the expression levels of 22 immune cell types in each sample using the CIBERSORT algorithm. CIBERSORT is a deconvolution algorithm known for its effectiveness in analyzing diverse immune cell subpopulations and quantifying the immune cell composition, thereby improving the precision of the analysis20. In this study, the selection of samples was based on a significance threshold of P < 0.05.
Correlation analysis between CRGs and immune cells and chromosomes
To further elucidate the association between CRGs and immune cell characteristics related to LDH, we employed the “corrplot” R package to examine the correlation coefficients between the expression levels of CRGs and the relative proportions of immune cells. Additionally, we utilized the “RCircos” package in R to visualize the chromosomal locations of CRGs.
Unsupervised clustering and gene set variance analysis of LDH patients
Initially, an unsupervised clustering analysis was conducted using the ConsensusClusterPlus software package21. The k-means algorithm was employed to partition all LDH samples into distinct clusters, with 1000 iterations performed. Subsequently, a comprehensive assessment of the maximum cluster number (k = 9) and the optimal cluster number was accomplished by integrating cumulative distribution function (CDF) curves, consistency matrices, and clustering consistency scores. Furthermore, the suitability of these genes for sample representation was evaluated through the utilization of a combination of PCA and t-distributed stochastic neighbor embedding (tSNE)22,23.
Re-consensus clustering based on the cross-genic clustering of CRGs
Using the genes acquired from unsupervised clustering of LDH patients, we employed the ConsensusClusterPlus and limma packages to reclassify LDH patients and discern distinct subgroups. Subsequently, we conducted an analysis on inter-cluster gene expression and immune features. To examine the expression differences of cuproptosis-related genes (CRGs) between the CRG cluster and other gene clusters, we utilized the ggpubr and reshape2 packages for the analysis.
Weighted gene co-expression network analysis
The utilization of the Weighted Gene Co-expression Network Analysis (WGCNA) method facilitates the examination of gene sets’ expression. In this particular investigation, the WGCNA R package was employed at various stages to establish and partition diverse gene networks into modules24. Initially, the most suitable soft threshold was determined to construct a weighted adjacency matrix. The module characteristic genes epitomize the comprehensive gene expression profiles within each module. The association between modules and disease status is conveyed through the module significance (MS), whereas the gene significance (GS) is delineated as the correlation between genes and clinical phenotypes25.
Gene set variation analysis (GSVA)
To further investigate the variation process of LDH in biological processes, we employed the GSVA package to conduct enrichment analysis. GSVA is a frequently utilized nonparametric and unsupervised method for estimating changes in pathway and biological process activity within expression data sets26. For our analysis, we acquired the “c2.cp.kegg.v6.2-symbols” and “c5.go.symbols” gene sets from the MSigDB database, which were subsequently utilized in the GSVA analysis. Generally, a corrected P-value of less than 0.05 (adjusted for the false discovery rate, FDR) is considered statistically significant and serves as the criterion for gene selection.
Functional enrichment analysis
The genes originating from the target modules were isolated from the network in order to conduct enrichment analysis, thereby facilitating a deeper exploration of the functions associated with each module. The enrichment analysis employed Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways27,28,29, employing a threshold of p value < 0.05 and an enrichment gene count > 2 to ascertain the outcomes of enrichment.
Constructing prediction models based on various machine learning methods
The Random Forest model (RF) is an ensemble machine learning technique that employs multiple autonomous decision trees for the purpose of classification or regression30. The Support Vector Machine model (SVM) is capable of generating a hyperplane with maximum margin in the feature space, thereby effectively discerning data points31. The Generalized Linear Model (GLM) is an extension of the multivariate linear regression model, enabling a more effective and flexible assessment of the association between normally distributed dependent features and categorical or continuous independent features32. The eXtreme Gradient Boosting (XGB) technique is a collection of ensemble trees that utilizes gradient boosting. It incorporates a robust algorithm that takes into account both classification error and model complexity33. In this study, we developed machine learning models using two distinct CRG clusters (RD, SVM, GLM, and XGB) through the utilization of the “caret” package in the R programming language. All models were executed with default parameters and assessed using fivefold cross-validation. To gain insights into the models' performance and interpretability, we employed the “DALEX” package in R to explain the four machine learning models and visualize their residual distribution and feature importance. The “pROC” package was employed to visually represent the area under the Receiver Operating Characteristic (ROC) curve (AUC). Ultimately, the optimal machine learning model was identified, and the top five genes were chosen as predictive variables for LDH.
Construction and validation of modal graph models
Construct a column line graph model utilizing the predictive genes derived from XGB and assess the LDH cluster employing the R package “rms”34. Each predictive variable is allocated a score, and the predictive capability of the Nomo model is evaluated through calibration curves and decision curve analysis (DCA).
Results
Identification and immune infiltration analysis of CRGs
By conducting an analysis of the GSE124272 and GSE150408 datasets, we examined the expression patterns of 14 CRGs in both LDH and non-LDH control groups. The findings revealed that 9 CRGs exhibited significant involvement in the differential expression associated with copper-induced cell death. Specifically, NLRP3, ATP7B, ATP7A, and MTF1 demonstrated significantly higher expression levels in LDH compared to non-LDH samples, whereas LIPT1, DLAT, PDHA1, GCSH, and DLST exhibited significantly lower expression levels in LDH relative to non-LDH samples (as depicted in Fig. 1A,B). Subsequently, we proceeded to examine the chromosomal positions of the 14 CRGs, as shown in Fig. 1C. Notably, a strong correlation was observed among 9 of these CRGs, prompting us to further investigate the relationship between variations in CRGs expression and their involvement in the development of LDH disease, as illustrated in Fig. 1D,E. Moreover, we conducted an analysis of immune infiltration in both LDH and non-LDH samples, as presented in Fig. 2. The findings revealed that LDH patients exhibited elevated levels of plasma cells, neutrophils, and monocytes, while the expression of γδ T cells was comparatively lower, as shown in Fig. 2A,B. This finding suggests a strong association between the development of LDH and the immune system. Subsequently, we performed an analysis of immune infiltration on these 9 CRGs that were differentially expressed. For example, PDHB demonstrated a notable positive correlation with monocytes, while displaying a significant negative correlation with activated mast cells and purified CD4 T cells. Similarly, NLRP3 displayed a significant positive correlation with neutrophils, but exhibited a significant negative correlation with activated CD4 memory T cells and monocytes. MTF1 exhibited a noteworthy positive association with monocytes and a positive association with purified CD4 T cells. LIPT1 displayed a positive association with resting mast cells and monocytes, while exhibiting a negative association with M0 macrophages, activated mast cells, and purified CD4 T cells (refer to Fig. 2C for detailed findings). These findings suggest that these CRGs potentially exert a crucial influence on LDH regulation and immune infiltration.

Correlation analysis of LDH and CRGs: (A) Boxplots showing the expression of 14 CRGs in LDH and non-LDH samples. (B) Heatmap presenting the differentially expressed CRGs. (C) Chromosomal locations of these 14 CRGs. (D) Correlation analysis of the 9 differentially expressed CRGs. (E) Circular plot depicting the correlation analysis results of the 9 differentially expressed CRGs, with red and green indicating positive and negative correlations, respectively. The size of the circles represents the magnitude of the correlation coefficients.

Analysis of LDH and CRG immune cell infiltrations. (A) Relative expression of LDH and non-LDH in 22 infiltrating immune cells. (B) Boxplot illustrating the differences in immune cell infiltrations between LDH and non-LDH. (C) Correlation analysis of the 9 differentially expressed CRGs with the infiltrating immune cells.
Unsupervised clustering identification and analysis of LDH
To analyze the expression of LDH and CRGs, we used a consensus clustering algorithm to group all LDH samples with 9 types of CRGs expression profiles. At k = 2, the observation of the maximum number of clusters is depicted in Fig. 3A. Subsequently, as the value of k increased to 4, a gradual decrease in the cumulative distribution function (CDF) values was observed, as illustrated in Figs. 3B–D. The 25 LDH samples were then categorized into two distinct groups, namely Cluster 1 (n = 17) and Cluster 2 (n = 8). Further analysis through principal component analysis (PCA) of these two clusters demonstrated significant dissimilarities between them, as depicted in Fig. 3E.

Identification of LDH copper death-associated gene molecular clusters. (A) Consensus clustering matrix, k = 2. (B–D) Representative cumulative distribution function (CDF) curve, CDF incremental area curve, and consensus clustering score. (E) Visualization analysis of two clustering distributions.
CRG cluster expression profile and immune infiltration characteristics
To examine the distinctive features of CRG clusters, we conducted an analysis on the differential expression of 9 CRGs between CRG cluster C1 and CRG cluster C2 (Fig. 4A). CRG cluster C1 exhibited elevated expression levels of LIPT1, DLAT, PDHB, DLST, and GCSH, whereas CRG cluster C2 displayed heightened expression levels of NLRP3, ATP7B, and MTF1 (Fig. 4B). Furthermore, we also examined the immune infiltration in CRG cluster C1 and CRG cluster C2 (Fig. 4C). The findings indicate that gene cluster C1 exhibited elevated levels of Macrophages M0, whereas CRG cluster C2 demonstrated heightened expression in T cells CD4 naive, Macrophages M0, Mast cells activated, and Neutrophils (Fig. 4D). In conclusion, the association between CRG cluster C2 and LDH may surpass that of CRGcluster C1.

Expression and immune profiling analysis between two molecular clusters. (A) Heatmap of the expression of 9 CRGs between the two molecular clusters. (B) Boxplot of the expression of 9 CRGs between the two molecular clusters. (C) Relative abundance of the two molecular clusters in 22 infiltrating immune cells. (D) Boxplot of immune infiltration between the two molecular clusters.
GSVA analysis reveals the biological function of CRGs clusters in LDH
The GSVA method is a non-parametric, unsupervised approach commonly employed to evaluate gene set enrichment in gene expression microarray and RNA-sequencing (RNA-seq) data35. This versatile method has been utilized in various studies investigating gene mechanisms related to survival outcomes, including those focused on breast cancer, colon cancer, bladder cancer, and other malignancies36. In our study, we performed GSVA enrichment analysis for KEGG and GO to explore the biological characteristics of different clusters. The results of GSVA KEGG enrichment indicated the widespread involvement of pathways such as oxidative phosphorylation, butanoate metabolism, p53 signaling pathway, protein export, and MAPK signaling pathway in the pathogenesis of LDH (Fig. 5A). The GSVA GO enrichment analysis demonstrated the presence of processes like chemokine (C-X3-C motif) binding, snRNA catabolic process, transcription export complex 2, ribosomal small subunit binding, and other related processes (Fig. 5B). These results indicate the significant involvement of immune response mechanisms in LDH.

GSVA analysis reveals the biological function of CRGs clusters in LDH. (A) GSVA for KEGG pathways analysis. (B) GSVA for biological processes analysis.
Co-expression network construction of the CRGs molecular cluster
The WGCNA algorithm was employed to analyze the gene modules closely associated with CRG cluster. Initially, sample clustering was conducted using the Pearson correlation coefficient, and a sample clustering tree was constructed (Fig. 6A) after eliminating evident outliers. Subsequently, four modules were identified using the dynamic tree-cut method, as illustrated in Fig. 6B. An unweighted network (Fig. 6C) was then established based on the criterion of R2 = 0.9. Upon linking the modules with phenotypic traits, a strong correlation was observed in the disease states (Cluster1 and Cluster2) traits, as depicted in Fig. 6D. The blue module, turquoise module, brown module, and yellow module were chosen for further analysis based on the criterion of clinical significance (p < 0.05). Upon analyzing the relationships between modules and clinical features, it was found that the genes in the ME Blue module exhibited a significantly high correlation of 0.66. The correlation analysis between genes in the ME Blue module and Cluster2 is depicted in Fig. 6E. Consequently, the ME Blue module was selected for subsequent analysis.

Weighted network analysis between two molecular clusters. (A) Dendrogram of co-expression modules. Different colors represent the determination of soft threshold power for different co-expression modules. (B) Detection of co-expression clusters with corresponding color distribution using dendrogram and module-feature relationship graph. (C) Determination of soft threshold power. (D) Correlation analysis between module feature genes and clinical states. (E) Scatter plot of differentially significant genes in the MEblue module and Cluster 2.
Functional analysis of the key module
We performed GO and KEGG enrichment analysis on the key modules of LDH and pathway annotation of genes within these modules to uncover the potential molecular biological processes associated with LDH. Regarding the key clinical modules associated with Lumbar Disc Herniation (LDH), the Gene Ontology (GO) terms in molecular function (MF) indicated significant enrichment in areas such as “structural constituent of ribosome,” “peptide binding,” and “amide binding.” In terms of cellular components, the genes were predominantly enriched in “ribosome,” “cytosolic ribosome,” “ribosomal subunit,” and “large ribosomal subunit.” In relation to biological processes, the genes exhibited significant enrichment in various aspects including “cytoplasmic translation,” “monosaccharide biosynthetic process,” “DNA damage response,” and “signal transduction by p53 class” (Fig. 7A,B). Additionally, the KEGG terms demonstrated enrichment in pathways such as “ribosome,” “pentose phosphate pathway,” “oxidative phosphorylation,” “coronavirus disease-COVID-19,” and “diabetic cardiomyopathy” (Fig. 7C-D). It is noteworthy that the enrichment in “coronavirus disease-COVID-19” and “diabetic cardiomyopathy” may not be directly related to LDH and could be due to the broad nature of the gene sets involved.

Key module function analysis results. (A–B) Gene Ontology (GO) enrichment analysis. (C–D) Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis. BP: Biological process; CC: Cell component; MF: Molecular function.
Construction and evaluation of machine learning models
To enhance the identification of genes possessing significant diagnostic potential, we developed four machine learning models, namely RF, SVM, GLM, and XGB, utilizing the MEblue module hub genes. The “DALEX” package was employed to compare these models and evaluate the residual distribution of each. The findings indicated that the XGB model exhibited the lowest residual value (Fig. 8A,B).Based on the outcomes of fivefold cross-validation, we generated ROC curves for the four models to comprehensively assess their discriminatory performance. The AUC values for these models were as follows: RF: AUC = 0.694; SVM: AUC = 0.714; XGB: AUC = 0.715; GLM: AUC = 0.571. Subsequently, we ranked the top 10 significant genes for each model based on the root mean square error (RMSE) (Fig. 8C). In summary, our findings indicate that the XGB model exhibits superior ability in distinguishing between different patient groups. The XGB model successfully identified five genes (CKS2, LRG1, RAB43, DYSF, and G6PD) as significant predictors for subsequent analysis. To further assess the predictive performance of the XGB model, we generated a bar-line plot (Fig. 9A). Additionally, we employed a combination of the calibration curve and decision curve analysis (DCA) to evaluate the prediction accuracy of the constructed line plot model. Notably, the calibration curve demonstrated minimal discrepancy between the observed and predicted risks of LDH clustering (Fig. 9B–C).

Shows the construction and evaluation of RF, SVM, GLM, and XGB machine learning models. (A) Box plots of residuals for the four machine learning models are displayed. The red dots represent the root mean square error (RMSE) of the residuals. (B) ROC curves of the four machine learning models based on a fivefold cross-validation using the test dataset. (C) Important functions of the four machine learning models.

Validation analysis of XGB predicted genes. (A) Nomogram model for predicting RA risk based on 5 genes in XGB. (B) Construction of calibration curve. (C) Construction of DCA.
Discussion
Lumbar intervertebral disc herniation (LDH) is a prevalent musculoskeletal disorder that leads to low back pain, productivity loss, and disability, imposing substantial socioeconomic burdens37,38. Given the multitude of high-risk factors implicated, the pathogenesis of lumbar disc herniation is notably intricate39. Consequently, timely and precise diagnosis, treatment, and management are of paramount importance for effective management of LDH. In recent decades, considerable advancements have been achieved in the symptomatic alleviation and mitigation of this condition40. The present study reveals that6 the observed instances of copper-dependent cell death are predominantly attributed to the accumulation of lipid acylation-related proteins, depletion of iron-sulfur cluster proteins, and a range of additional stress responses. These cascading events ultimately culminate in cellular demise and exhibit diverse connections with LDH16. However, there is currently a dearth of research investigating the precise mechanisms and impacts of copper in the modulation of cell death in diverse diseases. Consequently, the objective of this study is to elucidate the correlation between CRGs and LDH phenotype, examine their distinct functions in the immune microenvironment, and employ CRGs to forecast LDH subtypes.
This study presents a comprehensive analysis of differentially expressed CRGs in patients with LDH and healthy controls, marking the first investigation of its kind. The findings reveal the identification of nine distinct CRGs that exhibit differential expression. Notably, NLRP3, ATP7B, ATP7A, and MTF1 demonstrate significant upregulation in LDH compared to non-LDH levels, while LIPT1, DLAT, PDHA1, GCSH, and DLST exhibit significant downregulation in LDH compared to non-LDH levels. These results suggest a strong correlation between CRGs and the onset and progression of LDH. Following this, we conducted an immune infiltration analysis on samples from individuals with LDH and those without LDH. The findings revealed elevated levels of plasma cells, neutrophils, and monocytes infiltrating LDH patients, whereas the expression of γδT cells was diminished. This suggests a significant association between the progression of LDH and the immune system. Based on this, we proceeded to examine the correlation between CRGs and expounded upon the connection between CRGs and LDH. In CRG cluster C2, the genes LIPT1, DLAT, PDHB, DLST, and GCSH exhibited elevated expression levels, whereas in CRG cluster C1, NLRP3, ATP7B, and MTF1 displayed high expression levels. Cluster C1 demonstrated higher levels of macrophages M0, while CRGcluster C2 exhibited increased expression in CD4 naive T cells, activated macrophages M0, activated mast cells, and neutrophils. Our GSVA analysis revealed that the CRGclustes associated with cell death play a role in the regulation of immune response in LDH. This study employed the WGCNA algorithm to analyze the gene modules closely associated with CRG cluster, aiming to deepen the understanding of its biological role in LDH. Four gene modules, namely the blue module, turquoise module, brown module, and yellow module, were identified. Notably, the ME Blue module exhibited the highest gene significance, warranting additional investigation. The GO enrichment analysis demonstrated that genes within the blue module exhibited enrichment in various terms, including structural constituent of ribosome, peptide binding, amide binding, ribosome, cytosolic ribosome, ribosomal subunit, large ribosomal subunit, cytoplasmic translation, monosaccharide biosynthetic, among others (Fig. 7A). Moreover, based on the findings of the KEGG enrichment analysis, it was observed that genes within the blue module exhibited significant enrichment in various biological processes, including ribosome biogenesis, the pentose phosphate pathway, oxidative phosphorylation, as well as diseases such as coronavirus disease (COVID-19) and diabetic cardiomyopathy. Additionally, the pentose phosphate pathway was identified as another notable enrichment within this module (Fig. 7B).
In recent times, machine learning (ML) has gained significant traction in the clinical domain and is recognized as a crucial instrument in healthcare41. In contrast to conventional statistical models, the extensive analysis facilitated by machine learning guarantees the resilience of models and enhances prediction accuracy through iterative algorithms42,43. This study undertakes a comparative evaluation of the predictive capabilities of four machine learning models (RF, SVM, GLM, and XGB). These models are constructed based on 61 genes that exhibit a close association with the CRG cluster within the MEblue module of the GSE124272 and GSE150408 datasets. The XGBoost-based prediction model was developed and yielded the most accurate predictive outcome, with an AUC value of 0.715. Consequently, a total of five significant predictive genes (CKS2, LRG1, RAB43, DYSF, and G6PD) were identified through the utilization of XGB. CKS2, specifically, is a small, highly conserved cyclin-dependent kinase (CDK) interaction protein (10 kDa). As a crucial member of the CKS family, CKS2 plays a pivotal role in human embryogenesis, regulation of the cell cycle, somatic cell division, and apoptosis regulation. Moreover, CKS2 exhibits abnormal expression in various malignant tumor tissues and is closely linked to the biological characteristics of tumor initiation, progression, and metastasis44. Prior research has extensively examined CKS2, indicating its ability to augment the expression of cell cycle proteins, namely cyclin A, cyclin B1, and CDK1, thereby facilitating the proliferation of cancer cells. Moreover, multiple investigations have underscored the significance of CKS2 in preserving the functionality of hematopoietic stem cells45. Furthermore, evidence suggests that46 CKS2 might contribute to the advancement of rheumatoid arthritis. Nonetheless, the precise role and underlying mechanisms of CKS2 remain elusive. The findings of our research indicate that CKS2 could potentially serve as a novel functional regulator in the progression of LDH.
Leucine-rich α-2 glycoprotein 1 (LRG1) is a secreted member of the leucine-rich repeat (LRR) protein family, which has been identified as a pattern recognition system in the innate immune system. It is capable of recognizing motifs and engaging in protein–protein interactions, thereby contributing to various biological processes47. Notably, LRG1 has been found to be significantly upregulated in cancer and diabetes, indicating its potential involvement in these pathologies. Moreover, LRG1 exhibits multifunctionality as a pathogenic signaling molecule, with notable expression in a range of diseases including infection, cardiovascular, renal, pulmonary, neural, and autoimmune disorders. While the correlation between LRG1 levels and diseases does not establish causality, substantial evidence exists indicating that heightened levels or aberrant expression of LRG1 actively contribute to pathological alterations in various diseases48. Prior studies have identified49 serum LRG1 as a significant diagnostic indicator for monitoring autoimmune conditions such as lupus nephritis, psoriasis, rheumatoid arthritis, and vasculitis50,51,52,53. Research conducted by Codina R et al. has indicated that54 LRG1 functions as an acute-phase protein, exhibiting a swift rise in serum levels following microbial infection and other inflammatory triggers. Furthermore, LRG1 plays a crucial role in facilitating immune cell participation at sites of inflammation, promoting the extravasation and activation of neutrophils, and augmenting the differentiation of naïve CD4pos T cells into pro-inflammatory Th17 lymphocytes49. The aforementioned findings contribute to the existing body of evidence that establishes a correlation between the overexpression of LRG1 and various inflammatory conditions. To summarize, LRG1 assumes a significant role in immune cell signaling, immune responses, and the inflammatory process.
Ras-related GTP-binding protein 43 (RAB43) is a constituent of the Ras superfamily, predominantly localized within the endoplasmic reticulum and Golgi apparatus. It serves as a pivotal regulatory element in membrane transport, vesicle mobility, signal transduction, and tethering occurrences55. Prior investigations have demonstrated56 the involvement of RAB43 in the modulation of diverse signal transduction pathways linked to cellular invasion, apoptosis, and immune response. In their study, Li et al. made the discovery that an elevated expression of RAB43 is indicative of a poor prognosis and is associated with the occurrence of epithelial-mesenchymal transition in glioblastoma57. The Rab43 GTPase plays a critical role in facilitating the post-synaptic transport and neuron-specific sorting of G protein-coupled receptors58. Another research investigation has provided evidence that the administration of HMGB1 can impede the forward transport of CD91, which is regulated by Rab43, from the endoplasmic reticulum to the cell surface. This inhibition subsequently suppresses BMDM-mediated phagocytosis and delays the resolution of inflammation59. Notably, both the endoplasmic reticulum and immune response play pivotal roles in the pathogenesis of LDH and are intricately linked to Ras-related GTP-binding protein 43 (RAB43)60. Consequently, it is hypothesized that RAB43 may be closely associated with LDH by modulating various signal transduction pathways implicated in the endoplasmic reticulum and immune response.
Dysferlin, also known as Dysferlin or DYSF, is a transmembrane glycoprotein belonging to the ferlin family. It is predominantly found in skeletal muscle and cardiac tissue, where it is situated on the plasma membrane. Dysferlin interacts with Caveolin 3 (Cav3) and Mitsgumin 53 (MG53), key components of the membrane repair system, and is essential for vesicle fusion during the process of plasma membrane repair61. In the absence of Dysferlin, muscle tissues exhibit inflammatory foci, infiltration of monocytes, and heightened activation of the NFκB signaling pathway62. The dysregulation of DYSF expression has been found to be closely linked to various hereditary muscle diseases and autoimmune disorders. In a study conducted by Xiao et al.63,64, the significant role of DYSF in the progression of two subgroups of idiopathic inflammatory myopathies (IIM), namely dermatomyositis (DM) and polymyositis (PM), was reported. In addition, DYSF is one of the cuproptosis-related asthma diagnostic genes65. Dysferlin has the potential to regulate LDH by influencing the secretion of multiple inflammatory factors and the metabolism of mononuclear cells. This suggests that dysferlin could play a role in the immune response and cellular processes associated with LDH.
The primary role of Glucose-6-phosphate dehydrogenase (G6PD) is to facilitate the production of ribose and the reducing equivalent nicotinamide adenine dinucleotide phosphate (NADPH) via the pentose phosphate pathway (PPP)66. Plays a critical role in the synthesis of various biomolecules, including nucleic acids and fatty acids. Inadequate G6PD activity can result in hindered cell growth and increased mortality rates. Profound G6PD deficiency can impede embryonic development and hinder the growth of organisms. The present study reveals that alterations in G6PD activity are linked to autophagy, insulin resistance, infection, inflammation, and the pathophysiology of diabetes, and hypertension. Furthermore, abnormal activation of G6PD in various types of cancer results in increased cell proliferation and improved adaptability67. Moreover, G6PD may play a role in viral replication and the regulation of vascular smooth muscle function68. G6PD represents a newly identified gene signature linked to cuproptosis, with significant implications for the prognosis, clinical management, and potential immunotherapeutic strategies in hepatocellular carcinoma69. Notably, our findings demonstrate that G6PD exhibits a heightened diagnostic significance.
This study represents the initial exploration of the role of CRGs in LDH, drawing upon existing knowledge. Although promising findings have been obtained, it is crucial to acknowledge the limitations inherent in our research, which primarily relies on bioinformatics analysis. To establish a more robust understanding of the relationship between candidate genes implicated in copper-induced cell death and LDH, future investigations should encompass more extensive clinical or experimental studies.
Conclusions
In summary, our study represents the discriminatory potential of CRGs in LDH. Models constructed using machine learning methods and five genes (CKS2, LRG1, RAB43, DYSF, and G6PD) demonstrated strong predictive capabilities. These results offer insights into the pathogenesis and therapeutic research of LDH moving forward.
Data availability
The data can be found in the GEO database. Further inquiries can be directed to the corresponding author.
References
Hu, B. et al. Determining the minimum effective concentration of ropivacaine in epidural anesthesia for tolerable pain in transforaminal percutaneous endoscopic lumbar discectomy to avoid nerve injury: A double-blind study using a biased-coin design. Drug Des. Dev. Ther. 16, 315–323. https://doi.org/10.2147/dddt.S334605 (2022).
Wang, Y., Gao, F. & Zou, H. Numbness and weakness recovered at a less extent in patients with lumbar disc herniation after percutaneous endoscopic lumbar discectomy. Pain Res. Manag. 2019, 4642701. https://doi.org/10.1155/2019/4642701 (2019).
Hao, L. et al. Recurrent disc herniation following percutaneous endoscopic lumbar discectomy preferentially occurs when Modic changes are present. J. Orthop. Surg. Res. 15, 176. https://doi.org/10.1186/s13018-020-01695-6 (2020).
Zhang, Y. et al. Comparison of the application value of two commonly used minimally invasive spinal surgery in the treatment of lumbar disc herniation. Exp. Ther. Med. 21, 299. https://doi.org/10.3892/etm.2021.9730 (2021).
Suzuki, S. et al. Excessive reactive oxygen species are therapeutic targets for intervertebral disc degeneration. Arthritis Res. Ther. 17, 316. https://doi.org/10.1186/s13075-015-0834-8 (2015).
Li, K. et al. Computational analysis of the immune infiltration pattern and candidate diagnostic biomarkers in lumbar disc herniation. Front. Mol. Neurosci. 15, 846554. https://doi.org/10.3389/fnmol.2022.846554 (2022).
Tsang, T., Davis, C. I. & Brady, D. C. Copper biology. Curr. Biol. CB 31, R421-r427. https://doi.org/10.1016/j.cub.2021.03.054 (2021).
Cui, X. et al. The molecular mechanisms of defective copper metabolism in diabetic cardiomyopathy. Oxid. Med. Cell. Longev. 2022, 5418376. https://doi.org/10.1155/2022/5418376 (2022).
Oliveri, V. Selective targeting of cancer cells by copper ionophores: An overview. Front. Mol. Biosci. 9, 841814. https://doi.org/10.3389/fmolb.2022.841814 (2022).
Tang, D., Chen, X. & Kroemer, G. Cuproptosis: A copper-triggered modality of mitochondrial cell death. Cell Res. 32, 417–418. https://doi.org/10.1038/s41422-022-00653-7 (2022).
Soto, I. C., Fontanesi, F., Liu, J. & Barrientos, A. Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim. Biophys. Acta 883–897, 2012. https://doi.org/10.1016/j.bbabio.2011.09.005 (1817).
Dennerlein, S. & Rehling, P. Human mitochondrial COX1 assembly into cytochrome c oxidase at a glance. J. Cell Sci. 128, 833–837. https://doi.org/10.1242/jcs.161729 (2015).
Wang, Z. et al. Exosomes secreted by macrophages upon copper ion stimulation can promote angiogenesis. Mater. Sci. Eng. C Mater. Biol. Appl. 123, 111981. https://doi.org/10.1016/j.msec.2021.111981 (2021).
Chen, J. et al. The molecular mechanisms of copper metabolism and its roles in human diseases. Pflug. Arch. Eur. J. Physiol. 472, 1415–1429. https://doi.org/10.1007/s00424-020-02412-2 (2020).
Chen, L., Min, J. & Wang, F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct. Target. Therapy 7, 378. https://doi.org/10.1038/s41392-022-01229-y (2022).
Park, J. B., Lee, J. K., Park, S. J., Kim, K. W. & Riew, K. D. Mitochondrial involvement in fas-mediated apoptosis of human lumbar disc cells. J. Bone Joint Surg. 87, 1338–1342. https://doi.org/10.2106/jbjs.D.02527 (2005).
Zou, Y. et al. Leveraging diverse cell-death patterns to predict the prognosis and drug sensitivity of triple-negative breast cancer patients after surgery. Int. J. Surg. (London, England) 107, 106936. https://doi.org/10.1016/j.ijsu.2022.106936 (2022).
Wang, Y. et al. Transcriptome signatures reveal candidate key genes in the whole blood of patients with lumbar disc prolapse. Exp. Ther. Med. 18, 4591–4602. https://doi.org/10.3892/etm.2019.8137 (2019).
Tsvetkov, P. et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science (New York, N.Y.)375 , 1254-1261. https://doi.org/10.1126/science.abf0529 (2022).
Newman, A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782. https://doi.org/10.1038/s41587-019-0114-2 (2019).
Wilkerson, M. D. & Hayes, D. N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics (Oxford, England) 26, 1572–1573. https://doi.org/10.1093/bioinformatics/btq170 (2010).
Nguyen, T. S. & Fernando, B. Effective multimodal encoding for image paragraph captioning. IEEE Trans. Image Process. Publ. IEEE Signal Process. Soc. 31, 6381–6395. https://doi.org/10.1109/tip.2022.3211467 (2022).
Jiang, Z. et al. A novel risk score model of lactate metabolism for predicting over survival and immune signature in lung adenocarcinoma. Cancers https://doi.org/10.3390/cancers14153727 (2022).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Lai, Y. et al. Identification and immunological characterization of cuproptosis-related molecular clusters in Alzheimer’s disease. Front. Aging Neurosci. 14, 932676. https://doi.org/10.3389/fnagi.2022.932676 (2022).
Ma, C., Kang, W., Yu, L., Yang, Z. & Ding, T. AUNIP expression is correlated with immune infiltration and is a candidate diagnostic and prognostic biomarker for hepatocellular carcinoma and lung adenocarcinoma. Front. Oncol. 10, 590006. https://doi.org/10.3389/fonc.2020.590006 (2020).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. Publ. Protein Soc. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587-d592. https://doi.org/10.1093/nar/gkac963 (2023).
Li, W. et al. Machine learning-based prediction of lymph node metastasis among osteosarcoma patients. Front. Oncol. 12, 797103. https://doi.org/10.3389/fonc.2022.797103 (2022).
Lee, Y. W., Choi, J. W. & Shin, E. H. Machine learning model for predicting malaria using clinical information. Comput. Biol. Med. 129, 104151. https://doi.org/10.1016/j.compbiomed.2020.104151 (2021).
Hurtado, P. J. & Richards, C. Building mean field ODE models using the generalized linear chain trick & Markov chain theory. J. Biol. Dyn. 15, S248-s272. https://doi.org/10.1080/17513758.2021.1912418 (2021).
Li, W. et al. An external-validated prediction model to predict lung metastasis among osteosarcoma: A multicenter analysis based on machine learning. Comput. Intell. Neurosci. 2022, 2220527. https://doi.org/10.1155/2022/2220527 (2022).
Kong, D., Zhao, J., Tang, S., Shen, W. & Lee, H. K. Logarithmic data processing can be used justifiably in the plotting of a calibration curve. Anal. Chem. 93, 12156–12161. https://doi.org/10.1021/acs.analchem.1c02011 (2021).
Zhou, Z. et al. Hematopoietic gene expression regulation through m(6)A methylation predicts prognosis in stage iii colorectal cancer. Front. Oncol. 10, 572708. https://doi.org/10.3389/fonc.2020.572708 (2020).
Zhong, Z. et al. Identification of specific cervical cancer subtypes and prognostic gene sets in tumor and nontumor tissues based on GSVA analysis. J. Oncol. 2022, 6951885. https://doi.org/10.1155/2022/6951885 (2022).
Feng, Y., Gao, Y., Yang, W. & Feng, T. Reduction in nerve root compression by the nucleus pulposus after Feng’s spinal manipulation. Neural Regen. Res. 8, 1139–1145. https://doi.org/10.3969/j.issn.1673-5374.2013.12.009 (2013).
Zhou, M., Theologis, A. A. & O’Connell, G. D. Understanding the etiopathogenesis of lumbar intervertebral disc herniation: From clinical evidence to basic scientific research. JOR Spine 7, e1289. https://doi.org/10.1002/jsp2.1289 (2024).
Wang, L., Fan, W., Yu, C., Lang, M. & Sun, G. Clinical effects of electrical stimulation therapy on lumbar disc herniation-induced sciatica and its influence on peripheral ROS level. J. Musculoskelet. Neuronal Interact. 18, 393–398 (2018).
Xie, Z. et al. TNFAIP3 alleviates pain in lumbar disc herniation rats by inhibiting the NF-κB pathway. Ann. Transl. Med. 10, 80. https://doi.org/10.21037/atm-21-6499 (2022).
Dong, S. et al. Development and validation of a novel predictive model and web calculator for evaluating transfusion risk after spinal fusion for spinal tuberculosis: A retrospective cohort study. BMC Musculoskelet. Disord. 22, 825. https://doi.org/10.1186/s12891-021-04715-6 (2021).
Peiffer-Smadja, N. et al. Machine learning for clinical decision support in infectious diseases: A narrative review of current applications. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 26, 584–595. https://doi.org/10.1016/j.cmi.2019.09.009 (2020).
Choy, G. et al. Current applications and future impact of machine learning in radiology. Radiology 288, 318–328. https://doi.org/10.1148/radiol.2018171820 (2018).
Feng, F., Zhao, Z., Cai, X., Heng, X. & Ma, X. Cyclin-dependent kinase subunit2 (CKS2) promotes malignant phenotypes and epithelial-mesenchymal transition-like process in glioma by activating TGFβ/SMAD signaling. Cancer Med. 12, 5889–5907. https://doi.org/10.1002/cam4.5381 (2023).
Grey, W. et al. The CKS1/CKS2 proteostasis axis is crucial to maintain hematopoietic stem cell function. HemaSphere 7, e853. https://doi.org/10.1097/hs9.0000000000000853 (2023).
Jiang, F., Zhou, H. & Shen, H. Identification of critical biomarkers and immune infiltration in rheumatoid arthritis based on WGCNA and LASSO Algorithm. Front. Immunol. 13, 925695. https://doi.org/10.3389/fimmu.2022.925695 (2022).
Haupt, H. & Baudner, S. Isolation and characterization of an unknown, leucine-rich 3.1-S-alpha2-glycoprotein from human serum (author’s transl). Hoppe-Seyler’s Z. Physiol. Chem. 358, 639–646 (1977).
Ng, A. & Xavier, R. J. Leucine-rich repeat (LRR) proteins: Integrators of pattern recognition and signaling in immunity. Autophagy 7, 1082–1084. https://doi.org/10.4161/auto.7.9.16464 (2011).
Camilli, C., Hoeh, A. E., De Rossi, G., Moss, S. E. & Greenwood, J. LRG1: An emerging player in disease pathogenesis. J. Biomed. Sci. 29, 6. https://doi.org/10.1186/s12929-022-00790-6 (2022).
Yang, Y. et al. Leucine-rich α2-glycoprotein-1 upregulation in plasma and kidney of patients with lupus nephritis. BMC Nephrol. 21, 122. https://doi.org/10.1186/s12882-020-01782-0 (2020).
Rioux, G. et al. The tissue-engineered human psoriatic skin substitute: A valuable in vitro model to identify genes with altered expression in lesional psoriasis. Int. J. Mol. Sci. https://doi.org/10.3390/ijms19102923 (2018).
Fujimoto, M. et al. Leucine-rich α2 -glycoprotein as a potential biomarker for joint inflammation during anti-interleukin-6 biologic therapy in rheumatoid arthritis. Arthritis Rheumatol. (Hoboken, N.J.) 67, 2056–2060. https://doi.org/10.1002/art.39164 (2015).
Ishizaki, J. et al. Targeted proteomics reveals promising biomarkers of disease activity and organ involvement in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Res. Therapy 19, 218. https://doi.org/10.1186/s13075-017-1429-3 (2017).
Codina, R., Vanasse, A., Kelekar, A., Vezys, V. & Jemmerson, R. Cytochrome c-induced lymphocyte death from the outside in: Inhibition by serum leucine-rich alpha-2-glycoprotein-1. Apoptosis Int. J. Program. Cell Death 15, 139–152. https://doi.org/10.1007/s10495-009-0412-0 (2010).
Dejgaard, S. Y. et al. Rab18 and Rab43 have key roles in ER-Golgi trafficking. J. Cell Sci. 121, 2768–2781. https://doi.org/10.1242/jcs.021808 (2008).
Wang, R. et al. Analysis of 52 Rab GTPases from channel catfish and their involvement in immune responses after bacterial infections. Dev. Comp. Immunol. 45, 21–34. https://doi.org/10.1016/j.dci.2014.01.026 (2014).
Han, M. Z. et al. High expression of RAB43 predicts poor prognosis and is associated with epithelial-mesenchymal transition in gliomas. Oncol. Rep. 37, 903–912. https://doi.org/10.3892/or.2017.5349 (2017).
Wei, Z. et al. Rab43 GTPase directs postsynaptic trafficking and neuron-specific sorting of G protein-coupled receptors. J. Biol. Chem. 296, 100517. https://doi.org/10.1016/j.jbc.2021.100517 (2021).
Wang, Y. et al. Extracellular HMGB1 impairs macrophage-mediated efferocytosis by suppressing the Rab43-controlled cell surface transport of CD91. Front. Immunol. 13, 767630. https://doi.org/10.3389/fimmu.2022.767630 (2022).
Wang, H. et al. Role of death receptor, mitochondrial and endoplasmic reticulum pathways in different stages of degenerative human lumbar disc. Apoptosis Int. J. Program. Cell Death 16, 990–1003. https://doi.org/10.1007/s10495-011-0644-7 (2011).
Spadafora, P. et al. A novel homozygous variant in DYSF gene is associated with autosomal recessive limb girdle muscular dystrophy R2/2B. Int. J. Mol. Sci. https://doi.org/10.3390/ijms23168932 (2022).
Cohen, T. V. et al. Upregulated IL-1β in dysferlin-deficient muscle attenuates regeneration by blunting the response to pro-inflammatory macrophages. Skelet. Muscle 5, 24. https://doi.org/10.1186/s13395-015-0048-4 (2015).
Cacciottolo, M., Nogalska, A., D’Agostino, C., Engel, W. K. & Askanas, V. Dysferlin is a newly identified binding partner of AβPP and it co-aggregates with amyloid-β42 within sporadic inclusion-body myositis (s-IBM) muscle fibers. Acta Neuropathol. 126, 781–783. https://doi.org/10.1007/s00401-013-1186-6 (2013).
Xiao, Y. et al. Global analysis of protein expression in muscle tissues of dermatomyositis/polymyosisits patients demonstrated an association between dysferlin and human leucocyte antigen A. Rheumatology (Oxford England) https://doi.org/10.1093/rheumatology/kez085 (2019).
Wang, F., Su, Q. & Li, C. Identification of cuproptosis-related asthma diagnostic genes by WGCNA analysis and machine learning. J. Asthma Off. J. Assoc. Care Asthma 60, 2052–2063. https://doi.org/10.1080/02770903.2023.2213334 (2023).
Yang, H. C., Wu, Y. H., Liu, H. Y., Stern, A. & Chiu, D. T. What has passed is prolog: New cellular and physiological roles of G6PD. Free Radic. Res. 50, 1047–1064. https://doi.org/10.1080/10715762.2016.1223296 (2016).
Cai, T. et al. Glucose-6-phosphate dehydrogenase and NADPH oxidase 4 control STAT3 activity in melanoma cells through a pathway involving reactive oxygen species, c-SRC and SHP2. Am. J. Cancer Res. 5, 1610–1620 (2015).
Meng, Q. et al. Recent findings in the regulation of G6PD and its role in diseases. Front. Pharmacol. 13, 932154. https://doi.org/10.3389/fphar.2022.932154 (2022).
He, F. et al. Identification and validation of a novel cuproptosis-related genes signature associated with prognosis, clinical implications and immunotherapy of hepatocellular carcinoma. Front. Pharmacol. 14, 1088993. https://doi.org/10.3389/fphar.2023.1088993 (2023).
Acknowledgements
This work was supported by Natural Science Foundation of Zhejiang Province (Grant Numbers: LQ22H270007 and LQ24H270014), Zhejiang Chinese Medicine University 2023 affiliated hospital research project (Grant Number: 2023FSYYZQ06) and Zhejiang Province Traditional Chinese Medicine Science and Technology Project (Grant Number: 2022ZB121).
Author information
Authors and Affiliations
Contributions
H.P.X. and Y.H.J. were involved in writing, draft preparation, and contributed equally to this work as co-first authors. Y.W. and Q.L. were responsible for reviewing the concept design, article writing, and proofreading. Professor H.G.D. and J.X. conceived the study designing, writing, acquisition, analysis or interpretation of data. All authors critically revised the manuscript and approved its final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, H., Jiang, Y., Wen, Y. et al. Identification of copper death-associated molecular clusters and immunological profiles for lumbar disc herniation based on the machine learning. Sci Rep 14, 19294 (2024). https://doi.org/10.1038/s41598-024-69700-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-69700-0
