
Abstract
Bruton tyrosine kinase inhibitor (BTKi) combined with rituximab-based chemotherapy benefits diffuse large B-cell lymphoma (DLBCL) patients. However, drug resistance is the major cause of relapse and death of DLBCL. In this study, we conducted a comprehensive analysis BTKi-resistance related genes (BRRGs) and established a 10-gene (CARD16, TRIP13, PSRC1, CASP1, PLBD1, CARD6, CAPG, CACNA1A, CDH15, and NDUFA4) signature for early identifying high-risk DLBCL patients. The resistance scores based on the BRRGs signature were associated with prognosis. Furthermore, we developed a nomogram incorporating the BRRGs signature, which demonstrated excellent performance in predicting the prognosis of DLBCL patients. Notably, tumor immune microenvironment, biological pathways, and chemotherapy sensitivity were different between high- and low-resistance score groups. Additionally, we identified TRIP13 as a key gene in our model. TRIP13 was found to be overexpressed in DLBCL and BTKi-resistant DLBCL cell lines, knocking down TRIP13 suppresses cell proliferation, promotes cell apoptosis, and enhances the apoptosis effect of BTKi on DLBCL cells by regulating the Wnt/β-catenin pathway. In conclusion, our study presents a novel BRRGs signature that could serve as a promising prognostic marker in DLBCL, and TRIP13 might be a potential therapeutic target for resistant DLBCL.
Similar content being viewed by others


Introduction
Lymphoma is a group of heterogeneous malignancies caused by the clonal proliferation of lymphocytes, which are classified into Hodgkin’s lymphoma (HL) and non-Hodgkin’s lymphoma (NHL)1. Diffuse large B-cell lymphoma (DLBCL) is one of the most common NHL subtypes, accounting for approximately 30–40% of newly diagnosed cases2. Currently, rituximab combined with cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) immunochemotherapy regimen has greatly improved the remission rate of DLBCL. However, 30–40% of patients still experience drug resistance or relapse, these relapsed/refractory patients have few treatment options and poor prognosis, and the 2-year overall survival (OS) is only 20–40%3. The development of drug resistance is a complex process, with gene expression disorders playing a crucial role. Therefore, it is necessary to screen for key genes associated with drug resistance. Additionally, analyzing the impact of these genes on prognosis and drug sensitivity is an effective strategy to identify new therapeutic targets to reverse drug resistance and improve prognosis.
Bruton's tyrosine kinase inhibitor (BTKi) was approved by the US Food and Drug Administration (FDA) for the treatment of several B-cell lymphomas4. BTKi mainly inhibits tumor cell proliferation, adhesion, and migration by blocking the activity of the B-cell receptor (BCR) and downstream signaling cascades, and it exerts an inhibitory effect on stromal cells in the tumor microenvironment5,6,7. Recent clinical trials have shown that BTKi combined with rituximab-based chemotherapy benefits part subtype of DLBCL and is recommended for the treatment of relapsed/refractory non-germinal center B (non-GCB) DLBCL patients8. It also has some efficacy in patients with rare and aggressive primary central nervous system lymphoma, which in 90% of cases is DLBCL9,10,11. However, some patients do not benefit from this regimen due to primary and acquired resistance 12. Overall, the generation of BTKi resistance is related to target mutations, gene expression disorders, activation of bypass pathways, and the tumor immune microenvironment (TiME)13. Currently, research on BTKi resistance is limited, and few studies have investigated the important role of BTKi-resistance related genes (BRRGs) in patient prognosis and drug sensitivity of DLBCL.
Thyroid hormone receptor interactor 13 (TRIP13) belongs to ATPases family, which plays a role in promoting promotes tumor progression by influencing various biological activities such as chromosome synapsis, checkpoint signaling, and DNA break formation and recombination14. Previous studies have indicated that TRIP13 is overexpressed in various tumors, including lung cancer15, hepatocellular carcinoma16, colorectal cancer17, and multiple myeloma18. However, the exact role of TRIP13 in DLBCL development and drug resistance remains poorly understood.
With the rapid development of genomics, bioinformatics technology has shown outstanding advantages in analyzing large-scale clinical data and discovering new key targets and biomarkers. In this study, we comprehensively analyzed the BRRGs and constructed a novel signature that showed good predictive power for prognosis in DLBCL patients. We further analyzed the differences between TiME, chemosensitivity, and biological pathways in DLBCL patients with different resistance score groups. Furthermore, we elucidated the substantial impact and mechanism of the model key gene TRIP13 in promoting the DLBCL development and BTKi resistance via regulating the Wnt/β-catenin pathway. Taken together, our study presents a novel BRRGs signature that could serve as a promising prognostic marker and identifies TRIP13 might be a potential therapeutic target for resistant DLBCL.
Materials and methods
Data collection
The transcriptome data and corresponding clinical data were downloaded from the Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) database. All transcriptome data in Fragments Per Kilobase Million (FPKM) format underwent conversion to Transcripts Per Kilobase Million (TPM) normalized format for subsequent analysis using RStudio software. After excluding cases with missing prognostic information, the GSE31312 dataset comprised 471 cases and served as the training cohort for the prognostic model. Additionally, the GSE87371 and GSE10846 datasets, with 121 and 233 cases respectively, were utilized as external validation cohorts. The baseline characteristics of DLBCL patients from different cohorts are summarized in Table 1.
Identification of differentially expressed BRRGs
To identify key genes associated with ibrutinib resistance, we downloaded the transcriptome data of ibrutinib-resistant and non-resistant DLBCL cell lines (GSE138126) from the GEO database. All cell line was cultured with a medium containing ibrutinib for over 8 months, and resistant cell clones were selected. The ‘limma’ package (version 3.5.1) was used to identify differentially expressed BRRGs (DEBRRGs), and DEGs threshold was set as follows: |log2-fold change (FC)|> 2 and an adjusted P-value < 0.05. Volcano Plot and heatmap were conducted with the ‘pheatmap’ package for visualizing the gene expression differences.
Functional enrichment analysis
The ‘cluster Profiler’ package was used to gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of DEBRRGs involved in disease progression. The annotated gene sets of GSEA were selected, c2.cp.kegg.v2023.1.Hs.entrez and c5.go.bp.v2023.1.Hs.entrez sets from the Molecular Signature Database (MSigDB) (https://www.gseamsigdb.org/gsea/msigdb/index.jsp). The number of permutations was set to 1,000. The criteria for screening statistically significant pathways were set as adjusted P-value less than 0.05. Visualization was performed using the ‘enrichplot’ package.
Establishment and validation of BRRGs prognostic signature
The prognostic signature was constructed using the GSE31312 dataset as the training cohort, with the GSE87371 and GSE10846 datasets serving as external validation cohorts. Univariate and LASSO regression analysis was utilized to screen the risk model containing the BRRGs, and the risk model was defined as the BRRGs signature. The resistance score for each patient in the training cohort was computed using the following formula: \(\text{Resistance score}={\sum }_{i=1}^{N}(\text{exp}\times coef)\), where N is the number of model genes; exp represents the gene expression value of each gene; coef represents the coefficient index.
To validate the prognostic model, the resistance score for each individual was calculated through the regression coefficients and their expression. All cohorts of DLBCL patients were divided into high- and low-resistance score groups by the median resistance score. The disparities in overall survival (OS) among the groups were assessed utilizing the Kaplan–Meier survival analysis. The ‘timeROC’ package was utilized to construct the time-dependent receiver operating characteristic (ROC) curve and determine the area under the curve (AUC) of the 2-year, 3-year, and 5-year OS in the DLBCL patients. The predictive efficacy of the model was evaluated based on the AUC.
Immune infiltration assessment
The ‘GSVA’ package was used to perform a gene set enrichment analysis ssGSEA algorithm to unambiguously present the infiltrating score of 29 tumor-infiltrating immune cells and pathways in each sample (aDCs, APC co-inhibition, APC co-stimulation, B cells, CCR, CD8+ T cells, Check-point, Cytolytic activity, DCs, HLA, iDCs, Inflammation-promoting, Macrophages, Mast cells, MHC class I, Neutrophils, NK cells, Parainflammation, pDCs, T cell co-inhibition, T cell co-stimulation, T helper cells, Tfh, Th1 cells, Th2 cells, TIL, Treg, Type I IFN Response, and Type II IFN Response). Furthermore, we further analyzed the differences between the high- and low-resistance score groups.
Chemotherapeutic response prediction
The ‘pRRophetic’ package (version 0.5) was used to predict chemosensitivity between different groups, primarily mainly construct ridge regression model to infer half-maximal inhibitory concentration (IC50) values based on gene expression levels through ten-fold cross-validation19,20. The dataset within the ‘pRRophetic’ package is derived from the “cgp2016” initiative, encompassing gene expression matrices and drug treatment information. We analyzed common chemotherapy drugs and used boxplots to illustrate differences in drug sensitivity between the two groups.
Weighted gene co‑expression network analysis (WGCNA)
The ‘WGCNA’ package was used to identify BTKi-resistance gene clusters highly associated with DLBCL based on the GSE138126 dataset. The weighted adjacency matrix was converted into a topological overlap matrix (TOM) according to the optimal soft threshold (β = 10), and then hierarchical clustering analysis was performed to detect the correlation between gene modules (minmodulesize = 100; mergecutheight = 0.2). Interaction strength was assessed using the heatmap toolkit, and gene significance (GS) and module membership (MM) were calculated to assess the relationship between module and resistance characteristics.
Statements, patient samples and cell culture
All experiments were performed in accordance with relevant guidelines and regulations. 20 DLBCL tissues and 5 lymph node reactive hyperplasia tissues were obtained from the Second Affiliated Hospital of Anhui Medical University. All DLBCL patient’s baseline characteristics were summarized in Supplementary Table 1. This study was approved by the Ethics Committee of the Second Hospital of Anhui Medical University, and informed consent was obtained from the patients. Human DLBCL cell lines SU-DHL-2 (RRID:CVCL_9550) and SU-DHL-4 (RRID:CVCL_0539) were purchased from the Institute of Biochemistry and Cell Biology of the Chinese Academy of Science. Cells were cultured in RPMI-1640 (Hyclone, Logan, UT, USA) containing 10% FBS at 37 °C cell incubators with 5% CO2.
SiRNA transfection
Cells were transfected with TRIP13 siRNA and negative control siRNA (GenePharma, China) using LipofectamineTM 3000 (Invitrogen, USA) reagent following the manufacturer's instructions. Briefly, 4 × 105 cells were seeded in a 24-well plate, dilute 4 µL siRNA (20 µM) with 50 µL Opti-MEM, and dilute 1uL LipofectamineTM 3000 with 50 µL Opti-MEM. Mix the transfection reagent and siRNA diluent and add 24-well plate, continue to incubate for 6 h and then replace the medium, after 24 h of transfection, verify the expression of TRIP13 through subsequent assays. The sequences of siRNAs are listed in Supplementary Table 2.
RNA extraction and quantitative PCR
Total RNA was extracted using Trizol reagent (sangon Biotech, Shanghai) according to the manufacturer’s protocol, and cDNA was synthesized using RevertAid First Strand cDNA Synthesis Kit (Thermofisher, USA). mRNA levels were measured by qPCR using an ABI 7500 System (Life Technologies, USA). The relative expression was determined using the 2−ΔΔCt method, with GAPDH as an endogenous control. The primers are listed in Supplementary Table 3.
Western blot
Total protein was obtained as previously described21. Proteins were separated by SDS-PAGE and transferred to NC membranes. Incubate the membrane overnight with one of the antibodies listed in Supplementary Table 4. Then incubate with corresponding HRP-conjugated secondary antibodies. To save costs and reduce antibody loss, PVDF membranes were cut based on the molecular size of the target protein. Original blots are presented in Supplementary Fig. 1. Finally, immunoreactive bands were detected with WesternBright ECL kit (Advansta, USA). Grayscale analysis of WB bands was performed using ImageJ software.
Cell viability assay
Cell viability was detected using CCK8 reagent (Beyotime, China) according to the manufacturer’s protocol. The absorbance of the cells was measured using multiscan spectroscopy at a wavelength of 450 nm.
Chemosensitivity assay in vitro
Human DLBCL cell lines SU-DHL-2 and SU-DHL-4 were seeded into 96-well plates at a density of 1 × 104 cells per well after siRNA transfection. The cells were treated with various concentrations (0, 0.01, 0.1, 1, 2, 5, 10, 20 µg/mL) of ibrutinib (Aladdin) for 24 h. Cell viability was subsequently assessed using the CCK8 assay. The log(inhibitor) versus response model in GraphPad Prism 6 was employed to generate the fitted curve and calculate the half-maximal inhibitory concentration (IC50).
Apoptosis analysis
Apoptosis was detected by Annexin V-APC/PI Apoptosis Detection Kit (BestBio, China). The fluorescence of at least 5,000 cells per sample was measured on a Cytomics FC 500 flow cytometer (Beckman Coulter, USA) for further calculations.
Xenograft mouse model
Female BALB/c nude mice were purchased from Nanjing Jicui Yaokang Biotechnology Co., Ltd. This experiment was undertaken according to the guidelines for the Animal Care and Use Committee of the Anhui Medical University. The in vivo experiments were authorized by the Animal Care and Use Committee of the Anhui Medical University. All animal procedures were performed in specific pathogen-free (SPF) barrier facilities. We used sodium pentobarbital anesthesia to minimize the pain of nude mice during the experiment. 2 × 107 TRIP13 knockdown and control SU-DHL-4 cell lines were injected into subcutaneous of 5-week-old female BALB/c nude mice (n = 3 for each group). The tumor volume was measured every 3 days after injection. After 30 days, the mice were sacrificed under deep anesthesia induced by intraperitoneal injection of sodium pentobarbital, and the tumor weights were recorded.
immunohistochemistry testing
The experiment of immunohistochemistry (IHC) was executed as previously described22. All antibodies used in the study are listed in Supplementary Table 4. Typical images (40×) were captured using a microscope system (ZEISS, Germany).
Statistical analysis
All statistical analyses were performed using the R software (v.4.2.1). Student's t-tests were used to compare the differences between the two groups. The results were presented as the mean ± standard deviation (SD) of at least three independent experiments. P < 0.05 was considered statistically significant.
Results
Identification and functional annotation of DEBRRGs
To identify BTKi-resistance related genes in DLBCL, we downloaded and analyzed the transcriptome data of the parental and ibrutinib-resistant clonal cell line (GSE138126). With a threshold |log2FC| > 2 and adjusted P-value < 0.05, the volcano plot revealed a total of 1186 DEGs, with 552 upregulated genes and 634 downregulated genes in the ibrutinib-resistant group. The heatmap presents DEGs that differ between non-resistant and resistant groups (Fig. 1A,B) (Supplementary Table 5). GO analysis results revealed that the biological processes involved in these BRRGs were enriched mainly in cell–cell adhesion, T cell activation, etc. KEGG analysis results revealed that these BRRGs were enriched mainly in C-type lectin receptor signaling pathway, FoxO signaling pathway, and B-cell receptor signaling pathway (Fig. 1C,D). The above results indicate these biological processes and oncogenic pathways may be involved in BTKi resistance.

Identification and functional annotation of DEBRRGs. (A,B) Volcano plot and heatmap of DEBRRGs. (C) GO analysis the biological processes involved in DEBRRGs. (D) KEGG pathway enrichment analysis DEBRRGs.
Establishment of BRRGs prognostic signature
To investigate the relationship between BRRGs and prognosis in DLBCL and further establish a BRRGs prognostic signature. The GSE31312 dataset was used as the training cohort. Univariate Cox regression results showed that 247 BRRGs were related to OS (Supplementary Table 6). LASSO regression and tenfold cross-validation further identified 10 BRRGs to construct the model, as well as their regression coefficients in prognostic signature (Fig. 2A,B, Table2). The resistance score formula was obtained based on 10-BRRGs = (0.2242 * CARD16) + (0.0064 * TRIP13) + (0.0174 * PSRC1) + (0.0276 * CASP1) + (0.0118 * PLBD1) + (0.0627 * CARD6) + (-0.0394 * CAPG) + (− 0.0661 * CACNA1A) + (− 0.3578 * CDH15) + (0.6447 * NDUFA4). Based on the calculated resistance score of each patient, patients were divided into high- and low-resistance score groups by median resistance score. As shown in Fig. 2C, more deaths were observed in DLBCL patients in the high-resistance score group compared with the low-resistance score group. Kaplan–Meier survival analysis showed that high-resistance score group patients were associated with poor OS compared to low-resistance score group patients in the training cohort (HR = 2.855, 95%CI 2.243–3.634) (Fig. 2D). PCA analysis results showed that the resistance score was able to accurately distinguish patients (Fig. 2E). The AUCs of the 2-, 3-, and 5-year ROC curves were 0.731, 0.761, and 0.777 respectively, indicating that the BRRGs signature exhibited high efficacy in predicting the outcome of training cohort (Fig. 2F).

Establishment of BRRGs prognostic signature. (A,B) LASSO regression analysis for most suitable λ. (C) The distribution of resistance scores and survival status of DLBCL in training cohort, the dots represent the resistance score, survival time, and outcome for each patient. (D) Kaplan–Meier survival curve of high- and low-resistance score groups in training cohort. (E) PCA plot in the training cohort. The red dots represent high-resistance score group patients and blue dots represent low-resistance score group patients. (F) ROC curve of 2-, 3-, and 5-year survival prediction in the training cohort.
Validation of BRRGs prognostic signature
To further verify the reliability of the BRRGs prognostic signature, we selected two datasets (GSE87371 and GSE10846) as external validation cohorts. Based on the same calculation formula as the training cohort, patients in the two validation cohorts were divided into high- and low-resistance score groups, respectively. Consistent with the results of the training cohort, more deaths were observed in the high-resistance score group (Fig. 3A,B), and the BRRGs signature was able to accurately distinguish patients (Fig. 3C,D). Kaplan–Meier survival analysis showed that high-resistance score group was associated with worse OS in the GSE87371 validation cohort (HR = 1.69, 95%CI 1.07–2.65) and GSE10846 validation cohort (HR = 2.14, 95%CI 1.02–4.50) (Fig. 3E,F). In addition, meta-analysis results of three cohorts showed an important association between BRRGs signature and prognosis in DLBCL (HR = 2.51, 95%CI 2.04–3.08) (Fig. 3G). Also, the prediction of outcomes in the two validation cohorts exhibited high predictive power. In the GSE87371 validation cohort, the AUC at the 2-, 3-, and 5-year ROC curve were 0.654, 0.752, 0.610 (Fig. 3H). In the GSE10846 validation cohort, the AUC at the 2-, 3-, and 5-year ROC curve were 0.606, 0.603, 0.608 (Fig. 3I), which suggests the robustness and reliability of the BRRGs signature. In addition, we constructed a nomogram based on BRRGs signature (Fig. 3J). The calibration curve and C-index further verified the accuracy of BRRGs prognostic signature in predicting 2-, 3-, and 5-year survival rates, and the results showed high consistency with the nomogram predictions (Fig. 3K).

Validation of BRRGs prognostic signature. (A,B) The distribution of resistance scores and survival status of DLBCL in the validation cohort, the dots represent the resistance score, survival time, and outcome for each patient. (C,D) PCA plot in the validation cohort. The red dots represent high-resistance score group patients and blue dots represent low-resistance score group patients. (E,F) Kaplan–Meier survival curve of high- and low-resistance score groups in the validation cohort. (G) Meta-analysis of three datasets. (H,I) ROC curve of 2-, 3-, and 5-year survival prediction in the validation cohort. (J) Nomogram was constructed based on BRRGs signature. (K) Calibration curve of 2-, 3-, and 5- year OS.
Immune infiltration, biological pathways, and chemotherapeutic sensitivity in high- and low-resistance score groups
An increasing number of studies have shown that the types and functions of immune cells in TiME are closely related to tumor progression and chemotherapeutic response. We used ssGSEA algorithm to explore the differences in immune cell infiltration between the high- and low-resistance score groups patients. The results showed significant differences in 4 immune cell types, including CD8+ T cells, Th1 cells, TIL, and Treg, as well as 7 immune pathways, including APC co-stimulation, Cytolytic activity, inflammation-promoting, MHC class I, Parainflammation, T cell co-inhibition, and Type II IFN Response, between high- and low-resistance score groups (Fig. 4A,B). Inflammatory and immunosuppressive factors were significantly increased in high-resistance score group patients, suggesting the presence of an immunosuppressive microenvironment in these patients.

Immune infiltration, biological pathways, and chemotherapeutic sensitivity in high- and low-resistance score groups. (A,B) Difference of immune cells and pathways in high- and low-resistance score groups. (C,D) GSEA KEGG analysis in high- and low-resistance score groups. (E,F) GSEA GO analysis in high- and low-resistance score groups. (G–L) Sensitivity to chemotherapy drugs in high- and low-resistance score groups.
Subsequently, we used the ‘limma’ package to identify the differentially expressed genes between the high- and low- resistance score groups (Supplementary Table 7), and GSEA was conducted to investigate the biological functions and pathways between two groups, As shown in Fig. 4C–F, the biological pathways in the high-resistance group are mainly enriched in cell cycle, cytoplasmic DNA sensing pathways, primary immune deficiencies, etc., biological processes are mainly enriched in the B-cell receptor signaling pathway, cell cycle, etc.
We further explored the response of high- and low-resistance score group patients to chemotherapeutic sensitivity. The lower the IC50, the more sensitive the patient is to chemotherapy drugs. The results showed that patients with high-resistance score were more sensitive to most chemotherapy drugs, including doxorubicin, etoposide, and gemcitabine (Fig. 4G–I). In addition, patients with high-resistance score are also more sensitive to other inhibitors, such as BCL2 inhibitors (Obatoclax Mesylate), PI3K inhibitors (ZSTK474), and HDAC inhibitors (MS-275) (Fig. 4J–L). These results suggested that high-resistance score group patients may benefit from these drugs, and the BRRGs signature may help patients select appropriate chemotherapy regimens.
Knocking down TRIP13 inhibits cell proliferation, promotes cell apoptosis, and enhances BTKi sensitivity
To further explore the biological function of BRRGs in DLBCL, we applied the WGCNA algorithm to identify the most critical genes and conducted experimental verification. As shown in Fig. 5A, when the optimal soft threshold β is 10, the power of the scale-free topology fitting index reaches 0.98. Next, 8 gene modules were identified, and brown modules were highly correlated with BTKi resistance (Fig. 5B,C). The Venn diagram further identified three hub genes, including CARD16, TRIP13, and PSRC1 (Fig. 5D). We verified the expression level in the GEPIA database (http://gepia2.cancer-pku.cn), and the results showed that TRIP13 was up-regulated in DLBCL (Fig. 5E). Furthermore, TRIP13 mRNA levels were also up-regulated in DLBCL tissues. In addition, TRIP13 also upregulated in the ibrutinib-resistant cell lines (Fig. 5F). The above results show that TRIP13 may be related to the DLBCL progression and BTKi resistance. To clarify the important role of TRIP13, we performed functional assays in TRIP13-silenced DLBCL cells, and the knockdown efficiency was assessed by qPCR and western blot (Fig. 5G,H). Cell viability assays revealed that knockdown of TRIP13 inhibited cell proliferation and resulted in lower resistance and IC50 values (Fig. 5I,J). In addition, knockdown of TRIP13 promoted cell apoptosis and enhanced the apoptotic effect after BTKi treatment (Fig. 5K). In vivo experiments showed that knockdown of TRIP13 significantly suppressed xenograft tumor growth, evidenced by the tumor images, sizes, and weight (Fig. 6E–G). IHC testing results showed that knockdown of TRIP13 decreased BCL2 expression and increased BAX expression (Fig. 6H). These results indicated that TRIP13 plays an important role in the DLBCL progression and BTKi resistance.

Knocking down TRIP13 inhibits cell proliferation, promotes cell apoptosis, and enhances BTKi sensitivity. (A) Soft threshold was identified by scale independence and mean connectivity. (B) Gene expression data was classified into different modules. (C) Association between the modules and clinical traits. (D) Venn diagram to identify overlapping genes from WGCNA and LASSO. (E) Expression level of hub genes in GEPIA database. (F) qPCR detect mRNA level of TRIP13 in tissue samples, FPKM of TRIP13 in non-resistant and resistant cell lines. (G,H) The efficiency of TRIP13 knockdown by siRNA was confirmed by qPCR and western blot. (I) SU-DHL-2 and SU-DHL-4 cells were transfected with control or TRIP13 siRNA for 24h, 48h, 72h. Cellular viability was analyzed by CCK8 assay. (J) SU-DHL-2 and SU-DHL-4 cells were transfected with control or TRIP13 siRNA for 48h before treatment with increasing concentrations of ibrutinib for 24h. Cellular viability was analyzed by CCK8 assay, and the IC50 values were calculated by a nonlinear regression model. (K) SU-DHL-2 and SU-DHL-4 cells were transfected with control or TRIP13 siRNA for 48h before treatment with 0 or 1 μg/mL ibrutinib for 24h. Cell apoptosis were detected by flow cytometry. Data are expressed as mean ± SD (*p < 0.05, **p < 0.01, *** < 0.001). Original blots are presented in Supplementary Fig. 1.

TRIP13 contributes to DLBCL progression and BTKi resistance by modulating the cell cycle through the regulation of the Wnt/β-catenin pathway. (A,B) single-gene GSEA analysis of TRIP13 high expression group. (C,D) Western blot was performed to detect Wnt1, β-catenin, cyclinD1, CDK4, BAX, and BCL2 protein levels in control or TRIP13 knockdown. (E) Images of subcutaneous tumors in different groups. (F) The volume of tumors in the different groups was measured every 3 days. (G) The weight of the tumors was measured at the time of resection. (H) The expression of TRIP13, BAX, and BCL2 in the tumors was detected by IHC. Data are expressed as mean ± SD (*p < 0.05, **p < 0.01, *** < 0.001). Original blots are presented in Supplementary Fig. 1.
TRIP13 contributes to DLBCL progression and BTKi resistance by modulating the cell cycle through the regulation of the Wnt/β-catenin pathway
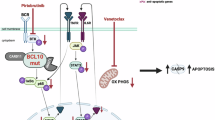
To explore the potential mechanism of TRIP13 in DLBCL, patients were divided into high- and low-expression groups based on the expression level of TRIP13 in the training cohort, and single-gene GSEA analysis was performed. As shown in Fig. 6A,B, cell cycle and DNA replication were significantly enriched in the high TRIP13 group. TRIP13, a member of the ATPase family, is involved in various cell biological processes, including the cell cycle. Moreover, TRIP13 regulates cell apoptosis and chemotherapy sensitivity through the Wnt/β-catenin pathway, particularly by activating the transcription of cell cycle proteins23,24. Therefore, we hypothesized that TRIP13 affects DLBCL progression and BTKi resistance by regulating the Wnt/β-catenin axis. We then assessed the expression of key proteins involved in cell cycle regulation, such as Wnt1, β-catenin, CyclinD1, and CDK4, as well as the expression of proteins involved in cell proliferation and apoptosis, such as BCL2 and BAX. Western blot showed that knocking down TRIP13 significantly reduced the expression of Wnt1, β-catenin, CyclinD1, CDK4, and BCL2 proteins, while increased the expression of BAX protein. Furthermore, this effect was enhanced by BTKi upon TRIP13 knockdown (Fig. 6C,D). Collectively, these results revealed that TRIP13 contributes to DLBCL progression and BTKi resistance by modulating the cell cycle through the regulation of the Wnt/β-catenin pathway.
Discussion
DLBCL is the commonest NHL histologic subtype, accounting for roughly 30%-40% of NHL cases25. The current first-line treatment for DLBCL is a combination of the anti-CD20 monoclonal antibody rituximab and chemotherapy. Second-line treatment options for relapsed/refractory patients include anti-CD19 chimeric antigen receptor (CAR) T cell therapy and targeted drugs such as BTKi8,26. Ibrutinib, as a typical Bruton’s tyrosine kinase inhibitor, combined with R-CHOP regimen benefits DLBCL patients and has broad therapeutic application prospects. However, the development of BTKi resistance remains a significant challenge during treatment. Therefore, elucidating the impact of BRRGs on prognosis and drug sensitivity, and further identifying new therapeutic targets, are effective strategies to reverse drug resistance and improve prognosis. In this study, we comprehensively analyzed the BRRGs and constructed a robust and feasible signature that could early distinguish high-risk patients with potential resistance, aid in the selection of alternative treatments, and predict the prognosis of DLBCL. In addition, we found that TRIP13 is a potential therapeutic target that regulates the cell cycle through the Wnt/β-catenin pathway and is involved in DLBCL malignant progression and BTKi resistance.
In this study, we first analyzed the transcriptome data of BTKi-resistant and non-resistant DLBCL cell lines and identified 1186 DEGs. These DEGs were mainly enriched in cell–cell adhesion, T cell activation, and pathways such as the C-type lectin receptor signaling pathway, FoxO signaling pathway, and B cell receptor signaling pathway. This result aligns with published literature, emphasizing that BTK-mediated B cell proliferation and survival occur through the B cell receptor signaling pathway and the FoxO signaling pathway27,28,29. These studies suggest that the biological processes and pathways involved may also be key mediators of BTKi resistance.
Numerous prognostic signatures based on immune-related and metabolism-related gene sets have been developed for DLBCL30,31, revealing the important role of various gene clusters and enriching the understanding of DLBCL pathogenesis. Resistance genes play an important role in the progression and relapse of DLBCL, and elucidating their function is of great significance19,32,33. In this study, we further found that 247 genes were associated with the prognosis of DLBCL. Using univariate Cox and Lasso regression analyses, we identified 10 key genes and constructed a new prognostic model. Based on the score of this model, patients with high resistance score were associated with worse OS. Furthermore, the BRRGs prognostic signature has shown consistency and good predictive performance in multiple independent cohorts, demonstrating its robustness and accuracy. Nevertheless, the signature still needs further testing in the real world.
As more research is performed to clarify the crucial role of TiME in DLBCL progression34, we further analyzed the types and functions of infiltrating immune cells between different resistance score groups. The results showed that inflammatory and immunosuppressive factors were significantly increased in patients with high-resistance score group, suggesting the presence of an immunosuppressive microenvironment. This finding aligns with previous studies suggesting that an inflammation and immunosuppressive microenvironment promotes drug resistance and relapse in DLBCL33. Mutations in BTKi or its downstream targets leading to persistent activation of BCR signaling, which is a crucial factor in drug resistance and relapse35. Our results consistently showed that the B-cell receptor signaling pathway and cell cycle were enriched in the high-resistance score group. Therefore, screening candidate drugs for BTKi-resistant patients is crucial. The most promising drugs include BCL2 inhibitors, PI3K inhibitors, and HDAC inhibitors36,37,38. We further explored the chemotherapeutic response to these drugs in patients with high- and low-resistance score groups. The results showed that patients with high-resistance score were more sensitive to these inhibitors, such as BCL2 inhibitors (Obatoclax Mesylate), PI3K inhibitors (ZSTK474), and HDAC inhibitors (MS-275). These results suggest that these drugs may be potential alternatives for the treatment of BTKi resistant patients. It should be noted that these findings are based on predictive results, and the effectiveness and adverse reactions of these drugs still need to be further explored in vivo and in vitro.
The functions and mechanisms of genes related to BTKi resistance deserve further exploration. We used the WGCNA algorithm and identified the key gene TRIP13. TRIP13 has been implicated in carcinogenesis and is overexpressed in several tumors, such as lung cancer, hepatocellular carcinoma, colorectal cancer, prostate cancer, and multiple myeloma, and is associated with poor prognosis15,16,17,18,39. Recent studies have shown that high levels of TRIP13 promote both nonhomologous end joining and homology-directed repair of double-strand breaks, potentially leading to chromosomal instability, cancer cell survival, and enhanced drug resistance40,41. In B cell malignant tumors, TRIP13 modulates protein deubiquitination and accelerates tumor progression and increases resistance to proteasome inhibition42. Furthermore, abnormal activation of the TRIP13-EZH2 signaling axis was associated with the maintenance of the stem phenotype of myeloma and resistance to bortezomib43. Similar to these studies, our findings consistently indicate that TRIP13 may be an important oncogene that is highly expressed in DLBCL and ibrutinib-resistant DLBCL cell lines and is associated with poor prognosis. Further experiments also showed that knocking down TRIP13 could inhibit cell proliferation, promote cell apoptosis, and enhance ibrutinib sensitivity. This suggests that targeting TRIP13 may be a potential treatment option for resistant DLBCL.
Aberrant Wnt signaling activation is a hallmark of many epithelial tumors and is widely involved in important biological processes, including cell proliferation, stemness formation, and multi-drug resistance44,45,46. Various studies have shown increased Wnt signaling and nuclear β-catenin protein levels in some DLBCL patients, correlating with poor prognosis47,48,49. In addition, Wnt signaling is crucial for maintaining the balance among DLBCL clonal populations, with its inhibition leading to reduced cell growth and colony formation50, while its activation promotes DLBCL cell growth by regulating β-catenin nuclear translocation51. Furthermore, in mantle cell lymphoma, studies have demonstrated that Wnt signaling activation and β-catenin deregulation are involved in chemotherapy resistance, and blocking this pathway enhances sensitivity to chemotherapy drugs52,53,54. In other tumors, TRIP13 regulates cell apoptosis and chemotherapy sensitivity through the Wnt/β-catenin pathway, particularly by activating the transcription of cell cycle proteins23,24. In this study, GSEA analysis revealed that cell cycle and DNA replication are associated with high TRIP13 expression. Subsequent western blot showed that knocking down TRIP13 significantly reduced the expression of Wnt1, β-catenin, Cyclin D1, CDK4, and BCL2 proteins, while increased the expression of BAX protein. Furthermore, this effect was enhanced by BTKi upon TRIP13 knockdown. These results indicate that TRIP13 contributes to DLBCL progression and BTKi resistance by modulating the cell cycle through the Wnt/β-catenin pathway.
In this study, we provide the first evidence that TRIP13 promotes progression and BTKi resistance in DLBCL, which has potential clinical significance. However, there are several limitations. First, the limited clinical sample size may impact the accuracy of the results, highlighting the need for a larger sample size to validate and enhance the reliability of the prognostic signature. Second, we only explored the biological functions and mechanisms of TRIP13 and did not conduct further research on other genes in DLBCL.
Conclusion
We comprehensively analyzed BTKi-resistance related genes and established a feasible gene signature that can early distinguish high-risk patients, aid in the selection of alternative treatments, and predict the prognosis of DLBCL. Additionally, we identified the key gene TRIP13 as a potential therapeutic target and elucidated its important mechanism in promoting DLBCL progression and BTKi resistance.
Data availability
Transcriptome data of ibrutinib-resistant and non-resistant DLBCL cell lines (GSE138126), as well as data of DLBCL training and validation cohorts GSE31312 (n = 471), GSE87371 (n = 121), and GSE10846 (n = 233) were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo). Other raw data are available from the corresponding author upon reasonable request.
References
Sabattini, E., Bacci, F., Sagramoso, C. & Pileri, S. A. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008: An overview. Pathologica 102(3), 83–87 (2010).
Tilly, H. et al. Diffuse large B-cell lymphoma (DLBCL): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol 26(Suppl 5), v116–v125 (2015).
Klener, P. & Klanova, M. Drug resistance in non-Hodgkin lymphomas. Int. J. Mol. Sci. 21(6), 2081 (2020).
Pal Singh, S., Dammeijer, F. & Hendriks, R. W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 17(1), 57 (2018).
Cheng, S. et al. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia 28(3), 649–657 (2014).
de Rooij, M. F. et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 119(11), 2590–2594 (2012).
Niemann, C. U. et al. Disruption of in vivo chronic lymphocytic leukemia tumor-microenvironment interactions by ibrutinib-findings from an investigator-initiated phase II study. Clin. Cancer Res. 22(7), 1572–1582 (2016).
Younes, A. et al. Randomized phase III trial of ibrutinib and rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in non-germinal center B-Cell diffuse large B-Cell lymphoma. J. Clin. Oncol. 37(15), 1285–1295 (2019).
Adams, S. V., Newcomb, P. A. & Shustov, A. R. Racial patterns of peripheral T-Cell lymphoma incidence and survival in the United States. J. Clin. Oncol. 34(9), 963–971 (2016).
Camilleri-Broet, S. et al. A uniform activated B-cell-like immunophenotype might explain the poor prognosis of primary central nervous system lymphomas: Analysis of 83 cases. Blood 107(1), 190–196 (2006).
Lionakis, M. S. et al. Inhibition of B cell receptor signaling by ibrutinib in primary CNS lymphoma. Cancer Cell 31(6), 833-843.e5 (2017).
Nakhoda, S., Vistarop, A. & Wang, Y. L. Resistance to Bruton tyrosine kinase inhibition in chronic lymphocytic leukaemia and non-Hodgkin lymphoma. Br. J. Haematol. 200(2), 137–149 (2023).
He, M. Y. & Kridel, R. Treatment resistance in diffuse large B-cell lymphoma. Leukemia 35(8), 2151–2165 (2021).
Lu, S., Qian, J., Guo, M., Gu, C. & Yang, Y. Insights into a crucial role of TRIP13 in human cancer. Comput. Struct. Biotechnol. J. 17, 854–861 (2019).
Cai, W., Ni, W., Jin, Y. & Li, Y. TRIP13 promotes lung cancer cell growth and metastasis through AKT/mTORC1/c-Myc signaling. Cancer Biomark. 30(2), 237–248 (2021).
Xue, J., Wu, H., Shi, Y. & Li, Z. TRIP13 overexpression in hepatocellular carcinoma: Implications for poor prognosis and immune cell infiltration. Discov. Oncol. 14(1), 176 (2023).
Sheng, N. et al. TRIP13 promotes tumor growth and is associated with poor prognosis in colorectal cancer. Cell Death Dis. 9(3), 402 (2018).
Tao, Y. et al. TRIP13 impairs mitotic checkpoint surveillance and is associated with poor prognosis in multiple myeloma. Oncotarget 8(16), 26718–26731 (2017).
Geeleher, P., Cox, N. J. & Huang, R. S. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 15(3), R47 (2014).
Geeleher, P., Cox, N. & Huang, R. S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 9(9), e107468 (2014).
Ding, C. et al. Tumor endothelial marker 8 promotes proliferation and metastasis via the Wnt/beta-catenin signaling pathway in lung adenocarcinoma. Front. Oncol. 11, 712371 (2021).
Guo, J. et al. SRRM2 may be a potential biomarker and immunotherapy target for multiple myeloma: A real-world study based on flow cytometry detection. Clin. Exp. Med. 24(1), 28 (2024).
Afaq, F. et al. Targeting of oncogenic AAA-ATPase TRIP13 reduces progression of pancreatic ductal adenocarcinoma. Neoplasia 47, 100951 (2024).
Kobayashi, S. et al. Transcriptional profiling identifies cyclin D1 as a critical downstream effector of mutant epidermal growth factor receptor signaling. Cancer Res. 66(23), 11389–11398 (2006).
Teras, L. R. et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J. Clin. 66(6), 443–459 (2016).
Schuster, S. J. et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 377(26), 2545–2554 (2017).
Hinman, R. M., Bushanam, J. N., Nichols, W. A. & Satterthwaite, A. B. B cell receptor signaling down-regulates forkhead box transcription factor class O 1 mRNA expression via phosphatidylinositol 3-kinase and Bruton’s tyrosine kinase. J. Immunol. 178(2), 740–747 (2007).
Wang, X., Kokabee, L., Kokabee, M. & Conklin, D. S. Bruton’s tyrosine kinase and its isoforms in cancer. Front. Cell Dev. Biol. 9, 668996 (2021).
Mihoub, I. et al. Identification of the axis beta-catenin-BTK in the dynamic adhesion of chronic lymphocytic leukemia cells to their microenvironment. Int. J. Mol. Sci. 24(24), 17623 (2023).
Jiang, Y. et al. Immunophenotypic landscape and prognosis-related mRNA signature in diffuse large B Cell lymphoma. Front. Genet. 13, 872001 (2022).
He, J. et al. Identification of molecular subtypes and a novel prognostic model of diffuse large B-cell lymphoma based on a metabolism-associated gene signature. J. Transl. Med. 20(1), 186 (2022).
He, M. Y. & Kayamori, K. Genome-wide CRISPR screening uncovers potential targets and mechanisms of vincristine resistance in DLBCL. Br. J. Haematol. 202(4), 720–721 (2023).
Wang, L. & Li, L. R. R-CHOP resistance in diffuse large B-cell lymphoma: Biological and molecular mechanisms. Chin. Med. J. 134(3), 253–260 (2020).
Cioroianu, A. I. et al. Tumor microenvironment in diffuse large B-cell lymphoma: Role and prognosis. Anal. Cell Pathol. 2019, 8586354 (2019).
Cheng, S. et al. Functional characterization of BTK(C481S) mutation that confers ibrutinib resistance: Exploration of alternative kinase inhibitors. Leukemia 29(4), 895–900 (2015).
Kuo, H. P. et al. Combination of ibrutinib and ABT-199 in diffuse large B-cell lymphoma and follicular lymphoma. Mol. Cancer Ther. 16(7), 1246–1256 (2017).
Jain, N. et al. Targeting phosphatidylinositol 3 kinase-beta and -delta for Bruton tyrosine kinase resistance in diffuse large B-cell lymphoma. Blood Adv. 4(18), 4382–4392 (2020).
Guo, H. et al. Dual inhibition of PI3K signaling and histone deacetylation halts proliferation and induces lethality in mantle cell lymphoma. Oncogene 38(11), 1802–1814 (2019).
Dong, L. et al. TRIP13 is a predictor for poor prognosis and regulates cell proliferation, migration and invasion in prostate cancer. Int. J. Biol. Macromol. 121, 200–206 (2019).
Banerjee, R. et al. TRIP13 promotes error-prone nonhomologous end joining and induces chemoresistance in head and neck cancer. Nat. Commun. 5, 4527 (2014).
Clairmont, C. S. et al. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat. Cell Biol. 22(1), 87–96 (2020).
Li, C. et al. TRIP13 modulates protein deubiquitination and accelerates tumor development and progression of B cell malignancies. J. Clin. Investig. https://doi.org/10.1172/JCI146893 (2021).
Xu, L. et al. Aberrant activation of TRIP13-EZH2 signaling axis promotes stemness and therapy resistance in multiple myeloma. Leukemia 37(7), 1576–1579 (2023).
Chiarini, F., Paganelli, F., Martelli, A. M. & Evangelisti, C. The role played by Wnt/beta-catenin signaling pathway in acute lymphoblastic leukemia. Int. J. Mol. Sci. 21(3), 1098 (2020).
Laranjeira, A. B. & Yang, S. X. Therapeutic target discovery and drug development in cancer stem cells for leukemia and lymphoma: From bench to the clinic. Expert Opin. Drug Discov. 11(11), 1071–1080 (2016).
Takebe, N., Harris, P. J., Warren, R. Q. & Ivy, S. P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 8(2), 97–106 (2011).
Ge, X., Lv, X., Feng, L., Liu, X. & Wang, X. High expression and nuclear localization of beta-catenin in diffuse large B-cell lymphoma. Mol. Med. Rep. 5(6), 1433–1437 (2012).
Reya, T. et al. Wnt signaling regulates B lymphocyte proliferation through a LEF-1 dependent mechanism. Immunity 13(1), 15–24 (2000).
Qiang, Y. W., Endo, Y., Rubin, J. S. & Rudikoff, S. Wnt signaling in B-cell neoplasia. Oncogene 22(10), 1536–1545 (2003).
Koch, R. et al. Populational equilibrium through exosome-mediated Wnt signaling in tumor progression of diffuse large B-cell lymphoma. Blood 123(14), 2189–2198 (2014).
Shi, X. et al. LncRNA FIRRE is activated by MYC and promotes the development of diffuse large B-cell lymphoma via Wnt/beta-catenin signaling pathway. Biochem. Biophys. Res. Commun. 510(4), 594–600 (2019).
Ge, X. & Wang, X. Role of Wnt canonical pathway in hematological malignancies. J. Hematol. Oncol. 3, 33 (2010).
Sanchez-Tillo, E. et al. The EMT activator ZEB1 promotes tumor growth and determines differential response to chemotherapy in mantle cell lymphoma. Cell Death Differ. 21(2), 247–257 (2014).
Mathur, R. et al. Targeting Wnt pathway in mantle cell lymphoma-initiating cells. J. Hematol. Oncol. 8, 63 (2015).
Acknowledgements
We thank the hematology staff for their contribution to the completion of this study.
Funding
This study was funded by the National Natural Science Foundation of China: [81670179]; Major Subject of Science and Technology of Anhui Province: [201903a07020030]; Key Research and Development Plan of Anhui Province: [201904a07020058]; and the Foundation of Anhui Medical University: [2019xkj134]; Anhui Medical University Postgraduate Research and Practical Innovation Project: [YJS20230135].
Author information
Authors and Affiliations
Contributions
Conceptualization: ZMZ, SDX, YYD; data analysis and visualization: YYD, KKH, CS; investigation: JLZ, XYJ, YL; methodology: ZZM, SDX, YYD; validation: KKH, ZLL, JJG, XJF; writing-original draft: YYD, CS; writing-review and editing: CHZ, ZMZ, SDX, YYD; all authors read and approved the final manuscript. Each author approved the manuscript before submission for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
All the animal experiments were approved by the Animal Care and Use Committee of the Anhui Medical University. This research was approved by the institutional ethics Committee of The Second Affiliated Hospital of Anhui Medical University, informed consent was obtained from all patients, and were performed in accordance with the Helsinki Declaration. The study is reported in accordance with ARRIVE guidelines (https://arriveguidelines.org).
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ding, Y., Huang, K., Sun, C. et al. A Bruton tyrosine kinase inhibitor-resistance gene signature predicts prognosis and identifies TRIP13 as a potential therapeutic target in diffuse large B-cell lymphoma. Sci Rep 14, 21184 (2024). https://doi.org/10.1038/s41598-024-72121-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-72121-8
Keywords
This article is cited by
-
CBX7 regulates chemotherapy-induced senescence-like growth arrest in multiple myeloma via the ERK/STAT3/PIM1 axis
Journal of Translational Medicine (2025)
