
Abstract
This study integrates pharmacology databases with bulk RNA-seq and scRNA-seq to reveal the latent anti-PDAC capacities of BBR. Target genes of BBR were sifted through TargetNet, CTD, SwissTargetPrediction, and Binding Database. Based on the GSE183795 dataset, DEG analysis, GSEA, and WGCNA were sequentially run to build a disease network. Through sub-network filtration acquired PDAC-related hub genes. A PPI network was established using the shared genes. Degree algorithm from cytoHubba screened the key cluster in the network. Analysis of differential mRNA expression and ROC curves gauged the diagnostic performance of clustered genes. CYBERSORT uncovered the potential role of the key cluster on PDAC immunomodulation. ScRNA-seq analysis evaluated the distribution and expression profile of the key cluster at the single-cell level, assessing enrichment within annotated cell subpopulations to delineate the target distribution of BBR in PDAC. We identified 425 drug target genes and 771 disease target genes, using 57 intersecting genes to construct the PPI network. CytoHubba anchored the top 10 highest contributing genes to be the key cluster. mRNA expression levels and ROC curves confirmed that these genes showed good robustness for PDAC. CYBERSORT revealed that the key cluster influenced immune pathways predominantly associated with Macrophages M0, CD8 T cells, and naïve B cells. ScRNA-seq analysis clarified that BBR mainly acted on epithelial cells and macrophages in PDAC tissues. BBR potentially targets CDK1, CCNB1, CTNNB1, CDK2, TOP2A, MCM2, RUNX2, MYC, PLK1, and AURKA to exert therapeutic effects on PDAC. The mechanisms of action appear to significantly involve macrophage polarization-related immunological responses.
Similar content being viewed by others


Introduction
Pancreatic cancer is one of the most fatal malignancies in human gastrointestinal and bilio-pancreatic tract. As reported in 2020 global cancer statistics, the number of new diagnoses of pancreatic cancer was 496,000, nearly equal to the number of deaths which was as high as 466,0001. Known as the main pathological type of pancreatic cancer, pancreatic ductal adenocarcinoma (PDAC) presented an overall 5-year survival rate dismally low at 5–9%2. Insidious onset, high invasiveness, poor prognosis, lack of precise diagnosis and treatment have made PDAC a huge challenge to global public health. An ideal therapeutic option has far been elusive. The efficacy of gemcitabine, the first-line chemotherapeutic standard of care for PDAC, is heavily limited by drug resistance. Modification of existing regimens, identification of potential therapeutic targets, discovery and development of new agents remain urgent needs.
Coptidis Rhizoma is a traditional Chinese medicine with a time-honored history and rich clinical applications. Berberine (BBR) is an isoquinoline alkaloid extracted from Coptidis Rhizoma, and it has exhibited effects on glucolipid metabolism regulation, antimicrobial, anti-inflammatory, antioxidant stress, and anticancer. The curative effects of BBR in various pancreatic diseases have been widely documented, including severe acute pancreatitis3, chronic pancreatitis4, type-2 diabetes5, and its complications6. In addition, it has demonstrated promising anticancer potential in a range of malignant solid tumors7.
Despite the paucity of extant researches, the abilities of both BBR and chemically modified analogs in inhibiting the proliferation and metastasis of PDAC cells have been uncovered8. It may be related to regulating mitochondrial citric acid cycle metabolites to block fatty acid synthesis9; intercalating DNA to increase DNA damage and repressing topoisomerase activity, jointly inducing DNA double-strand breaks10; preventing the abnormal production and secretion of extracellular vesicles11. Since a diverse spectrum of gene mutations provides PDAC with remarkable pathological heterogeneity, the anti-PDAC effect of BBR might also tend to be sophisticated, and a further elucidation of its detailed mechanisms is still required.
For the past few years, advances in high-throughput transcriptome sequencing technology and the construction of comprehensive pharmacology databases have enabled insights into disease development and treatment to be gradually extended to the gene layer. The integrated analysis of multi-omics sequencing data and drug databases brings fresh ideas for therapeutic target mining, existing drug repurposing, new drug design and development, and is able to shed more light on the fundamental molecular mechanisms behind the pharmacological effects. Conventional bulk RNA sequencing (RNA-seq) focuses on averaged gene expression profiles and transcriptional regulatory properties in tissue-level, but fails to reveal cellular heterogeneity, whereas single-cell RNA sequencing (scRNA-seq) could fill this gap. By measuring the structure of transcriptomic landscape in individual cells, scRNA-seq offers an in-depth understanding of cell abundance and cell-type specific gene expression patterns. Therefore, this study aims to combine pharmacology databases with bulk RNA-seq and scRNA-seq, in order to reveal the latent anti-PDAC capacities of BBR through thorough bioinformatics methods.
Materials and methods
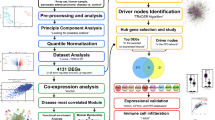
The flowchart of this study is illustrated in Fig. 1.

Flowchart.
Target acquisition of BBR and enrichment analysis
Canonical SMILES of BBR was obtained from PubChem. The molecular targets and corresponding target genes of BBR were acquired from TargetNet, Comparative Toxicogenomics Database (CTD), SwissTargetPrediction, and Binding Database using the following search parameters: The search strategy in CTD employed the keyword “berberine”. For the remaining databases, the canonical SMILES of BBR was used as the search query. In Binding Database, the search type was set to similarity with a threshold of 0.85. In TargetNet, models with an AUC ≥ 0.70 were included, and the fingerprint type was specified as ECFP4 fingerprints. All targets were integrated with duplicates removed, then annotated with disease ontology (DO) terms by using DOSE R package12, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) functional annotations13,14,15 were performed by EnrichR16. The R package enrichplot visualized annotation results.
Target acquisition of PDAC and enrichment analysis
Expression matrix construction of PDAC and differential expression analysis
While available public PDAC RNA-seq datasets with sufficient matched tumor and adjacent normal tissue samples were lacking for comprehensive analysis, the GSE183795 dataset, which contains microarray-based gene expression profiles of 139 pancreatic tumor tissues and 102 adjacent non-tumor pancreatic tissues from PDAC patients, was chosen to ensure the reliability and comparability of the study results. The raw data were preprocessed in R language for standardization and normalization, probe re-annotation, construction of expression matrix. During this process, probes without matched gene symbols were removed and replicate probes were averaged. The matrix formed by clinical phenotypes and gene expression data was conducted differential expression analysis by limma package17 in R. P-values were corrected through Benjamin-Hochberg method to control the false discovery rate (FDR) below 5%, and only genes meet the criteria of |log2fold change| > 1 were screened. Identified differentially expressed genes (DEGs) with expression values were visualized according to clinical phenotypes. The volcano plot was drawn via ggplot218.
Gene Set Enrichment analysis of DEGs
Gene set enrichment analysis (GSEA)19 was utilized to find general tendencies in the defined set of DEGs, as well as statistically significant differences between the two clinical traits. DEGs were rank ordered in line with descending fold change. Through KEGG enrichment process, the markedly up- and down-regulated gene pathways were mapped, and the enrichplot package visualized the results. The following thresholds were applied in the analysis: normal p-value < 0.05, FDR-corrected p-value < 0.05, and |normalized enrichment score (NES) |>1.
Weighted correlation network analysis of PDAC
Weighted correlation network analysis (WGCNA) has the capability of discovering clustered modules of genes that are strongly correlated in terms of expression properties and demonstrating correlation networks among them, describing complex interrelationships between modular gene expression and clinical phenotypes of diseases, and is extensively employed for the identification of biomarkers and the search for potential therapeutic targets. The weighted correlation network was constructed by the WGCNA R package20. The pickSoftThreshold function obtained the optimal weighted parameter of adjacency matrix, also referred to as soft threshold, for network topology analysis. The adjacency matrix was calculated and subsequently transformed into a topological overlap matrix (TOM). Based on the hierarchical clustering of a dissimilarity measure (1-TOM) refined gene co-expression modules. As the first principal component from each single module, module eigengenes summarized modular expression patterns. Module significance and gene significance within the module were computed to build gene module-trait relationships. Using Pearson’s product-moment correlation, the module with the highest correlation coefficient with PDAC was selected as the key module. To capture the broad spectrum of genes potentially relevant to the phenotype or pathology of PDAC, a Venn diagram was utilized to merge and illustrate the integration of gene lists from the key WGCNA module and DEGs. These genes were further incorporated into a complex disease network via STRINGdb21. The edge betweenness algorithm of igraph22 screened out nodes with above-average betweenness centrality in the network to be disease targets. Enrichment analysis was performed the same as described in part 2.1.
Protein-protein interaction (PPI) network construction, key cluster identification and enrichment analysis
Drug targets and disease targets were imported into the STRING database, the minimum required interaction score was set at medium confidence (0.400), and the PPI network was constructed with isolated nodes removed. Degree topological analysis provided by cytoHubba in Cytoscape (v3.9.1) was conducted to extract the key cluster composed of the Top 10 tightly linked core genes in the network. Enrichment analysis was performed the same as described in part 2.1.
Receiver operating characteristic curve analysis
To assess the association of clustered genes with PDAC, an external disease-control validation model was constructed grounded on another cohort (GSE62452) which involved 69 PDAC samples and 61 adjacent non-tumor samples. The mRNA level of critical genes was verified by the Wilcoxon rank-sum test. In the use of the mRNA expression data, the receiver operating characteristic (ROC) curves were plotted, and the area under the curve (AUC) was calculated with pROC package23. Genes with AUC > 0.6 have acquired the capacity to distinguish PDAC and non-tumor tissues, and those with AUC ≥ 0.75 would be considered more diagnostically discriminant24.
Immune infiltration analysis
Since PDAC is among the most immune-resistant tumor types, we adopted the CYBERSORT algorithm25 to transposed-convolutionally speculate on the cell types in bulk tissues in an attempt to uncover its immune micro-landscapes. The proportion of infiltrated immune cells in GSE183795 expression matrix was calculated and applied to study the relationship between immune cells and clinical phenotypes. To investigate the potential role of the key cluster in PDAC immunomodulation, Spearman correlation analysis was performed to clarify which immunity pathways might be regulated by the important targets.
ScRNA-seq data processing and analysis
The raw dataset GSE212966 which encompassed scRNA-seq data from the tumor biopsy specimens of 6 PDAC patients was retrieved for analysis. For quality control, Seurat26 preprocessed the raw dataset by filtering low-quality cells, cell doublets, multiplets, and cells with high levels of mitochondrial contamination. Data normalization was processed in the method of global scaling normalization. Highly variable genes were detected by the FindVariableFeatures function, and a total of 3000 features were returned and scaled to run principle component analysis for linear dimensionality reduction. After the data integration by Harmony27, heatmaps of principal components (PCs) and the elbow plot, which displayed the variance percentage explained by each PC, were combined to determine the appropriate number of PCs for downstream analysis. The K-nearest neighbor Machine learning algorithm then classified clusters of cells based on similar characteristic expression patterns. Various resolutions were set to observe cellular clustering effects, and at the optimal resolution employed the t-distributed Stochastic Neighbor Embedding (t-SNE) to reduce the dimensionality non-linearly. Cell type annotation was performed using SingleR28 to identify distinct cellular subpopulations. The enrichment of the key cluster, comprising crucial genes from prior topological network analysis, was assessed within the annotated subpopulations using irGSEA. Enrichment scores were calculated to determine specific cell types showing significant interactions with BBR, thereby clarifying the target distribution of BBR in the complex environment of PDAC.
Results
Target genes of BBR
Canonical SMILES of BBR was derived from PubChem (Fig. 2A). Through TargetNet, CTD, SwissTargetPrediction, and Binding Database, 425 drug targets were obtained (Fig. 2B). DO enrichment analysis showed that the drug targets were mainly enriched in breast carcinoma, cell type benign neoplasm, ischemia, renal cell carcinoma, hepatitis, arteriosclerosis, myocardial infarction, stomach cancer, and colon cancer (Fig. 2C). GO enrichment analysis indicated that biological process (GOBP) of response to xenobiotic stimulus, cellular component (GOCC) of protein-DNA complex, and molecular function (GOMF) of protein heterodimerization activity were primary GO terms that drug targets enriched (Fig. 2E). KEGG analysis revealed major enriching pathways such as viral carcinogenesis, PI3K-Akt signaling pathway, neutrophil extracellular trap formation, lipid and atherosclerosis, and alcoholism (Fig. 2D).
Target genes of PDAC
Identification of DEGs in PDAC
After preprocessing the GSE183795 dataset with R (Fig. 3A), the expression profiles of 17,164 genes were obtained. Filtered by the thresholds of |log2fold change| > 1 and FDR < 0.05, 266 DEGs were finally acquired, of which 158 were up-regulated genes and 108 were down-regulated genes, and the results were displayed in a volcano plot (Fig. 3B).
GSEA
Pathway enrichment of DEGs using GSEA highlighted that 2-oxo carboxylic acid metabolism, fat digestion and absorption, maturity onset diabetes of the young, pancreatic secretion, protein export were probably most concerned with the development of PDAC (Fig. 3C, D; Supplementary Table S1).
WGCNA
Genes with mean FPKM values in the top 50% were selected to create gene co-expression modules. Taking the correlation coefficient R2 > 0.9 as the screening criterion, pickSoftThreshold function discerned that when the minimum candidate soft-thresholding power was picked β = 11 (Fig. 4A), the constructed gene expression network could be closer to a scale-free network on the premise of preserving the mean connectivity as much as possible. The adjacency matrix, topological overlap matrix, dissimilarity matrix and hierarchical clustering tree were calculated and built stepwise (Fig. 4B). Branches of the clustered dendrogram were defined as modules and merged similar ones. Among acquired 15 gene modules, the grey60 module had the highest correlation with PDAC (cor = 0.64, P = 5e-28) (Fig. 4C, D), and the correlation coefficient between gene significance and module membership within the module was calculated as 0.91 (P < 1e-200) (Fig. 4E), indicating that this group of genes was strongly correlated with not only the disease state but also the module itself. Thus, the MEGrey60 module was definitively identified as the centermost module associated with PDAC, containing a total of 2,849 genes (Supplementary Table S2).

Target acquisition of Berberine (BBR) and enrichment analysis. (A) Chemical structure of BBR. (B) Venn diagram of BBR-targeted genes. (C) Disease Ontology (DO) annotation of BBR targets. (D) Pathway enrichment analysis of BBR targets with the method of Kyoto Encyclopedia of Genes and Genomes (KEGG). (E) Gene Ontology (GO) annotation of BBR targets.

Identification of differentially expressed genes (DEGs) in pancreatic ductal adenocarcinoma (PDAC) and gene set enrichment analysis (GSEA). (A) Before and after GSE183795 dataset standardization and normalization. (B) Volcano plot of DEGs. (C, D) Up- and down-regulated gene pathways mapped by GSEA.
Combining the key module genes from WGCNA and DEGs (Fig. 5A), 3002 disease targets were gained to structure the protein-protein interaction network of disease. To simplify this extensive network, we filtered it based on the betweenness centrality, selecting a sub-network of 771 nodes that exceeded the average value (Fig. 5B). GO enrichment revealed that the 771 disease targets primarily participated in GOBP of mitotic cell cycle phase transition, GOCC of chromosomal region, and GOMF of ATP hydrolysis activity (Fig. 5C). KEGG analysis indicated that the targets were predominantly enriched in pathways related to neurodegeneration-multiple diseases, Cell cycle, Alzheimer disease, Amyotrophic lateral sclerosis, and Parkinson disease (Fig. 5D).

Weighted correlation network analysis (WGCNA) based on GSE18379. A Determination of the minimum candidate soft-thresholding power. B Heatmap of topological overlap matrix (TOM). C, D Relationships of module eigengenes and two clinical traits. E Correlation of eigengenes in grey60 modules with the normal and PDAC traits.

Disease network construction, sub-network filtration and enrichment analysis. (A) Venn diagram of PDAC-related genes combining DEGs and WGCNA results. (B) Construction of PDAC network and filtration of the most contributing sub-network. Targets above the average value of the betweenness centrality were colored red. (C) GO annotation of PDAC targets. (D) Pathway enrichment analysis of PDAC targets with KEGG method.
PPI network construction
Followed by painting the Venn diagram of the relationship between molecular drug targets and disease targets (Fig. 6A), 57 intersecting targets were imported into the STRING online database to construct a PPI network, and imported into Cytoscape (v3.9.1) with the outlier protein nodes removed. In the network, the degree values of nodes were represented by varying shades of color, with darker colors signified higher degree values. The Degree algorithm from cytoHubba anchored a network formed by top 10 highest contributing genes, CDK1, CCNB1, CTNNB1, CDK2, TOP2A, MCM2, RUNX2, MYC, PLK1, and AURKA to be the key cluster (Fig. 6B, C). The results of DO enrichment analysis consisted predominantly of cancers with different systemic origins, as shown in Fig. 6D. GO terms were most notably related to mitotic cell cycle phase transition in GOBP; chromosomal region in GOCC; and protein serine kinase activity in GOMF (Fig. 6F). KEGG showed that DEGs were primarily enriched in cellular senescence, P13K-Akt signaling pathway, cell cycle, human T-cell leukemia virus 1 infection, as well as lipid and atherosclerosis (Fig. 6E).

PPI network construction, key cluster identification and enrichment analysis. (A) Venn diagram of BBR and PDAC targets. (B, C) Construction of PPI network and identification of key cluster formed by top 10 highest contributing genes. (D-F) GO, KEGG, and DO annotations for enrichment analysis of the key cluster.
ROC curve analysis
As demonstrated in the box plots, mRNA expression levels of the key cluster exhibited significant differences between the PDAC group and the control group (P < 0.001). ROC curves further confirmed that all genes showed good robustness for PDAC (AUC > 0.6). Among them, CDK1, PLK1, TOP2A, CTNNB1, CCNB1, RUNX2, and MYC proved to be highly diagnostically discriminant (AUC > 0.75) (Fig. 7A, B).
Immune infiltration analysis
CYBERSORT algorithm deconvolutionally calculated different infiltrating proportions of 22 immune cell types and showcased the immune landscape of GSE183795 dataset (Fig. 7C). Deviations of immune abundance in the distinct disease phenotypes were reflected by box plots (Fig. 7D). Notably, macrophages M0 (P < 0.001), macrophages M1, resting dendritic cells, activated dendritic cells, and regulatory T cells showed significantly increased infiltration in the tumor phenotype (P < 0.05). Conversely, neutrophils, naïve B cells, and CD8 T cells showed significantly lower infiltration compared to the normal phenotype (P < 0.05). Immune heatmaps showed that the key cluster primarily influences the immune pathways of Macrophages M0, CD8 T cells, and naïve B cells, indicating these genes’s roles in regulating the immune function of PDAC (Fig. 7E). This finding corroborates results from previous studies29,30.

Receiver operative characteristic curve (ROC) analysis and immune cell infiltration analysis of the key cluster. (A, B) Association assessment of clustered genes with PDAC in an external disease-control validation cohort (GSE62452). (C) The proportion of infiltrated immune cells in GSE183795 expression matrix. (D) Relationship between immune cells and two clinical phenotypes. (E) Relationship between the key cluster and immune cells.
ScRNA-seq analysis
ScRNA-seq of tumor tissue biopsy from 6 PDAC patients in GSE212966 dataset were preprocessed by the Seurat package to clarify the distribution and interrelationships of diversified cellular features. We found that the sum of all gene expressions detected in the cells was strongly and positively correlated with the number of unique genes detected in each cell, but not with the percentage of mitochondria (Fig. 8A, B). Based on these, the quality control filtering criteria were set to percent.mt < 20% and 200 < nFeature_RNA < 8000. PCA linear dimensionality reduction used a scaled matrix of 3000 highly variable genes, the relationships among the distinct PCs were displayed in Fig. 8C. Harmony’s soft clustering corrected batch effects (Fig. 8D). Combining the outcomes of elbow plot and principal component heatmap to select the 17 PCs that contributed most to cell clustering (Fig. 8E, F). Subsequently, the cluster tree was scaled to a resolution of 1.2, and 29 clusters were classified according to the clustering results (Fig. 9A, B). t-SNE reduced the data into a two-dimensional graph, which illustrates the distribution of cell expression, with cells colored by cluster (Fig. 9C). SingleR annotated the cell types, while the AUCell function calculated the enrichment scores of the key cluster, identifying BBR’s primary targets in PDAC as epithelial cells and macrophages (Fig. 9D, E). This finding aligns with the results of the immune infiltration analysis.

Preprocessing of GSE212966 single-cell dataset. (A, B) Determination of quality control filtering criteria. (C) Principle Component Analysis for linear dimensionality reduction. (D) Harmony’s soft clustering to correct batch effects. (E, F) Combining the elbow plot and principal component (PC) heatmap to determine the optimum PCs for downstream analysis.

Clusters classification, annotation, and BBR action pathways prediction. (A) Cluster tree partitioned diversified resolutions and clusters. (B) Heatmap of 29 clusters. (C) t-distributed Stochastic Neighbor Embedding (t-SNE) for linear dimensionality reduction, demonstrating the cell distribution and coloring the cells by clusters. (D) Cell type annotation of different clusters. (E) Target distribution of BBR on cellular subpopulations in PDAC.
Discussion
PDAC has the highest mortality rate among any solid malignancy31 and is notorious as one of the most lethal cancers. A high degree of heterogeneity driven by genomic mutations is considered an essential feature of PDAC. This heterogeneity can either impair therapeutic targeting, leading to poor or no response to therapy, or define transcriptomic and phenotypic profiles that promote malignant progression by continuously remodeling the tumor microenvironment32. This is also the pivotal reason why the eventual clinical outcomes in a vast majority of malignant tumors are closely linked to drug resistance and metastasis. Therefore, deciphering the heterogeneity of PDAC and the cellular composition of its tumor microenvironment is pertinent. Although BULK genomics has largely established our fundamental comprehension of the transcriptome and epigenetic profiles of tumor tissues, it does not fully capture the inter- and intra-tumor heterogeneity, that is, the heterogeneity exists both amongst different patients and within a single tumor. In contrast, single-cell genomics allows for the reconstruction of intricate tumor ecosystems33, revealing hidden and previously unknown cellular components. Consequently, single-cell transcriptome analysis of biopsy tissues from different PDAC patients can more effectively reveal cell-type-specific gene regulatory programs obscured by BULK omics techniques34.
Curative-intent resection followed by adjuvant chemotherapy continues to be the front-line regimen for PDAC. Although surgical treatment provides a survival benefit of at least 6 months35, only about 15% of patients can undergo surgery because most are diagnosed at an unresectable stage due to local advancement or distant metastasis36. Even with surgical resection, more than 80% of patients may experience recurrence within 2 years37. Given the limited therapeutic options, gemcitabine-based chemotherapy, either a single agent or in combination, will remain the optimal choice for most PDAC patients for the foreseeable future. Unfortunately, the insensitivity and resistance to gemcitabine in PDAC are not rare and hard to overcome38. Therefore, it is imperative to refine existing therapies or develop novel alternative approaches.
As a natural isoquinoline quaternary alkaloid, berberine possesses multiple pharmacological activities. In recent years, its antitumor effects have garnered increasing attention. BBR induces cancer cell death by activating autophagy, as well as intrinsic and extrinsic apoptosis. It prevents cancer cell proliferation by arresting cell cycle through suppressing the expression of cell cycle regulatory molecules. Additionally, it interferes with signaling in various oncogenic pathways39,40. In addition, studies have preliminarily revealed the anticancer or adjuvant anticancer potential of BBR in PDAC. BBR reduced the proportion of side population cells and down-regulated the expression of stem cell-related genes such as SOX2, POU5F1, and NANOG in PANC-1 and MIA-PaCa2 cell lines41. Both were able to induce cell cycle block, while gemcitabine focused mainly on the S phase, and BBR focused on the G1 phase42. In vivo studies showed that orthotopic PDAC-bearing mice orally administered with BBR had reduced tumor sizes, less invasive liver proliferation, and prolonged survival compared to the control group43. BBR-combined gemcitabine treatment promoted cytostasis and apoptosis in gemcitabine-tolerant PDAC cells via Rap1/PI3K/AKT axis, thereby mediating chemosensitization44. Synergizing BBR with the E3 ubiquitin ligase MDM2 inhibitor, nutlin-3a, significantly enhanced its potency in suppressing proliferation in WT-P53-introduced PDAC cell lines45. However, up until now, the anti-PDAC effects of BBR remain in an exploratory stage, and the targets and mechanisms of action have yet to be fully elucidated.
Pharmacological databases are capable of mapping drugs in clinical trials, approved drugs, and active compounds to their targeted human proteins and coding genes, facilitating the validation of gene-drug and gene-active compound interrelationships, as well as the expansion of indications for existing drugs46. Furthermore, Pharmacological databases act as a bridge between potential drug targets and disease transcriptomic information. This link allows for the identification of the distributional abundance of drug targets in pathological biopsy specimens, with precision down to single-cell resolution47.
In this study, the key therapeutic targets of BBR for the treatment of PDAC were identified, including CDK1, CCNB1, CTNNB1, CDK2, TOP2A, MCM2, RUNX2, MYC, PLK1, and AURKA. Hyperactivation of CDK1 and CDK2 drove the development of mutant KRAS-dependent PDAC48. Conversely, inactivation of both intercepted the immune escape in PDAC cells triggered by IFN-γ, through dual mechanisms of blocking immune checkpoint expression and propelling immunogenic cell death49. In particular, CDK1 inhibition specifically targeted the PDAC stem cell population50, and its expression level was proved to correlate with tumor size and histological grading in PDAC patients51, with the overexpression of CDK1 tending to portend a poor prognosis and a short survival period52. CCNB1, a B-type cyclin, is a master regulator of the cell cycle. It promoted the progression of PDAC cells from G2/S phase to G1/M phase by forming a complex with CDK1 to turn on substrate phosphorylation53,54. When CCNB1 was silenced, the cell cycle period ratios exhibited a decrease in S phase and an increase in G0/G1 phase. Simultaneously, the activation of p53 signaling pathway drove proliferation inhibition and induced senescence in PDAC cells55. CTNNB1 overexpression was considered a negative prognostic factor for progression-free survival and recurrence-free survival in human PDAC cohorts, and its mechanism might be associated with the regulation of cell-cell junctions dynamics to enhance the metastatic capacity of PDAC circulating tumor cells with stem-like properties56. Acting as a co-activator of β-cetanin, TOP2A engaged in the miR-139/TOP2A/β-cetanin axis, accelerating the malignant progression of PDAC57. TOP2A, along with its transcriptional activators SP1 and HMGB2, was overexpressed in human PDAC tissues, sensitizing PDAC cells with knocked-down TOP2A to chemotherapy drugs58. MCM2 assembles the hexameric helicase MCM complex, which is crucial for initiating DNA replication. When MCM expression was suppressed under gemcitabine exposure, it inhibited the restoration of DNA replication capacity in PDAC cell lines, demonstrating that MCM inhibition can synergize with chemotherapeutic agents to mediate replication blockade59. RUNX2 ameliorated the PDAC tumor microenvironment by regulating the extracellular matrix, affecting the transcription of SPARC and MMP160. Activation of the MYC signaling pathway and amplification of MYC expression supported PDAC metastasis by fostering the reprogramming of glucose metabolism61. Furthermore, and the clinical potential of MYC as a therapeutic target for aerobic glycolysis in PDAC is currently under investigation62. PLK1 depletion or downregulation was able to stimulate anti-tumor immune responses, upregulating PD-L1 expression to sensitize PDAC to immune checkpoint therapy63. Targeted delivery of PLK1 inhibitors into PDAC cells effectively slowed tumor growth by silencing PLK164. AURKA repression triggered necrosis-like apoptosis in PDAC cells by reducing necrosome-induced phosphorylation of downstream proteins65.
CYBERSORT revealed the key cluster predominantly regulates the immune responses of Macrophages M0, CD8 T cells, and naïve B cells. Analysis of scRNA-seq suggested the epithelial cells and macrophages in PDAC were more responsive to the key cluster. Remarkably, the two co-emphasized the modulatory role of BBR on macrophages in PDAC. The PDAC stroma exhibits extensive macrophage infiltration, contributing not only to the immunosuppression of the tumor microenvironment66, but also to gemcitabine tolerance67. Moreover, macrophages are capable of shielding tumor cells from complement-dependent cytotoxicity68. However, inhibiting the upstream encoding gene, PI3Kγ, can reprogram macrophages to activate CD8 + T cells, interfering with metastasis and proliferation of tumor cells69. Early-stage M0 macrophages, which possess high plasticity, can promote anti-tumor activity through increased secretion of TNF-α70.
Previously, researchers have gleaned significant anticancer insights from medicinal plants, such as paclitaxel, vincristine, and camptothecin. The strength of this study lies in the fact that through deeply mining the bioinformatics data of pharmacological databases in conjunction with BULK and single-cell genomics, combined with the existing research base, we initially elucidated the potential targets and molecular mechanisms of the anti-PDAC action of BBR, which may elucidate potential strategies for the development of anticancer drugs derived from natural ingredients. The present study has its shortcomings. Firstly, it lacks experimental validation. Confirming the bioinformatic results with real-world evidence is crucial for establishing a more comprehensive research framework. Secondly, the clinical application of BBR was restricted by its low oral bioavailability, inadequate tumor site delivery, and non-specificity. Further exploration into the optimization of its chemical structure and the development of novel drug delivery systems is necessary.
Conclusion
This study explored the multiple bioinformatics mechanisms of BBR against PDAC through integrated multi-omics profiling. BBR potentially targets CDK1, CCNB1, CTNNB1, CDK2, TOP2A, MCM2, RUNX2, MYC, PLK1, and AURKA to exert therapeutic effects on PDAC. CYBERSORT and scRNA-seq analyses collectively suggested that the process primarily involved macrophage polarization-related immunological responses. In subsequent research, we will thoroughly validate these findings through detailed molecular biology experiments.
Data availability
This study analyzed open-source data from publicly available databases, and all datasets could be retrieved from GEO, including GSE183795, GSE62452, and GSE212966.
References
Sung, H. et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J. Clin.71 (3), 209–249 (2021).
Mizrahi, J. D., Surana, R., Valle, J. W. & Shroff, R. T. Pancreatic cancer. Lancet. 395 (10242), 2008–2020 (2020).
Liang, H. Y., Chen, T., Yan, H. T., Huang, Z. & Tang, L. J. Berberine ameliorates severe acute pancreatitis–induced intestinal barrier dysfunction via a myosin light chain phosphorylation–dependent pathway. Mol. Med. Rep.9 (5), 1827–1833 (2014).
Bansod, S., Doijad, N. & Godugu, C. Berberine attenuates severity of chronic pancreatitis and fibrosis via AMPK-mediated inhibition of TGF-β1/Smad signaling and M2 polarization. Toxicol. Appl. Pharmacol.403, 115162 (2020).
Yin, J., Xing, H. & Ye, J. Efficacy of berberine in patients with type 2 diabetes mellitus. Metabolism. 57 (5), 712–717 (2008).
Yerra, V. G., Kalvala, A. K., Sherkhane, B., Areti, A. & Kumar, A. Adenosine monophosphate-activated protein kinase modulation by berberine attenuates mitochondrial deficits and redox imbalance in experimental diabetic neuropathy. Neuropharmacology. 131, 256–270 (2018).
Goel, A. Current understanding and future prospects on Berberine for anticancer therapy. Chem. Biol. Drug Des.102 (1), 177–200 (2023).
Abrams, S. L. et al. Abilities of berberine and chemically modified berberines to inhibit proliferation of pancreatic cancer cells. Adv. Biol. Regul.71, 172–182 (2019).
Liu, J., Luo, X., Guo, R., Jing, W. & Lu, H. Cell Metabolomics reveals Berberine-inhibited pancreatic Cancer cell viability and metastasis by regulating citrate metabolism. J. Proteome Res.19 (9), 3825–3836 (2020).
Pinto-Garcia, L., Efferth, T., Torres, A., Hoheisel, J. D. & Youns, M. Berberine inhibits cell growth and mediates caspase-independent cell death in human pancreatic cancer cells. Planta Med.76 (11), 1155–1161 (2010).
Gu, S. et al. Berberine inhibits cancer cells growth by suppressing fatty acid synthesis and biogenesis of extracellular vesicles. Life Sci.257, 118122 (2020).
Yu, G., Wang, L. G., Yan, G. R. & He, Q. Y. DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics. 31 (4), 608–609 (2015).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res.28 (1), 27–30 (2000).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res.51 (D1), D587–D592 (2023).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci.28 (11), 1947–1951 (2019).
Xie, Z. et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc.1 (3), e90 (2021).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res.43 (7), e47 (2015).
Gustavsson, E. K., Zhang, D., Reynolds, R. H., Garcia-Ruiz, S. & Ryten, M. Ggtranscript: an R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics. 38 (15), 3844–3846 (2022).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 102 (43), 15545–15550 (2005).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform.9, 559 (2008).
Szklarczyk, D. et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res.51 (D1), D638–D646 (2023).
Csardi, G. & Nepusz, T. The igraph software package for complex network research. InterJournal Complex. Syst.1695, 1–9 (2005).
Robin, X. et al. pROC: an open-source package for R and S + to analyze and compare ROC curves. BMC Bioinform.12, 77 (2011).
Alba, A. C. et al. Discrimination and calibration of clinical prediction models: users’ guides to the Medical Literature. JAMA. 318 (14), 1377 (2017).
Chen, B., Khodadoust, M. S., Liu, C. L., Newman, A. M. & Alizadeh, A. A. Profiling Tumor infiltrating Immune cells with CIBERSORT. Methods Mol. Biol.1711, 243–259 (2018).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell. 184 (13), 3573–3587e29 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 16 (12), 1289–1296 (2019).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol.20 (2), 163–172 (2019).
Zhu, Y. et al. Tissue-Resident macrophages in Pancreatic Ductal Adenocarcinoma Originate from embryonic hematopoiesis and promote Tumor Progression. Immunity. 47 (2), 323–338e6 (2017).
Helm, O. et al. Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis: role of macrophages in pancreatic cancer. Int. J. Cancer. 135 (4), 843–861 (2014).
Christenson, E. S., Jaffee, E. & Azad, N. S. Current and emerging therapies for patients with advanced pancreatic ductal adenocarcinoma: a bright future. Lancet Oncol.21 (3), e135–e145 (2020).
Zhang, A., Miao, K., Sun, H. & Deng, C. X. Tumor heterogeneity reshapes the tumor microenvironment to influence drug resistance. Int. J. Biol. Sci.18 (7), 3019–3033 (2022).
Suvà, M. L., Tirosh, I. & Single-Cell, R. N. A. Sequencing in Cancer: lessons learned and Emerging challenges. Mol. Cell.75 (1), 7–12 (2019).
Preissl, S., Gaulton, K. J. & Ren, B. Characterizing cis-regulatory elements using single-cell epigenomics. Nat. Rev. Genet.24 (1), 21–43 (2023).
Labori, K. J. et al. Impact of early disease progression and surgical complications on adjuvant chemotherapy completion rates and survival in patients undergoing the surgery first approach for resectable pancreatic ductal adenocarcinoma – a population-based cohort study. Acta Oncol.55 (3), 265–277 (2016).
Ansari, D., Gustafsson, A. & Andersson, R. Update on the management of pancreatic cancer: surgery is not enough. World J. Gastroenterol.21 (11), 3157–3165 (2015).
Daamen, L. A. et al. Postoperative surveillance of pancreatic cancer patients. Eur. J. Surg. Oncol.45 (10), 1770–1777 (2019).
Andersson, R. et al. Gemcitabine chemoresistance in pancreatic cancer: molecular mechanisms and potential solutions. Scand. J. Gastroenterol.44 (7), 782–786 (2009).
Samadi, P. et al. Berberine: a novel therapeutic strategy for cancer. IUBMB Life. 72 (10), 2065–2079 (2020).
Devarajan, N. et al. Berberine—A potent chemosensitizer and chemoprotector to conventional cancer therapies. Phytother. Res.35 (6), 3059–3077 (2021).
Park, S. H., Sung, J. H. & Chung, N. Berberine diminishes side population and down-regulates stem cell-associated genes in the pancreatic cancer cell lines PANC-1 and MIA PaCa-2. Mol. Cell. Biochem.394 (1–2), 209–215 (2014).
Park, S. H., Sung, J. H., Kim, E. J. & Chung, N. Berberine induces apoptosis via ROS generation in PANC-1 and MIA-PaCa2 pancreatic cell lines. Braz J. Med. Biol. Res.48 (2), 111–119 (2015).
Cheng, C. S. et al. Functional inhibition of lactate dehydrogenase suppresses pancreatic adenocarcinoma progression. Clin. Transl Med.11 (6), e467 (2021).
Okuno, K. et al. Berberine overcomes Gemcitabine-Associated Chemoresistance through Regulation of Rap1/PI3K-Akt signaling in pancreatic ductal adenocarcinoma. Pharmaceuticals. 15 (10), 1199 (2022).
Abrams, S. L. et al. Effects of the MDM2 inhibitor Nutlin-3a on sensitivity of pancreatic cancer cells to berberine and modified berberines in the presence and absence of WT-TP53. Adv. Biol. Regul.83, 100840 (2022).
Spreafico, R., Soriaga, L. B., Grosse, J., Virgin, H. W. & Telenti, A. Advances in Genomics for Drug Development. Genes (Basel). 11 (8), 942 (2020).
Heath, J. R., Ribas, A. & Mischel, P. S. Single-cell analysis tools for drug discovery and development. Nat. Rev. Drug Discov. 15 (3), 204–216 (2016).
Kazi, A. et al. Global Phosphoproteomics Reveal CDK suppression as a vulnerability to KRas Addiction in Pancreatic Cancer. Clin. Cancer Res.27 (14), 4012–4024 (2021).
Huang, J. et al. CDK1/2/5 inhibition overcomes IFNG-mediated adaptive immune resistance in pancreatic cancer. Gut. 70 (5), 890–899 (2021).
Wijnen, R. et al. Cyclin dependent Kinase-1 (CDK-1) inhibition as a Novel Therapeutic Strategy against Pancreatic Ductal Adenocarcinoma (PDAC). Cancers (Basel). 13 (17), 4389 (2021).
Piao, J. et al. High expression of CDK1 and BUB1 predicts poor prognosis of pancreatic ductal adenocarcinoma. Gene. 701, 15–22 (2019).
Dong, S., Huang, F., Zhang, H. & Chen, Q. Overexpression of BUB1B, CCNA2, CDC20, and CDK1 in tumor tissues predicts poor survival in pancreatic ductal adenocarcinoma. Biosci. Rep.39 (2), BSR20182306 (2019).
Fang, L., Du, W. W., Awan, F. M., Dong, J. & Yang, B. B. The circular RNA circ-Ccnb1 dissociates Ccnb1/Cdk1 complex suppressing cell invasion and tumorigenesis. Cancer Lett.459, 216–226 (2019).
Sano, M. et al. Induction of cell death in pancreatic ductal adenocarcinoma by indirubin 3’-oxime and 5-methoxyindirubin 3’-oxime in vitro and in vivo. Cancer Lett.397, 72–82 (2017).
Zhang, H. et al. Effect of CCNB1 silencing on cell cycle, senescence, and apoptosis through the p53 signaling pathway in pancreatic cancer. J. Cell. Physiol.234 (1), 619–631 (2018).
Zhu, L. et al. Characterization of stem-like circulating tumor cells in pancreatic Cancer. Diagnostics (Basel). 10 (5), 305 (2020).
Pei, Y. F., Yin, X. M. & Liu, X. Q. TOP2A induces malignant character of pancreatic cancer through activating β-catenin signaling pathway. Biochim. Biophys. Acta Mol. Basis Dis.1864 (1), 197–207 (2018).
Tanaka, T. et al. Molecular pathogenesis of pancreatic ductal adenocarcinoma: impact of miR-30c-5p and miR-30c-2-3p regulation on oncogenic genes. Cancers (Basel). 12 (10), 2731 (2020).
Bryant, V. L., Elias, R. M., McCarthy, S. M., Yeatman, T. J. & Alexandrow, M. G. Suppression of Reserve MCM Complexes Chemosensitizes to Gemcitabine and 5-Fluorouracil. Mol. Cancer Res.13 (9), 1296–1305 (2015).
Kayed, H. et al. Regulation and functional role of the runt-related transcription factor-2 in pancreatic cancer. Br. J. Cancer. 97 (8), 1106–1115 (2007).
Chen, M. et al. NUSAP1-LDHA-Glycolysis-lactate feedforward loop promotes Warburg effect and metastasis in pancreatic ductal adenocarcinoma. Cancer Lett.567, 216285 (2023).
Zhang, Z. & Zhang, H. J. Glycometabolic rearrangements-aerobic glycolysis in pancreatic ductal adenocarcinoma (PDAC): roles, regulatory networks, and therapeutic potential. Expert Opin. Ther. Targets. 25 (12), 1077–1093 (2021).
Zhang, Z. et al. Targeting Plk1 sensitizes pancreatic Cancer to Immune Checkpoint Therapy. Cancer Res.82 (19), 3532–3548 (2022).
Mahajan, U. M. et al. Tumour-specific delivery of siRNA-coupled superparamagnetic iron oxide nanoparticles, targeted against PLK1, stops progression of pancreatic cancer. Gut. 65 (11), 1838–1849 (2016).
Gustin, P., Lekeux, P., Lomba, F. & Clercx, C. Mechanical properties of excised calf lungs. Res. Vet. Sci.42 (3), 272–276 (1987).
Yang, J., Li, Y., Sun, Z. & Zhan, H. Macrophages in pancreatic cancer: an immunometabolic perspective. Cancer Lett.498, 188–200 (2021).
Spek, C. A., Aberson, H. L. & Duitman, J. Macrophage C/EBPδ drives Gemcitabine, but not 5-FU or Paclitaxel, Resistance of Pancreatic Cancer cells in a deoxycytidine-dependent manner. Biomedicines. 10 (2), 219 (2022).
Zhang, R. et al. Pancreatic cancer-educated macrophages protect cancer cells from complement-dependent cytotoxicity by up-regulation of CD59. Cell. Death Dis.10 (11), 836 (2019).
Kaneda, M. M. et al. Macrophage PI3Kγ drives pancreatic ductal adenocarcinoma progression. Cancer Discov. 6 (8), 870–885 (2016).
Tekin, C., Aberson, H. L., Bijlsma, M. F. & Spek, C. A. Early macrophage infiltrates impair pancreatic cancer cell growth by TNF-α secretion. BMC Cancer. 20 (1), 1183 (2020).
Acknowledgements
We are deeply indebted to the GEO database for open-source clinical data.
Funding
The manuscript was supported by grants from The National Natural Science Funds of China (No.81102567); Project of Shanghai Municipal Health Commission (2014LP055A / 201440508).
Author information
Authors and Affiliations
Contributions
JY designed the study and drafted the manuscript. JY, TX and HW made contributions to the data analysis. LW and YC revised the manuscript. YC acquired fundings. Before submitting, all authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, J., Xu, T., Wang, H. et al. Mechanisms of Berberine in anti-pancreatic ductal adenocarcinoma revealed by integrated multi-omics profiling. Sci Rep 14, 22929 (2024). https://doi.org/10.1038/s41598-024-74943-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-74943-y
